Embed Size (px)
Citation preview

molecules
Article
Ab Initio Dot Structures Beyond the Lewis Picture
Michael A. Heuer , Leonard Reuter and Arne Lüchow *
Citation: Heuer, M.A.; Reuter, L.;
Lüchow, A. Ab Initio Dot Structures
Beyond the Lewis Picture. Molecules
2021, 26, 911. https://doi.org/
10.3390/molecules26040911
Academic Editor: Ángel Martín
Pendás
Received: 31 December 2020
Accepted: 4 February 2021
Published: 9 February 2021
Publisher’s Note: MDPI stays neu-
tral with regard to jurisdictional clai-
ms in published maps and institutio-
nal affiliations.
Copyright: © 2021 by the authors. Li-
censee MDPI, Basel, Switzerland.
This article is an open access article
distributed under the terms and con-
ditions of the Creative Commons At-
tribution (CC BY) license (https://
creativecommons.org/licenses/by/
4.0/).
Institute of Physical Chemistry, RWTH Aachen University, Landoltweg 2, 52056 Aachen, Germany;[email protected] (M.A.H.); [email protected] (L.R.)* Correspondence: [email protected]
Abstract: The empirical Lewis picture of the chemical bond dominates the view chemists have ofmolecules, of their stability and reactivity. Within the mathematical framework of quantum mechan-ics, all this chemical information is hidden in the many-particle wave function Ψ. Thus, to reveal andunderstand it, there is great interest in enhancing the Lewis model and connecting it to computablequantities. As has previously been shown, the Lewis picture can often be recovered from the proba-bility density |Ψ|2 with probabilities in agreement with valence bond weights: the structures appearas most likely positions in the all-electron configuration space. Here, we systematically expand thistopological probability density analysis to molecules with multiple bonds and lone pairs, employingcorrelated Slater-Jastrow wave functions. In contrast to earlier studies, non-Lewis structures areobtained that disagree with the prevalent picture and have a potentially better predictive capability.While functional groups are still recovered with these ab initio structures, the boundary betweenbonds and lone pairs is mostly blurred or non-existent. In order to understand the newly foundstructures, the Lewis electron pairs are replaced with spin-coupled electron motifs as the fundamentalelectronic fragment. These electron motifs—which coincide with Lewis’ electron pairs for manysingle bonds—arise naturally from the generally applicable analysis presented. An attempt is madeto rationalize the geometry of the newly-found structures by considering the Coulomb force and thePauli repulsion.
Keywords: probability density analysis; electronic structure; Lewis structures; chemical bonding;similarity; clustering; spin coupling
1. Introduction
Structural formulas are such an important tool for chemists that they can justifiablybe labeled the ‘script of chemistry’. They not only show the geometry of moleculesbut also capture parts of the electronic structure by depicting bond orders and formalcharges. These sketches of the electronic properties were conceived by G.N. Lewis basedon empirical observations [1] and are usually referred to as ‘dot structures’. For non-radical systems, the depiction of individual electrons as dots is usually omitted. Instead,the structures are built from electron pairs which are drawn as lines. Compared to pure dotstructures, the concept of electron pairs adds information on the quantized spin: two pairedelectrons are spin-coupled. After the dawn of quantum mechanics, the concept of Lewisstructures was taken up in valence bond (VB) theory [2,3]. Within the VB interpretation,a system is no longer described by only one dot structure, but by multiple structures withdifferent weights.
Artmann [4] and, later, Zimmermann and Van Rysselberghe [5] were the first toinvestigate the maxima of the probability density |Ψ|2. Note that this probability density is,in principle, measurable [6]. Based on their analyses, Linnett developed the—therefore,semi-empirical—double quartet theory [7], attributing an important role to atom-centeredtetrahedrons and triangles of same-spin electrons when sketching his own spin-resolveddot structures. These early works were picked up by Scemama and coworkers, whoperformed a topological investigation with quantum Monte Carlo [8]. Recently, probability
Molecules 2021, 26, 911. https://doi.org/10.3390/molecules26040911 https://www.mdpi.com/journal/molecules

Molecules 2021, 26, 911 2 of 21
density analysis (PDA) has been introduced [9–12] as the all-electron equivalent of thequantum theory of atoms in molecules (QTAIM) [13,14] focusing on the one-electrondensity. The question arises about what can be gained from all-electron in comparison tothe one-electron methods that can be broadly categorized into electron density methods(e.g., QTAIM, electron localization function (ELF) [15]) and orbital localization methods(e.g., natural bond orbital (NBO) analysis [16]). Concerning electron density methods,many-body information of a chemical system is implicitly contained in the exact groundstate density [17], but its retrieval remains unclear, especially for approximate solutions.Orbital localization methods, on the other hand, lose this information due to the inherentmean-field view. In contrast, all-electron approaches offer an explicit description of many-body effects. However, this comes with the cost of additional complexity.
With PDA, ab initio dot structures are obtained as most likely electron configurations,i.e., local maxima of |Ψ|2. Their number increases rapidly with O(n!), which is mainlycaused by the permutational symmetry of |Ψ|2. These structures often agree with Lewisstructures, and their importance can be evaluated by integrating the probability densityover the respective maxima’s basins of attraction [11]. The resulting probabilities, which canbe obtained for any wave function, have shown to be in good agreement with the weightsof VB theory [12]. As such, these dot structures depict electronic structure informationhidden in the many-particle wave function in condensed form.
In this work, we describe the comprehensive clustering of the local maxima. Thisallows for introducing additional spin information to the representative dot structures of acluster revealing ‘electron motifs’ as characteristic spatial arrangements of spin-coupledelectrons. As such, these motifs are both a generalization of the electron pair and related toLinnett’s tetrahedrons. While the term ‘electron motif’ or ’motif’ has already been usedin the literature to describe arrangements of related electrons found in the electronicstructure [11,18–20], we propose an ab initio definition in this work. Although thesestructures are not intended to replace Lewis dot structures, direct insight into the electronicstructure is obtained from this generally applicable approach.
By systematic investigation of correlated Slater-Jastrow wave functions of 29 mole-cules, we identify at most four characteristic motifs per bond accounting for >99% ofthe total probability. Many of these motifs cannot be reconciled with the Lewis pictureof electron pairs. Whereas the individual probabilities differ significantly from those ofVB or complete active space (CAS) wave functions (due to the poor treatment of staticcorrelation), the shapes of the structures can be deemed highly accurate (due to the explicittreatment of dynamic correlation) for the chosen ansatz. Lastly, we show the interactionbetween motifs in acetic acid and their transferability in dehydroalanine (DHA).
It should be noted that a range of methods has been developed to also connect molec-ular orbital theory with the Lewis picture. These methods are mostly variants of localizedmolecular orbitals [21,22]: natural bond orbitals [23], quasi-atomic orbitals [24], intrinsicbond orbitals [25], and the correlation theory of the chemical bond [26,27]. Furthermore,other real-space analyses have been developed that focus on partitioning the 3-dimensionalspace instead of the full configuration space: QTAIM [13,14], the electron localization func-tion [28], and the electron distribution functions [29,30]. The tiling of the many-electronwave function by Schmidt and coworkers [18] is another topological analysis of |Ψ|2.
2. Methods
In this section, the probability density analysis is described, including the definitionsof electron motifs and ab initio dot structures that have been added in this work.
2.1. The Probability Density
The all-electron probability density p is obtained as the square of the normalized 3n-dimensional electronic wave function Ψ(R) for n electrons: p(R) = |Ψ(R)|2. Its argument

