Embed Size (px)
Citation preview

GENE THERAPY
A novel human gamma-globin gene vector for genetic correction of sickle cellanemia in a humanized sickle mouse model: critical determinants for successfulcorrection*Ajay Perumbeti,1,2 *Tomoyasu Higashimoto,1 Fabrizia Urbinati,1 Robert Franco,3 Herbert J. Meiselman,4 David Witte,5 andPunam Malik1,2
Divisions of 1Experimental Hematology/Cancer Biology and 2Hematology-Oncology, Cincinnati Children’s Research Foundation, Cincinnati Children’s HospitalMedical Center (CCHMC), OH; 3Division of Hematology-Oncology, University of Cincinnati College of Medicine, OH; 4Department of Physiology, Keck School ofMedicine, University of Southern California, Los Angeles; and 5Division of Pathology, University of Cincinnati College of Medicine, OH
We show that lentiviral delivery of human�-globin gene under �-globin regulatorycontrol elements in hematopoietic stemcells (HSCs) results in sufficient postna-tal fetal hemoglobin (HbF) expression tocorrect sickle cell anemia (SCA) in theBerkeley “humanized” sickle mouse.Upon de-escalating the amount of trans-duced HSCs in transplant recipients, us-ing reduced-intensity conditioning andvarying gene transfer efficiency and vec-tor copy number, we assessed critical
parameters needed for correction. A sys-tematic quantification of functional andhematologic red blood cell (RBC) indices,organ pathology, and life span was usedto determine the minimal amount of HbF,F cells, HbF/F-cell, and gene-modifiedHSCs required for correcting the sicklephenotype. We show that long-term ame-lioration of disease occurred (1) whenHbF exceeded 10%, F cells constitutedtwo-thirds of the circulating RBCs, andHbF/F cell was one-third of the total hemo-
globin in sickle RBCs; and (2) when ap-proximately 20% gene-modified HSCs re-populated the marrow. Moreover, we showa novel model using reduced-intensityconditioning to determine genetically cor-rected HSC threshold that corrects a he-matopoietic disease. These studies pro-vide a strong preclinical model for what itwould take to genetically correct SCA andare a foundation for the use of this vectorin a human clinical trial. (Blood. 2009;114:1174-1185)
Introduction
Sickle cell anemia (SCA) results from a point mutation in the�-globin gene (�S), resulting in sickle hemoglobin (HbS). HbSpolymerizes upon deoxygenation resulting in sickle-shaped RBCsthat occlude microvasculature. Patients with SCA have intermittentacute vascular occlusions and cumulative organ damage, reducingthe life span to 42 to 58.5 years.1,2 Besides sickling, excessivehemolysis and a state of chronic inflammation exist. SCA patientsaccount for approximately 75 000 hospitalizations per year, result-ing in an estimated annual expenditure of $475 million dollars inthe United States alone.3 Worldwide, SCA is second only tothalassemia in incidence of monogenic disorders, with more than200 000 children born annually in Africa.4
Current therapies include supportive care for episodic sickling,chronic transfusions with iron chelation, and hydroxyurea to inducefetal hemoglobin (HbF). These therapies impact disease morbidity,but their effectiveness is variable and dependent on compliance toan indefinite treatment regimen. A matched allogeneic hematopoi-etic stem cell (HSC) transplantation is curative, but restricted bythe availability of matched related donors5 and has potential seriouscomplications.6 A meta-analysis of 187 SCA transplantationsshows 6% to 7% conditioning-related peritransplantation mortality,7% to 10% acute rejection, and 13% to 20% chronic graft-versus-host disease (GVHD) in recipients.7
Gene therapy of autologous HSCs followed by transplantationcould result in a one-time cure, avoid adverse immunologic
consequences, and not be limited by availability of donors; it mayalso not require myeloablative-conditioning regimens, and therebyhave lower toxicity. The amount of HbF/anti–sickling globinrequired to correct SCA via a transgene is unknown. Recently,2 groups have used HIV-1–based lentivirus vectors carryingrecombinant/mutated �-globin genes designed to have antisicklingproperties to show correction of the disease in mouse models ofSCA when high levels of the mutant �-globin are expressed.8,9
Expression of HbF postnatally can be therapeutic, as is evidentby the protective effect of HbF in neonatal sickle RBCs and inpatients with hereditary persistence of HbF and SCA. The propor-tion of genetically corrected HSCs, the amount of exogenouslyexpressed HbF, and the proportion of F cells that will correct thepathophysiology are unknown. We have previously shown com-plete correction of human thalassemia major in vitro, and inxenografted mice in vivo, with a lentivirus vector carrying the�-globin gene and locus control region (LCR) elements.10 In thisreport, we modified this �-globin lentivirus vector to encode�-globin exons and transduced murine sickle HSCs. We firstcharacterized functional correction with a careful and detailedquantification of RBC sickling, half-life, and deformability, withsickle to normal transplantations and high HbF production to defineparameters of correction. Next, using reduced-intensity condition-ing and varying the percentage of transduced HSCs, we performedtransplantations on sickle mice with significant organ damage and
Submitted January 29, 2009; accepted May 18, 2009. Prepublished online asBlood First Edition paper, May 27, 2009; DOI 10.1182/blood-2009-01-201863.
*A.P. and T.H. contributed equally to this work.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page chargepayment. Therefore, and solely to indicate this fact, this article is herebymarked ‘‘advertisement’’ in accordance with 18 USC section 1734.
© 2009 by The American Society of Hematology
1174 BLOOD, 6 AUGUST 2009 � VOLUME 114, NUMBER 6
For personal use only.on May 9, 2016. by guest www.bloodjournal.orgFrom

demonstrate the proportions of (1) genetically corrected HSCs,(2) HbF, and (3) F cells, and (4) percentage of HbF/F cell requiredfor correction of the sickle RBC and amelioration of organdamage in SCA.
Methods
Detailed methodology is provided in supplemental methods (available onthe Blood website; see the Supplemental Materials link at the top of theonline article).
Vector
We have shown that a �-�-globin hybrid gene carrying lentivirus vector,I8H�/�W,11 expresses high �-globin mRNA in erythroid cells expressing“adultlike” globins.12 All �-globin coding sequences were changed to�-globin using site-directed mutagenesis and the �-�-globin hybrid gene,and LCR elements were cloned in reverse orientation to the viraltranscriptional unit to generate sGbG lentivirus vector.13 Virus was madewith cotransfection of 293T cells.10,13
Murine HSC enrichment
Bone marrow from 6- to 20-week-old BERK sickle mice14 was harvestedand lineage depleted with biotinylated CD5, CD8, B220, Mac-1, CD11b,Gr-1, and TER-119 antibodies and magnetic beads. The bead-free cellswere stained with antibodies to Sca-1, c-kit. Cells that were 7-AAD�,lineage�, c-kit� then Sca-1� (LSK cells) were sorted on FACSVantage (BDBiosciences). All experiments using Berkeley transgenic sickle mice andC57/BL6 mice were performed according to protocols approved by theCincinnati Children’s Hospital Medical Center.
Gene transfer and bone marrow transplantation
Myeloablative transplantations were performed from BERK3C57Bl/6mice because of ease of transplantation and ready availability of normalrecipients (9.5 � 0.6 weeks old) after 11.75 Gy radiation. Radiation controlexperiments showed that BERK mice receiving 8 to 9 Gy radiationsurvived without receiving LSK cells; and the lethal dose was lower than inC57Bl/6 mice. BERK mice receiving more than 10.5 Gy died when no LSKcells were given; those given LSK rescue survived long term. BERKmice are difficult to breed in large numbers at a given time, therefore2 mice/radiation dose level were to determine the sublethal dose. All BERKrecipients (12.9 � 0.4 weeks old) received 3 peritransplantation RBCtransfusions (days 1-7). Organ pathology in BERK recipients 1 year aftertransplantation was compared with 12-week-old BERK mice that did notundergo transplantation. The radiation was higher than classical reduced-intensity radiation dose of 4 Gy to allow a large degree of donor HSCchimerism. A range of MOI was used to vary the proportion of transduceddonor HSCs in the graft. LSK cells were prestimulated overnight13 andtransduced twice at an MOI of 30 for BERK3C57BL/6 transplantsand MOI of 30 to 100 for BERK3BERK transplants for 22 to 24 hours;10 000 to 24 000 LSK cells and untransduced LK cells were cotransplantedinto recipient C57BL/6 or BERK mice.
Copy number analysis
Copy number analysis was done on genomic DNA by real-time polymerasechain reaction using primers and probes described previously.13
Hematologic analysis
Hematologic analysis was obtained on Hemavet 950FS (Drew Scientific)under mouse settings. Reticulocyte analysis was performed as follows:0.1 �L blood and 200 �L BD Retic-COUNT Reagent were mixed (BectonDickinson), incubated at room temperature for 30 minutes, and analyzed byfluorescence-activated cell sorting (FACS).
Hemoglobin analysis
Hemoglobin electrophoresis was performed on cellulose acetate plates, asdescribed previously.13 Ion exchange high-performance liquid chromatogra-phy (HPLC) was performed with an Alliance 2690 HPLC machine (Waters)using a PolyCAT A column (item no. 3.54CT0510; Poly LC Inc).
