Embed Size (px)
Citation preview

Limnol. Oceanogr., 38(7), 1993, 1438-1451 0 1993, by the American Society of Limnology and Oceanography, Inc.
Anoxic and oxic degradation of 14C-labeled chloropigments and a 14C-labeled diatom in Long Island Sound sediments
Ming- Yi Sun,’ Cindy Lee, and Robert C. Aller Marine Sciences Research Center, SUNY, Stony Brook, New York 11794-5000
Abstract
Anoxic and oxic degradation pathways of sedimentary chloropigments were examined by spiking marine sediment with 14C-labeled algal cells and purified chloropigments from the diatom Skeletonema costatum. These experiments suggest that Chl a degrades through multiple pathways. Under oxic conditions, most bulk sedimentary Chl a degraded to various colorless compounds and only a minor fraction degraded to pheophytin a; added 14C-labeled Chl a also degraded quickly, but 30-40% of this Chl a was converted to pheophytin a. Under anoxic conditions, only a small fraction of bulk Chl a degraded, but added 14C- labeled Chl a continuously degraded and -30-40% of it was converted to pheophytin a, Pheophytin a is relatively stable under anoxic conditions but degrades under oxic conditions, thus it is a potential end product of chloropigmcnt degradation in anoxic environments.
Degradation pathways are likely dependent on the relative proportion of unassociated Chl a to chlo- rophyll complexes present in the sediment. Only unassociated Chl a appears to be available for anoxic decomposition. Under oxic conditions, some colorless products were further degraded and solubilized, none of the 14C label added as purified pigments was lost under anoxic conditions during the l-month incubation. About 80% of the acetone-extractable “C in labeled cells was lost in 1 month from sediments under oxic conditions and -30% under anoxic conditions.
Chlorophyll a and its primary degradation products, the pheopigments, are the most abundant photosynthetic pigments found in highly productive coastal sediments (Furlong and Carpenter 1988; Montagna et al. 1989; Sun et al. 1991). These tetrapyrrole com- pounds can be quickly degraded by diagenetic processes in the water column and surface sed- iments. However, a significant amount of chlorins, which are believed to originate from
Anoxic conditions may also be a critical factor affecting chloropigment preservation (Sun et al. 1993).
Some pheopigments and other nonpolar chlorophyll degradation products are pro- duced from grazing of planktonic algae by zoo- planktonic herbivores and are preserved in sediments. It is not clear how Chl a and labile pheopigments reaching the sediments trans- form or degrade into stable porphyrins or
chlorophyll, can be preserved in recent sedi- whether they are completely remineralized. In ments and even in older sediments (e.g. a Mio- marine sediments of Long Island Sound (LIS),
Chl a concentrations often exhibit an expo- cene oil shale: Prowse and Maxwell 199 1). There are several possible explanations for nential decrease with depth to a very low back- chlorin preservation in sediments: degradation to more stable pheopigments (Keely et al. 1990; Keely and Maxwell 199 l), incorporation into humic matter (Furlong and Carpenter 1988), and ester&cation to nonpolar chlorophyll deg- radation products (King and Repeta 199 1).
1 Present address: Skidaway lnstitute of Oceanography, P.O. Box 13687, Savannah, Georgia 31416.
ground level; total identified pheopigments also decrease with depth, although there are some differences in profile shapes and background levels for individual components (Sun 1992). The degradation rates of Chl a in LIS sedi- ments can be measured by monitoring changes in Chl a concentration with time during in- cubation experiments. However, examination of Chl a degradation pathways is hindered by the following: diversity of Chl a degradation rates and mechanisms due to structural char-
Acknowledgments acteristics (association with protein or other We thank E. Cospcr who provided access to cells and
culturing equipment. We are grateful to D. Rcpeta for complexes) or external environmental condi-
discussions and suggestions. tions (e.g. oxic vs. anoxic sediments, presence
This research was supported by grants from the National of benthic organisms, and sources of enzy- Science Foundation (OCE 90- 159 15 to C. L. and 90-O 1397 matic or hydrolytic reactions); multiple sources to RCA.). of pheopigments, including sinking particles
1438

