Embed Size (px)
Citation preview

Bioinspired Core-Crosslinked Micelles from Thymine-Functionalized
Amphiphilic Block Copolymers: Hydrogen Bonding and
Photo-Crosslinking Study
Gagan Kaur,1 Shery L. Y. Chang,2 Toby D. M. Bell,3 Milton T. W. Hearn,1 Kei Saito1
1Centre for Green Chemistry, Monash University, Clayton, Victoria 3800, Australia2Monash Centre for Electron Microscopy and School of Chemistry, Monash University, Clayton, Victoria 3800, Australia3School of Chemistry, Monash University, Clayton, Victoria 3800, Australia
Correspondence to: K. Saito (E-mail: [email protected])
Received 10 February 2011; accepted 17 June 2011; published online 12 July 2011
DOI: 10.1002/pola.24853
ABSTRACT: Bioinspired core-bound polymeric micelles, based
on hydrogen bonding and photo-crosslinking, of thymine have
been prepared from poly(vinylbenzylthymine)-b-poly(vinylben-
zyltriethylammonium chloride). The amphiphilic block copoly-
mer was synthesized by 2,2-tetramethylpiperidin-1-oxyl-
mediated living radical polymerization in water/ethylene glycol
solution. Micelle characterization and critical micelle concentra-
tion measurements demonstrated that the hydrogen bonding
of the attached thymine units stabilizes the micelles. Further,
core-crosslinked polymeric micelles were formed by ultraviolet
(UV) radiation showing that the stability of the micelle could
be controlled by the UV crosslinking of the attached thymines.
VC 2011 Wiley Periodicals, Inc. J Polym Sci Part A: Polym Chem
49: 4121–4128, 2011
KEYWORDS: biomimetic; block copolymer; core-shell polymers
INTRODUCTION Bioinspired mechanisms are used to createalternative materials based on the principles of green chem-istry.1,2 Nature provides abundant and elegant examples ofthe synthesis of molecules in terms of both atom economyand energy required. Thymine, one of the nucleic bases inDNA, has the ability to form both relatively strong hydrogenbonds and to be photo-crosslinked.3–5 The hydrogen bondingcapabilities of thymine are well known to stabilize the doublehelix structure of DNA. The photodimerization of thymine, aultraviolet (UV)-induced 2p þ 2p photocyclization, results inthe covalent dimerization of adjacent thymines, leading to thedisruption of the helical structure of DNA. This process hasbeen linked with the development of certain forms of skincancer. This photo-crosslinking of thymine occurs when irradi-ated at wavelengths >270 nm UV. The 2p electron system ofthe 5 and 6 carbons of two adjacent thymines form a cyclobu-tane crosslink. Crosslinking can be reversed either by irradia-tion at <249 nm or enzymatically. By using these mecha-nisms, thymine-functionalized chemical compounds can bephoto-crosslinked and photodecrosslinked.6–10
Amphiphilic block copolymers form nanoscopic self-assem-bling core/shell structures called polymeric micelles in thebulk aqueous medium. During the formation of thesemicelles, the hydrophobic segment forms the core while thehydrophilic segment forms the shell. The hydrophilic shell
stabilizes the hydrophobic core by acting as an interfacebetween the aqueous phase and the hydrophobic region.This unique assembly makes polymeric micelles suitable can-didates as chemical capsules for various applications such asindustrial surfactants, nanocapsules for biomedical usage,electronic pollution control devices, removal agents for or-ganic contaminates in water, and in other nanofabricatedmaterials.11–17 Chemicals may be loaded into the core byphysical entrapment, conjugation, or complexation.15,18–23 Fur-thermore, the controlled release of encapsulated guest materi-als from the polymeric micelles can be triggered by changesin temperature and pH, by chemical stimuli, by enzymes, orby application of light, ultrasound or magnetic field.24–28
For polymeric micelles to have broad application use, it iscritical that the amphiphilic block copolymer micelles mustbe stable. Critical micelle concentration (CMC) provides ameasure of stability of polymeric micelles. CMC is the thresh-old concentration at or above which the micelles form.Lower CMC values favor stable micelles even in dilute solu-tions. Stability of the polymeric micelles is affected by thenature of interactions between chains in the core.29,30 Cross-linking has been used by researchers to increase the struc-tural stability of polymeric micelles in dilute solutions andeven in organic solvents. Crosslinking may be induced in theshell or in the core. Shell crosslinked micelles can be
Additional Supporting Information may be found in the online version of this article.
VC 2011Wiley Periodicals, Inc.
