Embed Size (px)
Citation preview

Toxicology 170 (2002) 173–185
Acrolein-induced cytotoxicity in cultured human bronchialepithelial cells. Modulation by alpha-tocopherol and
ascorbic acid
Mirella Nardini a,b,*, E.I. Finkelstein b, S. Reddy b, G. Valacchi b, M. Traber c,C.E. Cross b, A. van der Vliet b
a Istituto Nazionale di Ricerca per gli Alimenti e la Nutrizione, Via Ardeatina 546, 00178 Rome, Italyb Department of Internal Medicine, School of Medicine, Center for Comparati�e Respiratory Biology and Medicine,
Uni�ersity of California, Da�is, CA, USAc Linus Pauling Institute, Oregon State Uni�ersity, Cor�allis, OR 97331, USA
Received 27 July 2001; received in revised form 9 October 2001; accepted 9 October 2001
Abstract
Acrolein is a highly reactive unsaturated hazardous air pollutant of human health concern, particularly as acomponent of cigarette smoke. In this study, the mechanisms of acrolein-induced cytotoxicity in human bronchialepithelial cells (HBE1) and the modulating effects of antioxidants were examined. Our results show that acroleininduces a cell death pathway in human bronchial epithelial cells, which retain key features of apoptosis, as indicatedby phosphatidylserine (PS) externalization and DNA fragmentation. Acrolein-induced apoptosis was associated withdepletion of cellular GSH and intracellular generation of oxidants. Supplementation of cells with either alpha-toco-pherol or ascorbic acid was found to strongly inhibit acrolein-induced apoptosis and to prevent the increase in thegeneration of intracellular oxidants, although GSH depletion was unaffected. Moreover, recovery of cellular GSHlevels after acrolein exposure was enhanced following either alpha-tocopherol or ascorbic acid supplementation. Theintracellular generation of oxidants following acrolein exposure seems to be an important event triggering theapoptotic response in this model system. © 2002 Elsevier Science Ireland Ltd. All rights reserved.
Keywords: Acrolein; Apoptosis; Glutathione; Reactive-oxygen species; Antioxidants
www.elsevier.com/locate/toxicol
Abbre�iations: AA, ascorbic acid; DCF, 2�,7�-dichlorofluorescein; DCF-DA, 2�,7�-dichlorofluorescin diacetate; DCFH, 2�,7�-dichlorofluorescin; FITC, fluorescein isothiocyanate; FITC-dUTP, FITC-labeled deoxyuridine triphosphate nucleaotides; GSH,glutathione; LDH, lactate dehydrogenase; MANN, D-mannitol; NAC, N-acetyl cysteine; PI, propidium iodide; PS, phosphatidylser-ine; ROS, reactive-oxygen species; TdT, terminal deoxynucleotidyl transferase; TOC, alpha-tocopherol; TUNEL, terminal deoxynu-cleotidyl transferase (TdT)-mediated dUTP nick end-labeling.
* Corresponding author. Tel.: +39-06-503-2412; fax: +39-06-503-1592.E-mail address: [email protected] (M. Nardini).
0300-483X/02/$ - see front matter © 2002 Elsevier Science Ireland Ltd. All rights reserved.
PII: S 0300 -483X(01 )00540 -6

M. Nardini et al. / Toxicology 170 (2002) 173–185174
1. Introduction
Acrolein is a highly reactive, volatile �,�-unsat-urated aldehyde that is generated by incompletecombustion or pyrolysis of organic materials suchas wood, fuels, food and tobacco (Astry andJakab, 1983; World Health Organization, 1991).It has been identified as an hazardous air pollu-tant of important human concern, particularly asa component of photochemical smog andcigarette smoke (Caldwell et al., 1998; Ayer andYeager, 1982; Altschuler and Mc Pherson, 1963).Toxic and genotoxic effects of this chemical havebeen extensively described (International Agencyfor Research on Cancer, 1995). Cigarette smokecauses widespread respiratory epithelial cell dam-age and apoptosis (Lannan et al., 1994), GSHdepletion and free radical species generation.Smoking one cigarette per m3 air of room-space in10–13 min (10 puffs) generates acrolein levels upto 0.84 mg/m3 (Li and Holian, 1998). Moreover,acrolein is estimated to reach concentrations up to80 �M in respiratory tract lining fluids as a resultof smoking (Eiserich et al., 1995).
Acrolein can also be generated endogeneouslyas a metabolic product of the anticancer-drugcyclophosphamide, during conditions of lipid oxi-dation (Fraiser et al., 1991; Uchida et al., 1998a;Uchida, 1999a), and during oxidation ofthreonine by neutrophil myeloperoxidase at sitesof inflammation (Anderson et al., 1997). Indeed,acrolein-protein adducts have been demonstratedin patients with chronic disease believed to be, atleast in part, related to oxidative stress, such asdiabetic nephropathy (Suzuki and Miyata, 1999),Alzheimer’s disease (Calingasan et al., 1999), andin plaque deposits in atherosclerosis (Uchida etal., 1998b). When inhaled, acrolein is a highlyselective respiratory tract toxicant (Astry andJakab, 1983; World Health Organization, 1991;Aranyi et al., 1986) inducing both upper andlower respiratory tract lesions (Beauchamp et al.1985). Acrolein exposure has also been reportedto affect inflammatory-immune processes (Li etal., 1997, 1999; Nguyen et al., 2001; Finkelstein etal., 2001) contributing to the reported deficienciesin lung defense against infections in smokers(Green, 1985; Jakab, 1977).
Acrolein reacts rapidly with cellular nucle-ophiles, particularly GSH (Kehrer and Biswal,2000). In addition, acrolein has been found toinduce the generation of oxygen radicals, such assuperoxide anion and hydroxyl radical, and ini-tiates lipid peroxidation (Adams and Klaidman,1993; Patel, 1987). Administration of acrolein torats decreased lung and liver levels of glutathione,ascorbic acid (AA) and alpha-tocopherol (Aru-mugam et al. 1999a,b).
In spite of extensive toxicology literature onacrolein, there is an incomplete understanding ofthe molecular basis of acrolein-induced cytotoxic-ity and the ability of different antioxidants tocounteract the adverse effects of this compound.In this study we investigate the mechanisms ofacrolein-induced cell death in human bronchialepithelial cells and the modulatory effect of alpha-tocopherol and AA.