Molecules 2021, 26, 911 3 of 21
is an electron configuration R ∈ R, which is an indexed set of n spinpositions x withindices i ∈ 1 . . . n:
R = (xi). (1)
A spinposition is a tuple of a position vector r ∈ R3 and a magnetic spin quantumnumber ms ∈ −1/2, +1/2:
x = (r, ms). (2)
An electron configuration can be regarded as a sample point from the spatial probabil-ity distribution. Since the electrons are indistinguishable, a permutation of the indices doesnot alter p(R).
2.2. Maxima, Basins of Attraction, and Probabilities
With PDA, the local maxima R∗ of p(R) are investigated. They partition the con-figuration space R into their basins of attraction ΩR∗ [9–12]. As such, a maximum is arepresentative configuration for the entire distribution of electron configurations constitut-ing its surrounding basin. The probability PR∗ of measuring the electron configuration R ina basin of attraction of a certain maximum R∗ is obtained by
PR∗ =∫
ΩR∗p(R)dR. (3)
Note that this purely topological analysis is identical to QTAIM for one-electronsystems (e.g., H +
2 ). While numerical integration of such 3n-dimensional integrals onreal-space grids becomes unfeasible already for small n, it is possible with Monte Carlointegration via importance sampling, which is a standard procedure in any quantum MonteCarlo code.
At this point, the molecule H2 should be briefly discussed. The protons are positionedat (0, 0, HA) and (0, 0, HB). The position vectors of both electrons are at these nuclei posi-tions for all four local maxima R∗; see Figure 1a. Here, it is already apparent that the threeLewis structures of H2 (Figure 1b) are reproduced: there are two ionic maxima (both elec-trons at the same proton) and two covalent maxima (one electron at each proton). Covalentand ionic maxima structures have been subject of discussion in previous works [11,31]. Welabel the two covalent maxima as equivalent, since they differ only in the spin coordinatesand they have equal shape and value due to spin symmetry. In contrast, the ionic maximaare not labeled as equivalent, since their equal shape and value is only due to spatial sym-metry. For a CAS(2,2) wave function in the minimal basis of 1s orbitals, the probabilitiesare 2× 49% for the two covalent and 2× 1.3% for the two ionic maxima [12].
For molecules with heavier atoms, valence electrons in the maxima are no longer atcore positions. While there are still equivalent maxima and qualitatively different maxima,others have very similar overall arrangements, with small displacements of the individualelectrons positions (see Figure A1 in Appendix A). These small differences arise from spinpermutations, the number of which increases with n! [11]. Accordingly, to allow theirchemical interpretation, it is crucial to group equivalent and similar maxima into clusters.

Molecules 2021, 26, 911 4 of 21
R∗covalent
ionic
basin Ω
(a)
HH(97.4%)
HH(1.3%)
HH(1.3%)
(b)
Figure 1. Probability density analysis for a CAS(2,2) wave function of H2. (a) Cross section of the 3n-dimensional probabilitydensity p(R) with both electrons on the z-axis. The two z-coordinates of the electrons are labeled z1 and z2 with the quantumnumbers ms,1 = −1/2 and ms,2 = +1/2. Boundaries of the basins of attraction are indicated by green lines. (b) Sketchesof a covalent (top) and two ionic (mid & bottom) ab initio dot structures. Cluster probabilities and corresponding Lewisstructures are given.
2.3. Clustering of Maxima
A distance d between two spinpositions a and b is defined simply as the differencebetween the two position vectors:
d(a, b) = |ra − rb|. (4)
In order to compare two electron configurations A′ and B, the best-match permutation(permuting A′ to A) is identified [9,10] that minimizes the cost function
∑i
d(ai, bi). (5)
We then denote A as assigned to B. After this assignment, the distance between bothconfigurations is calculated by the metric
D(A, B) = maxi
d(ai, bi) (6)
as the maximal single-electron distance. This metric has two advantages: first, it is indepen-dent on the electron number; second, it discriminates small deviations in many electronsfrom a large deviation in a single electron. Note that other metric choices are possible, buttheir discussion exceeds the scope of this work.
A cluster C is defined as a set of assigned maxima. At the beginning, each maximumis a cluster itself. Two clusters A and B can be merged, if their distance
D(A,B) = minA∈A,B∈B
D(A, B) (7)
is less than a similarity threshold dsim. This distance is the linkage function employed insingle-linkage cluster analysis [32,33]. Once the distances between all pairs of remainingclusters is larger than dsim, the clustering is complete.

Molecules 2021, 26, 911 5 of 21
Then, the cluster probability of a cluster C
PC = ∑R∗∈C
PR∗ (8)
is the total probability of its elements (i.e., its maxima). These probabilities compare well tothe weights of valence bond theory [12].
For larger systems, it is often advantageous to restrict the elements a ∈ A in (5) and (6)to a subset A ⊂ A (i.e., a ∈ A). For instance, the subset A can be defined by the distance ofits elements to the center of a bond, if only this bond is to be investigated. This yields localbasin and cluster probabilities PR∗ and PC , respectively. By means of this ‘local clustering’,the combinatorial explosion of the number of clusters for large molecules is tamed.
If the four maxima of H2 (Figure 1a) are clustered with this metric D and a thresholdless than the proton-proton distance (i.e., dsim < d(HA, HB)), the two covalent maximaare clustered together, while the two ionic maxima remain separated. With this approach,the three Lewis structures are reproduced; see Figure 1b.
For methanol and a threshold of dsim = 0.2 a0 (Bohr radius), two significant clustersare obtained that are depicted in Figure 2.
(a) covalent structure (52%) (b) ionic structure (46%)
Figure 2. Representative maxima of the two most probable clusters in methanol and their cluster probabilities PC obtainedfor dsim = 0.2 a0 . α and β electrons are visualized in semi-transparent red and blue so that coinciding opposite-spin electronsappear as violet. Black lines connect spin-coupled electrons cCij = −1 forming electron motifs. The nucleus positions arerepresented as transparent spheres, bond lines are added for clarity. The insets show the overlay of all assigned maxima ofthe respective cluster. Electrons with the same index have the same color.
Their probabilities sum up to 98%. The remaining 2% are constituted by several minorclusters with low probabilities showing the same motifs at the C O and O H bonds butwith an ionic motif at one or multiple C H bonds. If only the C O bond is of interest,local cluster probabilities of 53% for the covalent and 47% for the ionic cluster are obtained.
The insets in the figures show all maxima within the covalent (Figure 2a) and theionic cluster (Figure 2b). In these insets, the electrons are color-coded according by theirindex and show the positional spread of the maxima, which is a result of opposite-spinpermutations (cf. Figure A1 in Appendix A). It is apparent that the positional differences ofthe lone pair electrons across the maxima of one cluster are much larger than for the bondelectrons, which has already been discussed in previous work for methylamine [11]. Still,all maxima within one cluster are structurally similar.
Based on these observations and with the goal of keeping the overall bias intro-duced to our analysis minimal, we adhere to the following guideline for the choice ofdsim, the single parameter of the proposed clustering: choose dsim so that maxima with

Molecules 2021, 26, 911 6 of 21
different formal charges (i.e., different number of electrons in the vicinity of atoms) areseparated but structurally similar maxima originating solely from spin-permutations areclustered together.
Next, we utilize the spin information contained in the clustered maxima. This willlead to the recovery of the lines connecting the dots in the Lewis structures and beyond.
2.4. Spin Correlation, Electron Motifs, and Ab Initio Dot Structures
After the maxima clustering, a representative maximum R∗C can be chosen for eachcluster (e.g., the one with the highest function value p(R)). Additionally, for each pair ofelectron indices in a cluster C, a spin correlation cCij value can be calculated as the clusteraverage 〈 · 〉C
cCij = 4 〈ms,i ms,j〉C =4
PC∑
R∗∈CPR∗ mR∗
s,i mR∗s,j , (9)
weighted by the probabilities PR∗ and normalized by 4/PC to the interval [−1,+1]. Notethat this definition is equivalent to the statistical correlation for singlet systems, because〈ms,i〉C = 0. Furthermore, the spin correlation is also equivalent to the identically namedquantity in the Ising model [34].
We denote electron indices with cCij = −1 (i.e., pairs in which the spins are alwaysopposite) as ‘spin coupled’. Here, we define a group of spin-coupled electrons as an‘electron motif’. If the motif contains more than two electrons, values of cCij = +1 betweennext-neighboring electrons arise as a consequence. If one spin flips (comparing twomaxima) in such an electron motif, all other spins of the motif flip as well.
The distribution of spin-correlation values cCij over a range of molecules is displayed inFigure A2 in Appendix B and shows values of 0, −1, and +1 with much higher frequencycompared to all other, intermediary values. The latter, partial spin-coupling values areclosely and evenly distributed around cCij = 0 and will be subject of future works.
Finally, and in order to visualize the electron motif information obtained for a cluster ina condensed and comprehensible way, we visualize it in a representative maxima structureR∗C by black lines connecting the spin-coupled electrons (see Figures 1b for H2 and Figure 2for methanol). This finalizes the ab initio dot structure.
3. Computational Approach3.1. Wave Function Generation
Throughout this work, we employed a correlated Slater-Jastrow wave function ansatz.This compact ansatz offers the explicit description of dynamic correlation [35]. Note thatCAS and VB wave functions have recently been studied by two of the authors [12].
Gaussian 16 [36] was used for all density functional theory (DFT) calculations. Allgeometries were optimized with DFT (B3LYP [37–39] with the VWN(III) local correlationenergy [40]) using the aug-cc-pVTZ basis [41]. For the subsequent single-point calculations,each function of the Slater-type triple-ζ TZPae basis [42] was expanded into 14 primitiveGaussian-type functions [43,44]. The Kohn-Sham orbitals for the Slater part of the wavefunction were obtained in this TZPae basis. Note that the basis set dependence of PDAhas not yet been systematically investigated. This dependence is however—in principle—smaller than for non-real-space methods (e.g., valence bond theory), since the resultingwave function alone is investigated. Basis sets that give the same wave function also givethe same PDA results.
Subsequently, an sm444 Schmidt-Moskowitz type Jastrow factor [35,45,46] was addedto the Kohn-Sham determinant and energy minimized [47] with the open-source quantumMonte Carlo package Amolqc [48]. Together with the Slater-type basis set, this ensures acorrect description of two-particle cusps [35,49].