WWW.MATERIALSVIEWS.COM JOURNAL OF POLYMER SCIENCE PART A: POLYMER CHEMISTRY 2011, 49, 4121–4128 4121
WWW.POLYMERCHEMISTRY.ORG ARTICLE

prepared by UV irradiation, by addition of multifunctionalcrosslinking reagents, or by the direct reaction between thepolymer chains.31–36 Core crosslinked micelles have beenprepared by photo-crosslinking, free radical polymerizationwithin the core, sol–gel chemistry, and condensation reac-tions.16,37–39 Photo-crosslinking is the method of choice as itis non toxic, economical, and does not produce any by-prod-ucts. Furthermore, suitable photo-crosslinking does not affectthe micellar shape and size.40 Recently, our group has inves-tigated core-crosslinked micelles containing thymine groupsas photo-crosslinkable units.41,42 We have reported thatenhanced stability can be achieved with nano polymericmicellar aggregates derived from amphiphilic block copoly-mers of vinylbenzylthymine (VBT) and styrenesulfonate(VPS).41 In this previous work, we reported the synthesis ofa block copolymer of VBT and VPS and characterization ofthe micelles formed from it.41 However, the hydrogen bond-ing in the core of the micelles and their thermostability werenot investigated in detail in our previous work.
In this article, we report the synthesis of a related bioins-pired core-shell thymine-containing polymeric micellar sys-tem derived from the amphiphilic block copolymerization ofVBT and vinylbenzyltriethylammonium chloride (VBA). Thehydrogen bonding and photo-crosslinking attributes (Fig. 1)of these new polymer micelles are documented focusing par-ticularly on hydrogen bonding in the micelle core at highertemperature. Copolymers of VBA with 1-pyrenylmethyl meth-acrylates, (4-vinylbenzyl)-cinnamate, and VBT have beenreported in literature,43–47 but amphiphilic block copolymersof VBT and VBA have never been synthesized before. Wechose VBA as the hydrophilic block because polymers withquaternary ammonium groups are known to possess highantimicrobial activities.48–53
The amphiphilic block copolymers, poly(vinylbenzylthy-mine)-b-poly(vinylbenzyltriethylammonium chloride) (poly-(VBT-b-VBA)) were synthesized by 2,2-tetramethylpiperidin-1-oxyl (TEMPO)-mediated living radical polymerization inwater/ethylene glycol solution. Hydrogen bonded micelles,core-photo-crosslinked micelles, and nonhydrogen bondedmicelles were prepared from these block copolymers andtheir morphology and stability examined by dynamic lightscattering (DLS), transmission electron microscopy (TEM),and CMC measurements. The stability of micelles was furtherinvestigated by measuring the CMC values at higher temper-
ature (60 �C) and also from in situ TEM observations of mor-phological changes of micelles while heating the micelle sam-ples (from room temperature to 200 �C). These highertemperature studies have been used to examine the effect ofhydrogen bonding and photo-crosslinking on the thermo-stability of the micelles.
EXPERIMENTAL
MaterialsAll reagents and materials were purchased from Sigma-Aldrich in the purest form available and used as received.VBT and methylated vinylbenzylthymine (VMT) were synthe-sized from thymine and vinylbenzyl chloride as describedpreviously.41
Synthesis of VBAIn a round bottom flask, vinylbenzylchloride (12.5 mL, 0.08mol) and triethylamine (12.5 g, 0.125 mol) were dissolved inmethanol (15 mL). The reaction mixture was stirred at 30 �Covernight. The reaction mixture was then poured into excessof dry ether, which resulted in formation of white solid. Thesolid was recovered by filtration and the white product isdried under vacuum. The white solid obtained was thenrecrystallized in 2-propanol.
Yield: 99%. 1H NMR: (400 MHz, CDCl3) d1.30 (t, 9H, ACH3),3.16 (q, J ¼ 7.2 Hz, 6H, ANACH2A), 4.46 (s, 2H, ACH2A),5.38 (d, J ¼ 10.8 Hz, 1H, vinyl CAH), 5.95 (d, J ¼ 17.2 Hz,1H, vinyl CAH), 6.798 (dd, J ¼ 6.8 Hz, 1H, vinyl CAH), 7.49(d, J ¼ 8 Hz, 2H, aromatic CAH), 7.60 (d, J ¼ 8 Hz, 2H,aromatic CAH).
Synthesis of Poly(VBT-b-VBA)VBA (1.0 g, 3.94 mmol) was dissolved in 50% (v/v) ethyleneglycol/water (10 mL) and TEMPO (62 mg, 0.39 mmol) wasadded to the solution. The reaction mixture was stirred at60 �C until the TEMPO completely dissolved giving the solu-tion an orange color. Na2S2O5 (50 mg, 0.26 mmol) andK2S2O8 (60 mg, 0.22 mmol) were then added to the solutionand stirring continued at 60 �C under nitrogen until thereaction mixture became clear. The reaction mixture wasthen heated at 125 �C for 20 h under nitrogen to preparehomo VBA polymer. VBT (0.95 g, 3.94 mmol) was thenadded to the homo-VBA polymer solution and heating con-tinued for further 24 h at 125 �C under nitrogen with stir-ring. The polymerized product was purified by adding the
FIGURE 1 Core bound thymine-functionalized polymeric micelle.
ARTICLE WWW.POLYMERCHEMISTRY.ORG
4122 JOURNAL OF POLYMER SCIENCE PART A: POLYMER CHEMISTRY 2011, 49, 4121–4128

reaction mixture drop wise to acetone (300 mL) while stir-ring. The resulting solids were collected by filtration anddried under vacuum.
