Embed Size (px)
Citation preview

UNIVERSITY OF CALIFORNIA, SAN DIEGO
Bypass suppression defines critical genomic regulation by the NuA4 acetyltransferase complex
A dissertation submitted in partial satisfaction of the requirements for the degree Doctor of Philosophy
in
Biomedical Sciences
by
Naomi Searle
Committee in Charge:
Professor Lorraine Pillus, Chair Professor Dong-Er Zhang, Co-Chair Professor Dong Wang Professor Eugene Yeo Professor Huilin Zhou
2017

Copyright
Naomi Searle, 2017
All rights reserved.

iii
Chair
Signature Page
The Dissertation of Naomi Searle is approved, and it is
acceptable in quality and form for publication on microfilm and
electronically:
Co-Chair
University of California, San Diego
2017

iv
DEDICATION Dedication
This dissertation is dedicated to my family, for all of the support, love,
guidance, and inspiration.

v
TABLE OF CONTENTS Table of Contents
Signature Page .................................................................................................. iii Dedication ......................................................................................................... iv Table of Contents .............................................................................................. v List of Abbreviations ........................................................................................ viii List of Figures ................................................................................................... ix List of Tables .................................................................................................... xii Acknowledgements ......................................................................................... xiii Vita .................................................................................................................. xv Abstract ........................................................................................................... xvi Chapter 1. Introduction ...................................................................................... 1 Tip60 is the catalytic subunit of the NuA4 acetyltransferase complex. ............ 2 Enhancer of Polycomb is broadly conserved .................................................. 4 Comparative studies between model organisms promote functional definition ........................................................................................................................ 6 From basic function to translational significance ........................................... 12 Bypass suppression is a powerful genetic tool .............................................. 15 Promoting a balanced acetylation state allows for the bypass of ESA1 ........ 16 Bypass suppression to define key functions of chromatin modifying proteins17 Acknowledgements ....................................................................................... 20 References .................................................................................................... 21 Chapter 2. Chromatin regulation by the NuA4 acetyltransferase complex is mediated by essential interactions between Enhancer of Polycomb (Epl1) and Esa1 ................................................................................................................ 30 Abstract ......................................................................................................... 31 Introduction .................................................................................................... 31 Materials and Methods .................................................................................. 32 Yeast Strains and Plasmids ......................................................................... 32 Growth Assays ............................................................................................ 32 Flow Cytometry ............................................................................................ 32 Lysate preparation ....................................................................................... 33 Immunoprecipitation .................................................................................... 33

vi
Immunoblots ................................................................................................ 33 RNA-seq sample preparation and analysis ................................................. 33 qPCR validation ........................................................................................... 33 Data availability ........................................................................................... 34 Results ........................................................................................................... 34 Bypass and function of essential piccolo-NuA4 subunits ............................ 34 ESA1 and EPL1 bypass strains have nearly identical gene expression profiles ......................................................................................................... 35 Epl1 promotes the chromatin association of Esa1 ...................................... 36 Defining the critical regions of Epl1 in vivo .................................................. 37 Physical association between Esa1 and Epl1 is required for activity .......... 38 Discussion ..................................................................................................... 39 Acknowledgements ....................................................................................... 42 Literature Cited .............................................................................................. 43 Supplemental Materials ................................................................................. 44 Supplemental References ........................................................................... 48 Acknowledgements ....................................................................................... 49 Chapter 3. Bypass suppression defines chromatin dynamics between NuA4 and the Hda1 deacetylase ............................................................................... 50 Introduction .................................................................................................... 51 Class I deacetylases .................................................................................... 51 Class II deacetylases ................................................................................... 54 Class III deacetylases .................................................................................. 55 Opposing activities of NuA4 and the HDACs .............................................. 56 Results ........................................................................................................... 57 NuA4 has distinct interactions with histone deacetylases ........................... 57 Phenotypes of epl1∆ sds3∆ are selectively suppressed by deletion of HDACs. ........................................................................................................ 59 Epl1 is opposed by Hda1 independent of Rpd3L ........................................ 66 Discussion ..................................................................................................... 70 Hos1 and Hos3 may act in opposition to NuA4 at the rDNA locus and spindle pole body, respectively .................................................................... 71 Epl1’s interactions with Hst2 and Sir2 are distinct from Esa1’s genetic Interactions .................................................................................................. 72 Functional antagonism between Rpd3 and Hos2 ........................................ 74 The Hda1-Rpd3-NuA4 functional axis ......................................................... 75 References .................................................................................................... 79 Chapter 4. Defining the Esa1 Transcriptional Regulome ................................ 85 Introduction .................................................................................................... 86 Esa1 is important for global transcription .................................................... 86 Esa1 is involved in regulating the expression of ribosomal protein genes .. 87 Esa1 in ribosome biogenesis ....................................................................... 88

vii
Bypass suppression to define the Esa1 transcriptional ‘regulome.’ ............ 89 Results ........................................................................................................... 90 Differential expression analysis of ESA1 bypass mutant to define Esa1- regulated gene expression .......................................................................... 90 Ribosome biogenesis genes are regulated by Esa1 ................................... 94 A transcriptional basis for the hda1∆-mediated rescue of esa1∆ sds3∆ mutant phenotypes .................................................................................... 100 Discussion ................................................................................................... 109 Transcriptome analysis of ESA1 bypass ................................................... 109 Understanding the significance of Esa1 in ribosome biogenesis .............. 110 The role of Hda1 in balancing regulation of ribosome biogenesis ............. 112 References .................................................................................................. 123 Chapter 5. Conclusions and Future Directions .............................................. 127 Epl1 is a critical Esa1-cofactor .................................................................... 128 Loss of Hda1 suppresses epl1∆ sds3∆ phenotypes, and underscores a NuA4-Rpd3L-Hda1 regulatory axis .............................................................. 131 Esa1 is critical for proper regulation of ribosome biogenesis ...................... 136 Summary ..................................................................................................... 138 Acknowledgements ..................................................................................... 140 References .................................................................................................. 141 Appendix A. Experimental Methods .............................................................. 144 Halo Assays ................................................................................................. 145 Polysome profiling ....................................................................................... 146 References .................................................................................................. 147

viii
LIST OF ABBREVIATIONS List of Abbreviations
5-FOA 5-fluoroorotic acid Ade Adenine Arg Arginine Can Canavanine CEN Centromeric ChIP Chromatin immunoprecipitation CPT Camptothecin DMSO Dimethyl sulfoxide DNA Deoxyribonucleic acid EPC Enhancer of Polycomb GO Gene ontology HAT/KAT Histone acetyltranferase/ Lysine acetytransferase HDAC/KDAC Histone deacetylase/ Lysine deacetylase His Histidine HU Hydroxyurea log2FC Log2 Fold Change MMS Methyl methanesulfonate NAD+ Nicotinamide adenine dinucleotide p-adj adjusted p-value Pol I polymerase I Pol II polymerase II Pol III polymerase III qPCR quantiative polymerase chain reaction rDNA ribosomal DNA RNA Ribonucleic acid RNA-seq Ribonucleic acid sequencing RPKM Reads Per Kilobase Million rRNA ribosomal RNA SC Synthetic Complete sORF short open reading frame TFIID Transcription Factor II D Trp Tryptophan Ura Uracil UV Ultraviolet WT Wild type YPAD Yeast peptone dextrose supplemented with adenine

ix
LIST OF FIGURES List of Figures
Figure 1-1. Posttranslational modification of histone-tails by acetylation .......... 3 Figure 1-2. Tip60 is frequently altered in cancer ............................................... 5 Figure 1-3. Epl1 domain structure in S. cerevisiae ............................................ 8 Figure 1-4. EPC has widespread functions as evidenced by multimeric complex diversity and physical interactors ........................................................ 9 Figure 2-1. The requirement for two essential NuA4 subunits is bypassed by disassembly of Rpd3L ..................................................................................... 33 Figure 2-2. Bypass of EPL1 is phenotypically akin to esa1∆ sds3∆ ................ 34 Figure 2-3. ESA1 and EPL1 bypass strains have nearly identical gene expression profiles ........................................................................................... 35 Figure 2-4. Epl1 is required for stable chromatin association of Esa1 ............ 36 Figure 2-5. Defining functional regions of Epl1 in vivo .................................... 37 Figure 2-6. Subunit interaction domains are critical for Epl1 function in vivo .. 38 Figure 2-7. Viability of epl1 mutants is linked to stable Epl1-Esa1 interaction, not chromatin association ................................................................................ 39 Figure 2-8. Model: Epl1 is a core NuA4 regulator in tandem with Esa1 .......... 41 Figure 2-S1. Bypass of ESA1 and EPL1 occurs at both 24˚ and 30˚ .............. 44 Figure 2-S2. Validation of differentially expressed transcripts by qPCR ......... 44 Figure 2-S3. Crosslinking stabilizes Esa1 in the chromatin in epl1∆ sds3∆ .... 45 Figure 2-S4. Eaf1 is required for epl1-NP∆ viability ........................................ 45 Figure 3-1. Three classes of histone deacetylases, based on sequence homology and/or co-factor dependence .......................................................... 52 Figure 3-2. NuA4 has distinct interactions with difference HDACs .................. 58 Figure 3-3. Loss of HDA1 enhances temperature-dependent fitness ............. 60

x
Figure 3-4. Loss of HDA1 results in robust response to DNA damage in epl1∆ sds3∆ ............................................................................................................... 62 Figure 3-5. The epl1∆ sds3∆ cell cycle delay is partially suppressed by hda1∆ . ......................................................................................................................... 64 Figure 3-6. Low histone H4 acetylation is suppressed in epl1∆ sds3∆ hda1∆ 65 Figure 3-7. Oxidative stress sensitivity is rescued by hda1∆ in epl1∆ sds3∆ .. 67 Figure 3-8. rDNA silencing defect in epl1∆ sds3∆ is suppressed by hda1∆ .... 69 Figure 3-9. The Hda1-Rpd3-NuA4 functional axis ........................................... 77 Figure 4-1. Defining the ESA1 transcriptional ‘regulome’ through differential expression analysis of a bypass mutant .......................................................... 92 Figure 4-2. Distinct bimodal pattern of gene expression regulation by the Esa1 transcriptional regulome .................................................................................. 93 Figure 4-3. Metabolic process-related genes are downregulated upon loss of ESA1 ............................................................................................................... 95 Figure 4-4. Ribosome biogenesis-related genes are upregulated upon loss of ESA1 ............................................................................................................... 96 Figure 4-5. Summary of gene ontology highlights ribosome biogenesis ......... 97 Figure 4-6. Co-regulation of ribosome biogenesis gene expression ............... 99 Figure 4-7. Loss of ESA1 results in a distinct polysome profile ..................... 101 Figure 4-8. Defining the transcriptional rescue of esa1∆ sds3∆ by hda1∆ .... 103 Figure 4-9. There are 138 transcripts defining gene expression rescue by esa1∆ sds3∆ hda1∆ ....................................................................................... 104 Figure 4-10. Cell wall biogenesis gene expression is involved in HDA1-mediated rescue ............................................................................................ 106 Figure 4-11. Cell wall biogenesis genes are both up and downregulated in rescue ............................................................................................................ 107

xi
Figure 4-12. Osmotic support facilitates improved growth of esa1∆ sds3∆ but is distinct from DNA damage sensitivity ........................................................ 108 Figure 5-1. The Hda1-Rpd3-NuA4 functional axis includes H2A.Z ............... 134

xii
LIST OF TABLES List of Tables
Table 1-1. Enhancer of Polycomb orthologs ................................................... 18 Table 1-2. EPC interactors and effectors referenced in text ............................ 19 Table 2-S1. Yeast strains ................................................................................ 46 Table 2-S2. Plasmids ...................................................................................... 47 Table 2-S3. Oligonucleotides .......................................................................... 48 Table 3-1. Strains used in Chapter 3 ............................................................... 78 Table 4-1. Strains used in Chapter 4 ............................................................. 116 Table 4-2. Top 100 transcripts downregulated in esa1∆ sds3∆ relative to WT ... ....................................................................................................................... 117 Table 4-3. Transcripts 101-200 downregulated in esa1∆ sds3∆ relative to WT ....................................................................................................................... 118 Table 4-4. Transcripts 201-277 downregulated in esa1∆ sds3∆ relative to WT ....................................................................................................................... 119 Table 4-5. Top 100 transcripts upregulated in esa1∆ sds3∆ relative to WT ........ ....................................................................................................................... 120 Table 4-6. Transcripts 101-200 upregulated in esa1∆ sds3∆ relative to WT ....... ....................................................................................................................... 121 Table 4-7. Transcripts 201-300 upregulated in esa1∆ sds3∆ relative to WT ....... ....................................................................................................................... 122 Table 4-8. Transcripts 301-312 upregulated in esa1∆ sds3∆ relative to WT ....... ....................................................................................................................... 123

xiii
ACKNOWLEDGEMENTS Acknowledgements
I first would like to thank Lorraine for her unwavering, supportive, and
positive mentorship since welcoming me into the lab four years ago. She
provided exceptional training, and her excitement for science nurtured an
invigorating atmosphere. She taught me many valuable skills, both scientific
and professional, that I will undoubtedly carry with me for a lifetime.
I thank Ana Lilia Torres-Machorro for launching the bypass project, and
passing the torch on to me. The mentorship that she provided in the year that
we shared in the Pillus lab, and continues to provide from afar has been
invaluable. I particularly enjoyed discussing new ideas with her and owe a vast
amount of my yeast genetics experimental knowledge to her.
Other members of the Pillus lab and 2nd floor Pac Hall-ers, both past
and present, have been particularly wonderful colleagues. In particular, I owe
a great deal of thanks to Emily Petty. Her friendship, support, and patience
have truly shaped my graduate school experience in the Pillus lab and made
the entire experience overwhelmingly positive. Thank you as well for all of the
constructive feedback over the years on presentations, papers, and of course,
on this thesis and for the office chatter during breaks. Additional thanks to
Christie Chang and Michael Hayes for feedback on Chapter 2 of this thesis
that helped strengthen the Epl1 manuscript for publication.
I thank members of the Yeo lab, including Brian Yee, Julia Nussbacher,
and Gabe Pratt for their guidance with the initial processing of the RNA-seq

xiv
data, and to my former colleague Erin Smith for teaching me many of the
fundamentals of bioinformatics analysis in R. Thanks to Brian Zid and Joseph
Simpson for guidance on the polysome profiling portion of Chapter 4.
Additional thanks to my committee members for critical feedback
throughout the course of my thesis research, and for the enthusiasm that they
shared for my project that was particularly motivating.
Finally, I owe much thanks to my family: to my parents, and siblings for
the support, encouragement, guidance. An extra thanks to my parents for
inspiring me to pursue my dreams. Perhaps most of all, thank you to my
husband, David – for believing in me, encouraging me, lifting me up during the
lows, celebrating with me during the highs, and of course, for making sure I
was well fed after long days. Your unwavering support over the last 6 years
always drove me to think deeper and work harder, all while having fun doing it.
Chapter 1 and Chapter 5, in part, contain material as it appears in
Searle NE, Pillus L. 2017. Critical genomic regulation mediated by Enhancer
of Polycomb. Current Genetics. In-press. The dissertation author was the
primary investigator and first author of this paper.
Chapter 2, in full, is a reprint of the material as it appears in Searle NE,
Torres-Machorro AL, Pillus L. 2017. Chromatin Regulation by the NuA4
Acetyltransferase Complex Is Mediated by Essential Interactions Between
Enhancer of Polycomb (Epl1) and Esa1. Genetics 205: 1125-1137. The
dissertation author was the primary investigator and first author of this paper.

xv
VITA
2008-2010 Academic Peer Mentor and Advisor
College of Chemical and Life Sciences
University of Maryland, College Park
2009 Summer Research Fellow
National Institutes of Health, Bethesda, Maryland
2009-2010 Undergraduate Research Assistant
University of Maryland, College Park
2010 B.S., Biology – Neurobiology/Physiology
University of Maryland, College Park
2010-2011 Post-Baccalaureate Cancer Research Fellow
National Cancer Institute, Bethesda, Maryland
2015 Teaching Assistant
Department of Biology, University of California, San Diego
2017 Ph.D. Biomedical Sciences
University of California, San Diego
PUBLICATIONS
Searle NE, Torres-Machorro A. L., Pillus L., 2017 Chromatin Regulation by the NuA4 Acetyltransferase Complex Is Mediated by Essential Interactions Between Enhancer of Polycomb (Epl1) and Esa1. Genetics 205: 1125–1137. Searle NE, 2017 Critical genomic regulation mediated by Enhancer of Polycomb. Current Genetics. In-press.

xvi
Abstract of the Dissertation
ABSTRACT OF THE DISSERTATION
Bypass suppression defines critical genomic regulation by the NuA4 acetyltransferase complex
by
Naomi Searle
Doctor of Philosophy in Biomedical Sciences
University of California, San Diego, 2017
Professor Lorraine Pillus, Chair
Professor Dong-Er Zhang, Co-Chair

xvii
The organization and regulation of eukaryotic DNA is a critical genomic
process that is controlled by many key enzymes and multimeric complexes.
One mode of regulation involves the acetylation of histones, as coordinated by
the opposing enzymatic activities of histone acetyltransferases and
deacetylases. Some of the enzymatic and regulatory subunits involved in
chromatin regulation are essential, and thus present additional challenges to
comprehensively study their function. Subunits within the NuA4 complex, such
as the catalytic subunit Esa1, and non-catalytic Epl1 subunit, are examples of
essential chromatin-modifying components. In this thesis, the powerful genetic
tool of bypass suppression, is used to study null ESA1 and EPL1 mutants by
concurrent deletion of the Rpd3L deacetylase complex, promoting a more
balanced cellular acetylation state. Upon bypass, critical functions of Epl1
were defined for the first time in vivo, and implicated Epl1 as a key co-factor
required for Esa1 catalytic activity. Here, Epl1 is also found to be important for
targeting Esa1 to chromatin, thereby supporting Esa1’s critical activity as an
acetyltransferase. Furthermore, bypass suppression of NuA4 revealed a key
interaction network between NuA4 and the Rpd3L and Hda1 histone
deacetylases. Despite deleting an essential acetyltransferase and two
important deacetylases, growth of the epl1∆ sds3∆ hda1∆ mutant is
significantly more robust than the epl1∆ sds3∆ mutant and can withstand
various stresses such as high temperature and exposure to genotoxic agents.
The strong genetic interaction between NuA4, Rpd3L, and Hda1 underscores

xviii
the importance of understanding how acetyltransferases and deacetylases act
both in opposition and cooperatively. Finally, bypass suppression of ESA1
enabled the first transcriptomics study of esa1∆ cells, defining Esa1 as a
critical regulator of ribosome biogenesis. Together, the studies presented and
discussed in this thesis highlight the power of bypass suppression and define
key functions and regulatory targets of critical chromatin-modifiers in
Saccharomyces cerevisiae.

1
Introduction Chapter 1
Chapter 1.
Introduction

2
Throughout the course of evolution, the eukaryotic genome evolved into a
highly compacted and organized structure. The eukaryotic genome is packaged into
chromatin, which is composed of the basic nucleosome unit containing 147 bp of
DNA tightly wrapped 1.7 times around a histone octamer. The histone octamer
contains two copies of each of the four canonical histones, H3, H4, H2A, and H2B.
These dynamic chromatin components dictate the accessibility of DNA to crucial
machinery, and therefore have many established cellular roles, including functions in
recombination, DNA damage repair, and transcription [reviewed in (Kornberg and
Lorch 1999; Felsenfeld and Groudine 2003; Allis and Jenuwein 2016)].
The nucleosomes themselves are regulated by two primary mechanisms:
nucleosome positioning and mobilization, catalyzed by ATP-dependent chromatin
remodeling machineries, and post-translational modification (PTM) of histones
[reviewed in (Kouzarides 2007)]. Acetylation is one such key PTM that regulates gene
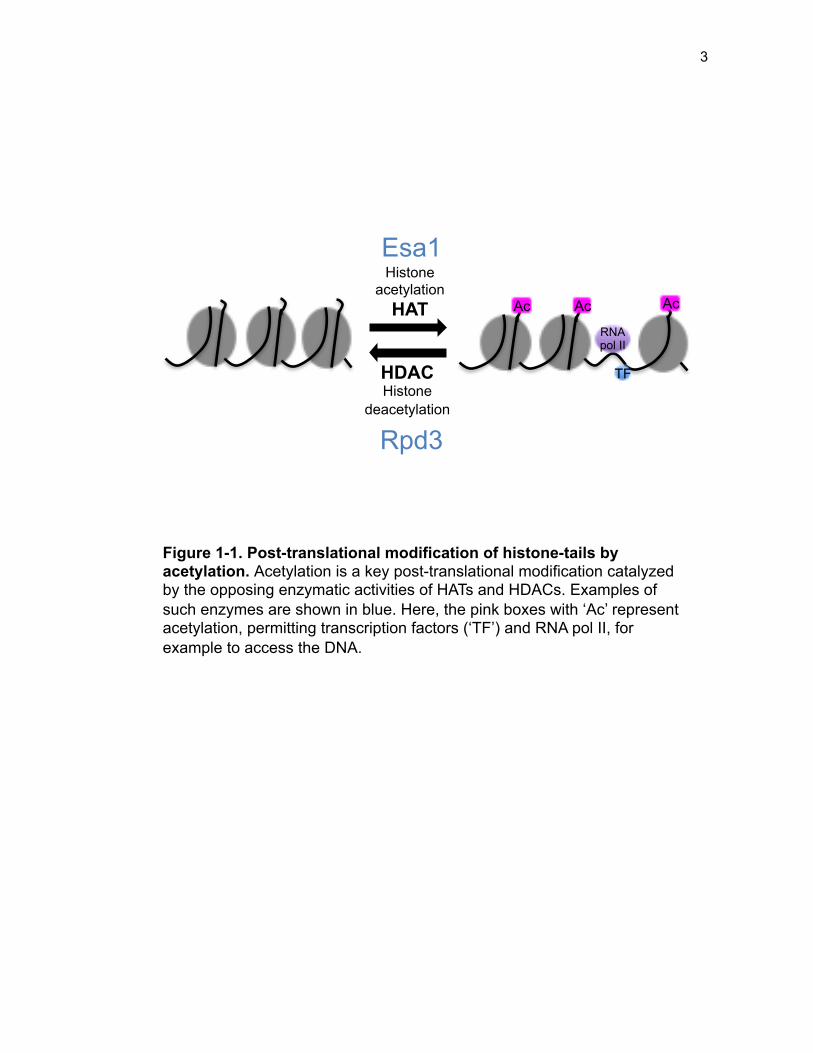
expression by modifying lysine residues on the N-terminal histone tails (Figure 1-1).
Histone/Lysine acetyltransferases (HATs/KATs) and deacetylases
(HDACs/KDACs) dynamically regulate acetylation, catalyzing the transfer or removal
of the acetyl group from acetyl-CoA to the ε-amino group of lysine residues in
histones. Histone acetylation neutralizes the positive charge on lysine residues,
promoting an open chromatin structure, thereby facilitating polymerase and
transcription factor accessibility. The majority of lysine acetylation studies have
focused on histone targets, yet KATs also act on many non-histone substrates (Lin et
al. 2009; Mitchell et al. 2013a; Downey et al. 2015).
Tip60 is the catalytic subunit of the NuA4 acetyltransferase complex. In
humans, Tip60 is one such KAT that is the essential catalytic subunit of a larger
3
Histone acetylation
HAT
HDAC Histone
deacetylation
Esa1
Rpd3
Ac Ac Ac
RNA pol II
TF
Figure 1-1. Post-translational modification of histone-tails by acetylation. Acetylation is a key post-translational modification catalyzed by the opposing enzymatic activities of HATs and HDACs. Examples of such enzymes are shown in blue. Here, the pink boxes with ‘Ac’ represent acetylation, permitting transcription factors (‘TF’) and RNA pol II, for example to access the DNA.

4
macromolecular complex, NuA4. Both Tip60 and the NuA4 holocomplex are deeply
conserved, with Esa1 acting as the catalytic subunit in budding yeast (Doyon et al.
2004; Lafon et al. 2007). Tip60 is an essential MYST-family acetyltransferase and
has several well-established roles in biology and human health, including extensive
roles in cell cycle progression, DNA damage repair, and gene regulation (Doyon and
Côté 2004; Squatrito et al. 2006). Importantly, Tip60 is involved in several human
carcinomas: Tip60 was identified as a haploinsufficient tumor suppressor with roles in
oncogene induced DNA damage response (Gorrini et al. 2007). Interestingly, Tip60 is
both up- and down-regulated in human cancers: it is downregulated in breast (Gorrini
et al. 2007) and colorectal (Mattera et al. 2009) cancers, yet is up-regulated in
prostate cancer (Shiota et al. 2010), along with in several other cancers as
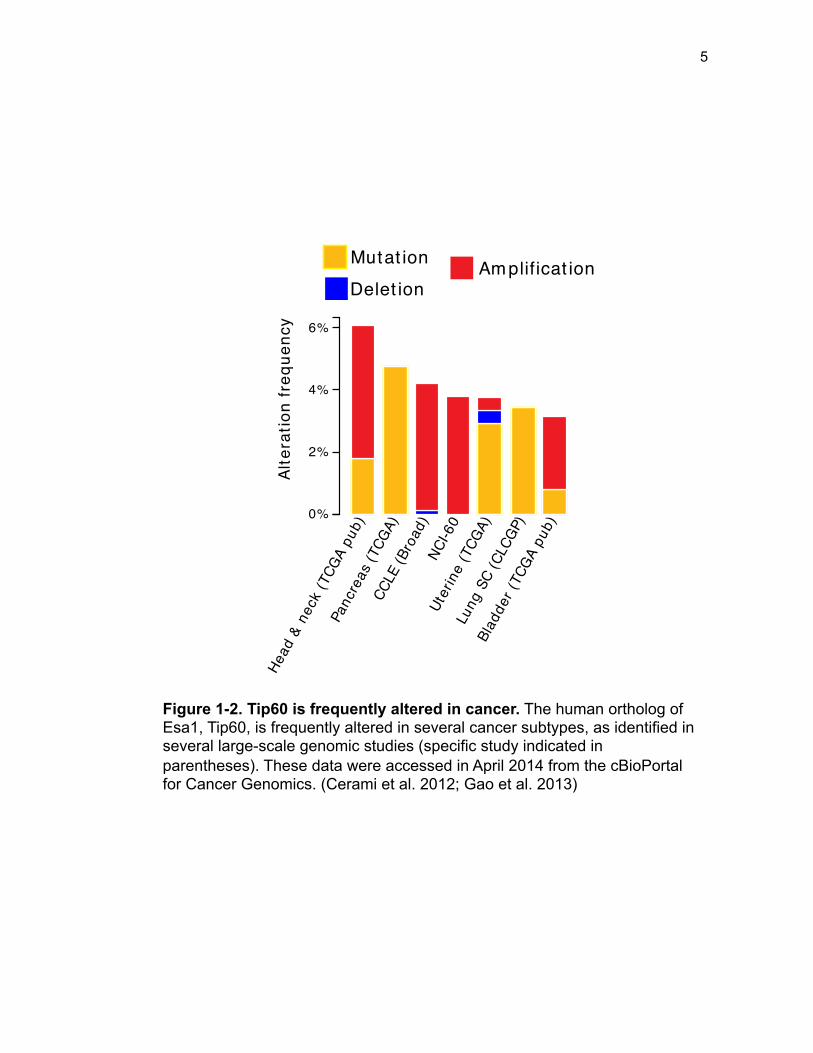
highlighted in large-scale cancer genomic studies [Figure 1-2; (Cerami et al. 2012;
Gao et al. 2013)]. These basic biological roles and disease connections highlight the
importance of tightly controlling the enzymatic activity of Tip60, and gaining a deeper
understanding for how Tip60 is regulated.
Enhancer of Polycomb is broadly conserved. In addition to Tip60, the
NuA4/TIP60 complex contains many other conserved and essential subunits (Doyon
and Côté 2004). As is the case with many enzymatic complexes, defining the function
of noncatalytic subunits is often challenging, despite the potential for crucial
contributions within or beyond the holocomplex. Establishing these functions often
require integrated genetic, genomic, and biochemical analyses. For subunits that are
broadly conserved, these efforts can be aided by the compilation of studies from
many organisms. Enhancer of polycomb (EPC) is one such NuA4/TIP60 subunit,
central to this thesis, which has no known catalytic activity, though it has been
5
Head
& n
eck
(TCG
A pu
b)Pa
ncre
as (T
CGA)
CCLE
(Bro
ad)
NCI-6
0Ut
erin
e (T
CGA)
Lung
SC
(CLC
GP)
Blad
der (
TCGA
pub
)0%
2%
4%
6%
Alte
ratio
n fr
eque
ncy
Mutat ionDelet ion
Am plificat ion
Figure 1-2. Tip60 is frequently altered in cancer. The human ortholog of Esa1, Tip60, is frequently altered in several cancer subtypes, as identified in several large-scale genomic studies (specific study indicated in parentheses). These data were accessed in April 2014 from the cBioPortal for Cancer Genomics. (Cerami et al. 2012; Gao et al. 2013)

6
annotated in more than 65 species (Aken et al. 2016). In this introduction, both key
recent and historical studies are highlighted that together provide a comprehensive
overview of EPC function. Specifically, studies that have contributed to the
understanding of EPC as an individual protein are the focus, rather than detailing the
diverse functions of the NuA4 complex as a whole.
EPC was first characterized as E(Pc) in Drosophila melanogaster, as a “new
enhancer of polycomb” (Sato et al. 1983). E(Pc) mutants did not have homeotic
phenotypes as did the Polycomb group mutants, which have defects in silencing HOX
gene expression (Kassis et al. 2017). Instead E(Pc) mutants acted as dominant
enhancers of Polycomb group mutants in adult flies, indicating unique underlying
genetic interactions; E(Pc)-/- flies however, are themselves embryonic lethal (Sato et
al. 1983; Cheng et al. 1994; Soto et al. 1995). Early phenotypic characterization of
E(Pc) also led to the observation that E(Pc) was a suppressor of position-effect
variegation, a phenotype generally associated with non-histone chromatin proteins
that influence the spread of heterochromatin (Clegg et al. 1998; Sinclair et al. 1998).
This was a timely observation, as several months later, orthologs of E(Pc) were
identified by sequence homology in yeast (Epl1), mammals (EPC1), and C. elegans
(Stankunas et al. 1998), with plant species soon to follow (Springer et al. 2002).
Within two years, yeast Epl1 was identified as an essential subunit in the NuA4
acetyltransferase complex (Galarneau et al. 2000). This early cross-species
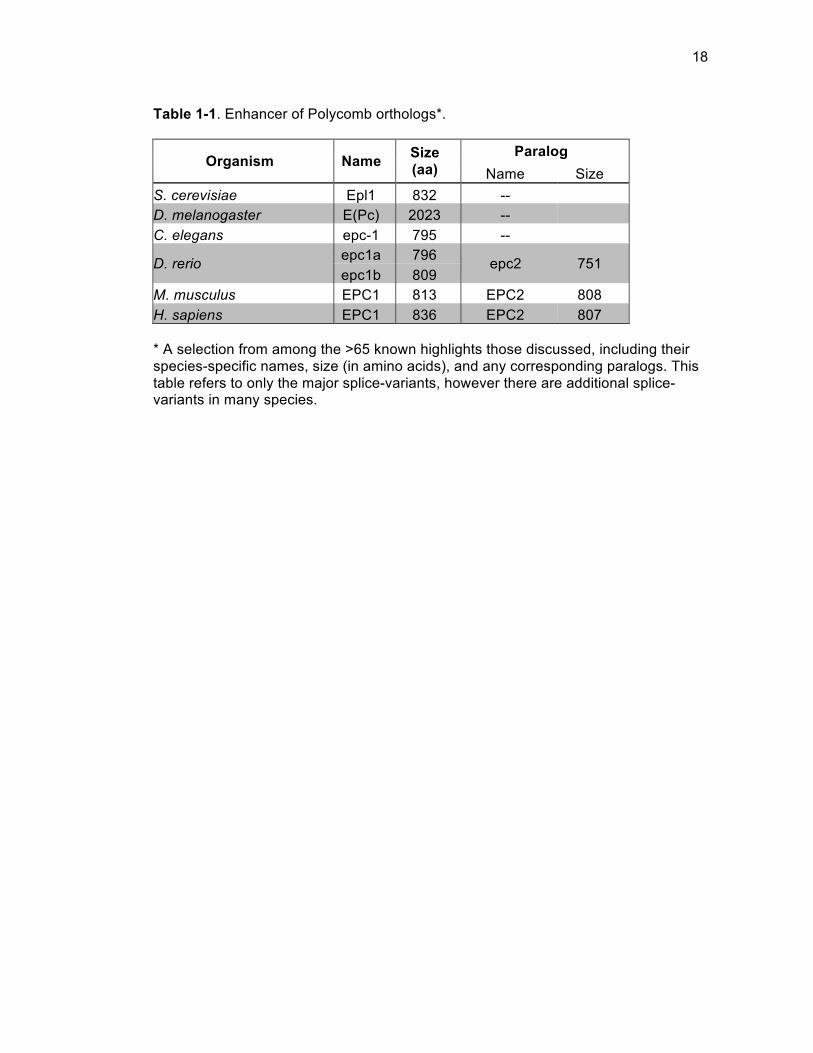
identification (Table 1-1) promoted concurrent multi-organism studies of EPC, and
overall, led to an enhanced understanding of function.
Comparative studies between model organisms promote functional
definition. Whereas the earliest studies of E(Pc) relied on Drosophila phenotypic

7
characterization, a deepened molecular understanding of E(Pc) was gained from
fundamental genetic and biochemical experiments in Saccharomyces cerevisiae.
Similar to Drosophila, yeast Epl1 was found to be essential for viability (Galarneau et
al. 2000), and low-dosage alleles of Epl1 indicated its importance in progression
through the cell cycle, response to DNA damage, histone H4 and H2A acetylation,
gene silencing, and in autophagy (Boudreault et al. 2003; Yi et al. 2012).
Many of Epl1’s functions have been defined based upon domain structure,
dividing Epl1 into a non-essential C-terminus and an essential N-terminus (Figure 1-
3). The C-terminus is quite variable in sequence among species, although it does
serve to tether piccolo-NuA4 subunits to the NuA4 holocomplex and targets the
acetyltransferase, Esa1, to chromatin (Boudreault et al. 2003; Searle et al. 2017). In
contrast, the conserved N-terminus of Epl1, known as the EPcA domain, physically
interacts with the NuA4 subunits Yng2, Eaf6, and the acetyltransferase, Esa1, which
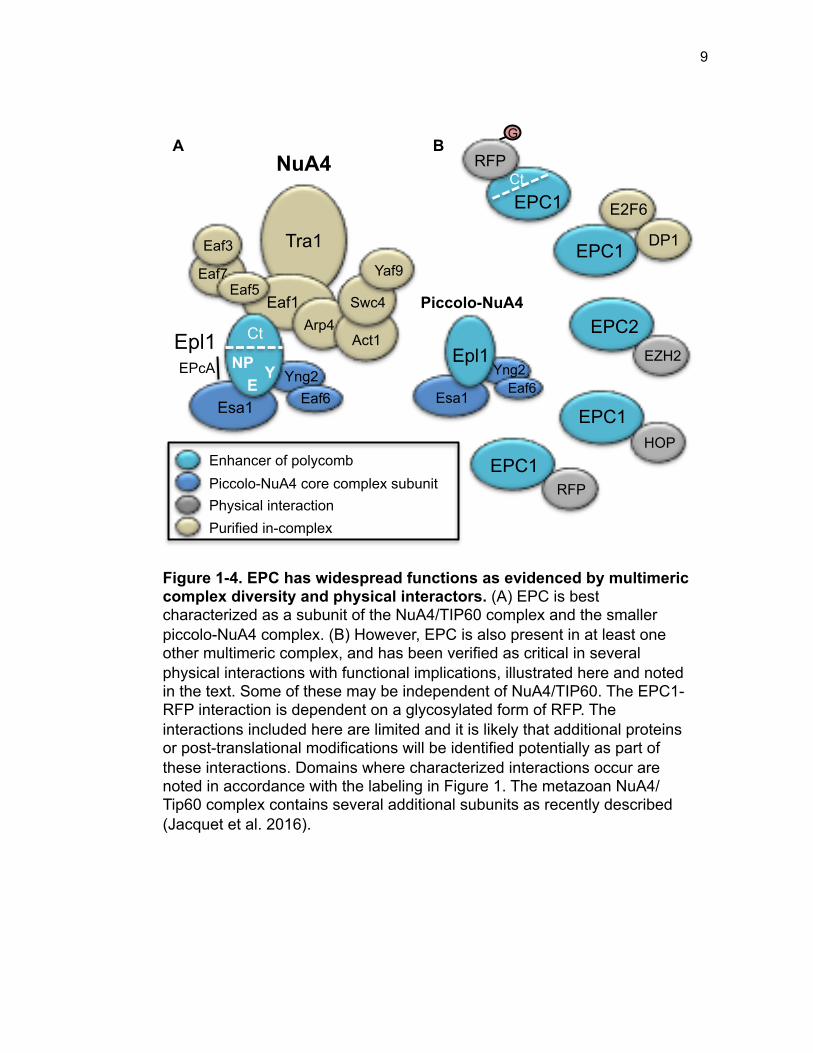
collectively with Epl1 are known as piccolo-NuA4 (Figure 1-4A) (Boudreault et al.
2003; Mitchell et al. 2008; Rossetto et al. 2014). Epl1, through its EPcA region, is
critical for Esa1 acetyltransferase activity, especially toward nucleosomes in vitro, and
further contributes to target specificity (Selleck et al. 2005; Berndsen et al. 2007;
Chittuluru et al. 2011; Huang and Tan 2013; Lalonde et al. 2013; Kuo et al. 2015).
Most recently, the structure of the EPcA domain was solved in complex with the other
piccolo-NuA4 subunits, and the first bypass mutant of EPL1 was identified using
powerful genetic suppression analysis that has been highly useful for the study of
many other critical proteins (Prelich 1999; Hughes 2016; van Leeuwen et al. 2017).
These suppression studies, building on earlier work with non-null alleles (Lin et al.
2008), implicated Epl1 as being a critical Esa1 co-factor, and highlighted the

8
N C
1 833
EPcA
NP E Y 50-125 149-200 250-375
C-terminus (Ct)
485
Epl1
Nucleosome core particle
Esa1 Yng2/Eaf6
Figure 1-3. Epl1 domain structure in S. cerevisiae. Epl1 contains the essential and conserved EpCA domain, broken down into 3 sub-domains, each named for their initial characterization based on physical interaction [as modified from (Boudreault et al. 2003; Chittuluru et al. 2011; Searle et al. 2017; Selleck et al. 2005)]. The C-terminus is highly variable among species, and accounts for the bulk of the differences in size of orthologs as listed in Table 1. Some metazoans do have shorter stretches of conserved residues within the C-terminus, referred to as EPc-B and EPc-C (Stankunas et al. 1998).
9
Tra1
Eaf1
Epl1
Esa1
Y NP EPcA
Ct
NuA4
E Eaf6
Yng2
Piccolo-NuA4
Epl1
Esa1
Yng2 Eaf6
Ct
EPC1
G
EPC1
E2F6
DP1
EPC2 EZH2
EPC1 HOP
EPC1 RFP
Enhancer of polycomb Piccolo-NuA4 core complex subunit Physical interaction Purified in-complex
Eaf3
Eaf7 Eaf5
Yaf9
Swc4 Arp4
Act1
RFP A B
Figure 1-4. EPC has widespread functions as evidenced by multimeric complex diversity and physical interactors. (A) EPC is best characterized as a subunit of the NuA4/TIP60 complex and the smaller piccolo-NuA4 complex. (B) However, EPC is also present in at least one other multimeric complex, and has been verified as critical in several physical interactions with functional implications, illustrated here and noted in the text. Some of these may be independent of NuA4/TIP60. The EPC1-RFP interaction is dependent on a glycosylated form of RFP. The interactions included here are limited and it is likely that additional proteins or post-translational modifications will be identified potentially as part of these interactions. Domains where characterized interactions occur are noted in accordance with the labeling in Figure 1. The metazoan NuA4/Tip60 complex contains several additional subunits as recently described (Jacquet et al. 2016).

10
importance of the physical Epl1-Esa1 interactions for acetyltransferase activity (Xu et
al. 2016; Searle et al. 2017).
Whereas yeast studies shed light on the basic cellular function of Epl1, and
demonstrated its importance in chromatin regulation, studies in multicellular
organisms allowed for expansion of these seminal results to understand how Epl1 is
involved in other cellular processes. Early studies of E(Pc) in Drosophila illustrated its
critical role in chromatin regulation and demonstrated that E(Pc) is also involved in
genomic imprint maintenance in Drosophila, likely through its role in heterochromatin
maintenance (Joanis and Lloyd 2002). E(Pc) has been further highlighted for its
important interactions (Table 1-2) with various genes and proteins involved in
apoptosis and chromatin regulation, such as with ISWI (Imitation SWI), His1 (Histone
H1), and Polycomb group genes (Ali and Bender 2004; Arancio et al. 2010; Fullard
and Baker 2015; Kavi et al. 2015). As in yeast, E(Pc) is important in the cell cycle in
Drosophila, where is it required during development for mitotic exit during the
transition to a post-mitotic state (Flegel et al. 2016). Additionally, there is evidence to
suggest that E(Pc) is also important in DNA damage repair, whereby mutation
increases the rate of homologous recombination (Holmes et al. 2006).
Developmental work in Drosophila revealed E(Pc)’s involvement in
differentiation and stem cell fate determination. E(Pc) is downregulated upon
activation of the JNK (Jun amino-terminal kinase) signaling pathway in imaginal disc
cells undergoing regeneration. This promotes wound healing, giving rise to most of
the major structures in the adult fly (Lee et al. 2005). In multipotent hematopoietic
progenitors, E(Pc) again acts downstream of JNK, here in combination with FoxO
(Forkhead box protein O transcription factor), to trigger cellular differentiation (Owusu-

11
Ansah and Banerjee 2009). E(Pc) was also identified as a regulator of cell fate and
differentiation in intestinal stem cells and germ cells in the testes, respectively (Zeng
et al. 2015; Feng et al. 2017). Related MAP-kinase signaling has also been linked to
heterochromatin formation in yeast, providing additional relevance to the EPC-JNK
relationship (Stone and Pillus 1996; Mazor and Kupiec 2009). Together, these
examples highlight the importance of the EPC-JNK regulation axis in fly development.
In addition to pioneering work in Drosophila and yeast, recent progress has
been made in studies of EPC in additional metazoans (Table 1-1), including C.
elegans and D. rerio. These studies began to hint at roles for EPC in oncogenesis,
perhaps not surprisingly, given its central role in chromatin regulation and in stem cell
identity. Knockdown of epc1 was found to decrease lifespan in a daf-16-dependent
manner in C. elegans, and was found to be a Ras antagonist in the regulation of cell
division and cell-fate determination (Ceol and Horvitz 2004; Kim and Sun 2007).
Additionally, analogous to Drosophila studies, epc2 was found to regulate
hematopoietic development in zebrafish, specifically in the development of primitive
erythroid cells. In this case knockdown of epc2 was consistent with a role in
mesodermal precursor differentiation in blood development via upregulation of scl,
gata1, and βe3-globin (Huang et al. 2013). These studies add further support for EPC
as a critical regulator of cellular processes, from early development through
subsequent aging and development of disease.
Human EPC was first purified in MCF7 and HeLa cell lines as a subunit of the
NuA4/TIP60 complex. Both splice variants and paralogs EPC1 and EPC2, were
concurrently identified (Doyon et al. 2004). EPC1 was found to tether MBTD1
(Malignant Brain Tumor Domain Containing 1) to the human TIP60 complex,

12
promoting TIP60-driven repair of DNA double stranded breaks by homologous
recombination (Jacquet et al. 2016).
Studies of EPC1 outside the NuA4/TIP60 complex in mammals have pointed
to roles for it and EPC2 that are independent of their canonical roles as NuA4
subunits. Beyond NuA4, EPC1/2 interacts with other proteins, supporting the
presence of additional novel functions in mice and humans (Figure 1-4B). For
example, a unique interaction between EPC1 and RFP (RET Finger Protein) was
identified in mice. Specifically, a glycosylated form of RFP was found to interact with
the C-terminus of EPC1 in repressive activities, whereas the EPcA domain of EPC1
was found to have transcriptional activating activities (Shimono et al. 2000; Tezel et
al. 2002). EPC1 was also identified as an E2F6 (E2F Transcription Factor 6) binding
partner, and furthermore was found to exist in a distinct stable complex in vitro and in
vivo with E2F6 and DP1. This complex was found to exist in proliferating normal and
transformed human cells and to co-elute with Sin3B to promote repressive activities
(Attwooll et al. 2005). Finally, the paralog EPC2 was shown to interact with EZH2 in
human colorectal cancer cells, with an involvement in transcriptional regulation (Guil
et al. 2012).
The diverse interactions of EPC1 and EPC2 begin to point toward specialized
roles for each paralog, hinting at cell-type and developmental-stage specific EPC-
containing complexes. This observation may be particularly noteworthy especially
considering the translational importance of EPC1 and EPC2 that has begun to be
defined in recent years.
From basic function to translational significance. With its well-established,
broadly critical genetic roles, it is not surprising that studies in more recent years have

13
also shed light on the clinical importance of EPC1 and EPC2, both in patient samples
and in murine models of human disease. EPC1 and EPC2 have been primarily
implicated in basic cancer biology and metastasis, and have also been found to
function in skeletal muscle differentiation. Many of these examples highlight EPC1
and EPC2 apart from NuA4/Tip60, and underscore the importance of EPC’s diverse
interacting partners.
EPC1 has been mechanistically implicated in metastasis. For example, it was
found that EPC1 activates E2F1 (E2F Transcription Factor 1), leading to the
upregulation of anti-apoptotic survival genes. This triggers a metastasis-related gene
signature that is prognostic of poor patient outcome. Cisplatin treatment of cancer cell
lines, such as SK-Mel-147 melanoma cells resulted in upregulation of EPC1, further
pointing towards EPC1 enabling survival of cancer cells. Accordingly, knockdown of
EPC1 led to increased DNA damage sensitivity and apoptosis in an E2F1 dependent
mechanism (Wang et al. 2016).
EPC is genetically altered in several cancers, including both hematological
cancers and solid tumors. Basic findings in zebrafish, illustrating a role for EPC in
blood development (Huang et al. 2013), may lead to further insights for multiple roles
of EPC in leukemia and other hematological conditions. For example, EPC1
expression is downregulated in leukemia cells as compared to its expression in
hematopoietic progenitor cells, and has been found as a breakpoint site in adults with
T-cell leukemia (Nakahata et al. 2009; Prasad et al. 2014). Both EPC1 and EPC2 are
required for acute myeloid leukemia cell proliferation; knockdown of EPC1 and/or
EPC2 leads to accumulation of MYC in acute myeloid leukemia cells, contributing to
selective apoptosis (Huang et al. 2014).