Molecules 2021, 26, 911 7 of 21
3.2. Monte Carlo Integration of the Probability Density
The 3n-dimensional basin probability (3) integrals were calculated by Monte Carlointegration. A sample of size N = 106 was drawn from the probability density p(R) forevery investigated molecule during a standard variational Monte Carlo run [50].
For each sampled electron configuration R, p(R) is locally maximized to identify thebasin ΩR∗ , it is found in. The probability of a local maximum R∗ can then be calculated as
PR∗ = limN→∞
NR∗
N, (10)
where NR∗ is the number of electron configurations found in the basin of attraction ΩR∗ .
3.3. Local Maximization
Instead of maximizing p, − ln p is minimized. Note that − ln p(R) decreases mono-tonically with p(R). Therefore, it has the same basins and critical points as the probabilitydensity, but its minimization is more stable.
A two-phase minimization algorithm was employed. In the first phase (during alatency period), a step-length limited steepest descent algorithm is used to ensure thelocality of the minimization. To avoid overshooting at the nuclear singularities, we employa correction in the vicinity of nuclei [51]. The latency period is reset each time an electronarrives at a singularity. After the latency period, we smoothly switch to an L-BFGS-Balgorithm [52], which moves the remaining valence electrons to their final positions in themaximum. The sampling of p(R), as well as the local minimization of the sampled electronconfigurations, were also conducted with Amolqc [48].
3.4. Clustering and Spin-Correlation Analysis
Clustering and spin-correlation analysis was conducted with the open-source softwarepackage inPsights [53].
The assignment problem (i.e., minimizing the cost function in (5)) is solved by theHungarian algorithm [54] in cubic time O(n3) [55]. If all N (potentially identical) maximastructures were compared with each other, the number of comparisons would grow withO(N2). To allow for sample sizes of N = 106, a pre-clustering strategy was employed toquickly reduce the number of structures N′ ≈ 101 to 103. For this, maxima were sorted bytheir function value and a greedy, spherical pre-clustering algorithm was employed witha sphere radius of r = 0.01 a0. The algorithm is described in Appendix C. The maximaclustering calculations were conducted on a standard desktop computer with a single CPU.Computation times ranged from minutes to several hours depending on the number ofelectrons and the complexity of the |Ψ|2 topology.
After the pre-clustering, we employed the DBSCAN clustering algorithm [56,57],where we set the minimal cluster size minPts = 1 to conduct single-linkage clustering.The dsim value was selected in the range of 0.1 a0 to 0.4 a0 with 0.2 a0 being the default.Acetic acid and DHA were clustered locally. All other molecules (i.e., molecules shown inTables 1–4) have been clustered considering all electrons, and marginal probabilities wereobtained by subsequent summation.
Infrequently, and for symmetrical molecules, spatially similar maxima are encounteredthat are exactly transformable into each other by rotation or reflection but for which a differ-ent distance-based assignment is found. If this different and arbitrary assignment leads toa spin flip, spin-correlation information is lost. In these cases, and to prevent this arbitraryassignment, the clustering dsim was decreased, and the clusters were assigned manually.

Molecules 2021, 26, 911 8 of 21
Table 1. Covalent ab initio dot structures found for homo- and heteroatomic single bonds between the elements H, B, C, N, O, and F with their associated local cluster probabilities inparentheses. Motifs of spin-coupled electrons within are indicated by black lines.
H B C N O F
H
1c: H H (96.5%)
H
H
2c: H BH2 (91.4%)
H
HH
3c: H CH3 (97.4%)
HH
4c: H NH2 (99.5%)H
5c: H OH (98.7%) † 6c: H F (43.9%)
B H
H
H
H
7c: H2B BH2 (99.7%)
H
HHH
H
8c: H2B CH3 (99.9%)
CH
HH H
HH
12c: H3C CH3 (99.9%)H
HH
HH
13c: H3C NH2 (81.9%)H
HHH
14c: H3C OH (52.6%)H
HH
15c: H3C F (0.3%)
NH
H
HH
16c: H2N NH2 (99.4%)
HH
H
17c: H2N OH (70.1%)
HH
18c: H2N F (53.1%)
OH
5c′: H OH (1.1%) †
H
H
19c: HO OH (60.6%) †
H
20c: HO F (58.0%) †
F
6c′ : H F (6.7%)
H
HH
15c′ : H3C F (0.3%)
HH
18c′ : H2N F (1.1%) 21c: F F (60.3%)
† extended correlations with O H electrons.

Molecules 2021, 26, 911 9 of 21
Table 2. Ionic ab initio dot structures found for homo- and heteroatomic single bonds between the elements H, B, C, N, O, and F with their associated local cluster probabilities inparentheses. Motifs of spin-coupled electrons within are indicated by black lines.
H B C N O F
H
1i: H H (2× 1.8%)
H
H
2i H BH2 (8.5%)
H
HH
3i: H CH3 (2.1%)
HH4i: H NH2 (0.5%) 6i: H F (49.4%)
B H
H
HH9i: H2B NH2 (99.9%) †
H
H
H10i: H2B OH (99.9%)
H
H
11i: H2B F (100%)
CH
HH
HH13i: H3C NH2 (18.0%)
H
HH H
14i H3C OH (47.4%)
H
HH
15i: H3C F (99.3%)
NH
H
HH16i: H2N NH2 (2× 0.1%) †
HH
H17i: H2N OH (29.8%) ‡
HH
18i: H2N F (45.9%)
O
H
H
19i: HO OH (2× 18.7%)
H
20i: HO F (32.2%) ‡
FH
20i’: HO F (9.8%) 21i: F F (2× 19.9%)
† extended correlations with N H electrons; ‡ extended correlations with O H electrons.
Molecules 2021, 26, 911 10 of 21
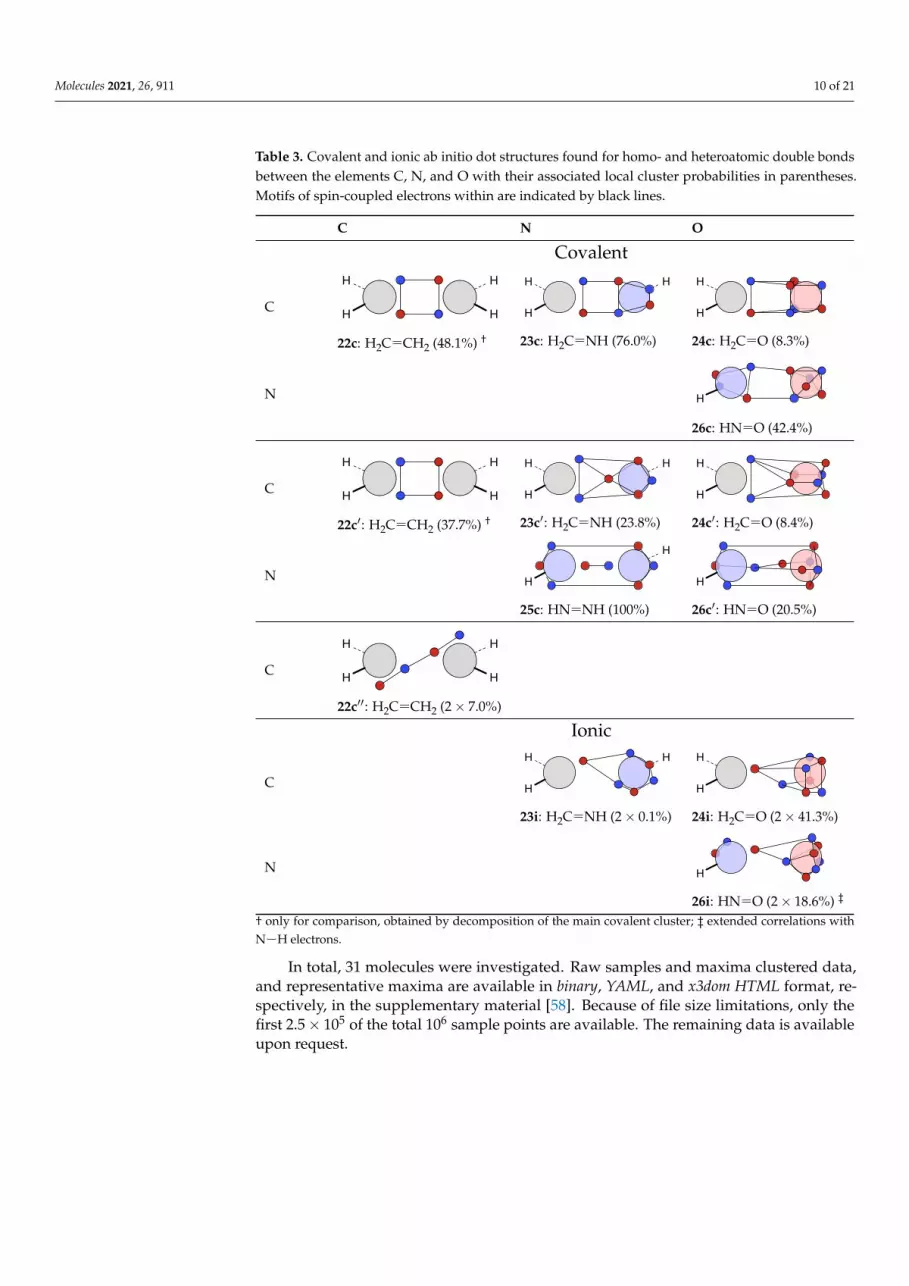
Table 3. Covalent and ionic ab initio dot structures found for homo- and heteroatomic double bondsbetween the elements C, N, and O with their associated local cluster probabilities in parentheses.Motifs of spin-coupled electrons within are indicated by black lines.
C N O
Covalent
C
H
H
H
H
22c: H2C CH2 (48.1%) †
HH
H
23c: H2C NH (76.0%)
H
H
24c: H2C O (8.3%)
N H
26c: HN O (42.4%)
C
H
H
H
H
22c′: H2C CH2 (37.7%) †
H
H
H
23c′: H2C NH (23.8%)
H
H
24c′: H2C O (8.4%)
N H
H
25c: HN NH (100%)
H
26c′: HN O (20.5%)
C
H
H
H
H
22c′′: H2C CH2 (2× 7.0%)
Ionic
C
HH
H
23i: H2C NH (2× 0.1%)
H
H
24i: H2C O (2× 41.3%)
N H
26i: HN O (2× 18.6%) ‡
† only for comparison, obtained by decomposition of the main covalent cluster; ‡ extended correlations withN H electrons.
In total, 31 molecules were investigated. Raw samples and maxima clustered data,and representative maxima are available in binary, YAML, and x3dom HTML format, re-spectively, in the supplementary material [58]. Because of file size limitations, only thefirst 2.5× 105 of the total 106 sample points are available. The remaining data is availableupon request.

