Embed Size (px)
Citation preview

Cellular Microbiology (2003)
5
(12), 957–971 doi:10.1046/j.1462-5822.2003.00339.x
© 2003 Blackwell Publishing Ltd
Blackwell Science, LtdOxford, UKCMICellular Microbiology 1462-5822Blackwell Publishing Ltd, 20035
12957971
Original Article
G. A. Grassl et al.Invasin triggers IL-8 production via Rac1 and MAPK cascades
Received 13 June, 2003; revised 18 August, 2003; accepted 22August, 2003. *For correspondence. E-mail [email protected]; Tel. (+49) 7071 29 5440; Fax (+49) 7071 295440.
Activation of NF-
κκκκ
B and IL-8 by
Yersinia enterocolitica
invasin protein is conferred by engagement of Rac1 and MAP kinase cascades
Guntram A. Grassl,
1
Michael Kracht,
2
Agnès Wiedemann,
3,5
Elke Hoffmann,
2
Martin Aepfelbacher,
3
Christoph von Eichel-Streiber,
4
Erwin Bohn
1
and Ingo B. Autenrieth
1
*
1
Institut für Medizinische Mikrobiologie und Krankenhaushygiene, Universitätsklinikum Tübingen, D-72076 Tübingen, Germany
2
Institut für Pharmakologie, Medizinische Hochschule Hannover D-30623 Hannover, Germany,
3
Max von Pettenkofer-Institut für Hygiene und Medizinische Mikrobiologie, Universität München, D-80336 München, Germany,
4
Institut für Medizinische Mikrobiologie und Hygiene, Universität Mainz, D-55101 Mainz, Germany
Keywords:
Yersinia
, NF-
κ
B, IL-8, MAP kinase, Rac,bacteria
Summary
Yersinia enterocolitica
triggers activation of thenuclear factor (NF)-
κκκκ
B and production of the proin-flammatory chemokine interleukin (IL)-8 in intestinalepithelial cells. This activation is due to adhesion ofthe bacteria via their outer membrane protein invasinto the host cells. Using
Clostridium difficile
toxinsthat specifically inactivate small GTPases, and trans-fection of inhibitory proteins of the Rho-GTPases, wedemonstrate that Rac1, but not Cdc42 or Rho, isrequired for activation of NF-
κκκκ
B by invasin. Invasinactivated the mitogen activated protein kinases(MAPK) p38 and c-Jun N-terminal protein kinase(JNK) but not extracellular signal regulated kinase(ERK). The functional relevance of these pathways forinvasin-mediated IL-8 expression was assessed byprotein kinase inhibitors and dominant-negativekinase mutants. While NF-
κκκκ
B and JNK contribute toIL-8 transcription, p38 MAPK also acts through stabi-lization of IL-8 mRNA, as confirmed by quantitativeRT-PCR and electrophoretic mobility shift assays.Transfection experiments with I-
κκκκ
B kinase (IKK)1 and
IKK2 mutants indicate that the release of NF-
κκκκ
B fromits cytoplasmic inhibitor I-
κκκκ
B and its translocationinto the nucleus is mediated by these kinases. Ourdata identify Rac1 as a key intermediate in invasin-triggered IL-8 synthesis and demonstrate that maxi-mum IL-8 induction involves several MAP kinasecascades.
Introduction
Besides operating as a mechanical barrier, intestinal epi-thelial cells function as the ‘watchdogs’ for the innate andadaptive immune system (Eckmann
et al
., 1995). Uponinfection with enteropathogenic bacteria such as
Salmo-nella
,
Shigella
,
Escherichia coli
(EPEC) or
Yersinia
, intes-tinal epithelial cells produce a variety of proinflammatorycytokines including interleukin-8 (IL-8) (Jung
et al
., 1995;Savkovic
et al
., 1997). IL-8 is an important chemoattrac-tant and activator of polymorphonuclear leukocytes(PMNs), which play an important although ambiguous rolein infection with the aforementioned pathogens. Largenumbers of PMNs are found in
Yersinia
-inducedabscesses in intestinal tissue.
Yersinia
is resistant tophagocytosis by PMNs, and recruitment of these cellstogether with concurrent tissue damage may ultimatelyfacilitate the dissemination of the bacteria rather than theirdestruction (Autenrieth
et al
., 1996; McCormick
et al
.,1997).
The three
Yersinia
species
Y. pestis
,
Y. pseudotuber-culosis
and
Y. enterocolitica
are pathogenic to humans.The latter two cause gastrointestinal disorders such asenteritis and enterocolitis, and also lymphadenitis,reactive arthritis, erythema nodosum, uveitis andsepticaemia (Bottone, 1977; Cover and Aber, 1989).Pathogenicity of
Yersinia
is determined by virulence fac-tors that are encoded either by genes of the bacterialchromosome such as invasin (Inv) and attachment-inva-sion locus (Ail) or by genes present on the pYV (
Yers-inia v
irulence) plasmid (Cornelis, 1994) such as
Yersinia
adhesin A (YadA) as well as the
Yersinia
outer proteins(Yops)(Cornelis, 1998). Yops are composed of structuralproteins for the type three secretion system, and at leastsix distinct effector molecules which are translocated tothe host cell cytoplasm and thereby interfere with hostsignaling pathways.

958
G. A. Grassl
et al.
© 2003 Blackwell Publishing Ltd,
Cellular Microbiology
,
5
, 957–971
The outer membrane protein invasin is important in theearly stages of infection (Pepe and Miller, 1993b) for effi-cient translocation of the bacteria through the M cells andcolonization of the Peyer’s patches (Schulte
et al
., 2000b).The invasin proteins of
Y. enterocolitica
and
Y. pseudotu-berculosis
are highly homologous except for a homomul-timerization domain which is absent in
Y. enterocolitica
invasin (Dersch and Isberg, 1999; Hamburger
et al
.,1999). The 192 and 195 carboxy-terminal amino acids of
Y. pseudotuberculosis
and
Y. enterocolitica
invasin,respectively, are necessary and sufficient for binding to
β
1-integrins and induction of phagocytosis (Leong
et al
.,1990; Isberg and Leong, 1990; Schulte
et al
., 2000a;Wiedemann
et al
., 2001).We previously demonstrated that a recombinant immo-
bilized fusion protein of GST and the C-terminal 195amino acids of invasin, induced IL-8 synthesis by trigger-ing the degradation of I-
κ
B
α
and subsequent translocationof NF-
κ
B p50/p65 and p65/p65 dimers to the nucleus(Schulte
et al
., 2000a). Adhesion of the bacteria to thehost cell without internalization is adequate for the induc-tion of IL-8 gene expression, suggesting that invasin actsas a ligand for a receptor-mediated signaling pathway.
Production of IL-8 is largely controlled at the transcrip-tional level. A sequence spanning nucleotides (nt)
−
133to
−
1 within the 5
′
flanking region of the IL-8 gene issufficient and essential for transcriptional regulation(reviewed in: Mukaida
et al
., 1994). This sequence con-tains AP-1, C/EBP and NF-
κ
B binding sites. (Mukaida
et al
., 1990; Matsusaka
et al
., 1993; Neff
et al
., 2001). TheNF-
κ
B element is crucial for transcriptional activationwhereas AP-1 and C/EBP sites may not be relevant invarious cell types (Yasumoto
et al
., 1992; Matsusaka
et al
., 1993; Kunsch
et al
., 1994; Wu
et al
., 1997). NF-
κ
Bis induced by large numbers of stimuli such as cytokines,doublestranded RNA, phorbol esters and infectious micro-organisms (Baeuerle, 1998b; Mercurio and Manning,1999). Recent work demonstrated that infection of hostcells with pathogenic bacteria such as
Helicobacter pylori
,
Salmonella typhimurium
, EPEC or
Yersinia enterocolitica
triggers activation of NF-
κ
B (Keates
et al
., 1997; Munzen-maier
et al
., 1997; Sharma
et al
., 1998; Eaves-Pyles
et al
., 1999; Elewaut
et al
., 1999; Schulte
et al
., 2000a).The signaling pathways leading to IL-8 production trig-
gered by the proinflammatory cytokines IL-1 and TNF
α
are well characterized. It was recently shown that IL-1-induced IL-8 production is regulated by a complex inter-play of different MAP kinase cascades at the transcrip-tional level as well as at the post-transcriptional levelregulating mRNA stability (Holtmann
et al
., 1999; Hoff-mann
et al
., 2002). Recent publications demonstrated theinvolvement of bacterial products in epithelial IL-8 produc-tion. The LPS-induced IL-8 production signaling events viatoll like receptor (TLR)4 have been described in detail
(Medzhitov, 2001). Uropathogenic
E. coli
activate IL-8 viaP-fimbriae (Hedlund
et al
., 1999; Wullt
et al
., 2001), andflagellin of different enteropathogenic bacteria activatesIL-8 via TLR5 (Gewirtz
et al
., 2001; Donnelly and Steiner,2002). However the molecular events for IL-8 productioninduced by other bacterial products and the host cellreceptors involved in recognition are less clear.
The purpose of this study was to elucidate the signalingnetwork that orchestrates activation of NF-
κ
B and pro-duction of IL-8 by invasin. We report that IL-8 inductionby invasin-expressing bacteria or recombinant invasinrequires the Rho-GTPase Rac1 but not Cdc42 or Rho.Furthermore we demonstrate that JNK and p38 MAPkinases are activated by invasin and contribute to maxi-mum induction of IL-8 transcription and IL-8 mRNAstabilization.