14
EPC1 is also a reported site of breakpoints in solid tumors, such as in
endometrial stromal sarcoma, though these EPC1-translocations account for a
minority of reported cases (Micci et al. 2006; Chiang et al. 2011). EPC is also altered
in sequence and in copy number, such as in early sporadic pancreatic ductal
adenocarcinoma, where EPC1, and to a lesser frequency, EPC2 are mutated and
have a loss of heterozygosity (Biankin et al. 2012). Finally, a site of common genetic
variation within the second intron of EPC2 was reported to elicit differential response
to gemcitabine, a common chemotherapeutic agent (Jarjanazi et al. 2008). This
points beyond the demonstrated importance of EPC in cancer biology, to triggering a
differential response to cancer therapy.
Discussion above illustrated the importance of EPC in cellular differentiation
and development. This is also highlighted in EPC1’s role in spermatogenesis in mice
(Dong et al. 2017), as well as in applied models of skeletal muscle differentiation.
EPC1 regulates skeletal muscle differentiation through interaction with HOP
(Homeodomain Only Protein) and also recruits Serum Response Factor (SRF) and
p300, in a manner that appears to be independent of NuA4/Tip60. Indeed, Tip60 is
undetectable in various muscle cell lines and tissues (Kee et al. 2007; Kim et al.
2009). The positive regulation of skeletal muscle differentiation by the EPC1-HOP
interaction is opposed by an interaction between EPC1 and RFP, whereby RFP
blocks the skeletal muscle differentiation that is induced by the collaboration of EPC1
and HOP (Kee et al. 2012). Knowledge of this role of EPC1 in muscle differentiation
may become directly applicable in a clinical setting. In a model of arterial injury, it was
found that local delivery of EPC1 reduced formation of scar tissue in smooth muscle
by promoting vascular smooth muscle cell differentiation (Joung et al. 2012).

15
Whereas translational studies of EPC are, to date, more limited than those of Tip60,
those discussed here underscore the role of EPC as a critical genomic regulator
perhaps ultimately bridging basic cellular functions to clinical significance.
Bypass suppression is a powerful genetic tool. The study of essential
genes creates additional challenges, as these critical genes cannot simply be deleted
from the genome without a loss of viability. Such studies, therefore, frequently rely on
hypomorphic alleles where key features, such as enzymatic activity are inhibited or
diminished conditionally, such as with elevated temperatures. Seminal studies of
Esa1 have been performed with such alleles, and led to a comprehensive
understanding of its functions (Clarke et al. 1999; Bird et al. 2002; Clarke et al. 2006).
However, such studies have limitations, in that they do not truly represent a complete
loss of gene function from the cell.
Another powerful tool to study essential genes is bypass suppression,
whereby the essential gene of interest is deleted in combination with an additional
gene, usually in a parallel pathway [reviewed in (Prelich 1999)]. The second deletion
promotes bypassing the requirement for the essential gene, often by counteracting a
key feature of the essential gene. Bypass suppression has led to key insights about
many crucial proteins (van Leeuwen et al. 2017). For example, deletion of SML1
bypasses the essential requirement for MEC1 and RAD53, and revealed key insights
into dNTP synthesis (Zhao et al. 1998). Most pertinent to this thesis however, was the
study that identified the first bypass suppressor within the NuA4 acetyltransferase
complex, and subsequently led to a greater understanding of essential activities
(Torres-Machorro and Pillus 2014).

16
Promoting a balanced acetylation state allows for the bypass of ESA1.
Previously, studies identified that the essential requirement for the conserved NuA4
HAT, Esa1, could be bypassed by loss of Rpd3L complex activity, through loss of
SDS3 (Torres-Machorro and Pillus 2014). In brief, though viable, esa1∆ sds3∆ cells
were not robust, displaying strong temperature and DNA damage sensitivity, along
with a G2/M cell cycle delay, and low global levels of H4 acetylation. It was
speculated that esa1∆ sds3∆ cells had impaired fitness because of their imbalanced
acetylation state, including one in which H4 acetylation levels were low relative to H3
acetylation, that remained relatively normal.
Various experiments “balancing” H4 acetylation relative to H3 improved the
fitness of esa1∆ sds3∆ cells. Mimicking H4 acetylation by mutating the acetylatable
lysines of H4 to glutamine improved esa1∆ sds3∆ fitness at high temperatures and in
the presence of DNA damage. Supporting the importance of an H3 and H4
acetylation balance, mutating key H3 lysine residues to arginine, mimicking an
unmodified lysine, resulted in increased fitness. Additionally, removal of key HDACs
improved the fitness of esa1∆ sds3∆ cells, perhaps through a similar mechanism as
the histone mutant strains. Specifically, deletion of HST1, HDA1, or SIR2 in esa1∆
sds3∆ cells each uniquely suppressed esa1∆ sds3∆ phenotypes. For example, esa1∆
sds3∆ hda1∆ strongly suppressed temperature sensitivity, cell cycle defects, and
rDNA silencing defects. Because manipulation of cellular acetylation levels
differentially affected the fitness of the bypass strain, it was proposed that the
essential function of Esa1 lies in its ability to promote balanced acetylation in the cell,
that were suggested, is likely to also include non-histone substrates (Torres-Machorro
and Pillus 2014).

17
Bypass suppression to define key functions of chromatin modifying
proteins. In this thesis, bypass suppression facilitated studies of key chromatin
proteins. Regulation of chromatin involves the careful coordination of many different
enzymes, regulatory proteins, and structural subunits. Oftentimes, due to their
essential roles, chromatin proteins are challenging to study. In Chapter 2, bypass
suppression enabled the first in vivo comprehensive characterization of the essential
NuA4 subunit, Epl1. Through biochemical, genomic, and mutational analyses, Epl1
was newly defined as a critical co-factor for the NuA4 catalytic subunit, Esa1. Chapter
3 utilizes the defined EPL1 bypass mutant to characterize important interactions
between NuA4 and HDACs, identifying Hda1 as crucial for the opposition of NuA4
activity, acting both in tandem and independently of the Rpd3L HDAC. Finally,
Chapter 4 utilizes the ESA1 bypass mutant to define the complete transcriptional
regulome of Esa1, and identifies Esa1 as a crucial transcriptional and functional
regulator of ribosome biogenesis. These findings are summarized and considered in
Chapter 5, with implications of these studies discussed and key areas of future
research proposed.
18
Table 1-1. Enhancer of Polycomb orthologs*.
Organism Name Size (aa)
Paralog Name Size
S. cerevisiae Epl1 832 -- D. melanogaster E(Pc) 2023 -- C. elegans epc-1 795 --
D. rerio epc1a 796 epc2 751 epc1b 809
M. musculus EPC1 813 EPC2 808 H. sapiens EPC1 836 EPC2 807 * A selection from among the >65 known highlights those discussed, including their species-specific names, size (in amino acids), and any corresponding paralogs. This table refers to only the major splice-variants, however there are additional splice-variants in many species.

19
Table 1-2. EPC interactors and effectors referenced in text**
Interactor Species Description
Iswi D. melanogaster ATPase member of chromatin remodeling complexes
His1 D. melanogaster Linker Histone H1
JNK D. melanogaster Jun amino-terminal kinase, a mitogen activated protein (MAP) kinase
FoxO D. melanogaster Forkhead box protein O transcription factor
RAS C. elegans Small GTP-ase signaling protein, with established oncogenic properties in mammals
scl D. rerio Transcription factor critical for hematopoetic development
gata1 D. rerio Erythroid-specific transcription factor
βe3-globin D. rerio hemoglobin beta embryonic-3,
RFP H. sapiens/ M. musculus
RET finger protein in the large B-box RING finger protein family
E2F6 H. sapiens E2F transcription factor, critical in cell cycle regulation
DP1 H. sapiens Transcription factor that heterodimerizes with E2F proteins to stimulate their transcription
Sin3B H. sapiens SIN3 transcriptional regulator
EZH2 H. sapiens Enhancer of Zeste homolog 2, histone methyltransferase activity
HOP H. sapiens/ M. musculus / R. norvegicus
Hsp70-Hsp90 organizing protein, a co-chaperone in the stress-inducible (STI) family of proteins
SRF H. sapiens/ M. musculus
Serum response factor, a master regulator transcription factor required for many processes, including cardiac development
p300 M. musculus/ R. norvegicus
Transcriptional co-activator with a histone acetyltransferase domain, bromodomain, and a PHD finger domain
** Additional interactions have been reported via genome-wide screens and other methods. This list and review highlight verified interactions of functional relevance, including the species for which the interaction was reported.

20
Acknowledgements This chapter, in part, contains material in-press for publication as it
appears in Searle NE, Pillus L. 2017. Critical genomic regulation mediated by
Enhancer of Polycomb. Current Genetics. In-press. The dissertation author
was the primary investigator and first author of this paper.

21
References Aken BL, Ayling S, Barrell D, Clarke L, Curwen V, Fairley S, Banet JF, Billis K,
Giron CG, Hourlier T, Howe K, Kahari A, Kokocinski F, Martin FJ, Murphy DN, Nag R, Ruffier M, Schuster M, Tang YA, Vogel JH, White S, Zadissa A, Flicek P, Searle SMJ. 2016. The Ensembl gene annotation system. Database-Oxford.
Ali JY, Bender W. 2004. Cross-regulation among the polycomb group genes in Drosophila melanogaster. Mol Cell Biol 24: 7737-7747.
Allis CD, Jenuwein T. 2016. The molecular hallmarks of epigenetic control. Nat Rev Genet 17: 487-500.
Arancio W, Onorati MC, Burgio G, Collesano M, Ingrassia AM, Genovese SI, Fanto M, Corona DF. 2010. The nucleosome remodeling factor ISWI functionally interacts with an evolutionarily conserved network of cellular factors. Genetics 185: 129-140.
Attwooll C, Oddi S, Cartwright P, Prosperini E, Agger K, Steensgaard P, Wagener C, Sardet C, Moroni MC, Helin K. 2005. A novel repressive E2F6 complex containing the polycomb group protein, EPC1, that interacts with EZH2 in a proliferation-specific manner. J Biol Chem 280: 1199-1208.
Berndsen CE, Selleck W, McBryant SJ, Hansen JC, Tan S, Denu JM. 2007. Nucleosome recognition by the Piccolo NuA4 histone acetyltransferase complex. Biochemistry 46: 2091-2099.
Biankin AV Waddell N Kassahn KS Gingras MC Muthuswamy LB Johns AL Miller DK Wilson PJ Patch AM Wu J Chang DK Cowley MJ Gardiner BB Song S Harliwong I Idrisoglu S Nourse C Nourbakhsh E Manning S Wani S Gongora M Pajic M Scarlett CJ Gill AJ Pinho AV Rooman I Anderson M Holmes O Leonard C Taylor D Wood S Xu Q Nones K Fink JL Christ A Bruxner T Cloonan N Kolle G Newell F Pinese M Mead RS Humphris JL Kaplan W Jones MD Colvin EK Nagrial AM Humphrey ES Chou A Chin VT Chantrill LA Mawson A Samra JS Kench JG Lovell JA Daly RJ Merrett ND Toon C Epari K Nguyen NQ Barbour A Zeps N Australian Pancreatic Cancer Genome I Kakkar N Zhao F Wu YQ Wang M Muzny DM Fisher WE Brunicardi FC Hodges SE Reid JG Drummond J Chang K Han Y Lewis LR Dinh H Buhay CJ Beck T Timms L Sam M Begley K Brown A Pai D Panchal A Buchner N De Borja R Denroche RE Yung CK Serra S Onetto N Mukhopadhyay D Tsao MS Shaw PA Petersen GM Gallinger S Hruban RH Maitra A Iacobuzio-Donahue CA Schulick RD Wolfgang CL Morgan RA Lawlor

22
RT Capelli P Corbo V Scardoni M Tortora G Tempero MA Mann KM Jenkins NA Perez-Mancera PA Adams DJ Largaespada DA Wessels LF Rust AG Stein LD Tuveson DA Copeland NG Musgrove EA Scarpa A Eshleman JR Hudson TJ Sutherland RL Wheeler DA Pearson JV McPherson JD Gibbs RA, Grimmond SM. 2012. Pancreatic cancer genomes reveal aberrations in axon guidance pathway genes. Nature 491: 399-405.
Bird AW, Yu DY, Pray-Grant MG, Qiu Q, Harmon KE, Megee PC, Grant PA, Smith MM, Christman MF. 2002. Acetylation of histone H4 by Esa1 is required for DNA double-strand break repair. Nature 419: 411-415.
Boudreault AA, Cronier D, Selleck W, Lacoste N, Utley RT, Allard S, Savard J, Lane WS, Tan S, Côté J. 2003. Yeast enhancer of polycomb defines global Esa1-dependent acetylation of chromatin. Genes Dev 17: 1415-1428.
Ceol CJ, Horvitz HR. 2004. A new class of C. elegans synMuv genes implicates a Tip60/NuA4-like HAT complex as a negative regulator of Ras signaling. Dev Cell 6: 563-576.
Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, Jacobsen A, Byrne CJ, Heuer ML, Larsson E, Antipin Y, Reva B, Goldberg AP, Sander C, Schultz N. 2012. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov 2: 401-404.
Cheng NN, Sinclair DA, Campbell RB, Brock HW. 1994. Interactions of polyhomeotic with Polycomb group genes of Drosophila melanogaster. Genetics 138: 1151-1162.
Chiang S, Ali R, Melnyk N, McAlpine JN, Huntsman DG, Gilks CB, Lee CH, Oliva E. 2011. Frequency of known gene rearrangements in endometrial stromal tumors. Am J Surg Pathol 35: 1364-1372.
Chittuluru JR, Chaban Y, Monnet-Saksouk J, Carrozza MJ, Sapountzi V, Selleck W, Huang J, Utley RT, Cramet M, Allard S, Cai G, Workman JL, Fried MG, Tan S, Côté J, Asturias FJ. 2011. Structure and nucleosome interaction of the yeast NuA4 and Piccolo-NuA4 histone acetyltransferase complexes. Nat Struct Mol Biol 18: 1196-1203.
Clarke AS, Lowell JE, Jacobson SJ, Pillus L. 1999. Esa1p is an essential histone acetyltransferase required for cell cycle progression. Molecular and Cellular Biology 19: 2515-2526.

23
Clarke AS, Samal E, Pillus L. 2006. Distinct roles for the essential MYST family HAT Esa1p in transcriptional silencing. Mol Biol Cell 17: 1744-1757.
Clegg NJ, Honda BM, Whitehead IP, Grigliatti TA, Wakimoto B, Brock HW, Lloyd VK, Sinclair DA. 1998. Suppressors of position-effect variegation in Drosophila melanogaster affect expression of the heterochromatic gene light in the absence of a chromosome rearrangement. Genome 41: 495-503.
Dong Y, Isono KI, Ohbo K, Endo TA, Ohara O, Maekawa M, Toyama Y, Ito C, Toshimori K, Helin K, Ogonuki N, Inoue K, Ogura A, Yamagata K, Kitabayashi I, Koseki H. 2017. EPC1/TIP60-mediated histone acetylation facilitates spermiogenesis in mice. Mol Cell Biol.
Downey M, Johnson JR, Davey NE, Newton BW, Johnson TL, Galaang S, Seller CA, Krogan N, Toczyski DP. 2015. Acetylome profiling reveals overlap in the regulation of diverse processes by sirtuins, gcn5, and esa1. Mol Cell Proteomics 14: 162-176.
Doyon Y, Côté J. 2004. The highly conserved and multifunctional NuA4 HAT complex. Curr Opin Genet Dev 14: 147-154.
Doyon Y, Selleck W, Lane WS, Tan S, Côté J. 2004. Structural and functional conservation of the NuA4 histone acetyltransferase complex from yeast to humans. Mol Cell Biol 24: 1884-1896.
Felsenfeld G, Groudine M. 2003. Controlling the double helix. Nature 421: 448-453.
Feng L, Shi Z, Chen X. 2017. Enhancer of polycomb coordinates multiple signaling pathways to promote both cyst and germline stem cell differentiation in the Drosophila adult testis. PLoS Genet 13: e1006571.
Flegel K, Grushko O, Bolin K, Griggs E, Buttitta L. 2016. Roles for the Histone Modifying and Exchange Complex NuA4 in Cell Cycle Progression in Drosophila melanogaster. Genetics 203: 1265-1281.
Fullard JF, Baker NE. 2015. Signaling by the engulfment receptor draper: a screen in Drosophila melanogaster implicates cytoskeletal regulators, Jun N-terminal Kinase, and Yorkie. Genetics 199: 117-134.
Galarneau L, Nourani A, Boudreault AA, Zhang Y, Heliot L, Allard S, Savard J, Lane WS, Stillman DJ, Côté J. 2000. Multiple links between the NuA4

24
histone acetyltransferase complex and epigenetic control of transcription. Mol Cell 5: 927-937.
Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, Sun Y, Jacobsen A, Sinha R, Larsson E, Cerami E, Sander C, Schultz N. 2013. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal 6: pl1.
Gorrini C, Squatrito M, Luise C, Syed N, Perna D, Wark L, Martinato F, Sardella D, Verrecchia A, Bennett S, Confalonieri S, Cesaroni M, Marchesi F, Gasco M, Scanziani E, Capra M, Mai S, Nuciforo P, Crook T, Lough J, Amati B. 2007. Tip60 is a haplo-insufficient tumour suppressor required for an oncogene-induced DNA damage response. Nature 448: 1063-1067.
Guil S, Soler M, Portela A, Carrere J, Fonalleras E, Gomez A, Villanueva A, Esteller M. 2012. Intronic RNAs mediate EZH2 regulation of epigenetic targets. Nat Struct Mol Biol 19: 664-670.
Holmes AM, Weedmark KA, Gloor GB. 2006. Mutations in the extra sex combs and Enhancer of Polycomb genes increase homologous recombination in somatic cells of Drosophila melanogaster. Genetics 172: 2367-2377.
Huang HT, Kathrein KL, Barton A, Gitlin Z, Huang YH, Ward TP, Hofmann O, Dibiase A, Song A, Tyekucheva S, Hide W, Zhou Y, Zon LI. 2013. A network of epigenetic regulators guides developmental haematopoiesis in vivo. Nat Cell Biol 15: 1516-1525.
Huang J, Tan S. 2013. Piccolo NuA4-catalyzed acetylation of nucleosomal histones: critical roles of an Esa1 Tudor/chromo barrel loop and an Epl1 enhancer of polycomb A (EPcA) basic region. Mol Cell Biol 33: 159-169.
Huang X, Spencer GJ, Lynch JT, Ciceri F, Somerville TD, Somervaille TC. 2014. Enhancers of Polycomb EPC1 and EPC2 sustain the oncogenic potential of MLL leukemia stem cells. Leukemia 28: 1081-1091.
Hughes D. 2016. Using the power of genetic suppressors to probe the essential functions of RNase E. Curr Genet 62: 53-57.
Jacquet K, Fradet-Turcotte A, Avvakumov N, Lambert JP, Roques C, Pandita RK, Paquet E, Herst P, Gingras AC, Pandita TK, Legube G, Doyon Y, Durocher D, Côté J. 2016. The TIP60 Complex Regulates Bivalent

25
Chromatin Recognition by 53BP1 through Direct H4K20me Binding and H2AK15 Acetylation. Mol Cell 62: 409-421.
Jarjanazi H, Kiefer J, Savas S, Briollais L, Tuzmen S, Pabalan N, Ibrahim-Zada I, Mousses S, Ozcelik H. 2008. Discovery of genetic profiles impacting response to chemotherapy: application to gemcitabine. Hum Mutat 29: 461-467.
Joanis V, Lloyd VK. 2002. Genomic imprinting in Drosophila is maintained by the products of Suppressor of variegation and trithorax group, but not Polycomb group, genes. Mol Genet Genomics 268: 103-112.
Joung H, Kwon JS, Kim JR, Shin S, Kang W, Ahn Y, Kook H, Kee HJ. 2012. Enhancer of polycomb1 lessens neointima formation by potentiation of myocardin-induced smooth muscle differentiation. Atherosclerosis 222: 84-91.
Kassis JA, Kennison JA, Tamkun JW. 2017. Polycomb and Trithorax Group Genes in Drosophila. Genetics 206: 1699-1725.
Kavi H, Lu X, Xu N, Bartholdy BA, Vershilova E, Skoultchi AI, Fyodorov DV. 2015. A genetic screen and transcript profiling reveal a shared regulatory program for Drosophila linker histone H1 and chromatin remodeler CHD1. G3 (Bethesda) 5: 677-687.
Kee HJ, Kim JR, Joung H, Choe N, Lee SE, Eom GH, Kim JC, Geyer SH, Jijiwa M, Kato T, Kawai K, Weninger WJ, Seo SB, Nam KI, Jeong MH, Takahashi M, Kook H. 2012. Ret finger protein inhibits muscle differentiation by modulating serum response factor and enhancer of polycomb1. Cell Death Differ 19: 121-131.
Kee HJ, Kim JR, Nam KI, Park HY, Shin S, Kim JC, Shimono Y, Takahashi M, Jeong MH, Kim N, Kim KK, Kook H. 2007. Enhancer of polycomb1, a novel homeodomain only protein-binding partner, induces skeletal muscle differentiation. J Biol Chem 282: 7700-7709.
Kim JR, Kee HJ, Kim JY, Joung H, Nam KI, Eom GH, Choe N, Kim HS, Kim JC, Kook H, Seo SB, Kook H. 2009. Enhancer of polycomb1 acts on serum response factor to regulate skeletal muscle differentiation. J Biol Chem 284: 16308-16316.
Kim Y, Sun H. 2007. Functional genomic approach to identify novel genes involved in the regulation of oxidative stress resistance and animal lifespan. Aging Cell 6: 489-503.

26
Kornberg RD, Lorch Y. 1999. Twenty-five years of the nucleosome, fundamental particle of the eukaryote chromosome. Cell 98: 285-294.
Kouzarides T. 2007. Chromatin modifications and their function. Cell 128: 693-705.
Kuo YM, Henry RA, Tan S, Côté J, Andrews AJ. 2015. Site specificity analysis of Piccolo NuA4-mediated acetylation for different histone complexes. Biochem J 472: 239-248.
Lafon A, Chang CS, Scott EM, Jacobson SJ, Pillus L. 2007. MYST opportunities for growth control: yeast genes illuminate human cancer gene functions. Oncogene 26: 5373-5384.
Lalonde ME, Avvakumov N, Glass KC, Joncas FH, Saksouk N, Holliday M, Paquet E, Yan K, Tong Q, Klein BJ, Tan S, Yang XJ, Kutateladze TG, Côté J. 2013. Exchange of associated factors directs a switch in HBO1 acetyltransferase histone tail specificity. Genes Dev 27: 2009-2024.
Lee N, Maurange C, Ringrose L, Paro R. 2005. Suppression of Polycomb group proteins by JNK signalling induces transdetermination in Drosophila imaginal discs. Nature 438: 234-237.
Lin YY, Lu JY, Zhang J, Walter W, Dang W, Wan J, Tao SC, Qian J, Zhao Y, Boeke JD, Berger SL, Zhu H. 2009. Protein acetylation microarray reveals that NuA4 controls key metabolic target regulating gluconeogenesis. Cell 136: 1073-1084.
Lin YY, Qi Y, Lu JY, Pan X, Yuan DS, Zhao Y, Bader JS, Boeke JD. 2008. A comprehensive synthetic genetic interaction network governing yeast histone acetylation and deacetylation. Genes Dev 22: 2062-2074.
Mattera L, Escaffit F, Pillaire MJ, Selves J, Tyteca S, Hoffmann JS, Gourraud PA, Chevillard-Briet M, Cazaux C, Trouche D. 2009. The p400/Tip60 ratio is critical for colorectal cancer cell proliferation through DNA damage response pathways. Oncogene 28: 1506-1517.
Mazor Y, Kupiec M. 2009. Developmentally regulated MAPK pathways modulate heterochromatin in Saccharomyces cerevisiae. Nucleic Acids Res 37: 4839-4849.
Micci F, Panagopoulos I, Bjerkehagen B, Heim S. 2006. Consistent rearrangement of chromosomal band 6p21 with generation of fusion genes JAZF1/PHF1 and EPC1/PHF1 in endometrial stromal sarcoma. Cancer Res 66: 107-112.

27
Mitchell L, Huard S, Cotrut M, Pourhanifeh-Lemeri R, Steunou AL, Hamza A, Lambert JP, Zhou H, Ning Z, Basu A, Cote J, Figeys DA, Baetz K. 2013. mChIP-KAT-MS, a method to map protein interactions and acetylation sites for lysine acetyltransferases. Proc Natl Acad Sci U S A 110: E1641-1650.
Mitchell L, Lambert JP, Gerdes M, Al-Madhoun AS, Skerjanc IS, Figeys D, Baetz K. 2008. Functional dissection of the NuA4 histone acetyltransferase reveals its role as a genetic hub and that Eaf1 is essential for complex integrity. Mol Cell Biol 28: 2244-2256.
Nakahata S, Saito Y, Hamasaki M, Hidaka T, Arai Y, Taki T, Taniwaki M, Morishita K. 2009. Alteration of enhancer of polycomb 1 at 10p11.2 is one of the genetic events leading to development of adult T-cell leukemia/lymphoma. Genes Chromosomes Cancer 48: 768-776.
Owusu-Ansah E, Banerjee U. 2009. Reactive oxygen species prime Drosophila haematopoietic progenitors for differentiation. Nature 461: 537-541.
Prasad P, Ronnerblad M, Arner E, Itoh M, Kawaji H, Lassmann T, Daub CO, Forrest AR, Lennartsson A, Ekwall K, consortium F. 2014. High-throughput transcription profiling identifies putative epigenetic regulators of hematopoiesis. Blood 123: e46-57.
Prelich G. 1999. Suppression mechanisms: themes from variations. Trends Genet 15: 261-266.
Rossetto D, Cramet M, Wang AY, Steunou AL, Lacoste N, Schulze JM, Cote V, Monnet-Saksouk J, Piquet S, Nourani A, Kobor MS, Côté J. 2014. Eaf5/7/3 form a functionally independent NuA4 submodule linked to RNA polymerase II-coupled nucleosome recycling. EMBO J 33: 1397-1415.
Sato T, Russell MA, Denell RE. 1983. Homoeosis in Drosophila: a new enhancer of polycomb and related homoeotic mutations. Genetics 105: 357-370.
Searle NE, Torres-Machorro AL, Pillus L. 2017. Chromatin Regulation by the NuA4 Acetyltransferase Complex Is Mediated by Essential Interactions Between Enhancer of Polycomb (Epl1) and Esa1. Genetics 205: 1125-1137.
Selleck W, Fortin I, Sermwittayawong D, Côté J, Tan S. 2005. The Saccharomyces cerevisiae Piccolo NuA4 histone acetyltransferase

28
complex requires the Enhancer of Polycomb A domain and chromodomain to acetylate nucleosomes. Mol Cell Biol 25: 5535-5542.
Shimono Y, Murakami H, Hasegawa Y, Takahashi M. 2000. RET finger protein is a transcriptional repressor and interacts with enhancer of polycomb that has dual transcriptional functions. J Biol Chem 275: 39411-39419.
Shiota M, Yokomizo A, Masubuchi D, Tada Y, Inokuchi J, Eto M, Uchiumi T, Fujimoto N, Naito S. 2010. Tip60 promotes prostate cancer cell proliferation by translocation of androgen receptor into the nucleus. Prostate 70: 540-554.
Sinclair DA, Clegg NJ, Antonchuk J, Milne TA, Stankunas K, Ruse C, Grigliatti TA, Kassis JA, Brock HW. 1998. Enhancer of Polycomb is a suppressor of position-effect variegation in Drosophila melanogaster. Genetics 148: 211-220.
Soto MC, Chou TB, Bender W. 1995. Comparison of germline mosaics of genes in the Polycomb group of Drosophila melanogaster. Genetics 140: 231-243.
Springer NM, Danilevskaya ON, Hermon P, Helentjaris TG, Phillips RL, Kaeppler HF, Kaeppler SM. 2002. Sequence relationships, conserved domains, and expression patterns for maize homologs of the polycomb group genes E(z), esc, and E(Pc). Plant Physiol 128: 1332-1345.
Squatrito M, Gorrini C, Amati B. 2006. Tip60 in DNA damage response and growth control: many tricks in one HAT. Trends Cell Biol 16: 433-442.
Stankunas K, Berger J, Ruse C, Sinclair DA, Randazzo F, Brock HW. 1998. The enhancer of polycomb gene of Drosophila encodes a chromatin protein conserved in yeast and mammals. Development 125: 4055-4066.
Stone EM, Pillus L. 1996. Activation of an MAP kinase cascade leads to Sir3p hyperphosphorylation and strengthens transcriptional silencing. J Cell Biol 135: 571-583.
Tezel G, Shimono Y, Murakumo Y, Kawai K, Fukuda T, Iwahashi N, Takahashi M. 2002. Role for O-glycosylation of RFP in the interaction with enhancer of polycomb. Biochem Biophys Res Commun 290: 409-414.
Torres-Machorro AL, Pillus L. 2014. Bypassing the requirement for an essential MYST acetyltransferase. Genetics 197: 851-863.

29
van Leeuwen J, Pons C, Boone C, Andrews BJ. 2017. Mechanisms of suppression: The wiring of genetic resilience. Bioessays 39.
Wang Y, Alla V, Goody D, Gupta SK, Spitschak A, Wolkenhauer O, Putzer BM, Engelmann D. 2016. Epigenetic factor EPC1 is a master regulator of DNA damage response by interacting with E2F1 to silence death and activate metastasis-related gene signatures. Nucleic Acids Res 44: 117-133.
Xu P, Li C, Chen Z, Jiang S, Fan S, Wang J, Dai J, Zhu P, Chen Z. 2016. The NuA4 Core Complex Acetylates Nucleosomal Histone H4 through a Double Recognition Mechanism. Mol Cell 63: 965-975.
Yi C, Ma M, Ran L, Zheng J, Tong J, Zhu J, Ma C, Sun Y, Zhang S, Feng W, Zhu L, Le Y, Gong X, Yan X, Hong B, Jiang FJ, Xie Z, Miao D, Deng H, Yu L. 2012. Function and molecular mechanism of acetylation in autophagy regulation. Science 336: 474-477.
Zeng X, Han L, Singh SR, Liu H, Neumuller RA, Yan D, Hu Y, Liu Y, Liu W, Lin X, Hou SX. 2015. Genome-wide RNAi screen identifies networks involved in intestinal stem cell regulation in Drosophila. Cell Rep 10: 1226-1238.
Zhao X, Muller EG, Rothstein R. 1998. A suppressor of two essential checkpoint genes identifies a novel protein that negatively affects dNTP pools. Mol Cell 2: 329-340.

30
Chapter 1 Chromatin regulation by the NuA4 acetyltransferase complex is mediated by essential interactions between Enhancer of Polycomb (Epl1) and Esa1
Chapter 2.
Chromatin regulation by the NuA4
acetyltransferase complex is
mediated by essential interactions
between Enhancer of Polycomb
(Epl1) and Esa1

31
HIGHLIGHTED ARTICLE| INVESTIGATION
Chromatin Regulation by the NuA4 AcetyltransferaseComplex Is Mediated by Essential Interactions
Between Enhancer of Polycomb (Epl1) and Esa1Naomi E. Searle,*,† Ana Lilia Torres-Machorro,†,1 and Lorraine Pillus†,2
*Biomedical Sciences, University of California, San Diego and †Section of Molecular Biology, Division of Biological Sciences,University of California, San Diego, UC San Diego Moores Cancer Center, La Jolla, California 92093
ORCID IDs: 0000-0002-5208-9700 (N.E.S.); 0000-0003-3100-6897 (A.L.T.-M.); 0000-0002-8818-5227 (L.P.)
ABSTRACT Enzymes that modify and remodel chromatin act in broadly conserved macromolecular complexes. One key modification is thedynamic acetylation of histones and other chromatin proteins by opposing activities of acetyltransferase and deacetylase complexes. Amongacetyltransferases, the NuA4 complex containing Tip60 or its Saccharomyces cerevisiae ortholog Esa1 is of particular significance because of itsroles in crucial genomic processes including DNA damage repair and transcription. The catalytic subunit Esa1 is essential, as are five noncatalyticNuA4 subunits. We found that of the noncatalytic subunits, deletion of Enhancer of polycomb (Epl1), but not the others, can be bypassed byloss of a major deacetylase complex, a property shared by Esa1. Noncatalytic complex subunits can be critical for complex assembly, stability,genomic targeting, substrate specificity, and regulation. Understanding the essential role of Epl1 has been previously limited, a limitation nowovercome by the discovery of its bypass suppression. Here, we present a comprehensive in vivo study of Epl1 using the powerful tool ofsuppression combined with transcriptional and mutational analyses. Our results highlight functional parallels between Epl1 and Esa1 andfurther illustrate that the structural role of Epl1 is important for promotion of Esa1 activity. This conclusion is strengthened by our dissection ofEpl1 domains required in vivo for interaction with specific NuA4 subunits, histone acetylation, and chromatin targeting. These results providenew insights for the conserved, essential nature of Epl1 and its homologs, such as EPC1/2 in humans, which is frequently altered in cancers.
KEYWORDS NuA4; EPL1; ESA1; chromatin; acetylation
EUKARYOTIC genomes are packaged into chromatin,which is composed of nucleosome units containing DNA
wrapped around a histone octamer (Kornberg and Lorch1999). Chromatin is subject to multiple, diverse modes ofpost-translational regulation that have many establishedroles, including functions in recombination, DNA damage re-pair, and transcription (Kouzarides 2007). Acetylation is onesuch post-translational modification that regulates chromatinfunction, mediated by the opposing enzymatic activities oflysine acetyltransferases (KATs/HATs) and deacetylases
(KDACs/HDACs) (Campos and Reinberg 2009). HATs oftenexist in large multimeric complexes, such as the deeply con-served NuA4 complex (Doyon et al. 2004).
In humans, the essential catalytic subunit of NuA4, KAT5/Tip60, along with additional essential subunits such as EPC1/2,are associatedwith several carcinomas (Avvakumov and Côté2007; Lafon et al. 2007; Nakahata et al. 2009; Biankin et al.2012; Huang et al. 2014), suggesting their importance forcontrolled cellular growth. Much of the basic understandingof NuA4 comes from studies performed in Saccharomycescerevisiae. NuA4 in yeast includes six essential subunits:Esa1 (Tip60 ortholog), Epl1 (EPC1/2 ortholog), Tra1,Arp4, Act1, and Swc4, all of which are broadly conserved.NuA4 primarily acetylates histones H4 and H2A in vivo(Smith et al. 1998; Clarke et al. 1999) along with nonca-nonical histones, such as H2A.Z (Keogh et al. 2006),and .250 nonhistone substrates (Lin et al. 2009; Yi et al.2012; Mitchell et al. 2013; Downey et al. 2015), including91 essential proteins.
Copyright © 2017 by the Genetics Society of Americadoi: 10.1534/genetics.116.197830Manuscript received November 8, 2016; accepted for publication January 16, 2017;published Early Online January 20, 2017.Supplemental material is available online at www.genetics.org/lookup/suppl/doi:10.1534/genetics.116.197830/-/DC1.1Present address: CONACYT, Instituto Nacional de Enfermedades Respiratorias,“Ismael Cosío Villegas,” Calzada de Tlalpan 4502, Colonia Sección XVI, Tlalpan,Mexico City, CP 14080.
2Corresponding author: 9500 Gilman Drive, La Jolla, CA 92093-0347.E-mail: [email protected]
Genetics, Vol. 205, 1125–1137 March 2017 1125

32
There are two distinct smaller complexes containing NuA4subunits: piccolo-NuA4, composed of Esa1, Epl1, Yng2, andEaf6 (Boudreault et al. 2003; Mitchell et al. 2008; Rossettoet al. 2014), and the TINTIN triad of Eaf5/7/3 (Cheng andCôté 2014; Rossetto et al. 2014). Piccolo-NuA4 is thought toalso exist alone (Ohba et al. 1999; Boudreault et al. 2003)and is sufficient for broad nucleosome acetylation in vitro,whereas the NuA4 holo-complex is required for more tar-geted NuA4 functions such as DNA damage repair and tran-scriptional activation (Figure 1A) (Bird et al. 2002; Boudreaultet al. 2003; Selleck et al. 2005; Friis et al. 2009).
Because Esa1 is essential, much of our early understand-ing of it came from studying hypomorphic alleles, where Esa1is only partially or conditionally functional (Clarke et al.1999; Decker et al. 2008). Recently, the first bypass suppres-sor of Esa1 was identified, where esa1D is rescued by loss ofthe Rpd3L HDAC complex (Torres-Machorro and Pillus2014). This bypass of Esa1 is promoted by establishing a rel-atively balanced cellular acetylation state. The discovery ofthis bypass allowed for the first studies in which cells werecompletely depleted of Esa1.
Among the six essential NuA4 subunits only Esa1 and Epl1are found in the very active smaller piccolo complex (Galarneauet al. 2000). Epl1 was first reported as the yeast ortholog ofDrosophila melanogaster Enhancer of Polycomb E(Pc),which can function as a suppressor of position-effect varie-gation and can increase the homeotic phenotype of Poly-comb group mutations (Sinclair et al. 1998; Stankunaset al. 1998). Epl1 and E(Pc) are broadly conserved andare orthologous to the EPC1/2 paralogs in humans(Shimono et al. 2000; Doyon et al. 2004).
It is noteworthy that despite its conservation and discov-ery nearly two decades ago, Epl1 function has beenonly minimally characterized, primarily based on low-dosagevariants, limited in vitro analyses, and most recently when itspartial structure bound to nucleosomeswas solved (Boudreaultet al. 2003; Selleck et al. 2005; Chittuluru et al. 2011; Huangand Tan 2012; Xu et al. 2016). Phenotypes of EPL1 depletionare quite similar to those of impaired ESA1. These include rolesin cell-cycle progression through G2/M, H4 acetylation, DNAdamage repair, telomeric silencing, and autophagy (Boudreaultet al. 2003; Yi et al. 2012).
Epl1 bridges Esa1 and the Yng2 and Eaf6 subunits to thelarger NuA4 complex (Boudreault et al. 2003; Mitchell et al.2008; Rossetto et al. 2014). The C-terminus of Epl1 contactsthe NuA4 holo-complex through Eaf1 (Auger et al. 2008), butonly the N-terminus (the EPcA domain) is essential for via-bility (Boudreault et al. 2003), suggesting that integrity ofpiccolo-NuA4 is crucial.
Despite progress made in earlier studies, the essentialfunction of Epl1 in vivo has remained unknown in S. cerevisiaeand metazoans alike. Here, we report that Epl1 can bebypassed by the same loss of the Rpd3L deacetylase complexobserved for Esa1 and present a comprehensive in vivoanalysis of Epl1 made possible only by its bypass suppression.Although Epl1 has no known catalytic activity, we find striking
phenotypic and transcriptional similarity between esa1D andepl1D mutant strains under bypass conditions, suggesting co-ordinated function and activity. Through mutational analysisof Epl1, we provide evidence that Epl1’s essential function isdirectly linked to physical contact with Esa1, such that withoutthe Epl1-Esa1 structural interaction, Esa1 is no longer fullyactive. These new findings thus help to illuminate the essentialcoordinated activity of aMYST-family acetyltransferase and itsbroadly conserved binding partner.
Materials and Methods
Yeast strains and plasmids
Strains, plasmids, and oligonucleotides are listed in Supple-mental Material, Tables S1–S3 in File S1. EPL1 and ESA1mutant strains were constructed initially with covering plas-mids (pLP3189 or pLP796). EPL1-13MYC-HISMX6 was de-rived from LPY21686 (QY237), and integrated at theendogenous EPL1 locus in LPY79 by amplification of the13MYC-HISMX6 tag with oLP2196 and oLP2172. EPL1-13MYC-HISMX6 was similarly cloned into pLP74, aspLP3337, by amplifying EPL1-13MYC-HISMX6 fromLPY21686 with oLP2169 and oLP2180, digested with HinDIII,and ligated into pLP74. Epl1mutant plasmidswere constructedusing NEB Q5 site-directed mutagenesis on pLP3337. Mutantswere tested for dominance by transforming into a wild-type(WT) strain. The mutations were then integrated at the EPL1locus in diploid WT W303 and dissected. Mutagenesis wasverified by sequencing both prior to and after integration.Strains were backcrossed prior to use.
Growth assays
Plate-based assays were performed using fivefold serial dilutionson standard media as described (Chang and Pillus 2009). Fortemperature and DNA damage assays, cultures were grown at24! in SC for 1–3 days and then plated with starting concentra-tions normalized to one A600 unit and imaged after 2–5 days.Methyl methanesulfonate (MMS) sensitivity was assayed at0.0075% in SC. Hydroxyurea (HU) sensitivity was assayedat 0.05 M in SC. Camptothecin (CPT) sensitivity was assayedat 7 mg/ml in SC (DMSO as vehicle control) prepared with100 mM phosphate buffer (pH 7.5). Cultures for 5-fluorooroticacid (5-FOA) assays were grown for 2 days at 30! to reachsaturation, normalized to starting dilutions of 5–7 A600 units,and imaged after 4–6days after plating. 5-FOA assays performedin the W303 background were plated on 10% glucose; all otherplate-based assays were performed with standard 2% glucose.
Flow cytometry
Strains grown at 24! in SC for 1–2 days were diluted andgrown to mid-log. One milliliter of exponentially growingcells (!3 3 107 cells) was fixed with cold 70% ethanol andprepared for flow cytometry, staining with propidium iodide(Chang et al. 2012). Thirty thousand cells were analyzedusing a BD Accuri C6 Flow Cytometer.
1126 N. E. Searle, A. L. Torres-Machorro, and L. Pillus