Molecules 2021, 26, 911 11 of 21
Table 4. Covalent and ionic ab initio dot structures found for homo- and heteroatomic triple bondsbetween the elements C and N with their associated local cluster probabilities in parentheses. Motifsof spin-coupled electrons within are indicated by black lines.
C N
Covalent
CH H
27c: HC CH (78.5%) †
H
28c: HC N (72.8%)
N
29c: N N (92.4%)
CHH
27c′: HC CH (0.1%) †
H
28c′: HC N (0.2%)
N
29c′: N N (0.7%)
CH H
27c′′: HC CH (15.5%)
H
28c′′: HC N (5.9%)
N
29c′′: N N (6.9%)
CH H
27c′′′: HC CH (5.8%)
Ionic
CH
28i: HC N (20.9%)† only for comparison, obtained by decomposition of the main covalent cluster.

Molecules 2021, 26, 911 12 of 21
4. Results and Discussion
First, a systematic study of the electron motifs found in the chemical bonds of smallmolecules including the elements H, B, C, N, O, and F is conducted. Second, we discuss themotif interaction in the carboxyl group of acetic acid. Lastly, the transferability of motifs isshown for the DHA molecule.
The probabilities presented are always local cluster probabilities for the depicted bond.These are either obtained directly from local clustering (cf. the discussion of methanol inSection 2.3) or by subsequent summation of cluster probabilities obtained from global clus-tering. Instead of the electron configuration of the representative maximum (cf. Figure 2),a schematic ab initio dot structure is shown for every cluster. Again, the electron positionsare colored (blue and red) with respect to the ms quantum number. Yet, this coloring onlyhas an implication within an electron motif of spin-coupled electrons (connected by blacklines) but not between different electron motifs. For any depicted electron motif, all colorscould be exchanged without changing the information of the structure.
4.1. Single Bonds
Small molecules exhibiting single bonds between the elements H, B, C, N, O, and Fhave been investigated. Based on the electron distribution among the two nuclei, covalent(c) and ionic structures can be identified and are represented by the ab initio dot structuresin the Tables 1 and 2, respectively. For the empty spots in the upper triangle of the Table 1,only ionic maxima are found, and vice versa.
For the series of homoatomic single bonds in dihydrogen (H H), diborane(4) D2d(H2B BH2), ethane (H3C CH3), hydrazine (H2N NH2), hydrogen peroxide (HO OH),and difluorine (F F), as expected, we find dominantly covalent structures 1c, 7c, 12c, 16c,19c, and 21c with high probabilities. Only for hydrogen peroxide and difluorine, ionic struc-tures (19i, 21i) have a significant probability, as well. This strong ionic contribution may berelated to the charge-shift character of these bonds, i.e., a large covalent–ionic resonanceinteraction energy in VB theory [59]. For heteroatomic single bonds, the probabilities of theionic structures follow the trend which is expected, given the bond polarities derived fromelectronegativity (see the columns and rows of Table 2). Note that the dominance of thecovalent structure is due to the correlated nature of the wave function. With a Hartree-Fockwave function, very similar electron positions are obtained for the homoatomic singlebonds but with an independent distribution of the two opposite spin electrons on the twopositions due to the lack of explicit Coulomb repulsion. Half the maxima, therefore, havetwo electrons at the same position, which is unphysical (except at the nuclei) and results inthe typical ionicity of 50% in Hartree-Fock.
While all polarity trends are described well, a wrong bond polarity is found for a fewmolecules: for methane (CH4) and ammonia (NH3), hydridic (i.e., H–) structures are found(4i and 5i) but not the expected protonic (i.e., H+) ones. This discrepancy is caused bythe poor treatment of static correlation in the wave function ansatz. Earlier work [12] hasshown that the probabilities are more reliable for VB and CAS wave functions. However,this work focuses on the shape of the structures, which is reliably predicted.
The dominant covalent structures of the single bonds are characterized by an electronpair shared between two atomic cores (see, for example, 12c in Figure 1). The respectiveprobability density maxima show one electron position outside each atomic core in directionto the other core. In contrast to the two core electron positions, this ‘bond position’ isdisplaced due to Pauli repulsion to the same-spin core electron and the Coulomb attractionto the second core (as discussed in detail in previous work [11]). Since hydrogen has nocore electrons, the maximum position is always located directly at the nucleus, like in H2as discussed in Section 2.2. The electrons identified at the two bond positions always haveopposite spin. Accordingly, the electron pair arises naturally as an electron motif.
Yet, the ab initio dot structures extend beyond the Lewis picture for several reasons.First, the ‘lone pair’ positions are all spin coupled for and cannot be divided into electronpairs without arbitrariness. Instead, they form intuitively understandable geometrical