Results
Inhibition of Rho-GTPases blocks Yersinia invasin-induced IL-8 production
Previous studies from this laboratory demonstrated that
Yersinia
invasin protein stimulates activation of NF-
κ
B pro-teins and expression of IL-8 mRNA (Kampik
et al
., 2000;Schulte
et al
., 2000a). Consistent with our previous obser-vations (Schulte
et al
., 1998; Kampik
et al
., 2000; Schulte
et al
., 2000a) we illustrate by three independentapproaches, namely plasmidless
Y. enterocolitica
(
Y.enterocolitica
pYV
–
),
E. coli
ectopically expressing invasin(
E. coli
HB101 pINV1914), or by recombinant invasin(GST-Inv195-coated beads), that invasin alone is clearlysufficient to induce IL-8 synthesis (Fig. 1A). Wildtype
Yers-inia
(
Y. enterocolitica
pYV
+
) did not induce IL-8 expres-sion, presumably because YopP proteins suppress hostcell signaling. In support of a specific induction of IL-8 byinvasin, an invasin-deficient
Yersinia
strain (
Y. enterocolit-ica
pYV
–
inv
–
),
E. coli
HB101 and GST-coated particleswere unable to trigger IL-8 production.
We showed that small GTPases play a key role intriggering phagocytosis of invasin-coated particles bymacrophages (Wiedemann
et al., 2001). Therefore, weinvestigated the role of GTPases in invasin-triggered sig-naling in epithelial cells leading to IL-8 induction. Weemployed C. difficile toxins TcdB10463, TcdB1470 and C.botulinum C3-transferase, which either specifically glyco-sylate or ADP-ribosylate and thereby inactivate distinctsmall GTPases. TcdB10463 inhibits Rho, Rac and Cdc42(Just et al., 1995), TcdB1470 targets Rac, Rap, Ral andR-Ras (Chaves-Olarte et al., 1997; Chaves-Olarte et al.,1999), and C3-transferase specifically inhibits Rho (Akto-ries et al., 1987; Sehr et al., 1998). HeLa cells were pre-treated with the different toxins and subsequently infectedwith invasin-expressing Y. enterocolitica pYV– or E. coli

Invasin triggers IL-8 production via Rac1 and MAPK cascades 959
© 2003 Blackwell Publishing Ltd, Cellular Microbiology, 5, 957–971
pINV1914 or stimulated with TNFα. IL-8 production wasdetermined by analysis of cell culture supernatants byELISA.
As shown in Fig. 1(B), TcdB1470 or TcdB10463 did notaffect TNFα-induced IL-8 secretion even at high concen-trations. In contrast, invasin-induced IL-8 production wasreduced in a dose-dependent manner in cells treated withvarious amounts of the toxins. To investigate whethertranslocation of NF-κB was affected, electromobility shiftassays (EMSAs) were performed. The data revealed thatin nuclear extracts from TcdB10463-treated cells infectedwith Y. enterocolitica pYV–, the binding activity to the IL-8 κB probe was reduced whereas in TcdB10463-treatedcells stimulated with TNFα, NF-κB binding activity wasunaffected (Fig. 1C). The data suggest that activation ofthe small GTPases is required for invasin-triggered NF-κBactivation. Treatment of the cells with C3-transferase (2–4 µg ml−1) did not alter the induced IL-8 levels (data notshown). Both TcdB10463 and TcdB1470 inhibit Rac, whichsuggests an important role for Rac in invasin-mediated IL-8 expression. However we cannot rule out that severaldistinct GTPases are utilized independently by invasin.
As the C. difficile toxins severely affect the host cellcytoskeleton we tested if adhesion of the bacteria to thehost cells was impaired by treatment with the toxins,which would explain the lower bacteria-triggered IL-8 pro-duction. HeLa cells were treated with TcdB10463 for 24 hand then infected with Y. enterocolitica pYV–. Figure 2 Ademonstrates that adhesion of Y. enterocolitica pYV– wasnot significantly affected by the presence of TcdB10463.This finding was verified by immunofluorescence micros-copy. Y. enterocolitica adhered to TcdB10463-treatedcells equally as well as to untreated cells (Fig. 2B). Incontrast, uptake into toxin-treated cells was blocked asdemonstrated by immunofluorescence microscopy(Fig. 2B).
Rac1 mediates invasin-induced activation of IL-8
To clarify the role of small GTPases in Yersinia triggeredIL-8 production, we cotransfected inhibitory constructs ofRac1 (N17Rac1) and Cdc42 (N17Cdc42) together with anIL-8 promoter-luciferase reporter construct and infectedthe cells with Y. enterocolitica pYV–. Activation of the IL-8
Fig. 1. Induction of IL-8 by Yersinia invasin protein. HeLa cells were infected with invasin-expressing bacteria or mutant bacterial strains or stimulated with TNFα (50 ng ml−1) or with protein coated beads. IL-8 in culture supernatants was determined by ELISA 4 h post stimulation. B–C. Inhibition of IL-8 synthesis in HeLa cells by C. difficile toxins. Cells were preincubated with TcdB1470 (inhibits Rac, Rap, Ral, R-Ras; B) or TcdB10463 (inhibits Rho, Rac, Cdc42; C) for 24 h prior to stimulation, and 4 h after stimulation IL-8 in culture supernatants was determined by ELISA. D. EMSA. Cells were treated with TcdB10463 as in (C) and 60 min after stimulation nuclear extracts were prepared and analyzed by EMSA with the IL-8 κB probe.

960 G. A. Grassl et al.
© 2003 Blackwell Publishing Ltd, Cellular Microbiology, 5, 957–971
promoter was impaired by N17Rac1 but not by N17Cdc42(Fig. 3A). This result confirms an important role of Rac1in invasin-induced IL-8 activation. Microinjection ofN17Rac1, N17Cdc42 and RhoRBD into HeLa cells andsubsequent infection with E. coli pINV1914 showed thatnuclear translocation of NF-κB was impaired in N17Rac1injected cells but not in cells injected with N17Cdc42 orRhoRBD (Fig. 3B-E and data not shown).
MAP kinases are activated by invasin
MAP kinases are necessary for IL-1- or TNFα-triggeredIL-8 induction (Holtmann et al., 1999) and they can beregulated by Rac1 (Mainiero et al., 2000). To determinethe role of p38 MAPK, JNK and ERK1/2 in invasin signal-ing, we first investigated whether MAPK were activatedby invasin. HeLa cells were infected with Y. enterocoliticapYV–, E. coli pINV1914 or E. coli, or were stimulated withTNFα. Whole HeLa cell lysates were separated by SDS–PAGE and immunoblots were performed with an antibody
against the activated, phosphorylated form of p38 MAPK.In cells stimulated with Y. enterocolitica pYV– or E. colipINV1914, p38 MAPK was rapidly and persistently phos-phorylated and was comparable to stimulation withTNFα. In control cells that were not stimulated or stimu-lated with E. coli, p38 MAPK was weakly and transientlyphosphorylated possibly due to the mechanical stressduring centrifugation of the cells at the time of infection(Fig. 4A).
In addition to p38 MAPK, we assessed the activation ofJNK and ERK protein kinases by specific immune com-plex protein kinase assays. Invasin-expressing E. colipINV1914 and Y. enterocolitica pYV– triggered strong JNKactivation similar to that upon stimulation with TNFα,whereas inv-deficient strains did not activate JNK(Fig. 4B). Interestingly, induction of JNK with Y. entero-colitica pYV– was delayed compared to E. coli pINV1914,which might be explained by the higher expression levelsof invasin by E. coli pINV1914. In contrast to JNK and p38,we did not detect any significant activation of ERK above
Fig. 2. Adhesion assay. HeLa cells were incubated with different amounts of TcdB10463 for 24 h and then infected with Y. enterocolitica pYV–. A. 15 min after infection, cells were washed, lysed and the number of adherent bacteria was assessed by plating serial dilutions. B. 15 min after infection cells were washed, fixed and analysed by double-immunofluorescence; red: cytoskeleton; blue: adherent extracellular bacteria; green: intracellular bacteria.
Adherent bacteria

Invasin triggers IL-8 production via Rac1 and MAPK cascades 961
© 2003 Blackwell Publishing Ltd, Cellular Microbiology, 5, 957–971
the levels found in untreated cells by immunoprecipitatingERK1 and 2 isoforms (Fig. 4C). We conclude that p38MAPK and JNK, but not ERK1/2, are activated by Yersiniainvasin protein.
MAPK are required for strong induction of IL-8 by invasin
Next we addressed the question whether MAPK signal-ing pathways actually contribute to invasin-induced IL-8production. HeLa cells were incubated with the inhibitorsSB202190 (p38), PD98059 (MEK1), and SP600125(JNK) or a mixture of the three inhibitors for 30 min andthen stimulated by invasin-expressing Y. enterocoliticapYV–. Preincubation with these inhibitors caused areduction of invasin-induced IL-8 production by approxi-mately 40% to 85% with the combination of the lowestconcentration of the inhibitors showing an additive effect(Fig. 5A). These data suggest that individual MAPKcascades contribute partially to invasin-mediated IL-8expression and they clearly demonstrate an importantrole for p38 MAPK, MEK1 and JNK in invasin-triggeredIL-8 production. Next we examined the effect of the MAPkinase inhibitors on the transcription of IL-8 promoter-,NF-κB- or AP-1-driven luciferase constructs. All threeinhibitors significantly impaired the transcription of IL-8promoter-, NF-κB- and the AP-1-luciferase constructs(Fig. 5B-D). This data imply that p38 MAPK, MEK1 andJNK operate in concert to activate invasin-triggered IL-8transcription.