Yield: 100%. 1H NMR: (400 MHz, DMSO-d6) d0.80–1.90 (br,18H, ANACH2ACH3, ACH3), 4.65 (br, 2H, ACH2A), 6.05–7.80 (br, 8H, aromatic CAH), 11.3 (br, 1H, NAH). IR: mNAH ¼3421, mC¼¼O ¼ 1684. Mn ¼ 1.16 � 104, Mw/Mn ¼ 1.8.
Synthesis of Poly(VMT-b-VBA)Poly(VMT-b-VBA) was synthesized by a similar method asused for synthesis of poly(VBT-b-VBA).
Yield: 100%. 1H NMR: (400 MHz, DMSO-d6) d0.70–2.10 (br,18H, ANACH2ACH3, ACH3), 3.16 (br, 3H, NACH3) 4.75 (br,2H, ACH2A), 6.10–8.30 (br, 8H, aromatic CAH). IR: mNAH ¼3405, mC¼¼O ¼ 1678. Mn ¼ 1.01 � 104, Mw/Mn ¼ 2.4.
Polymer CharacterizationNMR spectra were taken on a Bruker 400 MHz NMR spec-trometer. Molecular weights were determined by gel permea-tion chromatography (GPC) performed on a Tosoh EcosecHLC-8320GPC equipped with both refractive index and UVdetectors (UV-detection, k ¼ 280 nm) using Tosoh alpha4000 and 2500 columns. Dimethylformamide (DMF) contain-ing LiBr (10 mM) was used as the solvent. Calibration curveswere obtained using polystyrene standards. To determinemolecular weight of the hydrophilic block poly(VBA), aque-ous mobile phase was used (0.5 M aqueous NaBr [80% v/v]containing acetonitrile [20% v/v]). Calibration curves wereobtained using polyethylene glycol standards.
Micelle FormationA typical block copolymer (0.1 g) in DMSO (10 mL) wastransferred through a 0.45-lm filter into a preswollen semi-permeable membrane (Spectra/Por, SPEC-TRUM, molecularweight cutoff, 3500) and dialyzed against water (3 L) for72 h to form a micelle. The dialysate water was exchangedat 2, 5, and 8 h intervals.
Micelle CharacterizationThe polymeric micelles were characterized by DLS and TEM.The DLS measurements were performed using a Brookhavenparticle size analyzer equipped with a laser operating at awavelength of 659 nm at 25 �C. ZetaPlus particle sizing soft-ware version 4.04 was used to calculate the average effectivediameter of the micelles. Approximately 1.0 mg mL�1 blockcopolymer micelle solution was filtered and used for themeasurement. For the TEM specimen preparations, blockcopolymer micelle solutions were stained with 0.2% uranylacetate solution. All the measurements were done above theCMCs. The sample solution was drop cast on a copper TEMgrid coated with holey amorphous carbon film. The TEMimages were taken using a JEOL 2100F TEM operated at 200kV. For investigation of stability of micelles at elevated tem-perature, the micelle samples were heated in situ in JEOL2100F using a heating holder (Gatan Inc.). The specimenswere initially observed at room temperature and then heatedup to 200 �C with 20 �C intervals. At each step, the tempera-ture remained constant for at least 10 min.
Photo-Crosslinking of MicellesA solution of the block copolymer micelles (6.7 mg mL�1)was crosslinked in a quartz cuvette by irradiation in a shortwave (254 nm) crosslinker (CL-1000 Crosslinker, UVP Ltd.)with 4, 6.5, and 8 J cm�2. After photo-crosslinking, the UVabsorption spectrum of each solution was obtained usingHitachi U-1800 UV/Vis Spectrophotometer. The absorptionspectra were recorded for wavelength range of 220–350 nm.
Critical Micelle ConcentrationThe CMCs of the copolymers were estimated by fluorescencespectroscopy using the dye Nile Red as a probe. The micellarsolutions were diluted to a concentration range of 1.00 �10�5 to 1.00 mg mL�1. An acetone solution of nile red(1 mg mL�1) was prepared. Approximately 30 lL of dyesolution was added to 3 mL of each of the diluted solutions.The acetone was then evaporated from the solutions. Thefluorescence was then measured for each solution using aCary Eclipse Fluorescence Spectrophotometer. The measure-ments were carried out at 20 �C using an excitation wave-length of 550 nm and emission wavelengths in the range570–800 nm. To measure the CMC of the crosslinked mi-celle, the crosslinked micelle solutions were also diluted toa concentration range of 1.0 � 10�5 to 1.0 mg mL�1 andfluorescence was measured by the same procedure as theuncrosslinked micelles. The CMC measurements were alsocarried out at 60 �C for poly(VBT-b-VBA) micelles (beforeand after 8 J cm�2 dose of UV radiation) and poly(VMT-b-VBA) micelles.