33
Lysate preparation
Strains grown at 24! in SC were collected in mid-log forwhole-cell extract preparations by bead-beating as described(Clarke et al. 1999). Fractionation was performed usingspheroplasting, detergent-based lysis, and differential centri-fugation (Liang and Stillman 1997) to yield whole-cell ex-tract, and soluble and crude chromatin fractions. Lysateswere briefly sonicated prior to immunoblot analysis.
Immunoprecipitations
Strains were grown for 1–2 days in 3 ml of SC at 24!, ex-panded to 10 ml, then diluted into 200 ml for growth andcollected in mid-log phase. After pelleting and a phosphate-buffered saline (PBS) wash, cells were lysed by bead-beatingin 1 ml of cold immunoprecipitation (IP) lysis buffer per A600
OD of cells (50 mM HEPES-KOH pH 7.5, 100 mM NaCl,0.25% NP-40, 1 mM EDTA, 10% glycerol, and protease,
phosphatase, and deacetylase inhibitors). The lysate wascleared then incubated with rotation for 3 hr with 5 ml ofanti-Myc. IP mixtures were incubated for 50 min with 75 mlof Dynabeads Protein G (Thermo Fisher Scientific), prewashedwith lysis buffer. Protein–antibody–bead conjugates werewashed twice with lysis buffer and twice with wash buffer(50 mM HEPES-KOH pH 7.5, 150 mM NaCl, 1 mM EDTA)prior to elution by boiling for 10min in 40ml of sample loadingbuffer (250 mM Tris-HCL pH 6.8, 10% SDS, 30% glycerol, 5%b-mercaptoethanol, 0.02% bromophenol blue).
Immunoblots
Toevaluatehistones, proteinswere separatedusing15%SDS-PAGE, transferred to a 0.2-mm nitrocellulose membrane, andprobed with: anti-H4K8Ac (1:2000, EMD Millipore, Darm-stadt, Germany), anti-H4K5Ac (1:2000, Millipore), anti-H4K12Ac (1:2000, Active Motif), anti-H4 (1:2000, ActiveMotif), anti-H3K9/K14Ac (1:10,000, Upstate), and anti-H3(1:2500, Abcam). Other proteins were separated on 7.5, 8, or10% SDS-PAGE or for IP samples, 8–16% Novex WedgewellTris-Glycine gels (Thermo Fisher Scientific), transferred to a0.2-mm nitrocellulose membrane and probed with: anti-Myc(1:2500 for detection of Epl1, 1:5000 for detection of allother Myc-tagged proteins) (Evan et al. 1985), anti-HA(1:1000, Covance), anti-Yng2 (1:1000, graciously providedby S. Tan), anti-Sir2 (1:10,000) (Garcia and Pillus 2002),anti-Pgk1 (1:20,000), and anti-b-tubulin (1:20,000) (Bondet al. 1986).
RNA-seq sample preparation and analysis
RNA was prepared in biological triplicate using hot-phenolextraction from mid-log cells grown in SC at 24!. RNA wasDNase treated (Ambion). Quality was evaluated by gel elec-trophoresis and bioanalyzer (Agilent). Samples were de-pleted of rRNA (Ribo-zero Magnetic Gold Yeast, Epibio),and libraries were prepared (Tru-seq Stranded total RNA,Illumina). Twenty-four samples were sequenced with 50-bpsingle-reads on one lane of the HiSeq 2500 (Illumina), yield-ing a total of 287.52 million reads passing the quality filter.
Upon data generation, library adaptors were trimmedcomputationally with Cutadapt (Martin 2011), and readswere mapped to Repbase (Bao et al. 2015). Any reads map-ping to Repbase were excluded from further analysis. Theremaining reads were mapped to SacCer3 (Engel et al.2014) with STAR (Dobin et al. 2013). Differential expressionwas assessed with DESeq2 (Love et al. 2014), and transcriptswith a log2(fold change) $1 or #21 and P-adj #0.05 werecalled as differentially expressed. Further data analysis andvisualization was completed using R computing software(R Development Core Team 2015) and the ggplot2 package(Wickham 2009).
qPCR validation
Select transcripts were validated using RT-qPCR. Briefly,cDNA was synthesized in biological triplicate from the RNAsamples (TaqMan Reverse Transcriptase kit, Life Sciences)
Figure 1 The requirement for two essential NuA4 subunits is bypassed bydisassembly of Rpd3L. (A) The NuA4 histone acetyltransferase complexcontains six essential subunits (underlined). The NuA4 holo-complex hastargeted functions including roles in transcription and DNA damage re-pair, whereas the smaller piccolo-NuA4 complex is a broadly actingacetyltransferase complex. Piccolo-NuA4 contains the catalytic subunitEsa1, along with the essential subunit Epl1 and two nonessential sub-units. NuA4 complex schematic is based on: Boudreault et al. (2003);Bittner et al. (2004); Doyon et al. (2004); Mitchell et al. (2008);Chittuluru et al. (2011); and Rossetto et al. (2014). (B) A screen of allessential NuA4 subunits for bypass potential. Double mutant analysis ofesa1D sds3D (LPY20724), epl1D sds3D (LPY20609), act1D sds3D(LPY20974), arp4D sds3D (LPY20617), swc4D sds3D (LPY20611), andtra1D sds3D (LPY20443) revealed that only epl1D, like esa1D, could bebypassed by loss of SDS3, which encodes a central component of theRpd3L deacetylase complex. Serial dilutions on the plasmid counterselec-tive medium at 24! (and 30!, Figure S1 in File S1) illustrate that epl1D sds3D,like esa1D sds3D, survived without a plasmid-based copy of its correspondingessential gene.
Bypassing EPL1 Defines Key Functions 1127

34
and qPCR was performed using EvaGreen qPCR Master Mix(Lambdabio)onanMJResearchOpticon2 todetermine levelsrelative to the SCR1 control. Significance was tested andassigned based on P-values calculated by a Student’s t-test.
Data availability
Strains and plasmids are available upon request. Gene ex-pression data have been deposited in the Gene ExpressionOmnibus with accession number GSE92774.
Results
Bypass and function of essential piccolo-NuA4 subunits
The finding that the essential requirement for Esa1 could bebypassed by loss of the Rpd3L deacetylase due to deletion ofSDS3 (Torres-Machorro and Pillus 2014) was significant be-cause it marked the first condition where cellular viabilitywas maintained without an essential NuA4 subunit. Similarto bypass suppression of ESA1, identification of other NuA4bypass suppressors could facilitate in vivo analysis of theseessential chromatin factors.
To test the extent to which disruption of Rpd3L by sds3Dcould bypass loss of genes encoding the essential NuA4 sub-units (Esa1, Epl1, Act1, Arp4, Swc4, Tra1, underlined in Fig-ure 1A), double mutants were constructed. Initially, eachdouble mutant was recovered with a URA3-marked plasmidcarrying the corresponding wild-type NuA4 gene. The strainswere then challenged by plating on 5-FOA, which is toxic tocells expressing URA3. Growth on 5-FOA reveals mutant cellsthat can survive without the corresponding wild-type cover-ing plasmid. Of the five new double mutants tested, onlyepl1D could be bypassed by sds3D; all remaining essentialNuA4 subunits were still required for viability (Figure 1Band Figure S1 in File S1). The recovery of epl1D was of par-ticular interest because of Epl1’s limited in vivo characteriza-tion in any species and its close structural proximity to thecatalytic Esa1 in piccolo-NuA4/NuA4.
Previous in vitro and in vivo studies of Epl1 were reportedusing two hypomorphic alleles and repressible expression.Epl1 was shown to have roles similar to Esa1, such as inhistone H4 acetylation, DNA damage repair, and cell-cycleprogression (Boudreault et al. 2003). To evaluate potentialdistinctions between Epl1 and Esa1 function in vivo, we exam-ined phenotypes of the bypass strains. The esa1D epl1D sds3Dtriple mutant was viable (Figure 2A) and thus included in thephenotypic analysis.
TheNuA4bypass strainswere surveyed for growth across arange of temperatures: all showed extreme sensitivity to hightemperatures, and general growth defects at lower tempera-tures (Figure 2A). The epl1D sds3D and esa1D epl1D sds3Dmutants were sensitive to DNA-damaging agents (Figure 2B)as shown previously for esa1D sds3D (Torres-Machorro andPillus 2014). These strains were also sensitive to the vehiclecontrol for CPT, DMSO, which has been shown to broadlydecrease cellular proliferation (Kakolyri et al. 2016). This
sensitivity mirrors that which has been identified for mu-tants of other chromatin regulators (Gaytán et al. 2013;Sadowska-Bartosz et al. 2013). As illustrated by H4K8 andH4K12 acetylation, EPL1 bypass strains had low levels ofhistone H4 acetylation relative to WT and sds3D. By con-trast, H3 acetylation remained unaffected (Figure 2C). Fi-nally, loss of EPL1 resulted in a similar defect in cell-cycleprogression as loss of ESA1, characterized by a G2/M delay(Figure 2D).
Thus, loss of EPL1, despite not encoding acetyltransferaseactivity, had similar phenotypic and functional conse-quences as loss of ESA1. The observation that no distinctphenotypes were found when both ESA1 and EPL1 werelost, as compared to when only a single subunit wasbypassed, further emphasized a high degree of functionaloverlap.
Figure 2 Bypass of EPL1 is phenotypically akin to esa1D sds3D. (A) Theepl1D sds3D (LPY21299) bypass strain shared growth defect and temper-ature-sensitivity phenotypes of esa1D sds3D (LPY21631), relative to bothWT (LPY79) and sds3D (LPY20877). Likewise, in the triple mutant, loss ofboth EPL1 and ESA1 (LPY21751) had similar growth defects to loss ofeither essential subunit alone. (B) Bypass strains were surveyed at 24! forDNA damage, revealing sensitivity to all agents tested, relative to growthcontrol (A), and the DMSO-vehicle control for CPT. (C) Histone H4 acet-ylation is significantly reduced upon loss of ESA1 and/or EPL1 relative toWT and sds3D. Two acetylation isoforms were probed as representativesfor acetylation. Histone H3 acetylation remained unchanged relative toWT upon Esa1 or Epl1 mutation, highlighting the effect on histone H4acetylation as a NuA4 target rather than the H3–H4 tetramer. (D) Cellcycle profiles demonstrated that loss of Esa1 and Epl1 resulted in a G2/Mdelay. All experiments were completed in three or more independentassays. Representative results from each are shown here.
1128 N. E. Searle, A. L. Torres-Machorro, and L. Pillus
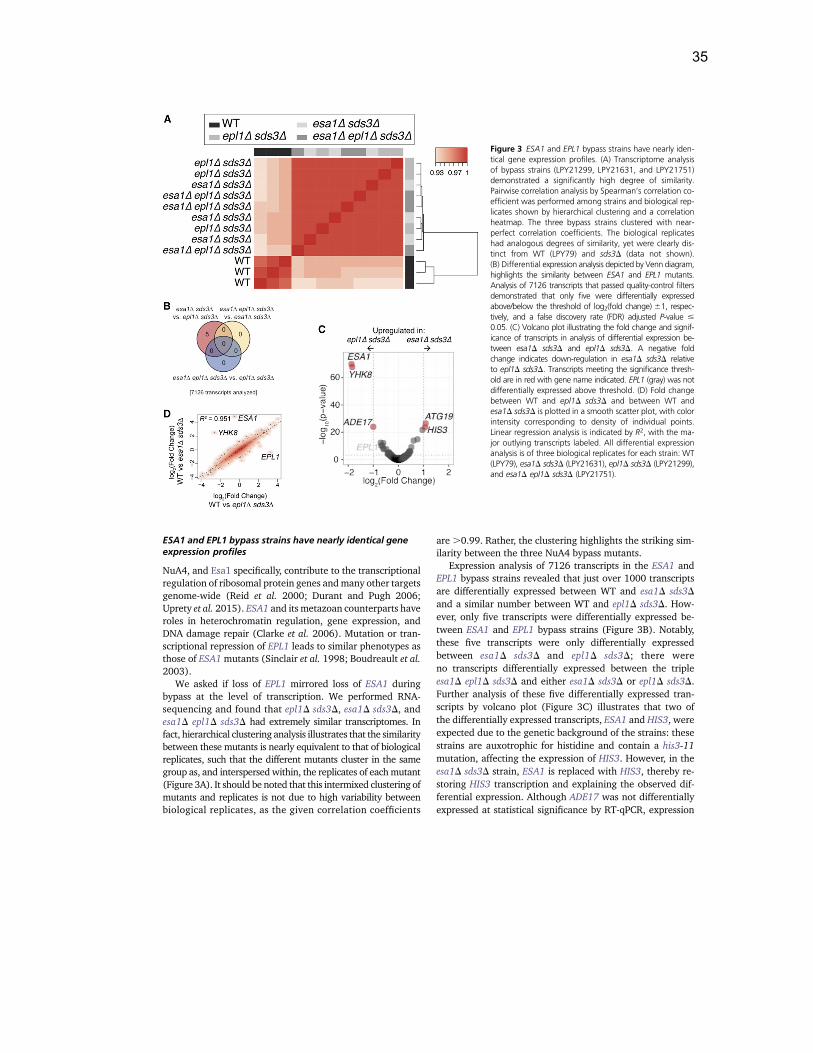
35
ESA1 and EPL1 bypass strains have nearly identical geneexpression profiles
NuA4, and Esa1 specifically, contribute to the transcriptionalregulation of ribosomal protein genes andmany other targetsgenome-wide (Reid et al. 2000; Durant and Pugh 2006;Uprety et al. 2015). ESA1 and its metazoan counterparts haveroles in heterochromatin regulation, gene expression, andDNA damage repair (Clarke et al. 2006). Mutation or tran-scriptional repression of EPL1 leads to similar phenotypes asthose of ESA1mutants (Sinclair et al. 1998; Boudreault et al.2003).
We asked if loss of EPL1 mirrored loss of ESA1 duringbypass at the level of transcription. We performed RNA-sequencing and found that epl1D sds3D, esa1D sds3D, andesa1D epl1D sds3D had extremely similar transcriptomes. Infact, hierarchical clustering analysis illustrates that the similaritybetween these mutants is nearly equivalent to that of biologicalreplicates, such that the different mutants cluster in the samegroup as, and interspersed within, the replicates of eachmutant(Figure 3A). It should be noted that this intermixed clustering ofmutants and replicates is not due to high variability betweenbiological replicates, as the given correlation coefficients
are .0.99. Rather, the clustering highlights the striking sim-ilarity between the three NuA4 bypass mutants.
Expression analysis of 7126 transcripts in the ESA1 andEPL1 bypass strains revealed that just over 1000 transcriptsare differentially expressed between WT and esa1D sds3Dand a similar number between WT and epl1D sds3D. How-ever, only five transcripts were differentially expressed be-tween ESA1 and EPL1 bypass strains (Figure 3B). Notably,these five transcripts were only differentially expressedbetween esa1D sds3D and epl1D sds3D; there wereno transcripts differentially expressed between the tripleesa1D epl1D sds3D and either esa1D sds3D or epl1D sds3D.Further analysis of these five differentially expressed tran-scripts by volcano plot (Figure 3C) illustrates that two ofthe differentially expressed transcripts, ESA1 and HIS3, wereexpected due to the genetic background of the strains: thesestrains are auxotrophic for histidine and contain a his3-11mutation, affecting the expression of HIS3. However, in theesa1D sds3D strain, ESA1 is replaced with HIS3, thereby re-storing HIS3 transcription and explaining the observed dif-ferential expression. Although ADE17 was not differentiallyexpressed at statistical significance by RT-qPCR, expression
Figure 3 ESA1 and EPL1 bypass strains have nearly iden-tical gene expression profiles. (A) Transcriptome analysisof bypass strains (LPY21299, LPY21631, and LPY21751)demonstrated a significantly high degree of similarity.Pairwise correlation analysis by Spearman’s correlation co-efficient was performed among strains and biological rep-licates shown by hierarchical clustering and a correlationheatmap. The three bypass strains clustered with near-perfect correlation coefficients. The biological replicateshad analogous degrees of similarity, yet were clearly dis-tinct from WT (LPY79) and sds3D (data not shown).(B) Differential expression analysis depicted by Venn diagram,highlights the similarity between ESA1 and EPL1 mutants.Analysis of 7126 transcripts that passed quality-control filtersdemonstrated that only five were differentially expressedabove/below the threshold of log2(fold change) 61, respec-tively, and a false discovery rate (FDR) adjusted P-value #0.05. (C) Volcano plot illustrating the fold change and signif-icance of transcripts in analysis of differential expression be-tween esa1D sds3D and epl1D sds3D. A negative foldchange indicates down-regulation in esa1D sds3D relativeto epl1D sds3D. Transcripts meeting the significance thresh-old are in red with gene name indicated. EPL1 (gray) was notdifferentially expressed above threshold. (D) Fold changebetween WT and epl1D sds3D and between WT andesa1D sds3D is plotted in a smooth scatter plot, with colorintensity corresponding to density of individual points.Linear regression analysis is indicated by R2, with the ma-jor outlying transcripts labeled. All differential expressionanalysis is of three biological replicates for each strain: WT(LPY79), esa1D sds3D (LPY21631), epl1D sds3D (LPY21299),and esa1D epl1D sds3D (LPY21751).
Bypassing EPL1 Defines Key Functions 1129

36
trended toward its down-regulation in esa1D sds3D as com-pared to epl1D sds3D. The ATG19 and YHK8 differential ex-pression was validated by RT-qPCR (Figure S2 in File S1),and in fact, YHK8, a largely uncharacterized open read-ing frame (ORF), is greater than sixfold up-regulated inepl1D sds3D as compared to esa1D sds3D by RT-qPCR. How-ever, all three of these transcripts are only differentiallyexpressed by one- to twofold by RNA-sequencing, and thereis no functional theme underlying and unifying their differ-ential expression, nor are the corresponding genes directlybound by Esa1 (Robert et al. 2004).
An analysis examining the differential expression of tran-scripts between WT and esa1D sds3D plotted against the differ-ential expression of transcripts between WT and epl1D sds3Dwas also telling. Plotting the log2 (fold change) of all tran-scripts relative to WT in esa1D sds3D vs. that in epl1D sds3Dillustrated a high correlation between differential expressionin esa1D sds3D and in epl1D sds3D, both relative to WT (Fig-ure 3D). As such, transcripts that differ between WT andesa1D sds3D also differ, and to a similar magnitude, betweenWT and epl1D sds3D. Thus, the transcriptional profiles ofEpl1 and Esa1 bypass conditions are virtually identical, de-spite their distinct noncatalytic and catalytic roles in NuA4.
Epl1 promotes the chromatin association of Esa1
Both phenotypic and transcriptional analyses of epl1D andesa1D emphasize their similarity, despite the overt differenceof Esa1’s catalytic activity. To further probe distinctions be-tween the roles of Epl1 and Esa1, and to determine the natureof EPL1’s essential function, we considered the in vitro char-acterization of Epl1, which reported that it associates withthe nucleosome core particle to promote Esa1’s enzymaticactivity (Chittuluru et al. 2011). We could ask for the firsttime if Epl1 drives Esa1’s chromatin association in vivo, andif Esa1 would remain chromatin associated in the absenceof Epl1.
To test the role of Epl1 in targeting Esa1 to chromatin,subcellular fractionation (Liang and Stillman 1997) and im-munoblotting were performed (Figure 4). Controls includedprobes for the chromatin-associated protein Sir2 and the gly-colytic enzyme Pgk1, a predominantly cytoplasmic protein.In WT, sds3D, and epl1D sds3D strains, Sir2 was primarilylocalized to the chromatin (C) fraction, whereas Pgk1 wasmore enriched in the soluble (S) fraction (Figure 4, A and B).In contrast, whereas Esa1 is largely localized to the chroma-tin fraction inWT and sds3D, it becomes shifted to the solublefraction upon loss of EPL1 and depleted from chromatin.Notably, this shift in association is specific for Esa1, as Sir2remains chromatin associated.
NuA4 contains subunits that have chromatin activity in-dependent of NuA4, including several that contain their ownchromatin targeting domains. We sought to determine if thenewly defined role for Epl1 in promoting chromatin associa-tion of Esa1 in vivowas extended to other NuA4 subunits, andtherefore, if its loss might have more widespread conse-quences. To test this possibility, we selected Swc4 for its
essential nature, dual-role in NuA4 and the Swc4 chroma-tin-remodeling complex, and its SANT domain (Krogan et al.2004). We found that Swc4 remained chromatin associatedin the absence of EPL1 (Figure 4C), demonstrating thatloss of Epl1 did not broadly affect all NuA4 subunits. LikeEsa1 localization, Swc4 is unaffected by sds3D alone, and
Figure 4 Epl1 is required for stable chromatin association of Esa1.(A) Subcellular fractionation assays reveal that in the absence of Epl1, afraction of Esa1 is released from chromatin. Cells were collected and lysedfor whole-cell extracts (W). Additional fractionation was performed toyield soluble (S) and crude chromatin (C) fractions. In WT cells(LPY21568), the majority of Esa1 is associated with the chromatin frac-tion, much like Sir2. However, in epl1D sds3D (LPY21596), Esa1 is shiftedto the soluble fraction, analogous to the Pgk1 control. A brief chemicalcross-link prior to lysis and fractionation was performed in parallel (FigureS3 in File S1). (B) The sds3D single mutant (LPY21579) alone does notalter Esa1 chromatin association, as illustrated by subcellular fractionationfollowed by immunoblotting for Esa1, and the Sir2 and Pgk1 controls.(C) Swc4 remains chromatin associated upon loss of EPL1. Subcellularfractionation demonstrates that Swc4, another essential NuA4 subunit,remains chromatin associated in epl1D sds3D (LPY21942), consistentwith WT (LPY22201) and much like the Sir2 control. (D) The sds3Dsingle mutant (LPY22202) alone also does not affect Swc4 chromatinassociation.
1130 N. E. Searle, A. L. Torres-Machorro, and L. Pillus
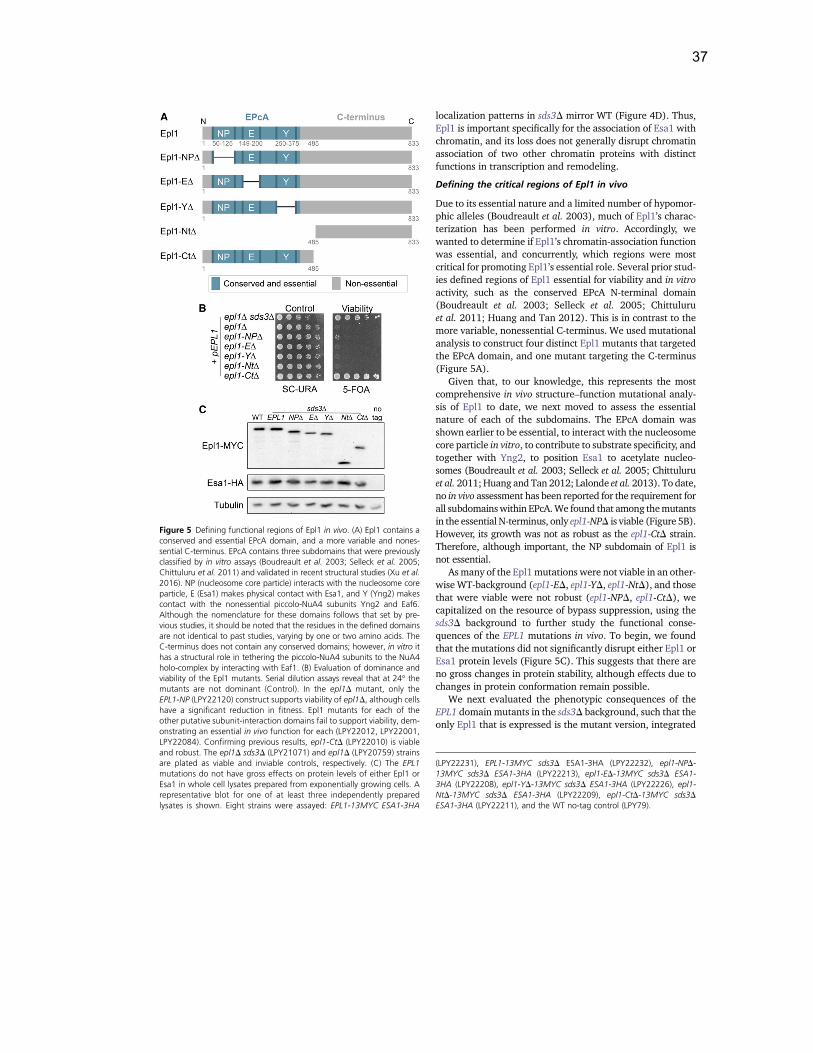
37
localization patterns in sds3D mirror WT (Figure 4D). Thus,Epl1 is important specifically for the association of Esa1 withchromatin, and its loss does not generally disrupt chromatinassociation of two other chromatin proteins with distinctfunctions in transcription and remodeling.
Defining the critical regions of Epl1 in vivo
Due to its essential nature and a limited number of hypomor-phic alleles (Boudreault et al. 2003), much of Epl1’s charac-terization has been performed in vitro. Accordingly, wewanted to determine if Epl1’s chromatin-association functionwas essential, and concurrently, which regions were mostcritical for promoting Epl1’s essential role. Several prior stud-ies defined regions of Epl1 essential for viability and in vitroactivity, such as the conserved EPcA N-terminal domain(Boudreault et al. 2003; Selleck et al. 2005; Chittuluruet al. 2011; Huang and Tan 2012). This is in contrast to themore variable, nonessential C-terminus. We used mutationalanalysis to construct four distinct Epl1 mutants that targetedthe EPcA domain, and one mutant targeting the C-terminus(Figure 5A).
Given that, to our knowledge, this represents the mostcomprehensive in vivo structure–function mutational analy-sis of Epl1 to date, we next moved to assess the essentialnature of each of the subdomains. The EPcA domain wasshown earlier to be essential, to interact with the nucleosomecore particle in vitro, to contribute to substrate specificity, andtogether with Yng2, to position Esa1 to acetylate nucleo-somes (Boudreault et al. 2003; Selleck et al. 2005; Chittuluruet al. 2011;Huang and Tan 2012; Lalonde et al. 2013). To date,no in vivo assessment has been reported for the requirement forall subdomainswithin EPcA.We found that among themutantsin the essential N-terminus, only epl1-NPD is viable (Figure 5B).However, its growth was not as robust as the epl1-CtD strain.Therefore, although important, the NP subdomain of Epl1 isnot essential.
As many of the Epl1mutations were not viable in an other-wise WT-background (epl1-ED, epl1-YD, epl1-NtD), and thosethat were viable were not robust (epl1-NPD, epl1-CtD), wecapitalized on the resource of bypass suppression, using thesds3D background to further study the functional conse-quences of the EPL1 mutations in vivo. To begin, we foundthat the mutations did not significantly disrupt either Epl1 orEsa1 protein levels (Figure 5C). This suggests that there areno gross changes in protein stability, although effects due tochanges in protein conformation remain possible.
We next evaluated the phenotypic consequences of theEPL1 domain mutants in the sds3D background, such that theonly Epl1 that is expressed is the mutant version, integrated
Figure 5 Defining functional regions of Epl1 in vivo. (A) Epl1 contains aconserved and essential EPcA domain, and a more variable and nones-sential C-terminus. EPcA contains three subdomains that were previouslyclassified by in vitro assays (Boudreault et al. 2003; Selleck et al. 2005;Chittuluru et al. 2011) and validated in recent structural studies (Xu et al.2016). NP (nucleosome core particle) interacts with the nucleosome coreparticle, E (Esa1) makes physical contact with Esa1, and Y (Yng2) makescontact with the nonessential piccolo-NuA4 subunits Yng2 and Eaf6.Although the nomenclature for these domains follows that set by pre-vious studies, it should be noted that the residues in the defined domainsare not identical to past studies, varying by one or two amino acids. TheC-terminus does not contain any conserved domains; however, in vitro ithas a structural role in tethering the piccolo-NuA4 subunits to the NuA4holo-complex by interacting with Eaf1. (B) Evaluation of dominance andviability of the Epl1 mutants. Serial dilution assays reveal that at 24! themutants are not dominant (Control). In the epl1D mutant, only theEPL1-NP (LPY22120) construct supports viability of epl1D, although cellshave a significant reduction in fitness. Epl1 mutants for each of theother putative subunit-interaction domains fail to support viability, dem-onstrating an essential in vivo function for each (LPY22012, LPY22001,LPY22084). Confirming previous results, epl1-CtD (LPY22010) is viableand robust. The epl1D sds3D (LPY21071) and epl1D (LPY20759) strainsare plated as viable and inviable controls, respectively. (C) The EPL1mutations do not have gross effects on protein levels of either Epl1 orEsa1 in whole cell lysates prepared from exponentially growing cells. Arepresentative blot for one of at least three independently preparedlysates is shown. Eight strains were assayed: EPL1-13MYC ESA1-3HA
(LPY22231), EPL1-13MYC sds3D ESA1-3HA (LPY22232), epl1-NPD-13MYC sds3D ESA1-3HA (LPY22213), epl1-ED-13MYC sds3D ESA1-3HA (LPY22208), epl1-YD-13MYC sds3D ESA1-3HA (LPY22226), epl1-NtD-13MYC sds3D ESA1-3HA (LPY22209), epl1-CtD-13MYC sds3DESA1-3HA (LPY22211), and the WT no-tag control (LPY79).
Bypassing EPL1 Defines Key Functions 1131

38
at the genomic locus. In these bypass conditions, mutants ofthe Epl1 subunit interaction domains (epl1-ED and epl1-YD)are sensitive to high temperature (Figure 6A), and DNA dam-age (Figure 6B). Accordingly, complete loss of EPL1 or loss ofthe entire essential EPcA domain (epl1-NtD) is phenotypicallysimilar to loss of either of the subunit interaction domains(epl1-ED and epl1-YD) alone. In contrast and consistent withepl1-NPD sufficiency for viability, this mutant has the mostrobust growth in bypass conditions when challenged withhigher temperatures and DNA-damaging agents. These re-sults suggest that the residues of Epl1 that interact with othersubunits in vitro (Epl1-E and Epl1-Y) aremost critical for bothviability and function in vivo, and that in bypass conditions,loss of either of these regions is as detrimental to cellularfitness as loss of the entire gene. In contrast, the domainpreviously defined as critical for nucleosome targeting(Epl1-NP) in vitro, although important, is less critical duringbypass suppression.
Because nucleosomal H4 acetylation by Esa1 in the pic-colo-NuA4 complex is one of its defining features(Boudreault et al. 2003), we evaluated H4 acetylation as aproxy for NuA4 catalytic activity in the EPL1mutants. For thelysines probed, we observed that mutants of all three EPcAsubdomains (epl1-NPD, epl1-ED, epl1-YD) were defective forH4 acetylation in the bypass state (Figure 6C). Accordingly,loss of the entire EPcA domain (epl1-NtD) leads to similarlylow levels of H4 acetylation. This is a striking distinctionfrom the growth assays where epl1-NPD was more robustthan epl1-ED or epl1-YD, thus pointing to the idea that sub-strates in addition to H4 may be critical for full biologicalfunction.
One of the keyfindings from the initial Epl1 bypass analysiswas that Epl1 promotes the stable chromatin association ofEsa1 (Figure 4A). Because our mutational studies revealedthat the most critical Epl1 residues (Epl1-E and Epl1-Y) wererequired for growth at high temperature, response to DNAdamage, and histone H4 acetylation, we initially hypothe-sized that these same residues might be important for pro-moting chromatin association. We performed fractionationassays as above, in this case with each of the Epl1 mutantsin the sds3D bypass background (Figure 7A). We found thateach of the mutants in the essential EPcA domain retainedchromatin association and, likewise, Esa1 remained chroma-tin associated in each of these mutants.
Previous in vitro studies suggested a key role for the Epl1-NPregion of the protein in nucleosomal binding (Chittuluruet al. 2011; Xu et al. 2016). However, the observed (Figure7A) epl1-NPD mutant protein associated with chromatinin vivo. In contrast, a small amount of the Epl1-CtD proteinshifted to the soluble pool (S), with a similar shift observedfor Esa1 in this background. Sir2 remained chromatin boundregardless of EPL1mutations. The shift to the soluble pool inepl1-CtD sds3D for both Epl1 and Esa1 supports the idea thatthe C-terminus acts to stabilize both Esa1 and Epl1 in chro-matin, thus defining a new role for this most divergent regionof Epl1 and its orthologs.
Physical association between Esa1 and Epl1 is requiredfor activity
From earlier in vitro studies, Epl1 was divided into two do-mains: the EPcA domain that physically interacts withthe Esa1, Yng2, and Eaf6 piccolo-NuA4 subunits and theC-terminus that tethers Epl1 and the piccolo subunits to theNuA4 holo-complex through Eaf1 (Boudreault et al. 2003;Auger et al. 2008; Rossetto et al. 2014). These regions hadnot yet been evaluated in vivo, so we sought to determinewhich are essential for the interaction with Esa1, and simul-taneously which are required for interaction with Yng2 and,by extension, Eaf6.
We immunoprecipitated Epl1 in WT, in each of the threeEPcA subdomain mutants, and in the C-terminal deletionmutant, and then immunoblotted for Esa1 and Yng2. Wefound that only Epl1-NPD and Epl1-CtD retained interactionwith both Esa1 and Yng2 (Figure 7B). This connection of
Figure 6 Subunit interaction domains are critical for Epl1 function in vivo.(A) Under sds3D bypass conditions, EPL1 mutants were surveyed forgrowth on SC medium: epl1-NPD (LPY22111), epl1-ED (LPY22017),epl1-YD (LPY22185), epl1-NtD (LPY22091), epl1-CtD (LPY22033).Growth at increasing temperatures is shown in comparison to WT(LPY22004) and sds3D (LPY22006). (B) Sensitivity to a spectrum ofDNA-damaging agents at 24!. The growth control 24! SC plate is shownin (A), and DMSO is included as the vehicle control for CPT. (C) HistoneH4 acetylation is low among mutants in the essential EPcA domainof EPL1 relative to WT, whereas H4 acetylation is at WT levels in theepl1-CtD mutant.
1132 N. E. Searle, A. L. Torres-Machorro, and L. Pillus

39
Epl1-NPD to the NuA4 holo-complex offers an explanationfor its ability to survive under nonbypass conditions, andupon DNA damage and high-temperature stress. Consis-tent with recent structural analysis (Xu et al. 2016), wefound that Epl1-YD lost physical interaction with Yng2in vivo. Interestingly, we found that Epl1-E and Epl1-Y,the two most critical domains of Epl1 defined phenotypi-cally, were both essential for robust physical interactionwith Esa1. Our results, in tandem with earlier in vitro stud-ies illustrating that HAT activity of Esa1 is directly aug-mented by Epl1 (Boudreault et al. 2003), support theidea that Epl1 is a critical NuA4 subunit due to a role asan Esa1-cofactor. Thus, Epl1 is a central regulator that is ascrucial for NuA4 complex function as the Esa1 enzymeitself.
Discussion
Defining the function of a noncatalytic component of a mac-romolecular complex can be a challenge, particularly whenthat component is essential forviability.Suchhasbeen thecaseforEnhancer of polycomb,originally identifiedasa suppressorof position-effect variegation in Drosophila (Sinclair et al.1998). Shortly after its genetic discovery, E(Pc) was clonedand found to be both deeply conserved from yeast (Epl1) tohumans (EPC1/2) (Stankunas et al. 1998; Doyon et al.2004) and to be essential for chromatin-directed functions.Specifically, Epl1 was identified as a critical subunit of theconserved MYST-family histone acetyltransferase NuA4
complex (Galarneau et al. 2000) and appears to be dedicatedto NuA4/piccolo-NuA4.
Progressmade toward understanding the functions of Epl1include identification of the essential EPcA domain of Epl1,characterization of essential in vitro functions, and most re-cently structural analysis of Epl1 as part of nucleosome-bound NuA4 core complex (Boudreault et al. 2003; Sellecket al. 2005; Chittuluru et al. 2011; Huang and Tan 2012; Xuet al. 2016). Despite this progress, analysis of Epl1 in vivo hasbeen relatively modest. The discovery reported here, that therequirement for EPL1 could be bypassed by deletion of acomponent of a histone deacetylase complex, provided aunique advantage for performing in vivo studies in epl1Dstrains.
We found that the essential requirement for Epl1 and Esa1could be bypassed by loss of the Rpd3L deacetylase, but notfor the other four essential subunits in NuA4 (Figure 1B).Earlier studies suggested that among the essential subunits,only Epl1 and Esa1 appear to be dedicated to NuA4/piccolo-NuA4, whereas the others participate in additional chromatin-modifying complexes or cellular structures. These includeTra1, an ATM-family cofactor, which serves as a recruitmentmodule in SAGA and SLIK/SALSA complexes (Grant et al.1998) and Swc4, Arp4, and Act1, which are components ofthe SWR1 chromatin-remodeling complex (Krogan et al.2004; Mizuguchi et al. 2004) and serve other cellular roles.Specifically, Arp4 and Act1 are also found in the INO80 ATP-dependent chromatin-remodeling complex (Shen et al.2000) and Act1 is an essential cytoskeletal protein (Shortle
Figure 7 Viability of epl1 mutants is linked to sta-ble Epl1–Esa1 interaction, not chromatin associa-tion. (A) Both Epl1 and Esa1 remain chromatinassociated like WT (LPY22231) in all mutants ofthe essential EPcA domain [epl1-NPD-13MYCsds3D ESA1-3HA (LPY22213), epl1-ED-13MYCsds3D ESA1-3HA (LPY22208), epl1-YD-13MYCsds3D ESA1-3HA (LPY22226), epl1-NtD-13MYCsds3D ESA1-3HA (LPY22209)]. However, upon lossof the C-terminus of Epl1 [epl1-CtD-13MYC sds3DESA1-3HA (LPY22211)], both Epl1 and Esa1 areshifted to occupy both soluble and chromatin-bound pools. The observed shift of Epl1 associa-tion in esa1D sds3D here also controls for apossibility of the MYC-tag causing unintended as-sociation. (B) The physical interaction of Esa1–Epl1is disrupted in the epl1-ED and epl1-YD mutants,but not in the epl1-NPD and epl1-CtD mu-tants. Immunoblots following immunoprecipitationillustrate the loss of the physical interaction inthe two essential domain mutants as seen in WT,epl1-NPD, and in epl1-CtD, and lack of nonspecificbinding at the relevant molecular weights in theno-tag control (LPY79). Additionally, physical inter-action with Yng2 is lost only in epl1-YD. ♦ markscross-reactivity with IgG-heavy chain of the anti-body. Whole-cell lysate was prepared for yng2D(LPY22421) in no-tag control background andis included as a negative control for the Yng2antibody.
Bypassing EPL1 Defines Key Functions 1133

40
et al. 1982). Given this context, the bypass suppression ofEpl1, but not the other essential subunits, underscores itsexclusive importance as a NuA4 subunit.
We have demonstrated that Epl1 is important for promot-ing the stable chromatin association of Esa1 through thenonessential C-terminus of Epl1. This was counter to expec-tations because Esa1 nucleosomal association was reportedpreviously to occur via the N-terminal EPcA subdomain(Epl1-NP), essential for H4 but not H2A acetylation(Boudreault et al. 2003; Selleck et al. 2005; Chittuluruet al. 2011; Huang and Tan 2012; Lalonde et al. 2013; Xuet al. 2016). An important distinction is that in contrast toin vitro experiments utilizing recombinant piccolo-NuA4components, epl1-NPD sds3D retains an assembled NuA4holo-complex. Thus, it is possible that whereas an isolat-ed Epl1 requires Epl1-NP for nucleosomal association, inepl1-NPD sds3D, other chromatin-interacting subunits inNuA4 that are still attached to Epl1 may efficiently targetEpl1 (and Esa1) to chromatin.
We found that without the Epl1 C-terminus (epl1-CtD), H4acetylation remained at WT levels. Additionally, in Epl1 mu-tants with disrupted Epl1-Esa1 physical interactions, Esa1remained chromatin-targeted. Whereas these findings mayat first appear to be at odds, there are several key consider-ations. It is possible that, by default, Esa1 is associated withchromatin.When loss of the C-terminus dissociates Epl1 fromchromatin, it may bring along Esa1. This possibility is sup-ported by the fact that in epl1-ED sds3D and epl1-YD sds3D,Esa1 remains chromatin associated (Figure 7A) despite notphysically interacting with Epl1 (Figure 7B). Even withoutEpl1, Esa1 may transiently associate with chromatin (FigureS3 in File S1), allowing the dynamic and rapid process ofacetylation to occur. It is also possible that the small amountof chromatin association that remains is sufficient for Esa1catalytic activity, especially in the bypass state, where Rpd3Ldoes not actively deacetylate histone H4. The NuA4 holo-complex may also be required for stable chromatin associa-tion, such that the interaction of Esa1 is facilitated by othersubunits, where loss of the C-terminus of Epl1 specificallyrepresents dissociation of piccolo-NuA4. Further studies in-volving the nonessential NuA4 Eaf1 subunit and the dynam-ics between Epl1 and Eaf1 in vivo may provide insight intothese possibilities.
Our results support a model in which Epl1 is physicallyrequired for promoting Esa1 enzymatic activity as a part ofthe piccolo-NuA4 and/or the NuA4 holo-complexes, muchlike Ada2 and Ada3, which act in a catalytic core to potentiatethe activity of the Gcn5 acetyltransferase (Balasubramanianet al. 2002). In WT or sds3D cells, Epl1 acts as an anchor forpiccolo-NuA4 subunits, including Esa1, and also tethers thesesubunits to the NuA4 holo-complex (Figure 8A). With bothNuA4 and piccolo-NuA4 intact, as they are in WT and insds3D, there are normal levels of acetylation. However, uponloss of the nonessential C-terminus (epl1-CtD sds3D), piccolo-NuA4 becomes untethered, and the NuA4 holo-complex isdisrupted, leaving only the broad nucleosomal HAT function
of piccolo-NuA4 (Figure 8B). This is sufficient for global, lesstargeted acetylation of histone H4 but perhaps not for acet-ylation of nonhistone substrates that contribute to fitness. Inepl1-NPD sds3D, all piccolo-NuA4 subunits, including Esa1and Epl1, still physically interact, with epl1-NPD still permit-ting both NuA4 and piccolo-NuA4 integrity; however, thismutation causes a reduction in acetylation relative to WTand sds3D (Figure 8C), perhaps due to inefficient nontar-geted chromatin binding.
Despite reduced acetylation levels in epl1-NPD, we hy-pothesize that the presence and targeted activities of theNuA4 holo-complex may be sufficient to promote cellularviability and in bypass conditions, response to cellularstresses such as DNA damage. In fact, we found that disrup-tion of the NuA4 holo-complex by eaf1D results in lethality ofepl1-NPD in nonbypass conditions (Figure S4 in File S1),highlighting the importance of the NuA4 holo-complex forgrowth in the epl1-NPD mutant background. Further, wefound that if Esa1 was simply disassociated from Epl1, suchthat it was no longer a component of NuA4 or piccolo-NuA4,as was the case in both epl1-ED sds3D and epl1-YD sds3D,cellular growth was severely compromised, with low levelsof acetylation, and death at elevated temperatures or withDNA damage (Figure 8, D and E). These findings comple-ment the recently published structural insights for piccolo-NuA4 (Xu et al. 2016), which illustrate that residues withinthose deleted in epl1-ED and epl1-YD were most critical forcontacting both Esa1 and the nucleosome.
Overall, our results support the concept that Epl1 is re-quired to function in tandem with Esa1, tethering Esa1 toother subunits for full and robust function. This concept issupported by in vitro experiments demonstrating negligi-ble HAT activity of Esa1 in the absence of the Epl1 andYng2 piccolo-NuA4 subunits (Boudreault et al. 2003). Wehave shown that if Epl1 is not present (epl1D sds3D), or un-able to physically interact with Esa1 (epl1-ED sds3D andepl1-YD sds3D), Esa1 becomes ineffectual.
The majority of the studies reported here have been per-formed where the requirement for Epl1 is conditionallybypassed by sds3D, serving to balance cellular acetylation,as in our studies of Esa1 (Torres-Machorro and Pillus2014). Historically, bypass suppression has promoted funda-mental understanding of multiple and diverse pathways. Thisincludes, for example, studies of cell-cycle checkpoints wheresuppression of mec1D and rad53D lethality is bypassed byconcurrent loss of SML1 (Zhao et al. 1998), and more recentstudies of transcription regulation, where the requirement forthe COMPASS methyltransferase subunit Swd2 is bypassedby set1D (Soares and Buratowski 2012). Likewise, bypasssuppression served as a powerful tool here that has allowedcomprehensive functional assessment of EPL1 in vivo. How-ever, the concurrent loss of a major deacetylase should bekept in mind in the interpretation of data. It is only in thiscontext that the relative comparisons between the mutantsin vivo can be made with the earlier biochemical analyses toprovide a deeper, valuable, and more holistic understanding
1134 N. E. Searle, A. L. Torres-Machorro, and L. Pillus

41
of an essential protein-modifying activity. Studies in Drosoph-ila and humans alike illustrate that Epl1 orthologs play keyroles in development and cancer, akin to Epl1’s essential rolein yeast. In addition to established roles in DNA damage(Figure 2B) (Boudreault et al. 2003), human EPC1 poten-tially has critical roles in DNA damage repair both withinand independently of NuA4 (Attwooll et al. 2005; Wanget al. 2016). Failures in DNA damage repair are associatedwith genomic instability, which is a major driving force incancer. The observation that mutational profiles of cancerpatients reflect frequent alterations in EPC1/2 highlightsthe importance of these proteins in human biology and dis-ease. Analysis of genomic cancer data illustrates, for exam-
ple, that EPC1 is frequently amplified in neuroendocrineprostate cancer, but deleted in prostate adenocarcinoma(Cerami et al. 2012; Gao et al. 2013). Additionally, indepen-dent analysis demonstrates that EPC1 and EPC2 are oftenmutated across the gene body in many cancer subtypes, in-cluding in the critical domains studied here (Forbes et al.2014). These frequent yet diverse alterations underscorethe importance of understanding the critical functions of spe-cific residues and domains of Epl1, along with the conse-quences of complete deletion of this essential gene. Ourresults provide new insights into both aspects of altered func-tion andwill be instrumental in deepening the understandingof Epl1 orthologs in development and disease.
Figure 8 Model: Epl1 is a core NuA4 regulator in tandem with Esa1. (A) Epl1 is a central component of NuA4 and piccolo-NuA4 (abbreviatedpic-NuA4, shown here only as a part of NuA4). In WT and sds3D, both complexes are intact and active, resulting in normal levels of acetylation, bothof histone H4, as shown in the tails of the H3–H4 tetramer, and of nonhistone substrates, with two representative substrates illustrated, out of over250 reported in proteomic studies. (B) Loss of the Epl1 C-terminus results in loss of the NuA4 holo-complex, but largely uncompromised pic-NuA4function, and slightly reduced acetylation levels of nonhistone substrates only in the sds3D background. (C) epl1-NPD keeps NuA4 intact, and theessential components of pic-NuA4 remain tethered, promoting robust fitness in the bypass state; however, low acetylation levels are present. Theassembly of the NuA4 holo-complex is critical here, such that upon loss of EAF1, epl1-NPD is no longer viable in nonbypass conditions (Figure S4 inFile S1). (D) Loss of the subunit-interaction domains [epl1-E here and epl1-Y in (E)] results in Esa1 no longer structurally bound to Epl1, causing bothNuA4 and pic-NuA4 to be compromised. This results in low acetylation and overwhelming loss of cellular fitness similar to epl1D sds3D. (E) Bycomparison, epl1-YD sds3D results in the similar loss of physical contact with Esa1, but also loss of physical interaction with Yng2, and therefore byextension, Eaf6. This mutant has the same severe fitness-deficits as epl1-ED sds3D and epl1D sds3D, underscoring the primary importance of theEsa1–Epl1 interaction. Although not illustrated in the model, epl1D sds3D would be similar to epl1-ED sds3D and epl1-YD sds3D, with low levels ofacetylation; however in the complete absence of Epl1, all piccolo-NuA4 subunits would be disassociated from the NuA4 holo-complex. Note that inour experiments, analysis of H4 acetylation is a proxy for NuA4/piccolo-NuA4 activity. Proteomic studies define many additional substrates for NuA4activity, some of which are likely to contribute to processes affected upon loss of Epl1 or Esa1 functions. Of note, both Yng2 and Epl1 were identifiedas substrates of Esa1, where acetylation has already been demonstrated to have a significant functional impact (Yi et al. 2012; Mitchell et al. 2013;Downey et al. 2015). Therefore, we believe that nonhistone substrates, though not specifically analyzed here, are a critical part of the observedphenotypes and model presented.
Bypassing EPL1 Defines Key Functions 1135

