Embed Size (px)
Citation preview

BALKAN PHYSICS LETTERS
©Bogazici University Press 09 February 2011
BPL, 19, 191015, pp. 137 – 152, (2011)
AB INITIO HARTREE-FOCK AND DENSITY FUNCTIONAL THEORY STUDY ON
CHARACTERIZATION OF 2-NITRO-N-(4- NITROPHENYL) BENZAMIDE
Y. ZALAOGLU
Physics Department, Osmaniye Korkut Ata University,
Osmaniye, TURKEY.
F.KARABOGA
Physics Department, Abant İzzet Baysal University,
Bolu, TURKEY.
G. YILDIRIM
Physics Department, Abant İzzet Baysal University,
Bolu, TURKEY.
C. TERZIOGLU
Physics Department, Abant İzzet Baysal University,
Bolu, TURKEY.
A. SEROL ERTURK
Chemistry Department, Adıyaman University,
Adıyaman, TURKEY.
and
A. TOLGA ULGEN
Electrical and Electronics Engineering Faculty, Sırnak University,
Sırnak, TURKEY.
Abstract. The optimized molecular structures including bond lengths and angels of 2-nitro-N-(4-nitrophenyl) benzamide
molecule were investigated by utilizing ab initio Hartree–Fock (HF) and Density Functional Theory (B3LYP) methods at 6–
31G(d,p) calculation level. All the calculated bond lengths and bond angles were observed to be in good agreement with each
other. Moreover, thermodynamic properties, atomic charges and ultraviolet visible (UV–Vis) spectra were determined and
interpreted for the characterization of the molecule. In addition, not only did we simulate frontier molecular orbitals (FMO)
and molecular electrostatic potential (MEP) but evaluated the transition state and energy band gap clearly.
Key Words: 2-nitro-N-(4-nitrophenyl)benzamide, B3LYP, HF, FMO, MEP

138 BALKAN PHYSICS LETTERS
1. Introduction
Benzamide obtained by the action of ammonia upon chloride of benzoyl, is the simplest aromatic
carboxylic amide and used in the synthesis of various organic compounds. Several kinds of benzamide
derivatives can be also synthesized from anacardic acid and the studies on these molecules increase day by day
owing to the fact that the increased interest in both the biological of these derivatives in the last decades stems
from its remarkable anthelmentic, antihistaminic, antifungal, and antibacterial [1–9]. 2-nitro-N-(4-nitrophenyl)
benzamide from benzamide derivatives undertakes some vital assignments to continue the biological activity
[10]. Thus, it is important to analyze the characterization of 2-nitro-N-(4-nitrophenyl) benzamide for future
studies. In order to support the experimental evidences, the scientists use computational methods which are
reliable to characterize the molecule because of their efficiency and accuracy with respect to the evaluation of a
number of molecular properties [11]. A suitable quantum chemical study is helpful to predict compound
properties economically and to clarify some experiment phenomena insightfully [12]. Hence theoretical studies
are either reliable or useful to identify the molecule. In this respect, the computational researches on compound
properties tend to increase [13–15]. In this study, we calculated the molecular structures using B3LYP/6–
31G(d,p) and HF/6–31G(d,p) basis sets and compared with the experimental data [10,16]. Comparison of
theoretical and experimental data exhibit well correlation confirming the reliability of the methods employed in
this work. Furthermore; after the frontier orbitals and molecular electrostatic potential were visualized, transition
states and energy band gap were determined and interpreted. Thermodynamic properties, UV-Vis spectra and
atomic charges were also mentioned for the molecule. The aim of this study is to not only investigate the
agreement between theoretical data and experimental results but also clarify the characterization of 2-nitro-N-(4-
nitrophenyl) benzamide and show the way to future studies of this molecule.
2. Computational Details
The optimized molecular structures, UV-Vis spectra, atomic charges, thermodynamic properties and
translation energy (HOMO–LUMO) and molecular electrostatic potential (MEP) of the 2-nitro-N-(4-
nitrophenyl) benzamide molecule were investigated using HF [17] and B3LYP [18] methods at 6–31G(d,p) [19]
calculation level. All the computations were performed by using Gaussian 09 program package program with
molecular visualization program [20,21] on the personal computer.
3. Result and Discussion
We determined the molecular geometry, thermodynamic properties, UV-Vis spectra, atomic charges,
potential energy distributions and frontier orbitals for the characterization of 2-nitro-N-(4-nitrophenyl)
benzamide.
3.1. Molecular Geometry

