Embed Size (px)
Citation preview

The EMBO Journal vol.10 no.9 pp.2489-2495, 1991
Authentic reverse transcriptase is coded by jockey, a
mobile Drosophila element related to mammalian LINEs
Vladimir A.lvanovl, Anatoly A.Melnikov2,Alexander V.Siunov2, lstvan l.Fodor2 andYurii V.llyin3
'Institute of Cell Biophysics, 2Institute of Biochemistry and Physiologyof Microorganisms, Academy of Sciences, 142292 Poustchino,Moscow Region and 3V.A.Engelghart Institute of Molecular Biology,Academy of Sciences of the USSR, 117984 Moscow, USSR
Communicated by G.P.Georgiev
The mobile element jockey is similar in structuralorganization and coding potential to the LINEs of variousorganisms. It is transcribed at different stages ofDrosophila ontogenesis. The Drosophila LINE familyincludes active transposable elements. Current models forthe mechanism of transposition involve reverse transcrip-tion of an RNA intermediate and utilization of element-encoded proteins. As demonstrated here, a 2.23 kb DNAfragment from the region ofjockey encoding the putativereverse transcriptase was stably introduced into anexpression system under inducible control of theEscherichia coli lac regulatory elements. We describe theexpression of the 92 kDa protein and identify thispolypeptide alone as the authentic jockey reversetranscriptase based on some of its physical and enzymicproperties. The jockey polymerase demonstrates RNAand DNA-directed DNA polymerase activities but lacksdetectable RNase H, has a temperature optimum at 260C,requires Mg2+ or Mn2+ as a cofactor and is inactivatedby sulphydryl reagent. The enzyme prefers poly(rC) andpoly(rA) as template and 'activated' DNA is not effective.Key words: E. coli expression/LINE-like element/proteinpurification/recombinant DNA/retrotransposon reversetranscriptase
IntroductionLINE elements occupying a special place among long,moderately repeated DNA sequences, are interspersed in alleukaryotic genomes so far studied and appear to include afew active transposable elements and a multitude ofnonfunctional copies (Hutchison et al., 1989). TheDrosophila genome harbours a large number of LINE-relatedretrotransposon families. Complete copies of elementsbelonging to each group potentially encode two proteins. Oneof these proteins [open reading frame 2 (ORF2)] showshomology to the LINE-1 type of putative reverse trans-criptase (RT) while the other (ORFI) features cysteine-richmotifs typical for retroviral gag polyproteins (Xiong andEickbush, 1988; Doolittle et al., 1989). The DrosophilaLINE elements constitute attractive model systems forinvestigating the life-style of LINEs. They are organized intorelatively small families; within each of these, completefunctional members have been identified by genetic means.
Moreover, transposition of at least some of them can beexperimentally induced (Pritchard et al., 1988; Chaboissieret al., 1990). Current models for the mechanism oftransposition of LINE-like elements suggest intermediateformation of full-length transcripts followed by reversetranscription catalysed by an element-encoded RT andinsertion into new genome loci (Rogers, 1985).We cloned the original copy ofjockey while studying the
molecular basis of the ctM PP mutation (Mizrokhi et al.,1985) and later reported that two classes ofjockey copiescan be detected in the genome: full-sized copies (-5 kb)and copies with deletions in the internal region (-2.5 kb)(Mizrokhi et al., 1988; Priimagi et al., 1988). Accordingto its characteristics, jockey is a typical LINE element: itcauses duplication of the host DNA at its integration site,lacks terminal repeats and is terminated with an oligo(dA)sequence of variable length. Its two long reading frames werefound to contain amino acid sequences typical of nucleic acidbinding proteins from replication-competent retroviruses andconserved regions in reverse transcriptases. Two poly-adenylated jockey transcripts detected at different stages ofDrosophila ontogenesis and in cell cultures had the samelength as genomic copies ofjockey and corresponded to thestrand containing ORFs. a-amanitin experiments indicate thatjockey is transcribed by RNA polymerase II. The analysisof both the expression of CAT constructions and the initiationof transcription injockey genomic and transfected copies hasshown that jockey transcription is controlled by an internalpromoter. Inward location of the promoter allows it to bepreserved in the course of replication via reverse transcrip-tion and accounts for the distribution ofjockey and probablyother LINEs throughout the genome (Mizrokhi et al., 1988).Functions of LINEs (if any) in gene expression, genomeorganization and evolution, and the mechanism for the spreadof LINEs in a wide range of organisms, are general questionsto be answered in the future. In the field, investigations offunctional activity of proteins coded by LINEs are ofessential significance. Endogenous RT activity has beenrecently found in human pluripotential embryonal carcinomacells NTera 2D1, and researchers suggested that the activityidentified could originate from LINE-I (Deragon et al.,1990). However, direct evidence for coding of authentic RTby a nonviral retrotransposon and the study of the enzymefeatures are necessary. Recently we have cloned and express-ed in E. coli enzymatically active Rous sarcoma virus (RSV)RT and investigated its properties (Melnikov et al., 1988;Molnar et al., 1988; Chernov et al., 1990, 1991). Wesubsequently attempted to use the cloning strategy to studythat of the jockey element.
In the present study we cloned a fragment from the regionof jockey encoding the putative RT (pol gene) into thebacterial expression vector and obtained the expression ofan RT activity. Protein possessing the enzymatic activity hasbeen isolated from transfected cells and purified to near
© Oxford University Press 2489

V.A.Ivanov et al.
electrophoretic homogeneity. The protein was identified asthe jockey RT based on some of its physical and enzymaticproperties. In this paper we are the first to demonstratestrictly that a mobile Drosophila element related tomammalian LINEs encodes the authentic RT, and to describefeatures of the recombinant enzyme.