2. Materials and methods
2.1. Materials
The following materials were obtained from theindicated sources: dexamethasone, apo-transfer-rin, insulin, bromobimane, L-AA, CHAPS, 2�,7�-dichlorofluorescin diacetate (DCFH-DA), bovineerythrocytes superoxide dismutase (3940 U/mg),bovine liver catalase (17 000 U/mg), horseradishperoxidase type 1 (120 U/mg) (HRP), N-acetylcysteine, deferoxamine and D-mannitol fromSigma (St. Louis, MO, USA); F12 medium, peni-cillin, streptomycin and gentamicin from GibcoBRL (Gaithersburg, MD); epidermal growth fac-tor from Upstate (Lake Placid, NY, USA);cholera toxin from List Biological Laboratories(Campbell, CA, USA); all rac-alpha-tocopherolwas from Fluka (Buchs, Switzerland).
2.2. Cell culture
Experiments were performed using the humanbronchial epithelial cell line HBE1. These cellswere isolated from normal donor and immortal-ized with papilloma virus (HPV18) genes 6 andE7 (Yankaskas et al., 1993). Immortalized HBE1

M. Nardini et al. / Toxicology 170 (2002) 173–185 175
line express differentiated phenotypic propertiesthat approximate those of respiratory tract nativeepithelium. Cells were grown as monolayer cul-tures in F12 medium supplemented with penicillin(60 U/ml), streptomycin (60 �g/ml), gentamicin(50 �g/ml), insulin (5.0 �g/ml), transferrin (5 �g/ml), epidermal growth factor (20 ng/ml), dexam-ethasone (0.1 �M), cholera toxin (20 ng/ml) andbovine hypothalamus extract (30 �g/ml) at 37 °Cin an atmosphere of 95% air and 5% CO2 as morecompletely described elsewhere (Robinson andWu, 1991).
2.3. Antioxidant supplementation
Cells were supplemented with all rac-alpha-to-copherol (TOC), dissolved in ethanol, 48 h beforeacrolein treatment. The maximum final concentra-tion of ethanol used (0.025%) had no effect onany of the variables reported herein. A 48 hpreincubation time with all rac-TOC was selectedon the basis of uptake experiments performed onHBE1 cells (data not shown). Supplementationwith 25 �M all rac-TOC resulted in cell levels of4.3�0.7 nmoles alpha-tocopherol per mg proteinafter 48 h. Ascorbic acid (AA), dissolved in PBS,was added to the culture medium 7 h beforeacrolein treatment, based on loading data re-ported in the literature (Savini et al., 2000). Sup-plementation with 100 �M AA resulted in cellularlevels of 180.5�10.5 ng AA per mg protein after7 h. N-acetyl cysteine (NAC), dissolved in PBSand neutralized with sodium hydroxide, wasadded to the culture medium 7 h before acroleinexposure.
2.4. Acrolein exposure
All experiments were performed with confluentmonolayers grown in 12-well plates (approxi-mately 1.2×105 cells per cm2). Before acroleinexposure, cells were cultured in F12 medium with-out growth factors for 12 h. Growth factors depri-vation (up to 48 h) did not result in anysignificant loss of cell viability, as measured bylactate dehydrogenase (LDH) leakage (Section2.9). For antioxidants supplementation, TOC andAA were added to the medium at appropriate
times to reach the total preincubation times re-ported above (48 h for TOC, 7 h for AA). Beforeexposure, cells were washed three times with PBSto eliminate antioxidants from the medium, thentreated for 30 min with acrolein in Krebs–Ringer’s buffer (137 mM NaCl, 4.9 mM KCl, 0.5mM CaCl2, 1.2 mM MgSO4, 5.7 mM sodiumphosphate, 5.5 mM glucose, pH 7.4). In all exper-iments parallel controls were performed withoutacrolein. After acrolein treatment, cells werewashed three times with PBS and incubated inF12 medium without growth factors, in the pres-ence or in the absence of added antioxidants.
2.5. Glutathione assay
GSH measurements were performed on celllysates by high performance liquid chromatogra-phy (HPLC) coupled with fluorimetric detectionafter derivatization with monobromobimane, aspreviously described (van der Vliet et al., 1999).Glutathione content was quantified as nmoles glu-tathione per mg protein. Protein content was mea-sured by the method of Lowry (Lowry et al.,1951).
2.6. Analysis of intracellular oxidants generation
The generation of intracellular oxidants wasdetermined spectrofluorometrically following theformation of a fluorescent derivative of 2�-7�-dichlorofluorescin (DCFH) (Slater et al., 1995).Briefly, at various times after acrolein exposure,cells were incubated with 25 �M DCFH-DA,(previously dissolved in ethanol, 0.01% ethanolfinal concentration) for 30 min at 37 °C. At theend of the incubation, cells were washed threetimes with PBS, then lysed in 0.5% Triton X-100,0.1% CHAPS, 0.1 mM EDTA in PBS, pH 7.0 for15 min and scraped from the plate. Cell lysateswere centrifuged at 15 000×g for 10 min at 4 °C,and the fluorescence intensity of the supernatantwas measured (excitation at 485 nm, emission at520 nm). Cellular oxidants level was expressed asrelative DCF fluorescence per �g of protein. Toassess for possible interferences in DCF fluores-cence by alpha-tocopherol, AA and acrolein, oxi-dation of DCFH was induced in solution by

M. Nardini et al. / Toxicology 170 (2002) 173–185176
hydrogen peroxide in a reaction catalyzed byhorseradish peroxidase (HRP). The fluorescenceinduced by hydrogen peroxide (0–40 �M) in thepresence of DCFH-DA (0.5 mM) and HRP (60U) was measured in the absence and in the pres-ence of alpha-tocopherol (25 �M), AA (up to 100�M) or acrolein (25 �M).
In another set of experiments, intracellular oxi-dants generation was measured by the ferrous ionoxidation-xylenol orange method (Nourooz-Zadeh et al., 1994) using a commercial kit(Bioxytech, Portland, OR). Briefly, at the end ofthe incubation, cells were washed three times withPBS, then lysed in 0.5% Triton X-100, 4 mMBHT in PBS. Analysis was performed on cellextracts according to manufacturer’s instruction.