Y.ZALAOGLU et. al.: AB INITIO HARTREE-FOCK... 139
The molecular structure of 2-nitro-N-(4-nitrophenyl) benzamidemolecule was depicted in Fig. 1 along with
labeling and symbolizing by means of schema. Geometric properties of the structure were depicted and
compared with experimental parameters obtained from the X-ray structure analyses of in Table 1. The
experimental results were found to be in good agreement with the theoretical determines for bond lengths and
bond angles. The largest differences of the calculated geometries from the experimental parameters were noted
to be about 0.02 Å (N23–C29) at B3LYP/6–31G(d,p), 0.04 Å (N22–O26) at HF/6–31G(d,p) calculation level for the
bond lengths; 1.140 (C2–N23–C29) at B3LYP/6–31G(d,p) and 1.23
0 (O24–N21–O25) at HF/6–31G(d,p) respectively
for the bond angles. Moreover, errors between the experimental and calculated bond lengths and bond angles of
2-nitro-N-(4-nitrophenyl) benzamide molecule are given in the same table. As can be seen that computations of
DFT level of theory are superior to that of HF method.
3.2. Thermodynamic Properties
Several thermodynamic parameters were performed by using HF and B3LYP with 6–31G(d,p) basis set and
given in Table 2. Scale factors were recommended [22] for an accurate prediction in determining the zero–point
vibration energies and rotational constants. The total energies and the change in the total entropy of the molecule
at room temperature at different theoretical methods were presented. Table 2 demonstrates several
thermodynamic parameters of the molecule without experimental determinations. As seen from the table,
calculations of HF/6–31G(d,p) basis set for energy parameters and rotational constants are close to that of
B3LYP/6–31G(d,p). Although translational and rotational computations are coincide with the entropies and heat
capacities, HF data are generally smaller than DFT ones.
3.3. HOMO and LUMO Analysis
Highest Occupied Molecular Orbital (HOMO) and Lowest Unoccupied Molecular Orbital (LUMO) from
frontier molecular orbitals, very important parameters for quantum chemistry, play important role in the electric
and optical properties [23]. We can determine the way the molecule interacts with other species; hence, they are
called the frontier orbitals. HOMO, which can be thought the outermost orbital containing electrons, tends to
give these electrons such as an electron donor. On the other hand; LUMO can be thought the innermost orbital
containing free places to accept electrons [24]. Owing to the interaction between HOMO and LUMO orbital of a
structure, transition state transition of π–π* type is observed with regard to the molecular orbital theory [25].
Therefore, while the energy of the HOMO is directly related to the ionization potential, LUMO energy is directly
related to the electron affinity. Energy difference between HOMO and LUMO orbital is called as energy gap that
is an important stability for structures [26] and given by using four different methods in Table 3. Further, 3D
plots of highest occupied molecular orbitals (HOMOs), lowest unoccupied molecular orbitals (LUMOs),
HOMO–1, HOMO–2, LUMO+1, and LUMO+2 were shown in Fig. 2. According to B3LYP/6–31G(d,p)
calculation level, the energy band gap │(ΔE)│ (translation from HOMO to LUMO) of the title molecule was