ResultsCloning and expression of jockey reversetranscriptase in E.coliWe have previously reported that jockey elements aretranscribed in vivo and that polyadenylated jockey transcripts,detected at different stages of Drosophila ontogenesis andin cell cultures, have the same length as genomic copies ofjockey and correspond to the strand containing ORFs(Mizrokhi et al., 1988). Our subsequent strategy was toclone the putative jockey pol gene into an appropriateexpression vector. Plasmid pJF1 consisted of an 8.1 kbBglHlBamHI-EcoRI fragment (Fl) of genomic copyincluding the full-length jockey element Jl (see Priimagiet al., 1988), which lacks a 350 bp portion of the 5' end,and plasmid pUC 19. We made specific deletions in the Flfragments inserted into pUC19 to remove all sequencesexcept those encoding the putative RT. This was achievedby a series of manipulations described in detail in Materialsand methods and illustrated in Figure 1. As a result of thecloning strategy, the final construct, pJPOL, contained a2228 bp fragment of the jockey element ORF2 defined asthe pol ORF (Priimagi et al., 1988). ORF2 lacks 210 and
307 bp portions of the 5' and 3' ends, respectively. Thecomparative analysis of the protein-coding jockey sequenceand those of other LINEs indicating a region of homologywith the conservative regions typical for retroviral RTdemonstrates that no regions are excised in the pol genefragment (see Priimagi et al., 1988). E.coli cells (DHl) weretransformed with the pJPOL construct shown in Figure lA,induced with lactose, and the extracts were prepared. Thecontrol extract was transformed with pUC9 (containing noRT sequences).
Purification of jockey RT expressed in E.coliTo detect RT activity, we measured the incorporation of[3H]dTTP or dGTP by using synthetic homopolymertemplates in the presence of short oligodeoxynucleotideprimers. DNA polymerase activity was readily detectablein both sample and control E. coli crude extracts in an assaywith 100 ,ig/ml poly(rA)/oligo(dT) or poly(rC)/oligo(dG)as template/primer (not shown). This result was notunexpected. E.coli DNA polymerase I is known to becapable of copying the all-ribonucleotide templates withrelatively high efficiency, at least, under carefully chosenconditions in vitro (Lee-Huang and Cavalieri, 1964;Chamberlin, 1965; Karkas, 1973; Loeb et al., 1973).However, the polymerase distinctly differs from viral RTsin its chromatographic behaviour (Richardson et al., 1964;Jovin et al., 1969; Verma, 1975; Hizi and Joklik, 1977).We could not use an E. coli strain containing, for example,a ts poll would give an extremely low background poly-merase activity in crude extracts (Larder et al., 1987). Thus,
*.
Fig. 1. Construction of an inducible expression system for synthesis of jockey reverse transcriptase in E. coli. (A) Diagram of cloning a pol genefragment into a pUC9 vector. The jockey ORF2 (pol gene) is shown at the top of the figure illustrating the position of a number of restrictionenzyme sites present within the pol gene region. Plasmid pJFI consisted of an 8.1 kb BglIlI/BamHI-EcoRI fragment of genomic copy including thefull-length jockey element J1 which lacks a portion 350 bp of the 5' end, and plasmid pUC19 (see Results). The initial clone, pJPOL, containing a2228 bp fragment from the jockey pol gene region was made as described in Materials and methods. (B) Analysis of plasmids by restrictionendonuclease mapping: (a) plasmid pJF1; (b) PstI digestion of pJFl; (c) PstI-PstI pol gene fragment; (d) vector pUC9; (e) PstI restriction of pUC9;(f) PstI restriction of pJPOL; (g) initial clone, pJPOL. The resulting 2228 bp pol gene fragment was recovered from agarose gel by the technique ofDretzen et al. (1981). Electrophoresis was carried in a 0.9% agarose gel at 20°C for 8 h at 5 V/cm. Arrows on the left refer to the positions oflinear marker polynucleotides run on the same gel. Fragments were obtained by digestion of phage X DNA (in nucleotides): 1, 4361; 2, 2322; 3,2037. Arrows on the right refer to the positions of marker plasmids run on the same gel: 1*, pBR325; 2*, pBR322; 3*, pUCl9.