2.7. Apoptosis detection by annexin V bindingassay
The assay was performed using FITC-labeledannexin V apoptosis detection kit (Pharmingen,San Diego, CA) according to the manufacturer’sdirections. Briefly, after trypsinization, cells werewashed in cold PBS and resuspended in assaybuffer. One hundred micro liter aliquots (corre-sponding to ca. 1×105 cells) were withdrawn foranalysis. After annexin V-FITC and propidiumiodide (PI) staining, samples were analyzed on aBecton–Dickinson (San Jose, CA) FACScan flowcytometer. Four cell populations were identified,according to the usual interpretation: control pop-ulation in lower-left quadrant (FITC−/PI−); earlyapoptotic population in lower-right quadrant(FITC+/PI−); necrotic population in upper-leftquadrant (FITC−/PI+); late apoptotic populationin upper-right quadrant (FITC+/PI+). In thiswork results are reported as percentage of totalapoptotic cells (early plus late apoptotic cells),which was obtained as the sum of cells in thelower-right and upper-right quadrants. The resultsobtained were qualitatively similar if only earlyapoptotic cells were considered.
2.8. Analysis of DNA fragmentation by TUNELassay
DNA fragmentation was analyzed by a termi-
nal deoxynucleotidyl transferase (TdT)-mediateddUTP Nick End-labeling (TUNEL) assay kit ac-cording to the manufacturer’s instructions (Apo-direct kit, Pharmingen, San Diego, CA). Aftertrypsinization, cells were washed in cold PBS andfixed in 1% (w/v) paraformaldehyde in PBS, andtreated according to the manufacturer’s proce-dure. Cells were treated with FITC-dUTP in thepresence of TdT enzyme that incorporates FITC-dUTP into the 3-OH-DNA ends found in apop-totic cells. Cells are then stained with PI for totalDNA and analyzed by flow cytometry on a Bec-ton Dickinson (San Jose, CA) FACScan flowcytometer.
2.9. Cell �iability assay
Cell viability was measured by LDH leakagefrom damaged cells into the culture medium, andexpressed as a percentage of total cellular activity.LDH activity was determined at 30 °C as thechange in absorbance at 340 nm, using 0.18 mMNADH and 0.72 mM pyruvate as substrates in 50mM K-phosphate buffer, pH 7.4.
2.10. Statistical analysis and data presentation
Data are presented as mean�S.D. of at leastthree separate experiments, as indicated. Statisti-cal analyses were performed using a one-factor analysis of variance (ANOVA) andScheffe’s method for multiple comparison. Proba-bility of P�0.05 was considered statistically sig-nificant.
3. Results
3.1. Acrolein induces apoptotic cell death inHBE1 cells
Apoptosis is characterized by a variety of mor-phological features. Changes in the plasma mem-brane are one of the earliest of these features. Inapoptotic cells, the membrane phospholipid PS istranslocated from the inner to the outer leaflet ofthe plasma membrane, thereby exposing PS to theexternal cellular environment. Annexin V, a phos-

M. Nardini et al. / Toxicology 170 (2002) 173–185 177
pholipid-binding protein with high affinity for PS,binds to cells with exposed PS. Annexin V conju-gated to the fluorochrome FITC was used todetect apoptosis in HBE1 cells. Fig. 1 showstypical diagrams of FITC-annexin V/PI flow cy-tometry obtained after acrolein exposure. Indeed,phosphatidyl serine externalization is a feature ofacrolein induced cell death in HBE1. A dose-de-pendent increase in early apoptotic population
(lower right quadrant, annexin V positive cells)was observed after 24 h incubation. Fig. 2Ashows that acrolein concentrations �10 �M in-duced a significant apoptotic response, as mea-sured by annexin V binding assay. Following 25�M acrolein-treatment, apoptosis was unde-tectable at 12 h, while increasing apoptosis levelswere observed at 18 and 24 h (Fig. 2B). Cellviability measurements indicated the absence of
Fig. 1. Contour diagram of FITC-annexin V/PI flow cytometry of HBE1 cells after acrolein treatment. HBE1 control cells wereexposed to acrolein for 30 min. At 24-h incubation after acrolein treatment, apoptosis was measured by annexin V binding assayusing a FACScan flow cytometer. (A) No acrolein; (B) +acrolein 10 �M; (C) +acrolein 25 �M. One representative experiment outof at least three is presented. Living cells (FITC−/PI−), lower left quadrant; early apoptotic cells (FITC+/PI−), lower rightquadrant; late apoptotic cells (FITC+/PI+), upper right quadrant; necrotic cells (FITC−/PI+), upper left quadrant.

M. Nardini et al. / Toxicology 170 (2002) 173–185178
Fig. 2. Dose–response and time course of acrolein-inducedapoptosis measured by annexin V flow cytometric assay. (A)HBE1 cells were exposed to 5–25 �M acrolein for 30 min. At24 h incubation after acrolein treatment, apoptosis was mea-sured by Annexin V binding assay with a FACScan flowcytometer. B, cells were exposed to 25 �M acrolein for 30 minand apoptosis was measured by Annexin V binding assay atthe indicated times after acrolein treatment. *P�0.05 com-pared with untreated cells. Data are mean�S.D. of threeindependent experiments.