MAP kinases do not affect NF-κB or AP-1 binding activity to the IL-8 promoter
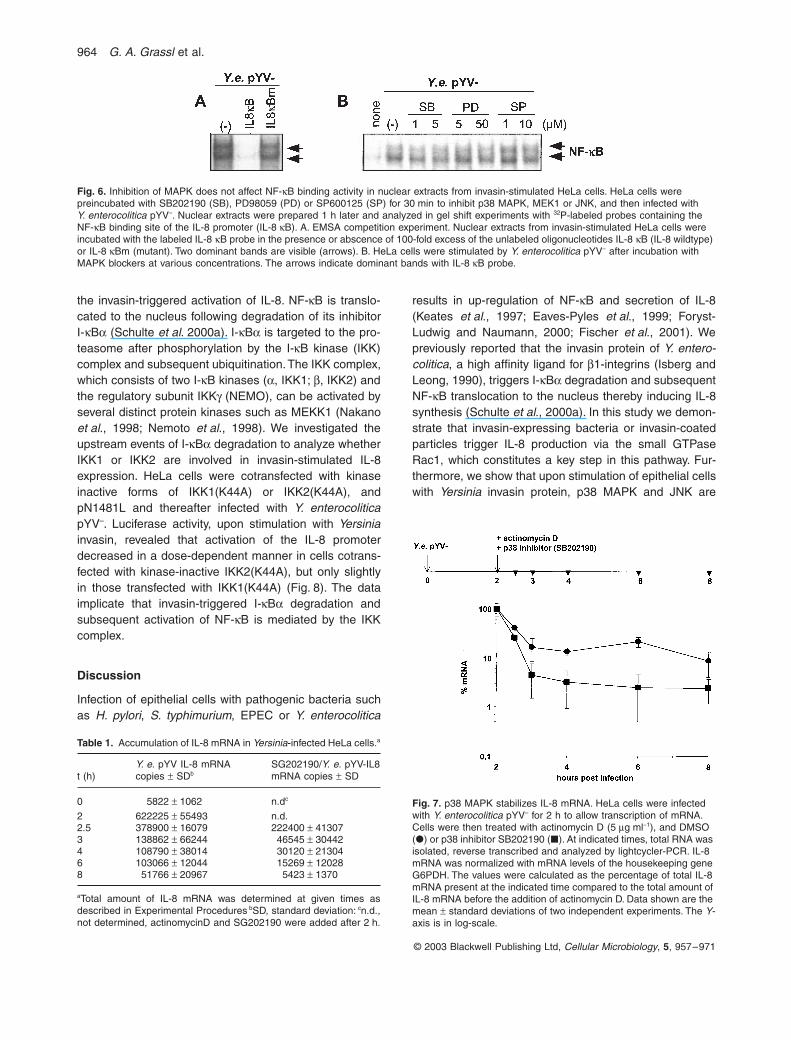
IKK and MAPK cascades are known to regulate theactivity of numerous transcription factors during infectionand inflammation (Kracht and Saklatvala, 2002). Part ofthis affects binding of AP-1 and NF-κB proteins to spe-cific cis-elements. Therefore, we investigated whetherthe MAPK cascades triggered by Yersinia invasin proteinexert a direct effect on transcription factor binding to theIL-8 promoter. HeLa cells were preincubated with MAPkinase inhibitors and subsequently infected with invasin-expressing bacteria. Nuclear extracts were preparedand analyzed by EMSA. The results in Fig. 6A demon-strate that two dominant bands are visible representingNF-κB p50/p65 hetero- and p65/p65 homodimers(Schulte et al., 2000a). These bands were specific, asthe addition of non-labeled wildtype probe IL-8 κB, butnot mutated IL-8 κBm probe, abolished the complexes.Treatment of HeLa cells with MAPK inhibitors(SB202190, PD98059 and SP600125) did not signifi-cantly alter binding of NF-κB proteins to the IL-8 pro-moter probe in vitro (Fig. 6B).
In agreement with the constitutive binding of AP-1proteins to their cognate DNA elements (Whitmarsh andDavis, 1996; Karin et al., 1997), no significant increasein AP-1 binding activity to IL-8 promoter DNA wasobserved after infection of HeLa cells with Y. enterocolit-ica pYV– (data not shown). EMSA supershift experi-
Fig. 3. Activation of IL-8 and NF-κB by invasin is mediated by Rac1. A. HeLa cells were cotransfected with inhibitory construct of small GTPases N17Rac1 and N17Cdc42 together with 100 ng pN1481L (IL-8 promoter driven luciferase construct). 48 h after transfection the cells were infected with Y. enterocolitica pYV–. Cell extracts were prepared for determination of luciferase activity at 6 h post infection. Data are expressed as relative light units. ctrl., cells that were trans-fected but left uninfected. B. non-infected cells. C. Cells infected with E. coli pINV1914. D–E. HeLa cells were microinjected with N17Rac1 together with Dextran-FITC and then infected with E. coli pINV1914. After 1 h cells were fixed and stained with an anti-NF-κB p65 antibody and a secondary TRITC-labeled antibody. D. green channel: cell microinjected with N17Rac1/Dextran-FITC; E. red channel: NF-κB p65.
A

962 G. A. Grassl et al.
© 2003 Blackwell Publishing Ltd, Cellular Microbiology, 5, 957–971
ments revealed that the proteins which boundconstitutively to the IL-8(AP-1) element comprised pro-teins from both Fos and Jun families and this bindingwas not affected by incubation with SB202190 andPD98059 (data not shown).
IL-8 mRNA is stabilized by p38 MAPK
IL-8 mRNA has a short half life. It was recently shown thatp38 MAPK enhances IL-1-induced IL-8 expression by sta-bilizing IL-8 mRNA (Holtmann et al., 1999; Winzen et al.,
Fig. 4. Activation of MAP kinases by Yersinia invasin. HeLa cells were infected with bacteria or stimulated with beads or TNFα as indicated. A. phosphorylation of p38 MAPK. At the indicated times whole-cell extracts were prepared and analyzed by Western blot with an anti-phospho-p38 MAPK-specific antibody (Pp38). The blots were stripped and re-probed with an anti-p38 MAPK antibody (p38). B,C. JNK (B) and ERK (C) kinase activity from cell extracts were assayed using GST-Jun or MBP, respectively, and [γ-32P]-ATP as substrates. The reaction products were resolved by SDS–PAGE. Phosphorylation was visualized by autoradiography. See Experimental Procedures for details.

Invasin triggers IL-8 production via Rac1 and MAPK cascades 963
© 2003 Blackwell Publishing Ltd, Cellular Microbiology, 5, 957–971
1999). We examined whether this was also the case withinvasin-triggered IL-8 production. Cells were infected withY. enterocolitica pYV– for 2 h to allow accumulation of IL-8 mRNA. Subsequently, cells were treated with actinomy-cin D (5 µg ml−1) to block transcription, and then treatedwith SB202190 to inhibit p38 MAPK. After transcriptionwas blocked, IL-8 mRNA was determined by quantitativeRT-PCR. Table 1 shows the actual accumulation of IL-8mRNA after 2 h and the subsequent decrease. Figure 7illustrates the rapid decay of IL-8 mRNA in cells treated
with the p38 inhibitor. In contrast, IL-8 mRNA of untreatedcells was significantly more stable compared to the mRNAof cells treated with the p38 inhibitor. These data suggestthat the p38 MAPK mediates transcription as well asmRNA stabilization of the IL-8 gene.
IKK1 and IKK2 are important for invasin-triggered IL-8 induction
We previously demonstrated the requirement of NF-κB for
Fig. 5. Reduction of IL-8 synthesis by MAPK inhibitors. HeLa cells were treated with SB202190, PD98059 or SP600125 for 30 min prior to infection with invasin-expressing Y. enterocolitica pYV–. A. IL-8 production was determined by ELISA in supernatants collected 6 h after infection. ctrl, non-infected cells. B-D. Activation of luciferase reporter constructs. HeLa cells were transfected with 100 ng of various promoter luciferase contructs. 48 h after transfection the cells were infected with Y. enterocolitica pYV–. Cell extracts were prepared for determination of luciferase activity at 6 h post infection. Data are expressed as relative light units. B. pN1481; C. NF-κB consensus; D. AP-1 consensus. ctrl., cells that were transfected with the corresponding plasmid but not infected.
964 G. A. Grassl et al.
© 2003 Blackwell Publishing Ltd, Cellular Microbiology, 5, 957–971
the invasin-triggered activation of IL-8. NF-κB is translo-cated to the nucleus following degradation of its inhibitorI-κBα (Schulte et al. 2000a). I-κBα is targeted to the pro-teasome after phosphorylation by the I-κB kinase (IKK)complex and subsequent ubiquitination. The IKK complex,which consists of two I-κB kinases (α, IKK1; β, IKK2) andthe regulatory subunit IKKγ (NEMO), can be activated byseveral distinct protein kinases such as MEKK1 (Nakanoet al., 1998; Nemoto et al., 1998). We investigated theupstream events of I-κBα degradation to analyze whetherIKK1 or IKK2 are involved in invasin-stimulated IL-8expression. HeLa cells were cotransfected with kinaseinactive forms of IKK1(K44A) or IKK2(K44A), andpN1481L and thereafter infected with Y. enterocoliticapYV–. Luciferase activity, upon stimulation with Yersiniainvasin, revealed that activation of the IL-8 promoterdecreased in a dose-dependent manner in cells cotrans-fected with kinase-inactive IKK2(K44A), but only slightlyin those transfected with IKK1(K44A) (Fig. 8). The dataimplicate that invasin-triggered I-κBα degradation andsubsequent activation of NF-κB is mediated by the IKKcomplex.