2490
Reverse transcriptase of LINE-like element
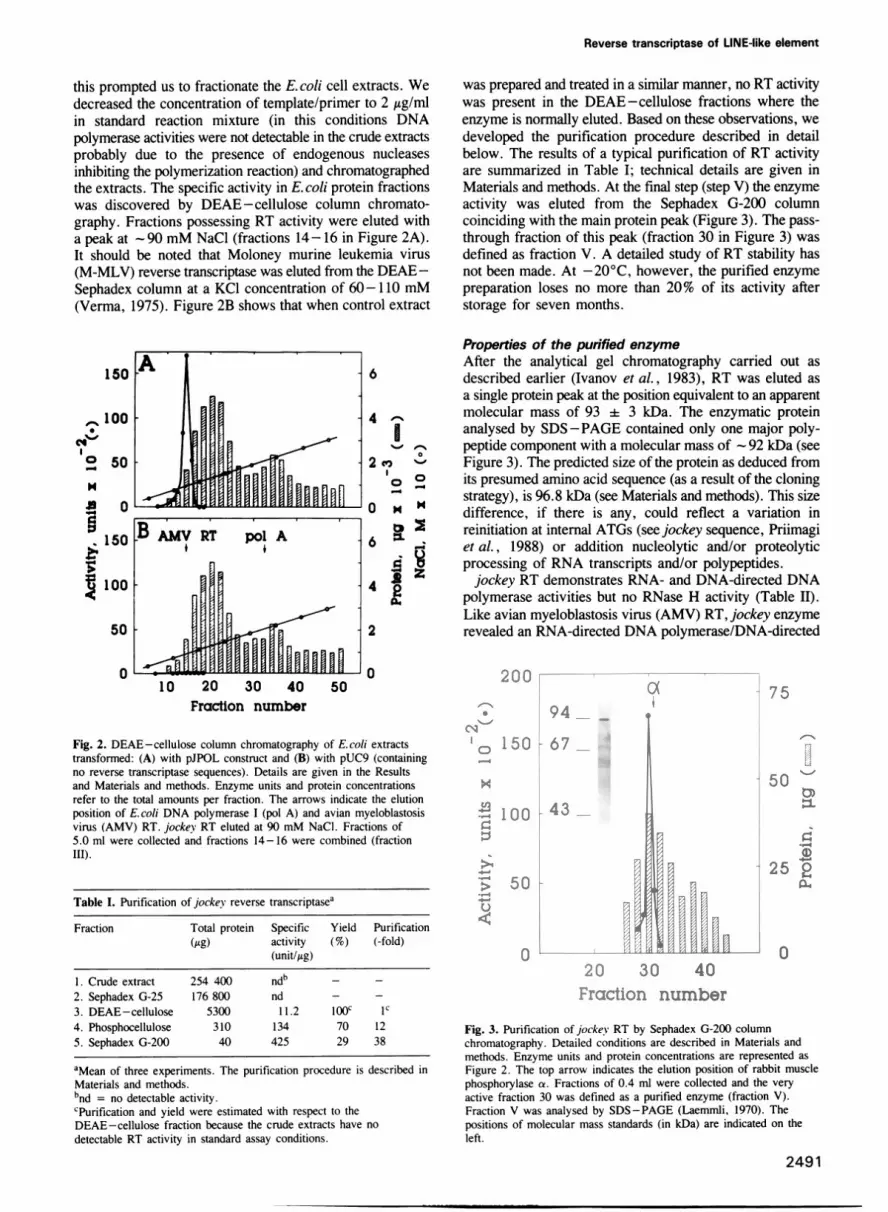
this prompted us to fractionate the E. coli cell extracts. Wedecreased the concentration of template/primer to 2 Itg/mlin standard reaction mixture (in this conditions DNApolymerase activities were not detectable in the crude extractsprobably due to the presence of endogenous nucleasesinhibiting the polymerization reaction) and chromatographedthe extracts. The specific activity in E. coli protein fractionswas discovered by DEAE-cellulose column chromato-graphy. Fractions possessing RT activity were eluted witha peak at - 90 mM NaCl (fractions 14-16 in Figure 2A).It should be noted that Moloney murine leukemia virus(M-MLV) reverse transcriptase was eluted from the DEAE -Sephadex column at a KCI concentration of 60-110 mM(Verma, 1975). Figure 2B shows that when control extract
was prepared and treated in a similar manner, no RT activitywas present in the DEAE -cellulose fractions where theenzyme is normally eluted. Based on these observations, wedeveloped the purification procedure described in detailbelow. The results of a typical purification of RT activityare summarized in Table I; technical details are given inMaterials and methods. At the final step (step V) the enzymeactivity was eluted from the Sephadex G-200 columncoinciding with the main protein peak (Figure 3). The pass-through fraction of this peak (fraction 30 in Figure 3) wasdefined as fraction V. A detailed study of RT stability hasnot been made. At -20°C, however, the purified enzymepreparation loses no more than 20% of its activity afterstorage for seven months.
150
100
° 50
O
150
100
50
0
BAMVRT pol AI
10 20 30 40 50Fraction number
Fig. 2. DEAE-cellulose column chromatography of E.coli extractstransformed: (A) with pJPOL construct and (B) with pUC9 (containingno reverse transcriptase sequences). Details are given in the Resultsand Materials and methods. Enzyme units and protein concentrationsrefer to the total amounts per fraction. The arrows indicate the elutionposition of E.coli DNA polymerase I (pol A) and avian myeloblastosisvirus (AMV) RT. jockey RT eluted at 90 mM NaCl. Fractions of5.0 ml were collected and fractions 14-16 were combined (fractionIII).
6
4 ^-I
6 0
2£e2
0 4.-
_1
8 CZ
4 a
2
0
Properties of the purified enzymeAfter the analytical gel chromatography carried out asdescribed earlier (Ivanov et al., 1983), RT was eluted asa single protein peak at the position equivalent to an apparentmolecular mass of 93 i 3 kDa. The enzymatic proteinanalysed by SDS-PAGE contained only one major poly-peptide component with a molecular mass of - 92 kDa (seeFigure 3). The predicted size of the protein as deduced fromits presumed amino acid sequence (as a result of the cloningstrategy), is 96.8 kDa (see Materials and methods). This sizedifference, if there is any, could reflect a variation inreinitiation at internal ATGs (see jockey sequence, Priimagiet al., 1988) or addition nucleolytic and/or proteolyticprocessing of RNA transcripts and/or polypeptides.jockey RT demonstrates RNA- and DNA-directed DNA
polymerase activities but no RNase H activity (Table II).Like avian myeloblastosis virus (AMV) RT, jockey enzymerevealed an RNA-directed DNA polymerase/DNA-directed
2075
94
(o i 50---
iOO.t-- 100
67
43
50Table I. Purification of jockey reverse transcriptasea
Fraction Total protein Specific Yield Purification(jg) activity (%) (-fold)
(unit/Ag)
1. Crude extract 254 400 ndb _ _2. Sephadex G-25 176 800 nd -
3. DEAE-cellulose 5300 11.2 lOOC lc4. Phosphocellulose 310 134 70 125. Sephadex G-200 40 425 29 38
aMean of three experiments. The purification procedure is described inMaterials and methods.bnd = no detectable activity.CPurification and yield were estimated with respect to theDEAE-cellulose fraction because the crude extracts have no
detectable RT activity in standard assay conditions.