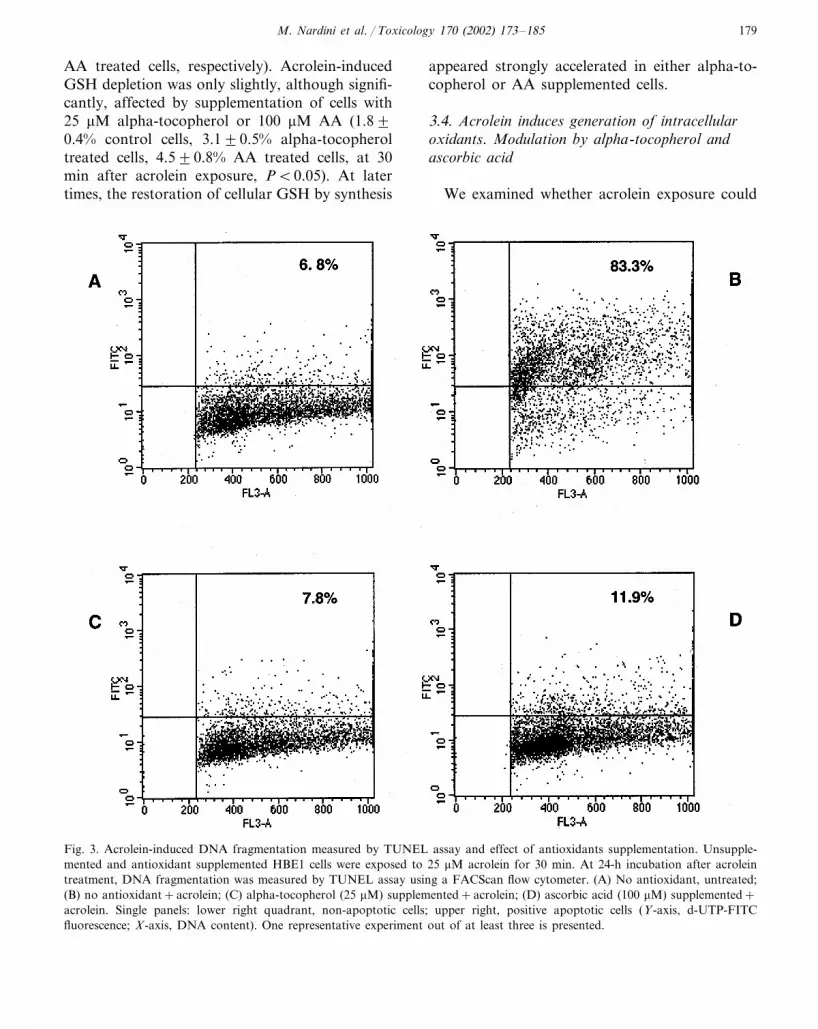
In the presence of TdT enzyme, apoptotic cellsincorporate FITC-dUTP into the 3-OH-DNAends. Acrolein treatment induced extensive DNAfragmentation, measured 24 h after acrolein expo-sure, as shown by the marked incorporation ofFITC (Fig. 3, panel B) in respect to untreatedcells (panel A).
3.2. Effects of alpha-tocopherol and ascorbic acidon acrolein-induced apoptosis
Supplementation of HBE1 cells with either al-pha-tocopherol or AA strongly inhibited acrolein-induced apoptosis, measured by annexin Vbinding (Fig. 4), in a dose-dependent manner. At25 �M alpha-tocopherol and 100 �M AA, theinhibition was almost complete. The strong in-hibitory effect of alpha-tocopherol and AA onacrolein-induced apoptosis was further confirmedby DNA fragmentation analyses by TUNEL as-say (Fig. 3, panels C and D). At 24 h incubationafter acrolein treatment, 25 �M alpha-tocopheroland 100 �M AA inhibited DNA fragmentation by88.4�3.0 and 87.3�4.5%, respectively.
3.3. Effect of acrolein on intracellular GSHle�els: modulation by alpha-tocopherol andascorbic acid
Acrolein is a potent electrophile that causes amarked GSH depletion (Kehrer and Biswal, 2000;Ku and Billings, 1986). To examine the effect ofacrolein treatment on the intracellular redoxstatus (as reflected by GSH levels), intracellularGSH levels were measured at different times afteracrolein exposure. As shown in Fig. 5, only 1.8�0.4% of initial GSH level was detected 30 minafter acrolein treatment. Oxidized GSH was unde-tectable, indicating that GSH depletion was notcoupled with increased levels of GSSG. Intracellu-lar GSH levels were partially restored in non-sup-plemented cells to 11.9�1.7% of initial levelsafter 60 min and 29.0�2.0% after 2 h. Supple-mentation with media containing either 25 �Malpha-tocopherol or 100 �M AA did not signifi-cantly affect basal intracellular GSH levels(13.6�0.6, 12.9�0.5, 13.8�0.9 nmoles/mgprotein for control, alpha-tocopherol or 100 �M
significant LDH leakage in culture medium up to22 h incubation after treatment with acrolein inthe range 5–25 �M. Significant LDH release wasobserved at high acrolein concentrations (�50�M) (42.6�1.7% release at 9 h incubation aftertreatment with 50 �M acrolein). Based on theseresults, 25 �M acrolein (corresponding to approx-imately 0.1 picomol acrolein/cell) was used in ourexperiments in the remainder of the study.
The apoptotic response of HBE 1 cells to acro-lein exposure was confirmed by analysis of DNAfragmentation by TUNEL assay. The assay wasperformed using a FITC-labeled deoxyuridinetriphosphate nucleotides (FITC-dUTP) for label-ing DNA breaks and PI for staining total DNA.
M. Nardini et al. / Toxicology 170 (2002) 173–185 179
AA treated cells, respectively). Acrolein-inducedGSH depletion was only slightly, although signifi-cantly, affected by supplementation of cells with25 �M alpha-tocopherol or 100 �M AA (1.8�0.4% control cells, 3.1�0.5% alpha-tocopheroltreated cells, 4.5�0.8% AA treated cells, at 30min after acrolein exposure, P�0.05). At latertimes, the restoration of cellular GSH by synthesis
appeared strongly accelerated in either alpha-to-copherol or AA supplemented cells.
3.4. Acrolein induces generation of intracellularoxidants. Modulation by alpha-tocopherol andascorbic acid
We examined whether acrolein exposure could
Fig. 3. Acrolein-induced DNA fragmentation measured by TUNEL assay and effect of antioxidants supplementation. Unsupple-mented and antioxidant supplemented HBE1 cells were exposed to 25 �M acrolein for 30 min. At 24-h incubation after acroleintreatment, DNA fragmentation was measured by TUNEL assay using a FACScan flow cytometer. (A) No antioxidant, untreated;(B) no antioxidant+acrolein; (C) alpha-tocopherol (25 �M) supplemented+acrolein; (D) ascorbic acid (100 �M) supplemented+acrolein. Single panels: lower right quadrant, non-apoptotic cells; upper right, positive apoptotic cells (Y-axis, d-UTP-FITCfluorescence; X-axis, DNA content). One representative experiment out of at least three is presented.