Red blood cell functional analysis
Irreversibly sickled cells (ISCs) were enumerated by scoring 500 RBCs inconsecutive fields. Graded deoxygenation was performed using tonometry.RBC deformability was determined using a laser-assisted optical rotationalcell analyzer (LORCA; RR Mechatronics).
RBC half-life
Mice were injected with 3 mg Sulfo-NHS biotin (Sigma) in 300 �L PBS as2 separate injections 1 hour apart; 2 to 5 �L blood was drawn at serialtimes, and stained with APC-Cy7–conjugated streptavidin.
Histology
Spleen, liver, bones, brain, and kidney were harvested and placed in 5 mL of10% formalin. Paraffin blocks were sectioned and stained with hematoxylinand eosin.
Results
High HbF after gene therapy and myeloablative transplantationcorrects SCA
The sGbG vector carries �-globin exons and �-globin noncodingand regulatory regions (supplemental Figure 1). Based upon ourpreviously studied sBG vector, which expresses high levels ofhuman �-globin,13 sGbG-transduced LSK cells from Berkeleysickle (BERK) mice14,15 were transplanted into lethally irradiated(myeloablated) normal C57Bl/6J mice (termed GbG mice). Mocktransductions on BERK LSK cells from the same bone marrowpool followed by transplantation resulted in mice with SCA(termed mock mice; supplemental Figure 2B). The majority ofRBCs in GbG mice expressed HbF (supplemental Figure 2A-D).Only GbG mice with 100% donor (HbS�) RBCs, with no evidenceof residual recipient murine hemoglobin by electrophoresis andHPLC (supplemental Figure 2B-C), were analyzed for hemato-logic, functional, and pathologic analysis. GbG mice with a smallproportion of recipient murine RBCs (supplemental Figure 2B)were used only to assess HbF/vector copy and frequency oftransduced HSCs.
The percentage of HbF (HbF/HbS�HbF) in blood, quantifiedby FACS, was approximately 40% in primary mice followed for6 months and in secondary recipients followed for 7.5 months(Figure 1A). Two-thirds of RBCs were F cells; their proportion wasalso stable in primary and secondary recipients (Figure 1B). Theproportion of F cells and vector copies correlated with HbF (Figure1C-D). Taken together, these data show significant HbF expressionfrom the sGbG vector in the majority of RBC with stable long-termexpression.
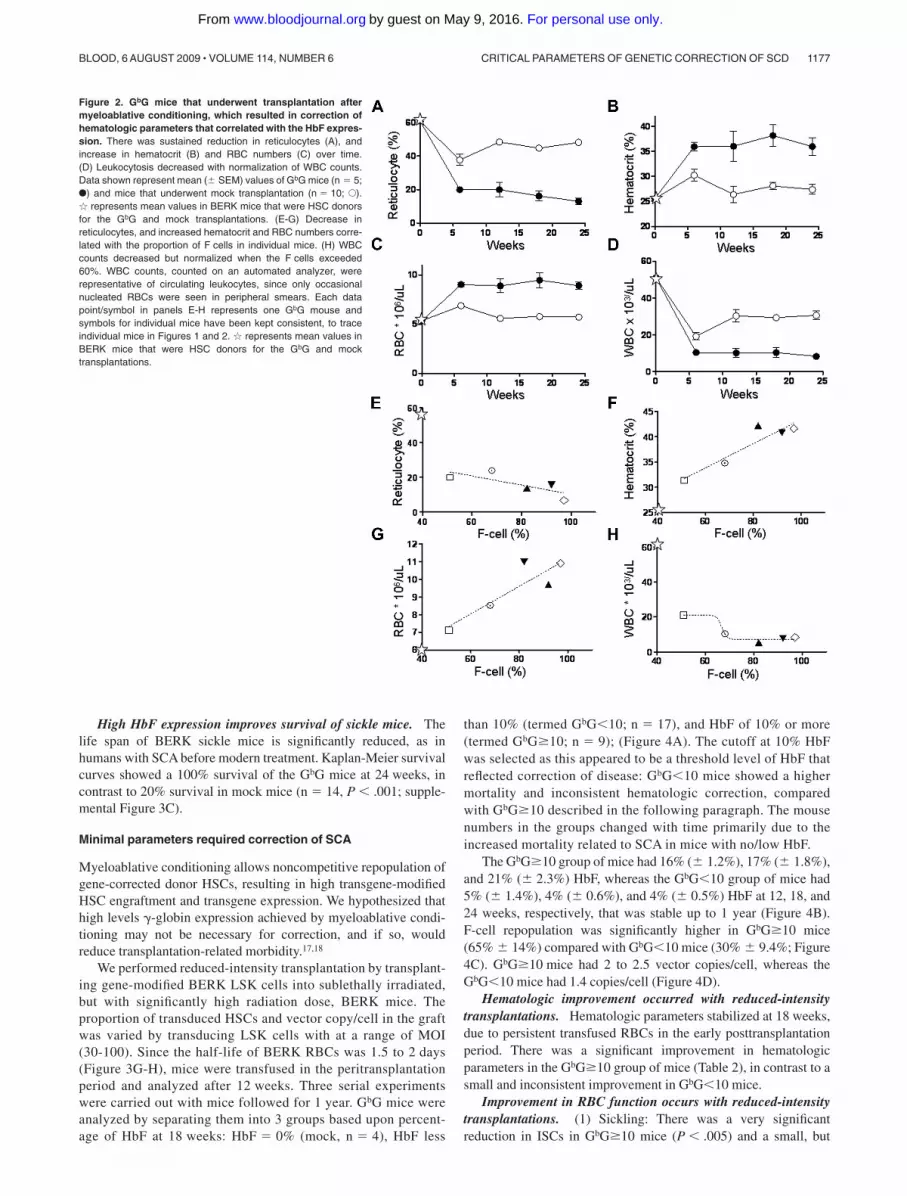
High levels of HbF result in sustained hematologic correction.Table 1 shows improvement of hematologic parameters in GbGmice. The proportion of reticulocytes decreased from approxi-mately 50% in mock mice to approximately 15% in GbG mice(P � .005; Figure 2A). There was correction of anemia by12 weeks, which persisted throughout the posttransplantation pe-riod (Figure 2B-C).
CRITICAL PARAMETERS OF GENETIC CORRECTION OF SCD 1175BLOOD, 6 AUGUST 2009 � VOLUME 114, NUMBER 6
For personal use only.on May 9, 2016. by guest www.bloodjournal.orgFrom

High white blood cell (WBC) counts in humans with SCA andBERK mice reflect the baseline inflammation in this disease. WBCreturned to normal levels in GbG mice (Figure 1D; Table 1).Notably, WBC counts were lower in the mock mice compared withBERK mice that did not undergo transplantation, likely because inthe former, sickle HSCs were transplanted into a normal “nonin-flamed” C57/BL6 background. Indeed, 6 weeks after transplanta-tion, WBC counts in mock group of mice were nearly normal, thengradually rose to high levels seen in SCA (Figure 2D). Overall,hematologic parameters showed marked improvement to nearnormal levels, and improvement was stable over a prolongedperiod in primary and secondary GbG mice. The degree ofcorrection correlated with the proportion of F cells (Figure 2E-H)and HbF (data not shown).
High levels of HbF improve the functional parameters of RBCsin sickle mice. (1) Sickling: The irreversibly sickled cells (ISCs)were significantly reduced to 2.3% plus or minus 0.7% in GbGmice, compared with 12% plus or minus 0.8% in BERK controlsand 10.2% plus or minus 0.3% in mock mice (Figure 3A-B).Deoxygenation of blood from a representative GbG mouse shows adramatic reduction in sickling (Figure 3C). A systematic quantifica-tion showed a marked decrease in the proportion of sickle RBCs inGbG mice with increasing hypoxia (Figure 3D). (2) RBC mem-brane deformability: Normal RBCs deform readily at low shearstress (3 Pascals [Pa]), representative of shear stress in smallvessels.16 Sickle RBCs have relatively rigid membranes withremarkably reduced deformability even at high shear stress (28 Pa;representative of shear stress in large vessels). There was markedly
improved deformability of RBCs of GbG mice, although it did notachieve normal levels (Figure 3E). This may reflect the proportionof circulating sickle RBCs that did not contain HbF. (3) RBCsurvival: Survival of human sickle RBCs is an order of magnitudeless than normal RBCs. We measured the time to 50% reduction(half-life) in GbG and mock/BERK sickle mice. The overallsurvival of the GbG RBCs was markedly improved, with the time to50% reduction approximately 4 times longer in RBCs from GbGmice compared with BERK or mock mice (Figure 3F). (4) RBChemolysis: RBC hemolysis detected by measuring lactate dehydro-genase (LDH) in blood was reduced from 2706 plus orminus 148 mg/dL in mock mice to 1286 plus or minus 345 mg/mLin GbG mice (n � 5; P � .004).
High levels of HbF prevent chronic organ damage associatedwith SCA. Bone marrow, spleen, liver, and kidneys at 24 weeksshowed complete prevention of organ pathology. There wasreduced erythroid hyperplasia in bone marrow and spleen, de-creased spleen size, and preservation of the splenic folliculararchitecture, compared with obliterated follicular architecture fromthe severe erythroid hyperplasia in mock mice. The focal tubularatrophy and segmental glomerular infarction seen in mock micewere absent in the GbG mouse kidneys. Infarctions and extramedul-lary hematopoiesis seen in livers of mock mice were absent inlivers of GbG mice (supplemental Figure 3 shows representativesections; Table 2 summarizes the data in all groups of mice).Overall, except for a mild erythroid hyperplasia no organ pathologywas observed in the GbG mice.