II'1-3030 40
50
-or_i
25 °
-- O
iction number
Fig. 3. Purification of jockev RT by Sephadex G-200 columnchromatography. Detailed conditions are described in Materials andmethods. Enzyme units and protein concentrations are represented asFigure 2. The top arrow indicates the elution position of rabbit musclephosphorylase ct. Fractions of 0.4 ml were collected and the veryactive fraction 30 was defined as a purified enzyme (fraction V).Fraction V was analysed by SDS-PAGE (Laemmli, 1970). Thepositions of molecular mass standards (in kDa) are indicated on theleft.
2491

V.A.Ivanov et al.
Table II. Enzymatic potential of jockey RT
Activitya Incorporated nucleotides (pmol/4g protein)
jockey RT AMV RT E.coli DNA polymerase I
RNA-directedDNA polymerase 423 1943 6455DNA-directedDNA polymerase 189 1252 11 129RNA-directed/DNA-directed 2.2 1.6 0.58
Hydrolysis of [3H]poly(rA)/oligo(dT)(pmol/4g) protein
RNase H 0 215 NDb
aRNA- and DNA-directed DNA polymerase reactions were performed in a standard assay mixture with 150 ng poly(rA)/oligo(dT) and 250 ngpoly(dA)/oligo(dT) as template/primer, respectively, in the presence of either 50 U jockey RT, 125 ng avian myeloblastosis virus (AMV) RT or10 ng Ecoli DNA polymerase I, as described in Materials and methods. The RNA-directed/DNA-directed ratio reflects the preference of DNApolymerases for either indicated activity. The reaction conditions for RNase H activity were as described in Materials and methods. Specific activitiesof all the enzymes are expressed in equivalent units.bND = not determined.
DNA polymerase activity ratio of > 1 (in contrast to thoseof E. coli DNA polymerase I of <1) in the reactionconditions described. Thejockey RT exhibited linear kineticsof polymerization for the time intervals of 0-15 min instandard assay mixture (Figure 4).The enzyme was active between 15 and 42°C with a
distinct temperature optimum at 26°C. At 22 and 30°C therates of polymerization were only 40 and 80% of themaximal rate, respectively (Figure 5). It should be notedthat a Ty element reverse transcriptase had maximum activityat 20°C (Garfinkel et al., 1985). The jockey polymerasepossessed a broad pH optimum between pH 7.5 and pH 8.7with maximum activity observed at pH 8.5. Divalent cationsMg2+ or Mn2+ were necessar for activity (Table III). Theoptimal concentration of Mg + was 1.0 mM and that ofMn2+ was 0.1 mM. The latter increased the rates ofpolymerization.The salt stimulated the activity at KCl concentration
(optimum = 30 mM), but jockey RT was sensitive to highionic strength: at 50 mM KCl and in optimal assay conditionsthe activity was decreased two-fold, and at 100 mM KClthe activity was not observed (Table III). Without KCl theconcentration of Nonidet P-40 required for detectable RTactivity was - 0.2 %, and the activity fell sharply to 0 whenthis detergent was used at < 0.1 %. Exposure to a sulphydrylreagent (0.5 mM p-chloromercuribenzoate) completelyinactivated the enzyme ( <2% remaining activity). RifamicinSV and novobiocin suppressed the RT polymerase reaction(complete inhibition at 2.0 and 1.0 mM, respectively). Theaddition of ethanol to the standard assay mixture alsoinhibited the enzyme (Table III).
Retroviral polymerase shows a preference for certainsynthetic template/primer combinations. Its transcription isalso specifically dependent on a divalent cation, saltconcentration, incubation temperature and other conditionsof reaction (Verma and Baltimore, 1974; Leis and Hurwitz,1974; Verma, 1977; Hoffman et al., 1985). We haveinvestigated jockey RT activity with 'activated' DNA andsome ribo- and deoxyribo-polymer templates primed witholigodeoxynucleotides. AMV RT and E. coli DNApolymerase I activities were also determined as controls forreaction conditions (Table IV). Like AMV enzyme, jockeyRT prefers poly(rC) and poly(rA) as template and 'activated'DNA is not effective.
2492
g 50!.40r30
20k
.5 lor-
E.0d 0 20 40 60 80 100 120
Time, min
Fig. 4. Kinetics of jockey RT polymerase reaction. The reactionconditions for the RT were as described in Materials and methods,except that 120 U enzyme and 360 ng poly(rA)/oligo(dT) (0) or 6 yg'activated' DNA (0) as template/primer were used and the reactionvolume was increased to 180 Al. 15 A1 aliquots were withdrawn atdifferent time intervals, spotted onto a Whatman DE81 filter paper andprocessed as described (Mellor et al., 1985). The dTTP incorporationshown is the average for four determinations.