Chloropigment degradation 1439
from the overlying water and production in the sediment due to Chl a degradation; in situ degradation of pheopigments; and technical limitations such as difficulty in detection due to loss of color of some degradation products.
Thus, to understand Chl a degradation path- ways in sediments requires a technique that can chemically separate various chloropig- ments and specifically indicate the relationship between precursor and products. High perfor- mance liquid chromatography (HPLC) tech- niques meet the requirement for separation and have been widely used to determine Chl a and its degradation product distributions in vari- ous natural environments. Radiocarbon tracer techniques are highly specific and sensitive and have been used to examine biological pro- cesses involving C cycling. In this study, we use both 14C-labeled algae and individual 14C- labeled chloropigments as tracers to explore Chl a degradation pathways and factors af- fecting chloropigment preservation in LIS sed- iments.
Methods
Preparation of 14C-labeled algal cells- A bacteria-free culture of the diatom Skeleto- nema costatum was grown in a closed flask with Enriched Instant Ocean media (EI0/2) at room temperature under continuous fluores- cent light (9 1.4 PEinst m-2 s-l). Two subsam- ples (3 ml each, 1 x lo5 cells ml-‘) of the culture were diluted to 800 ml with EIO me- dium, but one was spiked with 2 mCi H14C0,- in aqueous solution (2 ml, 50 mCi mM-I). Cell numbers were counted daily in the culture without 14C in the medium and 14C uptake rate was estimated by counting radio- activity in supernatant (no cells present) from the spiked medium every day. The pH of the medium was kept constant (- 8.2) by adding a trace amount of HCl when necessary. The proportion of 14C to 12C in the initial bicar- bonate present was -2%. The cultures were mixed twice every day. After 13 d (about eight cell divisions), the cells were harvested by cen- trifugation. Some of the 14C-labeled cells were first rinsed twice with seawater to remove any H14C0,- and then used to spike fresh sedi- ments in incubation experiments. The re- maining 14C-labeled cells were extracted twice with 100% acetone (5 ml each time) and the
extract was used to obtain purified 14C-labekd chloropigments.
PuriJication of 14C-labeled chloropigments- The acetone extracts from 14C-labeled cells (- 2 x 108) were combined and filtered through 0.2~pm Zetapor membrane filters and the vol- ume reduced to -3 ml under N2. The 14C- labeled chloropigments in the extract were first isolated by ion-pairing reversed-phase HPLC (Mantoura and Llewellyn 1983; Sun et al. 199 1). The HPLC system consisted of a Beck- man model 420 gradient controller with Beck- man 110 pumps and an Alltech 5-pm C- 18 Adsorbosphere column (250 mm X lo-mm i.d.). The solvent system was the same as in earlier work (Sun et al. 1991), ramping a pri- mary eluant (80% methanol; 20% aqueous so- lution of 0.5 mM tetrabutyl ammonium ace- tate and 10 mM ammonium acetate) against a secondary eluant (20% acetone in methanol) from 100% to 0% in 15 min with a 60-min hold at 0%. Chloropigments were detected with a Kratos FS 970 fluorometer having an exci- tation wavelength of 405 nm and measuring emission at >580 nm and with a Kratos UV absorbance detector having a wavelength set at 440 nm. After injection of 250 ~1 of the extract, Chl a and pheophytin a were collected with a Gilson 203 fraction collector at l-ml intervals.
Goericke (1990) showed that the Chl a iso-
lated by HPLC could contain unknown col- orless compounds (- 10% of the Chl a weight) that coelute with Chl a and can contain lo- 70% of the radioactivity in the Chl a fraction. Thus, to further purify 14C-labeled Chl a and pheophytin a, we used two-dimensional thin- layer chromatography (TLC) similar in prin- ciple to methods used by Redalje and Laws (198 1) and Welschmeyer and Lorenzen (1984).
The combined Chl a fractions (plus a few microliters of pyridine to avoid acidification of Chl a) and pheophytin a fractions collected from the HPLC were concentrated to - 1 ml under N2. Then, - 160 ~1 were spotted onto TLC plates and separated two dimensionally: first with 20 ml of methanol in 80 ml of acetone and second with 3 ml of pyridine in 97 ml of acetone. The TLC plates were Whatman KC- 18 reversed-phase ODS-bonded silica plates (20 X 20 cm and 200~ym layer thick). The Chl a and pheophytin a spots were easily recog- nized by color and by comparison of &values

1440 Sun et al.
with standards. The 14C-labeled chloropig- ments were allowed to dry on the plate for 30 min, and the ODS-bonded silica was then scraped off the plates. The silica was gently ground to a powder and stored in the freezer (- 17°C) until its use in the degradation ex- periments. Purity of 14C-labeled Chl a after 2-D TLC separation was examined by con- verting 14C-labeled Chl a into 14C-labeled pheophytin a. The conversion was carried out by acidifying the Chl a with a few drops of HCl, waiting 1 h, and then neutralizing with an equivalent amount of NaOH. Shifts in both HPLC Chl a concentration and in 14C activity of the Chl a peak were monitored to follow the conversion of 14C-labeled Chl a into 14C-la- beled pheophytin a.
Degradation experiments-Surface (top 1 cm) sediment samples for use in anoxic and oxic decomposition experiments were taken from intertidal sediments adjacent to Spartina marsh areas in Flax Pond, a shallow embay- ment on the north shore of Long Island (Woodwell and Pecan 1973). Mean organic C content in Flax Pond sediments is -2.8% and the salinity of sediment pore waters ranges from 23 to 27%0 (Wang and Lee 1990). Flax Pond sediments where samples were taken consisted primarily of sandy mud, were anoxic below the upper few millimeters, and had a pH range from - 6.5 (surface) to 7.1 (Swider and Mackin 1989). Seasonal organic matter decomposition rates in Flax Pond sediments encompass near- ly the entire range of rates commonly found in nearshore sediments and marshes (Mackin and Swider 1989).
Surface sediments were first sieved (0.5-mm mesh, no water added) to remove macrofauna and any coarse debris. Then, 14C-labeled cells or chloropigments (on silica) were spiked into wet sediments, with the added organic C con- tent from 14C-labeled cells composing < 1% of the initial sediment organic C content. Added 14C-labeled Chl a was <3% of natural Chl a concentrations, while 14C-labeled pheophytin a accounted for -26% of the natural pheo- phytin a in these sediments.
The 14C-labeled cells or chloropigments were mixed with wet sediments in a Vortex mixer for 5 min; a nearly homogeneous distribution of the tracers was reached (relative SD gen- erally <5%, tested by duplicate extractions). Sediments were placed in thin plugs (1.5 mm
thick, 2.3-cm i.d.) for use in incubation ex- periments (Aller and Mackin 1989; Sun et al. 1993). About 20% of the pore water was first removed by gentle centrifugation (500 rpm) to make preparation of plugs easier (final wt% H20, -60%). No 14C activity was found in the removed pore water. The sediment plugs con- taining 14C tracers were incubated in “open” anoxic and oxic systems. In open systems, the surface of a sediment slice was exposed to a relatively large (- 6 liters) water reservoir, and thus solute exchange between sediment and the water reservoir could occur easily. Anoxic or oxic conditions were controlled by contin- ual purging of the water reservoir with NJCO, or with air. Incubation experiments were car- ried out in the dark and temperature was kept constant (17.5”C). Techniques of extraction and HPLC analysis of Chl a and its degradation products were described earlier by Sun et al. (1991).
Radioactivity in extracts and HPLC eluate fractions was measured with a Packard 1600CA liquid scintillation counter. Generally, part of the acetone extract (100 ~1) of the incubated sediments was directly added to 3 ml of OPTI- fluor scintillation cocktail and 14C counted to 1% (SD) for 5 min. When higher sensitivity was required, the acetone extracts were con- centrated (5 ml to 1 ml) under N2 and then injected into the HPLC. Each fraction (1 ml) eluted was counted (0.8 ml of eluate per 3 ml of scintillation fluor) and a radiochromato- gram obtained.
Concentrations reported here are based on fluorometric peak area (converted to units of nmol 8-l dry sediment), while radioactivities in both acetone extracts and column eluates are reported in units of dpm g-l dry sediment.
Results PuriJication of 14C-labeled Chl a-Due to
their lack of solubility in water, chloropig- ments can be difficult to manipulate in exper- iments determining chlorophyll breakdown in sediments. The TLC technique used here offers a highly selective separation of 14C-labeled pig- ments and has the advantage that ODS-bond- ed silica acts as an excellent carrier for trans- port of pigment tracer into sediments. Recovery of Chl a from TLC plates was measured by extracting standard Chl a from the scraped ODS-bonded silica with acetone and found to