Figure 1. GbG mice that underwent transplantation aftermyeloablative conditioning have high HbF production that isstable and sustained in primary and secondary mice. GbGmice that were fully chimeric for donor RBCs were analyzed atdifferent time points. The proportion of HbF (A) and F cells (B) inblood of individual mice, as determined by ion-exchange HPLCand FACS analysis, respectively, is shown at different time pointsafter primary and secondary transplantations. (C) The amountof HbF in blood directly correlated with the proportion of F cells.(D) The amount of HbF produced was directly in proportion to thevector copy number in bone marrow. Each symbol represents onemouse (and consistently depicts the same particular mouse in allthe panels).
Table 1. Hematologic parameters of GbG mice that underwent transplantation after myeloablative conditioning
Mouse type No. WBC, 103/�L RBC, 106/�L Hb, g/dL MCV, fL MCH, pg RDW, % Plt, 103/�L Reticulocytes, %
BERK 5 56.8 � 5.4 5.3 � 0.4 5.8 � 0.5 48.2 � 1.05 10.7 � 0.5 35.3 � 1.6 733 � 80 60.8 � 5.0
GbG pri 5 10.6 � 3.1 9.4 � 0.8 10.0 � 0.8 40.7 � 1.3 10.4 � 0.6 27.6 � 1.1 733 � 82 15.8 � 3.2
Mock pri 10 29.7 � 1.4 5.8 � 0.4 7.6 � 0.7 48.5 � 1.8 10.7 � 0.2 32.0 � 0.9 921 � 50 40.0 � 3.0
P* .001 .007 .03 .001 .9 .009 .06 .006
GbG sec 6 6.8 � 1.4 8.9 � 0.4 10.1 � 0.5 40.5 � 1.6 11.4 � 0.5 28.3 � 1.4 658 � 33 13.8 � 2.9
Mock sec 1 31.7 5.2 6.4 47.6 12.2 32.1 923 49
Hb indicates hemoglobin; MCV, mean corpuscular volume; MCH, mean corpuscular hemoglobin; RDW, red cell distribution width; Plt, platelets; pri, primary mice; and sec,secondary mice.
*P values represent comparison of primary mock mice with the GbG group. Statistical comparisons of secondary mice were not made as only one secondary mock mousewas alive at the time of analysis.
1176 PERUMBETI et al BLOOD, 6 AUGUST 2009 � VOLUME 114, NUMBER 6
For personal use only.on May 9, 2016. by guest www.bloodjournal.orgFrom
High HbF expression improves survival of sickle mice. Thelife span of BERK sickle mice is significantly reduced, as inhumans with SCA before modern treatment. Kaplan-Meier survivalcurves showed a 100% survival of the GbG mice at 24 weeks, incontrast to 20% survival in mock mice (n � 14, P � .001; supple-mental Figure 3C).
Minimal parameters required correction of SCA
Myeloablative conditioning allows noncompetitive repopulation ofgene-corrected donor HSCs, resulting in high transgene-modifiedHSC engraftment and transgene expression. We hypothesized thathigh levels �-globin expression achieved by myeloablative condi-tioning may not be necessary for correction, and if so, wouldreduce transplantation-related morbidity.17,18
We performed reduced-intensity transplantation by transplant-ing gene-modified BERK LSK cells into sublethally irradiated,but with significantly high radiation dose, BERK mice. Theproportion of transduced HSCs and vector copy/cell in the graftwas varied by transducing LSK cells with at a range of MOI(30-100). Since the half-life of BERK RBCs was 1.5 to 2 days(Figure 3G-H), mice were transfused in the peritransplantationperiod and analyzed after 12 weeks. Three serial experimentswere carried out with mice followed for 1 year. GbG mice wereanalyzed by separating them into 3 groups based upon percent-age of HbF at 18 weeks: HbF � 0% (mock, n � 4), HbF less
than 10% (termed GbG�10; n � 17), and HbF of 10% or more(termed GbG�10; n � 9); (Figure 4A). The cutoff at 10% HbFwas selected as this appeared to be a threshold level of HbF thatreflected correction of disease: GbG�10 mice showed a highermortality and inconsistent hematologic correction, comparedwith GbG�10 described in the following paragraph. The mousenumbers in the groups changed with time primarily due to theincreased mortality related to SCA in mice with no/low HbF.
The GbG�10 group of mice had 16% (� 1.2%), 17% (� 1.8%),and 21% (� 2.3%) HbF, whereas the GbG�10 group of mice had5% (� 1.4%), 4% (� 0.6%), and 4% (� 0.5%) HbF at 12, 18, and24 weeks, respectively, that was stable up to 1 year (Figure 4B).F-cell repopulation was significantly higher in GbG�10 mice(65% � 14%) compared with GbG�10 mice (30% � 9.4%; Figure4C). GbG�10 mice had 2 to 2.5 vector copies/cell, whereas theGbG�10 mice had 1.4 copies/cell (Figure 4D).
Hematologic improvement occurred with reduced-intensitytransplantations. Hematologic parameters stabilized at 18 weeks,due to persistent transfused RBCs in the early posttransplantationperiod. There was a significant improvement in hematologicparameters in the GbG�10 group of mice (Table 2), in contrast to asmall and inconsistent improvement in GbG�10 mice.
Improvement in RBC function occurs with reduced-intensitytransplantations. (1) Sickling: There was a very significantreduction in ISCs in GbG�10 mice (P � .005) and a small, but
Figure 2. GbG mice that underwent transplantation aftermyeloablative conditioning, which resulted in correction ofhematologic parameters that correlated with the HbF expres-sion. There was sustained reduction in reticulocytes (A), andincrease in hematocrit (B) and RBC numbers (C) over time.(D) Leukocytosis decreased with normalization of WBC counts.Data shown represent mean (� SEM) values of GbG mice (n � 5;F) and mice that underwent mock transplantation (n � 10; E).� represents mean values in BERK mice that were HSC donorsfor the GbG and mock transplantations. (E-G) Decrease inreticulocytes, and increased hematocrit and RBC numbers corre-lated with the proportion of F cells in individual mice. (H) WBCcounts decreased but normalized when the F cells exceeded60%. WBC counts, counted on an automated analyzer, wererepresentative of circulating leukocytes, since only occasionalnucleated RBCs were seen in peripheral smears. Each datapoint/symbol in panels E-H represents one GbG mouse andsymbols for individual mice have been kept consistent, to traceindividual mice in Figures 1 and 2. � represents mean values inBERK mice that were HSC donors for the GbG and mocktransplantations.
CRITICAL PARAMETERS OF GENETIC CORRECTION OF SCD 1177BLOOD, 6 AUGUST 2009 � VOLUME 114, NUMBER 6
For personal use only.on May 9, 2016. by guest www.bloodjournal.orgFrom

significant reduction in ISCs in GbG�10 mice compared withmock/BERK controls (P � .05, Figure 4E). RBCs from GbG�10mice showed reduced sickling when exposed to graded hypoxia,compared with RBCs from GbG�10 or mock/BERK mice (n � 20,P � .01; Figure 4F). In contrast, there was no significant differencein sickling between GbG�10 and mock/BERK mice. (2) RBCmembrane deformability: Surprisingly, despite similar degree ofsickling with hypoxia in RBCs from GbG�10 mice and mock/BERK mice, there was slight improvement in RBC deformabilityin the GbG�10 mice. However, these differences were notstatistically significant from the mock/BERK mice due to the highvariance (Figure 4G). In contrast, there was a consistent significantimprovement in RBC deformability in GbG�10 mice (P � .001,Figure 4H). The deformability pattern suggested improved RBCflow through large vessels and microvessels.19 (3) RBC survival:RBC half-life of BERK mice was 1.5 days. RBCs of GbG mice with1%, 3%, and 7% HbF had a slightly higher half-life (2 days). TwoGbG mice with 18% HbF showed an RBC half-life of 6 days, a4-fold increase, similar to that seen in mice carrying 40% HbF inthe myeloablative transplantation model.
Taken together, the sGbG vector resulted in significant andconsistent hematologic and functional correction of SCA, when theHbF production exceeded 10% of the total hemoglobin. Notably,the improvement in phenotype was comparable with that achievedwith myeloablative conditioning.
Remarkable improvement in organ pathology when HbFconcentrations exceed 10%. The unique feature of thisBERK3BERK transplantation model was presence of signifi-cant sickle pathology in recipients at the time of transplantation(determined using BERK controls of comparable age as recipi-ent mice when they underwent transplantation). Therefore, thepotential for reversal of organ pathology after gene therapycould be assessed. Organ pathology in the surviving mice atapproximately 50 weeks after transplantation was comparedwith 3-month-old BERK mice that did not undergo transplanta-tion (Figure 5A; Table 3).