42
Acknowledgments
We thank the Amberg, Cole, Côté, Tan, Kobor, and Thornerlaboratories for providing strains, plasmids, and reagents,Dong Wang (University of California, San Diego [UCSD]Pharmaceutical Sciences) for structural biology perspectiveand guidance on Epl1 mutant design, and Gene Yeo (UCSDCellular and Molecular Medicine) and laboratory membersG. Pratt, B. Yee, and J. Nussbacher for guidance in processingand analyzing the RNA-seq data. RNA library preparation andsequencing was conducted at the IGM Genomics Center, Uni-versity of California, San Diego, La Jolla, CAwith core supportof National Institutes of Health grant P30CA023100. Wethank current and past members of the Pillus laboratory forthoughtful discussion and critical feedback throughout thecourse of this study and during the preparation of this man-uscript, and members of the Hampton laboratory for use ofequipment and technical advice. N.E.S. was supported by NIHpredoctoral training award T32 GM008666, the UCSD Fron-tiers of Innovation Scholars Program Graduate Fellowship,and the UCSD Division of Molecular Biology Cancer Fellow-ship. A.L.T.-M. was supported by the University of CaliforniaInstitute for Mexico and the United States (UCMEXUS) andby the National Council of Science and Technology of Mexico(CONACYT, Consejo Nacional de Ciencia y Tecnología).
Literature Cited
Attwooll, C., S. Oddi, P. Cartwright, E. Prosperini, K. Agger et al.,2005 A novel repressive E2F6 complex containing the poly-comb group protein, EPC1, that interacts with EZH2 in a pro-liferation-specific manner. J. Biol. Chem. 280: 1199–1208.
Auger, A., L. Galarneau, M. Altaf, A. Nourani, Y. Doyon et al.,2008 Eaf1 is the platform for NuA4 molecular assembly thatevolutionarily links chromatin acetylation to ATP-dependent ex-change of histone H2A variants. Mol. Cell. Biol. 28: 2257–2270.
Avvakumov, N., and J. Côté, 2007 The MYST family of histoneacetyltransferases and their intimate links to cancer. Oncogene26: 5395–5407.
Balasubramanian, R., M. G. Pray-Grant, W. Selleck, P. A. Grant, andS. Tan, 2002 Role of the Ada2 and Ada3 transcriptional coac-tivators in histone acetylation. J. Biol. Chem. 277: 7989–7995.
Bao, W., K. K. Kojima, and O. Kohany, 2015 Repbase update, a data-base of repetitive elements in eukaryotic genomes. Mob. DNA 6: 11.
Biankin, A. V., N. Waddell, K. S. Kassahn, M.-C. Gingras, L. B.Muthuswamy et al., 2012 Pancreatic cancer genomes revealaberrations in axon guidance pathway genes. Nature 491:399–405.
Bird, A. W., D. Y. Yu, M. G. Pray-Grant, Q. Qiu, K. E. Harmon et al.,2002 Acetylation of histone H4 by Esa1 is required for DNAdouble-strand break repair. Nature 419: 411–415.
Bittner, C. B., D. T. Zeisig, B. B. Zeisig, and R. K. Slany, 2004 Directphysical and functional interaction of the NuA4 complex compo-nents Yaf9p and Swc4p. Eukaryot. Cell 3: 976–983.
Bond, J. F., J. L. Fridovich-Keil, L. Pillus, R. C. Mulligan, and F.Solomon, 1986 A chicken-yeast chimeric beta-tubulin proteinis incorporated into mouse microtubules in vivo. Cell 44: 461–468.
Boudreault, A. A., D. Cronier, W. Selleck, N. Lacoste, R. T. Utleyet al., 2003 Yeast enhancer of polycomb defines global Esa1-dependent acetylation of chromatin. Genes Dev. 17: 1415–1428.
Campos, E. I., and D. Reinberg, 2009 Histones: annotating chro-matin. Annu. Rev. Genet. 43: 559–599.
Cerami, E., J. Gao, U. Dogrusoz, B. E. Gross, S. O. Sumer et al.,2012 The cBio cancer genomics portal: an open platform forexploring multidimensional cancer genomics data. Cancer Dis-cov. 2: 401–404.
Chang, C. S., and L. Pillus, 2009 Collaboration between the es-sential Esa1 acetyltransferase and the Rpd3 deacetylase is me-diated by H4K12 histone acetylation in Saccharomyces cerevisiae.Genetics 183: 149–160.
Chang, C. S., A. Clarke, and L. Pillus, 2012 Suppression analysis ofesa1 mutants in Saccharomyces cerevisiae links NAB3 to transcrip-tional silencing and nucleolar functions. G3 2: 1223–1232.
Cheng, X., and J. Côté, 2014 A new companion of elongating RNAPolymerase II: TINTIN, an independent sub-module of NuA4/TIP60 for nucleosome transactions. Transcription 5: e995571.
Chittuluru, J. R., Y. Chaban, J. Monnet-Saksouk, M. J. Carrozza, V.Sapountzi et al., 2011 Structure and nucleosome interaction ofthe yeast NuA4 and Piccolo-NuA4 histone acetyltransferasecomplexes. Nat. Struct. Mol. Biol. 18: 1196–1203.
Clarke, A. S., J. E. Lowell, S. J. Jacobson, and L. Pillus,1999 Esa1p is an essential histone acetyltransferase requiredfor cell cycle progression. Mol. Cell. Biol. 19: 2515–2526.
Clarke, A. S., E. Samal, and L. Pillus, 2006 Distinct roles for theessential MYST family HAT Esa1p in transcriptional silencing.Mol. Biol. Cell 17: 1744–1757.
Decker, P. V., D. Y. Yu, M. Iizuka, Q. Qiu, and M. M. Smith,2008 Catalytic-site mutations in the MYST family histone ace-tyltransferase Esa1. Genetics 178: 1209–1220.
Dobin, A., C. A. Davis, F. Schlesinger, J. Drenkow, C. Zaleski et al.,2013 STAR: ultrafast universal RNA-seq aligner. Bioinfor-matics 29: 15–21.
Downey, M., J. R. Johnson, N. E. Davey, B. W. Newton, T. L. Johnsonet al., 2015 Acetylome profiling reveals overlap in the regulationof diverse processes by sirtuins, gcn5, and esa1. MCP 14: 162–176.
Doyon, Y., W. Selleck, W. S. Lane, S. Tan, and J. Côté,2004 Structural and functional conservation of the NuA4 his-tone acetyltransferase complex from yeast to humans. Mol. Cell.Biol. 24: 1884–1896.
Durant, M., and B. F. Pugh, 2006 Genome-wide relationships be-tween TAF1 and histone acetyltransferases in Saccharomycescerevisiae. Mol. Cell. Biol. 26: 2791–2802.
Engel, S. R., F. S. Dietrich, D. G. Fisk, G. Binkley, R. Balakrishnanet al., 2014 The reference genome sequence of Saccharomycescerevisiae: then and now. G3 4: 389–398.
Evan, G. I., G. K. Lewis, G. Ramsay, and J. M. Bishop,1985 Isolation of monoclonal antibodies specific for humanc-myc proto-oncogene product. Mol. Cell. Biol. 5: 3610–3616.
Forbes, S., D. Beare, K. Leung, N. Bindal, S. Bamford et al.,2014 COSMIC: exploring novel cancer biomarkers. Eur.J. Cancer 50: S111.
Friis, R. M., B. P. Wu, S. N. Reinke, D. J. Hockman, B. D. Sykeset al., 2009 A glycolytic burst drives glucose induction ofglobal histone acetylation by picNuA4 and SAGA. Nucleic AcidsRes. 37: 3969–3980.
Galarneau, L., A. Nourani, A. A. Boudreault, Y. Zhang, L. Héliotet al., 2000 Multiple links between the NuA4 histone acetyl-transferase complex and epigenetic control of transcription.Mol. Cell 5: 927–937.
Gao, J., B. A. Aksoy, U. Dogrusoz, G. Dresdner, B. Gross et al.,2013 Integrative analysis of complex cancer genomics andclinical profiles using the cBioPortal. Sci. Signal. 6: pl1.
Garcia, S. N., and L. Pillus, 2002 A unique class of conditional sir2mutants displays distinct silencing defects in Saccharomyces cer-evisiae. Genetics 162: 721–736.
Gaytán, B. D., A. V. Loguinov, V. Y. De La Rosa, J.-M. Lerot, and C.D. Vulpe, 2013 Functional genomics indicates yeast requires
1136 N. E. Searle, A. L. Torres-Machorro, and L. Pillus

43
Golgi/ER transport, chromatin remodeling, and DNA repair forlow dose DMSO tolerance. Front. Genet. 4: 154.
Grant, P. A., D. Schieltz, M. G. Pray-Grant, I. Yates, R. John et al.,1998 The ATM-related cofactor Tra1 is a component of thepurified SAGA complex. Mol. Cell 2: 863–867.
Huang, J., and S. Tan, 2012 Piccolo NuA4-catalyzed acetylationof nucleosomal histones: critical roles of an Esa1 tudor/chromobarrel loop and an Epl1 Enhancer of Polycomb A (EPcA) basicregion. Mol. Cell. Biol. 33: 159–169.
Huang, X., G. J. Spencer, J. T. Lynch, F. Ciceri, T. D. D. Somerville et al.,2014 Enhancers of Polycomb EPC1 and EPC2 sustain the oncogenicpotential of MLL leukemia stem cells. Leukemia 28: 1081–1091.
Kakolyri, M., A. Margaritou, and E. Tiligada, 2016 Dimethyl sulph-oxide modifies growth and senescence and induces the non-revertible petite phenotype in yeast. FEMS Yeast Res. 16: fow008.
Keogh, M.-C., T. A. Mennella, C. Sawa, S. Berthelet, N. J. Kroganet al., 2006 The Saccharomyces cerevisiae histone H2A variantHtz1 is acetylated by NuA4. Genes Dev. 20: 660–665.
Kornberg, R. D., and Y. Lorch, 1999 Twenty-five years of thenucleosome, fundamental particle of the eukaryote chromo-some. Cell 98: 285–294.
Kouzarides, T., 2007 Chromatin modifications and their function.Cell 128: 693–705.
Krogan, N. J., K. Baetz, M.-C. Keogh, N. Datta, C. Sawa et al.,2004 Regulation of chromosome stability by the histone H2Avariant Htz1, the Swr1 chromatin remodeling complex, and thehistone acetyltransferase NuA4. Proc. Natl. Acad. Sci. USA 101:13513–13518.
Lafon, A., C. S. Chang, E. M. Scott, S. J. Jacobson, and L. Pillus,2007 MYST opportunities for growth control: yeast genes illu-minate human cancer gene functions. Oncogene 26: 5373–5384.
Lalonde, M. E., N. Avvakumov, K. C. Glass, F. H. Joncas, N. Saksouket al., 2013 Exchange of associated factors directs a switch in HBO1acetyltransferase histone tail specificity. Genes Dev. 27: 2009–2024.
Liang, C., and B. Stillman, 1997 Persistent initiation of DNA rep-lication and chromatin-bound MCM proteins during the cellcycle in cdc6 mutants. Genes Dev. 11: 3375–3386.
Lin, Y.-y., J.-y. Lu, J. Zhang, W. Walter, W. Dang et al., 2009 Proteinacetylation microarray reveals that NuA4 controls key metabolictarget regulating gluconeogenesis. Cell 136: 1073–1084.
Love, M. I., W. Huber, and S. Anders, 2014 Moderated estimationof fold change and dispersion for RNA-seq data with DESeq2.Genome Biol. 15: 550.
Martin, M., 2011 Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet.journal 17: 10–12.
Mitchell, L., J.-P. Lambert, M. Gerdes, A. S. Al-Madhoun, I. S. Skerjancet al., 2008 Functional dissection of the NuA4 histone acetyl-transferase reveals its role as a genetic hub and that Eaf1 is essen-tial for complex integrity. Mol. Cell. Biol. 28: 2244–2256.
Mitchell, L., S. Huard, M. Cotrut, R. Pourhanifeh-Lemeri, A.-L. Steunouet al., 2013 mChIP-KAT-MS, a method to map protein interac-tions and acetylation sites for lysine acetyltransferases. Proc. Natl.Acad. Sci. USA 110: E1641–E1650.
Mizuguchi, G., X. Shen, J. Landry, W.-H. Wu, S. Sen et al.,2004 ATP-driven exchange of histone H2AZ variant catalyzedby SWR1 chromatin remodeling complex. Science 303: 343–348.
Nakahata, S., Y. Saito, M. Hamasaki, T. Hidaka, Y. Arai et al.,2009 Alteration of enhancer of polycomb 1 at 10p11.2 isone of the genetic events leading to development of adultT-cell leukemia/lymphoma. Gene Chromosomes Cancer 48:768–776.
Ohba, R., D. J. Steger, J. E. Brownell, C. A. Mizzen, R. G. Cook et al.,1999 A novel H2A/H4 nucleosomal histone acetyltransferasein Tetrahymena thermophila. Mol. Cell. Biol. 19: 2061–2068.
R Development Core Team, 2015 R: A Language and Environmentfor Statistical Computing. R Foundation for Statistical Comput-ing, Vienna, Austria.
Reid, J. L., V. R. Iyer, P. O. Brown, and K. Struhl, 2000 Coordinateregulation of yeast ribosomal protein genes is associated with tar-geted recruitment of Esa1 histone acetylase. Mol. Cell 6: 1297–1307.
Robert, F., D. K. Pokholok, N. M. Hannett, N. J. Rinaldi, M. Chandyet al., 2004 Global position and recruitment of HATs andHDACs in the yeast genome. Mol. Cell 16: 199–209.
Rossetto, D., M. Cramet, A. Y. Wang, A. L. Steunou, N. Lacosteet al., 2014 Eaf5/7/3 form a functionally independent NuA4submodule linked to RNA polymerase II-coupled nucleosomerecycling. EMBO J. 33: 1397–1415.
Sadowska-Bartosz, I., A. Pączka, M. Mołoń, and G. Bartosz,2013 Dimethyl sulfoxide induces oxidative stress in the yeastSaccharomyces cerevisiae. FEMS Yeast Res. 13: 820–830.
Selleck, W., I. Fortin, D. Sermwittayawong, J. Côté, and S. Tan,2005 The Saccharomyces cerevisiae piccolo NuA4 histone ace-tyltransferase complex requires the enhancer of polycomb Adomain and chromodomain to acetylate nucleosomes. Mol. Cell.Biol. 25: 5535–5542.
Shen, X., G. Mizuguchi, A. Hamiche, and C. Wu, 2000 A chroma-tin remodelling complex involved in transcription and DNA pro-cessing. Nature 406: 541–544.
Shimono, Y., H. Murakami, Y. Hasegawa, and M. Takahashi,2000 RET finger protein is a transcriptional repressor and in-teracts with enhancer of polycomb that has dual transcriptionalfunctions. J. Biol. Chem. 275: 39411–39419.
Shortle, D., J. E. Haber, and D. Botstein, 1982 Lethal disruption ofthe yeast actin gene by integrative DNA transformation. Science217: 371–373.
Sinclair, D. A., N. J. Clegg, J. Antonchuk, T. A. Milne, K. Stankunaset al., 1998 Enhancer of Polycomb is a suppressor of position-effectvariegation in Drosophila melanogaster. Genetics 148: 211–220.
Smith, E. R., A. Eisen, W. Gu, M. Sattah, A. Pannuti et al.,1998 ESA1 is a histone acetyltransferase that is essential forgrowth in yeast. Proc. Natl. Acad. Sci. USA 95: 3561–3565.
Soares, L. M., and S. Buratowski, 2012 Yeast Swd2 is essentialbecause of antagonism between Set1 histone methyltransferasecomplex and APT (Associated with Pta1) termination factor.J. Biol. Chem. 287: 15219–15231.
Stankunas, K., J. Berger, C. Ruse, D. A. Sinclair, F. Randazzo et al.,1998 The enhancer of polycomb gene of Drosophila encodes achromatin protein conserved in yeast and mammals. Develop-ment 125: 4055–4066.
Torres-Machorro, A. L., and L. Pillus, 2014 Bypassing the require-ment for an essential MYST acetyltransferase. Genetics 197:851–863.
Uprety, B., R. Sen, and S. R. Bhaumik, 2015 Eaf1p is required forrecruitment of NuA4 in targeting TFIID to the promoters of theribosomal protein genes for transcriptional initiation in vivo.Mol. Cell. Biol. 35: 2947–2964.
Wang, Y., V. Alla, D. Goody, S. K. Gupta, A. Spitschak et al.,2016 Epigenetic factor EPC1 is a master regulator of DNAdamage response by interacting with E2F1 to silence deathand activate metastasis-related gene signatures. Nucleic AcidsRes. 44: 117–133.
Wickham, H., 2009 ggplot2: Elegant Graphics for Data Analysis.Springer-Verlag, New York.
Xu, P., C. Li, Z. Chen, S. Jiang, S. Fan et al., 2016 The NuA4 corecomplex acetylates nucleosomal histone H4 through a doublerecognition mechanism. Mol. Cell 63: 965–975.
Yi, C., M. Ma, L. Ran, J. Zheng, J. Tong et al., 2012 Function andmolecular mechanism of acetylation in autophagy regulation.Science 336: 474–477.
Zhao, X. L., E. G. D. Muller, and R. Rothstein, 1998 A suppressorof two essential checkpoint genes identifies a novel protein thatnegatively affects dNTP pools. Mol. Cell 2: 329–340.
Communicating editor: M. Hampsey
Bypassing EPL1 Defines Key Functions 1137
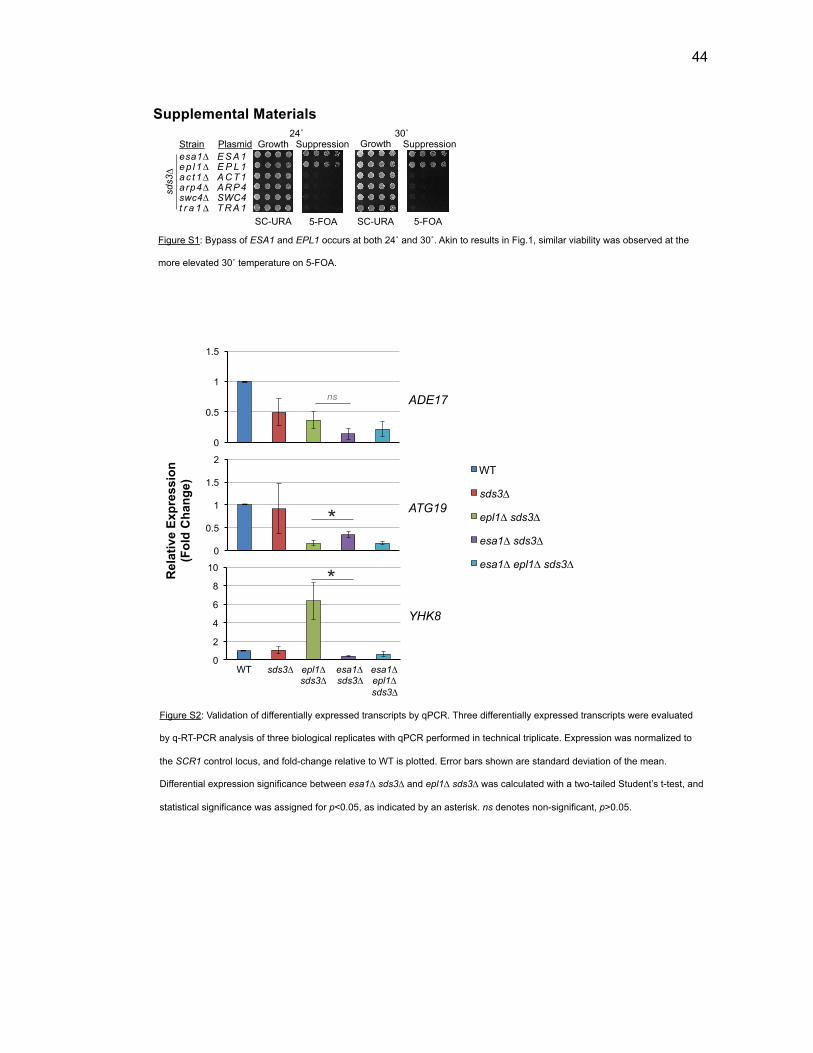
44
Supplemental Materials24˚ 30˚
Strain
esa1∆ e p l 1∆ a c t 1∆ a rp4∆swc4∆t r a 1 ∆
Growth Growth Suppression
SC-URA 5-FOA SC-URA 5-FOA
Suppression
E S A 1 E P L 1 A C T 1 ARP4 SWC4 T R A 1
Plasmid
sds3∆
Supplemental Materials Searle et al. 2016
0123456789
WT
WT
sds3∆
epl1∆ sds3∆
esa1∆ sds3∆
esa1∆ epl1∆ sds3∆
Rel
ativ
e E
xpre
ssio
n (F
old
Cha
nge)
0
0.5
1
1.5
0
0.5
1
1.5
2
0
2
4
6
8
10
YHK8
ATG19
ADE17
*
*
ns
WT sds3∆ epl1∆ esa1∆ esa1∆ sds3∆ sds3∆ epl1∆ sds3∆
Figure S1: Bypass of ESA1 and EPL1 occurs at both 24˚ and 30˚. Akin to results in Fig.1, similar viability was observed at the
more elevated 30˚ temperature on 5-FOA.
Figure S2: Validation of differentially expressed transcripts by qPCR. Three differentially expressed transcripts were evaluated
by q-RT-PCR analysis of three biological replicates with qPCR performed in technical triplicate. Expression was normalized to
the SCR1 control locus, and fold-change relative to WT is plotted. Error bars shown are standard deviation of the mean.
Differential expression significance between esa1∆ sds3∆ and epl1∆ sds3∆ was calculated with a two-tailed Student’s t-test, and
statistical significance was assigned for p<0.05, as indicated by an asterisk. ns denotes non-significant, p>0.05.

45
WT
sds3∆
Esa1-MYC
Sir2
Pgk1
epl1∆ sds3∆ W S C W S C W S C
SC-URA 5-FOA
epl1
-NP∆
+ pE
PL1
EAF1 eaf1∆ eaf1∆ sds3∆ EAF1 sds3∆
Control Viability
Supplemental Materials Searle et al. 2016
Figure S3: Crosslinking stabilizes Esa1 in the chromatin in epl1∆ sds3∆. Subcellular fractionation assays, as in Figure 4, were
performed after a brief 15-minute crosslink with 37% formaldehyde prior to cell lysis and differential centrifugation. With the
addition of the fixation step, Esa1 remained in the whole cell extract (W) and the crude chromatin (C) fractions, but was depleted
from the soluble fraction (S) in WT (LPY21568), sds3∆ (LPY21579), and in epl1∆ sds3∆ (LPY21596).
Figure S4: Eaf1 is required for epl1-NP∆ viability. Strains including the epl1-NP∆ mutant were mated to eaf1∆ to test for ability to
survive without a WT copy of EPL1. Loss of EAF1 serves as a proxy for loss of the NuA4-holocomplex. As compared to
epl1-NP∆ (LPY22120), epl1-NP∆ eaf1∆ (LPY22443) was unable to survive without a plasmid-borne non-mutant EPL1. Likewise,
under bypass conditions, eaf1∆ resulted in loss of fitness of epl1-NP∆ sds3∆ (LPY22000) as compared to wild-type EAF1
(LPY22440), overall indicating the importance of the NuA4-holocomplex in supporting the viability and fitness of epl1-NP∆, in
SDS3 and in sds3∆ bypass conditions, respectively.
46
TABLE S1 – YEAST STRAINS Strain Genotype Reference
LPY20724 MATa his3∆1 leu2∆0 met15∆0 ura3∆0 esa1∆::kanMX sds3∆::kanMX + pLP796
LPY20609 MATa his3∆1 leu2∆0 ura3∆0 epl1∆::kanMX sds3∆::natMX + pLP3189
LPY20974 MATa his3∆1 leu2∆0 ura3∆0 act1∆::natMX sds3∆::kanMX + pKFW29
act1∆::NatMX provided by D. Amberg (SVY12) (HAARER et al. 2007)
LPY20617 MATa his3∆1 leu2∆0 ura3∆0 arp4∆::kanMX sds3∆::natMX + pLP3183
LPY20611 MATa his3∆1 leu2∆0 ura3∆0 swc4∆::kanMX sds3∆::natMX + pLP3190
LPY20443 MATa ade2-1/2-101 can1-100/CAN1 his3-11,15/his3∆200 leu2-3,112 trp1-1 ura3-1/ura3-52 tra1∆::HIS3 sds3∆::kanMX + pTRA1 (URA3) *
tra1∆::HIS3 provided by M. Cole
LPY79 MATα W303 (ade2-1 can1-100 his3-11 leu2,3,112 trp1-1 ura3-1)
LPY20877 MATα W303 sds3∆::natMX
LPY21631 MATα W303 esa1∆::HIS3 sds3∆::natMX LPY21299 MATα W303 epl1∆::kanMX sds3∆::natMX
LPY21751 MATα W303 esa1∆::^HIS3 epl1∆::kanMX sds3∆::natMX
LPY21568 MATα W303 ESA1-13Myc-kanMX
LPY21579 MATα W303 ESA1-13Myc-kanMX sds3∆::natMX LPY21596 MATα W303 ESA1-13Myc-kanMX sds3∆::natMX epl1∆::kanMX
LPY22201 MATα W303 Swc4-3HA::kanMX Provided by M. Kobor
LPY22202 MATα W303 Swc4-3HA::kanMX sds3∆::natMX
LPY21942 MATα W303 epl1∆::kanMX sds3∆::natMX Swc4-3HA::kanMX
LPY21071 MATα W303 epl1∆::kanMX sds3∆::natMX + pLP3189 LPY20759 MATα W303 epl1∆::kanMX + pLP3189
LPY21686 MATa his3∆1 leu2∆0 met15∆0 ura3∆0 EPL1-13MYC-HISMX6
Provided by J. Côté (QY237) (ROSSETTO et al. 2014)
LPY22120 MATα W303 epl1-NP∆-13MYC-HISMX6 + pLP3189 LPY22012 MATα W303 epl1-E∆-13MYC-HISMX6 + pLP3189
LPY22001 MATα W303 epl1-Y∆-13MYC-HISMX6 + pLP3189
LPY22084 MATα W303 epl1-Nt∆-13MYC-HISMX6 + pLP3189
LPY22010 MATα W303 epl1-Ct∆-13MYC-HISMX6 + pLP3189 LPY22204 MATα W303 EPL1-13MYC-HISMX6
LPY22206 MATα W303 EPL1-13MYC-HISMX6 sds3∆::natMX
LPY22111 MATα W303 epl1-NP∆-13MYC-HISMX6 sds3∆::natMX
LPY22017 MATα W303 epl1-E∆-13MYC-HISMX6 sds3∆::natMX LPY22185 MATα W303 epl1-Y∆-13MYC-HISMX6 sds3∆::natMX

47
Searle et al., 2016
* Mixed strain background TABLE S2 – PLASMIDS Plasmid Information Reference
pLP796 ESA1, 2µ URA3 (in YEp352) pLP3183 ARP4, 2µ, URA3 (in pRS426) pLP3189 EPL1, 2µ, URA3 (in pRS426) pLP3190 SWC4, 2µ, URA3 (in pRS426) pKFW29 ACT1, CEN, URA3 D. Amberg (HAARER et al. 2007) pLP74 pKS+ (Bluescript) (ALTING-MEES AND SHORT 1989) pLP3337 EPL1-13MYC-HISMX6 in pLP74 * pLP3340 epl1-NP∆-13MYC-HISMX6 in pLP74 pLP3341 epl1-Y∆-13MYC-HISMX6 in pLP74 pLP3344 epl1-E∆-13MYC-HISMX6 in pLP74 pLP3345 epl1-Ct∆-13MYC-HISMX6 in pLP74 pLP3346 epl1-Nt∆-13MYC-HISMX6 in pLP74
* The genomic DNA that is contained in this plasmid originates from the S288C strain background, which is known to contain several polymorphisms relative to the W303 strain background in the EPL1 ORF (RALSER et al. 2012; ENGEL et al. 2014). At this time, no function has been assigned to the various strain-specific polymorphisms.
LPY22091 MATα W303 epl1-Nt∆-13MYC-HISMX6 sds3∆::natMX
LPY22033 MATα W303 epl1-Ct∆-13MYC-HISMX6 sds3∆::natMX
LPY22231 MATα W303 EPL1-13MYC-HISMX6 Esa1-3HA-kanMX
LPY22232 MATα W303 EPL1-13MYC-HISMX6 sds3∆::natMX Esa1-3HA-kanMX
LPY22213 MATα W303 epl1-NP∆-13MYC-HISMX6 sds3∆::natMX ESA1-3HA-kanMX
LPY22208 MATα W303 epl1-E∆-13MYC-HISMX6 sds3∆::natMX ESA1-3HA-kanMX
LPY22226 MATα W303 epl1-Y∆-13MYC-HISMX6 sds3∆::natMX ESA1-3HA-kanMX
LPY22209 MATα W303 epl1-Nt∆-13MYC-HISMX6 sds3∆::natMX ESA1-3HA-kanMX
LPY22211 MATα W303 epl1-Ct∆-13MYC-HISMX6 sds3∆::natMX ESA1-3HA-kanMX
LPY22421 MATα W303 yng2∆::URA3
LPY22000 MATα W303 epl1-NP∆-13MYC-HISMX6 sds3∆::natMX+ pLP3189
LPY22440 MATα W303 epl1-NP∆-13MYC-HISMX6 sds3∆::natMX eaf1∆::kanMX + pLP3189
LPY22443 MATα W303 epl1-NP∆-13MYC-HISMX6 eaf1∆::kanMX + pLP3189
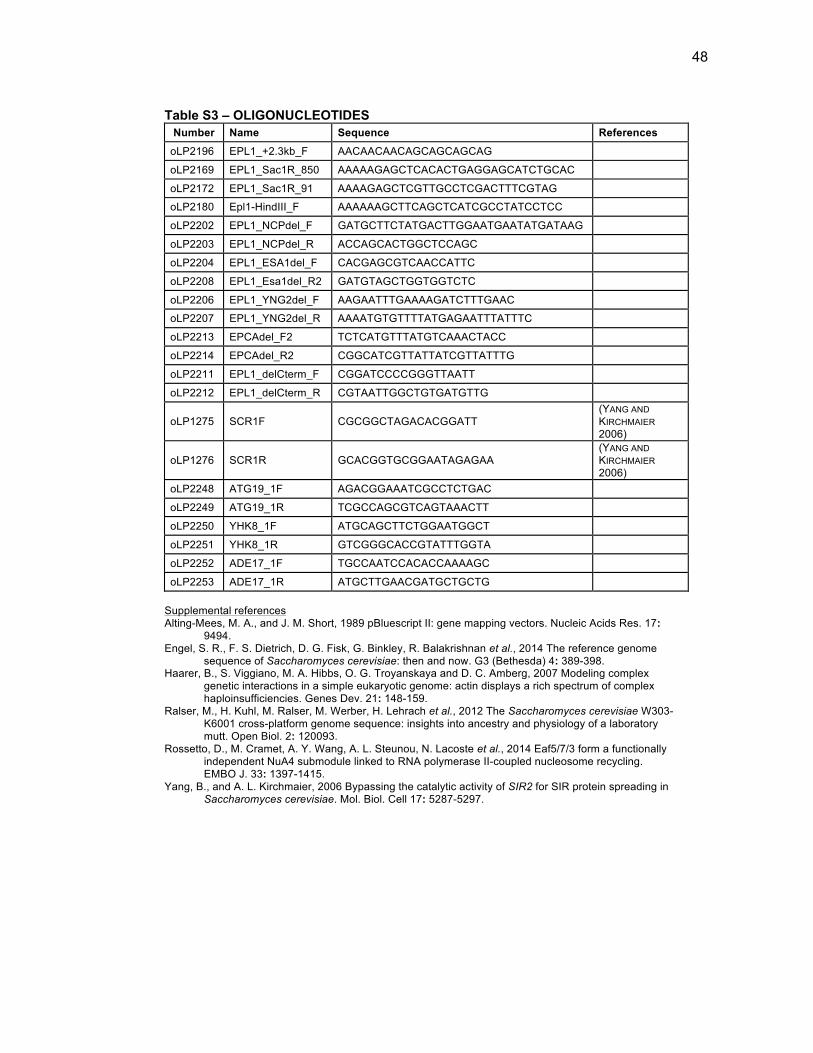
48
Searle et al., 2016
Table S3 – OLIGONUCLEOTIDES Number Name Sequence References
oLP2196 EPL1_+2.3kb_F AACAACAACAGCAGCAGCAG
oLP2169 EPL1_Sac1R_850 AAAAAGAGCTCACACTGAGGAGCATCTGCAC
oLP2172 EPL1_Sac1R_91 AAAAGAGCTCGTTGCCTCGACTTTCGTAG
oLP2180 Epl1-HindIII_F AAAAAAGCTTCAGCTCATCGCCTATCCTCC
oLP2202 EPL1_NCPdel_F GATGCTTCTATGACTTGGAATGAATATGATAAG
oLP2203 EPL1_NCPdel_R ACCAGCACTGGCTCCAGC
oLP2204 EPL1_ESA1del_F CACGAGCGTCAACCATTC
oLP2208 EPL1_Esa1del_R2 GATGTAGCTGGTGGTCTC
oLP2206 EPL1_YNG2del_F AAGAATTTGAAAAGATCTTTGAAC
oLP2207 EPL1_YNG2del_R AAAATGTGTTTTATGAGAATTTATTTC
oLP2213 EPCAdel_F2 TCTCATGTTTATGTCAAACTACC
oLP2214 EPCAdel_R2 CGGCATCGTTATTATCGTTATTTG
oLP2211 EPL1_delCterm_F CGGATCCCCGGGTTAATT
oLP2212 EPL1_delCterm_R CGTAATTGGCTGTGATGTTG
oLP1275 SCR1F CGCGGCTAGACACGGATT (YANG AND KIRCHMAIER 2006)
oLP1276 SCR1R GCACGGTGCGGAATAGAGAA (YANG AND KIRCHMAIER 2006)
oLP2248 ATG19_1F AGACGGAAATCGCCTCTGAC
oLP2249 ATG19_1R TCGCCAGCGTCAGTAAACTT
oLP2250 YHK8_1F ATGCAGCTTCTGGAATGGCT
oLP2251 YHK8_1R GTCGGGCACCGTATTTGGTA
oLP2252 ADE17_1F TGCCAATCCACACCAAAAGC
oLP2253 ADE17_1R ATGCTTGAACGATGCTGCTG Supplemental references Alting-Mees, M. A., and J. M. Short, 1989 pBluescript II: gene mapping vectors. Nucleic Acids Res. 17:
9494. Engel, S. R., F. S. Dietrich, D. G. Fisk, G. Binkley, R. Balakrishnan et al., 2014 The reference genome
sequence of Saccharomyces cerevisiae: then and now. G3 (Bethesda) 4: 389-398. Haarer, B., S. Viggiano, M. A. Hibbs, O. G. Troyanskaya and D. C. Amberg, 2007 Modeling complex
genetic interactions in a simple eukaryotic genome: actin displays a rich spectrum of complex haploinsufficiencies. Genes Dev. 21: 148-159.
Ralser, M., H. Kuhl, M. Ralser, M. Werber, H. Lehrach et al., 2012 The Saccharomyces cerevisiae W303-K6001 cross-platform genome sequence: insights into ancestry and physiology of a laboratory mutt. Open Biol. 2: 120093.
Rossetto, D., M. Cramet, A. Y. Wang, A. L. Steunou, N. Lacoste et al., 2014 Eaf5/7/3 form a functionally independent NuA4 submodule linked to RNA polymerase II-coupled nucleosome recycling. EMBO J. 33: 1397-1415.
Yang, B., and A. L. Kirchmaier, 2006 Bypassing the catalytic activity of SIR2 for SIR protein spreading in Saccharomyces cerevisiae. Mol. Biol. Cell 17: 5287-5297.

49
Acknowledgements This chapter, in full, is a reprint of the material as it appears in Searle
NE, Torres-Machorro AL, Pillus L. 2017. Chromatin Regulation by the NuA4
Acetyltransferase Complex Is Mediated by Essential Interactions Between
Enhancer of Polycomb (Epl1) and Esa1. Genetics 205: 1125-1137. The
dissertation author was the primary investigator and first author of this paper.

50
Chapter 2 Bypass suppression defines key chromatin dynamics between NuA4 and the Hda1 deacetylase
Chapter 3.
Bypass suppression defines key
chromatin dynamics between NuA4
and the Hda1 deacetylase

51
Introduction
The enzymatic activity of histone acetyltransferase (HAT) complexes,
such as NuA4, is opposed by that of histone deacetylase (HDAC) complexes,
another major class of chromatin regulators. There are three primary classes
of histone deacetylases in Saccharomyces cerevisiae, classified by sequence
homology or co-factor dependence (Figure 3-1). Class I includes Rpd3, Hos1,
and Hos2. Class II contains Hda1 and Hos3. Finally, class III (the sirtuins),
includes five NAD+ dependent HDACs, Sir2, Hst1, Hst2, Hst3, and Hst4. The
HDACs are not essential for viability in budding yeast, and generally have less
targeted activity than the HATs. However, studies have illustrated that each
HDAC has a specialized set of functions, and like HATs, are often part of
multimeric complexes, containing additional subunits necessary for full
enzymatic activity [reviewed in (Kurdistani and Grunstein 2003)].
Class I deacetylases. Rpd3 is the founding member of the class I
deacetylases. Rpd3 is generally considered to be one of the main
deacetylases in S. cerevisiae, affecting at least one-quarter of promoters
genome-wide (Kurdistani et al. 2002; Robyr et al. 2002). The Rpd3
deacetylase is primarily found in two distinct complexes, both of which
deacetylate histones H3, H4, H2A, and H2B, along with non-histone
substrates (De Nadal et al. 2004; Carrozza et al. 2005a; Keogh et al. 2005).
The Rpd3-containing complexes differ by subunit composition and genomic
localization. Rpd3S and Rpd3L complexes contain a core set of subunits:

52
Class Deacetylase
I Rpd3 (HDAC1) Hos1 (HDAC8) Hos2 (HDAC3)
II Hda1 (HDAC4) Hos3 (HDAC6)
III (NAD-dependent)
Sir2 Hst1 Hst2 Hst3 Hst4 (SIRT)
Figure 3-1. Three classes of histone deacetylases, based on sequence homology and/or co-factor dependence. There are three major classes of histone deacetylases. The S. cerevisiae HDACs are listed with the primary corresponding mammalian ortholog listed in parentheses. The third class of HDACs, known as the sirtuins, encode NAD-dependent enzymes, whereas the first two classes are classified by sequence homology.

53
Rpd3, Ume1, and Sin3. Rpd3L has additional subunits including Sds3 and
Dep1, responsible for maintaining complex integrity, and the Ume6 and Ash1
subunits that recruit Rpd3L to gene promoters (Carrozza et al. 2005a). In
addition to the core subunits, the Rpd3S complex contains Rco1, involved in
complex stability, and Eaf3, which is shared with the NuA4 complex. In
contrast to Rpd3L, Rpd3S is recruited by Eaf3, meditated by RNA polymerase
II, to H3K36me (Govind et al. 2010) at the 3’ end of coding sequences. Here,
Rpd3S plays a role in repression of cryptic transcription (Carrozza et al.
2005b).
A second class I deacetylase is Hos2, known to have a role in creating
a more open chromatin state, thereby promoting induction of transcription
(Wang et al. 2002). Hos2 is an enzymatic subunit of the Set3 complex, which
also contains the Hst1 deacetylase. The Set3 complex regulates the
expression of meiosis-specific genes that are a part of the mid/late sporulation
gene program, and is also important for initiating gene expression in the stress
response (Pijnappel et al. 2001; Sharma et al. 2007; Kim et al. 2012). Hos2 is
also important for the deacetylation of ribosomal protein genes (Robyr et al.
2002). Recently, it was found that Hos2 is critical in opposing Esa1 in DNA
damage repair, by rescuing defects in the induction of repair genes in esa1
mutants (Torres-Machorro et al. 2015).
The final class I deacetylase encoded by HOS1 deacetylates chromatin
at the ribosomal DNA locus (Robyr et al. 2002). Hos1 also deacetylates the

54
Smc3 protein, to promote chromosome cohesion (Borges et al. 2010). Apart
from these two main targets, little else is known about Hos1 and its functions.
Class II deacetylases. The founding member of the class II
deacetylases is Hda1. Hda1 acts in a small complex that includes Hda2 and
Hda3 (Wu et al. 2001a). Hda1 is recruited to target promoters by Tup1 and
deacetylates the histone variant H2A.Z, encoded by HTZ1, in addition to the
core histones H2B and H3 (Wu et al. 2001b; Lin et al. 2008). Along with Rpd3,
Hda1 is considered to be the other major yeast deacetylase, and they act on
nearly all gene promoters with little overlap (Robyr et al. 2002). Despite being
in different HDAC classes, as based upon general sequence divergence,
including the location of the deacetylase domain, Rpd3 and Hda1 still share
some sequence similarity and function yet have distinct targets (Rundlett et al.
1996; Kurdistani et al. 2002; Robyr et al. 2002). Specifically, Hda1
deacetylates large chromosomal regions known as HAST (Hda1-affected
subtelomeric) domains, found adjacent to telomeric heterochromatin, at the
end of most chromosomes (Robyr et al. 2002). These HAST domains are
specifically enriched for stress response genes, such as those involved in
osmotic shock, and are repressed by Hda1 in nutrient-rich conditions.
The second member of class II is Hos3, a unique deacetylase in that it
does not require additional subunits for full deacetylase activity (Carmen et al.
1999). Hos3 is also unique in that it is specifically targeted to the bud-neck of
the mother cell and to a single focus in the daughter cell, where it is involved in

55
the spindle position checkpoint, acting as a sensor of spindle orientation
defects (Wang and Collins 2014). Consistent with this highly specialized role,
hos3∆ does not change global histone acetylation, nor does it result in any
apparent phenotypes (Carmen et al. 1999; Robyr et al. 2002).
Class III deacetylases. The final class of deacetylases is comprised of
the sirtuins, some of the most thoroughly studied among yeast and many other
organisms [reviewed in (Jing and Lin 2015)]. The sirtuins are classified for
their NAD+ co-factor dependence coupled with lysine deacetylation, allowing
for potential regulation by cellular energy balance (Imai et al. 2000; Sauve et
al. 2006). The founding member of the sirtuins, Sir2, deacetylates H4K16, and
is required for silencing the mating type loci, HML and HMR as well as rDNA
and telomere-proximal genes [reviewed in (Blander and Guarente 2004)].
There are four paralogs of Sir2 (Homolog of Sirtuin): Hst1, Hst2, Hst3,
and Hst4, with Hst1, Hst3, and Hst4 thought to have similar gene silencing
functions (Brachmann et al. 1995). Hst1 is part of the Set3 complex, sharing
targets with Hos2, and is part of the Sum1-Rfm1-Hst1 complex, important for
replication initiation by H4K5 deacetylation (Weber et al. 2008). However, Hst1
is the most similar homolog to Sir2, and the two are thought to have partially
overlapping functions (Bedalov et al. 2003). Hst2 is the only primarily
cytoplasmic sirtuin, though does shuttle between the nucleus and the
cytoplasm for functional regulation (Perrod et al. 2001; Wilson et al. 2006).
Interestingly, overexpression of HST2 was found to partially suppress the

56
silencing defects of sir2∆ (Perrod et al. 2001). Hst3 and Hst4 deacetylate
H3K56 in a cell-cycle dependent manner (Maas et al. 2006). The careful
balance of H3K56 acetylation, as partially regulated by Hst3 and Hst4, is
critical for modulating sensitivity to genotoxic agents (Celic et al. 2006; Maas
et al. 2006).
Opposing activities of NuA4 and the HDACs. A key line of inquiry is
to understand the balance of acetylation as coordinated by the opposing
activities of histone deacetylases and acetyltransferases. Whereas the bypass
of ESA1 and EPL1 by concurrent loss of Rpd3L underscores the Rpd3L-NuA4
relationship (Torres-Machorro and Pillus 2014; Searle et al. 2017), other
studies have found that Hda1 is also relevant in opposing Esa1 enzymatic
activity (Vogelauer et al. 2000; Lin et al. 2008; Chang and Pillus 2009).
Interestingly, hda1∆ does not bypass the requirement for ESA1, however it
drastically improved the fitness after bypass of ESA1 in esa1∆ sds3∆ (Torres-
Machorro and Pillus 2014).
This chapter primarily focuses on understanding the specificity of
HDACs, and how they functionally and specifically oppose the NuA4
acetyltransferase upon bypass of EPL1. The previous chapter illustrated the
bypass of EPL1 by sds3∆, but demonstrated that this bypass strain, although
viable, is not robust. Accordingly, other deacetylases were deleted in the epl1∆
sds3∆ background to test if after bypass, cellular acetylation could be further
balanced, and likewise promote enhanced fitness. Other phenotypes, such as

57
sensitivity to stress agents, and rDNA silencing, for example, were also
examined to test if and how each epl1∆ sds3∆ HDAC∆ mutant suppressed the
epl1∆ sds3∆ phenotypic defect. Deletion of HDA1 was overwhelmingly the
most productive in terms of enhancing growth and fitness of epl1∆ sds3∆ and
rescuing many defects, such as rDNA silencing, progression through the cell
cycle, and response to damage and stress agents. This suggests the
combined importance of Hda1 and Rpd3L in opposing NuA4 enzymatic
activity.
Results
NuA4 has distinct interactions with histone deacetylases. Initial
studies of NuA4 bypass conditions revealed that individual HDACs have
distinct interactions with Esa1, as demonstrated by genetic interactions and
phenotypic studies (Torres-Machorro and Pillus 2014). To determine if these
genetic interactions were retained upon EPL1 bypass, an expanded set of
HDAC mutants was generated in the epl1∆ sds3∆ background. These studies
surveyed, for the first time, mutations of all of the sirtuin (class III) genes in a
NuA4 bypass background.
The ability to grow without an EPL1 plasmid in epl1∆ sds3∆ on the
counter-selective 5-FOA revealed that deletion of specific HDACs, such as
HDA1, likely further balanced cellular acetylation, and these activities acted in
opposition to Epl1 and NuA4 (Figure 3-2). Likewise, failure, or near inability to

58
SC-URA 5-FOA
hos2∆ hda1∆ s i r 2∆ hst1∆ hst2∆ hst3∆ hst4∆ ep
l1∆
sds3∆
+ pE
PL1
Growth Suppression
Figure 3-2. NuA4 has distinct interactions with different HDACs. Loss of deacetylases can either enhance or exacerbate the bypass of epl1∆ by concurrent loss of SDS3. Triple mutants of epl1∆ sds3∆ (LPY21071) with hos2∆ (LPY21292), hda1∆ (LPY21381), sir2∆ (LPY22115), hst1∆ (LPY21186), hst2∆ (LPY21236), hst3∆ (LPY21342), or hst4∆ (LPY22187) were constructed. Their ability to survive without a covering EPL1 plasmid was surveyed by growth on the counterselective 5-FOA, as compared to the growth control that selects for the EPL1 plasmid, at 24˚. Results ranged from lethality with hos2∆, to enhanced viability with hda1∆.