Chloropigment degradation 1441
be -97%. After spiking the tracer into sedi- ments, the immediate recovery of labeled Chl a was -93%.
Measurements of Chl a concentration and radioactivity in extracts purified by HPLC and 2-D TLC suggested that the Chl a was -90% pure. Other isomers apparently formed during extraction of the pigment from ODS-bonded silica. Specific activity of 14C-labeled Chl a was estimated to be 59.5 mCi mM-’ based on mea- sured Chl a concentration and integrated ac- tivity in the Chl a fraction. If all carbon atoms in Chl a were 14C, the maximum specific ac- tivity would be 3,465 mCi mM-I. Thus, the ratio of 14C atoms to total C atoms in our labeled Chl a was estimated to be 1.72%, sim- ilar to the proportion (2%) of 14C to total C in culture medium containing added H14C03-. This ratio suggests that there is only one 14C atom in each Chl a molecule (5 5 atoms of C per molecule) used in this study. We assume that 14C atoms were incorporated uniformly into each Chl a molecule, because the cells were grown through eight divisions. According to Welschmeyer and Lorenzen’s (1984) iso- tope-labeling model, > 99% of the cells are la- beled after eight divisions.
During the test for purity of the labeled Chl a, acidification of labeled Chl a purified through HPLC and 2-D TLC separation resulted in the conversion of >95% of the labeled Chl a into labeled pheophytin a (as measured both by chemical concentration and radioactivity). Af- ter HPLC purification alone, acidification of the Chl a extract resulted in the conversion of all the Chl a into pheophytin a but only -60% of the 14C activity into pheophytin a, suggest- ing that much of the material in the HPLC eluate was due to colorless impurities. Thus, the further purification by 2-D TLC was nec- essary.
Pheophytin a was prepared through the same 2-D TLC procedure as for Chl a rather than by acidification of labeled Chl a because both pigments could be collected by HPLC at the same time. The specific activity of labeled pheophytin a was estimated to be 47.6 mCi mm-‘, -20% lower than that of labeled Chl a. This decrease of specific activity may be caused by the formation of pheophytin a dur- ing early growth stages (early division) of the culture.
Incubation of 14C-labeled diatoms-Open
? P $ ‘on El
aa lo
0 t:
0 5 10 15 20 25 30
Time (day)
Fig. 1. Changes in total radioactivity in acetone ex- tracts of sediments spiked with 14C-labeled diatom cells during oxic and anoxic incubations. The data were fitted with a “multiple-G” model (expressed by curves, see dis- cussion).
incubations of sediments spiked with 14C-la- beled diatom cells showed that organic C de- rived from the cells (expressed by total 14C activity in extract, Fig. 1) was degraded more slowly under anoxic than oxic conditions. About 30% of the total 14C in cells was lost under anoxic conditions; it was probably re- mineralized to 14C0, and lost through diffu- sion and exchange between the pore waters and the overlying water. In contrast, - 80% of total 14C was lost under oxic conditions. Chl a deg- radation with time in this experiment dis- played a pattern approximately similar to pre- vious work (Sun et al. 1993). The previous study showed that concentrations of extract- able Chl a can increase initially due to release from a bound pool; only a slight increase oc- curred here,
Incubation of 14C-labeled Chl a-Open in- cubations of sediments spiked with labeled Chl a under anoxic and oxic conditions resulted in the loss of Chl a with time. HPLC chromat- ograms and radiochromatograms at different times during the incubation are illustrated in Fig. 2 (oxic) and Fig. 3 (anoxic). Changes in HPLC peak sizes show the loss of total Chl a (natural plus added radiolabeled) and the ap- pearance of degradation products, while the variations in corresponding radiochromato- grams reflect the loss of labeled Chl a and the appearance of radiolabeled degradation prod- ucts. Total radioactivities in acetone extracts under oxic and anoxic conditions (Fig. 4A)

1442 Sun et al.
2 3 100000
1 t = 0 days
1 !’
-J lfi
-J I b&
4
LA-
u t -‘U
L I!!, c, _ b
80000 - 3
I ,
60000 -
0 10 20 30 40 50 60
Oxic, t = 7 days
4
100000
80000
60000
40000
20000
0 0 10 20 30 40 50 60
100000
80000 1 Oxic, t = 21 days
60000 1
40000
i u 4
2ooo:: 0 10 20 30 40 50 6-O
HPLC elution time (min)
Fig. 2. Chromatograms and radiochromatograms of extracts from sediments spiked with 14C-labeled Chl a during oxic incubations. 1 -Pheophorbide a; 2-pyropheophorbide a; 3 -Chl a; 4-pheophytin a; U-unknown chlorin compound.