25
20 F
15
lo0
S14 18 22 26 30 34 38 42
Incubation tmperature, 0C
Fig. 5. The effect of temperature on the jockey RT activity. Reactionswere performed in a standard assay mixture as described in Materialsand methods at the following temperatures: 15, 22, 26, 30, 37 and42°C. Each assay sample contained 20 U enzyme and 60 ngpoly(rA)/oligo(dT) as template/primer. The values represent the meanof four determinations. The bars indicate the range of values ofdetermined activity.

Reverse transcriptase of LINE-like element
Table III. The effect of different reagents on the RT activity
Componentsa Concentration Relative activityb(%)
Standard assay mixture - 100
Mg2+ 0 mM 00.5 393.0 755.0 6110.0 10
Mn2+ 0.05 mM 1390.1 2630.3 1971.0 2
KCI 10.0 mM 4520.0 7550.0 58100.0 <2
+pCMB 0.05 mM 930.1 800.3 270.5 <2
+Rifamicin SV 0.1 mM 870.5 571.0 382.0 3
+ Novobiocin 0.1 mM 820.25 600.5 191.0 2
+ Ethanol 5 % 7710 1820 4
aThe reagents listed were either changed in their concentrations in thestandard assay mixture, or were supplied in addition (marked +). TheMn2 + replaced Mg2bThe enzyme activity was determined as described in Materials andmethods in the presence of the indicated reagents in the concentrationsgiven. The results are given as percentages of the activity measuredunder the standard assay conditions.
DiscussionWe have cloned and for the first time expressed in E. colia DNA polymerase coded by a mobile Drosophila element,jockey, related to mammalian LINEs. We have isolated andpurified the recombinant jockey polymerase and investigatedsome properties of the enzyme. The enzyme was identifiedas a reverse transcriptase by its specific enzyme propertiesand absence in control E. coli extracts. Various 'normal'DNA polymerases are known to be capable, at least underspecific conditions in vitro of transcribing the ribo-polymerof suitable template/primer combinations (Karkas, 1973;Chandra and Steel, 1977). For this reason, we havecompared the properties of jockey RT and E. coli DNApolymerases. This analysis shows that cellular E. coli DNApolymerases are easily distinguished fromjockey RT activityby their template specificity, molecular mass, pH optimum,ion requirements and responses to inhibitors. For example,jockey polymerase has a molecular mass of 92 000,
- 17 000 less than that of DNA polymerase I (Richardsonet al., 1964; see Appendix by Baldwin; Jovin et al., 1969).Unlike the latter, jockey RT is inactive with 'activated' DNAas template, inhibited by -SH reagents, has an optimumof Mg2+ and KCl concentrations corresponding to 1.0 mMand 30 mM (in contrast to 7 mM and 100 mM, respectively).Minor E. coli DNA polymerases II and III can have a
molecular mass of - 90 000 as a result of isolationprocedures or after SDS -PAGE but, unlikejockey RT, theyare highly specific for natural template/primer, preferring'sheared' DNA at a pH optimum of 7.6 (Knippers, 1970;Kornberg and Gefter, 1971; Gefter et al., 1971; Wickneret al., 1973). Ethanol inhibits the activity ofjockey RT andDNA polymerase II to differing extents but stimulates DNApolymerase HI activities (see Kornberg, 1974). Besides, thejockey RT has chromatographic properties on theDEAE-cellulose and/or phosphocellulose clearly differingfrom those of E. coli DNA polymerases (Richardson et al.,1964; Knippers, 1970; Gefter et al., 1971; Kornberg andGefter, 1971; Campbell et al., 1972).Recombinant jockey RT can be likened to the type of
enzymes similar to retroviral RNA-directed DNA poly-merases, i.e. it prefers polyribonucleotides as template, isinhibited by -SH reagents, requires bivalent cations (Mg2+or Mn2+) and detergent and/or KCl as an ionic strengthcarrier. However, the jockey RT differs in a number ofcatalytic properties (Table V). jockey RT has a temperatureoptimum of 26°C, in contrast to retroviral RTs, which have
Table IV. Comparative template preference of jockey RTa
Template/primer Incorporated nucleotidesb (pmol)
jockey RT AMV RT E.coli DNA polymerase I
poly(rA)/oligo(dT1O) 101 98 114
poly(rC)/oligo(dG1O) 152 73 NDd
poly(dA)/oligo(dT1O) 45 60 197'activated' DNA 2.29 2.54 34
poly(rA)/oligo(dT1O)/ 44.1 38.6 3.4'activated' DNAC
aThe values represent the mean of 6-8 determinations.bReactions were performed in standard assay conditions as described in Materials and methods, except that either 100 units jockey RT, 50 ng AMV
RT or 15 ng Ecoli DNA polymerase I and either 300 ng poly(rA)/oligo(dT). 500 ng poly(rC)/oligo(dG) or S yg 'activated' DNA as template/primerwere used.'The poly(rA)/oligo(dT)/'activated' DNA ratio reflects the preference of DNA polymerase for either indicated template.dND = not determined.