59
grow without an EPL1 plasmid in these same conditions revealed that other
HDACs, such as HOS2 or HST2 respectively were more critical for growth in
the EPL1 bypass background. The distinct genetic interactions between EPL1
and the genes encoding each deacetylase underscore the importance of the
target specificity of each HDAC in opposing that of EPL1 in NuA4.
Phenotypes of epl1∆ sds3∆ are selectively suppressed by deletion
of HDACs. All viable HDAC mutants in the epl1∆ sds3∆ background were
subjected to additional phenotypic analysis to determine which phenotypes of
epl1∆ sds3∆ were suppressed or unchanged upon loss of each HDAC. Loss of
HDA1 overwhelmingly suppressed many of the epl1∆ sds3∆ phenotypes. The
temperature sensitive and slow-growing epl1∆ sds3∆ mutant was found to
tolerate growth in temperatures ranging from 24˚ to 37˚ with the concurrent
loss of HDA1 (Figure 3-3). Whereas the epl1∆ sds3∆ hda1∆ mutant did not
grow as robustly as the sds3∆ single mutant, it consistently grew about 10-15-
fold better than epl1∆ sds3∆ upon temperature evaluation. This improvement
was remarkable, especially considering that an essential gene is bypassed
and deleted.
The remaining HDAC deletions in the epl1∆ sds3∆ background, such as
sir2∆, hst1∆, hst2∆, hst3∆, and hst4∆ did not significantly alter temperature-
challenged growth. This observation implies that these HDACs either have
overlapping functions with Rpd3L, already compromised in epl1∆ sds3∆, or

60
EPL1 epl1∆ epl1∆ hda1∆ epl1∆ sir2∆ epl1∆ hst1∆ epl1∆ hst2∆ epl1∆ hst3∆ epl1∆ hst4∆
sds3∆
24˚ 30˚ 35˚ 37˚
Figure 3-3. Loss of HDA1 enhances temperature-dependent fitness. Loss of HDA1 (LPY21459) in epl1∆ sds3∆ (LPY21299) partially restores growth as compared to sds3∆ (LPY20877) across temperatures, from 24˚ to 37˚. Strains with deletions of encoding the additional deacetylases were viable in epl1∆ sds3∆, including sir2∆ (LPY22450), hst1∆ (LPY22447), hst2∆ (LPY22448), hst3∆ (LPY22449), and hst4∆ (LPY22452). These mutations had negligible effects on growth in contrast with the epl1∆ sds3∆ double mutant.

61
function more independently of Epl1 and NuA4, causing no additional
temperature sensitivity or general growth defects.
Assessment of sensitivity to DNA damaging challenges yielded similar
findings to the temperature growth assays. Again, HDA1 stands out as
selectively enhancing growth upon deletion in the epl1∆ sds3∆ strain (Figure
3-4). The exception was with sensitivity to the topoisomerase I inhibitor, CPT,
which at the 7µg/ml concentration used, does not permit growth of any of the
bypass strains, including epl1∆ sds3∆ hda1∆. However, even the sds3∆ single
mutant showed a degree of sensitivity to CPT, implying that evaluating lower
concentrations than the standard titrations might be useful in future studies.
As demonstrated in Chapter 2 and in previous studies, NuA4, and
specifically Epl1 and Esa1 are necessary for normal progression through the
cell cycle, since their compromised functions result in a G2/M delay (Clarke et
al. 1999; Boudreault et al. 2003; Torres-Machorro and Pillus 2014; Searle et
al. 2017). Independent evidence suggests that many of the HDACs are likely
to be important in regulating important cell cycle genes as well (Bernstein et al.
2000; Desfosses-Baron et al. 2016). It was shown previously that loss of
HDA1 in esa1∆ sds3∆ partially suppressed the esa1∆ sds3∆ G2/M delay
(Torres-Machorro and Pillus 2014).
Here, the same selective partial suppression of the epl1∆ sds3∆ cell
cycle delay by hda1∆ was observed; however, deletion of none of the other
deacetylases in combination with epl1∆ sds3∆ was able to rescue this defect
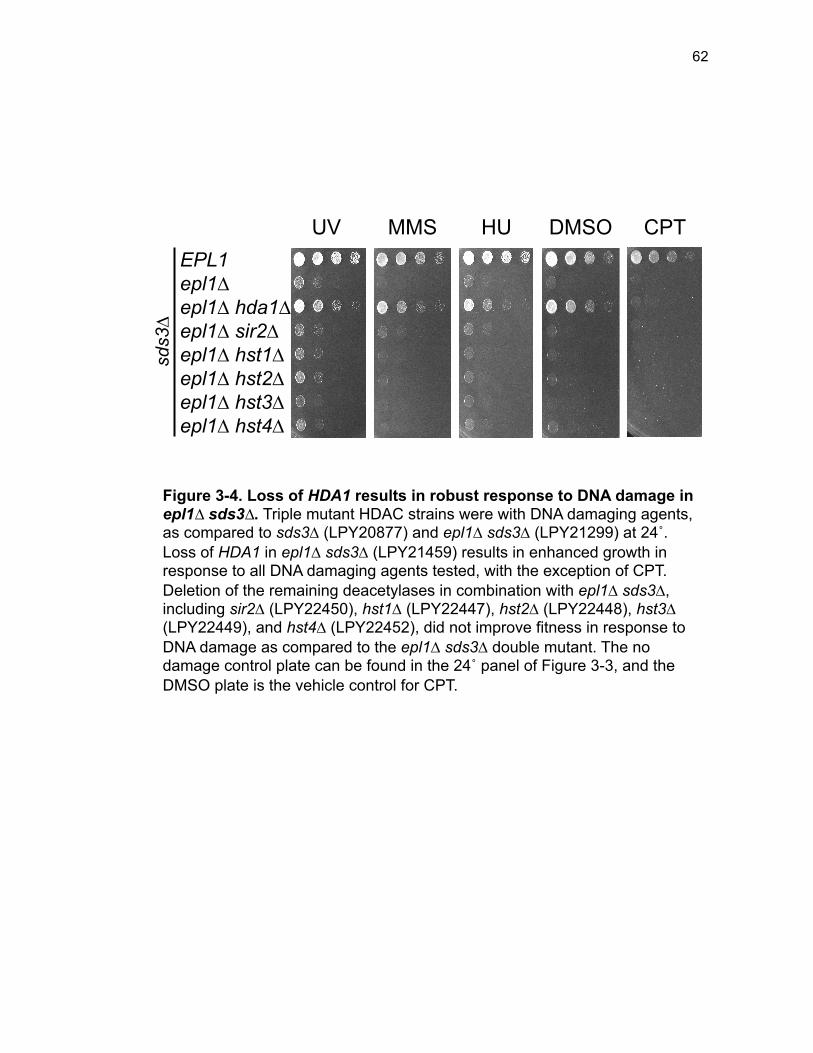
62
EPL1 epl1∆ epl1∆ hda1∆ epl1∆ sir2∆ epl1∆ hst1∆ epl1∆ hst2∆ epl1∆ hst3∆ epl1∆ hst4∆
sds3∆
UV MMS HU DMSO CPT
Figure 3-4. Loss of HDA1 results in robust response to DNA damage in epl1∆ sds3∆. Triple mutant HDAC strains were with DNA damaging agents, as compared to sds3∆ (LPY20877) and epl1∆ sds3∆ (LPY21299) at 24˚. Loss of HDA1 in epl1∆ sds3∆ (LPY21459) results in enhanced growth in response to all DNA damaging agents tested, with the exception of CPT. Deletion of the remaining deacetylases in combination with epl1∆ sds3∆, including sir2∆ (LPY22450), hst1∆ (LPY22447), hst2∆ (LPY22448), hst3∆ (LPY22449), and hst4∆ (LPY22452), did not improve fitness in response to DNA damage as compared to the epl1∆ sds3∆ double mutant. The no damage control plate can be found in the 24˚ panel of Figure 3-3, and the DMSO plate is the vehicle control for CPT.

63
(Figure 3-5). Although studies of Hda1 in the cell cycle have been relatively
limited, it is illustrated here that Hda1 acts in opposition to NuA4 and apart
from Rpd3L, either in deacetylating non-histone substrates that are important
for cell cycle regulation and progression, or though H3/H4 deacetylation.
Histone deacetylases with wide-ranging targets are likely to affect
histone acetylation in distinctly characteristic manners. If epl1∆ sds3∆ has a
primary defect in cellular acetylation, we would hypothesize that any mutant
that significantly enhances the fitness of this strain would suppress, to some
degree, the low histone H4 acetylation defect and that of other critical
substrates. Accordingly, two acetylatable-lysines in histone H4 were probed
for acetylation levels, and compared to both tubulin, as a loading control, and
acetylation at H3K9/K14, which is not a NuA4 target, but a target of many of
the deacetylases included in the study. Whereas epl1∆ sds3∆ had generally
reduced H4 acetylation levels, many of the triple mutants did indeed suppress
this defect to some degree, with epl1∆ sds3∆ hst3∆ being the exception here
(Figure 3-6). In line with the previously discussed phenotypes, epl1∆ sds3∆
hda1∆ can be highlighted, as this mutant demonstrates a robust rescue of H4
acetylation, along with rescue of the growth defects and temperature and DNA
damage sensitivities, as discussed earlier. Significantly however, rescue of H4
acetylation in many of the HDAC triple mutants highlights that histone
acetylation is not the only important factor in how NuA4 and these HDACs
interact. In many cases, suppression of the H4 acetylation defect did not
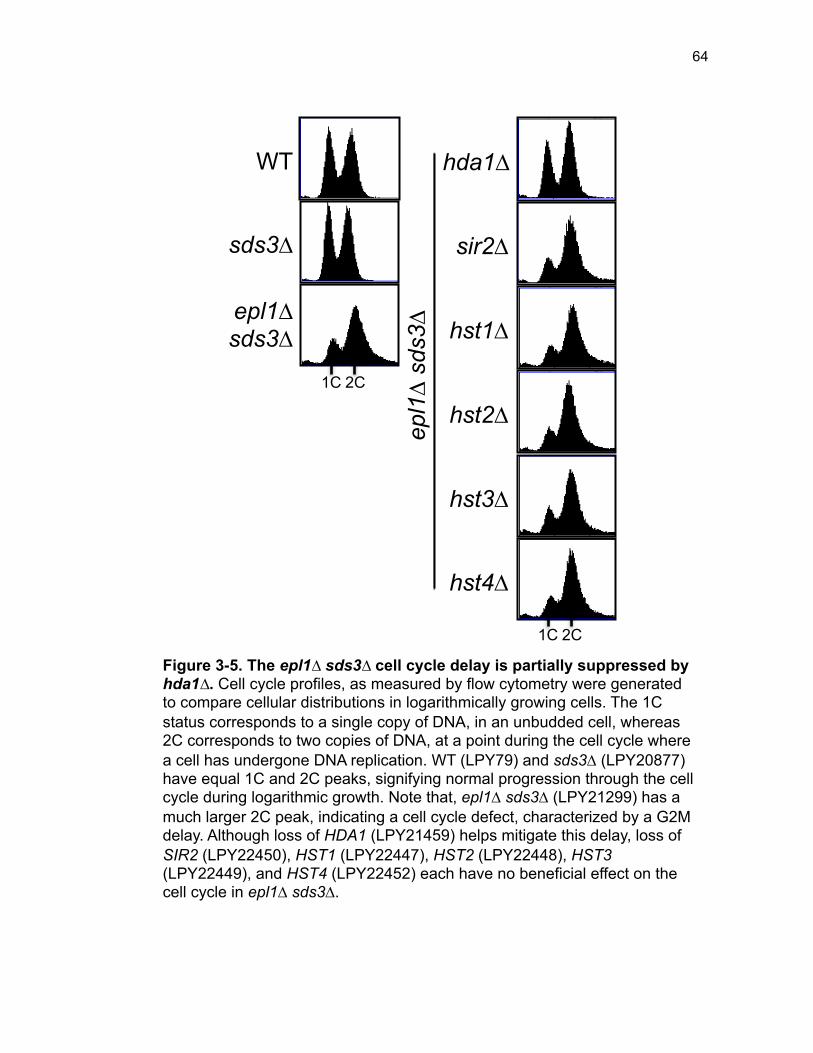
64
WT
sds3∆
epl1∆ sds3∆
1C 2C
hda1∆
1C 2C
sir2∆
hst1∆
hst2∆
hst3∆
hst4∆
epl1∆
sds3∆
Figure 3-5. The epl1∆ sds3∆ cell cycle delay is partially suppressed by hda1∆. Cell cycle profiles, as measured by flow cytometry were generated to compare cellular distributions in logarithmically growing cells. The 1C status corresponds to a single copy of DNA, in an unbudded cell, whereas 2C corresponds to two copies of DNA, at a point during the cell cycle where a cell has undergone DNA replication. WT (LPY79) and sds3∆ (LPY20877) have equal 1C and 2C peaks, signifying normal progression through the cell cycle during logarithmic growth. Note that, epl1∆ sds3∆ (LPY21299) has a much larger 2C peak, indicating a cell cycle defect, characterized by a G2M delay. Although loss of HDA1 (LPY21459) helps mitigate this delay, loss of SIR2 (LPY22450), HST1 (LPY22447), HST2 (LPY22448), HST3 (LPY22449), and HST4 (LPY22452) each have no beneficial effect on the cell cycle in epl1∆ sds3∆.
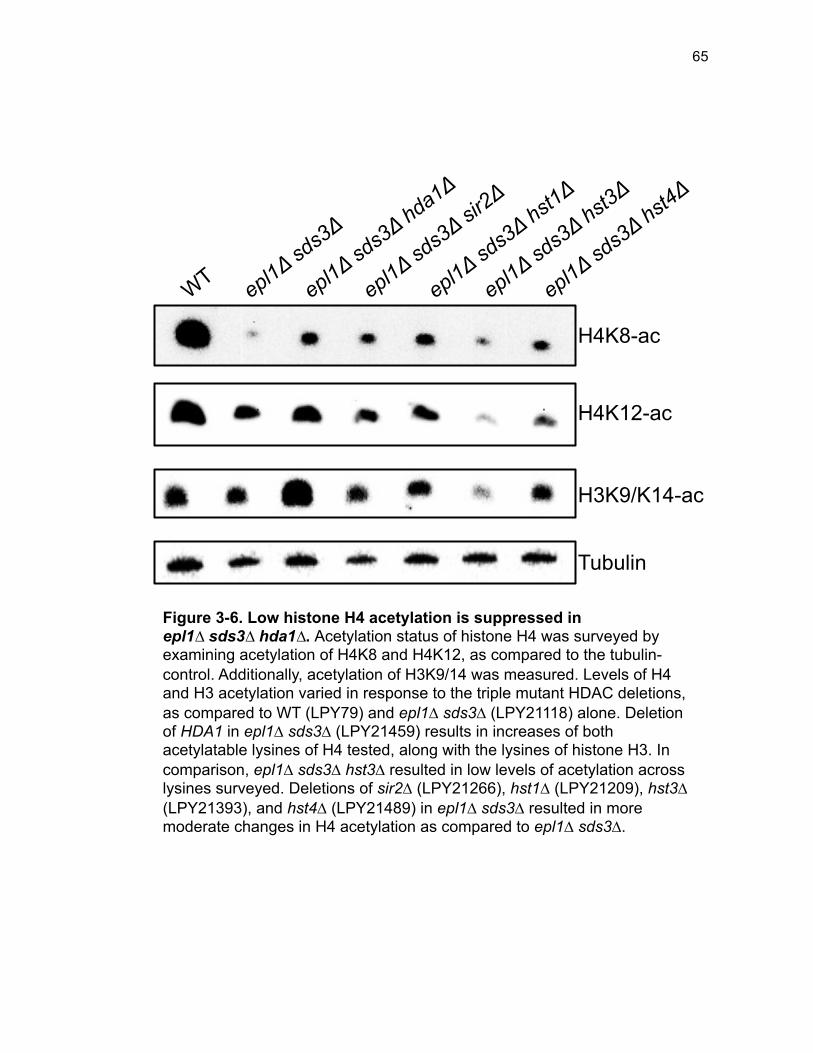
65
H3K9/K14-ac
H4K12-ac
H4K8-ac
Tubulin
WT epl1Δ
sds3Δ
epl1Δ sd
s3Δ hda1Δ
epl1Δ sd
s3Δ sir
2Δ
epl1Δ sd
s3Δ hst1
Δ
epl1Δ sd
s3Δ hst3
Δ
epl1Δ sd
s3Δ hst4
Δ
Figure 3-6. Low histone H4 acetylation is suppressed in epl1∆ sds3∆ hda1∆. Acetylation status of histone H4 was surveyed by examining acetylation of H4K8 and H4K12, as compared to the tubulin-control. Additionally, acetylation of H3K9/14 was measured. Levels of H4 and H3 acetylation varied in response to the triple mutant HDAC deletions, as compared to WT (LPY79) and epl1∆ sds3∆ (LPY21118) alone. Deletion of HDA1 in epl1∆ sds3∆ (LPY21459) results in increases of both acetylatable lysines of H4 tested, along with the lysines of histone H3. In comparison, epl1∆ sds3∆ hst3∆ resulted in low levels of acetylation across lysines surveyed. Deletions of sir2∆ (LPY21266), hst1∆ (LPY21209), hst3∆ (LPY21393), and hst4∆ (LPY21489) in epl1∆ sds3∆ resulted in more moderate changes in H4 acetylation as compared to epl1∆ sds3∆.

66
promote enhanced strain fitness. These results underscore the recognition
that even in HAT/HDAC mutants, response to other stresses is also critical for
determining general strain fitness.
Epl1 is opposed by Hda1 independent of Rpd3L. It has been
established that Epl1 is critical in autophagy where it promotes the acetylation
of a key autophagy protein, Atg3 (Yi et al. 2012). Autophagy is an important
cellular process that maintains cellular homeostasis by degrading and
recycling cellular organelles and proteins. The process of autophagy is
triggered by many cellular signals and stress conditions. Oxidative stress is
one such condition that specifically triggers the production of
autophagosomes, one of they key autophagy vesicles [reviewed in (Navarro-
Yepes et al. 2014)]. Given Epl1’s role in autophagy, and the link between
oxidative stress and autophagy, the oxidative stress tolerance of epl1∆ sds3∆
was evaluated. The assay selected was grounded in the observation that the
accumulation of reactive oxygen species, a hallmark of oxidative stress, is
critical for mediating apoptosis and programmed cell death [reviewed in
(Farrugia and Balzan 2012)]. The epl1∆ sds3∆ mutant was highly sensitive to
hydrogen peroxide (H2O2), an oxidative stress agent (Figure 3-7). Strikingly,
this sensitivity was suppressed by loss of HDA1. This suggests that whereas
Rpd3L opposes many of the most critical functions of NuA4, Hda1 directly
opposes many other challenge-specific roles.
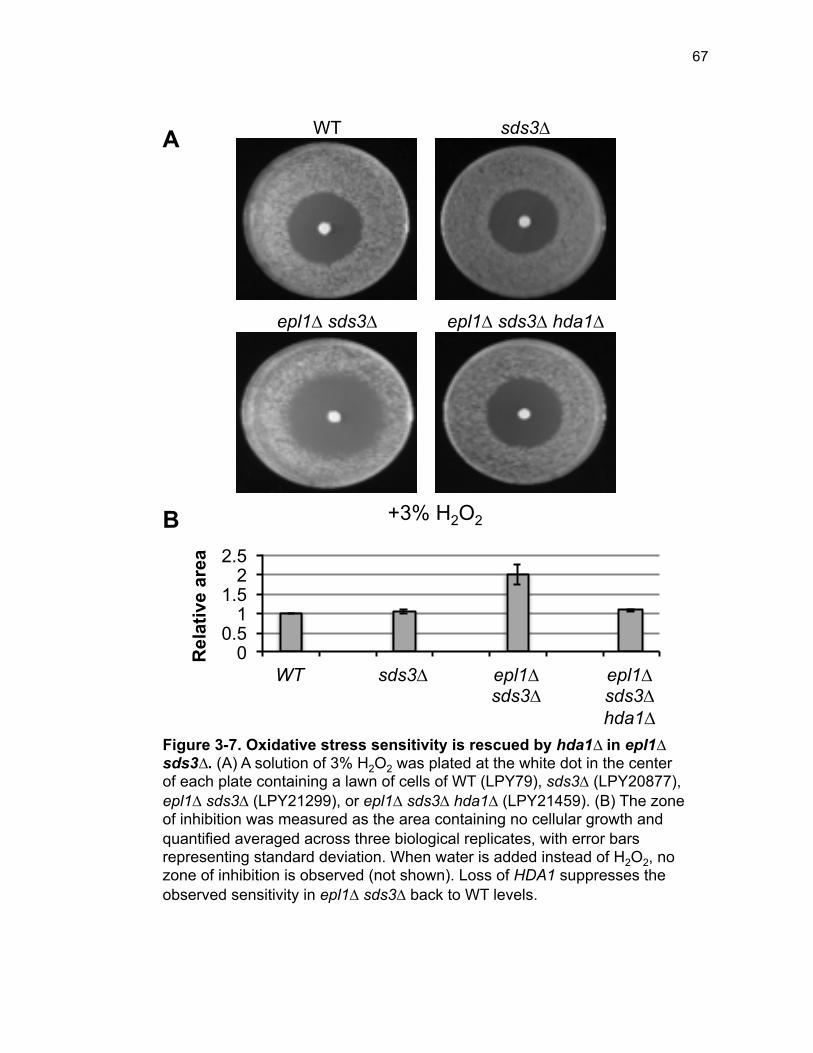
67
WT sds3∆
epl1∆ sds3∆ epl1∆ sds3∆ hda1∆
+3% H2O2
0 0.5
1 1.5
2 2.5
WT sds3∆ epl1∆ sds3∆
epl1∆ sds3∆ hda1∆
Rel
ativ
e ar
ea
A
B
Figure 3-7. Oxidative stress sensitivity is rescued by hda1∆ in epl1∆ sds3∆. (A) A solution of 3% H2O2 was plated at the white dot in the center of each plate containing a lawn of cells of WT (LPY79), sds3∆ (LPY20877), epl1∆ sds3∆ (LPY21299), or epl1∆ sds3∆ hda1∆ (LPY21459). (B) The zone of inhibition was measured as the area containing no cellular growth and quantified averaged across three biological replicates, with error bars representing standard deviation. When water is added instead of H2O2, no zone of inhibition is observed (not shown). Loss of HDA1 suppresses the observed sensitivity in epl1∆ sds3∆ back to WT levels.

68
It has been established that Esa1 regulates silencing of the rDNA locus
(Clarke et al. 2006), and that Epl1 also contributes to gene silencing
(Boudreault et al. 2003), underscoring the importance of NuA4/piccolo-NuA4
in these processes, a topic that is further explored in Chapter 4. An rDNA
silencing reporter was used to ask if Epl1 is involved in rDNA silencing, and if
Hda1 acts in direct opposition (Figure 3-8A). Here, esa1-414 was included as
a control known to be defective in rDNA silencing, and it was apparent that the
epl1∆ sds3∆ mutants and esa1∆ sds3∆ mutants were unable to silence the
reporter within the rDNA locus, indicative of a defect in rDNA silencing.
Notably, loss of HDA1 in either epl1∆ sds3∆ or esa1∆ sds3∆ suppressed this
rDNA silencing phenotype, demonstrating restoration of silencing at the rDNA
array (Figure 3-8B).
The results in this chapter bring the critical NuA4-Rpd3L-Hda1
relationship to focus. Previous studies have provided several lines of evidence
to suggest that Rpd3L and Hda1 are most critical for opposing NuA4 catalytic
activity (Vogelauer et al. 2000; Lin et al. 2008; Chang and Pillus 2009; Torres-
Machorro and Pillus 2014; Searle et al. 2017). However, many of these
studies have relied on hypomorphic alleles of ESA1 and EPL1, rather than the
complete deletion. This limitation is resolved by the bypass of EPL1,
facilitating the assessment of cellular status upon complete loss of EPL1.
Whereas the essential activities of EPL1 are bypassed by loss of Rpd3L, most
of the resulting mutant phenotypes are directly rescued by loss of HDA1. This
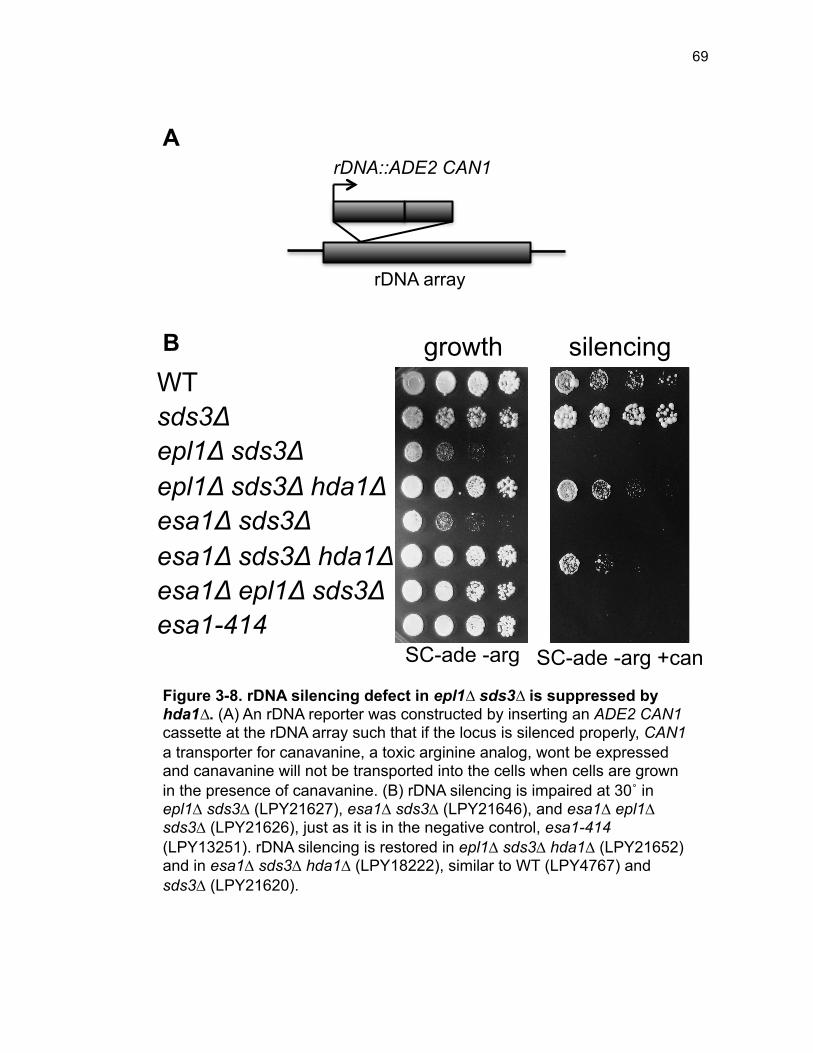
69
WT sds3Δ epl1Δ sds3Δ epl1Δ sds3Δ hda1Δ esa1Δ sds3Δ esa1Δ sds3Δ hda1Δ esa1Δ epl1Δ sds3Δ esa1-414
SC-ade -arg +can SC-ade -arg
growth silencing
rDNA::ADE2 CAN1
rDNA array
A
B
Figure 3-8. rDNA silencing defect in epl1∆ sds3∆ is suppressed by hda1∆. (A) An rDNA reporter was constructed by inserting an ADE2 CAN1 cassette at the rDNA array such that if the locus is silenced properly, CAN1 a transporter for canavanine, a toxic arginine analog, wont be expressed and canavanine will not be transported into the cells when cells are grown in the presence of canavanine. (B) rDNA silencing is impaired at 30˚ in epl1∆ sds3∆ (LPY21627), esa1∆ sds3∆ (LPY21646), and esa1∆ epl1∆ sds3∆ (LPY21626), just as it is in the negative control, esa1-414 (LPY13251). rDNA silencing is restored in epl1∆ sds3∆ hda1∆ (LPY21652) and in esa1∆ sds3∆ hda1∆ (LPY18222), similar to WT (LPY4767) and sds3∆ (LPY21620).

70
suggests that both Rpd3L and Hda1 directly oppose Epl1/NuA4. However,
several non-overlapping functions and/or targets of Rpd3L and Hda1 exist.
This was exemplified by different responses to genetic and pharmacological
challenges. These distinct functions are highly relevant in the context of NuA4
and specifically Epl1/Esa1.
Discussion
In Chapter 3, bypass suppression of Epl1 was used to explore the
functional relationship between NuA4 and genes encoding HDAC enzymes.
Previous studies relied on hypomorphic or low-dosage alleles of ESA1 and
EPL1 to diminish NuA4 activity and assess genetic interactions between NuA4
and HDAC genes (Lin et al. 2008; Chang and Pillus 2009). The work reported
here is significant because HDACs are generally considered to be less specific
than HATs, thereby acting on a wider range of targets. Understanding the
functional relationship between opposing enzyme classes leads to better
insight in how these critical enzymes promote tightly balanced cellular
acetylation, dynamically fine-tuned for regulatory control.
The results from this chapter illustrate the importance of Hda1 in
opposing the activity of NuA4. It is noteworthy that hda1∆ cannot bypass
esa1∆ (Torres-Machorro and Pillus 2014), especially given the fitness
enhancement and overwhelming phenotypic suppression of hda1∆ in both
esa1∆ sds3∆ and in epl1∆ sds3∆. In future studies, the epl1∆ hda1∆ strain will

71
be constructed in order to evaluate any potential Esa1/Epl1 differences, as
was observed with epl1∆ sds3∆ hst2∆ and esa1∆ sds3∆ hst2∆.
Hos1 and Hos3 may act in opposition to NuA4 at the rDNA locus
and spindle pole body, respectively. Eight HDACs (Rpd3L, Hos2, Hda1,
Sir2, Hst1, Hst2, Hst3, Hst4) were evaluated for genetic interactions with
NuA4. Rpd3, as it exists in the Rpd3L complex was comprehensively studied
in Chapter 2 and in previous studies, as the focus of the bypass studies of
ESA1 and EPL1 (Torres-Machorro and Pillus 2014). The remaining seven
HDACs were the focus of this chapter.
Both Hos1 and Hos3 are lesser characterized HDACs in yeast, and
have specialized functions, however neither Hos1 nor Hos3 were included in
this study. However, both could have interesting interactions with NuA4 that
remain to be characterized. For example, Hos1, known to deacetylate
chromatin at the rDNA (Robyr et al. 2002), might act in opposition to NuA4 at
the rDNA and could also relieve the rDNA silencing defects of epl1∆ sds3∆
and esa1∆ sds3∆, akin to hda1∆.
By contrast, it is possible that Hos3 might be relevant to NuA4 cell cycle
functions. It is known that NuA4 both physically interacts with and acetylates
several of the spindle pole body proteins (Mitchell et al. 2013a). In addition,
several NuA4 subunit mutants are sensitive to microtubule destabilization
assessed with benomyl (Le Masson et al. 2003). Rpd3 may play a role in
opposing NuA4 here as well, as rpd3∆ suppresses the benomyl sensitivity of

72
NuA4 mutants to varying degrees (Mitchell et al. 2013a). It is thus possible
that Hos3 might be a relevant NuA4 antagonist at the spindle pole body, given
its role in the spindle position checkpoint (Wang and Collins 2014).
Epl1’s interactions with Hst2 and Sir2 are distinct from Esa1’s
genetic interactions. The majority of the results presented here, in Chapter 2,
and in previous studies feature functional overlap between Epl1 and Esa1. For
example, growth in response to genotoxic agents, silencing at the rDNA, and
global gene expression in epl1∆ sds3∆ mirror that in esa1∆ sds3∆. However,
there are a few interesting differences between Epl1 and Esa1 with regard to
two specific genetic interactions. Previously, it was found that hst2∆ was
synthetic lethal with esa1∆ sds3∆ (Torres-Machorro and Pillus 2014). It is
therefore striking that epl1∆ sds3∆ hst2∆ is viable, as presented in this
chapter.
There are several possibilities to consider in understanding this
difference. For example when the esa1∆ sds3∆ hst2∆ +pESA1 strain was
grown on 5-FOA + 10% glucose (a concentration found to support enhanced
growth in some mutants, S. Holmes, personal communication), cells were able
to survive without the ESA1 covering plasmid (data not shown). This suggests
that 5-FOA counter-selection was a stressful event, and the supplementation
of extra glucose from 2 to 10% permitted growth. It is unclear why epl1∆ sds3∆
hst2∆ did not require this additional supplementation for viability. Another
confounding technical possibility involves the SDS3 knockout cassette.

73
Whereas SDS3 was replaced with a kanMX cassette in the esa1∆ mutants,
the epl1∆ mutants were constructed with sds3∆::natMX. Although these two
sds3∆ strains showed no apparent phenotypic differences, it is possible that
nuances between these largely isogenic strains are driving this difference in
viability.
In contrast, a more interesting possibility for the difference in EPL1 and
ESA1’s genetic interaction with HST2 involves functional differences between
epl1∆ sds3∆ and esa1∆ sds3∆. The results from Chapter 2 showed functional
overlap between Esa1 and Epl1 where loss of EPL1 was functionally and
phenotypically identical to loss of ESA1. There was one notable exception: In
epl1∆ sds3∆, Esa1 loses association with the chromatin. Since Hst2 encodes
a cytoplasmic sirtuin, but shuttles between the nucleus and the cytoplasm
(Perrod et al. 2001; Wilson et al. 2006), it is possible that in epl1∆ sds3∆, an
unstably associated Esa1 somehow permits loss of HST2. It remains to be
determined if the apparent discordance between viability of epl1∆ sds3∆ hst2∆
and lethality of esa1∆ sds3∆ hst2∆ has functional significance or is linked to
differences in markers. Construction of an esa1∆ sds3∆::natMX hst2∆ strain
can begin to resolve this question.
An additional other interesting divergence between the interaction of
HDACs with the ESA1 bypass mutant and with the EPL1 bypass mutant
comes from evaluation of SIR2. Whereas esa1∆ sds3∆ sir2∆ demonstrated
suppression of many esa1∆ sds3∆ DNA damage sensitivities, the same was

74
not true for epl1∆ sds3∆ sir2∆. The technical differences in strain construction
discussed for the hst2∆ strains above may be an explanatory factor here, or
may reflect a functional difference between Esa1 and Epl1. Such a difference
would be worthy or more thorough characterization, if it were indeed only
apparent in highly specific conditions. Other phenotypes, such as aging and
lifespan, hallmarks of Sir2 regulation [reviewed in (Steinkraus et al. 2008)] that
have not yet been assessed might reveal other nuances of this relationship. In
support of this possibility, it is known that Esa1 acetylates Pck1, a key
gluconeogenesis enzyme. Acetylation is crucial for Pck1 enzymatic activity
and an important determinant of yeast chronological life span, among other
processes (Lin et al. 2009).
Functional antagonism between Rpd3 and Hos2. Whereas NuA4 is
an essential acetyltransferase, none of the ten HDACs in yeast are essential
for viability. Of note, epl1∆ sds3∆ is synthetically lethal with hos2∆ (Figure 3-
2). However, this is likely not a reflection of a negative genetic interaction
between NuA4 and HOS2, but rather is likely one between Rpd3 and Hos2.
The epl1∆ sds3∆ hos2∆ mutant was sick and extremely slow growing even
when maintaining plasmid selection for pEPL1 (Figure 3-2). Additionally,
several previous studies have highlighted this synthetic growth defect of
hos2∆ with both sds3∆ and rpd3∆ (Collins et al. 2007; Cohen et al. 2008;
Costanzo et al. 2010; Costanzo et al. 2016). Therefore, although the synthetic
lethality of epl1∆ sds3∆ hos2∆ seems to stand out among the HDAC-NuA4

75
interactions, it is likely to signify more about an Rpd3-Hos2 relationship than a
Hos2-NuA4 negative genetic interaction.
The Hda1-Rpd3-NuA4 functional axis. The hda1∆ mutation
suppressed nearly every phenotype studied in epl1∆ sds3∆, and in fact, was
the only HDAC to suppress many of the epl1∆ sds3∆ phenotypes. The
significance and mechanism behind this rescue warrants additional
investigation, as discussed in depth in Chapter 5. This mechanism may
involve histone acetylation or acetylation of non-histone targets, such as
genes within the HAST domains, that may be driving this large-scale
suppression. It was possible that hda1∆ was rescuing aberrant global gene
expression in epl1∆ sds3∆. This possibility is explored in Chapter 4, where
transcriptional differences involved in the rescue by hda1∆ are discussed.
It is important to remember that these HATs/HDACs are not acting in
isolation. Rather, there is an important network between them, such that
modulation of one HAT/HDAC will likely affect the enzymatic activities of
others. An example of this was recently illustrated with Esa1 and the H2B/H3
HAT, Gcn5 at ribosomal protein genes. Loss of ESA1 shifted ribosomal protein
genes from Esa1/TFIID-dependency to Gcn5/SAGA dependence (Uprety et al.
2015). Therefore, a distinct point to consider is the genetic relationship
between HDA1 and RPD3. There is a documented synthetic sickness of
hda1∆ rpd3∆ that is rescued by esa1-531 (Lin et al. 2008). This implies that
the Rpd3-Hda1 interaction axis may be just as critical as the NuA4-Rpd3L and

76
NuA4-Hda1 axes. To fully assess this interaction triangle (Figure 3-9) between
NuA4, Rpd3L, and Hda1, the sds3∆ hda1∆ mutants should be studied and
phenotypically surveyed in more detail. Such analysis will serve to generate a
more complete picture of both how Rpd3L and Hda1 oppose NuA4, but also
how Rpd3L and Hda1 interact and functionally divide deacetylase activities in
the genome.
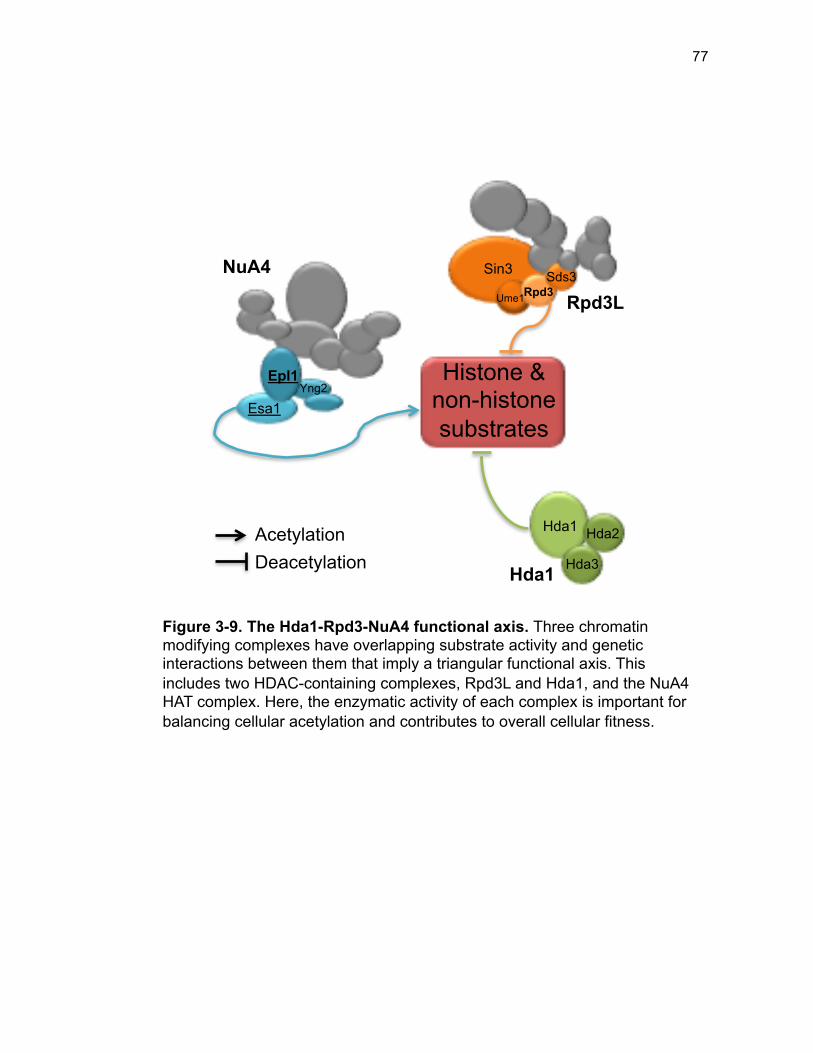
77
Figure 3-9. The Hda1-Rpd3-NuA4 functional axis. Three chromatin modifying complexes have overlapping substrate activity and genetic interactions between them that imply a triangular functional axis. This includes two HDAC-containing complexes, Rpd3L and Hda1, and the NuA4 HAT complex. Here, the enzymatic activity of each complex is important for balancing cellular acetylation and contributes to overall cellular fitness.
Epl1
Esa1
NuA4
Yng2
Sin3
Rpd3L Ume1 Rpd3 Sds3
Hda1 Hda2
Hda3 Hda1
Histone & non-histone substrates
Acetylation Deacetylation

78
Table 3-1. Strains used in Chapter 3. All were constructed for this thesis unless otherwise noted. Strain Genotype LPY79 MATα W303 (ade2-1 can1-100 his3-11 leu2,3,112 trp1-1 ura3-1) * LPY21071 MATα W303 epl1∆::kanMX sds3∆::natMX + pLP3189 LPY21292 MATα W303 epl1∆::kanMX sds3∆::natMX hos2∆::TRP1 + pLP3189 LPY21381 MATα W303 epl1∆::kanMX sds3∆::natMX hda1∆::TRP1 + pLP3189 LPY22115 MATα W303 epl1∆::kanMX sds3∆::natMX sir2∆::TRP1 + pLP3189 LPY21186 MATα W303 epl1∆::kanMX sds3∆::natMX hst1∆::LEU2 + pLP3189 LPY21236 MATα W303 epl1∆::kanMX sds3∆::natMX hst2∆::TRP1 + pLP3189 LPY21342 MATα W303 epl1∆::kanMX sds3∆::natMX hst3∆::kanMX + pLP3189 LPY22187 MATα W303 epl1∆::kanMX sds3∆::natMX hst4∆::kanXx + pLP3189 LPY20877 MATα W303 sds3∆::natMX LPY21299 MATα W303 epl1∆::kanMX sds3∆::natMX LPY21459 MATα W303 epl1∆::kanMX sds3∆::natMX hda1∆::TRP1 LPY22450 MATα W303 epl1∆::kanMX sds3∆::natMX sir2∆::TRP1 LPY22447 MATα W303 epl1∆::kanMX sds3∆::natMX hst1∆::LEU2 LPY22448 MATα W303 epl1∆::kanMX sds3∆::natMX hst2∆::TRP1 LPY22449 MATα W303 epl1∆::kanMX sds3∆::natMX hst3∆::kanMX LPY22452 MATα W303 epl1∆::kanMX sds3∆::natMX hst4∆::kanMX LPY21118 MATα W303 epl1∆::kanMX sds3∆::natMX LPY21266 MATα W303 epl1∆::kanMX sds3∆::natMX sir2∆::TRP1 LPY21209 MATα W303 epl1∆::kanMX sds3∆::natMX hst1∆::LEU2 LPY21393 MATα W303 epl1∆::kanMX sds3∆::natMX hst3∆::kanMX LPY21489 MATa W303 epl1∆::kanMX sds3∆::natMX hst4∆::kanMX LPY4767 MATα W303 rDNA::ADE2CAN1 ** LPY21620 MATα W303 rDNA::ADE2CAN1 sds3∆::natMX LPY21627 MATα W303 rDNA::ADE2CAN1epl1∆::kanMX sds3∆::natMX LPY21652 MATα W303 rDNA::ADE2CAN1epl1∆::kanMX sds3∆::natMX hda1∆::TRP LPY21646 MATα W303 rDNA::ADE2CAN1esa1∆::HIS3 sds3∆::natMX LPY18222 MATα W303 rDNA::ADE2CAN1esa1∆::HIS3 sds3∆::kanMX hda1∆::TRP1 *** LPY21626 MATα W303 rDNA::ADE2CAN1 sds3∆::natMX esa1∆::HIS3 epl1∆::kanMX LPY13251 MATα W303 rDNA::ADE2CAN1 esa1-414 ****
* (Thomas and Rothstein 1989) ** Pillus lab *** (Torres-Machorro and Pillus 2014) **** Pillus lab

79
References Bedalov A, Hirao M, Posakony J, Nelson M, Simon JA. 2003. NAD+-
dependent deacetylase Hst1p controls biosynthesis and cellular NAD+ levels in Saccharomyces cerevisiae. Mol Cell Biol 23: 7044-7054.
Bernstein BE, Tong JK, Schreiber SL. 2000. Genomewide studies of histone deacetylase function in yeast. P Natl Acad Sci USA 97: 13708-13713.
Blander G, Guarente L. 2004. The Sir2 family of protein deacetylases. Annu Rev Biochem 73: 417-435.
Borges V, Lehane C, Lopez-Serra L, Flynn H, Skehel M, Rolef Ben-Shahar T, Uhlmann F. 2010. Hos1 deacetylates Smc3 to close the cohesin acetylation cycle. Mol Cell 39: 677-688.
Boudreault AA, Cronier D, Selleck W, Lacoste N, Utley RT, Allard S, Savard J, Lane WS, Tan S, Côté J. 2003. Yeast enhancer of polycomb defines global Esa1-dependent acetylation of chromatin. Genes Dev 17: 1415-1428.
Brachmann CB, Sherman JM, Devine SE, Cameron EE, Pillus L, Boeke JD. 1995. The SIR2 gene family, conserved from bacteria to humans, functions in silencing, cell cycle progression, and chromosome stability. Genes Dev 9: 2888-2902.
Carmen AA, Griffin PR, Calaycay JR, Rundlett SE, Suka Y, Grunstein M. 1999. Yeast HOS3 forms a novel trichostatin A-insensitive homodimer with intrinsic histone deacetylase activity. Proc Natl Acad Sci U S A 96: 12356-12361.
Carrozza MJ, Florens L, Swanson SK, Shia WJ, Anderson S, Yates J, Washburn MP, Workman JL. 2005a. Stable incorporation of sequence specific repressors Ash1 and Ume6 into the Rpd3L complex. Biochim Biophys Acta 1731: 77-87; discussion 75-76.
Carrozza MJ, Li B, Florens L, Suganuma T, Swanson SK, Lee KK, Shia WJ, Anderson S, Yates J, Washburn MP, Workman JL. 2005b. Histone H3 methylation by Set2 directs deacetylation of coding regions by Rpd3S to suppress spurious intragenic transcription. Cell 123: 581-592.
Celic I, Masumoto H, Griffith WP, Meluh P, Cotter RJ, Boeke JD, Verreault A. 2006. The sirtuins hst3 and Hst4p preserve genome integrity by controlling histone h3 lysine 56 deacetylation. Curr Biol 16: 1280-1289.