Chloropigment degradation
3 100000 1 Anoxic, t = 7 days 2
80000
60000 I 4
40000- I U
1 4
-0 10 20 30 40 50 60
100000 Anoxic, t = 21 days
- ‘bo 80000
60000 4
1443
40000
10 20 30 40 50 60
100000 Anoxic, t = 35 days
80000
60000
40000 oad 20000
0 10 20 30 40 50 60
HPLC elution time (min) Fig. 3. As Fig. 2, but during anoxic incubations. C-colorless components.

Sun et al.
Fig. 4. A. Changes in total radioactivity in acetone extracts of sediment spiked with 14C-labeled Chl u. B. Changes in Chl a radioactivity and total concentration relative to initial values during oxic and anoxic incuba- tions m-concentration; O-radioactivity). Replicate con- centration measurements by HPLC varied by + 10%. The relative standard deviations in the activity of labeled trac- ers and total radioactivity were less than -+5%.
showed that, although the labeled Chl a quickly decayed in both oxic and anoxic systems, most 14C activity still remained in the acetone ex- tract (100% under anoxic conditions and - 77% under oxic conditions). This case was in con- trast to the incubation with radiolabeled dia- toms when most of the radioactivity was lost from the system (presumably as COz from mostly nonpigments).
Major degradation products of labeled Chl a were found to be pheophytin a (retention time, -45 min), an unknown chlorin com- pound (retention time, - 37 min), and several polar or less-polar (relative to Chl a) colorless components. Although the unknown chlorin at 37 min was seen in the radioisotope incu- bations, it has not been observed in natural sediments. Under anoxic conditions, polar colorless components apparently accumulated initially and then broke down further into oth- er components with different retention times. In contrast, polar components disappeared from acetone extracts under oxic conditions. With time, more components (including polar and less-polar) were produced under both an- oxic and oxic conditions. However, labeled pheophytin a formed during anoxic incubation seemed to be stable while it apparently de- graded under oxic conditions as shown by de- creases in both total concentration and radio- activity after 1 week of oxic incubation (Table 1).
Losses of total Chl a concentration and la- Incubation of 14C-labeled pheophytin a- beled Chl a activity with time (Fig. 4B) reveal Open oxic and anoxic incubations of sedi- differences between oxic and anoxic incuba- ments spiked with labeled pheophytin a are tions. Under oxic conditions, total Chl a con- illustrated in Figs. 5 and 6. Under oxic con- centration and labeled Chl a activity contin- ditions, both natural and labeled pheophytin uously decrease while only labeled Chl a a degraded over time although not as rapidly decreases substantially under anoxic condi- as Chl a; much (40%) of the pheophytin a re- tions. Meanwhile, pheophytin a was formed mained after 2 1 d (Fig. 7A). Under anoxic con- under both oxic and anoxic conditions (Table ditions, neither total nor labeled pheophytin a
Table 1. Changes in pheophytin a concentration and radioactivity during oxic and anoxic incubations of sedi- mcnt spiked with 14C-labeled Chl a.
Days
0 7
21 35
Concentration Radioactivity (nm01 g ‘) (IO5 dpm g I)
oxic Anoxic OXiC Anoxic
8.50 8.50 0 0 8.98 10.1 6.52 8.50 5.98 8.44 4.66 7.40
9.24 9.60
1); this formation was observed as an increase in the concentration of labeled pheophytin a. About 30-40% of added labeled Chl a (0.58- 0.77 nmol g-‘) was converted to labeled pheo- phytin a under both oxic and anoxic condi- tions even though the total pheophytin con- centration changed very little (l-2 nmol g-l pheophytin was produced when - 60 nmol g-l natural Chl a was degraded).