Molecules 2021, 26, 911 13 of 21
shapes avoiding other electrons (especially those of same spin) due to Coulomb and Paulirepulsion. Because they are all spin coupled, they form an electron motif that we willdenote as ‘lone motif’ in the following. Second, there are single bonds, where a lone motifis additionally spin coupled to the bonding electron pair (17c, 18c, 5c, 14c). Thus, there isa larger electron motif, which includes the lone positions, as well as the bond positions.However, the distinction between bond electrons and lone-pair electrons can still easilybe made. Last, this is no longer the case for the minor covalent structures (5c′, 6c′, 15c′′
and 18c′) found mostly for bonds with fluorine. Here, an electron pair on the bond can nolonger be identified. Instead, the border between bond and lone motif is blurred. Thesestructures may be compared to inverted bonds discussed in VB theory [60,61].
In the following, individual covalent structures will be discussed in more detail. Forhydrazine (16c), and similarly for methylamine (H3C NH2, 13c), and hydroxylamine(H2N OH, 17c), the lone pair electrons are aligned with the depicted bond. For ammonia(4c), the lone pair is aligned to any of the three N H bonds. For water (5c), hydroxylamine(17c), and hydrogen peroxide (19c), the lone motif is also coupled with the bond motifon the O H bond, which is not explicitly shown and indicated by a dagger in Table 1.For water and methanol, the bond pair is tilted off the bond axis, which has already beenobserved by Scemama et al. [8].
In the difluorine molecule (21c), the lone motif at each atom arranges in a hexagon ofalternating spins. In the depicted ab initio dot structure, the lone motif electron positionsare not in one plane. Instead, the electrons with same spin to the next bond electronare always further away from the bond, which is easily explained with Pauli repulsion.However, this system is a good example to discuss the impact of the clustering thresholddsim: because of the discussed distortion of the hexagon, a spin coupling is predictedbetween the lone motif and the bond pair for very small dsim. However, we chose a valuefor dsim, which is comparable to that for other systems, and treat the distortion of thehexagon like the small displacements discussed for methanol (Figure A1 in Appendix A).
Other ab initio dot structures of single bonds can clearly be identified as ionic struc-tures by the distribution of electrons to the cores (Table 2). For the homoatomic systems,only one of the two ionic structures is depicted. For all of the depicted dot structures,the ionic bond pair is always spin coupled to the lone motif and integrated into its geom-etry, forming a larger electron motif. Ionic fluorine structures (e.g., in hydrogen fluoride(H F, 6i) always show a cube motif, which is only slightly distorted and composed of twosame-spin tetrahedrons. As discussed previously for the neon atom [11], this fits perfectlyto both the cubic atom proposed by Lewis [1] and the double quartet theory by Linnett [7].
For first row atoms, there are two possible orientations for the ionic lone pair motifs:if an edge is directed towards the bond (e.g., for aminoborane (H2B NH2, 9i) and fluo-romethane (H3C F, 15i), a bond pair could still be identified. In this regard, the Lewispicture is recovered. This is no longer the case, if a vertex is directed towards the bond,e.g., for borinic acid (H2B OH, 10i) and fluoroborane (H2B F, 11i), as found only forboron- and hydrogen-containing bonds.
In summary, motifs found at single bonds are limited to at most three and exhibitclear structural regularities. Covalent structures are characterized by a bond pair that canbe coupled to lone motifs, where individual electron pairs cannot be identified. In ionicstructures, an additional pair of electrons is incorporated into a present lone motif. Thiswill be different for multiple bonds, where increased spin-coupling and structural varietyis observed.
4.2. Multiple Bonds
Small molecules exhibiting double and triple bonds between the elements C, N,and O have been investigated. Again, covalent and ionic structures are identified; seeTables 3 and 4.
Whereas the position of the bond pair electrons could be rationalized for single bondsby a combination of Coulomb force and Pauli repulsion, additional electrons complicate

Molecules 2021, 26, 911 14 of 21
the picture. In multiple bonds, there are two ways to accommodate these electrons. First,they can be added to the single bond structure, so that the bond pair motif stays relativelyunchanged. Second, all electron positions could be moved from the bond axis, so thatthere are more equivalent bond positions. Whereas the former is related to the σ-π pictureof multiple bonds, the latter fits to the τ picture, where the bond is decomposed intoequivalent ‘banana bonds’. These equivalent bonds fit best with the Lewis dot structuresand are also obtained as localized molecular orbitals with Edminston-Ruedenberg orFoster-Boys localization of molecular orbitals [62–64].
At first, the carbon-carbon bonds in ethylene (H2C C2H) and acetylene (HC CH) arediscussed. For both systems, a covalent structure is dominant; see Figure 3.
H
H
H
H
(a) ethylene (85.8%)
H H
(b) acetylene (78.6%)Figure 3. Ab initio dot structures of the most probable clusters of ethylene and acetylene with theircluster probabilities.
As discussed in earlier work [11,12], these structures fit with the τ picture. This is nolonger case the for the other systems in Tables 3 and 4. To compare the double bond inethylene to other double bonds, the main covalent cluster (Figure 3a) is divided into twoclusters, so that the two two-electron motifs are replaced with a rectangular four-electronmotif. This decomposition, which is again done only for comparison, leads to the twostructures with alternating spins (22c) and blocked spins (22c′). The same is done foracetylene: the main covalent structure (Figure 3b) is decomposed into 27c and 27c′.
For ethylene, we find two symmetric, diagonal structures (22c′′) in addition to theone presented in Figure 3a. These structures are in good agreement with VB theory for τbonds, which has already been discussed in earlier work [12]. Alternatively, they could bediscussed as a modified single bond structure, where the additional electrons are placedclose to the atoms and the single bond is only tilted off the bond axis.
For the double bond in methanimine (H2C NH), a rectangular bond motif (23c) isfound without decomposition. It is coupled with the lone motif to an extended motifin which the spins are alternating. For nitroxyl (HN O), a rectangular motif (26c) isalso recovered, albeit distorted and also coupled with the lone motifs on both atoms.Formaldehyde (H2C O) is dominated by an ionic structure (24i) reflecting the polarity ofthe bond but shows a variation of the rectangular motif (24c) with two edges of a triangularprism of six electrons on O directed towards a pair of electrons from C as a covalent motif.In this structure, the identification of four bond electrons is no longer possible. Thus, inthis aspect, it is comparable to the minor covalent structures (c′) found for single bonds.
The single structure for (E)-diimine (HN NH, 25c) can be related to both the rectan-gular motif and the diagonal motif: it can be described as a widened rectangle, to which acentral bond motif has been added. Alternatively, it is a diagonal motif, to which to lonepairs have been added. Note that the observed spin coupling does not justify the latter.The minor covalent structure for methanimine (23c′) is a combination of the ethylene andthe (E)-diimine structures 22c′ and 25c. In formaldehyde, the second covalent motif (24c′)is best described as a tetrahedron of electrons between the atoms coupled to four lonepair electrons.
The ab initio dot structures of acetylene, dinitrogen (N N), and hydrogen cyanide(HC N) are compared in Table 4. The larger number of bond electrons leads to electronarrangements around the favorable maximum positions along the bond axis or to electronsat or near these positions with other spin-coupled electrons.
For the lone-pair free acetylene, regular, triangular prism motifs (27c, 27c′) occur, whenthe dominant covalent structure (Figure 3b) is decomposed. Additionally, a triangular

Molecules 2021, 26, 911 15 of 21
antiprism (27c′′) and a rectangular bond motif, extended by two additional electrons to achair-like six-electron motif (26′′′), are found.
For hydrogen cyanide and dinitrogen, distorted (anti)prisms with one (28c′, 28c′′) ortwo electrons (29c′, 29c′′) at or near the single bond maximum positions along are observed,respectively. The dominant motif in dinitrogen (29c) has two electrons near the bondmaximum positions forming the tips of two pyramids of alternating spin. The dominantmotif in hydrogen cyanide (28c) is again a combination of the acetylene prism motif (27c)and the dominant dinitrogen motif (29c).
For hydrogen cyanide, a wedge-like ionic motif (28i) similar to that in formaldehyde(24c) in Table 3 is found. Again, the probability of finding the ionic motifs correlates withthe electronegativity difference.
In summary, and as for the single bonds, a small variety of only three and foursignificant motifs are found per double and triple bond, respectively, for the investigatedmolecules. Furthermore, clear structural regularities and relationships could be identified,thus making them characteristic for the respective bond type. In contrast to the singlebond motifs, the multiple bond motifs cannot be reconciled with the Lewis picture, as soonas lone pair electrons are involved. In this case, and with only one exception, all valenceelectrons are spin-coupled.
Overall, characteristic motifs were found for the two-center bonds investigated. Next,motif interactions at neighboring bonds are investigated at the example of acetic acid.
4.3. Motif Interaction in the Carboxyl Group of Acetic Acid
The electronic structure of molecules is predominantly local in the sense that functionalgroups exist with very similar properties in very different molecules. This locality is welldescribed by the electron density for parts of the molecule [14], by localized molecularorbitals and by VB theory. Here, we can show that a many-electron view to locality ispossible with the probability density analysis. In many cases, the ab initio dot structuresfound in small molecules containing a single functional group are directly transferable tomolecules with several functional groups where they appear with comparable probabilities.In other cases, the structures are found with significantly altered probabilities, while somemotifs or motif combinations are not found at all.
The carboxyl group (COOH) in acetic acid (H3C COOH) can be thought of as ahydroxy group (O H) adjacent to a carbonyl group (C O). Yet, chemically, the carboxylgroup differs substantially from alcohols and ketones. A substantial alteration of themaxima is, therefore, expected. For the carbonyl group in acetic acid, we recover the ionicdouble bond motif of formaldehyde 24i and the covalent double bond motif 24c′, but wealso find a distorted variant 24i′ of the ionic double bond motif 24i with two electronsalong the C O bond axis and six electrons arranged in a hexagon around the O nucleus.This motif is known from fluoromethane (15c; see Table 1).
The ionic motifs are found with a probability of 49% for 24i and 46% for 24i′ comparedto 83% for 24i in formaldehyde. The covalent motif 24c′ is recovered with 5% compared to8% in formaldehyde. The covalent motif 24c is found with < 1% in acetic acid compared to8% in formaldehyde.
For the C O H group, the covalent and ionic C O motifs (14c and 14i) are identified,both combined with a covalent O H bond motif (5c). In this context, these two C O Hstructures will be denoted 14c-5c and 14i-5c; see Figures 4a,b.
(a) 14c-5c (b) 14i-5c (c) 14c-5i (d) 14i-5iFigure 4. Combinations of C O and O H structures found at the C O H of acetic acid.