RESULTS AND DISCUSSION
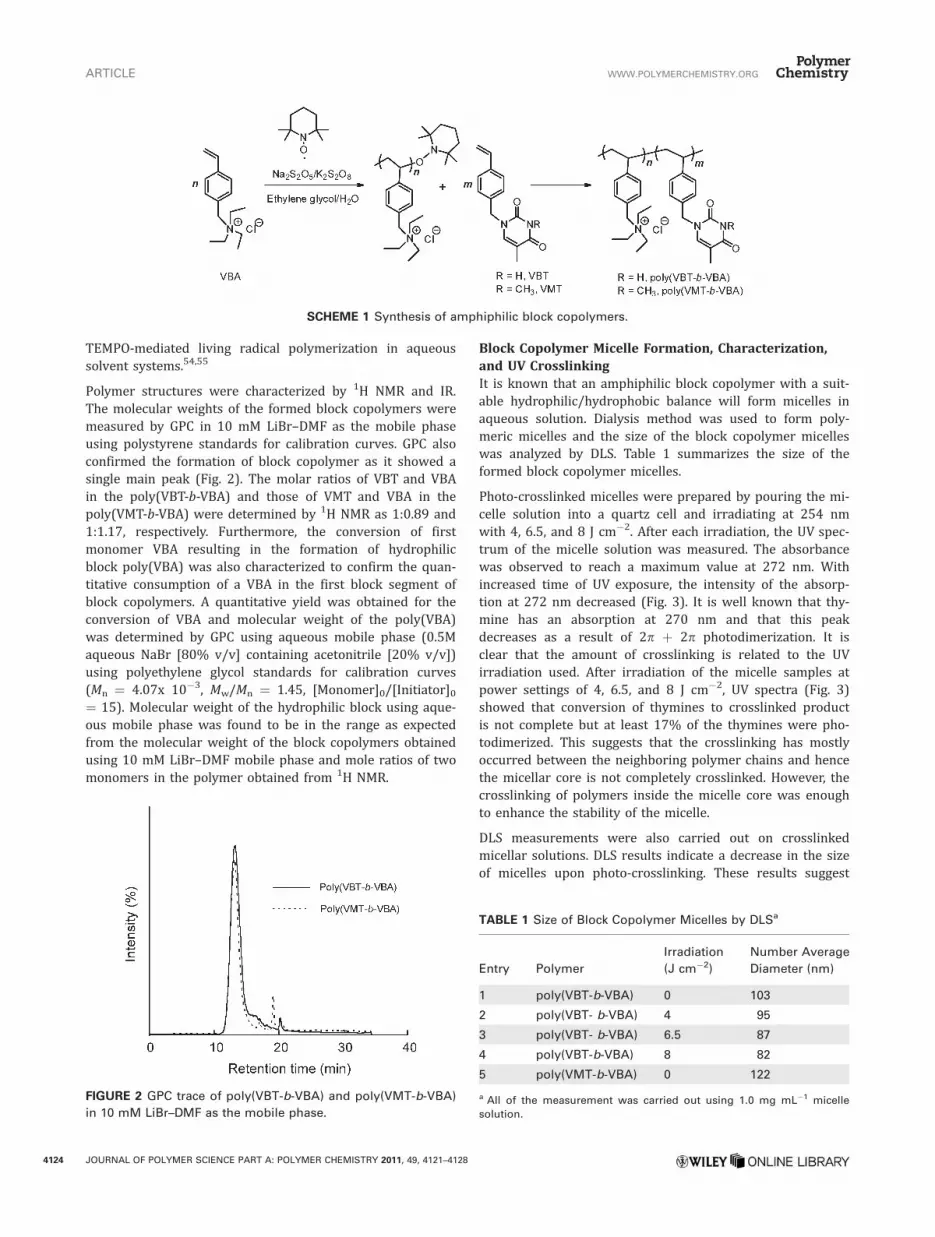
Poly(VBT-b-VBA) and Poly(VMT-b-VBA) SynthesisThe thymine-functionalized monomer VBT was synthesizedfrom p-vinylbenzylchloride and thymine in aqueous ethanolas reported previously.46 The amphiphilic block copolymersynthesis was carried out by a TEMPO-mediated living radi-cal polymerization system in a water/ethylene glycol mix-ture (Scheme 1). It is worth noting that this living radicalpolymerization system can be carried out as an aqueousmixture, a principle of green chemistry. The use of wateralso allows the polymerization of VBA without any addi-tional treatment.