Discussion
Infection of epithelial cells with pathogenic bacteria suchas H. pylori, S. typhimurium, EPEC or Y. enterocolitica
results in up-regulation of NF-κB and secretion of IL-8(Keates et al., 1997; Eaves-Pyles et al., 1999; Foryst-Ludwig and Naumann, 2000; Fischer et al., 2001). Wepreviously reported that the invasin protein of Y. entero-colitica, a high affinity ligand for β1-integrins (Isberg andLeong, 1990), triggers I-κBα degradation and subsequentNF-κB translocation to the nucleus thereby inducing IL-8synthesis (Schulte et al., 2000a). In this study we demon-strate that invasin-expressing bacteria or invasin-coatedparticles trigger IL-8 production via the small GTPaseRac1, which constitutes a key step in this pathway. Fur-thermore, we show that upon stimulation of epithelial cellswith Yersinia invasin protein, p38 MAPK and JNK are
Fig. 6. Inhibition of MAPK does not affect NF-κB binding activity in nuclear extracts from invasin-stimulated HeLa cells. HeLa cells were preincubated with SB202190 (SB), PD98059 (PD) or SP600125 (SP) for 30 min to inhibit p38 MAPK, MEK1 or JNK, and then infected with Y. enterocolitica pYV–. Nuclear extracts were prepared 1 h later and analyzed in gel shift experiments with 32P-labeled probes containing the NF-κB binding site of the IL-8 promoter (IL-8 κB). A. EMSA competition experiment. Nuclear extracts from invasin-stimulated HeLa cells were incubated with the labeled IL-8 κB probe in the presence or abscence of 100-fold excess of the unlabeled oligonucleotides IL-8 κB (IL-8 wildtype) or IL-8 κBm (mutant). Two dominant bands are visible (arrows). B. HeLa cells were stimulated by Y. enterocolitica pYV– after incubation with MAPK blockers at various concentrations. The arrows indicate dominant bands with IL-8 κB probe.
Table 1. Accumulation of IL-8 mRNA in Yersinia-infected HeLa cells.a
t (h)Y. e. pYV IL-8 mRNAcopies ± SDb
SG202190/Y. e. pYV-IL8mRNA copies ± SD
0 5822 ± 1062 n.dc
2 622225 ± 55493 n.d.2.5 378900 ± 16079 222400 ± 413073 138862 ± 66244 46545 ± 304424 108790 ± 38014 30120 ± 213046 103066 ± 12044 15269 ± 120288 51766 ± 20967 5423 ± 1370
aTotal amount of IL-8 mRNA was determined at given times asdescribed in Experimental Procedures bSD, standard deviation: cn.d.,not determined, actinomycinD and SG202190 were added after 2 h.
Fig. 7. p38 MAPK stabilizes IL-8 mRNA. HeLa cells were infected with Y. enterocolitica pYV– for 2 h to allow transcription of mRNA. Cells were then treated with actinomycin D (5 µg ml−1), and DMSO (�) or p38 inhibitor SB202190 (�). At indicated times, total RNA was isolated, reverse transcribed and analyzed by lightcycler-PCR. IL-8 mRNA was normalized with mRNA levels of the housekeeping gene G6PDH. The values were calculated as the percentage of total IL-8 mRNA present at the indicated time compared to the total amount of IL-8 mRNA before the addition of actinomycin D. Data shown are the mean ± standard deviations of two independent experiments. The Y-axis is in log-scale.

Invasin triggers IL-8 production via Rac1 and MAPK cascades 965
© 2003 Blackwell Publishing Ltd, Cellular Microbiology, 5, 957–971
activated and these kinases mediate together with MEK1the transcriptional activation of the IL-8 gene. Additionallyp38 MAPK stabilizes IL-8 mRNA and the IKK complex isalso required for activation of the IL-8 gene.
Rho GTPases play an important role in several pro-cesses in eukaryotic cells including organization of theactin cytoskeleton, gene transcription, cell cycle progres-sion and membrane trafficking (reviewed in: Van Aelstand D’Souza-Schorey, 1997; Schmitz et al., 2000).Recent work demonstrated that Rho, Rac1 and Cdc42are important for phagocytosis of Y. enterocoliticainvasin-coated particles in human macrophages. Micro-injection of the dominant-negative GTPase mutantsN17Cdc42 or N17Rac1, or of the Rho-specific C3-trans-ferase into macrophages, greatly attenuated invasin-induced formation of phagocytic cups into which WASPand the Arp2/3 complex were recruited (Wiedemannet al., 2001).
There are controversial reports about the uptake of Y.pseudotuberculosis invasin-coated particles. The RhoGTPase Rac1 and a functional N-WASP and Arp2/3 com-plex have been shown to be required for uptake of Y.pseudotuberculosis into HeLa cells (McGee et al., 2001),whereas a different group showed that phagocytosis of Y.pseudotuberculosis by COS-1 cells proceeds via Rac1and Arp2/3 bypassing N-WASP (Alrutz et al., 2001).Werner et al. demonstrated that RhoA, but not Rac orCdc42, is important for phagocytosis of invasin-coatedlatex beads by synovial fibroblasts (Werner et al., 2001).It is unclear whether the varying observations reflect theproperties of the various cell types, the different Yersinia
strains or administration of invasin either by bacteria orbound to latex beads.
In this study we demonstrated that Rac1 is an essen-tial mediator of invasin-triggered NF-κB activation andIL-8 production in epithelial cells, whereas Rho andCdc42 are not involved in the induction of this proinflam-matory signaling cascade. The C. difficile toxins weused in this study did not affect adhesion of the bacteriato the cells but they inhibited uptake of the bacteria. Inprevious studies we showed that uptake of Yersinia intohost cells and induction of IL-8 is accomplished by inde-pendent pathways (Schulte et al., 1998; Schulte et al.,2000a).
Rac and Cdc42 activate the MAP kinases p38 and JNKbut not ERK1/2 (Coso et al., 1995; Minden et al., 1995).Consistent with this we demonstrated that both p38 andJNK were activated by invasin-expressing bacteriawhereas invasin-deficient bacteria activated none of theMAP kinases. Additionally, wildtype Y. enterocolitica pYV+
inhibited JNK activation, most likely by translocation ofYopP, which has been shown to block MAP kinase path-ways (Orth et al., 1999; Ruckdeschel et al., 2001). It is yetunclear whether Rac is involved in invasin-induced p38and JNK activation. Others demonstrated that Rac mayactivate p38 MAPK and JNK via the p21 activated kinase(PAK)1 (Wery-Zennaro et al., 2000; del Pozo et al., 2000;Cotteret and Chernoff, 2002; Parrini et al., 2002). Autoin-hibited PAK1 homodimers dissociate upon binding to theactive forms of Cdc42 and Rac, GTP-Cdc42 and GTP-Rac (Parrini et al., 2002), and subsequently may activateMKK4, JNK (Almeida et al., 2000) and p38 MAPK (Wery-Zennaro et al., 2000).
All three MAPK inhibitors (SB202190, PD98059,SP600125) diminished NF-κB dependent luciferase activ-ity but did not significantly change NF-κB binding activity.This apparent discrepancy is due to the fact that NF-κBactivity is not only regulated by nuclear translocation ofthe NF-κB complexes but also by multiple phosphoryla-tions of the subunits. Inhibition of p38 MAPK withSB202190 resulted in a 60% – 75% decrease in the totalamounts of secreted IL-8. The inhibitor did not alter NF-κB or AP-1 binding activity, however, it significantlyreduced transcription of IL-8 promoter driven luciferaseconstructs. Additionally SB202190 significantly dimin-ished mRNA stability. This implicates that p38 MAPKdirectly effects transcription and also contributes to IL-8production by post-transcriptional mechanisms. The abilityof p38 MAPK to stabilize TNFα and IL-8 mRNA waspreviously reported by several groups (Winzen et al.,1999; Dean et al., 2001; Frevel et al., 2003). Additionallyp38 may contribute to IL-8 transcription by phosphoryla-tion and phosphoacetylation of histone H3 therebyenhancing the accessibility of the NF-κB site in the IL-8promoter (Saccani et al., 2002).
Fig. 8. IL-8 activity in invasin-stimulated HeLa cells modulated by transfection with dominant-negative forms of IKK1, and IKK2. HeLa cells were cotransfected with 250 ng of pN1481L and different amounts of IKK1(K44A) and IKK2(K44A) constructs. 16 h after trans-fection the cells were infected with Y. enterocolitica pYV–. Cell extracts were prepared for determination of luciferase activity at 6 h post infection. Data are expressed as relative light units. ctrl., cells that were transfected but left uninfected.

966 G. A. Grassl et al.
© 2003 Blackwell Publishing Ltd, Cellular Microbiology, 5, 957–971
The transcription factor AP-1 is composed of Fos andJun proteins which are regulated by JNK (Kyriakis andAvruch, 2001). Inhibition of JNK with SP600125decreased invasin-induced IL-8 production by about 60%indicating an important role for JNK in this pathway. UnlikeNF-κB, AP-1 complexes bind constitutively to the IL-8promoter, and the transcriptional activity is regulated bythe abundance and the phosphorylation status of the pro-teins (reviewed in:(Whitmarsh and Davis, 1996; Karinet al., 1997). Pretreatment of HeLa cells with PD98059,an inhibitor of MAPK/ERK (MEK)1, inhibited invasin-induced IL-8 production by 80%. MEK1 is a specific acti-vator of ERK1/2, however, we did not observe activationof ERK1/2 by invasin. This apparent discrepancy maybe due to the fact that PD98059 also inhibits MEK5,which activates ERK5, a recently identified MAP kinasewith largely unknown function (Kamakura et al., 1999;Cavanaugh et al., 2001).