140 BALKAN PHYSICS LETTERS
found to be about 0.145 (a.u.) while the band gap was obtained to be about 0.386 at HF/6–31G(d,p) level of
calculation. The highest occupied molecular orbitals were localized mainly on the ring–1 and vicinity of this
ring; in fact no translation appears on both the ring–2 and N21, O24 and O25 atoms. On the other hand, the lowest
unoccupied molecular orbitals were only localized on the ring–2 and slightly N23 and O28 atoms. Moreover, the
HOMO–1 and HOMO–2 orbitals were partially localized on different parts of the title molecule. The HOMO–1
orbitals are only delocalized on the benzen ring–1, while the HOMO–2 orbitals were delocalized on the benzene
ring–2 and O28 atom. Likewise, the LUMO+1 and LUMO+2 orbitals were partially delocalized on different
parts of the molecule. The LUMO+1 orbitals were delocalized on the molecule except for benzene ring–2,
whereas the LUMO+2 orbitals were delocalized on the benzene ring–2 and slightly delocalized on N22, O26 and
O27 atoms. In addition, Lowest MO Eigen value was calculated –19.20 and –20.62 (a.u.) at B3LYP/6–31G(d,p)
and HF/6–31G(d,p) basis sets, respectively. Highest MO Eigen value was also found to be about 4.83 (a.u.) at
B3LYP/6–31G(d,p) and 5.28 (a.u.) at HF/6–31G(d,p) calculation level. The HOMO–1, HOMO–2, LUMO+1
and LUMO+2 Eigen values of the molecule were also depicted in the same table.
3.4. UV-Vis Spectra Analysis
Electronic transitions are usually classified according to the orbitals engaged or to specific parts of the
molecule involved. Common types of electronic transitions in organic compounds are π–π*, n–π* and
π*(acceptor)–π (donor) [27]. In order to understand the electronic transitions of 2-nitro-N-(4-nitrophenyl)
benzamide, theoretical calculations on electronic absorption spectrum, capable of describing the spectral features
of the molecule, were performed in vacuum by TD [28] methods. The calculated visible absorption maxima of λ
which are a function of the electron availability were reported in Table 4. The visible absorption maxima of the
title molecule were corresponded to the electron transition between frontier orbitals (such as translation from
HOMO to LUMO; or HOMO–3 to LUMO+1) by using calculations of molecular orbital geometry. As can be
seen from the table, λmax were arranged in an order from 300 to 334 nm at TD–B3LYP/6–31G(d,p) whereas they
are obtained to change from 228 to 240 nm at TD–HF/6–31G(d,p) calculation level. The results show the
computations of the calculation levels were noted to be close to each other. In addition, oscillator strength values
depicted in the Table 4 were noticed to be close to each other for all translations at the basis sets. The less intense
band centered at 303 (237) nm at B3LYP (HF) basis set is due to the partly forbidden n–π* (HOMO-
3↔LUMO+1) transition. On the other hand, the more intense band (ascribed to an allowed π–π* (HOMO-
1↔LUMO) transition) observed at TD–B3LYP/6–31G(d,p) calculation level was obtained to be about 0.0117 at
300 nm while the maximum computation (the more intense band) at TD–HF/6–31G(d,p) basis set was calculated
to be 0.4669 at 228 nm.
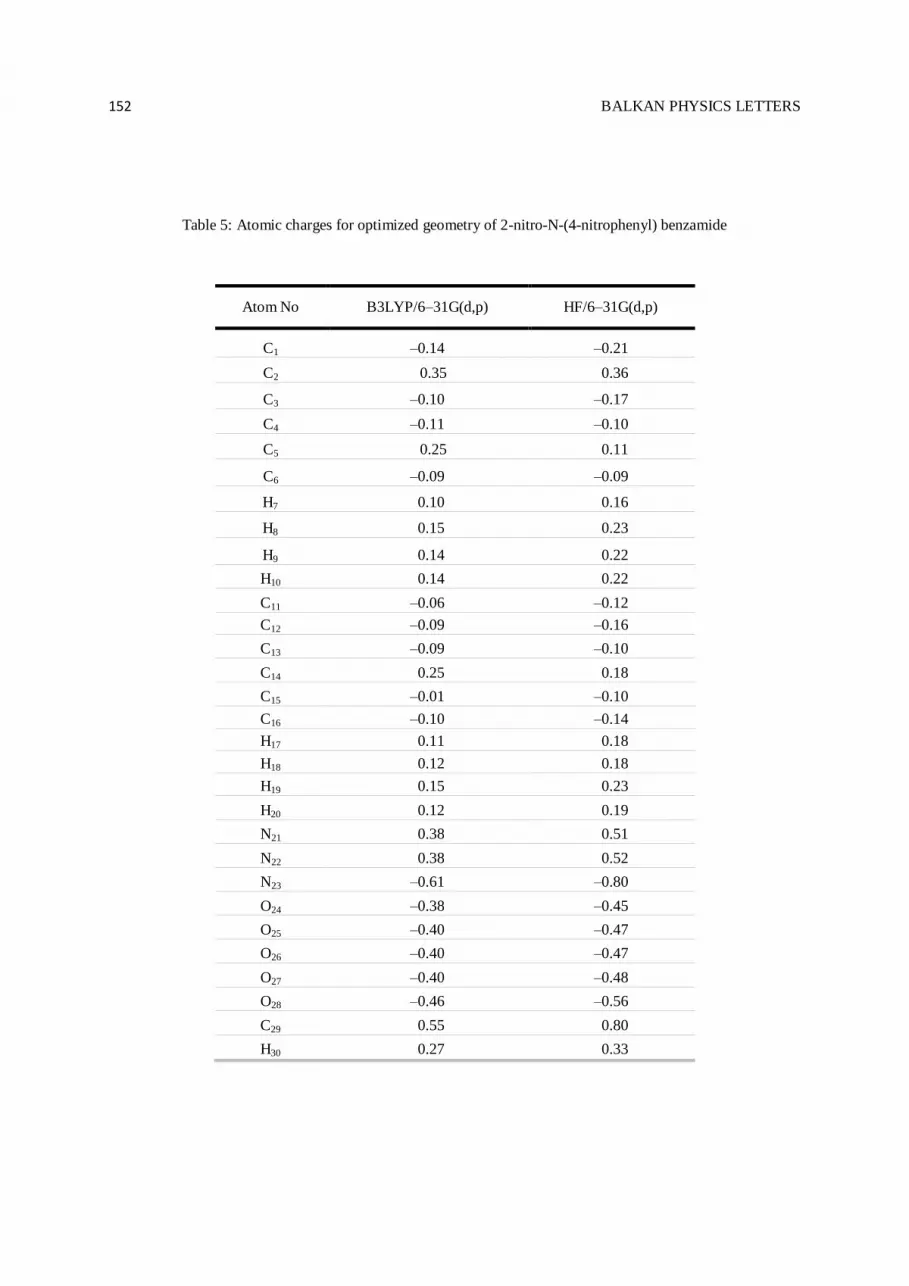
3.5. Atomic Charges