To achieve the synthesis of the block copolymers, the homo-polymer of VBA was prepared first and then VBT was addedto the reaction solution to prepare the block copolymer withequal mole ratios. In this manner, poly(VBT-b-VBA) (Mn ¼1.16 � 104, Mw/Mn ¼ 1.8) was prepared. In addition, 3-methyl-1-(4-vinylbenzyl)thymine (VMT) was prepared bymethylation of VBT as a surrogate molecule to block thehydrogen bond donating ability of the monomer. The amphi-philic block copolymer of VMT and VBA, poly(VMT-b-VBA)(Mn ¼ 1.01 � 104, Mw/Mn ¼ 2.4), was then synthesized toexamine the effect of hydrogen bonding on the stability ofmicelles. The polydispersity values of the block copolymersare high when compared with other TEMPO-mediated livingradical polymerizations.54 However, it is worth noting thatthe polydispersity values obtained are low when comparedwith previously reported block copolymer synthesis using
WWW.POLYMERCHEMISTRY.ORG ARTICLE
WWW.MATERIALSVIEWS.COM JOURNAL OF POLYMER SCIENCE PART A: POLYMER CHEMISTRY 2011, 49, 4121–4128 4123
TEMPO-mediated living radical polymerization in aqueoussolvent systems.54,55
Polymer structures were characterized by 1H NMR and IR.The molecular weights of the formed block copolymers weremeasured by GPC in 10 mM LiBr–DMF as the mobile phaseusing polystyrene standards for calibration curves. GPC alsoconfirmed the formation of block copolymer as it showed asingle main peak (Fig. 2). The molar ratios of VBT and VBAin the poly(VBT-b-VBA) and those of VMT and VBA in thepoly(VMT-b-VBA) were determined by 1H NMR as 1:0.89 and1:1.17, respectively. Furthermore, the conversion of firstmonomer VBA resulting in the formation of hydrophilicblock poly(VBA) was also characterized to confirm the quan-titative consumption of a VBA in the first block segment ofblock copolymers. A quantitative yield was obtained for theconversion of VBA and molecular weight of the poly(VBA)was determined by GPC using aqueous mobile phase (0.5Maqueous NaBr [80% v/v] containing acetonitrile [20% v/v])using polyethylene glycol standards for calibration curves(Mn ¼ 4.07x 10�3, Mw/Mn ¼ 1.45, [Monomer]0/[Initiator]0¼ 15). Molecular weight of the hydrophilic block using aque-ous mobile phase was found to be in the range as expectedfrom the molecular weight of the block copolymers obtainedusing 10 mM LiBr–DMF mobile phase and mole ratios of twomonomers in the polymer obtained from 1H NMR.
Block Copolymer Micelle Formation, Characterization,and UV CrosslinkingIt is known that an amphiphilic block copolymer with a suit-able hydrophilic/hydrophobic balance will form micelles inaqueous solution. Dialysis method was used to form poly-meric micelles and the size of the block copolymer micelleswas analyzed by DLS. Table 1 summarizes the size of theformed block copolymer micelles.
Photo-crosslinked micelles were prepared by pouring the mi-celle solution into a quartz cell and irradiating at 254 nmwith 4, 6.5, and 8 J cm�2. After each irradiation, the UV spec-trum of the micelle solution was measured. The absorbancewas observed to reach a maximum value at 272 nm. Withincreased time of UV exposure, the intensity of the absorp-tion at 272 nm decreased (Fig. 3). It is well known that thy-mine has an absorption at 270 nm and that this peakdecreases as a result of 2p þ 2p photodimerization. It isclear that the amount of crosslinking is related to the UVirradiation used. After irradiation of the micelle samples atpower settings of 4, 6.5, and 8 J cm�2, UV spectra (Fig. 3)showed that conversion of thymines to crosslinked productis not complete but at least 17% of the thymines were pho-todimerized. This suggests that the crosslinking has mostlyoccurred between the neighboring polymer chains and hencethe micellar core is not completely crosslinked. However, thecrosslinking of polymers inside the micelle core was enoughto enhance the stability of the micelle.
DLS measurements were also carried out on crosslinkedmicellar solutions. DLS results indicate a decrease in the sizeof micelles upon photo-crosslinking. These results suggest
SCHEME 1 Synthesis of amphiphilic block copolymers.
FIGURE 2 GPC trace of poly(VBT-b-VBA) and poly(VMT-b-VBA)
in 10 mM LiBr–DMF as the mobile phase.
TABLE 1 Size of Block Copolymer Micelles by DLSa
Entry Polymer
Irradiation
(J cm�2)
Number Average
Diameter (nm)
1 poly(VBT-b-VBA) 0 103
2 poly(VBT- b-VBA) 4 95
3 poly(VBT- b-VBA) 6.5 87
4 poly(VBT-b-VBA) 8 82
5 poly(VMT-b-VBA) 0 122
a All of the measurement was carried out using 1.0 mg mL�1 micelle
solution.
ARTICLE WWW.POLYMERCHEMISTRY.ORG
4124 JOURNAL OF POLYMER SCIENCE PART A: POLYMER CHEMISTRY 2011, 49, 4121–4128

that photo-crosslinking because of photodimerization of thy-mines between adjacent polymer chains brings the polymerchains closer in the core causing the micelles to shrink in size.Moreover, DLS results show a decrease in the size of micelles,confirming that photo-crosslinking is occurring in the core ofthe micelles and not between micelles which would haveresulted in increase in micelle size. Furthermore, the sizeremained constant after 8 J cm�2 of UV irradiation. This resultmight be due to the maximum limit of crosslinking reached.
Examination of the micelle solution by IR was carried out inan attempt to demonstrate the hydrogen-bonding of thymineunits but this was inconclusive because of the many broadpeaks around 3500–3000 cm�1 due to ANH bond in thymine.The bright-field TEM images (Fig. 4) of the stained poly(VBT-b-VBA) micelles were taken before and after crosslinking.
TEM images of the poly(VBT-b-VBA) micelle samples,‘‘before’’ and ‘‘after’’ photo-crosslinking, demonstrated largelyspherical objects in the range of 30–60 nm. These resultsalso indicate that crosslinking of attached thymine in these
block copolymer micelles proceeds only inside the core anddid not change the spherical morphology of the micelles.