M. Nardini et al. / Toxicology 170 (2002) 173–185180
Fig. 4. Effect of antioxidants on acrolein-induced apoptosis.Unsupplemented and antioxidant supplemented HBE1 cellswere exposed to 25 �M acrolein for 30 min. At 24-h incuba-tion after acrolein treatment, apoptosis was measured byannexin V binding assay using a FACScan flow cytometer andexpressed as percentage taken as 100% the value measured inacrolein-treated control cells, without antioxidants supplemen-tation. Data are mean�S.D. of three independent experi-ments. *P�0.05 compared with acrolein-treated cells withoutantioxidants.
or acrolein itself, we measured the fluorescenceinduced by hydrogen peroxide (under cell-freeconditions) in the presence of DCFH-DA andHRP, in the absence and in the presence of alpha-tocopherol, AA or acrolein. As shown in Fig. 6B,alpha-tocopherol (25 �M), AA (100 �M) andacrolein (25 �M) did not significantly affect DCFfluorescence in our system. In addition, the incu-bation of acrolein (25 �M) in Krebs–Ringer’s buffer for 30 min at 37 °C did notinduce any detectable increase in DCF fluores-cence, as assayed in the presence of DCFH-DAand HRP, indicating that acrolein does not gener-ate prooxidant species in the incubation mediumitself. Similar results were obtained using theFe3+ -xylenol orange complex assay to detect in-tracellular oxidants species generation (data notshown).
The involvement of oxidant species generationin acrolein-induced apoptosis was further exam-ined evaluating the ability of extracellular addi-tion of either catalase and superoxide dismutaseon acrolein-induced apoptosis. Hydrogen perox-ide is freely permeable through cell membranesand, therefore, it readily diffuses out of the cells ifgenerated intracellularly, while superoxide anionsgo through the membrane via anion selectivepores. Therefore, the action of extracellularlyadded catalase and superoxide dismutase is ex-pected to decrease the intracellular concentrationof these oxidant species. Indeed, the addition of
lead to the generation of intracellular prooxidantspecies in HBE1 cells. As shown in Fig. 6A,acrolein treatment was followed by a 5 to 6-foldincrease in DCF fluorescence during the first hourafter exposure, slightly higher increases occurringat longer incubation times of up to 6 h. Pre-incu-bation of cells with media containing alpha-toco-pherol (25 �M) or AA (100 �M) stronglyinhibited acrolein-induced generation of intracel-lular prooxidant species. To rule out possibleinterferences in DCF fluorescence by antioxidants
Fig. 5. Glutathione levels after acrolein treatment and effect of antioxidants. HBE1 cells supplemented without or with antioxidants(25 �M alpha-tocopherol; 100 �M ascorbic acid) were exposed to 25 �M acrolein for 30 min. At various times after exposure toacrolein, total GSH levels were quantified (nmoles glutathione per mg protein) and expressed relatively to those prior to acroleintreatment for each pretreatment group. Results are mean�S.D. of three separate experiments. *P�0.05 compared with untreatedcells at the same incubation time.

M. Nardini et al. / Toxicology 170 (2002) 173–185 181
Fig. 6. Intracellular oxidants generation after acrolein-treat-ment and effect of antioxidants. (A) HBE1 cells supplementedwithout or with antioxidants (25 �M alpha-tocopherol; 100�M ascorbic acid) were exposed to 25 �M acrolein for 30 min.Intracellular prooxidants generation was measured by DCFH-DA assay as described in Section 2. Data are mean�S.D. ofthree separate experiments. *P�0.05 compared with un-treated cells at the same incubation time. (B) Under cell-freeconditions, hydrogen peroxide at the indicated concentrationswas reacted with DCFH-DA (0.5 mM) and HRP (60 U) in theabsence and in the presence of alpha-tocopherol (25 �M),ascorbic acid (100 �M) or acrolein (25 �M) and fluorescencewas measured. Data are mean�S.D. of three independentdeterminations.
membrane, failed to inhibit acrolein-inducedapoptosis at any extent. Collectively, these datasuggest a role for intracellularly generated oxidantspecies in acrolein induced apoptotic response.
4. Discussion
In the present study, we report that acroleininduces a cell death pathway in human bronchialepithelial cells which displays several features ofapoptotic cell death. Moreover, our results illus-trate that this response seems to be related to theintracellular generation of oxidants, and can beattenuated by preloading cells with antioxidants.Indeed, we found that cell supplementation withalpha-tocopherol and AA at concentrations closeto those in human plasma, inhibited both thegeneration of pro-oxidant species and acrolein-re-lated apoptosis in human bronchial epithelialcells. In the same way, iron chelator (DEF) andoxidant scavengers such as CAT, SOD and NACsignificantly inhibited acrolein-induced apoptosis.
Fig. 7. Effect of superoxide dismutase, catalase, deferroxam-ine, D-mannitol and N-acetyl cysteine on acrolein-inducedapoptosis. HBE1 cells were pretreated separately with defer-roxamine (DEF) (90 min, 5 �M), D-mannitol (MANN) (90min, 20 mM), N-acetyl cysteine (NAC) (7 h, 1 mM), superox-ide dismutase (SOD) (30 min, 500 U/ml), catalase (CAT) (30min, 2000 U/ml), then exposed to 25 �M acrolein for 30 min,in the presence of the inhibitors. After 24-h incubation, with orwithout inhibitors, apoptosis was measured by annexin Vbinding assay using a FACScan flow cytometer and expressedas percentage of acrolein-treated control cells, without in-hibitors. Data are mean�S.D. of three independent experi-ments. *P�0.05 compared with acrolein treated cells withoutinhibitors.