Y.ZALAOGLU et. al.: AB INITIO HARTREE-FOCK... 141
Atomic charges of 2-nitro-N-(4-nitrophenyl)benzamide, calculated by Mulliken method [29,30] at the
B3LYP/6–31G(d,p) and HF/6–31G(d,p) levels of calculation, were given in Table 5. As can be seen from the
table, all the obtained magnitudes are in good agreement with each other. The magnitudes of the carbon atomic
charges were found to be either positive or negative at the basis sets. These magnitudes were obtained to change
between –0.21 and 0.80. Whereas each carbon atom connected to nitrogen atoms has the positive charge
magnitude, C29 connected to oxygen atom has the maximum charge magnitude (0.55 and 0.80 at B3LYP/6–
31G(d,p) and HF/6–31G(d,p) basis sets, respectively). The magnitudes of the other carbon atoms connected to
hydrogen atoms were calculated to be negative value. Moreover, the magnitudes of the hydrogen atomic charges
were arranged in an order from 0.10 to 0.27 at B3LYP/6–31G(d,p) and 0.16 to 0.33 at HF/6–31G(d,p)
calculation level. The maximum charge magnitude of hydrogen atoms was found for H30 atom connected to
nitrogen atom. Like carbon atoms, the magnitudes of nitrogen atomic charges found to be either positive or
negative at the calculation levels were noted to change from –0.61 to 0.38 at B3LYP/6–31G(d,p) and –0.80 to
0.51 at HF/6–31G(d,p) level of calculation, respectively. The minimum charge magnitude of nitrogen atoms was
noted for N23 atom. On the other hand, for oxygen atoms the magnitudes of charges were calculated to change
from –0.46 to –0.38 for DFT and from –0.56 to –45 HF methods. The minimum charge magnitude was obtained
for O28 atom. The results show that:
* All the hydrogen atoms in molecules lost electrons.
* All oxygen atoms in molecules accepted electrons.
* Charge migration to heavy atoms can be related to molecular interactions.
* HF/6–31G(d,p) basis set has more negative magnitudes than the others.
* Computations of B3LYP and HF calculation levels are in good agreement with each other.
3.6. Molecular Electrostatic Potential
At any given point r(x, y, z) in the vicinity of a molecule, the molecular electrostatic potential, V(r) is
defined in terms of the interaction energy between the electrical charge generated from the molecule electrons
and nuclei and a positive test charge (a proton) located at r [31,32]. The molecular electrostatic potential (MEP)
is related to the electronic density and a very useful descriptor for determining sites for electrophilic attack and
nucleophilic reactions as well as hydrogen–bonding interactions [33,34]. In Fig. 3, whereas electrophilic
reactivity was presented by negative (red) regions, nucleophilic reactivity was shown by the positive (blue)
regions of MEP. As seen from the figure, the red region was localized on the oxygen and vicinity of these atoms.
On the other hand, although the blue region was not seen clearly the nucleophilic reactivity of the molecule was
localized on the hydrogen atoms (especially H30 atom). In this respect, the compound is useful to both bond
metallically and interact intermolecularly. This result was also supported by the evidences of charge analyses
part.