The GbG�10 group of mice showed slight improvement inorgan pathology: There was a slight reduction in spleen weight(717 � 162 mg in GbG�10 vs 870 � 71 mg in BERK/mock mice;P value, NS). Bone marrow and spleens showed moderate to severe
Figure 3. GbG mice that underwent transplantation after myeloabla-tive conditioning, which resulted in correction of functional RBCparameters in primary and secondary mice. (A) Peripheral bloodsmears showing numerous irreversibly sickled cells (ISCs) in a mouse thatunderwent mock transplantation and a paucity of ISCs in a GbG mouse.(B) Quantification of ISCs in peripheral blood smears of BERK mice thatdid not undergo transplantation (n � 5), mock mice (n � 3), and GbG mice(n � 5). (*P � .05; **P � .01). (C) Deoxygenation of blood induces sick-ling of RBCs in a mock mouse; sickling is largely absent in a GbG mouse.(D) Quantification of sickle RBCs upon graded hypoxia (by tonometry) inthe GbG mice (F), compared with mock mice (E). (E) RBC deformability byLORCA analysis in GbG, mock, and normal mice (C57, ) analyzed at18 weeks in primary transplant recipients. Similar data were seen insecondary recipients (data not shown). Flow at low (3 Pa) and high(28 Pa) shear stress is represented by shaded areas. (F) RBC half-life(determined by in vivo biotin labeling) in the GbG mice, mock/BERK mice,and normal mice after primary transplantations. Similar results were seenin secondary recipients (data not shown).
Table 2. Correction of organ pathology in GbG mice that underwent transplantation after myeloablative conditioning
Kidney Liver Spleen Bone marrow
Mock mice, n � 5 Focal tubular atrophy (1/5), mild
congestion (3/5)
2� liver infarction (1/5), 3� liver infarction
(1/5), E-M hematopoiesis (2/5)
Weight 500 � 60 mg, severe
erythroid hyperplasia (5/5)
Severe erythroid hyperplasia
(5/5)
GbG mice, n � 5 Normal kidney (5/5) Normal liver (5/5) Weight 256 � 51 mg, mild
erythroid hyperplasia (5/5),
mild congestion (5/5)
Mild erythroid hyperplasia
(5/5)
2� liver infarction indicates 2 to 3 infarctions/section; 3� liver infarction, more than 3 infarctions/section; and E-M, extramedullary.Mild congestion of the spleen vessels with sickle RBCs is seen when splenic architecture is restored. This is not noted when the splenic architecture is effaced by
extramedullary erythropoiesis. Splenic erythroid hyperplasia: severe is complete obliteration of splenic follicles; moderate, more than 1 follicle present/section; and mild,preservation of follicles with evidence of erythroid islands.
Bone marrow: normal erythropoiesis indicates M/E � 5:2; mild erythroid hyperplasia, M/E � 2:1; moderate erythroid hyperplasia, M:E � 1:1; and severe erythroidhyperplasia, M/E � 1:3. Bone marrow erythropoiesis expressed as myeloid-erythroid ratio (M/E). Numbers in parentheses indicate the histologic feature seen in the number ofmice/total number of mice analyzed in that group.
1178 PERUMBETI et al BLOOD, 6 AUGUST 2009 � VOLUME 114, NUMBER 6
For personal use only.on May 9, 2016. by guest www.bloodjournal.orgFrom

erythroid hyperplasia; livers had infarctions and extramedullaryhematopoiesis; and the kidneys showed occasional focal segmentallesions, focal tubular atrophy, and vascular congestion (Table 4).
In contrast, a dramatic reversal of organ pathology was seen inGbG�10 mice: there was a 50% reduction in spleen weight to363 plus or minus 85 mg, preservation of splenic follicles, andmild erythroid hyperplasia in bone marrow and spleen. Remark-ably, no liver infarctions and no kidney pathology were detected,except in one mouse with a single focus of focal tubular atrophy.
Overall, GbG�10 mice showed correction of organ pathology.The lack of organ pathology in GbG mice at 15 months of agecompared with 3-month-old BERK controls demonstrates that genetherapy with the sGbG vector in a reduced-intensity transplantationsetting prevents any further organ damage, and the existent organdamage at the time of transplantation probably reverses fromregeneration.
Survival. There was a significant improvement in overallsurvival in the GbG�10 mice compared with GbG�10 or mockmice (Figure 5B; P � .05). Indeed, at 24 weeks, survival of theGbG�10 mice was comparable with survival in mice with approxi-mately 40% HbF in the myeloablative transplantation model thatwere followed for 24 weeks (supplemental Figure 3). There wassome improvement in early survival in GbG�10 mice comparedwith mock mice (P � .05). However, by 1 year, there was nodifference in survival of GbG�10 mice over mock mice.
F cells and HbF/F cell critical for improved RBC survival andcorrection of SCA
Using biotin surface labeling and intracellular HbF staining, westudied survival of F cells and non-F cells in the same animal,which allowed quantification of the HbF/F cell necessary forimproved sickle RBC survival and deformability. F cells showed aselective prolonged survival, as anticipated (Figure 6A). Theaverage HbF/F cell20 in GbG mice in the BERK3C57Bl/6 modelwas 64% (in these mice, HbF was 41% � 5%, F cells were64% � 6%). In the reduced-intensity transplantation model,GbG�10 mice had 32% HbF/F cell (in these mice HbF was21% � 2%, F cells were 65% � 14%), and GbG�10 mice had13% HbF/F cell (HbF, 4% � 0.1%; F cells, 30% � 9.4%). Notethat GbG mice in the myeloablative model and GbG�10 mice hadsimilar F-cell repopulation (64%-65%), suggesting that 32% HbF/F cellwas sufficient to correct the sickle phenotype. However GbG�10mice with 13% HbF/F cell and 30% F cells had inconsistent andinsignificant amelioration of the disease phenotype.
We therefore determined the half-life of F cells in mice groupedby the percentage of HbF/F cell. GbG mice with low (16%; n � 2),intermediate (33%; n � 4), and very high (89%; n � 2) HbF/F cellwas injected with biotin and followed by periodic blood sampling.We found mice with low HbF/F cell had no improvement inRBC half-life over BERK controls (Figure 6B), those with 33%
Figure 4. HbF expression and functional correction inGbG mice that underwent transplantation after reduced-intensity conditioning, separated into 2 groups: mice withHbF of 10% or more (GbG>10) and mice with HbF ofless than 10% (GbG<10). (A) HbF in individual BERK mice18 weeks after transplantation of sGbG-transduced BERKHSCs, after reduced-intensity conditioning. (B-C) Stable andhigh HbF expression and F-cell repopulation in long-termsurvivors analyzed at 11 months. (D) Box and whisker plotshowing vector copy numbers in GbG�10 and GbG�10 mice,with mean vector copy number denoted by the line. Symbolsin panels A through C represent mouse groups: E � mock(HbF 0%), ƒ � GbG�10 (HbF � 10%), and F � GbG�10(HbF � 10%). (E) The proportion of ISCs was reduced(P � .04) in GbG�10 mice, but was markedly reducedin GbG�10 mice (P � .001), compared with mock mice.(F) Graded deoxygenation via tonometry demonstrates signifi-cant reduction in sickling at physiologically relevant partialoxygen pressures (PO2) in GbG�10 mice, whereas GbG�10mice RBC sickled similar to controls. (G-H) RBC deformabilityshowed highly variable improvement in deformability inGbG�10 mice. In contrast, RBC deformability in GbG�10mice was highly significantly improved at low and high shearstress (P � .001). Symbols represent mouse groups: E, mock;ƒ, GbG�10; F, GbG�10; and , wild-type mice (C57BL/6).Gray shaded rectangles are representative of low and highshear stress through microvessels and large vessels, respec-tively. Error bars indicate SEM.
CRITICAL PARAMETERS OF GENETIC CORRECTION OF SCD 1179BLOOD, 6 AUGUST 2009 � VOLUME 114, NUMBER 6
For personal use only.on May 9, 2016. by guest www.bloodjournal.orgFrom

HbF/F cell had a 3- to 4-fold improvement in half-life, and micewith very high amounts of HbF/F cell showed RBC survivalsimilar to normal mice. These data demonstrate that if one-thirdof the hemoglobin within a sickle RBC is HbF, there issignificant improvement in RBC survival. Note that mice withthese levels of HbF/F cell showed approximately 65% F cells,more than 10% HbF.
To confirm the impact of percentage of circulating F cells onoverall RBC deformability, we grouped mice from both the
myeloablative and reduced-intensity experiments (n � 34) into3 groups: mice with less than 33% circulating F cells, 33% to 65%F cells, and 66% or more F cells and measured RBC deformability.Only data from the low (3 Pa) and high (28 Pa) shear rates areplotted in Figure 6C. Mice with more than 66% F cells had a highlysignificant improvement in RBC deformability at both high andlow shear stress (P � .01). Mice with 33% to 66% F cells hadsignificantly improved RBC deformability only at high shear stress(P � .05). Mice with less than 33% F cells showed inconsistentimprovement in RBC deformability at low or high shear stress,which was not significantly different from mock controls. Thesedata quantify the critical amount of HbF/F cell, the proportion ofF cells, and overall HbF that are necessary for correction of sicklecell disease.