2493

V.A.Ivanov et al.
optima of 37°C. [Strictly speaking, the temperature optimumof retroviral RT reactions appears to be > 37°C. Forexample, the subsequent increase of reaction temperatureup to 40.5°C stimulated the activity of RSV RT by 8.3%(Verma et al., 1974).] This is an important issue. It is knownthat temperature sensitivity of growth and transformation bycertain RSV mutants correlates with thermolability of theirvirion-associated DNA polymerase (Verma et al., 1974), i.e.the optimal (functional) reaction temperature may be due toan intrinsic property of RTs. It should be noted that optimaltemperature of Drosophila cultivation is 26°C. The optimaltemperature of the RT reaction may suggest that therecombinant RT is similar to Drosophila's own reverse
transcriptase. The optimal concentration of Mg2 + was
1.0 mM and that of Mn2 , 0.1 mM for jockey RT (incontrast to 10 mM for Mg2+ and 0.2 mM for Mn2+ forRSV RT, and 2 mM for Mg2+ and 1 mM for Mn2+ forM-MLV RT) (Table V). The Mg2+/Mn2+ ratio, whichreflects the preference ofjockey RT for either cation, is 0.38(in contrast to 1.01 for RSV RT, 0.28 for M-MLV RT and2.6 for AMV RT but with d(AT) copolymer as tem-plate/primer; for the latter see Leis and Hurwitz, 1974).
Novobiocin, an inhibitor of DNA replication and transcrip-tion, is known to suppress the varied polymerase activities(Sung, 1974; Mattern and Painter, 1979). It also inhibitsjockey RT. The concentration of novobiocin (1 mM) thatcompletely inhibits the reaction (2% remaining activity) isthe same concentration as that which is effective for genomicenzymes (Snyder et al., 1982; Ivanov, 1988). The lastessential feature of jockey RT is the absence of RNase Hactivity. This is expected because the jockey element containsno RNase H sequences (see Priimagi et al., 1988).
Properties of recombinant RSV RT and virion-associatedRT of the virus have a detailed accordance (see Table V).We suggest that the features ofjockey RT investigated hereare also similar to jockey RT expressed in vivo. The resultsof this work suggest that the RNA-directed DNA polymeraseencoded by jockey elements may be involved in thetransposition of the elements where the next step is catalysedby RNase H that is also detected in Drosophila cells as itsown distinct enzyme activity (DiFrancesco and Lehman,1985).
Materials and methods
Reagents and determinationsRestriction enzymes, T4 DNA ligase and E. coli DNA polymerase I were
obtained from Bethesda Research Laboratories and avian myeloblastosis
virus reverse transcriptase was from Boehringer Mannheim.Poly(rA)/oligo(dT10), poly(rC)/oligo(dG10), poly(rA)/oligo(dT10) werefrom Pharmacia; novobiocin, rifamycin SV and lysozyme were from Sigma,[3H]dTTP and [3H]dGTP from Amersham and [3H]poly(rA) from NewEngland Nuclear. Protein concentrations were determined by the techniqueof Bradford (1976). Proteins were analysed by SDS-PAGE (Laemmli,1970) on a continuous 5- 15% gradient gel. After electrophoresis, proteinswere visualized by Coomassie blue staining.
DNA preparationsCalf thymus DNA isolated by the procedure of Marmur (1961). was,activated' according to the method of (Aposhian and Kornberg, 1962).Plasmid DNA was isolated by the alkaline lysis method as described (Maniatiset al., 1982) with additional purification by centrifugation in a caesiumchloride gradient. DNA concentration was determined by the doublewavelength method (Spirin, 1958) as well as by a fluorescence method usingethidium bromide (Karsten and Wollenberger, 1972).
Plasmid constructionBg/II-EcoRI fragment in recombinant plasmid pJFI, which was clonedfrom the original copy of the jockey element (Mizrohki et al., 1985) andcomprised the JI jockey copy without 350 bp of the 5' end, was used toobtain a fragment of the reverse transcriptase coding region (pol gene). Theresulting 2228 bp pol gene fragment (fragment representing nucleotides2258-4485 of the full-length jockey sequence; see Priimagi et al., 1988)was excised from pJFl by digestion with PstI, identified by restrictionendonuclease mapping (using BgIII and HindlIl), recovered from agarose
gel by the technique of Dretzen et al. (1981) and then inserted into themultiple cloning site of pUC9. The plasmid obtained (pJPOL) containsregulatory elements for the initiation of transcription and translation derivedfrom the lacZ gene in pUC9 as well as the translation terminator of lacZ.As a result of the cloning strategy, the predicted N-terminal sequence ofthe recombinant reverse transcriptase contains eight additional amino acidresidues and C-terminal sequence an additional 54 residues (deduced fromthe nucleotide sequences of pUC9 and J, jockex'). The ligation mixture was
used to transform E.coli cells (strain DHl) yielding the clone pJPOL whichexpresses reverse transcriptase. Orientation of the pol segment with respectto the promoter in the vector was determined by restriction endonucleasemapping (using HindIII).
Assay of reverse transcriptase activityThe enzymatic activity was assayed by measuring the incorporation oflabelled nucleotide substrate using the synthetic template/primer: the standardmixture contained 50 mM Tris-HCI, pH 8.5, 30 ng poly(rA)/oligo(dT10)or 50 ng poly(rC)/oligo(dG10), 100 ,uM dTTP, 10 mM dithiothreitol, 1 mM
MgCI2, 30 mM KCI, 0.2% NP-40, 5 ,uCi [3H]dTTP or GTP and 10 Uof the reverse transcriptase in a final volume of 15 itl. After incubationat 26°C for 15 min, the samples were spotted onto a Whatman DE81 filterpaper and processed as described (Mellor et al., 1985). The radioactivityretained on the filter was counted in scintillation fluid. Background counts,determined for each experiment in a reaction with no sample added, weresubtracted. One unit of RT is defined as the amount of enzyme whichincorporates 1 pmol of nucleotides in 15 min under the conditions described.