Previous work conducted in our laboratory showed thatinvasin triggers the degradation of I-κBα (Schulte et al.,2000a). The release of NF-κB from its cytoplasmic inhib-itor I-κBα is a prerequisite for translocation to the nucleus(Henkel et al., 1993; Baeuerle, 1998a; Baeuerle, 1998b).Prior to degradation by the proteasome, I-κBα is phos-phorylated by the IKK complex and subsequently ubiquit-inated. The IKK complex is comprised of the catalyticsubunits IKK1 and IKK2 and the regulatory subunit IKKγ/NEMO (Malinin et al., 1997; Rothwarf and Karin, 1999).Our data indicate that invasin triggers NF-κB activationby activation of IKK1/2 and subsequent degradation ofI-κBα.
There is substantial evidence that invasin triggers theinternalization of Yersinia into host cells (Isberg andLeong, 1990; Dersch and Isberg, 1999; Wiedemannet al., 2001; McGee et al., 2001), and this event appearsto be important for translocation of Yersinia through the Mcells and thus for initiating the course of invasive infection(Pepe and Miller, 1993a; Pepe and Miller, 1993b; Schulteet al., 2000b). This study sheds new light on the role ofinvasin protein in Yersinia infections. Along with our pre-vious reports (Schulte and Autenrieth, 1998; Schulteet al., 1998; Kampik et al., 2000; Schulte et al., 2000a),the data indicate that invasin may promote proinflamma-tory host cell reactions including production of IL-8. Yers-inia invasin stimulates signaling pathways which appearto be linked to host cell responses involved in the controlof innate immunity. Therefore, it is tempting to speculatethat the signaling events during bacterial adhesion andinvasion processes are commonly linked to signalingpathways operating in innate immunity, thus representinga concerted defense strategy of the host. Interestingly,the signaling pathways leading to invasin-triggered inter-nalization and inflammation appear to be distinct andindependent of PI3K. The PI3K-inhibitors blocked phago-
cytosis of Y. enterocolitica but did not affect Yersinia-induced IL-8-production (Schulte et al., 1998; Schulteet al., 2000a). Recent work demonstrated that apart frominvasin, YopB of Y. pseudotuberculosis may stimulate IL-8 production in HeLa cells by a proinflammatory cascade(Viboud et al., 2003). The receptor and signaling path-ways engaged by YopB are not yet solved. At conditionsin which YopB is expressed, invasin seems to berepressed and vice versa. Thus both Inv and YopB mayconstitute complementary mechanisms in stimulatingproinflammatory host cell responses. Some plasmid-encoded Yops such as YopP and YopE counteract theproinflammatory signaling by acting on IKKβ, MAPkinases or small GTPases (Ruckdeschel et al., 2001;Andor et al., 2001). Nevertheless, during infection in vivoinflammatory reactions obviously occur very early andare operating despite the presence of plasmid-encodedfactors. Whether the production of IL-8 in vivo is accom-plished by activation of signaling pathways via Inv, YopB,LPS or all of these components is not known. Therefore,future studies will have to demonstrate which of thepathogenicity factors including invasin and Yops actuallycontribute to induction and modulation/suppression ofsignaling events leading to proinflammatory host cellreactions in vivo.
Experimental procedures
Bacterial strains and growth conditions
Wildtype Y. enterocolitica WA314 (pYV+), plasmid-cured Y.enterocolitica WA314 pYV– (Heesemann and Laufs, 1983), Y.enterocolitica WA314 pYV– inv– mutant (Ruckdeschel et al.,1996), non-invasive E. coli HB101, and E. coli HB101pINV1914 expressing the Y. enterocolitica inv (Schulte et al.,1998) were grown in Luria-Bertani broth (LB). For infectionexperiments, overnight cultures were diluted to an optical den-sity at 600 nm (OD600) of 0.2 in LB and incubated for 3 h at27°C or 37°C.
Cell culture and infection
Human HeLa cervical epithelial cells (ATCC CCL-2.1; AmericanType Culture Collection, Manassas, Va.) were maintained inRPMI 1640 and infected as described (Schulte et al., 2000a) withan MOI of 100 or as indicated. TNFα was a kind gift from G. Adolf(Bender, Vienna, Austria).
Adhesion assay
HeLa cells were seeded in 24 well plate and grown overnight.Cells were infected with bacteria with an MOI of 100 and centri-fuged for 5 min at 400× g. 15 min after infection, cells were exten-sively washed with PBS and lysed with 1% TritonX-100/PBS for5 min. To determine the number of adherent bacteria, serial dilu-tions were plated, incubated at 27°C and colonies were counted2 ds later.

Invasin triggers IL-8 production via Rac1 and MAPK cascades 967
© 2003 Blackwell Publishing Ltd, Cellular Microbiology, 5, 957–971
DNA constructs
Vector pN1481L containing nt −1481 to +44 of the IL-8 promoterfused to the luciferase reporter gene was kindly provided by RonCrystal (Cornell Medical Center, New York). pNF-κB-Luc andpAP-1-Luc were purchased from Stratagene (Amsterdam, Neth-erlands). The expression plasmid for glutathione-S-transferase(GST)-Jun (amino acids 1–135) was a generous gift of J. R.Woodgett (The Ontario Cancer Research Institute, Ontario, Can-ada). pCDNA3-IKK1(K44A) was a kind gift from L. Schmitz (Uni-versity of Bern, Bern, Switzerland) and pCDNA3-IKK2(K44A)was a kind gift from S. Ludwig (Institut fuer Medizinische Strahl-enkunde, Wuerzburg, Germany). N17Rac1 and N17Cdc42 werecloned into pHM6 (Roche, Mannheim).
Inhibitors
C. difficile toxins TcdB-10463 and TcdB-1740 were purified aspreviously described (Chaves-Olarte et al., 1997; Moos andEichel-Streiber, 2000). MAP kinase inhibitors SB202190 (inhibits:p38), PD98059 (inhibits: MEK1) and SP600125 (inhibits: JNK)were purchased from Calbiochem (Schwalbach, Germany) anddissolved in dimethylsulfoxide (DMSO). Actinomycin D was pur-chased from Calbiochem (Schwalbach).
Expression and purification of proteins
C-terminal 195 aa of Y. enterocolitica invasin protein (GST-Inv195)
was expressed and purified as described (Schulte et al., 2000a).
C3-transferase. The protein was dialyzed against microinjection
buffer (50 mM Tris-HCl pH 7.5, 150 mM NaCl, 5 mM MgCl2), con-
centrated in Centricon or Microcon (Amicon, Witten, Germany),
snap-frozen and stored at −80°C. Purity was checked by per-
forming SDS–PAGE and visualized with Coomassie staining.
Coating of proteins to latex beads
Beads were non-covalently coated by incubating with purifiedprotein dialyzed against PBS (pH 7.0). Approximately 109 latexbeads (1 µm diameter, sulphate-modified; Molecular Probes,Leiden, Netherlands) were washed with 1 ml PBS and resus-pended in 0.5 ml PBS. Purified GST or GST-Inv195 fusion protein(500 µg) was added and allowed to adsorb to the beads for 3 hat room temperature. Following the addition of 0.5 ml BSA(20 mg ml−1), the solution was incubated at room temperature fora further 1 h. The beads were then washed with PBS containing1 mg ml−1 BSA and stored at 4°C in PBS containing 0.2 mg ml−1
BSA. Integrity of coated GST-Inv195 fusion protein was con-trolled by Western blot analysis.
IL-8 ELISA
The amount of IL-8 secreted by HeLa cells into the supernatantwas determined as previously described (Schulte and Autenrieth,1998) using an enzyme-linked immunosorbent assay (ELISA)with optimal concentrations of a mouse antihuman IL-8 mono-clonal antibody (Mab) (G265-5; Pharmingen, San Diego, CA) anda biotinylated mouse antihuman IL-8 Mab (G265-8; Pharmingen)
as the detecting antibody. IL-8 concentrations were calculatedfrom the straight-line portion of a standard curve derived usingrecombinant human IL-8 (Pharmingen).
Quantitative RT-PCR
Total RNA of infected HeLa cells in 6-well plates was extractedusing 1 ml of TRIzol reagent (Life Technologies). Five microgramsof RNA were reverse transcribed as previously described(Schulte et al., 1998). PCR was performed in the LightCyclerapparatus using the LC-FastStart DNA Master SYBR Green I Kit(Roche). Thermocycling was performed in a final volume of 20 µlcontaining 2 µl of cDNA sample (or standard), 4 mM MgCl2,0.5 µM of each primer, as well as 1× ready-to-use reaction mixcontaining Taq DNA polymerase, reaction buffer, dNTP mix, andSYBR Green. Following 10 min of initial denaturation at 95°C, thecycling conditions of 35 cycles consisted of denaturation at 95°Cfor 15 s, annealing at 59°C for 5 s and elongation at 72°C for11 s. The LightCycler measured the fluorescence (SYBR Green)of each sample in every cycle at the end of the elongation step.The expression levels of IL-8 were normalized with the house-keeping gene glucose-6-phosphate dehydrogenase (G6PDH). Inaddition, melting curve analysis was performed according to themanufacturer’s instructions (Roche) to ensure the purity ofthe amplified PCR product. Negative controls were performedby omitting RNA from the cDNA synthesis and specific PCRamplifications.