Molecules 2021, 26, 911 16 of 21
Two new structures arise for the C O H group in acetic acid, both with ionic O Hgroups (i.e., with no electron at the proton). The first motif combines a covalent C O partcoupled with lone pairs and an ionic O H part, denoted 14c-5i, while the other is ionicboth in C O and O H (14i-5i); see Figures 4c,d. The combined probability of finding14c-5i or 14i-5i is 43% (see Table 5), while ionic O H maxima are not found in water ormethanol at all.
Table 5. Local probabilities of all significant (P > 1%) electronic motifs combinations in acetic acid.
C O H C O PC /%
14i-5c 24i′ 3014c-5i 24i 2614c-5c 24i 2214i-5i 24i′ 1614i-5c 24c′ 4
The substantially increased ionicity of the structures reflects the increased polarity ofthe O H bond, and thus acidity, in acetic acid, compared to water and alcohols. A shift toionic motifs is also found for the C O group. The covalent C O motif 14c is found with53% in methanol compared to 49% (14c-5c and 14c-5i) in acetic acid and the ionic motif 14iwith 47% in methanol and with 51% in acetic acid (14i-5c and 14i-5i); see Table 5.
In addition to new motifs found in the carboxyl group, we find that the C O andC O H motifs are not statistically independent. We do not find a spin coupling to specificcarboxyl motifs like the spin coupling of lone pair electrons to bond electrons but we dofind strong statistical coupling between the motifs. In Table 5, all observed combinationsof motifs and their probabilities are listed. The covalent C O motif 24c′ is found onlycombined with the ionic C O H motif 14i-5c, and the new motif 24i′ coexists only withthe ionic C O H motifs 14i-5c and 14i-5i.
With PDA, the carboxyl group in acetic acid is obtained as a combination of C O Hand C O motifs, in which its probabilities are substantially altered in comparison withH3C O H and H2C O. The motif probabilities show a strong shift towards ionic motifs.Furthermore, the C O H and C O motifs are not independent but strongly correlated.These findings correlate well with chemical and physical properties, such as acidity andinfrared group frequencies of the carboxyl group, in comparison to free hydroxyl andketone groups [65].
4.4. Motif Transferability in Dehydroalanine
The DHA molecule depicted in Figure 5 contains a C C double bond, a C N singlebond, and a carboxyl group.
Version December 23, 2020 submitted to Journal Not Specified 16 of 22
(a) 14c-5c (b) 14i-5c (c) 14c-5i (d) 14i-5iFigure 4. Combinations of C O and O H structures found at the C O H of acetic acid.
C O H C O PC / %
14i-5c 24i0 3014c-5i 24i 2614c-5c 24i 2214i-5i 24i0 1614i-5c 24c0 4
Table 5. Marginal probabilities of all significant (P > 1 %) electronic motifs combinations in acetic acidwith.
For the C O H group, the covalent and ionic C O motifs (14c and 14i) are identified, both381
combined with a covalent O H bond motif (5c). In this context, these two C O H structures will be382
denoted 14c-5c and 14i-5c, see Figure 4. Two new structures arise for the C O H group in acetic acid,383
both with ionic O H groups (i.e. with no electron at the proton). The first motif combines a covalent384
C O part coupled with lone pairs and an ionic O H part, denoted 14c-5i, while the other is ionic both385
in C O and O H (14i-5i), see Figure 4. The combined marginal probabilty of finding 14c-5i and 14i-5i386
is 43 % (see Table 5), while ionic O H maxima are not found in water or methanol at all.387
The substantially increased ionicity of the structures reflects the increased polarity of the O H388
bond, and thus acidity, in acetic acid, compared to water and alcohols. A shift to ionic motifs is also389
found for the C O group. The covalent C O motif 14c is found with 53 % in methanol compared to390
49 % (14c-5c + 14c-5i) in acetic acid and the ionic motif 14i with 47 % in methanol and with 51 % in391
acetic acid (14i-5c and 14i-5i), see Table 5.392
In addition to new motifs found in the carboxyl group, we find that the C O and C O H motifs393
are not statistically independent. In Table 5 all observed combinations of motifs and their marginal394
probabilties are listed. The covalent C O motif 24c0 is found only combined with the ionic C O H395
motif 14i-5c, and the new motif 24i0 coexists only with the ionic C O H motifs 14i-5c and 14i-5i.396
With PDA, the carboxyl group in acetic acid is obtained as a combination of C O H and C O397
motifs whose marginal probabilities are substantially altered in comparison with H3C O H and398
H2C O. The motif probabilities show a strong shift towards ionic motifs. Furthermore, the C O H399
and C O motifs are not independent but strongly correlated. These findings correlate well with400
chemical and physical properties such as acidity and infrared group frequencies of the carboxyl group401
in comparison to free hydroxyl and ketone groups.[62]402
How to cite multiple pages?403
4.4. Motif Transferability in Dehydroalanine404
The dehydroalanine molecule405
H2N
C
CH2
C
O
OH406
contains a C C double bond, a C N single bond, and a carboxyl group in the maxima. The electron407
motifs of all three groups and their statistical correlations are investigated.408
Figure 5. Dehydroalanine.
The electron motifs of all three groups and their statistical correlations are investigated.For DHA, the number of maxima and also the number of maxima clusters is substan-
tially increased in comparison to the small molecules discussed above. However, it is notnecessary to obtain and analyze all maxima clusters. Instead, we employ local clustering toidentify the motifs and their probabilities for each functional group. Statistical correlationsbetween neighboring functional groups can be analyzed by simultaneous local clusteringof two functional groups. This way, large numbers of maxima clusters do not occur at anystage of the analysis, and the size of the molecules to be investigated is limited only by thepossibility of running a variational Monte Carlo calculation.