142 BALKAN PHYSICS LETTERS
4. Conclusion
In this study, when clarifying the characterization of 2-nitro-N-(4-nitrophenyl) benzamide, we used ab
initio Hartree Fock and density functional theory methods at 6-31G(d,p) calculation level. Optimized geometric
were carried out using HF/6–31G(d,p) and B3LYP/6–31G(d,p) methods and compared with experimental
values. It was found that all the compared data were shown to have a good agreement with each other. This good
agreement is well within the accuracy of computational results. Moreover, after frontier orbitals and molecular
electrostatic potential were visualized, electronic structure and energy band gap of the title molecule were
investigated and interpreted. Atomic charges, thermodynamic properties and UV–Vis spectra were also
determined for the identification of the molecule. In conclusion, all the calculated data and simulations not only
show the way to the characterization of the molecule but also help for the fundamental researches in physics,
chemistry and biology in the future.
REFERENCES
[1] H. Mrozik, H. Jones, J. Friedman, G. Schwartzkopf, R. Schardt, A. Patchett, D. R. Holff, J. J. Yakstis, R. F.
Riek, D. A. Ostlind, G. A. Plischker, R. W. Butler, A. C. Cuckler and W. C. Campbell, Experientia 25, 883
1969.
[2] Japan Patent, 73, 37, 819, Chem. Abstr. 81 (1974) 73387 (1973).
[3] Braz Pedido PI N80 04, 641, Chem.Abstr. 95 (1981) 61812z (1981).
[4] G.A. White, Pestic. Biochem. Physiol. 34, 255 1989.
[5] I. Yalcin, B. K. Kaymakcioglu, I. Oren, E. Sener, O. Temiz, A. Akin and N. Altanlar, II Farmaco 52, 685
1997.
[6] K. J. Pradhan, P. S. Variyar and J. R. Bandekar, Lebensm. Wiss. Technol. 32, 121 1999.
[7] E. Aki-Sener, K. K. Bingol, I. Oren, O. Temiz-Arpaci, I. Yalcin and N. Altanlar, II Farmaco 55, 469 2000.
[8] E. Aki-Sener, K. K. Bingol, O. Temiz-Arpaci, I. Yalcin and N. Altanlar, II Farmaco 57, 451 2002.
[9] I. Yildiz-Oren, E. Aki-Sener, C. Ertas, O. Temiz-Arpaci, I. Yalcin and N. Altanlar, Turk. J. Chem. 28, 441
2004.
[10] A. Saeed, S. Hussain and U. Flörke, Turk J Chem. 32, 481 2008.
[11] C. Ravikumar, I. H. Joe and V. S. Jayakumar, Chem. Phys. Lett. 460, 552 2008.
[12] Y. X. Suna, Q. L. Haoa, L. D. Lua, X. Wanga and X. J. Yanga, Spectrochim. Acta A 75, 203 2010.

Y.ZALAOGLU et. al.: AB INITIO HARTREE-FOCK... 143
[13] C. Y. Panicker , H. T. Varghese, L. Ushakumari, T. Ertan , I. Yildiz , C. M. Granadeiro , H. I. S. Nogueira
and Y. S. Mary, J. Raman Spectrosc. 41, 381 2010.
[14] L. Ushakumari, H.T. Varghese, C.Y. Panicker, T. Ertan and I. Yidiz, J. Raman Spectrosc. 39, 1832 2008.
[15] H. A. Dabbagh , A. Teimouri , A.N. Chermahini and M. Shahraki , Spectrochim. Acta A 69, 449 2008.
[16] A. Saeed, S. Hussain, N. Abbas and M. Bolte, J. Chem. Crystallogr. 40, 919 2010.
[17] D. C. Young, Computational Chemistry: A Practical Guide for Applying Techniques to Real World
Problems, John Wiley & Sons Inc., New York, 2001.
[18] A. D. Becke, J. Chem. Phys. 98, 5648 1993.
[19] M. M. Francl, W. J. Pietro, W. J. Hehre, J. S. Binkley, M. S. Gordon, D. J. DeFrees, and J. A. Pople, J.
Chem. Phys. 77, 3654 1982.
[20] M. J. Frisch, et. al, Gaussian, Inc., Wallingford CT, 2009.
[21] W. Gaussview, A. E. Frisch, A. B. Nielsen, A. J. Holder, Gaussian Inc., Carnegie Office Park, Building 6,
Pittsburg, PA 15106, USA, 2003.
[22] J. B. Foresman, A. Frisch, Exploring Chemistry with Electronic Structure Methods, Gaussian Inc.,
Pittsburgh, PA, USA, 1996.
[23] I. Fleming, Frontier Orbitals and Organic Chemical Reactions, Wiley, London, 1976.
[24] G. Gece, Corros. Sci. 50, 2981 2008.
[25] K. Fukui, Theory of Orientation, Stereoselection, Springer-Verlag, Berlin, 1975.
[26] D. F. V. Lewis, C. Ioannides and D. V. Parke, Xenobiotica 24, 401 1994.
[27] D. A. Dhas, I. H. Joe, S. D. D. Roy and T. H. Freeda, Spectrochim. Acta A 77, 36 2010.
[28] R. Bauernschmitt and R.Ahlrichs, Chem. Phys. Lett. 256, 454 1996.
[29] T. U. Helgaker, H. J. A. Jensen and P. J. Jørgensen, Chem. Phys. 84, 6280 1986.
[30] H. Buyukuslu, M. Akdogan, G. Yildirim and C. Parlak, Spectrochim. Acta A 75, 1362 2010.
[31] N. Özdemir, B.Eren, M. Dinçer and Y. Bekdemir, Mol. Phys. 108, 13 2010.
[32] P. Politzer and J. S. Murray, Theor. Chem. Acc. 108, 134 2002.
[33] F. J. Luque, J. M. Lopez and M. Orozco, Theor. Chem. Acc. 103, 343 2000.
[34] N. Okulik and A. H. Joubert, Internet Electronic Journal of Molecular Design 4, 17 2005.