Proportion of transduced HSCs required for phenotypiccorrection
The proportion of HSCs transduced with sGbG in GbG mice wasanalyzed by the secondary spleen colony-forming unit (CFU-S)assay performed at 6 months in both models (Figure 7A-B). Bonemarrow aspirates were performed at 6 months in the BERK3BERKmice that were followed for 1 year. The proportion of transducedCFU-S’s was determined by HbF expression. We have previouslyshown that all vector-positive CFUs express the transgene in anidentical vector that encodes �-globin.13 GbG mice in the myeloab-lative conditioning group had 16% to 87% sGbG-transducedCFU-S’s (average HSC transduction was 50%), and those in thereduced-intensity group had 5% to 60% transduced HSCs (averageHSC transduction was 30%). It is to be noted that in thereduced-intensity model, HSC transduction is overestimated,secondary to the higher mortality of GbG�10% mice in the first6 months.
Importantly, 3 mice with 16%, 20%, and 22% transducedCFU-S’s had more than 10% HbF (HbF was 20%, 11%, and 18%,respectively) and showed complete phenotypic correction. A vectorcopy number analysis was performed concurrently at 24 weekson bone marrow cells and showed 1 to 3 copies/cell and 1 to2.5 copies/cell in GbG mice that underwent transplantation usingthe myeloablative conditioning and reduced-intensity conditioningmodels, respectively. When corrected for HSC transduction, therewere 1.5 to 5 vector copies/cell.
Transduction of human CD34� cells. The percentage ofgene-modified HSCs necessary for effective gene therapy is criticalin this disease. In vitro studies on SCA marrow can be done only ona small scale, and would read out correction in progenitors, not
Table 3. Hematologic parameters of GbG mice that underwent transplantation following reduced-intensity conditioning
Time point/mouse type n WBC, 103/�L RBC, 106/�L Hb, g/dL MCV, fL MCH, pg RDW, % Plt, 103/�L Reticulocyte, %
Week 12
Mock 6 21.7 � 3.8 6.1 � 0.7 7.9 � 0.7 46.2 � 2.3 12.5 � 0.2 27.0 � 1.4 839 � 86 53.2 � 3.9
GbG�10 14 32.3 � 4.6 7.4 � 0.3 7.9 � 0.4 41.7 � 0.7 10.7 � 0.3 29.1 � 0.5 737 � 36 20.2 � 2.5
P* .6 .16 .96 .12 .001 .2 .3 .001
Week 18
Mock 4 32.3 � 4.6 5.6 � 0.1 6.8 � 0.2 46.9 � 1.3 12.2 � 0.3 31.4 � 0.9 802 � 91 54.5 � 2.3
GbG�10 9 12.9 � 1.4 7.7 � 0.4 8.9 � 0.4 43.2 � 1.5 11.7 � 0.6 28.8 � 0.8 798 � 97 26 � 4.1
P† .01 .002 .001 .09 .57 .07 .97 .0001
Week 24
Mock 4 34.1 � 9.4 5.6 � 0.3 7.4 � 0.3 48.7 � 2.4 13.2 � .0.3 30.1 � 1.9 780 � 100 50.8 � 1.9
GbG�10 5 13.4 � 1.1 8.1 � 0.5 9.3 � 0.6 43.9 � 1.2 11.3 � 0.2 29.8 � 1.1 764 � 61 21.2 � 1.9
P‡ .07 .003 .04 .1 .002 .9 .9 .001
Hematologic parameters and abbreviations as stated in Table 1. P values represent comparisons of mock mice with GbG�10 at 12 (*), 18 (†), and 24 weeks (‡).
Figure 5. Correction of organ pathology in GbG>10 mice that underwenttransplantation after reduced-intensity conditioning and improved overallsurvival. (A) Representative hematoxylin-eosin–stained sections of a kidney, liver,and spleen of GbG�10 and GbG�10 mice 48 to 50 weeks after reduced-intensityconditioning transplantation and a 3-month-old BERK control. Image acquisitioninformation is available in supplemental data. (B) Kaplan-Meier survival curveshowed significantly improved survival of the GbG�10 mice compared with mock/GbG�10 mice at 50 weeks. Survival at 24 weeks is denoted by a dashed vertical lineto compare with survival of the GbG mice in the myeloablative transplantation model(supplemental Figure 3c).
1180 PERUMBETI et al BLOOD, 6 AUGUST 2009 � VOLUME 114, NUMBER 6
For personal use only.on May 9, 2016. by guest www.bloodjournal.orgFrom

HSCs. We have shown HSC correction in humanized models ofSCA with long-term analysis. The extremely limited numbers ofRBCs we were able to produce from injecting human thalassemiabone marrow CD34� cells10 are prohibitive for studies on sickling.Therefore, we optimized lentivirus transduction into normal humanCD34� cells for a preclinical scale-up, using a GFP lentivirusvector and the severe combined immunodeficient (SCID)–repopulating assay. Granulocyte colony-stimulating factor–mobi-lized peripheral blood CD34� cells transduced with a GFPlentivirus vector were transplanted into nonobese diabetic (NOD)/LtSz-scid IL2R�null (NOG) mice. Here, mock mice were those thatreceived a transplant of untransduced CD34� cells immediatelyafter selection, as controls for the effect of transduction onengraftment and clonogenicity. At 6 weeks, CFUs were plated from
bone marrow derived from NOG mice, and 36 individual CFUs/mouse were analyzed for the percentage of gene-marked colonies(supplemental Figure 4). The 18-hour transduction did not affectengraftment or clonogenicity (data not shown). We observed a 77%gene transfer on average in the SCID-repopulating cell assay,similar to our previous data in human thalassemia CD34� cells.10
Discussion
We show that lentiviral delivery human �-globin under �-globinregulatory control elements in HSCs results in sufficient postnatalHbF expression to correct SCA in mice. We then de-escalated theamount of HbF and transduced HSCs, using reduced-intensity
Table 4. Organ pathology in GbG mice that underwent transplantation after reduced-intensity transplantation
Mouse type n
Pathology
Kidney Liver Spleen Bone marrow
BERK/mock 7 Mesangial proliferation (2/7),
E-M hematopoiesis (7/7),
focal ischemic lesion (2/7),
cystic kidney dilation (1/7),
congestion* (7/7)
E-M hematopoiesis (7/7), 1� liver
infarction (3/7), 3� liver
infarction (4/7)
Weight 870 � 71 mg, severe
erythroid hyperplasia (6/7),
moderate erythroid
hyperplasia (1/7),
obliteration of lymphoid
follicles (7/7)
Severe erythroid hyperplasia
(6/7), moderate erythroid
hyperplasia (1/7)
GbG�10 4 Focal segmental lesion (1/4),
focal tubular atrophy (1/4),
congestion (4/4)
1� liver infarction (2/4), 2� liver
infarction (1/4), hemosiderosis
(1/4)
Weight 717 � 162 mg,
severe erythroid
hyperplasia (1/4),
moderate erythroid
hyperplasia (2/4), mild
erythroid hyperplasia (1/4),
occasional lymphoid
follicles (2/4)
Mild erythroid hyperplasia
(1/3), moderate erythroid
hyperplasia (2/3)
GbG�10 4 Mild congestion (1/4), focal
tubular atrophy (1/4),
normal kidney (2/4)
Normal liver (4/4) Weight 363 � 85 mg, mild
erythroid hyperplasia (4/4),
multiple lymphoid follicles
(4/4), congestion (4/4)
Mild erythroid hyperplasia
(2/2)
E-M indicates extramedullary; and 1� liver infarction, 1 infarction/section.*Congestion of vessels and presence of sickle RBC in vessels. Notably, congested vessels were visible in spleens only when erythroid hyperplasia effacing splenic
architecture was reduced. The terminology used to quantify organ pathology is the same as documented in Table 2.
Figure 6. Effect of HbF, F cells, and percentage HbF/F cell required for functional improvement in RBCsurvival and deformability. (A) RBC half-life. Left panelshows a representative GbG mouse injected with biotin,with biotin-labeled F cells (upper right quadrants) andnon-F cells (lower right quadrants) determined by FACS.Right panel shows survival of F cells ( ), compared withthe non-F cells ( ) in GbG mice (n � 4); wild-type mice(‚); and Berkeley mice (E). (B) A cohort of GbG miceanalyzed for RBC survival in vivo, based upon thepercentage of HbF/F cell. Each symbol represents amouse group with HbF percentage and number of micelisted in the adjacent table legend. (C) All GbG and mockmice (n � 34) that were analyzed for RBC deformabilitywere divided into groups based on proportion of F cells:0%, 1% to 33%, 33% to 66%, and more than 66%, anddeformability of total RBC in these mice was plotted at low(3 Pa, ‚) and high (28 Pa, ƒ) shear stress. Significantlyimproved deformability over mock controls is denoted by*(P � .05) and **(P � .01). Error bars indicate SEM.
CRITICAL PARAMETERS OF GENETIC CORRECTION OF SCD 1181BLOOD, 6 AUGUST 2009 � VOLUME 114, NUMBER 6
For personal use only.on May 9, 2016. by guest www.bloodjournal.orgFrom

conditioning and varying MOI, to assess critical parameters neededfor correction. A systematic quantification of functional andhematologic RBC indices, organ pathology, and life span werecritical to determine the minimal amount of HbF, F cells, HbF/F cell, and gene-modified HSCs required for reversing the sicklephenotype. We show the following: (1) Amelioration of diseaseoccurred when HbF exceeded 10%, F cells constituted two-thirdsof the circulating RBCs, and HbF/F cell was one-third of the totalhemoglobin in RBCs; and when approximately 20% sGbG-modified HSCs repopulated the marrow. (2) Genetic correction wassustained in primary or secondary transplant recipients followedlong-term. (3) There is a method of determining minimum HSCchimerism for correction of a hematopoietic disease in an in vivomodel, which would contribute to design of cell dose andconditioning regimens to achieve equivalent genetically correctedHSCs in human clinical trials.