Furthermore, the in situ heating TEM results indicated differ-ences between the micelle samples when heated up to200 �C (Fig. 5). It was observed that poly(VBT-b-VBA)micelles were stable to heating to 60 �C. However, the TEMimage of the micelle started to fade when the sample washeated above 60 �C. In comparison, the crosslinked poly(VBT-b-VBA) micelles were stable even at 200 �C, which supportsthe stability enhancement of the micelles by crosslinking. Thehigher temperature study indicated that the thermostableproperty of the micelles can be controlled by the irradiation.
CMC MeasurementsCMC measurements can be used to assess the stability ofmicelles. A lower CMC value indicates higher stability. TheCMCs of the block copolymer micelles were determined byfluorescence measurements in aqueous solution using NileRed as a probe (see Supporting Information). The CMC isidentified as the break point on a plot of fluorescence inten-sity versus concentration (Fig. 6). Nile Red has a strongfluorescence in a hydrophobic environment, but in a polarenvironment such as water, fluorescence quenching is greatlyenhanced, significantly reducing fluorescence emission. Thus,the CMC can be determined by measuring the concentrationpoint at which the fluorescence emission intensity of NileRed dye becomes constant. The CMCs of poly(VBT-b-VBA)micelles after UV crosslinking (irradiation doses: 0, 4, 6.5,and 8 J cm�2) were also measured to determine the effect ofphoto-crosslinking on the stability of the micelles. In addi-tion, the CMC of poly(VMT-b-VBA) micelles was measured toexamine the effect of hydrogen bonding on the stability ofthe micelles.
Data for the measured CMCs (at 20 �C) of the block copoly-mer micelles are summarized in Table 2. Most significantly,the poly(VMT-b-VBA) micelles, which are incapable of form-ing self-assembled hydrogen bond dimers in the corebecause of methylation, have a much higher CMC value when
FIGURE 3 The UV absorption spectra of poly(VBT-b-VBA) mi-
celle solution upon UV exposure with 0, 4, 6.5, and 8 J cm�2.
FIGURE 4 Bright-field TEM images of poly(VBT-b-VBA) micelles before and after UV crosslinking (8 J cm�2).
WWW.POLYMERCHEMISTRY.ORG ARTICLE
WWW.MATERIALSVIEWS.COM JOURNAL OF POLYMER SCIENCE PART A: POLYMER CHEMISTRY 2011, 49, 4121–4128 4125

compared with poly(VBT-b-VBA) micelles that are capable ofhydrogen bonding in the core. The CMC of poly(VMT-b-VBA)was found to be approximately 80% higher than poly(VBT-b-VBA). These observed differences in the CMC values of thepoly(VMT-b-VBA) micelles and the poly(VBT-b-VBA) micellesare consistent with a functional role for hydrogen bonding ofthe thymine units. In addition, these observations confirmthat hydrogen bonding of the attached thymines enhancesthe stability of the block copolymer micelles. The CMC of thepoly(VBT-b-VBA) micelles decreases with increased UVirradiation. With 8 J cm�2 of UV irradiation, the CMC of thepoly(VBT-b-VBA) micelle decreased by approximately 45%when compared with the uncrosslinked sample.
The CMC values were also measured at 60 �C (Table 3). At60 �C, poly(VMT-b-VBA) micelles have the greatest CMC
value indicating the lowest stability, whereas the crosslinkedpoly(VBT-b-VBA) micelles had the lowest CMC value indicat-ing highest stability. The CMC of crosslinked poly(VBT-b-VBA) micelles measured at 60 �C was still low (39 lg mL�1)when compared with the CMC of the uncrosslinked micelle(46 lg mL�1) consistent with the stability enhancement ofthe micelles due to crosslinking. Collectively, these resultsclearly indicate that the stability of the micelles is enhancedby the core-photo-crosslinking of the attached thymines andthe stability of the micelles can be modulated by the UVirradiation.
CONCLUSIONS
We have synthesized an amphiphilic block copolymer of VBTand VBA by TEMPO-mediated living radical polymerizationand used this novel material to prepare polymeric micellesin aqueous solution. DLS, TEM, and CMC measurementsdemonstrated that the amphiphilic block copolymers of VBTand VBA can form stability-enhanced self-assemblingmicelles based on hydrogen bonding of the attached thy-mines in the core and that this stability can be further
FIGURE 5 Bright-field TEM images of (a) poly(VBT-b-VBA) micelle at room temperature, 60, 100, and 200 �C as well as (b) core-
crosslinked poly(VBT-b-VBA) micelle at room temperature, 60, 100, and 200 �C.
FIGURE 6 Fluorescence intensity of Nile Red at its emission
maximum versus copolymer concentration.