144 BALKAN PHYSICS LETTERS
Figure Captions
Figure 1. The molecular structure of 2-nitro-N-(4-nitrophenyl)benzamide
Figure 2. 3D plots of a) the HOMO; b) LUMO; c) HOMO–1; d) HOMO–2; e) LUMO+1 and f) LUMO+2 of 2-
nitro-N-(4-nitrophenyl) benzamide obtained from DFT method (Red regions show the positive phase
while greens present the negative phase).
Figure 3. 3D plots of the molecular electrostatic potential map of 2-nitro-N-(4-nitrophenyl) benzamide obtained
from DFT method ( Red color shows the negative regions while blue color presents the positive
regions of MEP).
Figure 1: The molecular structure of 2-nitro-N-(4-nitrophenyl)benzamide

Y.ZALAOGLU et. al.: AB INITIO HARTREE-FOCK... 145
Figure 2: 3D plots of a) the HOMO; b) LUMO; c) HOMO–1; d) HOMO–2; e) LUMO+1 and f) LUMO+2 of 2-
nitro-N-(4-nitrophenyl) benzamide obtained from DFT method (Red regions show the positive phase while
greens present the negative phase).

146 BALKAN PHYSICS LETTERS
Figure 3: 3D plots of the molecular electrostatic potential map of 2-nitro-N-(4-nitrophenyl) benzamide obtained
from DFT method ( Red color shows the negative regions while blue color presents the positive regions of
MEP).
Table Captions
Table 1. Experimental values and theoretical optimized geometric parameters of 2-nitro-N-(4-
nitrophenyl)benzamide
Table 2. Theoretically computed energies (a.u.), zero–point vibrational energies (kcal mol–1), rotational
constants (GHz), entropies (cal mol–1 K–1) and dipole moment (Debye)
Table 3. Some of the calculated energy values of 2-nitro-N-(4-nitrophenyl) benzamide molecule in its ground
state with singlet symmetry at DFT and HF methods
Table 4. Theoretical electronic absorption spectra values of 2-nitro-N-(4-nitrophenyl) benzamide molecule
Table 5. Atomic charges for optimized geometry of 2-nitro-N-(4-nitrophenyl) benzamide