80
Chang CS, Pillus L. 2009. Collaboration Between the Essential Esa1 Acetyltransferase and the Rpd3 Deacetylase Is Mediated by H4K12 Histone Acetylation in Saccharomyces cerevisiae. Genetics 183: 149-160.
Clarke AS, Lowell JE, Jacobson SJ, Pillus L. 1999. Esa1p is an essential histone acetyltransferase required for cell cycle progression. Molecular and Cellular Biology 19: 2515-2526.
Clarke AS, Samal E, Pillus L. 2006. Distinct roles for the essential MYST family HAT Esa1p in transcriptional silencing. Mol Biol Cell 17: 1744-1757.
Cohen TJ, Mallory MJ, Strich R, Yao TP. 2008. Hos2p/Set3p deacetylase complex signals secretory stress through the Mpk1p cell integrity pathway. Eukaryot Cell 7: 1191-1199.
Collins SR, Miller KM, Maas NL, Roguev A, Fillingham J, Chu CS, Schuldiner M, Gebbia M, Recht J, Shales M, Ding H, Xu H, Han J, Ingvarsdottir K, Cheng B, Andrews B, Boone C, Berger SL, Hieter P, Zhang Z, Brown GW, Ingles CJ, Emili A, Allis CD, Toczyski DP, Weissman JS, Greenblatt JF, Krogan NJ. 2007. Functional dissection of protein complexes involved in yeast chromosome biology using a genetic interaction map. Nature 446: 806-810.
Costanzo M, Baryshnikova A, Bellay J, Kim Y, Spear ED, Sevier CS, Ding H, Koh JL, Toufighi K, Mostafavi S, Prinz J, St Onge RP, VanderSluis B, Makhnevych T, Vizeacoumar FJ, Alizadeh S, Bahr S, Brost RL, Chen Y, Cokol M, Deshpande R, Li Z, Lin ZY, Liang W, Marback M, Paw J, San Luis BJ, Shuteriqi E, Tong AH, van Dyk N, Wallace IM, Whitney JA, Weirauch MT, Zhong G, Zhu H, Houry WA, Brudno M, Ragibizadeh S, Papp B, Pal C, Roth FP, Giaever G, Nislow C, Troyanskaya OG, Bussey H, Bader GD, Gingras AC, Morris QD, Kim PM, Kaiser CA, Myers CL, Andrews BJ, Boone C. 2010. The genetic landscape of a cell. Science 327: 425-431.
Costanzo M, VanderSluis B, Koch EN, Baryshnikova A, Pons C, Tan G, Wang W, Usaj M, Hanchard J, Lee SD, Pelechano V, Styles EB, Billmann M, van Leeuwen J, van Dyk N, Lin ZY, Kuzmin E, Nelson J, Piotrowski JS, Srikumar T, Bahr S, Chen Y, Deshpande R, Kurat CF, Li SC, Li Z, Usaj MM, Okada H, Pascoe N, San Luis BJ, Sharifpoor S, Shuteriqi E, Simpkins SW, Snider J, Suresh HG, Tan Y, Zhu H, Malod-Dognin N, Janjic V, Przulj N, Troyanskaya OG, Stagljar I, Xia T, Ohya Y, Gingras AC, Raught B, Boutros M, Steinmetz LM, Moore CL, Rosebrock AP, Caudy AA, Myers CL, Andrews B, Boone C. 2016. A global genetic

81
interaction network maps a wiring diagram of cellular function. Science 353.
De Nadal E, Zapater M, Alepuz PM, Sumoy L, Mas G, Posas F. 2004. The MAPK Hog1 recruits Rpd3 histone deacetylase to activate osmoresponsive genes. Nature 427: 370-374.
Desfosses-Baron K, Hammond-Martel I, Simoneau A, Sellam A, Roberts S, Wurtele H. 2016. Valproate inhibits MAP kinase signalling and cell cycle progression in S. cerevisiae. Sci Rep-Uk 6.
Farrugia G, Balzan R. 2012. Oxidative stress and programmed cell death in yeast. Front Oncol 2: 64.
Govind CK, Qiu H, Ginsburg DS, Ruan C, Hofmeyer K, Hu C, Swaminathan V, Workman JL, Li B, Hinnebusch AG. 2010. Phosphorylated Pol II CTD recruits multiple HDACs, including Rpd3C(S), for methylation-dependent deacetylation of ORF nucleosomes. Mol Cell 39: 234-246.
Imai S, Armstrong CM, Kaeberlein M, Guarente L. 2000. Transcriptional silencing and longevity protein Sir2 is an NAD-dependent histone deacetylase. Nature 403: 795-800.
Jing H, Lin H. 2015. Sirtuins in epigenetic regulation. Chem Rev 115: 2350-2375.
Keogh MC, Kurdistani SK, Morris SA, Ahn SH, Podolny V, Collins SR, Schuldiner M, Chin K, Punna T, Thompson NJ, Boone C, Emili A, Weissman JS, Hughes TR, Strahl BD, Grunstein M, Greenblatt JF, Buratowski S, Krogan NJ. 2005. Cotranscriptional set2 methylation of histone H3 lysine 36 recruits a repressive Rpd3 complex. Cell 123: 593-605.
Kim T, Xu Z, Clauder-Munster S, Steinmetz LM, Buratowski S. 2012. Set3 HDAC mediates effects of overlapping noncoding transcription on gene induction kinetics. Cell 150: 1158-1169.
Kurdistani SK, Grunstein M. 2003. Histone acetylation and deacetylation in yeast. Nat Rev Mol Cell Biol 4: 276-284.
Kurdistani SK, Robyr D, Tavazoie S, Grunstein M. 2002. Genome-wide binding map of the histone deacetylase Rpd3 in yeast. Nat Genet 31: 248-254.
Le Masson I, Yu DY, Jensen K, Chevalier A, Courbeyrette R, Boulard Y, Smith MM, Mann C. 2003. Yaf9, a novel NuA4 histone acetyltransferase

82
subunit, is required for the cellular response to spindle stress in yeast. Mol Cell Biol 23: 6086-6102.
Lin YY, Lu JY, Zhang J, Walter W, Dang W, Wan J, Tao SC, Qian J, Zhao Y, Boeke JD, Berger SL, Zhu H. 2009. Protein acetylation microarray reveals that NuA4 controls key metabolic target regulating gluconeogenesis. Cell 136: 1073-1084.
Lin YY, Qi Y, Lu JY, Pan X, Yuan DS, Zhao Y, Bader JS, Boeke JD. 2008. A comprehensive synthetic genetic interaction network governing yeast histone acetylation and deacetylation. Genes Dev 22: 2062-2074.
Maas NL, Miller KM, DeFazio LG, Toczyski DP. 2006. Cell cycle and checkpoint regulation of histone H3 K56 acetylation by Hst3 and Hst4. Mol Cell 23: 109-119.
Mitchell L, Huard S, Cotrut M, Pourhanifeh-Lemeri R, Steunou AL, Hamza A, Lambert JP, Zhou H, Ning Z, Basu A, Cote J, Figeys DA, Baetz K. 2013. mChIP-KAT-MS, a method to map protein interactions and acetylation sites for lysine acetyltransferases. Proc Natl Acad Sci U S A 110: E1641-1650.
Navarro-Yepes J, Burns M, Anandhan A, Khalimonchuk O, del Razo LM, Quintanilla-Vega B, Pappa A, Panayiotidis MI, Franco R. 2014. Oxidative stress, redox signaling, and autophagy: cell death versus survival. Antioxid Redox Signal 21: 66-85.
Perrod S, Cockell MM, Laroche T, Renauld H, Ducrest AL, Bonnard C, Gasser SM. 2001. A cytosolic NAD-dependent deacetylase, Hst2p, can modulate nucleolar and telomeric silencing in yeast. EMBO J 20: 197-209.
Pijnappel WW, Schaft D, Roguev A, Shevchenko A, Tekotte H, Wilm M, Rigaut G, Seraphin B, Aasland R, Stewart AF. 2001. The S. cerevisiae SET3 complex includes two histone deacetylases, Hos2 and Hst1, and is a meiotic-specific repressor of the sporulation gene program. Genes Dev 15: 2991-3004.
Robyr D, Suka Y, Xenarios I, Kurdistani SK, Wang A, Suka N, Grunstein M. 2002. Microarray deacetylation maps determine genome-wide functions for yeast histone deacetylases. Cell 109: 437-446.
Rundlett SE, Carmen AA, Kobayashi R, Bavykin S, Turner BM, Grunstein M. 1996. HDA1 and RPD3 are members of distinct yeast histone

83
deacetylase complexes that regulate silencing and transcription. Proc Natl Acad Sci U S A 93: 14503-14508.
Sauve AA, Wolberger C, Schramm VL, Boeke JD. 2006. The biochemistry of sirtuins. Annu Rev Biochem 75: 435-465.
Searle NE, Torres-Machorro AL, Pillus L. 2017. Chromatin Regulation by the NuA4 Acetyltransferase Complex Is Mediated by Essential Interactions Between Enhancer of Polycomb (Epl1) and Esa1. Genetics 205: 1125-1137.
Sharma VM, Tomar RS, Dempsey AE, Reese JC. 2007. Histone deacetylases RPD3 and HOS2 regulate the transcriptional activation of DNA damage-inducible genes. Mol Cell Biol 27: 3199-3210.
Steinkraus KA, Kaeberlein M, Kennedy BK. 2008. Replicative aging in yeast: the means to the end. Annu Rev Cell Dev Biol 24: 29-54.
Thomas BJ, Rothstein R. 1989. Elevated recombination rates in transcriptionally active DNA. Cell 56: 619-630.
Torres-Machorro AL, Clark LG, Chang CS, Pillus L. 2015. The Set3 Complex Antagonizes the MYST Acetyltransferase Esa1 in the DNA Damage Response. Mol Cell Biol 35: 3714-3725.
Torres-Machorro AL, Pillus L. 2014. Bypassing the requirement for an essential MYST acetyltransferase. Genetics 197: 851-863.
Uprety B, Sen R, Bhaumik SR. 2015. Eaf1p Is Required for Recruitment of NuA4 in Targeting TFIID to the Promoters of the Ribosomal Protein Genes for Transcriptional Initiation In Vivo. Mol Cell Biol 35: 2947-2964.
Vogelauer M, Wu JS, Suka N, Grunstein M. 2000. Global histone acetylation and deacetylation in yeast. Nature 408: 495-498.
Wang A, Kurdistani SK, Grunstein M. 2002. Requirement of Hos2 histone deacetylase for gene activity in yeast. Science 298: 1412-1414.
Wang M, Collins RN. 2014. A lysine deacetylase Hos3 is targeted to the bud neck and involved in the spindle position checkpoint. Mol Biol Cell 25: 2720-2734.
Weber JM, Irlbacher H, Ehrenhofer-Murray AE. 2008. Control of replication initiation by the Sum1/Rfm1/Hst1 histone deacetylase. BMC Mol Biol 9: 100.

84
Wilson JM, Le VQ, Zimmerman C, Marmorstein R, Pillus L. 2006. Nuclear export modulates the cytoplasmic Sir2 homologue Hst2. EMBO Rep 7: 1247-1251.
Wu J, Carmen AA, Kobayashi R, Suka N, Grunstein M. 2001a. HDA2 and HDA3 are related proteins that interact with and are essential for the activity of the yeast histone deacetylase HDA1. Proc Natl Acad Sci U S A 98: 4391-4396.
Wu J, Suka N, Carlson M, Grunstein M. 2001b. TUP1 utilizes histone H3/H2B-specific HDA1 deacetylase to repress gene activity in yeast. Mol Cell 7: 117-126.
Yi C, Ma M, Ran L, Zheng J, Tong J, Zhu J, Ma C, Sun Y, Zhang S, Feng W, Zhu L, Le Y, Gong X, Yan X, Hong B, Jiang FJ, Xie Z, Miao D, Deng H, Yu L. 2012. Function and molecular mechanism of acetylation in autophagy regulation. Science 336: 474-477.

85
Chapter 4 Defining the Esa1 Transcriptional Regulome
Chapter 4.
Defining the Esa1 Transcriptional
Regulome

86
Introduction
Gene expression is both positively and negatively regulated by the
alteration of chromatin structure through histone modifications such as
acetylation, phosphorylation, and methylation [reviewed in (Kurdistani and
Grunstein 2003; Rando and Winston 2012)]. The acetylation of histone tails,
as mediated by histone acetyltransferases is associated with a more
accessible chromatin structure, and correspondingly is classically correlated
with increased gene expression and transcriptional activation. In contrast, the
opposing activity of histone deacetylation is generally coupled with decreased
gene expression and transcriptionally silent chromatin.
However, growing evidence suggests that this canonical dogma is
overly simplistic and that the biological reality is more nuanced. In many
cases, histone acetylation and deacetylation have each been shown to be
important in both active and silent chromatin, thereby supporting the need for
the initial conventions to be expanded (Iizuka and Smith 2003). For example,
loss of RPD3, which encodes the Rpd3 deacetylase, extends silencing into
normally transcribed genes proximal to telomeres (Zhou et al. 2009). For this
reason and other allied observations, it becomes even more important to
understand how specific chromatin modifying enzymes affect global and
targeted transcription.
Esa1 is important for global transcription. Esa1 has global and
specific transcriptional targets among eukaryotes. Most broadly, Esa1 is

87
recruited to the promoters of actively transcribed genes in yeast. In this case,
Esa1 occupancy is correlated with genome-wide transcription rates (Robert et
al. 2004). At the telomeres, Esa1 silences RNA polymerase II-transcribed
genes (Clarke et al. 2006).
Esa1 is also important for transcriptional elongation. Through H4
acetylation, Esa1 promotes histone eviction and occupancy of the RSC
chromatin-remodeling complex in the coding sequences of genes to stimulate
transcriptional elongation. Here, Esa1 acts in cooperation with the H3 HAT,
Gcn5 (Ginsburg et al. 2009). Thus, Esa1 appears to be important for global
transcriptional regulation.
Esa1 is involved in regulating the expression of ribosomal protein
genes. Additional studies have demonstrated the importance of Esa1 in the
expression of ribosomal protein genes. Specifically, Esa1 is recruited by the
DNA binding protein and transcriptional activator, Rap1, and is associated with
the promoters of ribosomal protein genes (Reid et al. 2000). Here, through its
focused acetyltransferase activity, Esa1 targets TFIID to ribosomal protein
gene promoters for transcription initiation (Uprety et al. 2015). Conversely,
upon loss of Esa1 enzymatic activity, ribosomal protein genes lose TFIID
dependency, and instead rely on the major co-transcriptional activator
complex, SAGA, for initiation. Whereas the latter observation suggests that
Esa1 may not be absolutely critical for expression of ribosomal protein genes,

88
it implies that there is an intricate mechanism regulating ribosome gene
expression worthy of additional study.
Esa1 in ribosome biogenesis. Esa1 and NuA4 are implicated in
many facets of the regulation of ribosome biogenesis, a metabolically intensive
process that is estimated to account for nearly 80% of total transcription in
yeast cells (Warner et al. 2001). Ribosome biogenesis involves the
coordination of the rDNA locus and over 100 ribosomal protein genes to
ultimately facilitate mRNA translation [reviewed in (Woolford and Baserga
2013)]. Esa1 is critically involved in regulating the ribosomal DNA (rDNA)
locus, and has been shown to be important for nucleolar morphology, the cell’s
organellar hub of ribosomal biogenesis (Clarke et al. 1999; Clarke et al. 2006).
In yeast, approximately 150 rDNA repeats are arranged in tandem on
chromosome XII. This is distinct from metazoans, where the rDNA repeats are
distributed among several chromosomes. Each rDNA repeat in yeast contains
the pol III-transcribed 5S rRNA gene and a 35S rRNA gene transcribed by pol
I, along with several cis-regulatory elements. Esa1 is enriched at rDNA and its
acetyltransferase activity is important for silencing the locus, promoting a
mechanism for controlling the rDNA transcription in response to growth
conditions (Clarke et al. 2006). Consistently, esa1 mutants have impaired
silencing of the rDNA locus (Clarke et al. 2006; Torres-Machorro and Pillus
2014). Aberrant rDNA silencing coincides with morphological changes in the
nucleolus. This was most obviously seen by electron microscopy in which

89
widespread localization of the dense heterochromatin material was observed
throughout the nucleus, vastly different from the typical concentration the
dense material focused in the crescent-shaped yeast nucleolus (Clarke et al.
1999). Nucleolar proteins, Nop1 and Sir2 also mislocalized upon shifting esa1
mutants to a restrictive temperature, as visualized by immunofluorescence
(Clarke et al. 2006).
Bypass suppression to define the Esa1 transcriptional ‘regulome.’
Whereas the majority of the studies characterizing Esa1’s transcriptional
targets and roles in ribosome biogenesis were performed using conditional
alleles of ESA1, in this chapter, the ESA1 bypass strain is used to characterize
its transcriptional “regulome” in the complete absence of ESA1. Since
transcription and ribosome biogenesis are both responsive to environmental
stimuli, and alleles of esa1 require a shift to a higher, restrictive temperature
for loss of Esa1-activity, which simultaneously results in an different high-
temperature transcriptional program, it becomes even more noteworthy to
assess Esa1 transcriptional targets and their roles in ribosome biogenesis at
lower permissive temperatures.
In Chapter 4, whole-genome RNA sequencing was performed to
characterize the transcriptional targets regulated by Esa1. These targets were
identified by differential expression analysis driven by the comparison of
profiles of esa1∆ sds3∆ relative to sds3∆ and WT strains to detect targets
specific to Esa1. Given Esa1’s reported, yet not fully elucidated role in

90
ribosome biogenesis, it was especially noteworthy that Esa1 was found to
regulate genes involved in ribosome biogenesis, but not ribosomal protein
genes exclusively. Esa1’s role in ribosome biogenesis was verified
functionally, as a distinct change in the polysome profile upon loss of ESA1.
Additionally, tying together findings from both Chapters 2 and 3, gene
expression analysis was used to determine if altered gene expression
underlies the hda1∆-mediated rescue of the bypass strain phenotypes, here
focusing on esa1∆ sds3∆. Whereas no large-scale changes in gene
expression were found to underlie the hda1∆ rescue, a new phenotype
involving cell wall-related stress of esa1∆ sds3∆ rescued by hda1∆ was
identified through gene expression analysis and was functionally validated.
The results from this chapter point toward the importance of Esa1-regulated
gene expression, and specifically implicate a critical role for Esa1 in ribosome
biogenesis.
Results
Differential expression analysis of ESA1 bypass mutant to define
Esa1-regulated gene expression. Bypass suppression of ESA1 made
possible the first studies whereby cells could be examined completely devoid
of ESA1. This was of great utility, but also presented challenges, since any
esa1∆ mutant also carries an sds3∆ mutation. In order to understand the
transcriptional targets of Esa1, a multi-level differential expression analysis

91
was necessary to define targets unique to Esa1, parsed from sds3∆
transcriptional affects.
The direct transcriptional targets of Esa1 were defined by differential
expression analysis of WT, sds3∆, and esa1∆ sds3∆. Esa1 targets were
distinguished from Sds3 targets by meeting the following criteria for differential
expression: (1) transcripts that differed between WT and esa1∆ sds3∆ and (2)
transcripts that did not differ between WT and sds3∆. As illustrated by Venn
diagram (Figure 4-1), these criteria correspond to two segments of the three-
way diagram, with the number of differentially expressed transcripts underlined
in bold, totaling 587 transcripts putatively regulated by Esa1.
Since determining the degree to which Esa1 is a positive or a negative
regulator of specific gene expression is an important goal, hierarchical
clustering and heatmap analysis was performed on the 587 Esa1-regulated
transcripts (Figure 4-2). It was found that nearly half of the transcripts are
upregulated relative to WT upon loss of ESA1 (Tables 4-5, 4-6, 4-7, 4-8)
whereas the other transcripts are downregulated (Tables 4-2, 4-3, 4-4). In
many cases, sds3∆ expression is at an intermediate level between WT and
esa1∆ sds3∆. Hierarchical clustering of the 587 transcripts results in esa1∆
sds3∆ clustering apart from WT and sds3∆, verifying that these transcripts are
distinct in esa1∆ sds3∆.
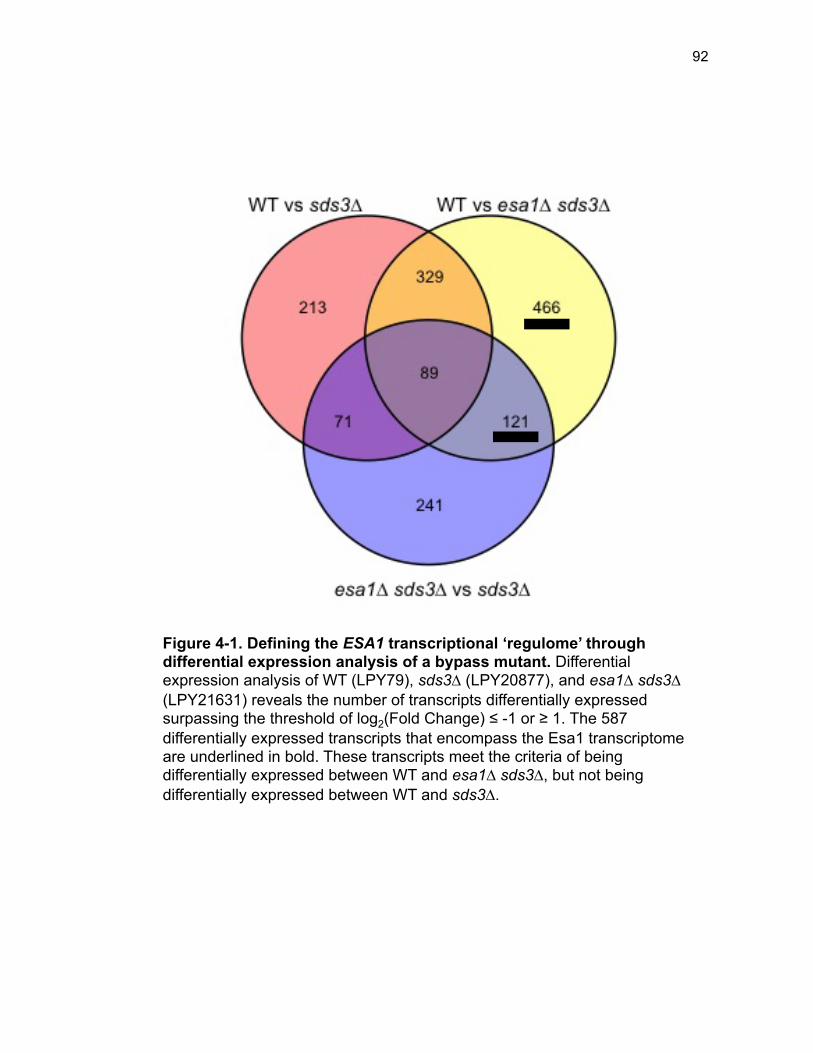
92
Figure 4-1. Defining the ESA1 transcriptional ‘regulome’ through differential expression analysis of a bypass mutant. Differential expression analysis of WT (LPY79), sds3∆ (LPY20877), and esa1∆ sds3∆ (LPY21631) reveals the number of transcripts differentially expressed surpassing the threshold of log2(Fold Change) ≤ -1 or ≥ 1. The 587 differentially expressed transcripts that encompass the Esa1 transcriptome are underlined in bold. These transcripts meet the criteria of being differentially expressed between WT and esa1∆ sds3∆, but not being differentially expressed between WT and sds3∆.
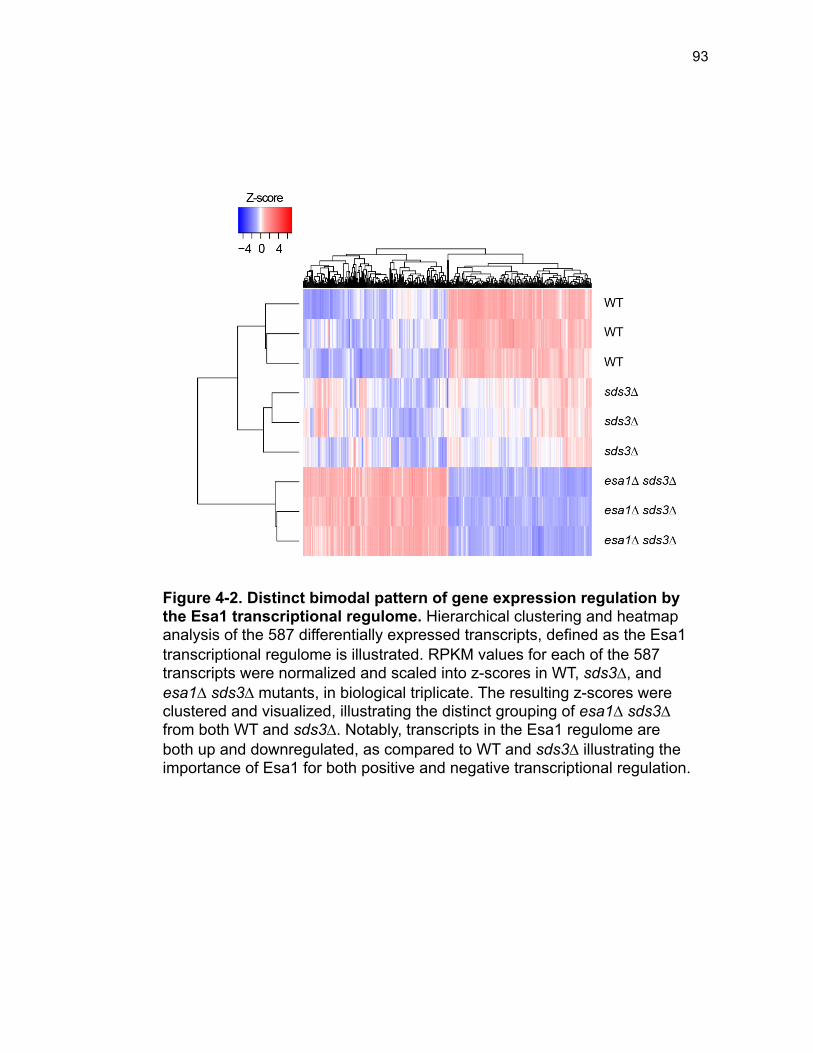
93
Figure 4-2. Distinct bimodal pattern of gene expression regulation by the Esa1 transcriptional regulome. Hierarchical clustering and heatmap analysis of the 587 differentially expressed transcripts, defined as the Esa1 transcriptional regulome is illustrated. RPKM values for each of the 587 transcripts were normalized and scaled into z-scores in WT, sds3∆, and esa1∆ sds3∆ mutants, in biological triplicate. The resulting z-scores were clustered and visualized, illustrating the distinct grouping of esa1∆ sds3∆ from both WT and sds3∆. Notably, transcripts in the Esa1 regulome are both up and downregulated, as compared to WT and sds3∆ illustrating the importance of Esa1 for both positive and negative transcriptional regulation.

94
Ribosome biogenesis genes are regulated by Esa1. To better
understand the broad identity of the Esa1-regulated transcripts, gene ontology
(GO) analysis was
performed. This revealed that several genes involved in metabolism were
downregulated upon loss of ESA1 (Figure 4-3). The enrichment of this gene
class, although significant, could be indicative of the slowed growth rate, and
corresponding reduced metabolic rate of the esa1∆ sds3∆ mutants. Because
the metabolism gene class may have been differentially expression due to
growth rate in the ESA1 mutants, analysis instead focused on a profound set
of genes downregulated upon loss of ESA1.
A striking enrichment was found for genes involved in ribosome
biogenesis, upregulated upon loss of ESA1 (Figure 4-4). With this upregulated
gene set, there were many significantly enriched GO terms, all involving
ribosome biogenesis or rRNA metabolism. These were visualized with a
REViGO plot in Figure 4-5 by collapsing highly similar or hierarchical terms in
a 2-dimensional space, where the area of each GO-term box is proportional to
the significance of its enrichment (Supek et al. 2011). Therefore, whereas
many different GO terms seemed to be enriched in the upregulated gene set,
the upregulated genes are overwhelmingly involved in facets of ribosome
biogenesis.
It is well documented that ribosome biogenesis genes are frequently co-
regulated as part of a gene set often referred to as the Ribi regulon that
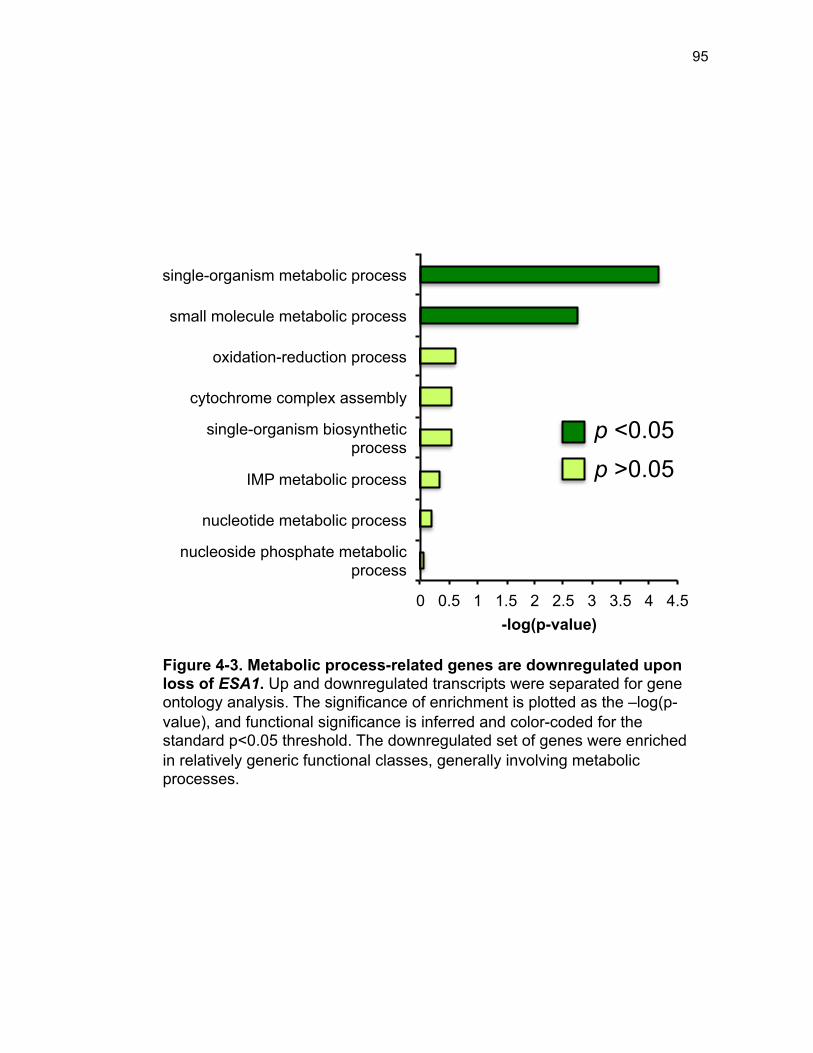
95
0 0.5 1 1.5 2 2.5 3 3.5 4 4.5
nucleoside phosphate metabolic process
nucleotide metabolic process
IMP metabolic process
single-organism biosynthetic process
cytochrome complex assembly
oxidation-reduction process
small molecule metabolic process
single-organism metabolic process
-log(p-value)
p <0.05
p >0.05
Figure 4-3. Metabolic process-related genes are downregulated upon loss of ESA1. Up and downregulated transcripts were separated for gene ontology analysis. The significance of enrichment is plotted as the –log(p-value), and functional significance is inferred and color-coded for the standard p<0.05 threshold. The downregulated set of genes were enriched in relatively generic functional classes, generally involving metabolic processes.
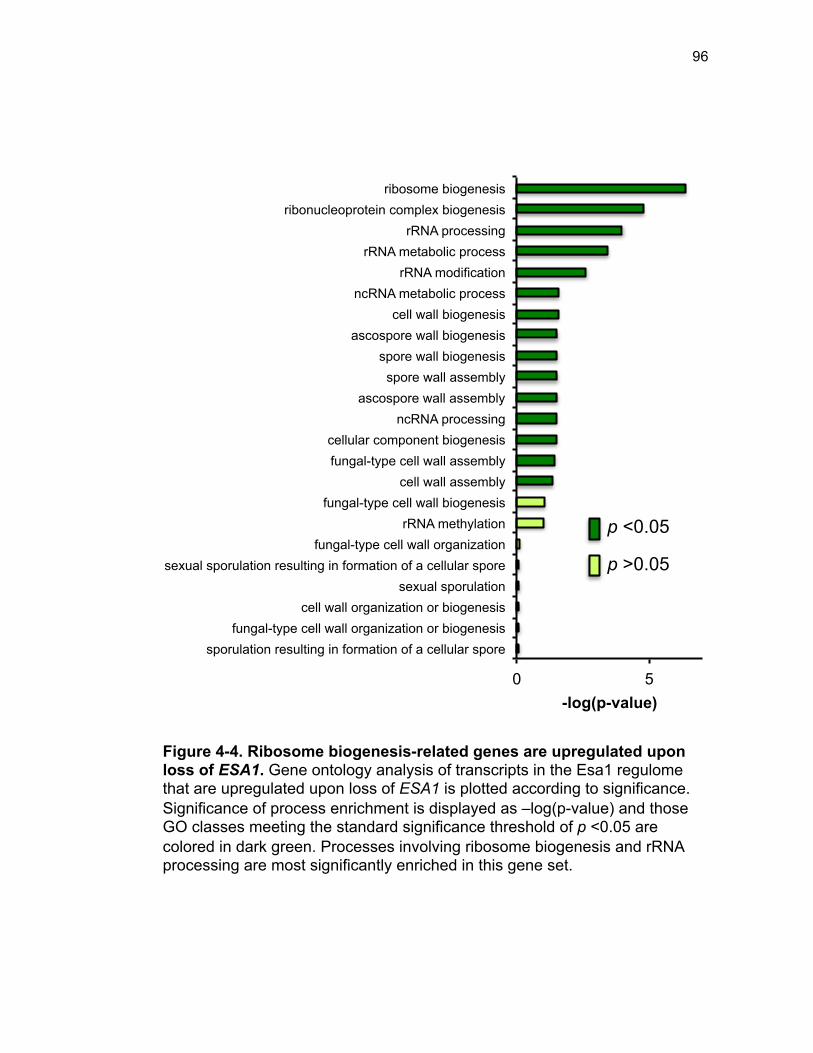
96
0 5
sporulation resulting in formation of a cellular spore fungal-type cell wall organization or biogenesis
cell wall organization or biogenesis sexual sporulation
sexual sporulation resulting in formation of a cellular spore fungal-type cell wall organization
rRNA methylation fungal-type cell wall biogenesis
cell wall assembly fungal-type cell wall assembly cellular component biogenesis
ncRNA processing ascospore wall assembly
spore wall assembly spore wall biogenesis
ascospore wall biogenesis cell wall biogenesis
ncRNA metabolic process rRNA modification
rRNA metabolic process rRNA processing
ribonucleoprotein complex biogenesis ribosome biogenesis
-log(p-value)
p <0.05
p >0.05
Figure 4-4. Ribosome biogenesis-related genes are upregulated upon loss of ESA1. Gene ontology analysis of transcripts in the Esa1 regulome that are upregulated upon loss of ESA1 is plotted according to significance. Significance of process enrichment is displayed as –log(p-value) and those GO classes meeting the standard significance threshold of p <0.05 are colored in dark green. Processes involving ribosome biogenesis and rRNA processing are most significantly enriched in this gene set.

97
Figure 4-5. Summary of gene ontology highlights ribosome biogenesis. To visualize the large set of gene ontology classes significantly overrepresented in the genes downregulated upon loss of ESA1, the REViGO algorithm (Supek et al. 2011) was used to visualize the major overrepresented gene ontology classes, by stacking hierarchical gene ontology terms. The area of each box is proportional to the significance by which the term is enriched. Individual gene ontology terms are in black, and the overarching more general gene ontology term that encompasses several terms is written in more transparent white font. The two major processes enriched in this gene set are ribosome biogenesis and rRNA metabolism.

98
responds similarly to genetic and environmental manipulations (Gasch et al.
2000; Wade et al. 2001; Jorgensen et al. 2002; Jorgensen et al. 2004; Wade
et al. 2006). Since the Ribi regulon is a seemingly ever-expanding set of
genes, rather than defining a direct overlay of the Esa1-ribosome biogenesis-
related genes with the reported Ribi regulon genes, STRING analysis (Jensen
et al. 2009) was used to evaluate the degree of co-expression de novo. As
depicted by the network diagram for the protein-coding ribosome biogenesis
genes upregulated upon loss of ESA1, the majority of these genes are co-
expressed (Figure 4-6). Additionally, this analysis reveals Esa1 involvement in
a small sub-network of co-regulated ribosomal protein genes towards the
upper part of the network diagram (Figure 4-6, dashed circle), but in general,
there is a high degree of interconnection among all genes queried, and no
enrichment of any one specific ribosomal component or biogenesis process.
Given the slow growth and impaired fitness of the esa1∆ sds3∆ strain, it
was initially surprising that genes involved in ribosome biogenesis were
upregulated upon loss of ESA1. The common notion is that slow growth would
be associated with a decrease in protein synthesis, as regulated by TOR-
pathway sensing, causing a decline in ribosome biogenesis [reviewed in
(Lempiainen and Shore 2009)]. However, it is the tight regulation of ribosome
biogenesis that is most critical, and dysregulation in either direction is likely to
be detrimental to cellular growth and fitness.

99
Figure 4-6. Co-regulation of ribosome biogenesis gene expression. STRING analysis (Jensen et al. 2009) was performed with the query set of 51 genes that were tagged as upregulated in the Esa1 regulome and specifically involved in ribosome biogenesis by gene ontology analysis. Several transcripts were queried but not ultimately included in STRING analysis, including 15 snRNAs and the 21S-rRNA transcript, since STRING analysis is limited to transcripts that encode proteins, only. The remaining 36 transcripts are represented by nodes in the diagram, with the lines representing reported co-expression. The blue-dashed circle highlights a network of co-regulated ribosomal protein genes. There is a high degree of co-expression in the upregulated ribosome biogenesis gene-set.