either catalase (1500 U/ml) and superoxide dismu-tase (500 U/ml) during incubation after acroleinexposure was found to strongly inhibit acrolein-induced apoptosis (75.1�5.8 and 73.4�4.1% in-hibition, respectively), as measured by annexin Vbinding assay (Fig. 7). Moreover, preloading cellswith the iron chelator deferroxamine (5 �M) re-sulted in 66.6�3.2% apoptosis inhibition. Cellsupplementation with the antioxidant N-acetylcysteine (NAC) (1 mM) also resulted in significantapoptosis inhibition. On the contrary, supplemen-tation with D-mannitol (20 mM), a hydroxyl radi-cal scavenger which does not cross the cell

M. Nardini et al. / Toxicology 170 (2002) 173–185182
However, the inhibition observed by NAC mightalso be due, at least partially, to its ability inscavenging aldehydes. Furthermore, we observedthat 1 h treatment with 250 �M hydrogen perox-ide also induced an apoptotic response (52.6�3.1% apoptotic cells) in HBE 1 cells, as measuredafter 24 h incubation by annexin V binding (un-published observation). Several previous studiesdemonstrated acrolein-induced intracellular gener-ation of free radicals and lipid peroxidation(Adams and Klaidman, 1993; Patel, 1987), eitherthrough metabolism of acrolein or its adduct withGSH (glutathionylpropionaldehyde) or by rapiddepletion of cellular GSH. GSH depletion com-promises the activity of GSH peroxidases, whichuse GSH as a cosubstrate to reduce hydrogenperoxide and lipid peroxide levels. Hydrogen per-oxide and lipid peroxides, in the presence of iron,can be cleaved to form free radical species. Al-though GSH has been suggested to play a role inapoptosis, whether changes in cellular levels ofthis tripeptide are a direct effector or a secondarymodulating influence has not been fully ascer-tained (Uchida et al., 1999b; Sato et al., 1995;Yan et al., 1995). Recently, GSH depletion hasbeen described to induce apoptosis through thegeneration of ROS and oxidative stress in rathepatocytes and lung fibroblasts (Domenicotti etal., 2000; Aoshiba et al., 1999). Our data appearto support this suggestion, as the increasedformation of intracellular oxidant species seems tobe an important event triggering the acrolein-in-duced apoptotic response. Also, more direct andspecific mechanisms might be involved in acrolein-induced apoptosis other than oxidants gen-eration, such as phosphatases inhibition. How-ever, we observed a detectable phosphatases inhi-bition only at high acrolein concentrations(100–200 �M) in human lung epithelial cells (un-published results).
Supplementation with either alpha-tocopherolor AA failed to prevent the fast acrolein-inducedloss of intracellular GSH, strongly suggesting thatthis depletion results primarily from direct reac-tion of acrolein with GSH (Ku and Billings,1986), which occurs spontaneously and rapidly atneutral pH (Adams and Klaidman, 1993).Moreover, GSH transferases have also been re-ported to catalyze the conjugation of acrolein
with reduced glutathione (Berhane and Manner-vick, 1990).
The acrolein-induced GSH depletion is fol-lowed by a recovery phase, likely due to de novosynthesis (Horton et al., 1997) and supplementa-tion with either alpha-tocopherol or AA positivelyaffected GSH recovery. Indeed, acrolein has beenobserved to induce gene expression of phase IIdetoxifying enzymes and to induce intracellularGSH synthesis, through enhanced gene expressionof �-glutamylcysteine synthetase, a rate-limitingenzyme of GSH biosynthesis (Uchida, 1999a). Asimilar effect of recovery after GSH depletion hasbeen reported for human lung adenocarcinomacells after acrolein exposure (Horton et al., 1997)and for rat liver epithelial cells after treatmentwith the highly reactive unsaturated aldehyde 4-hydroxy-2-nonenal (Uchida et al., 1999b). Thehigher GSH recovery in alpha-tocopherol and AAsupplemented cells may be due to the free radicalsscavenging activity of these antioxidants, resultingin a sparing effect of newly synthesized GSH.Further, cells undergoing apoptosis may blockGSH synthesis at a certain point of the cell deathprogram and this could explain the lower GSHrecovery observed in unsupplemented acrolein-treated apoptotic cells in respect to antioxidant-supplemented, acrolein treated non-apoptoticcells.
Acrolein is an ubiquitous pollutant in the envi-ronment reported to be highly toxic (InternationalAgency for Research on Cancer, 1995). It is alsothe major unsaturated aldehyde in gas-phasecigarette smoke. Increased damage to respiratorytract epithelial cells exposed to whole and gas-phase cigarette smoke has been reported, such asdecreased cell attachment and cell proliferation,increased apoptosis (Lannan et al., 1994; Hoshinoet al. 2001), associated with GSH depletion andfree radical species generation (Spencer et al.,1995). Moreover, lower plasma concentrations ofselected antioxidants have been reported in smok-ers in respect to non smokers, which may con-tribute to their higher risk of pulmonary andvascular diseases (Lakier, 1992; Thompson et al.,1992; Stampfer et al., 1993; Anderson, 1991). Areduced plasma concentration and an increasedturnover of AA have been reported in smokers

M. Nardini et al. / Toxicology 170 (2002) 173–185 183
compared with non smokers (Anderson, 1991;Cross et al., 1993; Kallner et al., 1981). The hereinreported ability of acrolein to induce apoptosis inhuman bronchial epithelial cells may be an impor-tant pathogenic mechanism of acrolein-relatedrespiratory tract toxicity and may contribute tothe development of a new strategy to preventacrolein-induced respiratory tract injury. Further,the present study shows evidence that antioxi-dants, particularly alpha-tocopherol and ascor-bate, can inhibit acrolein-induced oxidantsgeneration and apoptosis in cultured humanbronchial epithelial cells. Bronchial lining fluidcontains antioxidant molecules, which may coun-teract the cytotoxic effects of acrolein present insmog and smoke. However, this antioxidant poolis known to be decreased in disease-related condi-tions or by cigarette smoking (Carp and Janoff,1978). Thus, cigarette smoke may cause apoptosisof bronchial epithelial cells in subjects with infl-ammatory lung diseases and chronic smokers.Our results suggest that antioxidant supplementa-tion may be of therapeutic benefit in protectingthe respiratory tract from damaging effects ofacrolein contained in smog and cigarette smoke.Due to the emerging role of acrolein as a lipidperoxidation product and mediator of oxidativedamage in a variety of human diseases, themodulation of acrolein-induced apoptotic re-sponse by antioxidants, mainly alpha-tocopheroland AA, may possibly play a more general role inthe modulation and onset of those pathologicalconditions associated with oxidative stress. Al-though the inhibitory and protective effects ob-served for alpha-tocopherol and AA may beattributable to a general antioxidant action, nev-ertheless the contribution of more direct and spe-cific effects of these antioxidants in themodulation of acrolein-induced apoptosis cannotbe ruled out.
Finally, it should be noted that, in our experi-mental conditions, cells were depleted of growthfactors 12 h before acrolein exposure and might,therefore, show a susceptibility to apoptotic stim-uli not comparable to the in vivo situation.Hence, caution should be exercised in extrapolat-ing the results of this study to in vivo respiratorytract conditions.
Acknowledgements
The authors thank Dr Carol Oxford (Depart-ment of Medical Pathology, UC Davis, CA) forFACScan analyses. This work was supported bygrants from NIH (HL47528; HL07013 andHL60812) and the University of California To-bacco-Related Disease Research Program (7RT-0160 and 7RT-0167).