Y.ZALAOGLU et. al.: AB INITIO HARTREE-FOCK... 147
Table 1: Experimental values and theoretical optimized geometric parameters of
2-nitro-N-(4-nitrophenyl)benzamide
Bond length
(Å)
Calculated DFT
6–31G(d, p)
Calculated HF
6–31G(d, p) aExperimental
bExperimental
C2-N23 1.4033 ± 0.0139 1.4001 ± 0.0171 1.4092 1.4172
C5-N22 1.4646 ± 0.0184 1.4518 ± 0.0114 1.4632 1.4462
C14-N21 1.4734 ± 0.0041 1.4584 ± 0.0128 1.4712 1.4693
N21-O24 1.2281 ± 0.0099 1.1908 ± 0.0315 1.2182 1.2223
N21-O25 1.2311 ± 0.0069 1.1942 ± 0.0301 1.2242 1.2243
N22-O26 1.2317 ± 0.0094 1.1942 ± 0.0350 1.2292 1.2223
N22-O27 1.2326 ± 0.0083 1.1953 ± 0.0299 1.2252 1.2243
N23-C29 1.3779 ± 0.0914 1.3626 ± 0.1067 1.3612 1.4693
O28-C29 1.2176 ± 0.0044 1.1912 ± 0.0240 1.2132 1.2152
C1-C2 1.4074 ± 0.0241 1.3960 ± 0.0127 1.3833
C1-C6 1.3858 ± 0.0024 1.3758 ± 0.0076 1.3834
C1-H7 1.0868 ± 0.1368 1.0758 ± 0.1258 0.9500
C2-C3 1.4065 ± 0.0172 1.3930 ± 0.0037 1.3893
C3-C4 1.3898 ± 0.0015 1.3817 ± 0.0066 1.3883
C3-H8 1.0800 ± 0.2914 1.0681 ± 0.3033 1.3714
C4-C5 1.3934 ± 0.0021 1.3815 ± 0.0098 1.3913
C4-H9 1.0827 ± 0.1327 1.0716 ± 0.1216 0.9500
C5-C6 1.3945 ± 0.0058 1.3838 ± 0.0165 1.4003
C6-H10 1.0824 ± 0.1324 1.0714 ± 0.1214 0.9500
C11-C12 1.3953 ± 0.0030 1.3829 ± 0.0094 1.3923
C11-C16 1.3959 ± 0.0094 1.3867 ± 0.0186 1.4053
C11-H17 1.0854 ± 0.1354 1.0749 ± 0.1249 0.9500
C12-C13 1.3925 ± 0.0028 1.3838 ± 0.0115 1.3953
C12-H18 1.0849 ± 0.1349 1.0742 ± 0.1242 0.9500
C13-C14 1.3927 ± 0.0026 1.3806 ± 0.0147 1.3953
C13-H19 1.0828 ± 0.1328 1.0717 ± 0.1217 0.9500
C14-C15 1.4006 ± 0.4206 1.3889 ± 0.4089 0.9800
C15-C16 1.3991 ± 0.0038 1.3858 ± 0.0095 1.3953
C15-C29 1.5189 ± 0.0046 1.5150 ± 0.0007 1.5143
C16-H20 1.0857 ± 0.1057 1.0750 ± 0.095 0.9800
N23-H30 1.0107 0.9938

148 BALKAN PHYSICS LETTERS
a: Taken f rom Ref. [10] b: Taken f rom Ref. [34]
Bond Angle (0)
Calculated DFT
6–31G(d, p)
Calculated HF
6–31G(d, p) aExperimental
bExperimental
C1-C2-C3 119.94 ± 0.85 119.74 ± 0.65 120.38 119.09
C1-C2-N23 117.17 ± 0.82 116.67 ± 0.32 116.35 116.52
C3-C2-N23 123.29 ± 0.68 123.58 ± 0.39 123.27 123.97
C14-N21-O24 117.76 ± 0.46 117.75 ± 0.47 118.00 118.22
C14-N21-O25 117.89 ± 0.96 117.07 ± 1.78 118.07 118.85
O24-N21-O25 124.93 ± 2.01 125.16 ± 2.24 123.93 122.92
C5-N22-O26 117.68± 0.54 117.81 ± 0.41 117.85 118.22
C5-N22-O27 117.98 ± 0.87 117.67 ± 1.18 118.67 118.85
O26-N22-O27 124.52 ± 1.60 124.52 ± 1.60 123.48 122.92
C2-N23-C29 128.59 ± 1.14 128.40 ± 0.95 127.45
C2-C1-C6 120.67 ± 0.55 120.73 ± 0.61 120.12
C2-C1-H7 119.79 ± 0.21 119.93 ± 0.07 120.00
C6-C1-H7 119.54 ± 0.46 119.34 ± 0.66 120.00
C2-C3-C4 119.49 ± 0.55 119.43 ± 0.49 118.94
C2-C3-H8 119.59 ± 0.29 120.36 ± 1.06 119.30
C4-C3-H8 120.92 ± 0.12 120.20 ± 0.60 120.80
C3-C4-C5 119.86 ± 0.21 119.99 ± 0.34 119.65
C3-C4-H9 120.88 ± 0.68 120.25 ± 0.05 120.20
C5-C4-H9 119.25 ± 0.95 119.76 ± 0.44 120.20
C4-C5-C6 121.41 ± 0.99 121.26 ± 0.84 120.42
C4-C5-N22 119.47 ± 0.01 119.51 ± 0.03 119.48
C6-C5-N22 119.12 ± 1.15 119.23 ± 1.04 120.27
C1-C6-C5 118.83 ± 0.37 118.85 ± 0.39 118.46
C1-C6-H10 121.62 ± 0.92 121.02 ± 0.32 120.70
C6-C5-H10 119.55 ± 0.35 120.13 ± 0.23 119.90
C12-C11-C16 120.24 ± 0.60 120.34 ± 0.50 120.84
C12-C11-H17 120.13 ± 0.27 120.09 ± 0.31 120.40
C16-C11-H17 119.63 ± 0.33 119.56 ± 0.26 119.30
C11-C12-C13 119.83 ± 0.74 119.79 ± 0.70 119.09
C11-C12-H18 120.42 ± 0.62 120.47 ± 0.67 119.80
C13-C12-H18 119.75 ± 0.45 119.74 ± 0.46 120.20
C12-C13-C14 119.21 ± 0.44 119.13 ± 0.52 119.65
C12-C13-H19 121.95 ± 1.25 121.45 ± 0.75 120.70
C14-C13-H19 118.84 ± 0.66 119.42 ± 0.08 119.50
C13-C14-C15 122.17 ± 0.98 122.19 ± 1.00 121.19
C13-C14-N21 117.61 ± 0.51 117.63 ± 0.49 118.12
C15-C14-N21 120.19 ± 0.81 120.15 ± 0.85 121.00
C14-C15-C16 117.59 ± 0.39 117.78 ± 0.20 117.98
C14-C15-C29 123.90 ± 0.34 124.00 ± 0.24 124.24
C16-C15-C29 118.12 ± 0.34 117.95 ± 0.51 118.46
C11-C16-C15 120.96 ± 0.64 120.77 ± 0.45 120.32