Molecules 2021, 26, 911 17 of 21
For the individual functional groups, the same motifs are found as in the correspond-ing environments in the smaller molecules ethylene, methylamine, and acetic acid. Incontrast to the discussion of the carboxyl group, no additional motifs arise. This indicatesthat the electronic motifs are indeed characteristic for functional groups.
Whereas the electron motifs of the smaller molecules are recovered in DHA, the in-dividual probabilities of the motifs shift significantly. In Table 6, the probabilities for themotifs in DHA are compared with those of the corresponding smaller molecules.
Table 6. Local probabilities of motifs in dehydroalanine (DHA) compared to simple molecules (SM).
Group Motif PC(DHA)/% PC(SM)/%
C C 22c or 22c′ 93 86 a
C C 22c′′ 6 14 a
C N 13c 73 82 b
C N 13i 27 18 b
COOH 14i-5c and 24i′ 43 30 c
COOH 14c-5i and 24i 25 26 c
COOH 14c-5c and 24i 11 22 c
COOH 14i-5i and 24i′ 19 16 c
COOH 14i-5c and 24c′ 3 4 c
a ethene, b methylamine, c acetic acid.
These probability changes, once more, indicate an influence of the environment onthe electronic structure in a functional group and deserve further investigation whichis beyond the scope of this paper. Contrary to the situation in acetic acid, no statisticalcorrelation between individual motifs of any two functional groups is observed.
5. Conclusions
In this work, the probability density analysis based on the local maxima of the squaredwave function was presented. The existing analysis was expanded by a comprehensiveclustering of these maxima, which allows for the definition of a spin correlation and thegeneralization of the electron pair to the electron motif. As an alternative to the empiricalLewis structures, as well as to the semi-empirical Linnett structures, ab initio dot structureshave been introduced as the representation of a cluster and its spin information. Theseab initio structures have been presented and analyzed for single, double, and triple bondsin small first row molecules. For single bonds, the ionic-covalent resonance of VB theorycan be reproduced (covalent and ionic structures are identified), and the results mostly fitwith the Lewis picture. However, the concept of distinct lone pairs cannot be reconciledwith the findings by probability density analysis, so that they have been replaced with alone motif. For multiple bonds, the agreement with the Lewis picture vanished for manysystems, and extended electron motifs occur due to the coupling of the bond electrons withthe lone motifs. In general, only very few motifs are found for each bond or functionalgroup. In acetic acid, the motifs of the C O H group in methanol and the C O groupin formaldehyde are recovered, but additional motifs arise. Furthermore, the motifs ofthe two groups are strongly statistically correlated, reaffirming the status of the carboxylfunction as an independent functional group from an electronic structure perspective. InDHA, on the other hand, the motifs of the three functional groups are very similar to themotifs of the corresponding smaller molecules, and no significant statistical correlation isfound. This demonstrates the transferability of electron motifs corresponding to individualfunctional groups. Furthermore, the limited spatial extent of the motifs reflects the localityof chemistry at the level of real space many-electron analysis. This allows a probabilitydensity analysis for large molecules, in spite of the exponentially increasing number oflocal maxima, by employing a local clustering focused on only one or two bonds.
Author Contributions: Conceptualization, M.A.H., L.R., and A.L.; investigation, M.A.H.; software,M.A.H.; resources, A.L.; writing—original draft preparation, M.A.H.; writing—review and editing,

Molecules 2021, 26, 911 18 of 21
A.L., L.R.; visualization, M.A.H.; supervision, A.L. All authors have read and agreed to the publishedversion of the manuscript.
Funding: This research received no external funding.
Institutional Review Board Statement: Not applicable.
Informed Consent Statement: Not applicable.
Data Availability Statement: The data presented in this study are openly available in the OpenScience Framework at 10.17605/OSF.IO/6Z2J9, reference number 6Z2J9.
Acknowledgments: The authors thank Julian Hüffel for performing preliminary calculations.
Conflicts of Interest: The authors declare no conflict of interest.
Appendix A. Structural Similarity
Figure A1. Two similar maxima of methanol. The flipped spins at the CH3 group lead to smallpositional shifts of the valence electrons.
Appendix B. Spin-Correlation Distribution
Figure A2. Histogram with 101 bins of spin-correlation values cCij calculated from all clusters of the
molecules 1 to 29. Same-spin correlation values (cCij = +1) were only found within motifs composedof more than two electrons.
Appendix C. Pre-Clustering
To reduce the number of comparisons, we utilize that similar maxima also havesimilar probability density values p. Thus, by sorting the maxima and comparing onlythose within a window of values [− ln p,− ln p + ∆), the number of comparisons is oftendrastically reduced. We employ this in a spherical greedy pre-clustering algorithm, wherewe start with the first maximum as a cluster center and compare it with all maxima fromthe window. Maxima within a similarity sphere of radius r are then clustered together. The

Molecules 2021, 26, 911 19 of 21
first maximum being outside of r constitutes the center of a new cluster sphere to whichall subsequent maxima from the window must be compared. This is then repeated for allvalue windows. This algorithm is greedy because the creation of spherical cluster centers israndom and depends on the order of maxima. As long as the sphere radius is chosen withr ≤ dsim/2, the maxima in the pre-cluster spheres will be merged into the same clustersduring the main-clustering, as they would have been without the pre-clustering step.
References1. Lewis, G.N. The atom and the molecule. J. Am. Chem. Soc. 1916, 38, 762–785. [CrossRef]2. Heitler, W.; London, F. Wechselwirkung neutraler Atome und homöopolare Bindung nach der Quantenmechanik. [Interaction of
neutral atoms and homopolar bond according to quantum mechanics]. Z. Phys. 1927, 44, 455–472. [CrossRef]3. Pauling, L. The nature of the chemical bond. Applications of results obtained from the quantum mechanics and from a theory of
paramagnetic susceptibility to the structure of molecules. J. Am. Chem. Soc. 1931, 53, 1367–1400. [CrossRef]4. Artmann, K. Zur Quantentheorie der gewinkelten Valenz, I. Mitteilung: Eigenfunktion und Valenzbetätigung des Zentralatoms.
[On the quantum theory of angled valence, 1st report: Eigenfunctions and valence activity of the central atom]. Z. Naturforsch. A1946, 1, 426–432. [CrossRef]
5. Zimmerman, H.K.; Van Rysselberghe, P. Directed valence as a property of determinant wave functions. J. Chem. Phys. 1949,17, 598–602. [CrossRef]
6. Waitz, M.; Bello, R.Y.; Metz, D.; Lower, J.; Trinter, F.; Schober, C.; Keiling, M.; Lenz, U.; Pitzer, M.; Mertens, K.; et al. Imaging thesquare of the correlated two-electron wave function of a hydrogen molecule. Nat. Commun. 2017, 8, 2266. [CrossRef]
7. Linnett, J.W. A modification of the Lewis–Langmuir octet rule. J. Am. Chem. Soc. 1961, 83, 2643–2653. [CrossRef]8. Scemama, A.; Caffarel, M.; Savin, A. Maximum probability domains from quantum Monte Carlo calculations. J. Comput. Chem.
2007, 28, 442–454. [CrossRef] [PubMed]9. Lüchow, A.; Petz, R. Single electron densities: A new tool to analyze molecular wavefunctions. J. Comput. Chem. 2011,
32, 2619–2626. [CrossRef]10. Lüchow, A.; Petz, R. Single Electron Densities from Quantum Monte Carlo Simulations. In Advances in Quantum Monte Carlo;
American Chemical Society: Washington, DC, USA, 2012; pp. 65–75. [CrossRef]11. Lüchow, A. Maxima of |Ψ|2: A connection between quantum mechanics and Lewis structures. J. Comput. Chem. 2014, 35, 854–864.
[CrossRef]12. Reuter, L.; Lüchow, A. On the connection between probability density analysis, QTAIM, and VB theory. Phys. Chem. Chem. Phys.
2020, 22, 25892–25903. [CrossRef]13. Bader, R.F.W.; Beddall, P.M. Virial field relationship for molecular charge distributions and the spatial partitioning of molecular
properties. J. Chem. Phys. 1972, 56, 3320–3329. [CrossRef]14. Bader, R.F.W. A quantum theory of molecular structure and its applications. Chem. Rev. 1991, 91, 893–928. [CrossRef]15. Becke, A.D.; Edgecombe, K.E. A simple measure of electron localization in atomic and molecular systems. J. Chem. Phys. 1990,
92, 5397–5403. [CrossRef]16. Reed, A.E.; Weinhold, F. Natural bond orbital analysis of near-Hartree–Fock water dimer. J. Chem. Phys. 1983, 78, 4066–4073.
[CrossRef]17. Hohenberg, P.; Kohn, W. Inhomogeneous Electron Gas. Phys. Rev. 1964, 136, B864–B871. [CrossRef]18. Liu, Y.; Frankcombe, T.J.; Schmidt, T.W. Chemical bonding motifs from a tiling of the many-electron wavefunction. Phys. Chem.
Chem. Phys. 2016, 18, 13385–13394. [CrossRef] [PubMed]19. Hutchison, J.R.; Rzepa, H.S. Foliacenes: Ab Initio Modeling of Metallocomplexes Exhibiting a Unique Form of 16-Electron,
Metal-Induced Aromaticity. J. Am. Chem. Soc. 2004, 126, 14865–14870. [CrossRef] [PubMed]20. Peart, P.A.; Repka, L.M.; Tovar, J.D. Emerging Prospects for Unusual Aromaticity in Organic Electronic Materials: The Case for
Methano[10]annulene. Eur. J. Org. Chem. 2008, 2008, 2193–2206. [CrossRef]21. Lennard-Jones, J.E. The molecular orbital theory of chemical valency I. The determination of molecular orbitals. Proc. R. Soc.
Lond. A 1949, 198, 1–13. [CrossRef]22. England, W.; Salmon, L.S.; Ruedenberg, K. Localized molecular orbitals: A bridge between chemical intuition and molecular
quantum mechanics. In Molecular Orbitals; Springer: Berlin/Heidelberg, Germany, 1970; pp. 31–123. [CrossRef]23. Weinhold, F.; Landis, C.R. Discovering Chemistry with Natural Bond Orbitals; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2012.
[CrossRef]24. Lu, W.C.; Wang, C.Z.; Schmidt, M.W.; Bytautas, L.; Ho, K.M.; Ruedenberg, K. Molecule intrinsic minimal basis sets. I. Exact
resolution of ab initio optimized molecular orbitals in terms of deformed atomic minimal-basis orbitals. J. Chem. Phys. 2004,120, 2629–2637. [CrossRef]
25. Knizia, G. Intrinsic atomic orbitals: An unbiased bridge between quantum theory and chemical concepts. J. Chem. Theory Comput.2013, 9, 4834–4843. [CrossRef]
26. Szalay, S. Multipartite entanglement measures. Phys. Rev. A 2015, 92, 042329. [CrossRef]27. Szalay, S.; Barcza, G.; Szilvási, T.; Veis, L.; Legeza, Ö. The correlation theory of the chemical bond. Sci. Rep. 2017, 7, 2237.
[CrossRef] [PubMed]