100
Accordingly, it was important to see at a molecular level, if and what
affect this upregulation in ribosome biogenesis-related gene expression, that
appeared to be independent of TOR signaling, was having mechanistically.
Polysome profiling was used to assess the abundance of ribosomal
components in esa1∆ sds3∆ when compared to sds3∆. Upon analysis of the
polysome profiles, two phenotypes were most readily apparent (Figure 4-7).
Primarily, esa1∆ sds3∆ mutant cells had a greater proportion of 80S
monosomes than the sds3∆ single mutant. Additionally, in esa1∆ sds3∆ the
polysome fraction was significantly smaller and more collapsed relative to that
of sds3∆. The polysome collapse and concurrent increase in the 80S
monosome is traditionally associated with a large-scale reduction in translation
initiation, and is frequently observed upon stress-induction such as with
glucose starvation (Ashe et al. 2000). Therefore, despite increases in
transcription of genes associated with ribosome biogenesis, polysome profiling
illustrates that esa1∆ sds3∆ cells have impaired translation. This implies that
Esa1 is crucial for regulation and coordination of ribosome biogenesis.
Whereas loss of ESA1 results in upregulation of ribosome biogenesis-related
genes, this upregulation is consistent with dysregulation concomitant with
impaired cellular fitness.
A transcriptional basis for the hda1∆-mediated rescue of esa1∆
sds3∆ mutant phenotypes. Given that the expression of a sub-group of
ribosome biogenesis genes was dysregulated by loss of ESA1 (Figure 4-5),
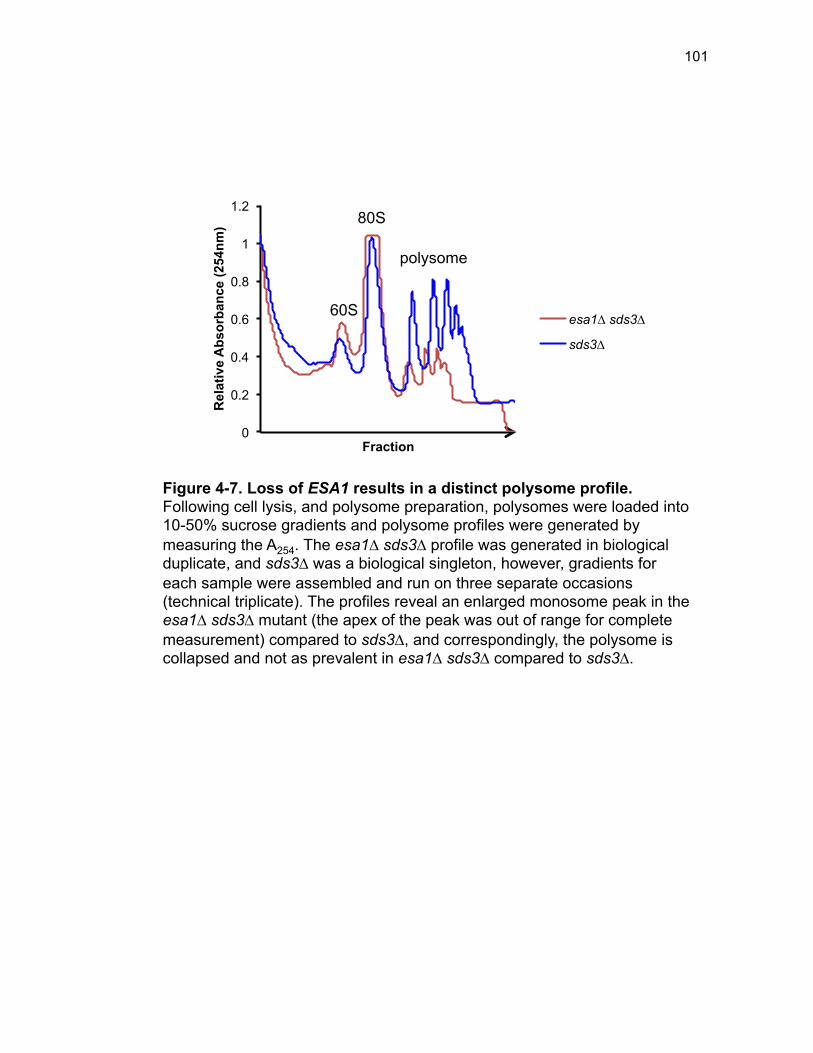
101
0
0.2
0.4
0.6
0.8
1
1.2 R
elat
ive
Abs
orba
nce
(254
nm)
Fraction
esa1∆ sds3∆
sds3∆
Figure 4-7. Loss of ESA1 results in a distinct polysome profile. Following cell lysis, and polysome preparation, polysomes were loaded into 10-50% sucrose gradients and polysome profiles were generated by measuring the A254. The esa1∆ sds3∆ profile was generated in biological duplicate, and sds3∆ was a biological singleton, however, gradients for each sample were assembled and run on three separate occasions (technical triplicate). The profiles reveal an enlarged monosome peak in the esa1∆ sds3∆ mutant (the apex of the peak was out of range for complete measurement) compared to sds3∆, and correspondingly, the polysome is collapsed and not as prevalent in esa1∆ sds3∆ compared to sds3∆.
80S
polysome
60S

102
and that esa1∆ sds3∆ mutants had aberrant polysome profiles (Figure 4-7)
and impaired rDNA silencing (Figure 3-8B) with the latter rescued by hda1∆,
gene expression analysis was performed on the esa1∆ sds3∆ hda1∆ mutant
to determine if this strain had suppressed the ribosome biogenesis gene
expression defect. Additionally, it was possible that other esa1∆ sds3∆ gene
expression signatures would be rescued by hda1∆. Therefore non-biased
global RNA-sequencing was performed on the esa1∆ sds3∆ hda1∆ mutant.
Gene expression analysis between WT, esa1∆ sds3∆, and esa1∆
sds3∆ hda1∆ revealed several additional transcripts differentially expressed in
the hda1∆ rescue triple mutant (Figure 4-8). To identify transcripts specifically
involved in the rescue of esa1∆ sds3∆ by hda1∆, transcripts that were
significantly different between WT and esa1∆ sds3∆ and that were also
different between esa1∆ sds3∆ and esa1∆ sds3∆ hda1∆ were identified.
These filtering criteria resulted in the identification of 138 esa1∆ sds3∆
transcripts that the hda1∆ mutation restored expression back to WT-levels.
To determine the degree to which these 138 hda1∆-rescue transcripts
are either up or downregulated in esa1∆ sds3∆ as compared to both WT and
esa1∆ sds3∆ hda1∆, hierarchical clustering and heatmap analysis of scaled-
expression values was performed (Figure 4-9). Again, there was a split, with
about 40% of the transcripts upregulated in esa1∆ sds3∆, and the remaining
60% downregulated relative to WT and esa1∆ sds3∆ hda1∆. To assess
functional relevance, this list of transcripts was overlaid with global Esa1
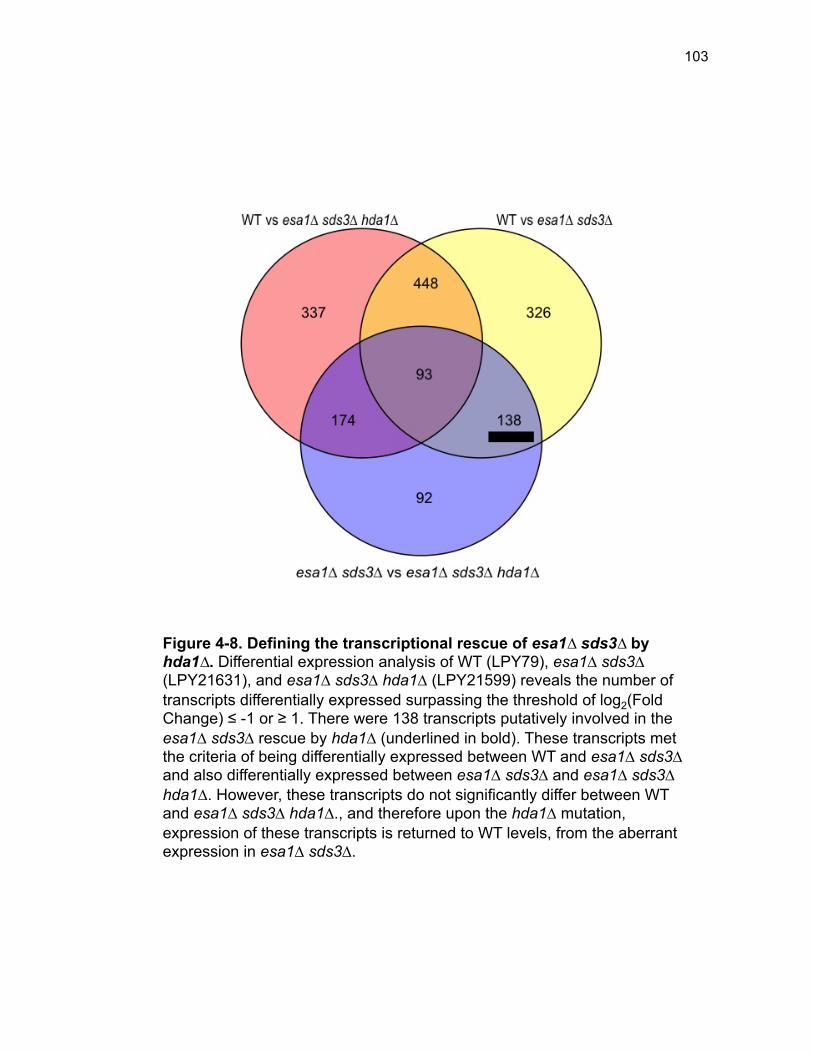
103
Figure 4-8. Defining the transcriptional rescue of esa1∆ sds3∆ by hda1∆. Differential expression analysis of WT (LPY79), esa1∆ sds3∆ (LPY21631), and esa1∆ sds3∆ hda1∆ (LPY21599) reveals the number of transcripts differentially expressed surpassing the threshold of log2(Fold Change) ≤ -1 or ≥ 1. There were 138 transcripts putatively involved in the esa1∆ sds3∆ rescue by hda1∆ (underlined in bold). These transcripts met the criteria of being differentially expressed between WT and esa1∆ sds3∆ and also differentially expressed between esa1∆ sds3∆ and esa1∆ sds3∆ hda1∆. However, these transcripts do not significantly differ between WT and esa1∆ sds3∆ hda1∆., and therefore upon the hda1∆ mutation, expression of these transcripts is returned to WT levels, from the aberrant expression in esa1∆ sds3∆.

104
Figure 4-9. There are 138 transcripts defining gene expression rescue by esa1∆ sds3∆ hda1∆. Hierarchical clustering and heatmap analysis was applied to scaled and normalized RPKM expression data for the 138 transcripts putatively involved in the hda1∆ rescue of esa1∆ sds3∆ across the biological triplicates for each mutant. There is a higher abundance of transcripts that are downregulated in esa1∆ sds3∆ that are a part of this subset, but still a substantial number are upregulated as well. Here, esa1∆ sds3∆ clusters apart from WT and esa1∆ sds3∆ hda1∆, as expected, suggesting that these transcripts bring esa1∆ sds3∆ hda1∆ closer to WT in expression values.

105
binding data from (Robert et al. 2004). No significant enrichment patterns were
identified in this gene set when compared with Esa1 binding. The failure to
identify enrichment of Esa1-regulated differentially expressed genes with
Esa1-bound genes could be due to differences in strain growth, and/or the
general correlation between transcription and Esa1 occupancy that was
reported (Robert et al. 2004) and discussed earlier in the introduction to this
chapter. Gene ontology analysis of the 138 differentially expressed transcripts
revealed that there was a modest yet statistically significant enrichment of
genes involved in cell wall biogenesis (Figure 4-10). Unlike the exclusively
downregulated ribosome biogenesis (Figure 4-4) or upregulated metabolism
(Figure 4-3) gene classes, these cell wall biogenesis genes were not
exclusively positively nor negatively regulated by esa1∆ sds3∆ (Figure 4-11).
Cell wall related phenotypes were not explicitly surveyed in either
esa1∆ sds3∆ (Torres-Machorro and Pillus 2014) or epl1∆ sds3∆ (Chapters 2
and 3). To address any cell wall defects in esa1∆ sds3∆ and the putative
rescue of such defects by hda1∆, as suggested by transcriptional evidence,
esa1∆ sds3∆ was grown in the absence and presence of sorbitol at 30˚ and at
an elevated temperature (Figure 4-12A). The presence of 1M sorbitol provides
enhanced osmotic support, and thus, any mutant strains with cell wall defects
would be expected to have improved growth upon relief of cell wall related
stress. The addition of sorbitol resulted in an ~10-fold increase in growth of
esa1∆ sds3∆ at both 30˚ and 37˚, demonstrating that loss of ESA1 results in a
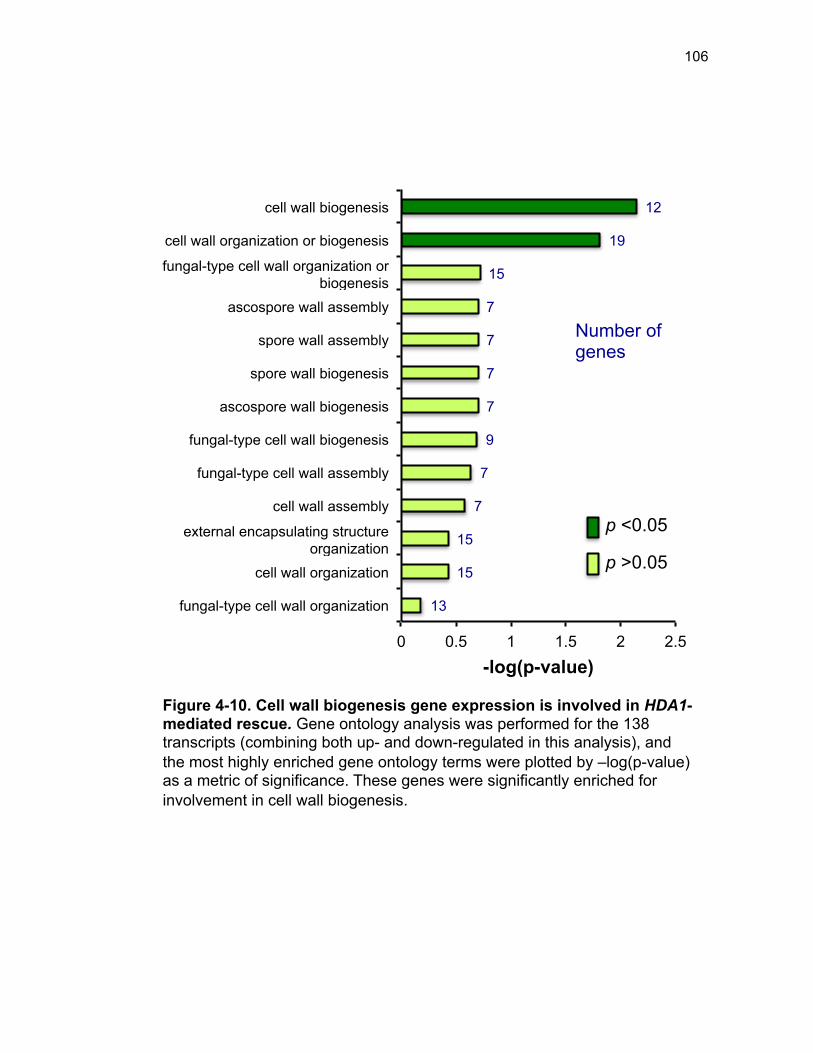
106
13
15
15
7
7
9
7
7
7
7
15
19
12
0 0.5 1 1.5 2 2.5
fungal-type cell wall organization
cell wall organization
external encapsulating structure organization
cell wall assembly
fungal-type cell wall assembly
fungal-type cell wall biogenesis
ascospore wall biogenesis
spore wall biogenesis
spore wall assembly
ascospore wall assembly
fungal-type cell wall organization or biogenesis
cell wall organization or biogenesis
cell wall biogenesis
-log(p-value)
Number of genes
Figure 4-10. Cell wall biogenesis gene expression is involved in HDA1-mediated rescue. Gene ontology analysis was performed for the 138 transcripts (combining both up- and down-regulated in this analysis), and the most highly enriched gene ontology terms were plotted by –log(p-value) as a metric of significance. These genes were significantly enriched for involvement in cell wall biogenesis.
p <0.05
p >0.05
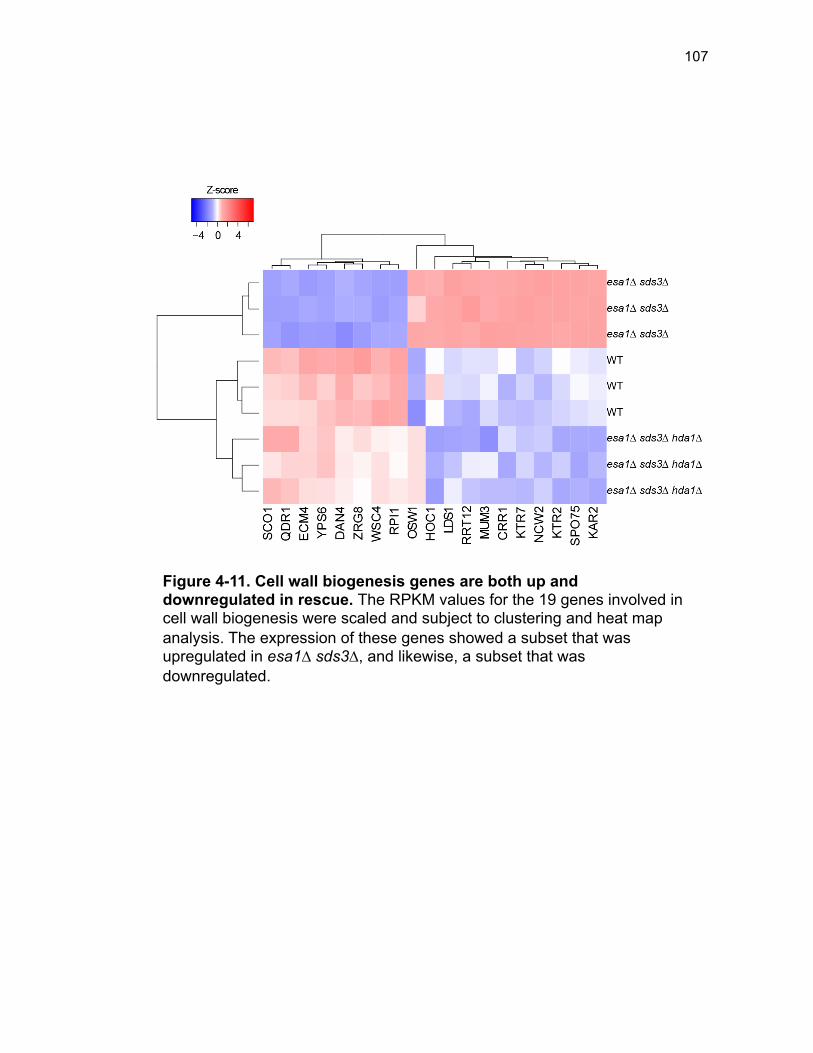
107
Figure 4-11. Cell wall biogenesis genes are both up and downregulated in rescue. The RPKM values for the 19 genes involved in cell wall biogenesis were scaled and subject to clustering and heat map analysis. The expression of these genes showed a subset that was upregulated in esa1∆ sds3∆, and likewise, a subset that was downregulated.

108
WT sds3∆ hda1∆
esa1∆ sds3∆ esa1∆ sds3∆ hda1∆
37˚ 30˚
YPAD + sorbitol
YPAD YPAD + sorbitol
YPAD
Figure 4-12. Osmotic support facilitates improved growth of esa1∆ sds3∆ but is distinct from DNA damage sensitivity. (A) The presence of a defect in the cell wall was probed with serial dilution assays with the osmoprotectant 1M sorbitol, to test if addition of osmotic support would lead to enhanced growth at either 30˚ or 37˚. WT (LPY79), sds3∆ (LPY20877), hda1∆ (LPY12885), esa1∆ sds3∆ (LPY21631), and esa1∆ sds3∆ hda1∆ (LPY21599) were grown in YPAD and plated in 5-fold serial dilutions on YPAD or YPAD + 1M sorbitol. The addition of sorbitol resulted in enhanced growth of esa1∆ sds3∆ at both temperatures, but also improved esa1∆ sds3∆ hda1∆ growth at 37˚. (B) The strains were assessed for the effect of osmotic support on DNA damage sensitivity, and plated on YPAD + MMS or HU +/- 1M sorbitol at 30˚. Here, the addition of sorbitol did not significantly augment growth in response to DNA damage.
WT sds3∆ hda1∆
esa1∆ sds3∆ esa1∆ sds3∆ hda1∆
MMS + sorbitol
MMS HU + sorbitol
HU
A
B 30˚

109
cell wall defect that is rescued with osmotic support. There was a modest
growth advantage conferred when esa1∆ sds3∆ hda1∆ was grown with
sorbitol, especially at elevated temperatures. This implies that whereas esa1∆
sds3∆ hda1∆ suppressed the esa1∆ sds3∆ cell wall defect, the suppression
was partial. This conclusion is also supported by the expression of the cell-wall
related genes that are not expressed at parallel levels between WT and esa1∆
sds3∆ hda1∆ (Figure 4-9).
Since the rescue of esa1∆ sds3∆ by hda1∆ revealed diverse
phenotypes, it was important to understand the degree to which the cell-wall
defect underlies many of the other observed phenotypic defects in
esa1∆ sds3∆ such as the sensitivity to genotoxic stresses. For example, if
esa1∆ sds3∆ were relieved of osmotic-stress, would it still be highly sensitive
to DNA damage?
To test if the cell-wall defect impairs response to DNA damage, esa1∆
sds3∆ was subject to DNA damage in the absence and presence of 1M
sorbitol. Even with the addition of osmotic support, esa1∆ sds3∆ was still
unable to grow in the presence of DNA damaging agents (Figure 4-12B). This
suggests that whereas global gene expression analysis identified cell wall
biogenesis as the major process transcriptionally rescued by hda1∆ under
normal growth conditions, the validated esa1∆ sds3∆ cell wall defect is not the
root cause of the large-scale rescue by hda1∆, but instead represents an
additional suppressed phenotype.

110
Discussion
In this chapter, global gene expression analysis was used to address
the question of how Esa1 is regulating transcription genome-wide, and
simultaneously to what extent the hda1∆-mediated rescue of esa1∆ sds3∆
occurs at the level of transcription. Previous studies had assessed the
transcriptional consequences of the loss of Esa1 enzymatic activity with
hypomorphic alleles of ESA1, rather than complete null mutants. Here, gene
expression profiling revealed that Esa1 is critical for the proper expression of
ribosome biogenesis networks. Correspondingly, the loss of ESA1 results in
several ribosome biogenesis-related phenotypes, some that were previously
characterized, and others that were examined here for the first time.
Interestingly, no widespread gene expression signature appears to underlie
the hda1∆ rescue of esa1∆ sds3∆, however, transcriptional analysis promoted
the identification of new a cell wall related phenotype in esa1∆ sds3∆ that is
rescued upon loss of HDA1.
Transcriptome analysis of ESA1 bypass. The identification of the
bypass suppression of ESA1 facilitated many additional studies to
characterize the implications of complete loss of ESA1. Here, the question of
transcriptional targets was addressed, however, with this rich dataset, many
additional questions can be raised. For example, the same expression data

111
can be used to ask if expression levels of any transcripts are specifically
important for the bypass of ESA1.
Additionally, it will be important to understand how esa1∆ sds3∆ is
divergent from the established hypomorphic alleles of ESA1 used in many
fundamental studies. These seminal studies of esa1 set the basis for our
understanding of Esa1 function, and it is important to acknowledge the
limitations of and differences between hypomorphic alleles and bypass
suppression. A simple differential expression analysis of the esa1∆ sds3∆ data
presented here as compared to esa1 expression data, such as has already
been generated in Resgen strains for the esa1-414 allele (van Bakel et al.
2013), would readily address this question.
Understanding the significance of Esa1 in ribosome biogenesis.
The most prominent result from this chapter is the involvement of Esa1 in
ribosome biogenesis. It was striking that loss of ESA1 resulted in an
upregulation of several genes involved in ribosome biogenesis, despite the
slow growth of the mutant strain, and also that no particular ribosomal
component was selectively affected. Whereas previous studies had implicated
Esa1 as important for expression of ribosomal protein genes (Reid et al.
2000), the results here support the more recent evidence suggesting that
ribosomal protein genes become SAGA-dependent upon loss of NuA4 (Uprety
et al. 2015). Additional validation studies using qPCR will be important to
validate the expression of candidates selected from among these targets.

112
However, since many ribosome biogenesis genes were differentially
expressed, and it is unlikely that high-throughput sequencing falsely identified
an entire set of genes, validation is not likely to vastly alter the interpretation of
results.
Furthermore, the TOR pathway, a primary ribosome biogenesis
regulatory pathway, responsive to environmental and nutritional conditions,
was not affected by esa1∆ sds3∆ [reviewed in (Loewith and Hall 2011)]. This
suggests that Esa1 regulates ribosome biogenesis independently of TOR-
signaling. However, the question remains of how TOR-signaling and the ESA1
bypass mutant intersect. Mutant alleles of esa1 and epl1 are highly sensitive
to rapamycin (Boudreault et al. 2003). However, the rapamycin-response and
sensitivity of the bypass mutants remains to be tested, and such direct
analysis would begin to address this question.
To assess the function of the altered ribosome biogenesis gene
expression, polysome profiling was performed. Polysome profiling revealed
that the 80S monosome fraction of esa1∆ sds3∆ was enriched as compared to
sds3∆ (Figure 4-7). Whereas it is presumed that this implies a defect in
translation (Ashe et al. 2000; Heyer and Moore 2016), it is also possible that
there are simply more assembled, but nonfunctional, ribosomes in esa1∆
sds3∆. Given that the ribosome genes were upregulated, it would be
interesting if this simply rendered more ribosomes, but not more translation. A
classic salt dissociation experiment of ribosomes would help reveal if

113
translation is stalled or if there are simply more ribosomes in esa1∆ sds3∆.
Ribosomes that are assembled, but not actively participating in translation will
be disassociated into the 40S and 60S subunits by 0.8M KCl (Martin and
Hartwell 1970), and will address this unresolved question.
Another possible explanation for the enrichment of the 80S monosome
peak in esa1∆ sds3∆ is enrichment of specific mRNAs that co-sediment with
the monosomes. Upon bypass of ESA1, many short open reading frames
(sORFs) are differentially expressed between WT and esa1∆ sds3∆ (Tables 4-
2 – Table 4-8). There are ongoing efforts to understand both the function and
significance of sORFs, as some are non-coding, and others code for short
regulatory peptides [reviewed in (Basrai et al. 1997; Andrews and Rothnagel
2014)]. The significance for the enrichment of these short transcripts in the
ESA1 bypass transcriptome remains to be determined, but it was previously
found that mRNAs with sORFs co-sediment with the 80S monocomes (Arava
et al. 2003). If the mRNA transcribed by these short ORFs is driving the large
80S peak, the monosome should remain intact upon KCl treatment, and can
begin to be assessed by the salt dissociation experiments as well. Further
assessing the molecular significance of the increased monosome peak will be
critical for understanding specifically how Esa1 regulates ribosome biogenesis.
The role of Hda1 in balancing regulation of ribosome biogenesis.
In Chapter 3, it was illustrated that hda1∆ suppresses the esa1∆ sds3∆ defect
in rDNA silencing. However, in this chapter, hda1∆ did not rescue the aberrant

114
ribosomal biogenesis gene expression signature of esa1∆ sds3∆ and instead
was found only to rescue an unrelated cell-wall phenotype. It is possible that
Hda1 is involved in regulation limited to rDNA, and not other ribosome-related
gene expression. It is also possible that hda1∆ is deeply involved in balancing
the ribosome biogenesis dysregulation in esa1∆ sds3∆, despite the gene
expression results.
To begin to address the role of Hda1 in opposing NuA4 regulation of
ribosome biogenesis, polysome profiling should be performed in esa1∆ sds3∆
hda1∆ cells to determine the relative monosome-polysome abundances. This
analysis would also provide insight into the significance and relevance of the
esa1∆ sds3∆ gene expression signature that appear to potentially be at odds
with the otherwise robust Hda1-mediated rescue. Nevertheless, whereas the
specific involvement of Hda1 in opposing NuA4 in ribosome biogenesis
regulation remains unclear, it is apparent that Esa1 and NuA4 are critical for
regulation of ribosome biogenesis, both at the rDNA and at the level of
ribosomal gene expression.
The results in this chapter present the striking finding that Esa1
regulates ribosome biogenesis gene expression. This regulation appears to be
independent of the canonical TOR-signaling and regulation, and has a
functional effect on ribosome composition in the cell. However, the mechanism
for how Esa1 regulates ribosome biogenesis remains to be fully elucidated.
Esa1-directed acetylation has only been reported for one of the ribosome

115
biogenesis differentially expressed genes (PRP43, encoding an RNA helicase
required for biogenesis of both small and large subunit rRNAs) (Lin et al. 2009;
Mitchell et al. 2013a; Downey et al. 2015). Interestingly, despite the slow-
growth of esa1∆ bypass mutants, Esa1-mediated regulation is important for
controlling levels of ribosome biogenesis; without ESA1 several ribosome
biogenesis genes are upregulated. Upregulation of ribosome biogenesis gene
expression is frequently associated with oncogenesis (Ruggero 2012; van
Sluis and McStay 2014), and thus underscores the importance of tight
regulation. Whereas esa1 hypomorphic alleles have led to conflicting reports
of Esa1’s role in ribosome biogenesis and regulation, the ESA1 bypass mutant
provides a new and interesting way to test and better understand how Esa1
regulates ribosome biology.