Transient transfection
HeLa cells (1 × 105) seeded in 12-well plates for 24 h werecotransfected with 100–250 ng of pN1481L and 100–1000 ng ofkinase constructs using ExGen 500 Transfection Reagent (MBI-Fermentas, St. Leon-Rot, Germany) according to the manufac-turer’s instructions, and incubated for 16–72 h at 37°C. Theconstructs were cotransfected with 250 ng of β-galactosidaseconstruct under the control of a constitutive promoter (pCMV-β-gal) for determination of transfection efficiency. HeLa cells wereinfected with bacteria at a MOI 100 and incubated at 37°C for2 h, then washed twice with PBS and incubated for an additional4 h in the presence of gentamicin. Supernatants were removed6 h after infection for determination of IL-8 by ELISA. Then HeLacells were washed twice with PBS, and lysed with luc lysis bufferaccording to the manufacturers instructions (Roche, Mannheim).Lysates were centrifuged and supernatants removed for deter-mination of protein, β-galactosidase and luciferase activity.Luciferase activity was normalized on beta-galactosidase activityand protein (relative light units).
Western blotting
HeLa cells (1 × 106) in 6-well plates were infected as describedabove. At various times the cells were washed twice with PBSand resuspended in 200 µl SDS sample buffer. Extracts weretransferred to Eppendorf tubes and boiled for 5 min before 10 µlsamples were separated by SDS–PAGE on a 10% polyacryla-mide gel and electrophoretically transferred to a nitrocellulosemembrane. p38 MAP kinase phosphorylation was evalutatedusing the phospho-p38 MAPK Kit (New England Biolabs, Frank-furt, Germany) according to the manufacturer’s instructions.

968 G. A. Grassl et al.
© 2003 Blackwell Publishing Ltd, Cellular Microbiology, 5, 957–971
Immune complex protein kinase assays
Whole cell extract (250–500 µg protein) was diluted in 500 µl of
ice-cold immunoprecipitation (IP) buffer A (20 mM Tris pH 7.4,
154 mM NaCl, 50 mM NaF, 1 mM Na3VO4, 1% Triton X-100).
Samples were incubated for 3 h with 2 µl of rabbit antibodies
made to synthetic peptides for the C-terminus of JNK2 (peptide
DSSLDASTGPLEGR, aa 409–423) or ERK MAPK (ERK2 (C-14);
Santa Cruz, CA) followed by the addition of 20 µl of a 50%
suspension of protein A-sepharose beads (Amersham Bio-
sciences) and incubation for 1–2 h at 4°C. Beads were spun
down, washed three times with 1 ml of IP buffer A and resus-
pended in 10 µl of the same buffer. One microgram of GST-
JUN1-135 fusion protein or MBP (Sigma) in 10 µl H2O and 10 µl of
kinase buffer (150 mM Tris pH 7.4, 30 mM MgCl2, 60 µM ATP,
4 µCi [γ-32P]ATP; Hartmann Analytics, Braunschweig, Germany)
were added. After 30 min at room temperature SDS–PAGE sam-
ple buffer was added and proteins were eluted from the beads
by boiling for 5 min. After centrifugation at 10 000× g for 5 min,
supernatants were separated by electrophoresis on a 10%- or
12.5%-polyacrylamide gel. Phosphorylated proteins were visual-
ized by autoradiography (Wolter et al. 2002).
Electrophoretic mobility shift assay (EMSA)
HeLa cells (5 × 106) were infected as described above. At variousintervals post infection, nuclear extracts were prepared as previ-ously described (Schreiber et al., 1989). Aliquots of the superna-tant containing nuclear proteins were stored at −80°C. Proteinconcentrations were determined by the Bradford assay. Theoligonucleotide probes described below were labeled with[γ32P]ATP (Amersham Biosciences) using T4-Polynucleotidkinase(New England Biolabs) and then purified on a NucTrap probepurification column (Stratagene). The following oligonucleotideswere used: IL-8-κB wildtype (IL-8 κB; 5′-ATCGTGGAATTTCCTCTGA-3′; Metabion, Munich, Germany), IL-8-κB mutant (IL-8 κBm; 5′-ATCcTGcAATgTCgTCTGA-3′; Metabion). Nuclearextracts (3–6 µg) were incubated with 30 000 cpm of the 32P-labeled oligonucleotide probe for 45 min on ice in a buffer con-taining 5% glycerol, 80 mM NaCl, 1 mM DTT, 1 mM EDTApH 8.0, 10 mM Tris-HCl pH 7.2 and 1 µg dIdC for NF-κB shifts.The same buffer containing 5 mM DTT was used for AP-1 shifts.Antibodies against p50 (sc-1190X), p65 (sc-372X), pan-Fos(sc-253X) and pan-Jun (sc-44X; Santa Cruz) were includedin the binding reaction for supershift analyses. Samples wereresolved on a 5% non-denaturating polyacrylamide gel using0.5× TBE (25 mM Tris-HCl, 25 mM boric acid, 0.5 mM EDTA)as running buffer. Gels were transferred to Whatman 3M paperand dried under vacuum. Protein binding was assessed viaautoradiography.
Immunofluorescence staining and confocal laser scanning microscopy
Immunofluorescence microscopy (CLSM) was performed asdescribed (Schulte et al. 2000a). In brief, after 3% paraformalde-hyde(Sigma) fixation of the monolayers, extracellular bacteria orbeads were stained with polyclonal rabbit anti-Yersinia antibod-
ies, followed by Cy-5-conjugated goat anti-rabbit antibodies(Dianova, Hamburg, Germany). After three washings, cells werepermeabilized with 2% Triton X-100 in PBS, washed, and intrac-ellular bacteria or beads were stained with anti-Yersinia antibod-ies, followed by FITC-conjugated goat antirabbit antibodies(Dianova). F-actin was stained by TRITC-conjugated phalloidin(Sigma). The fluorescence images were obtained with a confocallaser scanning microscope (Leica DM IRE2).
Statistics
Data shown in figures are from representative experiments.Comparable results were obtained in at least two additionalexperiments. Differences between mean values were analyzedusing the Student’s t-test. P < 0.05 was considered statisticallysignificant.
Acknowledgments
We thank Pierre A. Kyme for critical reading of the manuscript.This work was supported by a grant from the DeutscheForschungsgemeinschaft.
References
Aktories, K., Weller, U., and Chhatwal, G.S. (1987) Clostrid-ium botulinum type C produces a novel ADP-ribosyltrans-ferase distinct from botulinum C2 toxin. FEBS Lett 212:109–113.
Almeida, E.A., Ilic, D., Han, Q., Hauck, C.R., Jin, F.,Kawakatsu, H., Schlaepfer, D.D., and Damsky, C.H. (2000)Matrix survival signaling: from fibronectin via focal adhe-sion kinase to c-Jun NH (2) -terminal kinase. J Cell Biol149: 741–754.
Alrutz, M.A., Srivastava, A., Wong, K.W., D’Souza-Schorey,C., Tang, M., Ch’Ng, L.E., Snapper, S.B., and Isberg, R.R.(2001) Efficient uptake of Yersinia pseudotuberculosis viaintegrin receptors involves a Rac1-Arp 2/3 pathway thatbypasses N-WASP function. Mol Microbiol 42: 689–703.
Andor, A., Trulzsch, K., Essler, M., Roggenkamp, A., Wiede-mann, A., Heesemann, J., and Aepfelbacher, M. (2001)YopE of Yersinia, a GAP for Rho GTPases, selectivelymodulates Rac-dependent actin structures in endothelialcells. Cell Microbiol 3: 301–310.
Autenrieth, I.B., Kempf, V., Sprinz, T., Preger, S., andSchnell, A. (1996) Defence mechanisms in Peyer’s patchesand mesenteric lymph nodes against Yersinia enterocolit-ica involve integrins and cytokines. Infect Immun 64: 1357–1368.
Baeuerle, P.A. (1998a) IkappaB-NF-kappaB structures: atthe interface of inflammation control. Cell 95: 729–731.
Baeuerle, P.A. (1998b) Pro-inflammatory signaling: lastpieces in the NF-kappaB puzzle? Curr Biol 8: R19–R22.
Bottone, E.J. (1977) Yersinia enterocolitica: a panoramic viewof a charismatic microorganism. CRC Crit Rev Microbiol 5:211–241.
Cavanaugh, J.E., Ham, J., Hetman, M., Poser, S., Yan, C.,and Xia, Z. (2001) Differential regulation of mitogen-acti-vated protein kinases ERK1/2 and ERK5 by neurotrophins,neuronal activity, and cAMP in neurons. J Neurosci 21:434–443.

Invasin triggers IL-8 production via Rac1 and MAPK cascades 969
© 2003 Blackwell Publishing Ltd, Cellular Microbiology, 5, 957–971
Chaves-Olarte, E., Low, P., Freer, E., Norlin, T., Weidmann,M., Eichel-Streiber, C., and Thelestam, M. (1999) A novelcytotoxin from Clostridium difficile serogroup F is a func-tional hybrid between two other large clostridial cytotoxins.J Biol Chem 274: 11046–11052.
Chaves-Olarte, E., Weidmann, M., Eichel-Streiber, C., andThelestam, M. (1997) Toxins A and B from Clostridiumdifficile differ with respect to enzymatic potencies, cellularsubstrate specificities, and surface binding to culturedcells. J Clin Invest 100: 1734–1741.
Cornelis, G.R. (1994) Yersinia pathogenicity factors. Curr TopMicrobiol Immunol 192: 243–263.
Cornelis, G.R. (1998) The Yersinia deadly kiss. J Bacteriol180: 5495–5504.
Coso, O.A., Chiariello, M., Yu, J.C., Teramoto, H., Crespo,P., Xu, N., Miki, T., and Gutkind, J.S. (1995) The smallGTP-binding proteins Rac1 and Cdc42 regulate the activ-ity of the JNK/SAPK signaling pathway. Cell 81: 1137–1146.