Chloropigment degradation 1445
2 3 100000 1 Oxic, t = 0 days
80000-
60000-
-0 10 20 30 40 50 60
100000- Oxic, t = 7 dap
80000-
4
60000- I
40000-
C
20000-
O- . 0 10 20 30 40 50 60
100000
80000
60000
Oxic, t = 21 days
0 10 20 30 40 50 60
HPLC elution time (min) Fig. 5. Chromatograms and radiochromatograms of extracts from sediments spiked with 14C-labeled pheophytin
a during oxic incubation. 1 -Pheophorbide a; 2-pyropheophorbide a; 3-Chl a; 4-pheophytin a; C-colorless components.
1446
3 100000 1 Anoxic, t = 7 days
Sun et al.
80000- 4
60000-
40000-
20000-
0 10 20 30 40 50 60
Anoxic. t = 21 days
0 10 20 30 40 50 60
HPLC elution time (min)
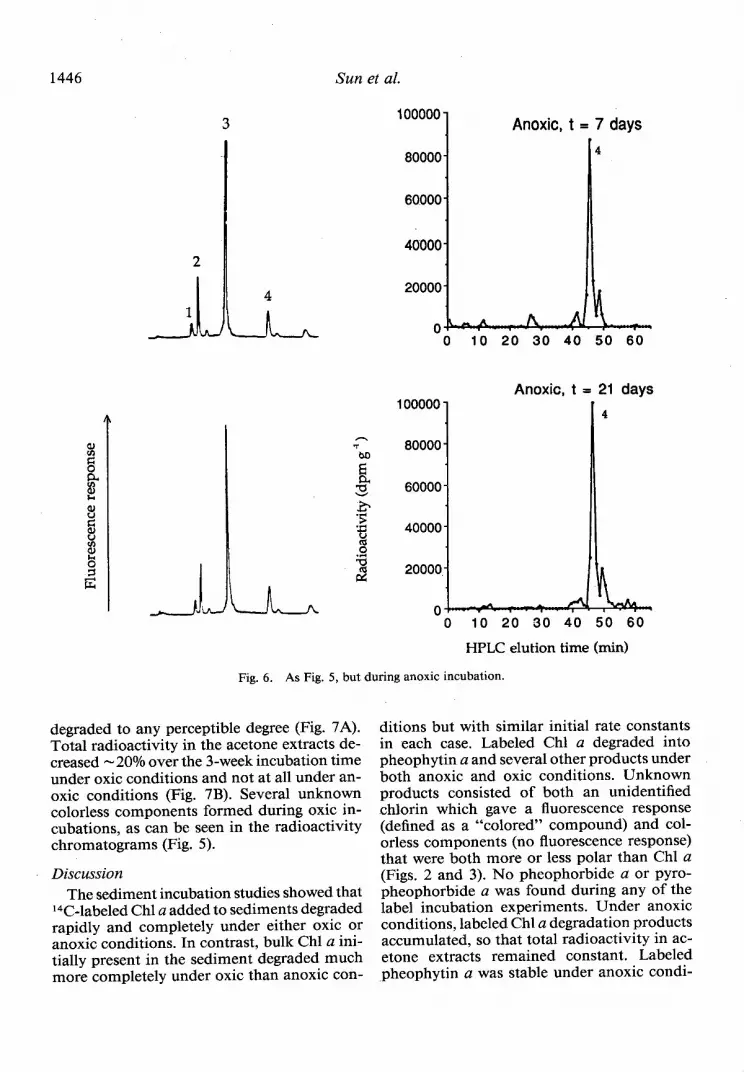
Fig. 6. As Fig. 5, but during anoxic incubation.
degraded to any perceptible degree (Fig. 7A). Total radioactivity in the acetone extracts de- creased - 20% over the 3-week incubation time under oxic conditions and not at all under an- oxic conditions (Fig. 7B). Several unknown colorless components formed during oxic in- cubations, as can be seen in the radioactivity chromatograms (Fig. 5).
Discussion The sediment incubation studies showed that
14C-labeled Chl a added to sediments degraded rapidly and completely under either oxic or anoxic conditions. In contrast, bulk Chl a ini- tially present in the sediment degraded much more completely under oxic than anoxic con-
ditions but with similar initial rate constants in each case. Labeled Chl a degraded into pheophytin a and several other products under both anoxic and oxic conditions. Unknown products consisted of both an unidentified chlorin which gave a fluorescence response (defined as a “colored” compound) and col- orless components (no fluorescence response) that were both more or less polar than Chl a (Figs. 2 and 3). No pheophorbide a or pyro- pheophorbide a was found during any of the label incubation experiments. Under anoxic conditions, labeled Chl a degradation products accumulated, so that total radioactivity in ac- etone extracts remained constant. Labeled -pheophytin a was stable under anoxic condi-

Chloropigment degradation 1447
tions for up to 3 weeks. In contrast, under oxic conditions, some of the colorless products were solubilized and total extractable radioactivity decreased by -20%. Bulk pheophytin a also degraded under oxic conditions, although more slowly than Chl a. Overall degradation path- ways of bulk and tracer Chl a are summarized in Fig. 8.
The differences in bulk Chl a and labeled Chl a degradation both within and between given redox states are likely due to combina- tions of differential reactivity of chlorophyll present in natural complexes and the relative rate of enzymatic and abiogenic reactions un- der oxic and anoxic conditions. For example, most chlorophyll in living cells is noncova- lently bound to various proteins (Larkum and Barrett 1983). The relative proportions of these pigment-protein complexes vary with the type of plants and species (Brown 1987). Larkum and Barrett (1983) found that different pig- ment-protein complexes have different reac- tivities to degradation agents such as enzymes and acids due to variations in size, solubility, and molecular association.
Differential reactivity of Chl a between oxic and anoxic systems may also be due to the activity of Chl-degrading enzymes. There are at least two different enzymes involved in the degradation of chlorophyll in marine phyto- plankton. Chlorophyllase is a “particulate” en- zyme located in the chloroplast (Gage and Aronoff 1956; Ardao and Vennesland 1960) and can catalyze the hydrolytic removal of phytol from chlorophyll in various phyto- plankton species (Barrett and Jeffrey 1979). The Mg-releasing enzyme is capable of cata- lyzing the removal of Mg2+ from Chl a or chlo- rophyllide a, leading to the formation of pheo- pigments (Owens and Falkowski 1982). Ziegler et al. (1988) also suggested that the formation of pyropheophorbide a during senescence of Chlorella fusca was caused by enzymatic re- actions.
Differential rates and pathways of Chl a de- cay may also be due to differences in the deg- radation mechanisms of heterotrophs rather than in the nature of the Chl a. For example, Chl a is degraded through several known bi- ological and chemical processes, e.g. bacterial, viral, or autolytic cell lysis, grazing, and en- zymatic or hydrolysis reactions. Grazing by zooplankton has been emphasized as a major
1.4
4 0.6
1 0.4 k
- oxic . . . . . . . m anoxic A
10 20 30
4 T
oxic . . . . . . . . . . anoxic B
3
w 0 10 20 30
Time (day)
Fig. 7. A. Changes in pheophytin a radioactivity and concentration relative to initial values m-concentration; O-radioactivity). B. Changes in total radioactivity in ac- etone extracts of sediments spiked with W-labeled pheo- phytin a during oxic and anoxic incubations.
pathway of Chl a degradation in. pelagic sys- tems, but which degradation products are formed depends on the source plant species and on the grazer community. Chl a is de- graded to pheophytin a (removal of Mg) as a result of grazing activities of waterfleas (Daley 1973), salps (Jeffrey 1980; Hallegraeff 198 l), and copepods (Hallegraeff 198 1). The removal of Mg may be achieved during digestion through the action of acid (Shuman and Loren- zen 1975) as well as enzymes. Hawkins et al. (1986) suggested that chlorophyll degradation occurred in the stomach of the mussel, Mytilus edulis, due to low pH (6.2). Pheophorbide a (removal of both Mg and the phytol chain) can be found after grazing of algae by copepods (Jeffrey 1980; Shuman and Lorenzen 1975). In sediments, benthic organisms can also convert Chl a into different degradation products. Bianchi et al. ( 1988) reported that macrofauna