The novel aspect of our study is that it addresses, for the firsttime, the gene dosage and the gene-modified hematopoietic stemcell dosage required for correction of a genetic defect. Expressing atremendous amount of fetal/antisickling hemoglobin will undoubt-edly correct disease, as has been shown by others,8,9,21 but is notpractically possible in a clinical setting. As an example, an initialgene therapy for adenosine deaminase (ADA) deficiency wasperformed using no conditioning, and was not therapeutic, eventhough few gene-marked stem cells engrafted, and a selectiveadvantage to gene-corrected lymphocytes was evident upon with-drawal of ADA.22 In a subsequent trial, 4 mg/kg busulfan was usedbefore transplantation, as conditioning, resulting in adequategene-corrected stem cell dose and gene-modified T cells.23,24
Although these pioneering studies provided us with invaluable informa-tion, they underscore the critical importance of determining thresholdsfor genetic correction before embarking on clinical studies.
Previous laboratories have shown genetic correction of SCAwith antisickling globins using myeloablative transplantation mod-els. Pawliuk et al showed correction of marine SCA in 3 mice usinga lentivirus encoding �T87Q, a recombinant antisickling �-globin.They demonstrated reduction in ISCs and spleen size after ge-netic correction in a BERK to C57/BL6 transplantation model.8
Levasseur et al further modified �-globin with 3 mutations (�AS3)and corrected disease in the Townes knockout-transgenic (KO-TG)to C57Bl/6 model.9 Townes KO-TG mice are extremely anemic butshowed increased hemoglobin to nearly 80 g/L (8 g/dL) with 2 to2.5 vector copies/cell, corrected organ pathology, and sustainedcorrection in secondary mice. Both of these studies showed asignificant antisickling effect of recombinant �-globins. Recently,Pestina et al showed correction of BERK mice with a �-globinlentivirus.21 Using the sickle to C57BL/6 transplantation model, we
show proof-of-concept studies of correction of SCA with a novel�/�-hybrid vector and determine functional and quantifiable assaysof disease correction. Although we and others correct disease at 1 to3 copies/cell, we show that the percentage of transduced stem cellsin this setting of lethal irradiation/transplantation is very high(average HSCs transduced are 50%, as analyzed by a stringentsecondary CFU-S assay). This level of HSC transduction wouldlikely not be achieved in the clinical setting unless myeloablation isperformed.
Therefore a novel model (BERK to BERK transplantation) wasdeveloped to address the minimal gene transfer needed, and answerquestions of correction of SCA in a mouse with significant sicklepathology at 12 weeks of life (Figure 5). Notably, a sickle to normalmyeloablative transplantation, used by other groups showingcorrection of SCD,8,9,21 is a disease prevention model, where therewas no underlying pathology at time of transplantation. Our studiesshow that repair of preexisting pathology can occur, if geneticcorrection results in more than 10% HbF.
A specific concern of using antisickling �-globins is a potentialto illicit an immune response. The long history of chronictransfusions suggests that globin proteins have low immunogenic-ity. However, development of antibodies against hemoglobin A hasbeen reported in 3 multiply transfused SCA patients, with theepitope directed to the sixth codon.25 This report suggests a singlebase pair difference between � and �S globin can be immunogenic.How immunogenic endogenous production of recombinant �-globins by erythroid progenitors occurs is an unknown, thoughgiven the overall low immunogenicity of globin proteins, immuneresponses against antisickling �-globins may not occur. In thisregard, production of the normally expressed �-globin would bedevoid of any immunologic sequelae. Recently, Samakoglu et alused a lentivirus encoding human �-globin and a small hairpinRNA (shRNA) against �S-globin and showed reduction in �S-globin mRNA in primary human cells in vitro.26 Whether thesechanges in MRNA expression resulted in changes in proteinexpression or functional improvement in sickle RBC was notdemonstrated.
Blouin et al generated SAD mice27 transgenic for human�-globin with 9% to 16% HbF, without significant hematologicimprovement.28 Fabry et al produced sickle mice that exclusivelyproduced HbS but varying amounts of HbF and observed signifi-cant improvement in hematologic/pathologic features only in thehigh HbF mice.29 It is to be noted that in transgenic mice, HbFexpression is pancellular, whereas in genetic correction of HSCs,there is heterocellular correction. The results from HbF-inducingagents such as hydroxyurea are not entirely applicable to HbFinduction via gene therapy, because stress erythropoiesis and RBC
Figure 7. Proportion of transduced HSCs in GbG mice. Propor-tions in the myeloablative (A) and reduced-intensity (B) transplanta-tion models are shown. The proportion of sGbG-transduced HSCswas determined by spleen colonies (30-36 colonies/mouse) byintracellular staining with HbF and HbS. Each bar represents anindividual mouse. (A) In the myeloablative transplantation model,symbols beneath each bar (representing one mouse) are consistentwith the symbols in mice labeled in Figures 1 and 2. (B) In thereduced-intensity group, bone marrow was successfully aspiratedfrom 8 mice at 24 weeks and mice were followed for an additional24 weeks. The HbF expression in peripheral blood by HPLC andbone marrow copy number of the respective mice at 24 weeks arelabeled under each bar.
1182 PERUMBETI et al BLOOD, 6 AUGUST 2009 � VOLUME 114, NUMBER 6
For personal use only.on May 9, 2016. by guest www.bloodjournal.orgFrom

macrocytosis occur with hydroxyurea and ameliorate sicklingpartly from a dilutional effect of HbS. Some comparisons on HSCchimerism can be drawn in patients with partial chimerism afterallogeneic transplantation, although these patients have normalRBCs that contain HbA, rather than HbF-containing sickle RBCsamid sickle RBCs.
BERK mice have some degree of thalassemia. Therefore oneconcern in using this model for genetic therapy studies for sicklecell anemia is that correction of thalassemia would obscureimprovements made by the antisickling effects of HbF. Surpris-ingly we saw no significant change in MCH in GbG�10 orGbG�10 mice (Table 3), including mice with HbF/F cell as high as89% (Figure 6). These results were surprising, but showed that thecorrection of sickling in RBCs was not secondary to correction ofthalassemia, as seen in murine thalassemia model,30 where increas-ing MCH was seen with increases in HbF of 4% or higher.30
Conceivably, HbF is produced at the expense of HbS.Although BERK mice exclusively carry human hemoglobin,
the total hemoglobin in the mouse RBCs is one-third of a humanRBC. Therefore, HbF and HbF/F cell were expressed as a percent-age, rather than in absolute amounts, to best compare murine datato human. An increase of HbF from 3.6% to 13.6% has been shownto reduce acute sickle events in patients on decitabine.31 Similarimprovement in sickle events occurs with 25% or more HbF/F cellin patients responsive to hydroxyurea.32,33 Our data showingimprovement with 33% HbF/F cell is concordant with thesereports, but more closely resemble RBCs in infants with SCA,where less than one-third HbF/F cell at 10 to 12 months isconsidered a threshold for intracellular sickle polymerization.34,35
The most remarkable effect of �-globin production with thesGbG vector was a dramatic absence of chronic organ damage andan improved survival of the sickle mice when HbF exceeded 10%.Patients with high HbF have an improved survival, confirmed bythe multicenter study on hydroxyurea.2,36 HbF expressed from thesGbG vector was comparable with, or even better than, effectivehydroxyurea treatment. The potential of a one-time correction,where responsiveness to hydroxyurea and compliance to dailylife-long administration would not be limiting factors, would be atremendous advantage of gene therapy. Indeed, we did not antici-pate we would get the same conclusion with gene therapy, asderived from collective knowledge on (1) transgenic mice, inwhich every RBC has the same amount of HbF although we wereimposing HbF on SS RBCs28; (2) chimeric transplantations, inwhich normal amounts of HbA-producing RBCs (AA RBC) arepresent mixed with SS RBCs17,37,38; or (3) SCD patients onhydroxyurea, in whom macrocytosis induced by hydroxyureawould dilute HbS and reduce the threshold for sickling. Weexpected a much higher threshold of genetically corrected sickleHSCs necessary for F-cell repopulation and correction of SCAphenotype, as we were exogenously imposing HbF into a sicklecell with normal amounts of HbS. We were surprised that despitethese distinct differences in transgenics/chimeras, our conclusionswere similar with exogenous �-globin expression: Indeed express-ing exogenous HbF in RBCs at concentrations from 33% to as highas 89% resulted in no significant increase in MCV or MCH, yetcorrected sickling. Although not formally tested, our data suggestthat genetic delivery of HbF decreases endogenous HbS.