References
Adams, J.D. Jr, Klaidman, L.K., 1993. Acrolein-induced oxy-gen radical formation. Free Radic. Biol. Med. 15, 187–193.
Altschuler, A.P., Mc Pherson, S.P., 1963. Spectrophotometricanalysis of aldehydes in the Los Angeles atmosphere. J. AirPollut. Control Assoc. 13, 109–111.
Anderson, R., 1991. Assessment of the roles of vitamin C,vitamin E, and beta-carotene in the modulation of oxidantstress mediated by cigarette smoke-activated phagocytes.Am. J. Clin. Nutr. 53, 358S–361S.
Anderson, M.M., Hazen, S.L., Hsu, F.F., Heinecke, J.W.,1997. Human neutrophils employ the myeloperoxidase-hy-drogen peroxide-chloride system to convert hydroxy-aminoacids into glycoaldehyde, 2-hydroxypropanal, and acrolein:a mechanism for the generation of highly reactive alpha-hydroxy and alpha,beta-unsaturated aldehydes by phagoc-ites at sites of inflammation. J. Clin. Invest. 99, 424–432.
Aoshiba, K., Yasui, S., Nishimura, K., Nagai, A., 1999. Thioldepletion induces apoptosis in cultured lung fibroblasts.Am. J. Resp. Cell Mol. Biol. 21, 54–64.
Aranyi, C., O’Shea, W.J., Graham, J.A., Miller, F.J., 1986.The effects of inhalation of organic chemical air contami-nants on murine lung host defenses. Fundam. Appl. Toxi-col. 6, 713–720.
Arumugam, N., Sivakumar, V., Thanislass, J., Pillai, K.S.,Devaraj, S.N., Devaraj, H., 1999a. Acute pulmonary toxic-ity of acrolein in rats-underlying mechanism. Toxicol. Lett.104, 189–194.
Arumugam, N., Thanislass, J., Ragunath, K., Devaraj, S.N.,Devaraj, H., 1999b. Acrolein-induced toxicity-defective mi-tochondrial function as a possible mechanism. Arch. Envi-ron. Contam. Toxicol. 36, 373–376.
Astry, C.L., Jakab, G.J., 1983. The effects of acrolein exposureon pulmonary antibacterial defenses. Toxicol. Appl. Phar-macol. 67, 49–54.
Ayer, H.E., Yeager, D.W., 1982. Irritants in cigarette smokeplumes. Am. J. Public Health 72, 1283–1285.
Beauchamp, R.O., Andjelkovich, D.A., Kligerman, A.D.,Morgan, K.T., Heck, H.D., 1985. A critical review of theliterature on acrolein toxicity. Crit. Rev. Toxicol. 14, 309–380.

M. Nardini et al. / Toxicology 170 (2002) 173–185184
Berhane, K., Mannervick, B., 1990. Inactivation of thegenotoxic aldehyde by human glutathione transferases ofclasses alpha, mu, and pi. Mol. Pharmacol. 37, 251–254.
Caldwell, J.C., Woodruff, R., Morello-Frosch, R., Axelrad,D.A., 1998. Application of health information to haz-ardous air pollutants modeled in EPA’s cumulative expo-sure project. Toxicol. Ind. Health 14, 429–454.
Calingasan, N.Y., Uchida, K., Gibson, G.E., 1999. Protein-bound acrolein: a novel marker of oxidative stress inAlzheimer’s disease. J. Neurochem. 72, 751–756.
Carp, H., Janoff, A., 1978. Possible mechanisms of emphy-sema in smokers. In vitro suppression of serum elastase-in-hibitory capacity by fresh cigarette smoke and itsprevention by antioxidants. Am. Rev. Resp. Dis. 118,617–621.
Cross, C.E., O’Neill, C.A., Reznick, A.Z., 1993. Cigarettesmoke oxidation of human plasma constituents. Ann. NewYork Acad. Sci. 686, 72–89.
Domenicotti, C., Dimitri, P., Vitali, A., Nitti, M., D’Abramo,C., Cottalasso, D., Maloberti, G., Biasi, F., Poli, G.,Chiarpotto, E., Marinari, U.M., Pronzato, M.A., 2000.Glutathione depletion induces apoptosis of rat hepatocytesthrough activation of protein kinase C novel isoforms anddependent increase in AP-1 nuclear binding. Free Radic.Biol. Chem. 29, 1280–1290.
Eiserich, J.P., van der Vliet, A., Handelman, G.J., Halliwell,B., Cross, C.E., 1995. Dietary antioxidants and cigarettesmoke-induced biomolecular damage: a complex interac-tion. Am. J. Clin. Nutr. 62, 1490S–1500S.
Finkelstein, E.I., Nardini, M., van der Vliet, A., 2001. Inhibi-tion of neutrophil apoptosis by acrolein: a contributingfactor in tobacco-related inflammatory lung disease? Am.J. Physiol. 281, 732–739.
Fraiser, L.H., Kanekal, S., Kehrer, J.P., 1991. Cyclophos-phamide toxicity. Characterising and avoiding the prob-lem. Drugs 42, 781–795.
Green, G.M., 1985. Mechanisms of tobacco smoke toxicity onpulmonary macrophage cells. Eur. J. Resp. Dis. Suppl.139, 82–85.
Horton, N.D., Mamiya, B.M., Kehrer, J.P., 1997. Relation-ships between cell density, glutathione and proliferation ofA549 human lung adenocarcinoma cells treated with acro-lein. Toxicology 122, 111–122.
Hoshino, Y., Mio, T., Nagai, S., Miki, H., Ito, I., Izumi, T.,2001. Cytotoxic effects of cigarette smoke extract on analveolar type II cell-derived cell line. Am. J. Physiol. LungCell Mol. Physiol. 281, L509L–516.
International Agency for Research on Cancer, 1995, Acrolein.IARC Monogr. Eval. Carcinog. Risks Hum. 63, 337–372.
Jakab, G.J., 1977. Adverse effect of a cigarette smoke compo-nent, acrolein, on pulmonary antibacterial defenses and onviral-bacterial interactions in the lung. Am. Rev. Resp.Dis. 115, 33–38.
Kallner, A.B., Harmann, D., Hornig, D.H., 1981. On therequirements of ascorbic acid in man: steady-state turnoverand body pool in smokers. Am. J. Clin. Nutr. 34, 1347–1355.