TABLE 2 Critical Micelles Concentration (CMC) of Block
Copolymer Micelles at 20 8C
Entry Polymer
Irradiation
(J cm�2)
CMC at 20 �C(lg mL�1)
1 poly(VMT-b-VBA) 0 62
2 poly(VBT-b-VBA) 0 35
3 poly(VBT-b-VBA) 4 27
4 poly(VBT-b-VBA) 6.5 25
5 poly(VBT-b-VBA) 8 19
ARTICLE WWW.POLYMERCHEMISTRY.ORG
4126 JOURNAL OF POLYMER SCIENCE PART A: POLYMER CHEMISTRY 2011, 49, 4121–4128

controlled by core-photo-crosslinking of the attached thy-mines. As such, polymeric micelles obtained from poly(VBT-b-VBA) have the potential to allow the encapsulation of guestmaterials by hydrogen bonding due to the attached thyminesin the core. Furthermore, the control of the CMC of the blockcopolymer micelles by UV photo-crosslinking provides a newopportunity for the controlled release of materials encapsu-lated in the micelles. Thus, based on a bioinspired mecha-nism, these investigations provide a novel approach for thedesign and use of stable and controllable nano micelles.
ACKNOWLEDGMENT
This work was supported by Monash University Faculty of Sci-ence Early Career Research Fund and Australian Research Coun-cil. The authors thank Roy Jackson, Tony Patti, and WarwickRaverty for fruitful discussions, and Monash Centre for ElectronMicroscopy for providing access to the JEOL 2100F TEM.
REFERENCES AND NOTES
1 Warner, J. C.; Cannon, A. S.; Dye, K. M. Environ Impact
Assess Rev 2004, 24, 775–799.
2 Anastas, P. T.; Warner, J. C. Green Chemistry: Theory and
Practice; Oxford University Press: London, 1998.
3 Watson, J. D.; Crick, F. H. C. Nature 1953, 171, 737–738.
4 Boal, A. K.; Ilhan, F.; Derouchey, J. E.; Thurn-Albrecht, T.;
Russell, T. P.; Rotello, V. M. Nature 2000, 404, 746–748.
5 Viswanathan, K.; Ozhalici, H.; Elkins, C. L.; Heisey, C.; Ward,
T. C.; Long, T. E. Langmuir 2006, 22, 1099–1105.
6 Cheng, C. M.; Egbe, M. I.; Grasshoff, J. M.; Guarrera, D. J.;
Pai, R. P.; Warner, J. C.; Taylor, L. D. J Polym Sci Part A: Polym
Chem 1995, 33, 2515–2519.
7 Grasshoff, J. M.; Taylor, L. D.; Warner, J. C. U.S. Patent
5,708,106, January 13, 1998.
8 Kiarie, C.; Bianchini, J.; Trakhtenberg, S.; Warner, J. C.
J Macromol Sci Pure Appl Chem 2005, 42A, 1489–1496.
9 Whitfield, J. R.; Morelli, A.; Warner, J. C. J Macromol Sci
Pure Appl Chem 2005, 42A, 1541–1546.
10 Trakhtenberg, S.; Hangun-Balkir, Y.; Warner, J. C.; Bruno, F.
F.; Kumar, J.; Nagarajan, R.; Samuelson, L. A. J Am Chem Soc
2005, 127, 9100–9104.
11 Hamley, I. W. In Encyclopedia of Polymer Science and Tech-
nology; Mark, H. F., Ed.; Wiley: New Jersey, 2003, pp 457–482.
12 Hadjichristidis, N.; Pispas, S.; Floudas, G. Block Copolymers;
Wiley: New Jersey, 2003.
13 Kataoka, K.; Harada, A.; Nagasaki, Y. Adv Drug Deliv Rev
2001, 47, 113–131.
14 Rosler, A.; Vandermeulen, G. W. M.; Klok, H. A. Adv Drug
Deliv Rev 2001, 53, 95–108.
15 Adams, M. L.; Lavasanifar, A.; Kwon, G. S. J Pharmaceut
Sci 2003, 92, 1343–1355.
16 Henselwood, F.; Liu, G. Macromolecules 1997, 30, 488–493.
17 Savic, R.; Luo, L.; Eisenberg, A.; Maysinger, D. Science
2003, 300, 615–618.
18 Kataoka, K.; Matsumoto, T.; Yokoyama, M.; Okano, T.;
Sakurai, Y.; Fukushima, S.; Okamoto, K.; Kwon, G. S. J Contr
Release 2000, 64, 143–153.
19 Gao, Z.; Lukyanov, A. N.; Singhal, A.; Torchilin, V. P. Nano
Lett 2002, 2, 979–982.
20 Lukyanov, A. N.; Gao, Z.; Torchilin, V. P. J Contr Release
2003, 91, 97–102.
21 Nishiyama, N.; Bae, Y.; Miyata, K.; Fukushima, S.; Kataoka,
K. Drug Discov Today Tech 2005, 2, 21–26.
22 Qiu, L.; Zheng, C.; Jin, Y.; Zhu, K. Expert Opin Ther Pat
2007, 17, 819–830.
23 Nishiyama, N.; Kato, Y.; Sugiyama, Y.; Kataoka, K. Pharma-
ceut Res 2001, 18, 1035–1041.
24 Nishiyama, N.; Kataoka, K. Pharmacol Therapeut 2006, 112,
630–648.
25 Dimitrov, I.; Trzebicka, B.; Muller, A. H. E.; Dworak, A.; Tsve-
tanov, C. B. Prog Polym Sci 2007, 32, 1275–1343.