1448 Sun et al.
Reaction time scale
Bulk Chl u:
<2%
Pheophytin u
Stable Chl II
Colorless - lo%, anoxic
Tracer [14ClChl a:
oxic/anoxic 3040%
Pheophytin u
Chla Unidentified chlorin
Colorless
anoxic
i
Stable
oxic Colorless
1
oxic
oxic
E
Solubilizeci
anoxic Colorless transformation
anoxic
I
Stable
Colorless OXlC
anoxic
I
Stable
Colorless
1
oxic
oxic Solubilized
Colorless transformation
Fig. 8. Representation of degradation pathways and reaction time scales of bulk and tracer Chl a in sediments.
produced significantly more pheophorbide a oxic conditions. Either redox-specific struc- than microfauna, which produced only pheo- tural associations must stabilize pheophytin or phytin a. Klein et al. (1986) documented that enzyme activity capable of catalyzing further when heterotrophic protozoans are a compo- degradation is inhibited under anoxic condi- nent of the grazer community, Chl a and ca- tions. About 30-40% of the labeled Chl a add- rotenoids can be degraded to colorless residues ed to the sediments was converted to pheo- while very little Chl a is degraded to pheo- phytin a under both oxic and anoxic conditions. phytin and none to pheophorbide. However, the pheophytin a derived from nat-
Pheophytin a formed during Chl a degra- ural Chl a degradation was only a minor frac- dation can further degrade under oxic condi- tion (<4% of the total Chl a lost). This differ- tions but appears to remain stable under an- ence may be related to the proportion between oxic conditions. This degradation behavior is various forms of associated Chl a (e.g. Chl a consistent with the incubation experiments in protein complexes and other polymeric as- with labeled pheophytin a (Fig. 7A). That la- sociations) and unassociated Chl a molecules. beled Chl a degrades while labeled pheophytin Most natural Chl a was converted to various a does not degrade in anoxic sediments implies colorless compounds under oxic conditions, that acidification from decreased pH may be but only a small fraction of total natural Chl a major cause for Chl a degradation under an- a could be degraded under anoxic conditions,