Kehrer, J.P., Biswal, S.S., 2000. The molecular effects ofacrolein. Toxicol. Sci. 57, 6–15.
Ku, R.H., Billings, R.E., 1986. The role of mitochondrialglutathione and cellular protein sulphydryls in formalde-hyde toxicity in glutathione-depleted rat hepatocytes. Arch.Biochem. Biophys. 247, 183–189.
Lakier, J.B., 1992. Smoking and cardiovascular disease. Am. J.Med. 93, 8S–12S.
Lannan, S., Donaldson, K., Brown, D., Mac Nee, W., 1994.Effect of cigarette smoke and its condensates on alveolarepithelial cell injury in vitro. Am. J. Physiol. 266, L92–L100.
Li, L., Holian, A., 1998. Acrolein: a respiratory toxin thatsuppresses pulmonary host defense. Rev. Environ. Health13, 99–108.
Li, L., Hamilton, R.F. Jr, Taylor, D.E., Holian, A., 1997.Acrolein-induced cell death in human alveolarmacrophages. Toxicol. Appl. Pharmacol. 145, 331–339.
Li, L., Hamilton, R.F. Jr, Holian, A., 1999. Effect of acroleinon human alveolar macrophage NF-�B activity. Am. J.Physiol. 277, L550–L557.
Lowry, O.H., Rosenbrough, N.J., Farr, A.L., Randall, R.J.,1951. Protein measurements with the Folin phenol reagent.J. Biol. Chem. 193, 265–275.
Nguyen, H., Finkelstein, E., Reznick, A., Cross, C., van derVliet, A., 2001. Cigarette smoke impairs neutrophil respira-tory burst activation by aldehyde-induced thiol modifica-tion. Toxicology 160, 207–217.
Nourooz-Zadeh, J., Tajaddini-Sarmadi, J., Wolff, S.P., 1994.Measurement of plasma hydroperoxide concentrations bythe ferrous oxidation-xylenol orange assay in conjuctionwith triphenylphosphine. Anal. Biochem. 220, 403–409.
Patel, J.M., 1987. Stimulation of cyclophosphamide-inducedpulmonary microsomal lipid peroxidation by oxygen. Tox-icology 45, 79–91.
Robinson, C.B., Wu, R., 1991. Culture of conducting airwayepithelial cells in serum-free medium. J. Tissue CultureMethods 13, 95–102.
Sato, N., Iwata, S., Nakamura, K., Hori, T., Mori, K., Yodoi,J., 1995. Thiol-mediated redox regulation of apoptosis.Possible roles of cellular thiols other than glutathione in Tcell apoptosis. J. Immunol. 154, 3194–3203.
Savini, I., Duflot, S., Avigliano, L., 2000. Dehydroascorbicacid uptake in a human keratinocyte cell line (HaCaT) isglutathione-independent. Biochem. J. 345, 665–672.
Slater, A.F.G., Nobel, S.I., Maellaro, E., Bustamante, J.,Kimland, M., Orrenius, S., 1995. Nitrone spin traps and anitroxide antioxidant inhibit a common pathway of thymo-cyte apoptosis. Biochem. J. 306, 771–778.
Spencer, J.P.E., Jenner, A., Chimel, K., Aruoma, O.I., Cross,C.E., Wu, R., Halliwell, B., 1995. DNA damage in humanrespiratory tract epithelial cells: damage by gas phasecigarette smoke apparently involves attack by reactivenitrogen species in addition to oxygen radicals. FEBS Lett.375, 179–182.
Stampfer, M.J., Hennekens, C.H., Manson, J.E., Colditz,G.A., Rosner, B., Willet, E.C., 1993. Vitamin E consump-

M. Nardini et al. / Toxicology 170 (2002) 173–185 185
tion and the risk of coronary disease in women. New Engl.J. Med. 328, 1444–1449.
Suzuki, D., Miyata, T., 1999. Carbonyl stress in the pathogen-esis of diabetic nephropathy. Intern. Med. 38, 309–314.
Thompson, R.L., Margetts, B.M., Wood, D.A., Jackson,A.A., 1992. Cigarette smoking and food and nutrientintakes in relation to coronary hearth disease. Nutr. Res.Rev. 5, 131–152.
Uchida, K., Kanematsu, M., Morimitsu, Y., Osawa, T.,Noguchi, N., Niki, E., 1998a. Acrolein is a product of lipidperoxidation reaction. J. Biol. Chem. 273, 16058–16066.
Uchida, K., Kanematsu, M., Sakai, K., Matsudam, T., Hat-tori, N., Mizuno, Y., Suzuki, D., Nogluchi Miyata, T.,Noguchi, N., Niki, E., Osawa, T., 1998b. Protein-boundacrolein: potential markers for oxidative stress. Proc. Natl.Acad. Sci. USA 95, 4882–4887.
Uchida, K., 1999a. Current status of acrolein as a lipidperoxidation product. Trends Cardiovasc. Med. 9, 109–113.
Uchida, K., Shiraishi, M., Naito, Y., Torii, Y., Nakamura, Y.,
Osawa, T., 1999b. Activation of stress signaling pathwaysby the end product of lipid peroxidation. J. Biol. Chem.274, 2234–2242.
van der Vliet, A., O’Neill, C.A., Cross, C.E., Koostra, J.M.,Volz, W.G., Halliwell, B., Louie, S., 1999. Determinationof low-molecular-mass antioxidant concentrations in hu-man respiratory tract lining fluids. Am. J. Physiol. LungCell Mol. Physiol. 276, 1289–1296.
World Health Organization, 1991. Environmental health crite-ria. In: Acrolein. Geneva: World Health Organization(publ. 127).
Yan, C.Y.I., Ferrari, G., Greene, L.A., 1995. N-acetylcysteine-promoted survival of PC 12 cells is glutathione-indepen-dent but transcription-dependent. J. Biol. Chem. 270,26827–26832.
Yankaskas, J.R., Haizlip, J.E., Conrad, M., Koval, D.,Lazarowsky, E., Paradiso, A.M., Rinehart, C.A., Sarkadi,B., Schlegel, R., Boucher, R., 1993. Papilloma virus im-mortalized tracheal epithelial cells retain a well-differenti-ated phenotype. Am. J. Physiol. 264, C1219–C1230.





