26 Oerlemans, C.; Bult, W.; Bos, M.; Storm, G.; Nijsen, J. F. W.;
Hennink, W. E. Pharmaceut Res 2010, 27, 2569–2589.
27 Bae, Y.; Kataoka, K. Adv Drug Deliv Rev 2009, 61, 768–784.
28 Alvarez-Lorenzo, C.; Concheiro, A. Mini Rev Med Chem
2008, 8, 1065–1074.
29 Jones, M. C.; Leroux, J. C. Eur J Pharmaceut Biopharmaceut
1999, 48, 101–111.
30 Haag, R. Angew Chem Int Ed 2004, 43, 278–282.
31 Zhang, Y.; Jiang, M.; Zhao, J.; Zhou, J.; Chen, D. Macromo-
lecules 2004, 37, 1537–1543.
32 Pilon, L. N.; Armes, S. P.; Findlay, P.; Rannard, S. P. Lang-
muir 2005, 21, 3808–3813.
33 Pilon, L. N.; Armes, S. P.; Findlay, P.; Rannard, S. P. Eur
Polym J 2006, 42, 1487–1498.
34 Jiang, X.; Luo, S.; Armes, S. P.; Shi, W.; Liu, S. Macromole-
cules 2006, 39, 5987–5994.
35 Liu, S.; Armes, S. P. J Am Chem Soc 2001, 123, 9910–
9911.
36 Butun, V.; Wang, X. S.; de Paz Banez, M. V.; Robinson, K. L.;
Billingham, N. C.; Armes, S. P.; Tuzar, Z. Macromolecules 1999,
33, 1–3.
37 Bronich, T. K.; Keifer, P. A.; Shlyakhtenko, L. S.; Kabanov, A.
V. J Am Chem Soc 2005, 127, 8236–8237.
38 Wang, G.; Henselwood, F.; Liu, G. Langmuir 1998, 14,
1554–1559.
39 Tao, J.; Stewart, S.; Liu, G.; Yang, M. Macromolecules 1997,
30, 2738–2745.
40 Chen, Y.; Tavakley, A. E.; Mathiason, T. M.; Taton, T. A.
J Polym Sci Part A Polym Chem 2006, 44, 2604–2614.
41 Saito, K.; Ingalls, L. R.; Lee, J.; Warner, J. C. Chem Commun
2007, 2503–2505.
42 Saito, K.; Warner, J. C. Green Chem Lett Rev 2009, 2, 71–76.
43 Tazuke, S.; Takasaki, R. J Polym Sci Polym Chem Ed 1983,
21, 1529–1534.
44 Cannon, A. S.; Warner, J. C. J Macromol Sci Pure Appl
Chem 2005, 42A, 1507–1514.
45 Martino, D. M.; Reyna, D.; Estenoz, D. A.; Trakhtenberg, S.;
Warner, J. C. J Phys Chem A 2008, 112, 4786–4792.
TABLE 3 Critical Micelles Concentration (CMC) of Block
Copolymer Micelles at 60 8C
Entry Polymer
Irradiation
(J cm�2)
CMC
(lg mL�1)
1 poly(VMT-b-VBA) 0 93
2 poly(VBT-b-VBA) 0 46
5 poly(VBT-b-VBA) 8 39
WWW.POLYMERCHEMISTRY.ORG ARTICLE
WWW.MATERIALSVIEWS.COM JOURNAL OF POLYMER SCIENCE PART A: POLYMER CHEMISTRY 2011, 49, 4121–4128 4127

46 Bianchini, J. R.; Saito, K.; Balin, T. B.; Dua, V.; Warner, J. C.
J Polym Sci Part A Polym Chem 2007, 45, 1296–1303.
47 Barbarini, A. L.; Estenoz, D. A.; Martino, D. M. Macromol
React Eng 2010, 4, 453–459.
48 El-Hayek, R. F.; Dye, K.; Warner, J. C. J Biomed Mater Res
Part A 2006, 79A, 874–881.
49 Senuma, M.; Tashiro, T.; Iwakura, M.; Kaeriyama, K.; Shi-
mura, Y. J Appl Polym Sci 1989, 37, 2837–2843.
50 Dizman, B.; Elasri, M. O.; Mathias, L. J. J Appl Polym Sci
2004, 94, 635–642.
51 Lu, G.; Wu, D.; Fu, R. React Funct Polym 2007, 67,
355–366.
52 Kenawy, E. R.; Worley, S. D.; Broughton, R. Biomacromole-
cules 2007, 8, 1359–1384.
53 Ikeda, T.; Tazuke, S.; Suzuki, Y. Die Makromol Chem 1984,
185, 869–876.
54 Shim, S. E.; Oh, S.; Chang, Y. H.; Jin, M.; Choe, S. Polymer
2004, 45, 4731–4739.
55 Desjardins, A.; Eisenberg, A. Macromolecules 1991, 24,
5779–5790.
ARTICLE WWW.POLYMERCHEMISTRY.ORG
4128 JOURNAL OF POLYMER SCIENCE PART A: POLYMER CHEMISTRY 2011, 49, 4121–4128





