Cotteret, S., and Chernoff, J. (2002) The evolutionary historyof effectors downstream of Cdc42 and Rac. Genome Biol3: REVIEWS0002.
Cover, T.L., and Aber, R.C. (1989) Yersinia enterocolitica. NEngl J Med 321: 16–24.
Dean, J.L., Wait, R., Mahtani, K.R., Sully, G., Clark, A.R., andSaklatvala, J. (2001) The 3′ untranslated region of tumornecrosis factor alpha mRNA is a target of the mRNA-stabilizing factor HuR. Mol Cell Biol 21: 721–730.
Dersch, P., and Isberg, R.R. (1999) A region of the Yersiniapseudotuberculosis invasin protein enhances integrin-mediated uptake into mammalian cells and promotes selfassociation. EMBO J 18: 1199–1213.
Donnelly, M.A., and Steiner, T.S. (2002) Two nonadjacentregions in enteroaggregative escherichia coli flagellin arerequired for activation of toll-like receptor 5. J BiolChem.
Eaves-Pyles, T., Szabo, C., and Salzman, A.L. (1999) Bac-terial invasion is not required for activation of NF-kappaBin enterocytes. Infect Immun 67: 800–804.
Eckmann, L., Kagnoff, M.F., and Fierer, J. (1995) Intestinalepithelial cells as watchdogs for the natural immune sys-tem. Trends Microbiol 3: 118–120.
Elewaut, D., DiDonato, J.A., Kim, J.M., Truong, F., Eckmann,L., and Kagnoff, M.F. (1999) NF-kappa B is a central reg-ulator of the intestinal epithelial cell innate immuneresponse induced by infection with enteroinvasive bacteria.J Immunol 163: 1457–1466.
Fischer, W., Puls, J., Buhrdorf, R., Gebert, B., Odenbreit, S.,and Haas, R. (2001) Systematic mutagenesis of the Heli-cobacter pylori cag pathogenicity island: essential genesfor CagA translocation in host cells and induction ofinterleukin-8. Mol Microbiol 42: 1337–1348.
Foryst-Ludwig, A., and Naumann, M. (2000) p21-activatedkinase 1 activates the nuclear factor kappa B (NF-kappaB) -inducing kinase-Ikappa B kinases NF-kappa B pathwayand proinflammatory cytokines in Helicobacter pylori infec-tion. J Biol Chem 275: 39779–39785.
Frevel, M.A., Bakheet, T., Silva, A.M., Hissong, J.G., Khabar,K.S., and Williams, B.R. (2003) p38 Mitogen-activated pro-tein kinase-dependent and – independent signaling ofmRNA stability of AU-rich element-containing transcripts.Mol Cell Biol 23: 425–436.
Gewirtz, A.T., Navas, T.A., Lyons, S., Godowski, P.J., andMadara, J.L. (2001) Cutting edge: bacterial flagellin acti-
vates basolaterally expressed TLR5 to induce epithelialproinflammatory gene expression. J Immunol 167: 1882–1885.
Hamburger, Z.A., Brown, M.S., Isberg, R.R., and Bjorkman,P.J. (1999) Crystal structure of invasin: a bacterial integrin-binding protein. Science 286: 291–295.
Hedlund, M., Wachtler, C., Johansson, E., Hang, L.,Somerville, J.E., Darveau, R.P., and Svanborg, C.(1999) P fimbriae-dependent, lipopolysaccharide-independent activation of epithelial cytokine responses.Mol Microbiol 33: 693–703.
Heesemann, J., and Laufs, R. (1983) Construction of a mobi-lizable Yersinia enterocolitica virulence plasmid. J Bacteriol155: 761–767.
Henkel, T., Machleidt, T., Alkalay, I., Kronke, M., Ben Neriah,Y., and Baeuerle, P.A. (1993) Rapid proteolysis of I kappaB-alpha is necessary for activation of transcription factorNF-kappa B. Nature 365: 182–185.
Hoffmann, E., Dittrich-Breiholz, O., Holtmann, H., and Kracht,M. (2002) Multiple control of interleukin-8 gene expression.J Leukoc Biol 72: 847–855.
Holtmann, H., Winzen, R., Holland, P., Eickemeier, S., Hoff-mann, E., Wallach, D., Malinin, N.L., Cooper, J.A., Resch,K., and Kracht, M. (1999) Induction of interleukin-8 synthe-sis integrates effects on transcription and mRNA degrada-tion from at least three different cytokine- or stress-activated signal transduction pathways. Mol Cell Biol 19:6742–6753.
Isberg, R.R., and Leong, J.M. (1990) Multiple beta 1 chainintegrins are receptors for invasin, a protein that promotesbacterial penetration into mammalian cells. Cell 60: 861–871.
Jung, H.C., Eckmann, L., Yang, S.K., Panja, A., Fierer, J.,Morzycka-Wroblewska, E., and Kagnoff, M.F. (1995) A dis-tinct array of proinflammatory cytokines is expressed inhuman colon epithelial cells in response to bacterial inva-sion. J Clin Invest 95: 55–65.
Just, I., Selzer, J., Wilm, M., Eichel-Streiber, C., Mann, M.,and Aktories, K. (1995) Glucosylation of Rho proteins byClostridium difficile toxin B. Nature 375: 500–503.
Kamakura, S., Moriguchi, T., and Nishida, E. (1999) Activa-tion of the protein kinase ERK5/BMK1 by receptor tyrosinekinases. Identification and characterization of a signalingpathway to the nucleus. J Biol Chem 274: 26563–26571.
Kampik, D., Schulte, R., and Autenrieth, I.B. (2000) Yersiniaenterocolitica invasin protein triggers differential productionof interleukin-1, interleukin-8, monocyte chemoattractantprotein 1, granulocyte-macrophage colony-stimulating fac-tor, and tumor necrosis factor alpha in epithelial cells: impli-cations for understanding the early cytokine network inYersinia infections. Infect Immun 68: 2484–2492.
Karin, M., Liu, Z., and Zandi, E. (1997) AP-1 function andregulation. Curr Opin Cell Biol 9: 240–246.
Keates, S., Hitti, Y.S., Upton, M., and Kelly, C.P. (1997)Helicobacter pylori infection activates NF-kappa B in gas-tric epithelial cells. Gastroenterology 113: 1099–1109.
Kracht, M., and Saklatvala, J. (2002) Transcriptional andpost-transcriptional control of gene expression in inflamma-tion. Cytokine 20: 91–106.
Kunsch, C., Lang, R.K., Rosen, C.A., and Shannon, M.F.(1994) Synergistic transcriptional activation of the IL-8gene by NF-kappa B p65 (RelA) and NF-IL-6. J Immunol153: 153–164.
Kyriakis, J.M., and Avruch, J. (2001) Mammalian mitogen-

970 G. A. Grassl et al.
© 2003 Blackwell Publishing Ltd, Cellular Microbiology, 5, 957–971
activated protein kinase signal transduction pathways acti-vated by stress and inflammation. Physiol Rev 81: 807–869.
Leong, J.M., Fournier, R.S., and Isberg, R.R. (1990) Identifi-cation of the integrin binding domain of the Yersiniapseudotuberculosis invasin protein. EMBO J 9: 1979–1989.
Mainiero, F., Soriani, A., Strippoli, R., Jacobelli, J., Gismondi,A., Piccoli, M., Frati, L., and Santoni, A. (2000) RAC1/P38MAPK signaling pathway controls beta1 integrin-inducedinterleukin-8 production in human natural killer cells. Immu-nity 12: 7–16.
Malinin, N.L., Boldin, M.P., Kovalenko, A.V., and Wallach, D.(1997) MAP3K-related kinase involved in NF-kappaBinduction by TNF, CD95 and IL-1. Nature 385: 540–544.
Matsusaka, T., Fujikawa, K., Nishio, Y., Mukaida, N., Mat-sushima, K., Kishimoto, T., and Akira, S. (1993) Transcrip-tion factors NF-IL6 and NF-kappa B synergistically activatetranscription of the inflammatory cytokines, interleukin 6and interleukin 8. Proc Natl Acad Sci U S A 90: 10193–10197.
McCormick, B.A., Nusrat, A., Parkos, C.A., D’Andrea, L.,Hofman, P.M., Carnes, D., Liang, T.W., and Madara, J.L.(1997) Unmasking of intestinal epithelial lateral mem-brane beta1 integrin consequent to transepithelial neutro-phil migration in vitro facilitates inv-mediated invasion byYersinia pseudotuberculosis. Infect Immun 65: 1414–1421.
McGee, K., Zettl, M., Way, M., and Fallman, M. (2001) A rolefor N-WASP in invasin-promoted internalisation. FEBS Lett509: 59–65.
Medzhitov, R. (2001) Toll-like receptors and innate immunity.Nat Rev Immunol 1: 135–145.
Mercurio, F., and Manning, A.M. (1999) Multiple signals con-verging on NF-kappaB. Curr Opin Cell Biol 11: 226–232.
Minden, A., Lin, A., Claret, F.X., Abo, A., and Karin, M. (1995)Selective activation of the JNK signaling cascade and c-Jun transcriptional activity by the small GTPases Rac andCdc42Hs. Cell 81: 1147–1157.
Moos, M., and Eichel-Streiber, C. (2000) Purification andevaluation of large clostridial cytotoxins that inhibit smallGTPases of Rho and Ras subfamilies. Meth Enzymol 325:114–125.
Mukaida, N., Mahe, Y., and Matsushima, K. (1990) Co-operative interaction of nuclear factor-kappa B- and cis-regulatory enhancer binding protein-like factor bindingelements in activating the interleukin-8 gene by pro-inflammatory cytokines. J Biol Chem 265: 21128–21133.