Chloropigment degradation 1449
implying that aerobic organisms (e.g. micro- fauna) may be responsible for converting Chl a to colorless compounds during oxic incu- bations.
There are two probable reasons for the ab- sence of pheophorbide production in our ex- perimental sediments. First, macrofauna were previously removed with a 0.5-mm sieve and thus only meiofauna and microfauna likely played a role in Chl a degradation. These or- ganisms usually convert Chl a into colorless compounds or pheophytin as mentioned ear- lier (Bianchi et al. 1988; Klein et al. 1986). Secondly, 14C-labeled Chl a was added in the free form (although absorbed to ODS-bonded silica), so that there was no active dephytoli- zation enzyme (chlorophyllase) present such as is commonly found in chloroplasts of algal cells.
The results of labeled Chl a and pheophytin a incubations suggest that pheophytin a is an early product of Chl a degradation and is well preserved under anoxic conditions. Pheophy- tin has been observed to be one of the most abundant chloropigments in several deposi- tional environments. For example, in Black Sea anoxic sediments, pheophytin a was found to be the most abundant pigment component (figure 1 of King and Repeta 199 1). Keely and Maxwell (199 1) reported pheophytin a and pyropheophytin a in the bottom sediment of a eutrophic lake (Priest Pot Lake, Cumbria, U.K.). In long-buried (Bronze Age) plant re- mains, Hendry et al. ( 19 8 7) found no pigments other than traces of pheophytin a (< 10 pmol g-l). In LIS, both pheophytin a and pheo- phorbide a are present in surface sediments (top 1 cm) at similar concentrations. Below 5 cm, however, pheophytin a is more concen- trated (> 10 x ) than pheophorbide a. More- over, the ratios of pheophytin a to Chl a gen- erally increase with depth, while the ratios of pheophorbide a to Chl a typically decrease with depth (Sun 1992).
Preservation of pheophytin in LIS sedi- ments relative to other chloropigments can probably be explained by the different degra- dation rates and mechanisms of the various chloropigments. First, the degradation rate of pheophytin may be intrinsically slower than those of Chl a and pheophorbide. This hy- pothesis was partially confirmed by oxic in- cubations of sediments spiked with labeled Chl
a and pheophytin a (Figs. 2 and 5). Secondly, pheophytin may be continuously produced during Chl a degradation. Thirdly, pheophytin may be more resistant to chemical reactions caused by decreases in pH under anoxic con- ditions because its Mg has been replaced by two atoms of H.
A more general picture of organic matter degradation is obtained by comparing the ra- dioactivity lost in acetone extracts during sed- iment incubations where 14C-labeled algal cells, 14C-labeled Chl a, and 14C-labeled pheophytin a were added (Figs. 1, 4A, and 7B). For ex- ample, after -30 d under oxic conditions, much more labeled organic C (-80% of total added 14C) was lost from the acetone-extract- able phase of sediment spiked with 14C-labeled diatoms than that spiked with 14C-labeled Chl a (- 20%) or 14C-labeled pheophytin a (5 20%), implying that 14C-labeled algal cells contain acetone-extractable components that decom- pose more quickly than the chloropigments. Henrichs and Doyle (1986) suggested that the soluble fraction of algal cells decomposed markedly faster than the particulate material. They found that at least 30-40% of the total 14C-labeled organic C they added to sediments was remineralized to 14C02 by aerobic oxi- dation.
A “multiple-G” degradation model (Berner 1980; Westrich and Berner 1984) fit to the oxic incubation data of sediment spiked with 14C- labeled algae (Fig. 1) suggests that two organic fractions with remarkably different first-order degradation rate constants are present. The re- active fraction Gl (- 76%) was quickly de- graded (rate constant k, = 0.25 d-l), and the less-reactive fraction G2 (-24%) was more slowly degraded (rate constant k2 = 0.0 15 d-l). Rough estimations (-0.03-0.09 d-l) of the oxic degradation rate constants of Chl a based on concentration and radioactivity changes are generally in the range of our previous results (Sun et al. 1993) and are consistent with other reported values (0.01-o. 1 d-l) of Chl a first- order kinetic degradation rate constants (e.g. Leavitt and Carpenter 1990; Bianchi and Findlay 1991). Carbon lost from the experi- mental sediments probably decomposed to CO2 or other small soluble products and was lost through diffusion and exchange between pore water and the overlying water. Under anoxic conditions, - 30% of the acetone-extractable