Molecules 2021, 26, 911 20 of 21
28. Savin, A.; Nesper, R.; Wengert, S.; Fässler, T.F. ELF: The electron localization function. Angew. Chem. Int. Ed. Engl. 1997,36, 1808–1832. [CrossRef]
29. Chamorro, E.; Fuentealba, P.; Savin, A. Electron probability distribution in AIM and ELF basins. J. Comput. Chem. 2003,24, 496–504. [CrossRef]
30. Martín Pendás, A.; Francisco, E.; Blanco, M.A. An electron number distribution view of chemical bonds in real space. Phys. Chem.Chem. Phys. 2007, 9, 1087–1092. [CrossRef] [PubMed]
31. Schulte, C.; Lüchow, A. Quantum Monte Carlo Calculations on the Anomeric Effect. Recent Prog. Quantum Monte Carlo 2016,1955, 89–105. [CrossRef]
32. Florek, K.; Łukaszewicz, J.; Perkal, J.; Steinhaus, H.; Zubrzycki, S. Sur la liaison et la division des points d’un ensemble fini. [Onthe connection and division of points of a finite set]. Colloq. Math. 1951, 2, 282–285. [CrossRef]
33. Gower, J.C.; Ross, G.J.S. Minimum spanning trees and single linkage cluster analysis. Appl. Stat. 1969, 18, 54. [CrossRef]34. Kaufman, B.; Onsager, L. Crystal statistics. III. Short-range order in a binary Ising lattice. Phys. Rev. 1949, 76, 1244–1252.
[CrossRef]35. Lüchow, A.; Sturm, A.; Schulte, C.; Haghighi Mood, K. Generic expansion of the Jastrow correlation factor in polynomials
satisfying symmetry and cusp conditions. J. Chem. Phys. 2015, 142, 084111. [CrossRef]36. Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.;
Nakatsuji, H.; et al. Gaussian 16 Revision C.01; Gaussian Inc.: Wallingford, CT, USA, 2016.37. Becke, A.D. Correlation energy of an inhomogeneous electron gas: A coordinate-space model. J. Chem. Phys. 1988, 88, 1053–1062.
[CrossRef]38. Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density.
Phys. Rev. B 1988, 37, 785–789. [CrossRef] [PubMed]39. Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [CrossRef]40. Vosko, S.H.; Wilk, L.; Nusair, M. Accurate spin-dependent electron liquid correlation energies for local spin density calculations:
A critical analysis. Can. J. Phys. 1980, 58, 1200–1211. [CrossRef]41. Dunning, T.H. Gaussian basis sets for use in correlated molecular calculations. I. The atoms boron through neon and hydrogen.
J. Chem. Phys. 1989, 90, 1007–1023. [CrossRef]42. Van Lenthe, E.; Baerends, E.J. Optimized Slater-type basis sets for the elements 1-118. J. Comput. Chem. 2003, 24, 1142–1156.
[CrossRef] [PubMed]43. O-ohata, K.; Taketa, H.; Huzinaga, S. Gaussian expansions of atomic orbitals. J. Phys. Soc. Jpn. 1966, 21, 2306–2313. [CrossRef]44. Petersson, G.A.; Zhong, S.; Montgomery, J.A.; Frisch, M.J. On the optimization of Gaussian basis sets. J. Chem. Phys. 2003,
118, 1101–1109. [CrossRef]45. Schmidt, K.E.; Moskowitz, J.W. Correlated Monte Carlo wave functions for the atoms He through Ne. J. Chem. Phys. 1990,
93, 4172–4178. [CrossRef]46. Güçlü, A.D.; Jeon, G.S.; Umrigar, C.J.; Jain, J.K. Quantum Monte Carlo study of composite fermions in quantum dots: The effect
of Landau-level mixing. Phys. Rev. B 2005, 72, 205327. [CrossRef]47. Toulouse, J.; Umrigar, C.J. Optimization of quantum Monte Carlo wave functions by energy minimization. J. Chem. Phys. 2007,
126, 084102. [CrossRef]48. Lüchow, A.; Manten, S.; Diedrich, C.; Bande, A.; Scott, T.C.; Schwarz, A.; Berner, R.; Petz, R.; Sturm, A.; Hermsen, M.; et al.
Amolqc. Zenodo 2021. [CrossRef]49. Kato, T. On the eigenfunctions of many-particle systems in quantum mechanics. Commun. Pure Appl. Math. 1957, 10, 151–177.
[CrossRef]50. Lüchow, A. Quantum Monte Carlo methods. WIREs Comput. Mol. Sci. 2011, 1, 388–402. [CrossRef]51. Umrigar, C.J.; Nightingale, M.P.; Runge, K.J. A diffusion Monte Carlo algorithm with very small time-step errors. J. Chem. Phys.
1993, 99, 2865–2890. [CrossRef]52. Liu, D.C.; Nocedal, J. On the limited memory BFGS method for large scale optimization. Math. Program. 1989, 45, 503–528.
[CrossRef]53. Heuer, M.A.; Reuter, L. InPsights. Zenodo 2020. [CrossRef]54. Munkres, J. Algorithms for the assignment and transportation problems. J. Soc. Ind. Appl. Math. 1957, 5, 32–38. [CrossRef]55. Wong, J.K. A new implementation of an algorithm for the optimal assignment problem: An improved version of Munkres’
algorithm. BIT Numer. Math. 1979, 19, 418–424. [CrossRef]56. Ester, M.; Kriegel, H.P.; Sander, J.; Xu, X. A density-based algorithm for discovering clusters in large spatial databases with noise.
In Proceedings of the 2nd International Conference on Knowledge Discovery and Data Mining, Portland, OR, USA, 2–4 August1996; pp. 226–231.
57. Schubert, E.; Sander, J.; Ester, M.; Kriegel, H.P.; Xu, X. DBSCAN revisited, revisited. ACM Trans. Database Syst. 2017, 42, 1–21.[CrossRef]
58. Heuer, M.A.; Reuter, L.; Lüchow, A. A Dataset for Probability Density Analysis Containing 30 Small Molecules and Dehydroala-nine. Available online: https://osf.io/6z2j9 (accessed on 31 December 2020).
59. Shaik, S.; Danovich, D.; Wu, W.; Hiberty, P.C. Charge-shift bonding and its manifestations in chemistry. Nat. Chem. 2009,1, 443–449. [CrossRef]

Molecules 2021, 26, 911 21 of 21
60. Wu, W.; Gu, J.; Song, J.; Shaik, S.; Hiberty, P.C. The inverted bond in [1.1.1]propellane is a charge-shift bond. Angew. Chem. 2009,121, 1435–1438. [CrossRef]
61. Shaik, S.; Danovich, D.; Wu, W.; Su, P.; Rzepa, H.S.; Hiberty, P.C. Quadruple bonding in C2 and analogous eight-valence electronspecies. Nat. Chem. 2012, 4, 195–200. [CrossRef]
62. Edmiston, C.; Ruedenberg, K. Localized atomic and molecular orbitals. Rev. Mod. Phys. 1963, 35, 457–464. [CrossRef]63. Foster, J.M.; Boys, S.F. A quantum variational calculation for HCHO. Rev. Mod. Phys. 1960, 32, 303–304. [CrossRef]64. Boys, S.F. Construction of some molecular orbitals to be approximately invariant for changes from one molecule to another.
Rev. Mod. Phys. 1960, 32, 296–299. [CrossRef]65. Socrates, G. Infrared and Raman Characteristic Group Frequencies: Tables and Charts, 3rd ed.; John Wiley & Sons: Hoboken, NJ, USA,
2004; pp. 94, 117, 125.





