We show that the percentage of transduced HSCs in the settingof lethal irradiation/transplantation is very high (50% on average,as analyzed by a stringent secondary CFU-S assay at 24 weeks), anumber that would be difficult to achieve in a clinical setting. TheBERK3BERK transplantation model, however, shows that 20%
autologous HSC correction may suffice for a significant ameliora-tion of sickling, organ damage, and survival. However, whetherthis percentage of gene-modified HSCs necessary for effectivegene therapy is achievable is critical to determine, since there is nosurvival advantage to the gene-modified HSCs in this disease. Highhuman HSC transduction has been a limitation of gene therapy withthe traditional gammaretrovirus vectors. Lentivirus vectors canovercome this barrier: a 20% long-term transduction has beenshown in adrenoleukodystrophy with a lentivirus vector.39 Weoptimized lentivirus transduction into human CD34� cells usingthe SCID-repopulating cell assay and can achieve approximately75% gene transfer in SCID-repopulating cell, on average, similar toour previous data in human thalassemia CD34� cells, where 70%transduction was seen 3 to 4 months after transplantation intoimmune-deficient mice.10 Notably, this level of gene transfer in theSCID mice is encouraging, and indeed higher than the gene transferobserved in NOD-SCID mice with the adrenoleukodystrophylentivirus vector in preclinical studies.40
Gene therapy using this approach could also overcome the toxicityand immunologic consequences of the traditional allogeneic bonemarrow transplantation/reduced-intensity transplantation.18,41,42 Mis-matched mixed chimerism of normal and sickle marrow in murinetransplantations shows that a near complete chimerism is typicallynecessary for correction of organ damage.39,43 It is encouraging that, in aclinical series, reduced-intensity conditioning (RIC) transplantation with8 mg/kg busulfan along with fludarabine, antithymocyte globulin, andtotal lymphoid irradiation in SCA patients has shown an averageallogeneic engraftment of 78% at 2 to 8.5 years after transplantation,with correction of SCAphenotype.44 This high level of donor chimerismeven in an allogeneic RIC setting, where immune rejection can occur,suggests that high gene transfer efficiency into autologous CD34� cellsfollowed by RIC may be a potentially safer alternative to myeloablativeconditioning. We show 77% gene transfer efficiency in human stem/progenitors using a NOD-SCID repopulating cell assay (supplementalFigure 4) and a correction of phenotype in mice with 1.3 to 1.5 copiesper cell and approximately 20% gene-marked CFU-Ss (Figure 7).However, whether RIC will be sufficient for genetic correction of SCAin humans remains to be tested in a clinical trial.
Of note, correction occurred at 1 to 3 vector copies per cell, aclinically achievable goal. Flanking the GbG virus with a chromatininsulator may increase HbF/vector copy by 2- to 4-fold.10,13 Inexperimental models, the insulator appears to reduce clonaldominance, although whether the insulator element lowers the riskof insertional oncogenesis is unknown.45,46 The risk of insertionaloncogenesis observed with randomly integrating vectors has beenshown to be lower with a lentivirus vector than a gammaretrovirusvector.47 It would be further lowered when the enhancer element isactive only in a restricted erythroid lineage.48
In summary, SCA is a common monogenic blood disorder withpotentially devastating consequences due to the chronic andepisodic disease; has an enormous impact on the health caresystem, and is associated with a significant reduction in life span.We show that gene therapy with a �-globin–expressing lentivirusvector offers the potential of a one-time cure and defines parame-ters needed to cure the disease. We further demonstrate a preclini-cal in vivo method of determining the minimal amount ofgenetically corrected hematopoietic stem cells that would berequired to correct disease, important in design of clinical genetherapy trials. We are currently studying whether this vectorcorrects the functional phenotype in human sickle RBCs aseffectively as it does in the humanized mouse models of SCA.
CRITICAL PARAMETERS OF GENETIC CORRECTION OF SCD 1183BLOOD, 6 AUGUST 2009 � VOLUME 114, NUMBER 6
For personal use only.on May 9, 2016. by guest www.bloodjournal.orgFrom

Acknowledgments
We gratefully acknowledge the help of Ping Xia with virusproduction; Anastacia Loberg, Kristy Lauderback, and GabrielEstavez-Pegani with mouse genotyping and experiments; Rosal-inda Wenby with deformability analysis; Lana Wenckbach, PatriciaAdkins, and Licheng Zheng with HPLC analysis; and Peter Ciarolowith tonometry. We owe special thanks to Dr Clinton H. Joiner for acritical review of the paper and helpful comments, and Dr TimothyM. Townes for generously providing the sickle knockin mice.
We thank Jamie Wilhelm and the Hematology Repository andElke Grassman and Diana Nordling in the Translational CoreLaboratories, Tom Leemhuis at the Hoxworth Blood Center, andCarrie Stevens in the Translational Research Trials Office for helpwith obtaining human samples, apheresis, CliniMacs selections,and gene transfer studies in human CD34� cells.
This work was supported by National Institutes of Health,National Heart, Lung, and Blood Institute grants U54-HL070595(P.M., H.J.M.), RO1-HL70135-01 (P.M.), and U54-HL06-008 (P.M.).
Authorship
Contribution: A.P. performed gene transfer and transplantationexperiments, analyzed the hematologic and functional parameters,analyzed the data, and wrote the paper; T.H. performed genetransfer and transplantation experiments, molecular analysis oncopy numbers, and CFU-S and the NOD-SCID experiments; F.U.cloned the vector backbone; R.F. supervised tonometry analysis;H.J.M. supervised RBC deformability analysis; D.W. analyzedhistopathology of mouse organs; and P.M. contributed to thehypothesis and overall study design and experiments, preparation,and editing of the paper.
Conflict-of-interest disclosure: The authors declare no compet-ing financial interests.
Correspondence: Punam Malik, TCHRF 6564, Division ofExperimental Hematology, Cincinnati Children’s Hospital MedicalCenter, ML 7013, 3333 Burnet Ave, Cincinnati, OH 45229; e-mail:[email protected].
References
1. Darbari DS, Kple-Faget P, Kwagyan J, Rana S,Gordeuk VR, Castro O. Circumstances of deathin adult sickle cell disease patients. Am J Hema-tol. 2006;81(11):858-863.
2. Platt OS, Brambilla DJ, Rosse WF, et al. Mortalityin sickle cell disease: life expectancy and risk fac-tors for early death. N Engl J Med. 1994;330(23):1639-1644.
3. Davis H, Schoendorf KC, Gergen PJ, Moore RMJr. National trends in the mortality of children withsickle cell disease, 1968 through 1992. Am JPublic Health. 1997;87(8):1317-1322.
4. World Health Organization. Sickle-Cell Anaemia.Fifty-Ninth World Health Assembly, ProvisionalAgenda Item 11.4. 2006;A59:1. http://www.who.int/gb/ebwha/pdf_files/WHA59/A59_9-en.pdf. Accessed June 13, 2006.
5. Walters MC, Patience M, Leisenring W, et al. Bar-riers to bone marrow transplantation for sickle cellanemia. Biol Blood Marrow Transplant. 1996;2(2):100-104.
6. Fitzhugh CD, Perl S, Hsieh MM. Late effects ofmyeloablative bone marrow transplantation(BMT) in sickle cell disease (SCD) [letter]. Blood.2008;111(3):1742-1743; author reply 1744.
7. Bernaudin F, Socie G, Kuentz M, et al. Long-termresults of related myeloablative stem-cell trans-plantation to cure sickle cell disease. Blood.2007;110(7):2749-2756.
8. Pawliuk R, Westerman KA, Fabry ME, et al.Correction of sickle cell disease in transgenicmouse models by gene therapy. Science. 2001;294(5550):2368-2371.
9. Levasseur DN, Ryan TM, Pawlik KM, TownesTM. Correction of a mouse model of sickle celldisease: lentiviral/antisickling beta-globin genetransduction of unmobilized, purified hematopoi-etic stem cells. Blood. 2003;102(13):4312-4319.
10. Puthenveetil G, Scholes J, Carbonell D, et al.Successful correction of the human beta-thalassemia major phenotype using a lentiviralvector. Blood. 2004;104(12):3445-3453.
11. Moreau-Gaudry F, Xia P, Jiang G, et al. High-levelerythroid-specific gene expression in primary hu-man and murine hematopoietic cells with self-inactivating lentiviral vectors. Blood. 2001;98(9):2664-2672.
12. Rubin JE, Pasceri P, Wu X, Leboulch P, Ellis J.Locus control region activity by 5HS3 requires afunctional interaction with beta-globin gene regu-latory elements: expression of novel beta/
gamma-globin hybrid transgenes. Blood. 2000;95(10):3242-3249.
13. Arumugam PI, Scholes J, Perelman N, Xia P, YeeJK, Malik P. Improved human beta-globin expres-sion from self-inactivating lentiviral vectors carry-ing the chicken hypersensitive site-4 (cHS4) insu-lator element. Mol Ther. 2007;15(10):1863-1871.
14. Paszty C, Brion CM, Manci E, et al. Transgenicknockout mice with exclusively human sickle he-moglobin and sickle cell disease. Science. 1997;278(5339):876-878.
15. Manci EA, Hillery CA, Bodian CA, Zhang ZG,Lutty GA, Coller BS. Pathology of Berkeley sicklecell mice: similarities and differences with humansickle cell disease. Blood. 2006;107(4):1651-1658.
16. Baskurt OK, Meiselman HJ. Blood rheology andhemodynamics. Semin Thromb Hemost. 2003;29(5):435-450.
17. Iannone R, Luznik L, Engstrom LW, et al. Effectsof mixed hematopoietic chimerism in a mousemodel of bone marrow transplantation for sicklecell anemia. Blood. 2001;97(12):3960-3965.