1450 Sun
radiolabeled organic C in algae was lost from the sediments, but there was almost no loss of labeled chloropigments from the sediments. This finding implies that the more reactive compounds in acetone extracts of algae de- compose into CO2 or soluble products that can be lost from sediments through diffusion and exchange, while the chloropigments (Chl a and pheophytin a in this case) degrade into com- ponents that are acetone extractable and are not lost from the sediments.
References ALLER, R. C., AND J. E. MACKIN. 1989. Open-incubation,
diffusion methods for measuring solute reaction rates in sediments. J. Mar. Res. 47: 41 l-440.
ARDAO, C., AND B. VENNESLAND. 1960. Chlorophyllase activity of spinach chloroplastin. Plant Physiol. 35: 368-371.
BARRETT, J., AND S. W. JEFFREY. 1979. A note on the occurrence of chlorophyllase in marine algae. J. Exp. Mar. Biol. Ecol. 7: 255-262.
BERNER, R. A. 1980. Early diagenesis: A theoretical ap- proach. Princeton.
BIANCHI, T. S., R. DAWSON, AND P. SAWANGWONG. 1988. The effects of macrobenthic deposit-feeding on the degradation of chloropigments in sandy sediments. J. Exp. Mar. Biol. Ecol. 122: 243-255.
- AND S. FINDLAY. 199 1. Decomposition of Hud- son estuary macrophytes: Photosynthetic pigment transformation and decay constants. Estuaries 14: 65- 73.
BROWN, J. S. 1987. Functional organization of chloro- phyll-a and carotenoids in the alga, Nannochloropsis salina. Plant Physiol. 83: 434-437.
DALEY, R. J. 1973. Experimental characterization of la- custrine chlorophyll diagenesis. 2. Bacterial, viral and herbivore grazing effects. Arch. Hydrobiol. 72: 409- 439.
FURLONG, E. T., AND R. CARPENTER. 1988. Pigment preservation and remineralization in oxic coastal ma- rine sediments. Geochim. Cosmochim. Acta 52: 87- 99.
GAGE, R. S., AND S. ARONOFF. 1956. Chlorophyllase in soybean. Plant Physiol. 31: 368-37 1.
GOERICKE, R. 1990. Pigments as ecological tracers for the study of abundance and growth of marine phy- toplankton. Ph.D. thesis, Harvard Univ. 4 18 p.
HALLEGRAEFF, G. M. 198 1. Seasonal study of photo- plankton pigments and species at a coastal station off Sydney: Importance of diatoms and the nanoplank- ton. Mar. Biol. 61: 107-l 18.
HAWKINS, A. J. S., B. L. BAYNE, R. F. C. MANTOURA, AND C. A. LLEWELLYN. 1986. Chlorophyll degradation and absorption throughout the digestive system of the blue mussel Mytilus edulis L. J. Exp. Mar. Biol. Ecol. 96: 213-223.
HENDRY, G. A. F., J. D. HOUGHTON, AND S. B. BROWN. 1987. The degradation of chlorophyll-a biological enigma. New Phytol. 107: 255-302.
HENRICHS, S. M., AND A. P. DOYLE. 1986. Decompo-
et al.
sition of ‘%-labeled organic substances in marine sed- iments. Limnol. Oceanogr. 31: 765-778.
JEFFREY, S. W. 1980. Algal pigment systems, p. 33-58. Zn Primary productivity in the sea. Brookhaven Symp. Biol. 3 1. Plenum.
KEELY, B. J., AND J. R. MAXWELL. 199 1. Structural char- acterization of the major chlorins in a Recent sedi- ment. Org. Geochem. 17: 663-669.
- W. G. PROWSE, AND J. R. MAXWELL. 1990. The Trkibs hypothesis: An evaluation based on structural studies. Energy Fuels 4: 628-634.
KING, L. L., AND D. J. REPETA. 199 1. Novel pyropheo- phorbide steryl esters in Black Sea sediments. Gco- chim. Cosmochim. Acta 55: 2067-2074.
KLEIN, B., W. W. C. GIESKES, AND G. G. KRAAY. 1986. Digestion of chlorophylls and carotenoids by the ma- rine protozoan Oxyrrhis marina studied by h.p.1.c. analysis of algal pigments. J. Plankton Res. 8: 827- 836.
LARKUM, A. W. D., AND J. BARRETT. 1983. Light-har- vesting processes in algae. Adv. Bot. Res. 10: 3-219.
LEAVITT, P. R., AND S. R. CARPENTER.. 1990. Aphotic pigment degradation in the hypolimnion: Implica- tions for sedimentation studies and palcolimnology. Limnol. Oceanogr. 35: 520-534.
MACKIN, J. E., AND K. T. SWIDER. 1989. Organic matter decomposition pathways and oxygen consumption in coastal marine sediments. .7. Mar. Res. 47: 681-716.
MANTOURA, R. F. C., AND C. A. LLEWELLYN. 1983. The rapid determination of algal chlorophyll and carot- enoid pigments and their breakdown products in nat- ural waters by reversed-phase high-performance liq- uid chromatography. Anal. Chim. Acta 151: 297-3 14.
MONTAGNA P. A., J. E. BAUER, D. HARDIN, AND R. B. SPLES. 1989. Vertical distribution of microbial and meiofaunal populations in sediments of a natural coastal hydrocarbon seep. J. Mar. Res. 47:657-680.
OWENS, T. G., AND P. G. FALKOWSKI. 1982. Enzymatic degradation of chlorophyll-a by marine phytoplank- ton in vitro. Phytochemistry 21: 979-984.
PROWSE, W. G., AND J. R. MAXWELL. 199 1. High mo- lecular weight chlorins in a lacustrine shale. Org. Geo- them. 17: 877-886.
REDALJE, D. G., AND E. A. LAWS. 198 1. A new method for estimating phytoplankton growth rates and carbon biomass. Mar. Biol. 62: 73-79.
SHUMAN, F. K., AND C. J. LORENZEN. 1975. Quantitative degradation of chlorophyll by a marine herbivore. Limnol. Oceanogr. 20: 580-586.
SW, M.-Y. 1992. Early diagenesis of chloropigments in coastal sediments. Ph.D. thesis, State University of New York at Stony Brook. 3 14 p.
-, R. C. ALLER, AND C. LEE. 199 1. Early diagenesis of chlorophyll-a in Long Island Sound sediments: A measure of carbon flux and particle reworking. J. Mar. Res. 49: 379-401.
-, C. LEE, AND R. C. ALLER. 1993. Laboratory stud- ies of oxic and anoxic degradation of chlorophyll-a in Long Island Sound sediments. Geochim. Cosmo- chim. Acta 57: 147-157.
SWIDER, K. T., AND J. E. MACIUN. 1989. Transforma- tions of sulfur compounds in marsh-flat sediments. Geochim. Cosmochim. Acta 53: 23 11-2323.
WANG, X.-C., AND C. LEE. 1990. The distribution and

Chloropigment degradatibn 1451
adsorption behavior of aliphatic amines in marine An estuarine marsh. Brookhaven Natl. Lab., BNL and lacustrine sediments. Geochim. Cosmochim. Acta 50397. 54: 2759-2774. ZIEGLER, R., A. BLAHETA, N. GUHA, AND B. SCH~NEGGE.
WELSCHMEYER, N. A., AND C. J. LORENZEN. 1984. Car- 1988. Enzymatic formation of pheophorbide and bon- 14 labeling of phytoplankton carbon and chlo- pyropheophorbide during chlorophyll degradation in rophyll a carbon: Determination of specific growth a mutant of Chlorella fusca Shihira et Kraus. J. Plant rates. Limnol. Oceanogr. 29: 135-145. Physiol. 132: 327-332.
WESTRICH, J. T., AND R. A. BERNER. 1984. The role of sedimentary organic matter in bacterial sulfate re- duction. Limnol. Oceanogr. 29: 236-249.
WOODWELL, G. M., AND E. V. PECAN. 1973. Flax Pond:
Submitted: 22 May 1992 Accepted: 27 January 1993 Revised: 24 February 1993



![Impact of carbon nanomaterials on the behaviour of 14C-phenanthrene and 14C-benzo-[a] pyrene in soil](https://img.pdfslide.net/doc/110x75/633305c7f00804055104ba85/impact-of-carbon-nanomaterials-on-the-behaviour-of-14c-phenanthrene-and-14c-benzo-a.jpg)








![Biologically stable [18F]-labeled benzylfluoride derivatives](https://img.pdfslide.net/doc/110x75/634b0a65e2b881b8bf01abb5/biologically-stable-18f-labeled-benzylfluoride-derivatives.jpg)
