Mukaida, N., Okamoto, S., Ishikawa, Y., and Matsushima, K.(1994) Molecular mechanism of interleukin-8 gene expres-sion. J Leukoc Biol 56: 554–558.
Munzenmaier, A., Lange, C., Glocker, E., Covacci, A., Moran,A., Bereswill, S., Baeuerle, P.A., Kist, M., and Pahl, H.L.(1997) A secreted/shed product of Helicobacter pylori acti-vates transcription factor nuclear factor-kappa B. J Immu-nol 159: 6140–6147.
Nakano, H., Shindo, M., Sakon, S., Nishinaka, S., Mihara,M., Yagita, H., and Okumura, K. (1998) Differential regula-tion of IkappaB kinase alpha and beta by two upstreamkinases, NF-kappaB-inducing kinase and mitogen-activated protein kinase/ERK kinase kinase-1. Proc NatlAcad Sci U S A 95: 3537–3542.
Neff, L., Zeisel, M., Sibilia, J., Scholler-Guinard, M., Klein,J.P., and Wachsmann, D. (2001) NF-kappaB and the MAP
kinases/AP-1 pathways are both involved in interleukin-6and interleukin-8 expression in fibroblast-like synoviocytesstimulated by protein I/II, a modulin from oral streptococci.Cell Microbiol 3: 703–712.
Nemoto, S., DiDonato, J.A., and Lin, A. (1998) Coordinateregulation of IkappaB kinases by mitogen-activated proteinkinase kinase kinase 1 and NF-kappaB-inducing kinase.Mol Cell Biol 18: 7336–7343.
Orth, K., Palmer, L.E., Bao, Z.Q., Stewart, S., Rudolph, A.E.,Bliska, J.B., and Dixon, J.E. (1999) Inhibition of themitogen-activated protein kinase kinase superfamily by aYersinia effector. Science 285: 1920–1923.
Parrini, M.C., Lei, M., Harrison, S.C., and Mayer, B.J. (2002)Pak1 kinase homodimers are autoinhibited in trans anddissociated upon activation by Cdc42 and Rac1. Mol Cell9: 73–83.
Pepe, J.C., and Miller, V.L. (1993a) The biological role ofinvasin during a Yersinia enterocolitica infection. InfectAgents Dis 2: 236–241.
Pepe, J.C., and Miller, V.L. (1993b) Yersinia enterocoliticainvasin: a primary role in the initiation of infection. Proc NatlAcad Sci U S A 90: 6473–6477.
del Pozo, M.A., Price, L.S., Alderson, N.B., Ren, X.D., andSchwartz, M.A. (2000) Adhesion to the extracellular matrixregulates the coupling of the small GTPase Rac to itseffector PAK. EMBO J 19: 2008–2014.
Rothwarf, D.M., and Karin, M. (1999) The NF-kappa B acti-vation pathway: a paradigm in information transfer frommembrane to nucleus. Sci STKE 1999: RE1.
Ruckdeschel, K., Mannel, O., Richter, K., Jacobi, C.A.,Trulzsch, K., Rouot, B., and Heesemann, J. (2001) Yersiniaouter protein P of Yersinia enterocolitica simultaneouslyblocks the nuclear factor-kappa B pathway and exploitslipopolysaccharide signaling to trigger apoptosis in mac-rophages. J Immunol 166: 1823–1831.
Ruckdeschel, K., Roggenkamp, A., Schubert, S., and Heese-mann, J. (1996) Differential contribution of Yersinia entero-colitica virulence factors to evasion of microbicidal actionof neutrophils. Infect Immun 64: 724–733.
Saccani, S., Pantano, S., and Natoli, G. (2002) p38-Dependent marking of inflammatory genes for increasedNF-kappa B recruitment. Nat Immunol 3: 69–75.
Savkovic, S.D., Koutsouris, A., and Hecht, G. (1997) Activa-tion of NF-kappaB in intestinal epithelial cells by entero-pathogenic Escherichia coli. Am J Physiol 273: C1160–C1167.
Schmitz, A.A., Govek, E.E., Bottner, B., and Van Aelst, L.(2000) Rho GTPases: signaling, migration, and invasion.Exp Cell Res 261: 1–12.
Schreiber, E., Matthias, P., Muller, M.M., and Schaffner, W.(1989) Rapid detection of octamer binding proteins with‘mini-extracts’, prepared from a small number of cells. NuclAcids Res 17: 6419.
Schulte, R., and Autenrieth, I.B. (1998) Yersinia enterocolit-ica-induced interleukin-8 secretion by human intestinal epi-thelial cells depends on cell differentiation. Infect Immun66: 1216–1224.
Schulte, R., Grassl, G.A., Preger, S., Fessele, S., Jacobi,C.A., Schaller, M., Nelson, P.J., and Autenrieth, I.B.(2000a) Yersinia enterocolitica invasin protein triggers IL-8production in epithelial cells via activation of Rel p65-p65homodimers. FASEB J 14: 1471–1484.
Schulte, R., Kerneis, S., Klinke, S., Bartels, H., Preger, S.,Kraehenbuhl, J.P., Pringault, E., and Autenrieth, I.B.

Invasin triggers IL-8 production via Rac1 and MAPK cascades 971
© 2003 Blackwell Publishing Ltd, Cellular Microbiology, 5, 957–971
(2000b) Translocation of Yersinia entrocolitica acrossreconstituted intestinal epithelial monolayers is triggered byYersinia invasin binding to beta1 integrins apicallyexpressed on M-like cells. Cell Microbiol 2: 173–185.
Schulte, R., Zumbihl, R., Kampik, D., Fauconnier, A.,and Autenrieth, I.B. (1998) Wortmannin blocks Yersiniainvasin-triggered internalization, but not interleukin-8 pro-duction by epithelial cells. Med Microbiol Immunol (Berl)187: 53–60.
Sehr, P., Joseph, G., Genth, H., Just, I., Pick, E., and Akto-ries, K. (1998) Glucosylation and ADP ribosylation of rhoproteins: effects on nucleotide binding, GTPase activity,and effector coupling. Biochemistry 37: 5296–5304.
Sharma, S.A., Tummuru, M.K., Blaser, M.J., and Kerr, L.D.(1998) Activation of IL-8 gene expression by Helicobacterpylori is regulated by transcription factor nuclear factor-kappa B in gastric epithelial cells. J Immunol 160: 2401–2407.
Van Aelst, L., and D’Souza-Schorey, C. (1997) Rho GTPasesand signaling networks. Genes Dev 11: 2295–2322.
Viboud, G.I., So, S.S., Ryndak, M.B., and Bliska, J.B. (2003)Proinflammatory signalling stimulated by the type III trans-location factor YopB is counteracted by multiple effectors inepithelial cells infected with Yersinia pseudotuberculosis.Mol Microbiol 47: 1305–1315.
Werner, E., Kheradmand, F., Isberg, R.R., and Werb, Z.(2001) Phagocytosis mediated by Yersinia invasin inducescollagenase-1 expression in rabbit synovial fibroblaststhrough a proinflammatory cascade. J Cell Sci 114: 3333–3343.
Wery-Zennaro, S., Zugaza, J.L., Letourneur, M., Bertoglio,J., and Pierre, J. (2000) IL-4 regulation of IL-6 productioninvolves Rac/Cdc42- and p38 MAPK-dependent pathwaysin keratinocytes. Oncogene 19: 1596–1604.
Whitmarsh, A.J., and Davis, R.J. (1996) Transcription factorAP-1 regulation by mitogen-activated protein kinase signaltransduction pathways. J Mol Med 74: 589–607.
Wiedemann, A., Linder, S., Grassl, G., Albert, M., Autenrieth,I., and Aepfelbacher, M. (2001) Yersinia enterocolitica inva-sin triggers phagocytosis via beta1 integrins, CDC42Hsand WASp in macrophages. Cell Microbiol 3: 693–702.
Winzen, R., Kracht, M., Ritter, B., Wilhelm, A., Chen, C.Y.,Shyu, A.B., Muller, M., Gaestel, M., Resch, K., and Holt-mann, H. (1999) The p38 MAP kinase pathway signals forcytokine-induced mRNA stabilization via MAP kinase-activated protein kinase 2 and an AU-rich region-targetedmechanism. EMBO J 18: 4969–4980.
Wolter, S., Mushinski, J.F., Saboori, A.M., Resch, K., andKracht, M. (2002) Inducible expression of a constitutivelyactive mutant of mitogen-activated protein kinase kinase 7specifically activates c-JUN NH2-terminal protein kinase,alters expression of at least nine genes, and inhibits cellproliferation. J Biol Chem 277: 3576–3584.
Wu, G.D., Lai, E.J., Huang, N., and Wen, X. (1997) Oct–1and CCAAT/enhancer-binding protein (C/EBP) bind tooverlapping elements within the interleukin-8 promoter.The role of Oct–1 as a transcriptional repressor. J BiolChem 272: 2396–2403.
Wullt, B., Bergsten, G., Connell, H., Rollano, P., Gebratsedik,N., Hang, L., and Svanborg, C. (2001) P-fimbriae triggermucosal responses to Escherichia coli in the human uri-nary tract. Cell Microbiol 3: 255–264.
Yasumoto, K., Okamoto, S., Mukaida, N., Murakami, S., Mai,M., and Matsushima, K. (1992) Tumor necrosis factor alphaand interferon gamma synergistically induce interleukin 8production in a human gastric cancer cell line throughacting concurrently on AP-1 and NF-kB-like binding sitesof the interleukin 8 gene. J Biol Chem 267: 22506–22511.





