18. Iannone R, Casella JF, Fuchs EJ, et al. Results ofminimally toxic nonmyeloablative transplantationin patients with sickle cell anemia and beta-thalassemia. Biol Blood Marrow Transplant. 2003;9(8):519-528.
19. Hardeman MR, Ince C. Clinical potential of invitro measured red cell deformability, a myth?Clin Hemorheol Microcirc. 1999;21(3-4):277-284.
20. Franco RS, Lohmann J, Silberstein EB, et al.Time-dependent changes in the density and he-moglobin F content of biotin-labeled sickle cells.J Clin Invest. 1998;101(12):2730-2740.
21. Pestina TI, Hargrove PW, Jay D, Gray JT, BoydKM, Persons DA. Correction of murine sickle celldisease using gamma-globin lentiviral vectors tomediate high-level expression of fetal hemoglo-bin. Mol Ther. 2009;17(2):245-252.
22. Kohn DB, Hershfield MS, Carbonaro D, et al. Tlymphocytes with a normal ADA gene accumulateafter transplantation of transduced autologousumbilical cord blood CD34� cells in ADA-defi-cient SCID neonates. Nat Med. 1998;4(7):775-780.
23. Aiuti A, Slavin S, Aker M, et al. Correction of ADA-SCID by stem cell gene therapy combined withnonmyeloablative conditioning. Science. 2002;296(5577):2410-2413.
24. Aiuti A, Cattaneo F, Galimberti S, et al. Gene
therapy for immunodeficiency due to adenosinedeaminase deficiency. N Engl J Med.2009;360(5):447-458.
25. Noronha PA, Vida LN, Park CL, Honig GR. He-moglobin-specific antibody in a multiply trans-fused patient with sickle cell disease. Blood.1997;89(6):2155-2158.
26. Samakoglu S, Lisowski L, Budak-Alpdogan T, etal. A genetic strategy to treat sickle cell anemia bycoregulating globin transgene expression andRNA interference. Nat Biotechnol. 2006;24(1):89-94.
27. Trudel M, De Paepe ME, Chretien N, et al. Sicklecell disease of transgenic SAD mice. Blood.1994;84(9):3189-3197.
28. Blouin MJ, Beauchemin H, Wright A, et al. Ge-netic correction of sickle cell disease: insightsusing transgenic mouse models. Nat Med. 2000;6(2):177-182.
29. Fabry ME, Suzuka SM, Weinberg RS, et al. Sec-ond generation knockout sickle mice: the effect ofHbF. Blood. 2001;97(2):410-418.
30. Persons DA, Hargrove PW, Allay ER, Hanawa H,Nienhuis AW. The degree of phenotypic correc-tion of murine beta-thalassemia intermedia fol-lowing lentiviral-mediated transfer of a humangamma-globin gene is influenced by chromo-somal position effects and vector copy number.Blood. 2003;101(6):2175-2183.
31. Koshy M, Dorn L, Bressler L, et al. 2-deoxy5-azacytidine and fetal hemoglobin induction insickle cell anemia. Blood. 2000;96(7):2379-2384.
32. Charache S, Dover GJ, Moyer MA, Moore JW.Hydroxyurea-induced augmentation of fetal he-moglobin production in patients with sickle cellanemia. Blood. 1987;69(1):109-116.
33. Maier-Redelsperger M, de Montalembert M,Flahault A, et al. Fetal hemoglobin and F-cell re-sponses to long-term hydroxyurea treatment inyoung sickle cell patients: The French StudyGroup on Sickle Cell Disease. Blood. 1998;91(12):4472-4479.
34. Marcus SJ, Ware RE. Physiologic decline in fetalhemoglobin parameters in infants with sickle celldisease: implications for pharmacological inter-vention. J Pediatr Hematol Oncol. 1999;21(5):407-411.
35. Maier-Redelsperger M, Noguchi CT,de Montalembert M, et al. Variation in fetal hemo-globin parameters and predicted hemoglobin Spolymerization in sickle cell children in the firsttwo years of life: Parisian Prospective Study on
1184 PERUMBETI et al BLOOD, 6 AUGUST 2009 � VOLUME 114, NUMBER 6
For personal use only.on May 9, 2016. by guest www.bloodjournal.orgFrom

Sickle Cell Disease. Blood. 1994;84(9):3182-3188.
36. Steinberg MH, Barton F, Castro O, et al. Effect ofhydroxyurea on mortality and morbidity in adultsickle cell anemia: risks and benefits up to 9years of treatment. JAMA. 2003;289(13):1645-1651.
37. Kean LS, Manci EA, Perry J, et al. Chimerismand cure: hematologic and pathologic correctionof murine sickle cell disease. Blood. 2003;102(13):4582-4593.
38. Walters MC, Patience M, Leisenring W, et al.Stable mixed hematopoietic chimerism after bonemarrow transplantation for sickle cell anemia. BiolBlood Marrow Transplant. 2001;7(12):665-673.
39. Cavazzana-Calvo M, Cartier N, Abina SH-B, et al.Hematopoietic stem cell gene therapy trial withlentiviral vector in X-linked adrenoleukodystrophy.Blood (ASH Annual Meeting Abstracts). 2008;(suppl)112. Abstract 821.
40. Benhamida S, Pflumio F, Dubart-KupperschmittA, et al. Transduced CD34� cells from adreno-
leukodystrophy patients with HIV-derived vectormediate long-term engraftment of NOD/SCIDmice. Mol Ther. 2003;7(3):317-324.
41. Horan JT, Liesveld JL, Fenton P, Blumberg N,Walters MC. Hematopoietic stem cell transplanta-tion for multiply transfused patients with sicklecell disease and thalassemia after low-dose totalbody irradiation, fludarabine, and rabbit anti-thymocyte globulin. Bone Marrow Transplant.2005;35(2):171-177.
42. Krishnamurti L, Blazar BR, Wagner JE. Bonemarrow transplantation without myeloablation forsickle cell disease. N Engl J Med. 2001;344(1):68.
43. Kean LS, Durham MM, Adams AB, et al. A curefor murine sickle cell disease through stablemixed chimerism and tolerance induction afternonmyeloablative conditioning and major histo-compatibility complex-mismatched bone marrowtransplantation. Blood. 2002;99(5):1840-1849.
44. Krishnamurti L, Kharbanda S, Biernacki MA, et al.Stable long-term donor engraftment following re-
duced-intensity hematopoietic cell transplantationfor sickle cell disease. Biol Blood Marrow Trans-plant. 2008;14(11):1270-1278.
45. Ryu BY, Evans-Galea MV, Gray JT, Bodine DM,Persons DA, Nienhuis AW. An experimental sys-tem for the evaluation of retroviral vector designto diminish the risk for proto-oncogene activation.Blood. 2008;111(4):1866-1875.
46. Ryu BY, Persons DA, Evans-Galea MV, Gray JT,Nienhuis AW. A chromatin insulator blocks inter-actions between globin regulatory elements andcellular promoters in erythroid cells. Blood CellsMol Dis. 2007;39(3):221-228.
47. Montini E, Cesana D, Schmidt M, et al. Hemato-poietic stem cell gene transfer in a tumor-pronemouse model uncovers low genotoxicity of lentivi-ral vector integration. Nat Biotechnol. 2006;24(6):687-696.
48. von Kalle C, Baum C, Williams DA. Lenti in red:progress in gene therapy for human hemoglobi-nopathies. J Clin Invest. 2004;114(7):889-891.
CRITICAL PARAMETERS OF GENETIC CORRECTION OF SCD 1185BLOOD, 6 AUGUST 2009 � VOLUME 114, NUMBER 6
For personal use only.on May 9, 2016. by guest www.bloodjournal.orgFrom

online May 27, 2009 originally publisheddoi:10.1182/blood-2009-01-201863
2009 114: 1174-1185
David Witte and Punam MalikAjay Perumbeti, Tomoyasu Higashimoto, Fabrizia Urbinati, Robert Franco, Herbert J. Meiselman, determinants for successful correctionsickle cell anemia in a humanized sickle mouse model: critical A novel human gamma-globin gene vector for genetic correction of
http://www.bloodjournal.org/content/114/6/1174.full.htmlUpdated information and services can be found at:
(712 articles)Red Cells, Iron, and Erythropoiesis (565 articles)Gene Therapy
Articles on similar topics can be found in the following Blood collections
http://www.bloodjournal.org/site/misc/rights.xhtml#repub_requestsInformation about reproducing this article in parts or in its entirety may be found online at:
http://www.bloodjournal.org/site/misc/rights.xhtml#reprintsInformation about ordering reprints may be found online at:
http://www.bloodjournal.org/site/subscriptions/index.xhtmlInformation about subscriptions and ASH membership may be found online at:
Copyright 2011 by The American Society of Hematology; all rights reserved.of Hematology, 2021 L St, NW, Suite 900, Washington DC 20036.Blood (print ISSN 0006-4971, online ISSN 1528-0020), is published weekly by the American Society
For personal use only.on May 9, 2016. by guest www.bloodjournal.orgFrom





