Y.ZALAOGLU et. al.: AB INITIO HARTREE-FOCK... 149
Table 2: Theoretically computed energies (a.u.), zero–point vibrational energies (kcal mol–1), rotational
constants (GHz), entropies (cal mol–1 K–1) and dipole moment (Debye)
Parameters B3LYP/6–31(d,p) HF/6–31(d,p)
Total energy -1041.011 -1034.9819
Zero–point energy 131.55 133.16
Rotational constant
0.86 0.88
0.12 0.13
0.12 0.12
Entropy
Total 139.361 135.295
Translational 42.861 42.861
Rotational 34.507 34.456
Vibrational 61.993 57.978
Heat Capacity
Total 64.288 59.419
Translational 2.981 2.981
Rotational 2.981 2.981
Vibrational 58.326 53.457
Dipole Moment 9.3664 10.3045

150 BALKAN PHYSICS LETTERS
Table 3: Some of the calculated energy values of 2-nitro-N-(4-nitrophenyl) benzamide molecule in its ground
state with singlet symmetry at DFT and HF method
Quantity Value
B3LYP/6–31G(d,p) HF/6–31G(d,p)
Lowest MO Eigen value (a.u.) -19.20 -20.62
Highest MO Eigen value (a.u.) 4.83 5.28
The virial (–V/T) 2.0091 2.0017
HOMO (a.u.) –0.251 –0.342
LUMO (a.u.) –0.106 0.044
HOMO–LUMO gap (a.u.), │ΔE │ 0.145 0.386
HOMO –1 (a.u.) –0.278 –0.369
LUMO + 1 (a.u.) –0.280 –0.387
HOMO –2 (a.u.) –0.086 0.062
LUMO + 2 (a.u.) –0.051 0.092

Y.ZALAOGLU et. al.: AB INITIO HARTREE-FOCK... 151
Table 4: Theoretical electronic absorption spectra values of 2-nitro-N-(4-nitrophenyl) benzamide molecule
Calculated, λcal (nm)
TD–B3LYP/6–31G(d,p) TD–HF/6–31G(d,p)
Wave Length
(nm)
Oscillator
Strength
Wave Length
(nm)
Oscillator
Strength Translation
334 0.0001 240 0.0089 HOMO↔LUMO
303 0.0000 237 0.0000 HOMO-3↔LUMO+1
300 0.0117 228 0.4669 HOMO-1↔LUMO
152 BALKAN PHYSICS LETTERS
Table 5: Atomic charges for optimized geometry of 2-nitro-N-(4-nitrophenyl) benzamide
Atom No B3LYP/6–31G(d,p) HF/6–31G(d,p)
C1 –0.14 –0.21
C2 0.35 0.36
C3 –0.10 –0.17
C4 –0.11 –0.10
C5 0.25 0.11
C6 –0.09 –0.09
H7 0.10 0.16
H8 0.15 0.23
H9 0.14 0.22
H10 0.14 0.22
C11 –0.06 –0.12
C12 –0.09 –0.16
C13 –0.09 –0.10
C14 0.25 0.18
C15 –0.01 –0.10
C16 –0.10 –0.14
H17 0.11 0.18
H18 0.12 0.18
H19 0.15 0.23
H20 0.12 0.19
N21 0.38 0.51
N22 0.38 0.52
N23 –0.61 –0.80
O24 –0.38 –0.45
O25 –0.40 –0.47
O26 –0.40 –0.47
O27 –0.40 –0.48
O28 –0.46 –0.56
C29 0.55 0.80
H30 0.27 0.33





