Purification of reverse transcriptaseAll operations were carried out at 0-4'C. The cells (5.2 g) were suspendedby gently vortexing in 25 ml buffer A (50 mM Tris-HCI, pH 8.18, mM
Table V. Features of jockey RT and retroviral DNA polymerasesa
Enzyme Number of Molecular Optimum Optimum Optimum Optimum [Mg2+]/[Mn2+1, Optimum [salt]b RNase H Referencessubunits subunit mass pH temperature [Mg2+ 1b [Mn2+Ib (mM) activity
(kDa) (0C) (mM) mM) (+ or -)
jockey RT 1 92 8.5 26 1.0 0.1 0.38 30 - This work
Moloney murineleukemia virus RT 80 8.3 37 2.0 1.0 0.28 60 + Verma (1975)Rous sarcomavirus RT 2 7.6 37 10.0 0.2 1.01 40 + Farus et al. (1972)63
Recombinant Rous 977.0-7.6 37 10.0 0.2 1.13 50 + Chemov et al. (1990)sarcoma virus RT 65
Chemov et al. (1991)
allI parameters refer to optimal reaction conditions for the polymerases listed with poly(rA)/oligo(dT) as template/primer.bThe optimal concentration of the indicated reagents.cThe Mg2+/Mn2+ ratio reflects the preference of the polymerase for either cation.
2494

Reverse transcriptase of LINE-like element
EDTA, 1 mM PMSF, 5 mM 2-mercaptoethanol, 10% sucrose). The cellmaterial was lysed by successive addition of 40 mg lysozyme, 1 ml 10%NP-40 and 10 ml 5 M NaCl to suspension with 10, 5 and 10 min incubationson ice, respectively, after each addition listed. The precipitate was removedby centrifugation for 30 min at 20 000 r.p.m. (JA-20 rotor, Beckman, J2-21)and protein solution (fraction I) was concentrated by dialysis against drypolyethyleneglycol and passed through a Sephadex G-25 column(1.6 x 20 cm) previously equilibrated with buffer B (50 mM Tris-HCI,pH 7.12, 1 mM EDTA, 5 mM 2-mercaptoethanol, 0.1 % NP-40, 20 mMNaCI, 5% glycerol). Low salt fraction II was applied to a DEAE-52 cellulosecolumn (1.2 x 43 cm) previously equilibrated with buffer B. The columnwas washed with 2 vol buffer and eluted with a linear NaCI gradient(20-700 mM) in 600 ml of the same buffer. Fractions possessing RT activity(5 ml fractions), eluting at - 90 mM NaCl, were combined and dialysedfor 15 h against buffer B without NaCl but containing 10% glycerol. Theprotein solution (fraction IH) was applied to a phosphocellulose P11 column(0.8 x 20 cm) previously equilibrated with buffer B. The column was washedwith 2 vol buffer and then the enzyme was eluted with a linear NaCl gradient(20-500 mM) in 100 ml of the same buffer. Active fractions (1 mnl) elutingbetween 240 and 260 mM NaCI were combined and concentrated to 0.2 mlby dialysis against dry polyethyleneglycol. The enzyme fraction (0.2 mlfraction IV) was applied to a Sephadex G-200 column (0.4 x 30 cm)previously equilibrated with buffer B containing 300 mM NaCl. The veryactive fraction of 0.4 ml was dialysed against glycerol in 50 mM Tris-HCI,pH 8.0, 0.2% NP-40 and stored in 40% glycerol at -20°C. The purifiedenzyme (fraction V) was used in further experiments.
Assay of interfering activitiesDNA-dependent DNA polymerase activity was determined by measuringthe incorporation of labelled [3H]dTTP using either poly(dA)/oligo(dT1O)or 'activated' DNA as template/primer. The reaction conditions were asdescribed above for reverse transcriptase assay, except that either 0.05 /Agpoly(dA)/oligo(dT1O) or 0.5 jig 'activated' DNA and 50 yM each of dATP,dCTP, dGTP and dTTP were used. After incubation at 26°C for 15 min,the samples were spotted onto a Whatman DE81 filter paper and processedas for reverse transcriptase determination.RNase H activity was determined in reaction conditions described for
RT activity, except that 0.05 ytg [3H]poly(rA)/oligo(dT1O) as substratereplacing template/primer, was used. After incubation at 26°C for 15 min,the residual radioactivity was determined on DE81 filter paper as mentionedabove.
References
Alexander,F., Leis,J., Soltis,D.A., Crowl,R.M., Danho,W., Poonian,M.S.,Pan,Y.-C.E. and Skalka,A.M. (1987) J. Virol., 61, 534-542.
Aposhian,H.V. and Kornberg,A. (1962) J. Biol. Chem., 237, 519-525.Bradford,M.M. (1976) Anal. Biochem., 72, 248-254.Campbell,J.L., Sall,L. and Richardson,C.C. (1972) Proc. Nat!. Acad. Sci.
USA, 69, 2090-2094.Chaboissier,M.-C., Busseau,I., Prosser,J., Finnegan,D.J. and Bucheton,A.
(1990) EMBO J., 9, 3557-3563.Chamberlin,M.J. (1965) Fed. Proc., 24, 1446-1457.Chandra,P. and Steel,L. (1977) Biochem. J., 167, 513-524.Chernov,A.P., Melnikov,A.A., Shmatchenko,V.V. and Fodor,I. (1990)
Biokhimiya, 55, 586-594.Chernov,A.P., Melnikov,A.A. and Fodor,I. (1991) Biomed. Sci., in press.Deragon,J.-M., Sinnett,D. and Labuda,D. (1990) EMBOJ., 9, 3363-3368.DiFrancesco,R.A. and Lehman,I.R. (1985) J. Biol. Chem., 260,
14764-14770.Doolittle,R.F., Feng,D.-F., Johnson,M.S. and McClure,M.A. (1989) Q.
Rev. Biol., 64, 1-30.Dretzen,G., Bellard,M., Sassone-Corsi,P. and Chambon,P. (1981) Anal.
Biochem., 112, 295-298.Farus,A.J., Taylor,J.M., McDonnell,J.P., Levinson,W.E. and Bishop,J.M.
(1972) Biochemistry, 11, 2334-2342.Garfinkel,D.J., Boeke,J.D. and Fink,G.R. (1985) Cell, 42, 507-517.Gefter,M.L., Hirota,Y., Kornberg,T., Wechsler,J.A. and Barnoux,C.
(1971) Proc. Natl. Acad. Sci. USA, 68, 3150-3153.Hizi,A. and Joklik,W.K. (1977) J. Biol. Chem., 252, 2281-2289.Hoffman,A.D., Banapour,B. and Levy,J.A. (1985) Virology, 147,
326-335.Hutchison,C.A., Hardies,S.C., Loeb,D.D., Shchee,W.R. and Edgell,M.H.
Ivanov,V.A., Gaziev,A.I. and Tretyak,T.M. (1983) Eur. J. Biochem., 137,517-522.
Jovin,T.M., Englund,P.T. and Bertson,L.L. (1969) J. Biol. Chem., 244,2996-3008.
Karkas,J.D. (1973) Proc. Natl. Acad. Sci. USA, 70, 3834-3838.Karsten,U. and Wollenberger,A. (1972) Anal. Biochem., 46, 135-146.Knippers,R. (1970) Nature, 228, 1050-1053.Kornberg,A. (1974) DNA Synthesis. W.H.Freeman and Co., San Francisco.Kornberg,T. and Gefter,M.L. (1971) Proc. Natl. Acad. Sci. USA, 68,
761 -764.Laemmli,U.K. (1970) Nature, 227, 680-685.Larder,B., Purifoy,D., Powell,K. and Darly,G. (1987) EMBO J., 6,
3133-3137.Lee-Huang,S. and Cavalieri,L.F. (1964) Proc. Natl. Acad. Sci. USA, 51,
1022-1028.Leis,J. and Hurwitz,J. (1974) Methods Enzymol. 29, 143-150.Loeb,L.A., Tartof,K.D. and Travaglini,E.O. (1973) Nature, 242, 66-69.Maniatis,I., Fritsch,E.F. and Sambrook,J. (1982) Molecular Cloning. A
Laboratory Manual. Cold Spring Harbor Laboratory Press, Cold SpringHarbor, NY.
Marmur,J. (1961) J. Mol. Biol., 3, 208-218.Mattern,M.R. and Painter,R.B. (1979) Biochim. Biophys. Acta, 563,
306-312.Mellor,J., Malim,M.H., Gull,K., Tuite,M.F., McCready,S.,
Dibbayawan,T., Kingsman,S.M. and Kingsman,A.J. (1985) Nature, 318,583-586.
Melnikov,A.A., Molnar,J., Horvath,P. and Fodor,I. (1988) Dokl. Acad.Nauk SSSR, 299, 486-489.
Mizrokhi,L.J., Obolenkova,L.A., Priimagi,A.F., Ilyin,Y.V.,Gerasimova,T.I. and Georgiev,G.P. (1985) EMBO J., 4, 3781-3787.
Mizrokhi,L.J., Georgieva,S.G. and Ilyin,Y.V. (1988) Cell, 54, 685-691.Molnar,J., Melnikov,A.A., Horvath,P., Tchernov,A.P., Dube,S. and
Fodor,I. (1988) Mol. Gen. (Life Sci. Adv.), 7, 27-31.Priimagi,A.F., Mizrokhi,L.J. and Ilyin,Y.V. (1988) Gene, 70, 253 -262.Pritchard,M.A., Dura,J.-M., Pelisson,A., Bucheton,A. and Finnegan,D.J.
(1988) Mol. Gen. Genet., 214, 533-540.Richardson,C.C., Schildkrant,C.L., Aposhian,H.V. and Kornberg,A. (1964)
J. Biol. Chem., 239, 222-232.Rogers,J.H. (1985) Int. Rev. Cytol., 93, 188-280.Snyder,R.D., Houten,B.V. and Regan,J.D. (1982) Nucleic Acids Res., 10,
6207-6219.Spirin,A.S. (1958) Biokhimiya, 23, 656-660.Sung,S.C. (1974) Biochim. Biophys. Acta, 361, 115-117.Verma,I.M. (1975) J. Virol., 15, 843-854.Verma,I.M. (1977) Biochim. Biophys. Acta, 473, 1-38.Venna,I.M. and Baltimore,D. (1974) Methods Enzymol., 29, 143-150.Verma,I.M., Mason,W.S., Drost,S.D. and Baltimore,D. (1974) Nature,
251, 27-31.Wickner,W., Schekman,R., Geider,K. and Kornberg,A. (1973) Proc. Natl.
Acad. Sci. USA, 70, 1764-1767.Xiong,Y. and Eickbuch,T.H. (1988) Mol. Biol. Evol., 5, 675-690.
Received on March 18, 1991; revised on June 5, 1991
(1989) In Berg,D.E. and Howe,M.M. (eds), Mobile DNA. AmericanSociety Microbiology, Washington DC, pp. 593-617.
Ivanov,V.A. (1988) Neurosci. Lett., 94, 99-103.
2495





