116
Table 4-1. Strains used in Chapter 4. All strains were constructed for this thesis unless otherwise noted. Strain Genotype LPY79 MATα W303 (ade2-1 can1-100 his3-11 leu2,3,112 trp1-1 ura3-1) * LPY20877 MATα W303 sds3∆::natMX LPY21631 MATα W303 esa1∆::HIS3 sds3∆::natMX LPY21599 MATα W303 esa1∆::HIS3 sds3∆::natMX hda1∆::TRP1 LPY12885 MATα W303 hda1∆::TRP1 * (Thomas and Rothstein 1989)
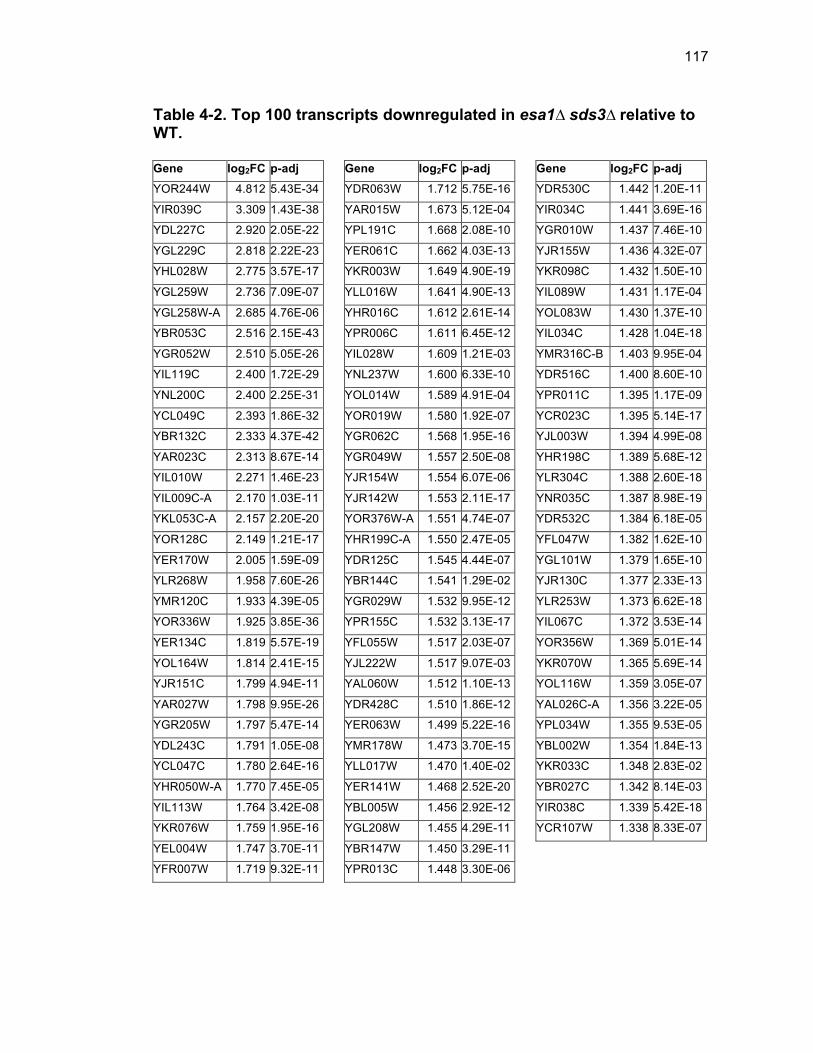
117
Table 4-2. Top 100 transcripts downregulated in esa1∆ sds3∆ relative to WT. Gene log2FC p-adj YOR244W 4.812 5.43E-34
YIR039C 3.309 1.43E-38
YDL227C 2.920 2.05E-22
YGL229C 2.818 2.22E-23
YHL028W 2.775 3.57E-17
YGL259W 2.736 7.09E-07
YGL258W-A 2.685 4.76E-06
YBR053C 2.516 2.15E-43
YGR052W 2.510 5.05E-26
YIL119C 2.400 1.72E-29
YNL200C 2.400 2.25E-31
YCL049C 2.393 1.86E-32
YBR132C 2.333 4.37E-42
YAR023C 2.313 8.67E-14
YIL010W 2.271 1.46E-23
YIL009C-A 2.170 1.03E-11
YKL053C-A 2.157 2.20E-20
YOR128C 2.149 1.21E-17
YER170W 2.005 1.59E-09
YLR268W 1.958 7.60E-26
YMR120C 1.933 4.39E-05
YOR336W 1.925 3.85E-36
YER134C 1.819 5.57E-19
YOL164W 1.814 2.41E-15
YJR151C 1.799 4.94E-11
YAR027W 1.798 9.95E-26
YGR205W 1.797 5.47E-14
YDL243C 1.791 1.05E-08
YCL047C 1.780 2.64E-16
YHR050W-A 1.770 7.45E-05
YIL113W 1.764 3.42E-08
YKR076W 1.759 1.95E-16
YEL004W 1.747 3.70E-11
YFR007W 1.719 9.32E-11
Gene log2FC p-adj YDR063W 1.712 5.75E-16
YAR015W 1.673 5.12E-04
YPL191C 1.668 2.08E-10
YER061C 1.662 4.03E-13
YKR003W 1.649 4.90E-19
YLL016W 1.641 4.90E-13
YHR016C 1.612 2.61E-14
YPR006C 1.611 6.45E-12
YIL028W 1.609 1.21E-03
YNL237W 1.600 6.33E-10
YOL014W 1.589 4.91E-04
YOR019W 1.580 1.92E-07
YGR062C 1.568 1.95E-16
YGR049W 1.557 2.50E-08
YJR154W 1.554 6.07E-06
YJR142W 1.553 2.11E-17
YOR376W-A 1.551 4.74E-07
YHR199C-A 1.550 2.47E-05
YDR125C 1.545 4.44E-07
YBR144C 1.541 1.29E-02
YGR029W 1.532 9.95E-12
YPR155C 1.532 3.13E-17
YFL055W 1.517 2.03E-07
YJL222W 1.517 9.07E-03
YAL060W 1.512 1.10E-13
YDR428C 1.510 1.86E-12
YER063W 1.499 5.22E-16
YMR178W 1.473 3.70E-15
YLL017W 1.470 1.40E-02
YER141W 1.468 2.52E-20
YBL005W 1.456 2.92E-12
YGL208W 1.455 4.29E-11
YBR147W 1.450 3.29E-11
YPR013C 1.448 3.30E-06
Gene log2FC p-adj YDR530C 1.442 1.20E-11
YIR034C 1.441 3.69E-16
YGR010W 1.437 7.46E-10
YJR155W 1.436 4.32E-07
YKR098C 1.432 1.50E-10
YIL089W 1.431 1.17E-04
YOL083W 1.430 1.37E-10
YIL034C 1.428 1.04E-18
YMR316C-B 1.403 9.95E-04
YDR516C 1.400 8.60E-10
YPR011C 1.395 1.17E-09
YCR023C 1.395 5.14E-17
YJL003W 1.394 4.99E-08
YHR198C 1.389 5.68E-12
YLR304C 1.388 2.60E-18
YNR035C 1.387 8.98E-19
YDR532C 1.384 6.18E-05
YFL047W 1.382 1.62E-10
YGL101W 1.379 1.65E-10
YJR130C 1.377 2.33E-13
YLR253W 1.373 6.62E-18
YIL067C 1.372 3.53E-14
YOR356W 1.369 5.01E-14
YKR070W 1.365 5.69E-14
YOL116W 1.359 3.05E-07
YAL026C-A 1.356 3.22E-05
YPL034W 1.355 9.53E-05
YBL002W 1.354 1.84E-13
YKR033C 1.348 2.83E-02
YBR027C 1.342 8.14E-03
YIR038C 1.339 5.42E-18
YCR107W 1.338 8.33E-07
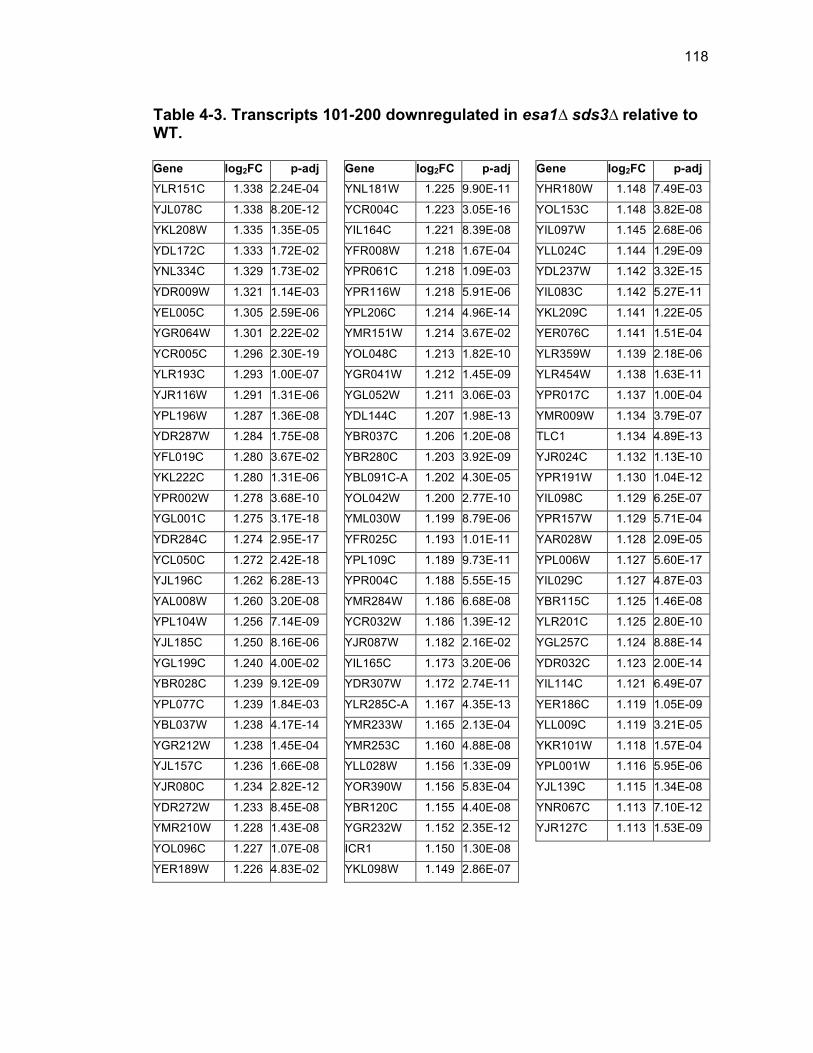
118
Table 4-3. Transcripts 101-200 downregulated in esa1∆ sds3∆ relative to WT. Gene log2FC p-adj YLR151C 1.338 2.24E-04
YJL078C 1.338 8.20E-12
YKL208W 1.335 1.35E-05
YDL172C 1.333 1.72E-02
YNL334C 1.329 1.73E-02
YDR009W 1.321 1.14E-03
YEL005C 1.305 2.59E-06
YGR064W 1.301 2.22E-02
YCR005C 1.296 2.30E-19
YLR193C 1.293 1.00E-07
YJR116W 1.291 1.31E-06
YPL196W 1.287 1.36E-08
YDR287W 1.284 1.75E-08
YFL019C 1.280 3.67E-02
YKL222C 1.280 1.31E-06
YPR002W 1.278 3.68E-10
YGL001C 1.275 3.17E-18
YDR284C 1.274 2.95E-17
YCL050C 1.272 2.42E-18
YJL196C 1.262 6.28E-13
YAL008W 1.260 3.20E-08
YPL104W 1.256 7.14E-09
YJL185C 1.250 8.16E-06
YGL199C 1.240 4.00E-02
YBR028C 1.239 9.12E-09
YPL077C 1.239 1.84E-03
YBL037W 1.238 4.17E-14
YGR212W 1.238 1.45E-04
YJL157C 1.236 1.66E-08
YJR080C 1.234 2.82E-12
YDR272W 1.233 8.45E-08
YMR210W 1.228 1.43E-08
YOL096C 1.227 1.07E-08
YER189W 1.226 4.83E-02
Gene log2FC p-adj YNL181W 1.225 9.90E-11
YCR004C 1.223 3.05E-16
YIL164C 1.221 8.39E-08
YFR008W 1.218 1.67E-04
YPR061C 1.218 1.09E-03
YPR116W 1.218 5.91E-06
YPL206C 1.214 4.96E-14
YMR151W 1.214 3.67E-02
YOL048C 1.213 1.82E-10
YGR041W 1.212 1.45E-09
YGL052W 1.211 3.06E-03
YDL144C 1.207 1.98E-13
YBR037C 1.206 1.20E-08
YBR280C 1.203 3.92E-09
YBL091C-A 1.202 4.30E-05
YOL042W 1.200 2.77E-10
YML030W 1.199 8.79E-06
YFR025C 1.193 1.01E-11
YPL109C 1.189 9.73E-11
YPR004C 1.188 5.55E-15
YMR284W 1.186 6.68E-08
YCR032W 1.186 1.39E-12
YJR087W 1.182 2.16E-02
YIL165C 1.173 3.20E-06
YDR307W 1.172 2.74E-11
YLR285C-A 1.167 4.35E-13
YMR233W 1.165 2.13E-04
YMR253C 1.160 4.88E-08
YLL028W 1.156 1.33E-09
YOR390W 1.156 5.83E-04
YBR120C 1.155 4.40E-08
YGR232W 1.152 2.35E-12
ICR1 1.150 1.30E-08
YKL098W 1.149 2.86E-07
Gene log2FC p-adj YHR180W 1.148 7.49E-03
YOL153C 1.148 3.82E-08
YIL097W 1.145 2.68E-06
YLL024C 1.144 1.29E-09
YDL237W 1.142 3.32E-15
YIL083C 1.142 5.27E-11
YKL209C 1.141 1.22E-05
YER076C 1.141 1.51E-04
YLR359W 1.139 2.18E-06
YLR454W 1.138 1.63E-11
YPR017C 1.137 1.00E-04
YMR009W 1.134 3.79E-07
TLC1 1.134 4.89E-13
YJR024C 1.132 1.13E-10
YPR191W 1.130 1.04E-12
YIL098C 1.129 6.25E-07
YPR157W 1.129 5.71E-04
YAR028W 1.128 2.09E-05
YPL006W 1.127 5.60E-17
YIL029C 1.127 4.87E-03
YBR115C 1.125 1.46E-08
YLR201C 1.125 2.80E-10
YGL257C 1.124 8.88E-14
YDR032C 1.123 2.00E-14
YIL114C 1.121 6.49E-07
YER186C 1.119 1.05E-09
YLL009C 1.119 3.21E-05
YKR101W 1.118 1.57E-04
YPL001W 1.116 5.95E-06
YJL139C 1.115 1.34E-08
YNR067C 1.113 7.10E-12
YJR127C 1.113 1.53E-09
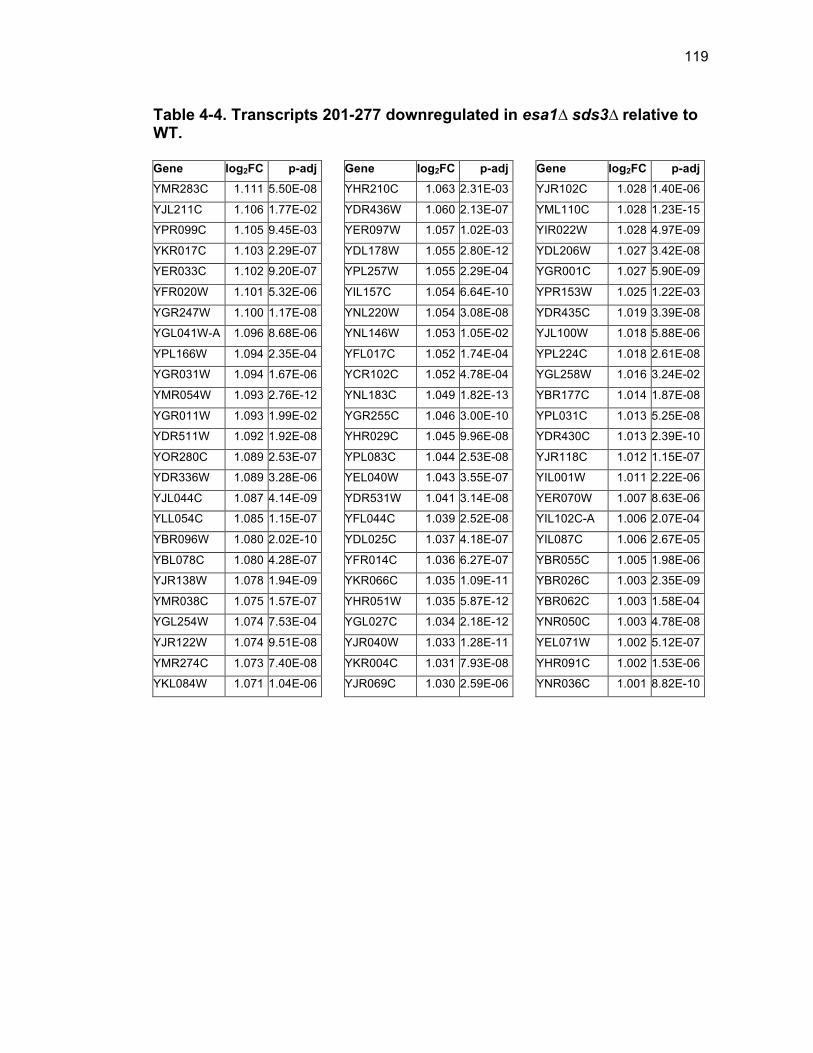
119
Table 4-4. Transcripts 201-277 downregulated in esa1∆ sds3∆ relative to WT. Gene log2FC p-adj YMR283C 1.111 5.50E-08
YJL211C 1.106 1.77E-02
YPR099C 1.105 9.45E-03
YKR017C 1.103 2.29E-07
YER033C 1.102 9.20E-07
YFR020W 1.101 5.32E-06
YGR247W 1.100 1.17E-08
YGL041W-A 1.096 8.68E-06
YPL166W 1.094 2.35E-04
YGR031W 1.094 1.67E-06
YMR054W 1.093 2.76E-12
YGR011W 1.093 1.99E-02
YDR511W 1.092 1.92E-08
YOR280C 1.089 2.53E-07
YDR336W 1.089 3.28E-06
YJL044C 1.087 4.14E-09
YLL054C 1.085 1.15E-07
YBR096W 1.080 2.02E-10
YBL078C 1.080 4.28E-07
YJR138W 1.078 1.94E-09
YMR038C 1.075 1.57E-07
YGL254W 1.074 7.53E-04
YJR122W 1.074 9.51E-08
YMR274C 1.073 7.40E-08
YKL084W 1.071 1.04E-06
Gene log2FC p-adj YHR210C 1.063 2.31E-03
YDR436W 1.060 2.13E-07
YER097W 1.057 1.02E-03
YDL178W 1.055 2.80E-12
YPL257W 1.055 2.29E-04
YIL157C 1.054 6.64E-10
YNL220W 1.054 3.08E-08
YNL146W 1.053 1.05E-02
YFL017C 1.052 1.74E-04
YCR102C 1.052 4.78E-04
YNL183C 1.049 1.82E-13
YGR255C 1.046 3.00E-10
YHR029C 1.045 9.96E-08
YPL083C 1.044 2.53E-08
YEL040W 1.043 3.55E-07
YDR531W 1.041 3.14E-08
YFL044C 1.039 2.52E-08
YDL025C 1.037 4.18E-07
YFR014C 1.036 6.27E-07
YKR066C 1.035 1.09E-11
YHR051W 1.035 5.87E-12
YGL027C 1.034 2.18E-12
YJR040W 1.033 1.28E-11
YKR004C 1.031 7.93E-08
YJR069C 1.030 2.59E-06
Gene log2FC p-adj YJR102C 1.028 1.40E-06
YML110C 1.028 1.23E-15
YIR022W 1.028 4.97E-09
YDL206W 1.027 3.42E-08
YGR001C 1.027 5.90E-09
YPR153W 1.025 1.22E-03
YDR435C 1.019 3.39E-08
YJL100W 1.018 5.88E-06
YPL224C 1.018 2.61E-08
YGL258W 1.016 3.24E-02
YBR177C 1.014 1.87E-08
YPL031C 1.013 5.25E-08
YDR430C 1.013 2.39E-10
YJR118C 1.012 1.15E-07
YIL001W 1.011 2.22E-06
YER070W 1.007 8.63E-06
YIL102C-A 1.006 2.07E-04
YIL087C 1.006 2.67E-05
YBR055C 1.005 1.98E-06
YBR026C 1.003 2.35E-09
YBR062C 1.003 1.58E-04
YNR050C 1.003 4.78E-08
YEL071W 1.002 5.12E-07
YHR091C 1.002 1.53E-06
YNR036C 1.001 8.82E-10
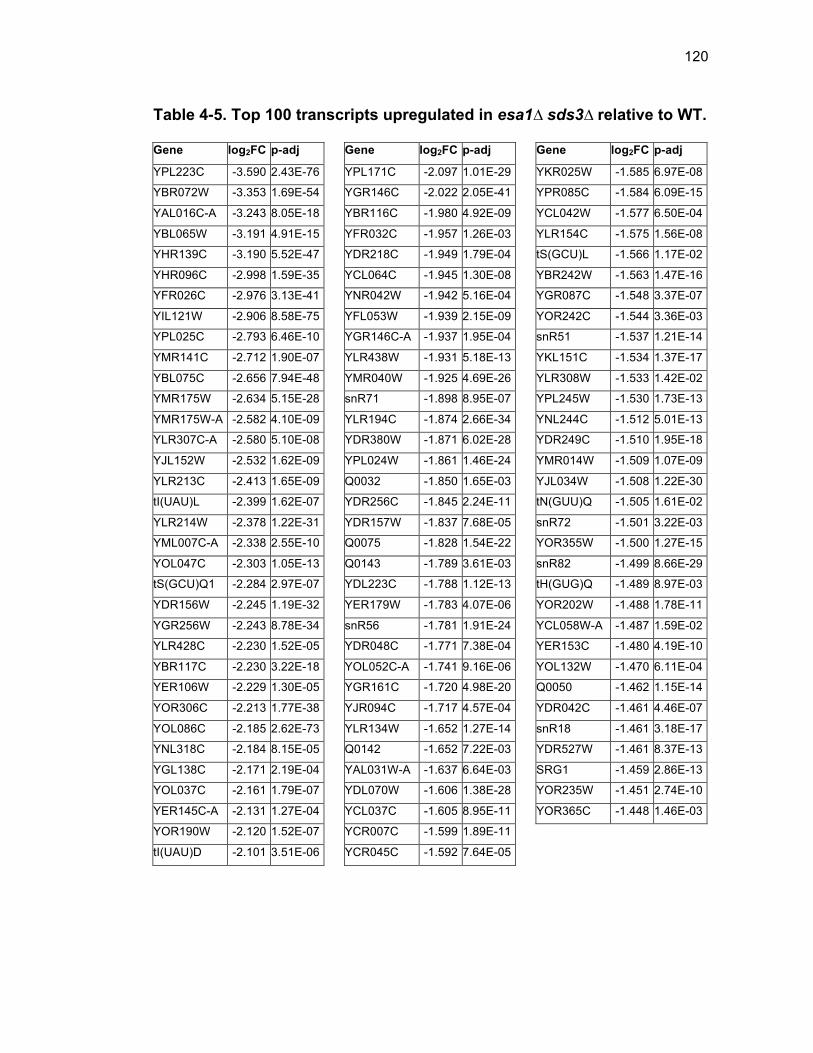
120
Table 4-5. Top 100 transcripts upregulated in esa1∆ sds3∆ relative to WT. Gene log2FC p-adj
YPL223C -3.590 2.43E-76
YBR072W -3.353 1.69E-54
YAL016C-A -3.243 8.05E-18
YBL065W -3.191 4.91E-15
YHR139C -3.190 5.52E-47
YHR096C -2.998 1.59E-35
YFR026C -2.976 3.13E-41
YIL121W -2.906 8.58E-75
YPL025C -2.793 6.46E-10
YMR141C -2.712 1.90E-07
YBL075C -2.656 7.94E-48
YMR175W -2.634 5.15E-28
YMR175W-A -2.582 4.10E-09
YLR307C-A -2.580 5.10E-08
YJL152W -2.532 1.62E-09
YLR213C -2.413 1.65E-09
tI(UAU)L -2.399 1.62E-07
YLR214W -2.378 1.22E-31
YML007C-A -2.338 2.55E-10
YOL047C -2.303 1.05E-13
tS(GCU)Q1 -2.284 2.97E-07
YDR156W -2.245 1.19E-32
YGR256W -2.243 8.78E-34
YLR428C -2.230 1.52E-05
YBR117C -2.230 3.22E-18
YER106W -2.229 1.30E-05
YOR306C -2.213 1.77E-38
YOL086C -2.185 2.62E-73
YNL318C -2.184 8.15E-05
YGL138C -2.171 2.19E-04
YOL037C -2.161 1.79E-07
YER145C-A -2.131 1.27E-04
YOR190W -2.120 1.52E-07
tI(UAU)D -2.101 3.51E-06
Gene log2FC p-adj
YPL171C -2.097 1.01E-29
YGR146C -2.022 2.05E-41
YBR116C -1.980 4.92E-09
YFR032C -1.957 1.26E-03
YDR218C -1.949 1.79E-04
YCL064C -1.945 1.30E-08
YNR042W -1.942 5.16E-04
YFL053W -1.939 2.15E-09
YGR146C-A -1.937 1.95E-04
YLR438W -1.931 5.18E-13
YMR040W -1.925 4.69E-26
snR71 -1.898 8.95E-07
YLR194C -1.874 2.66E-34
YDR380W -1.871 6.02E-28
YPL024W -1.861 1.46E-24
Q0032 -1.850 1.65E-03
YDR256C -1.845 2.24E-11
YDR157W -1.837 7.68E-05
Q0075 -1.828 1.54E-22
Q0143 -1.789 3.61E-03
YDL223C -1.788 1.12E-13
YER179W -1.783 4.07E-06
snR56 -1.781 1.91E-24
YDR048C -1.771 7.38E-04
YOL052C-A -1.741 9.16E-06
YGR161C -1.720 4.98E-20
YJR094C -1.717 4.57E-04
YLR134W -1.652 1.27E-14
Q0142 -1.652 7.22E-03
YAL031W-A -1.637 6.64E-03
YDL070W -1.606 1.38E-28
YCL037C -1.605 8.95E-11
YCR007C -1.599 1.89E-11
YCR045C -1.592 7.64E-05
Gene log2FC p-adj
YKR025W -1.585 6.97E-08
YPR085C -1.584 6.09E-15
YCL042W -1.577 6.50E-04
YLR154C -1.575 1.56E-08
tS(GCU)L -1.566 1.17E-02
YBR242W -1.563 1.47E-16
YGR087C -1.548 3.37E-07
YOR242C -1.544 3.36E-03
snR51 -1.537 1.21E-14
YKL151C -1.534 1.37E-17
YLR308W -1.533 1.42E-02
YPL245W -1.530 1.73E-13
YNL244C -1.512 5.01E-13
YDR249C -1.510 1.95E-18
YMR014W -1.509 1.07E-09
YJL034W -1.508 1.22E-30
tN(GUU)Q -1.505 1.61E-02
snR72 -1.501 3.22E-03
YOR355W -1.500 1.27E-15
snR82 -1.499 8.66E-29
tH(GUG)Q -1.489 8.97E-03
YOR202W -1.488 1.78E-11
YCL058W-A -1.487 1.59E-02
YER153C -1.480 4.19E-10
YOL132W -1.470 6.11E-04
Q0050 -1.462 1.15E-14
YDR042C -1.461 4.46E-07
snR18 -1.461 3.18E-17
YDR527W -1.461 8.37E-13
SRG1 -1.459 2.86E-13
YOR235W -1.451 2.74E-10
YOR365C -1.448 1.46E-03
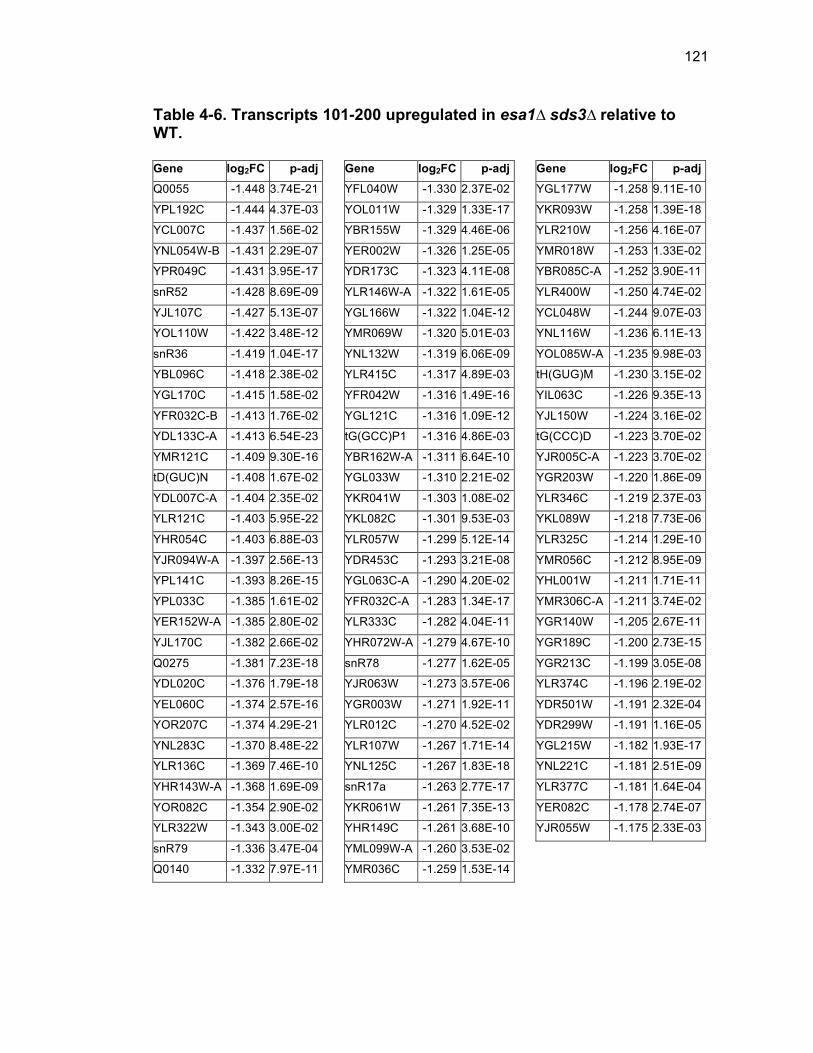
121
Table 4-6. Transcripts 101-200 upregulated in esa1∆ sds3∆ relative to WT. Gene log2FC p-adj Q0055 -1.448 3.74E-21
YPL192C -1.444 4.37E-03
YCL007C -1.437 1.56E-02
YNL054W-B -1.431 2.29E-07
YPR049C -1.431 3.95E-17
snR52 -1.428 8.69E-09
YJL107C -1.427 5.13E-07
YOL110W -1.422 3.48E-12
snR36 -1.419 1.04E-17
YBL096C -1.418 2.38E-02
YGL170C -1.415 1.58E-02
YFR032C-B -1.413 1.76E-02
YDL133C-A -1.413 6.54E-23
YMR121C -1.409 9.30E-16
tD(GUC)N -1.408 1.67E-02
YDL007C-A -1.404 2.35E-02
YLR121C -1.403 5.95E-22
YHR054C -1.403 6.88E-03
YJR094W-A -1.397 2.56E-13
YPL141C -1.393 8.26E-15
YPL033C -1.385 1.61E-02
YER152W-A -1.385 2.80E-02
YJL170C -1.382 2.66E-02
Q0275 -1.381 7.23E-18
YDL020C -1.376 1.79E-18
YEL060C -1.374 2.57E-16
YOR207C -1.374 4.29E-21
YNL283C -1.370 8.48E-22
YLR136C -1.369 7.46E-10
YHR143W-A -1.368 1.69E-09
YOR082C -1.354 2.90E-02
YLR322W -1.343 3.00E-02
snR79 -1.336 3.47E-04
Q0140 -1.332 7.97E-11
Gene log2FC p-adj YFL040W -1.330 2.37E-02
YOL011W -1.329 1.33E-17
YBR155W -1.329 4.46E-06
YER002W -1.326 1.25E-05
YDR173C -1.323 4.11E-08
YLR146W-A -1.322 1.61E-05
YGL166W -1.322 1.04E-12
YMR069W -1.320 5.01E-03
YNL132W -1.319 6.06E-09
YLR415C -1.317 4.89E-03
YFR042W -1.316 1.49E-16
YGL121C -1.316 1.09E-12
tG(GCC)P1 -1.316 4.86E-03
YBR162W-A -1.311 6.64E-10
YGL033W -1.310 2.21E-02
YKR041W -1.303 1.08E-02
YKL082C -1.301 9.53E-03
YLR057W -1.299 5.12E-14
YDR453C -1.293 3.21E-08
YGL063C-A -1.290 4.20E-02
YFR032C-A -1.283 1.34E-17
YLR333C -1.282 4.04E-11
YHR072W-A -1.279 4.67E-10
snR78 -1.277 1.62E-05
YJR063W -1.273 3.57E-06
YGR003W -1.271 1.92E-11
YLR012C -1.270 4.52E-02
YLR107W -1.267 1.71E-14
YNL125C -1.267 1.83E-18
snR17a -1.263 2.77E-17
YKR061W -1.261 7.35E-13
YHR149C -1.261 3.68E-10
YML099W-A -1.260 3.53E-02
YMR036C -1.259 1.53E-14
Gene log2FC p-adj YGL177W -1.258 9.11E-10
YKR093W -1.258 1.39E-18
YLR210W -1.256 4.16E-07
YMR018W -1.253 1.33E-02
YBR085C-A -1.252 3.90E-11
YLR400W -1.250 4.74E-02
YCL048W -1.244 9.07E-03
YNL116W -1.236 6.11E-13
YOL085W-A -1.235 9.98E-03
tH(GUG)M -1.230 3.15E-02
YIL063C -1.226 9.35E-13
YJL150W -1.224 3.16E-02
tG(CCC)D -1.223 3.70E-02
YJR005C-A -1.223 3.70E-02
YGR203W -1.220 1.86E-09
YLR346C -1.219 2.37E-03
YKL089W -1.218 7.73E-06
YLR325C -1.214 1.29E-10
YMR056C -1.212 8.95E-09
YHL001W -1.211 1.71E-11
YMR306C-A -1.211 3.74E-02
YGR140W -1.205 2.67E-11
YGR189C -1.200 2.73E-15
YGR213C -1.199 3.05E-08
YLR374C -1.196 2.19E-02
YDR501W -1.191 2.32E-04
YDR299W -1.191 1.16E-05
YGL215W -1.182 1.93E-17
YNL221C -1.181 2.51E-09
YLR377C -1.181 1.64E-04
YER082C -1.178 2.74E-07
YJR055W -1.175 2.33E-03
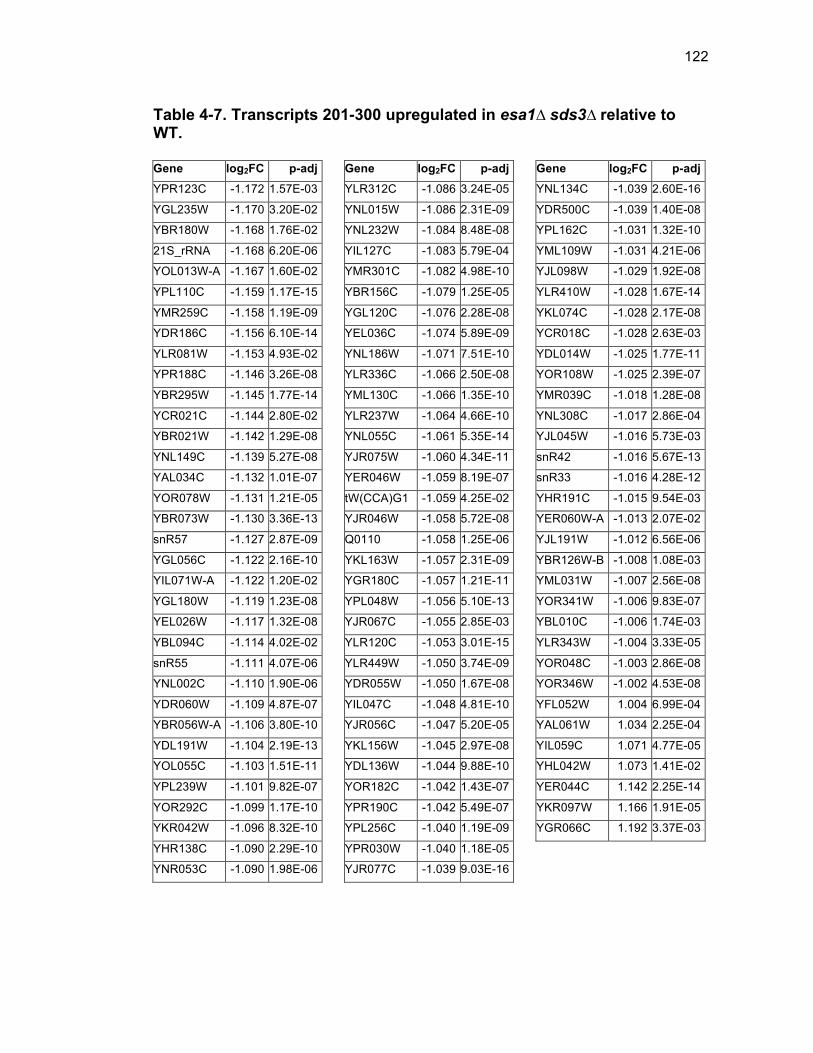
122
Table 4-7. Transcripts 201-300 upregulated in esa1∆ sds3∆ relative to WT. Gene log2FC p-adj YPR123C -1.172 1.57E-03
YGL235W -1.170 3.20E-02
YBR180W -1.168 1.76E-02
21S_rRNA -1.168 6.20E-06
YOL013W-A -1.167 1.60E-02
YPL110C -1.159 1.17E-15
YMR259C -1.158 1.19E-09
YDR186C -1.156 6.10E-14
YLR081W -1.153 4.93E-02
YPR188C -1.146 3.26E-08
YBR295W -1.145 1.77E-14
YCR021C -1.144 2.80E-02
YBR021W -1.142 1.29E-08
YNL149C -1.139 5.27E-08
YAL034C -1.132 1.01E-07
YOR078W -1.131 1.21E-05
YBR073W -1.130 3.36E-13
snR57 -1.127 2.87E-09
YGL056C -1.122 2.16E-10
YIL071W-A -1.122 1.20E-02
YGL180W -1.119 1.23E-08
YEL026W -1.117 1.32E-08
YBL094C -1.114 4.02E-02
snR55 -1.111 4.07E-06
YNL002C -1.110 1.90E-06
YDR060W -1.109 4.87E-07
YBR056W-A -1.106 3.80E-10
YDL191W -1.104 2.19E-13
YOL055C -1.103 1.51E-11
YPL239W -1.101 9.82E-07
YOR292C -1.099 1.17E-10
YKR042W -1.096 8.32E-10
YHR138C -1.090 2.29E-10
YNR053C -1.090 1.98E-06
Gene log2FC p-adj YLR312C -1.086 3.24E-05
YNL015W -1.086 2.31E-09
YNL232W -1.084 8.48E-08
YIL127C -1.083 5.79E-04
YMR301C -1.082 4.98E-10
YBR156C -1.079 1.25E-05
YGL120C -1.076 2.28E-08
YEL036C -1.074 5.89E-09
YNL186W -1.071 7.51E-10
YLR336C -1.066 2.50E-08
YML130C -1.066 1.35E-10
YLR237W -1.064 4.66E-10
YNL055C -1.061 5.35E-14
YJR075W -1.060 4.34E-11
YER046W -1.059 8.19E-07
tW(CCA)G1 -1.059 4.25E-02
YJR046W -1.058 5.72E-08
Q0110 -1.058 1.25E-06
YKL163W -1.057 2.31E-09
YGR180C -1.057 1.21E-11
YPL048W -1.056 5.10E-13
YJR067C -1.055 2.85E-03
YLR120C -1.053 3.01E-15
YLR449W -1.050 3.74E-09
YDR055W -1.050 1.67E-08
YIL047C -1.048 4.81E-10
YJR056C -1.047 5.20E-05
YKL156W -1.045 2.97E-08
YDL136W -1.044 9.88E-10
YOR182C -1.042 1.43E-07
YPR190C -1.042 5.49E-07
YPL256C -1.040 1.19E-09
YPR030W -1.040 1.18E-05
YJR077C -1.039 9.03E-16
Gene log2FC p-adj YNL134C -1.039 2.60E-16
YDR500C -1.039 1.40E-08
YPL162C -1.031 1.32E-10
YML109W -1.031 4.21E-06
YJL098W -1.029 1.92E-08
YLR410W -1.028 1.67E-14
YKL074C -1.028 2.17E-08
YCR018C -1.028 2.63E-03
YDL014W -1.025 1.77E-11
YOR108W -1.025 2.39E-07
YMR039C -1.018 1.28E-08
YNL308C -1.017 2.86E-04
YJL045W -1.016 5.73E-03
snR42 -1.016 5.67E-13
snR33 -1.016 4.28E-12
YHR191C -1.015 9.54E-03
YER060W-A -1.013 2.07E-02
YJL191W -1.012 6.56E-06
YBR126W-B -1.008 1.08E-03
YML031W -1.007 2.56E-08
YOR341W -1.006 9.83E-07
YBL010C -1.006 1.74E-03
YLR343W -1.004 3.33E-05
YOR048C -1.003 2.86E-08
YOR346W -1.002 4.53E-08
YFL052W 1.004 6.99E-04
YAL061W 1.034 2.25E-04
YIL059C 1.071 4.77E-05
YHL042W 1.073 1.41E-02
YER044C 1.142 2.25E-14
YKR097W 1.166 1.91E-05
YGR066C 1.192 3.37E-03
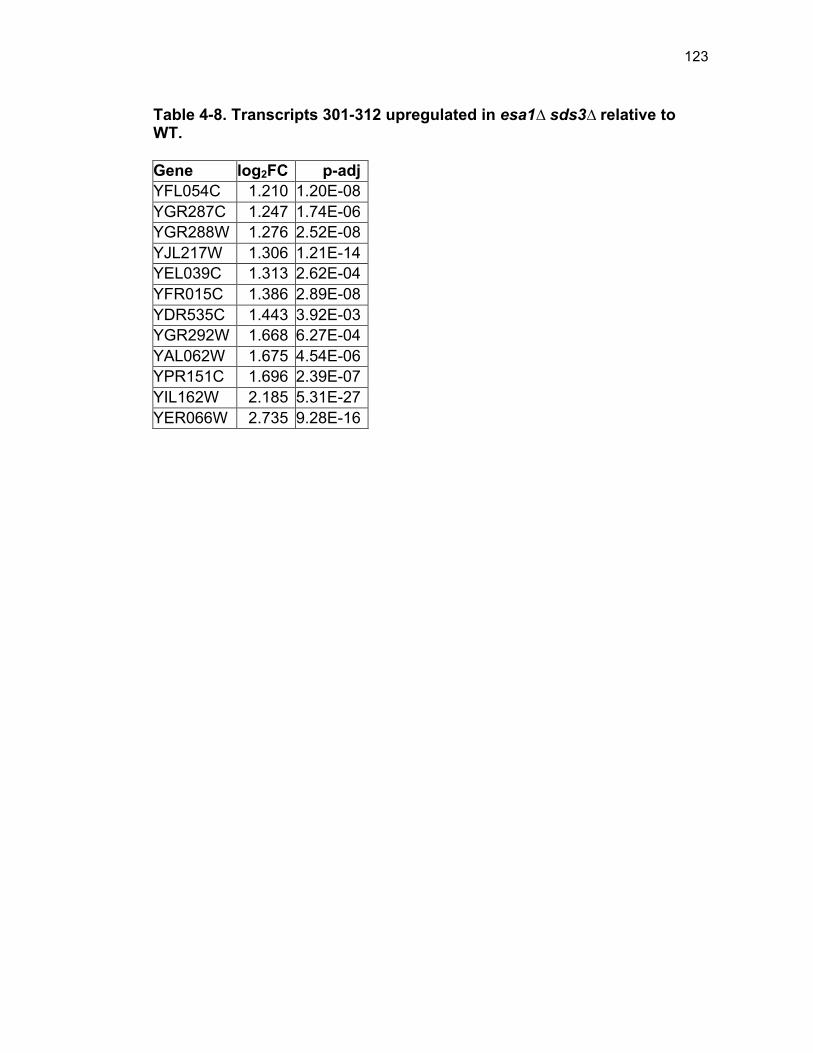
123
Table 4-8. Transcripts 301-312 upregulated in esa1∆ sds3∆ relative to WT. Gene log2FC p-adj YFL054C 1.210 1.20E-08 YGR287C 1.247 1.74E-06 YGR288W 1.276 2.52E-08 YJL217W 1.306 1.21E-14 YEL039C 1.313 2.62E-04 YFR015C 1.386 2.89E-08 YDR535C 1.443 3.92E-03 YGR292W 1.668 6.27E-04 YAL062W 1.675 4.54E-06 YPR151C 1.696 2.39E-07 YIL162W 2.185 5.31E-27 YER066W 2.735 9.28E-16

124
References Andrews SJ, Rothnagel JA. 2014. Emerging evidence for functional peptides
encoded by short open reading frames. Nat Rev Genet 15: 193-204.
Arava Y, Wang Y, Storey JD, Liu CL, Brown PO, Herschlag D. 2003. Genome-wide analysis of mRNA translation profiles in Saccharomyces cerevisiae. Proc Natl Acad Sci U S A 100: 3889-3894.
Ashe MP, De Long SK, Sachs AB. 2000. Glucose depletion rapidly inhibits translation initiation in yeast. Mol Biol Cell 11: 833-848.
Basrai MA, Hieter P, Boeke JD. 1997. Small open reading frames: beautiful needles in the haystack. Genome Res 7: 768-771.
Boudreault AA, Cronier D, Selleck W, Lacoste N, Utley RT, Allard S, Savard J, Lane WS, Tan S, Côté J. 2003. Yeast enhancer of polycomb defines global Esa1-dependent acetylation of chromatin. Genes Dev 17: 1415-1428.
Clarke AS, Lowell JE, Jacobson SJ, Pillus L. 1999. Esa1p is an essential histone acetyltransferase required for cell cycle progression. Molecular and Cellular Biology 19: 2515-2526.
Clarke AS, Samal E, Pillus L. 2006. Distinct roles for the essential MYST family HAT Esa1p in transcriptional silencing. Mol Biol Cell 17: 1744-1757.
Downey M, Johnson JR, Davey NE, Newton BW, Johnson TL, Galaang S, Seller CA, Krogan N, Toczyski DP. 2015. Acetylome profiling reveals overlap in the regulation of diverse processes by sirtuins, gcn5, and esa1. Mol Cell Proteomics 14: 162-176.
Gasch AP, Spellman PT, Kao CM, Carmel-Harel O, Eisen MB, Storz G, Botstein D, Brown PO. 2000. Genomic expression programs in the response of yeast cells to environmental changes. Mol Biol Cell 11: 4241-4257.
Ginsburg DS, Govind CK, Hinnebusch AG. 2009. NuA4 lysine acetyltransferase Esa1 is targeted to coding regions and stimulates transcription elongation with Gcn5. Mol Cell Biol 29: 6473-6487.
Heyer EE, Moore MJ. 2016. Redefining the Translational Status of 80S Monosomes. Cell 164: 757-769.
Iizuka M, Smith MM. 2003. Functional consequences of histone modifications. Curr Opin Genet Dev 13: 154-160.

125
Jensen LJ, Kuhn M, Stark M, Chaffron S, Creevey C, Muller J, Doerks T, Julien P, Roth A, Simonovic M, Bork P, von Mering C. 2009. STRING 8--a global view on proteins and their functional interactions in 630 organisms. Nucleic Acids Res 37: D412-416.
Jorgensen P, Nishikawa JL, Breitkreutz BJ, Tyers M. 2002. Systematic identification of pathways that couple cell growth and division in yeast. Science 297: 395-400.
Jorgensen P, Rupes I, Sharom JR, Schneper L, Broach JR, Tyers M. 2004. A dynamic transcriptional network communicates growth potential to ribosome synthesis and critical cell size. Genes Dev 18: 2491-2505.
Kurdistani SK, Grunstein M. 2003. Histone acetylation and deacetylation in yeast. Nat Rev Mol Cell Biol 4: 276-284.
Lempiainen H, Shore D. 2009. Growth control and ribosome biogenesis. Curr Opin Cell Biol 21: 855-863.
Lin YY, Lu JY, Zhang J, Walter W, Dang W, Wan J, Tao SC, Qian J, Zhao Y, Boeke JD, Berger SL, Zhu H. 2009. Protein acetylation microarray reveals that NuA4 controls key metabolic target regulating gluconeogenesis. Cell 136: 1073-1084.
Loewith R, Hall MN. 2011. Target of rapamycin (TOR) in nutrient signaling and growth control. Genetics 189: 1177-1201.
Martin TE, Hartwell LH. 1970. Resistance of active yeast ribosomes to dissociation by KCl. J Biol Chem 245: 1504-1506.
Mitchell L, Huard S, Cotrut M, Pourhanifeh-Lemeri R, Steunou AL, Hamza A, Lambert JP, Zhou H, Ning Z, Basu A, Cote J, Figeys DA, Baetz K. 2013. mChIP-KAT-MS, a method to map protein interactions and acetylation sites for lysine acetyltransferases. Proc Natl Acad Sci U S A 110: E1641-1650.
Rando OJ, Winston F. 2012. Chromatin and transcription in yeast. Genetics 190: 351-387.
Reid JL, Iyer VR, Brown PO, Struhl K. 2000. Coordinate regulation of yeast ribosomal protein genes is associated with targeted recruitment of Esa1 histone acetylase. Mol Cell 6: 1297-1307.
Robert F, Pokholok DK, Hannett NM, Rinaldi NJ, Chandy M, Rolfe A, Workman JL, Gifford DK, Young RA. 2004. Global position and

126
recruitment of HATs and HDACs in the yeast genome. Mol Cell 16: 199-209.
Ruggero D. 2012. Revisiting the nucleolus: from marker to dynamic integrator of cancer signaling. Sci Signal 5: pe38.
Supek F, Bosnjak M, Skunca N, Smuc T. 2011. REVIGO summarizes and visualizes long lists of gene ontology terms. PLoS One 6: e21800.
Thomas BJ, Rothstein R. 1989. Elevated recombination rates in transcriptionally active DNA. Cell 56: 619-630.
Torres-Machorro AL, Pillus L. 2014. Bypassing the requirement for an essential MYST acetyltransferase. Genetics 197: 851-863.
Uprety B, Sen R, Bhaumik SR. 2015. Eaf1p Is Required for Recruitment of NuA4 in Targeting TFIID to the Promoters of the Ribosomal Protein Genes for Transcriptional Initiation In Vivo. Mol Cell Biol 35: 2947-2964.
van Bakel H, Tsui K, Gebbia M, Mnaimneh S, Hughes TR, Nislow C. 2013. A compendium of nucleosome and transcript profiles reveals determinants of chromatin architecture and transcription. PLoS Genet 9: e1003479.
van Sluis M, McStay B. 2014. Ribosome biogenesis: Achilles heel of cancer? Genes Cancer 5: 152-153.
Wade C, Shea KA, Jensen RV, McAlear MA. 2001. EBP2 is a member of the yeast RRB regulon, a transcriptionally coregulated set of genes that are required for ribosome and rRNA biosynthesis. Mol Cell Biol 21: 8638-8650.
Wade CH, Umbarger MA, McAlear MA. 2006. The budding yeast rRNA and ribosome biosynthesis (RRB) regulon contains over 200 genes. Yeast 23: 293-306.
Warner JR, Vilardell J, Sohn JH. 2001. Economics of ribosome biosynthesis. Cold Spring Harb Symp Quant Biol 66: 567-574.
Woolford JL, Jr., Baserga SJ. 2013. Ribosome biogenesis in the yeast Saccharomyces cerevisiae. Genetics 195: 643-681.
Zhou J, Zhou BO, Lenzmeier BA, Zhou JQ. 2009. Histone deacetylase Rpd3 antagonizes Sir2-dependent silent chromatin propagation. Nucleic Acids Res 37: 3699-3713.

127
Chapter 5 Conclusions and Future Directions
Chapter 5.
Conclusions and Future Directions

128
This results presented in this thesis rely on the powerful tools of bypass
suppression and yeast genetics to study critical chromatin modifying
components. Through a combination of genetic, genomic, and biochemical
analyses, key features of essential chromatin-related protein subunits were
elucidated, and important interactions between different chromatin regulatory
subunits were highlighted. Whereas several of the major proteins studied in
this thesis have been central to other seminal studies, bypass suppression
facilitated a new type of analysis and promoted the discovery of new findings,
whereby functions of the essential components could be inferred when studied
via their complete cellular depletion.
Epl1 is a critical Esa1-cofactor. The initial results of this thesis, as
presented in Chapter 2 built upon earlier studies identifying the bypass
suppression of ESA1 by loss of the Rpd3L deacetylase complex (Torres-
Machorro and Pillus 2014). One additional bypass suppressor was identified
within NuA4, and fostered a comprehensive mutagenesis-based structure-
function study of this essential subunit, Epl1. Deletion of domains within Epl1
coupled with biochemical analysis led to the development of a model in which
Epl1 is critical for the catalytic activity of Esa1.
Similar studies for EPC in other organisms might prove fruitful as well.
For example, although the EPcA domain is highly conserved, and thought to
generally encompass a protein-binding domain in a selection of chromatin
proteins (Perry 2006), the C-terminus is much more variable. Some

129
metazoans, for example, have additional domains termed EPc-B and EPc-C
within the C-terminus of their Epl1-orthologs, however these are not as
ubiquitous as EPcA (Figure 1-3). Although some studies have assigned
domains required for interactions reported here (Table 1-1), a CRISPR-Cas9-
based approach would facilitate a more comprehensive functional assignment
for specific residues, and may correspondingly be used to test the significance
of cancer-associated mutations. Along these lines, a recent study reported
generation of a viable Epc1-/- mouse (Dong et al. 2017), whereas previous
studies demonstrated that homozygous deletion of Epc1 resulted in embryonic
lethality (Kim et al. 2009). The specific Epc1 residues involved in the disruption
might explain this apparent discrepancy in reports for the necessity for EPC1
in mice. The viable Epc1-/- mice were generated by disrupting exons 3-5 (Dong
et al. 2017) thereby leaving the EPcA domain largely intact, whereas the non-
viable homozygous null Epc1 knockout mice involved disruption within the first
exon (Kim et al. 2009). These differing results underscore the importance of
considering structure-function when assessing EPC.
Another important issue not yet thoroughly addressed is the
significance of the duplication of EPC1. Throughout the course of evolution, a
paralog of Enhancer of Polycomb, EPC2, arose by duplication in zebrafish,
rodents, and humans. Multiple studies have highlighted either EPC1 or EPC2
independently, yet it generally remains unclear under what conditions one
paralog may be preferentially critical. Neither EPC1 nor EPC2 can functionally

130
replace yeast Epl1 (Hamza et al. 2015). However, it is possible that
coexpression of EPC1 and EPC2 might promote viability in epl1∆, and shed
light on the importance of both paralogs.
How many distinct moonlighting roles might EPC have beyond
NuA4/Tip60 complexes? As noted, studies have described functions of EPC
appearing independent of NuA4/piccolo-NuA4, either as a part of a distinct
complex (Attwooll et al. 2005), or as presumed due to expression patterns
distinct from NuA4 (Kim et al. 2009). Further, several large-scale proteomic
and genomic screens returned EPC as a hit without identifying other NuA4
subunits, such as TIP60 (Kim and Sun 2007; Huang et al. 2013; Zeng et al.
2015). It is possible that EPC exists in additional multimeric complexes, yet to
be identified, which for example, may assemble upon specific stimuli or during
particular developmental states. EPC function may also be fine-tuned by splice
variants, many of which have been identified in metazoans (Table 1-2).
Finally, although several studies have pointed to EPC1/2 alterations in
cancer, few have yet to shed light on the functional significance of EPC
alterations in cancer biology. For example, in the case where EPC1 is
downregulated in leukemia (Prasad et al. 2014), is this downregulation EPC-
specific, or does it have more broad effects on NuA4 complex dynamics and
stoichiometry? One could speculate that in the latter case, if NuA4 activity
were altered, acetylation of histone-substrates would be disrupted. Non-
histone NuA4 substrates have also been identified in humans [reviewed in

131
(Squatrito et al. 2006; Avvakumov and Côté 2007; Lee and Workman 2007)]
and in yeast (Lin et al. 2009; Yi et al. 2012; Mitchell et al. 2013b; Downey et al.
2015). Pursuing the corresponding effect of EPC alteration on these
substrates will be an important direction for future research.
Loss of Hda1 suppresses epl1∆ sds3∆ phenotypes, and
underscores a NuA4-Rpd3L-Hda1 regulatory axis. In Chapter 3, genetic
interactions between NuA4 and HDACs were probed, using epl1∆ sds3∆ as a
proxy for the loss of NuA4. Whereas each deacetylase mutant examined
demonstrated distinct interactions with NuA4 and Epl1 specifically, the most
noteworthy was Hda1. Each phenotype assessed, such as temperature, DNA
damage, and oxidative stress sensitivities, as well as progression through the
cell cycle, histone H4 acetylation, and rDNA silencing, was suppressed by
hda1∆ in epl1∆ sds3∆.
The robust rescue of epl1∆ sds3∆ by hda1∆ brings the apparent
question of what is driving such a widespread, robust rescue? One
mechanism that may have been responsible for the rescue involved gene
expression. It was possible that bypass of NuA4 resulted in dysregulation of
widespread transcription, rescued upon loss of HDA1. This hypothesis was
addressed and tested in Chapter 4, but was found to be unsupported.
Whereas bypass of NuA4, using esa1∆ sds3∆ as a proxy, did cause
widespread transcription dysregulation, primarily focused on ribosome
biogenesis, esa1∆ sds3∆ hda1∆ did not promote a pervasive restoration of

132
gene expression. Additional analysis of the genes that were presumably
involved in the rescue of gene expression might provide further insights.
Whereas these differentially expressed genes were neither enriched for H4
acetylation, nor for Esa1 binding, it remains to be determined if they are in
fact, enriched for Hda1 binding, or contained within any kind of genomic
structural element, such as the Hda1-regulated HAST-domains. Such
additional analyses may inform on a new significance within the NuA4-Hda1-
regulated transcriptome.
Upon ruling out generalized widespread transcription under normal
growth conditions as the mode driving the Hda1-mediated rescue, it is perhaps
useful to consider that there may be a different broad-acting mechanism
behind the HDA1-driven rescue. A possible candidate behind the rescue is
H2A histone variant, H2A.Z, encoded in yeast by HTZ1. Whereas H2A.Z is not
essential in yeast, it has been implicated in many important functions including
formation of boundaries in silent chromatin (Meneghini et al. 2003), and is
widespread in its genomic localization, though primarily focused at promoters
of inactive genes (Guillemette et al. 2005; Millar et al. 2006). Additionally,
htz1∆ results in dysregulation of about 5% of yeast genes (Meneghini et al.
2003), and impairs splicing of intron-containing genes (Neves et al. 2017).
Most pertinent, however, is that H2A.Z is acetylated by Esa1 (Keogh et al.
2006) and deacetylated by Hda1 (Lin et al. 2008), placing it centrally in the

133
Esa1-Hda1 relationship (Figure 5-1) as a candidate for driving the hda1∆-
mediated rescue of epl1∆ sds3∆.
Directly testing the importance of HTZ1 in the Hda1-rescue mechanism
is not trivial. Rpd3L, the deletion of which allows survival of epl1∆ and esa1∆,
has a synthetic lethal genetic relationship with HTZ1 (Lin et al. 2008). To work
around this lethality and to simultaneously test the importance of acetylated
Htz1, lysine-tail point mutants of HTZ1 can begin to dissect the importance of
the NuA4-Hda1-Htz1 relationship. Previous experiments done by Ana Lilia
Torres-Machorro illustrated that mutating each of the four N-terminal lysines (K
3,8,10,14) of HTZ1 to alanine resulted in similarly strong temperature
sensitivity as esa1∆ sds3∆ (unpublished data), but when even three of the four
lysines were mutated in esa1∆ sds3∆, no phenotype was observed. This is
consistent with reported internal redundancies of N-terminal lysine acetylation
of Htz1 (Mehta et al. 2010). Additional experiments should be carried out to
test if acetylation-dead Htz1 interferes with the Hda1-mediated rescue of epl1∆
sds3∆, including evaluation of the nonacetylated K>R mutants and the
acetylation-mimetic K>Q mutants in both the epl1∆ sds3∆ and the epl1∆
sds3∆ hda1∆ backgrounds.
Initial analysis did not show significant enrichment of Htz1 binding sites
[Htz1-loci data obtained from (Guillemette et al. 2005)] with the dysregulated
loci in the Esa1-transcriptome (Chapter 4). However, the Htz1 binding data
analysis was a preliminary exploration, as it was performed in WT cells only,
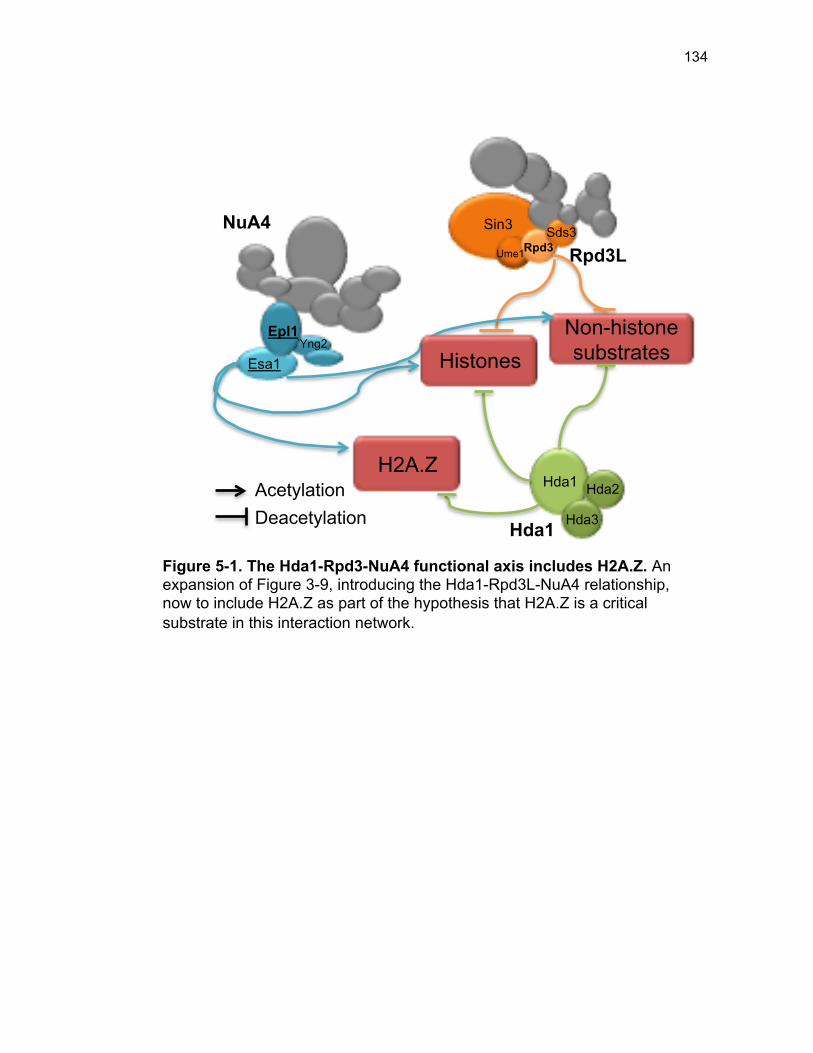
134
Figure 5-1. The Hda1-Rpd3-NuA4 functional axis includes H2A.Z. An expansion of Figure 3-9, introducing the Hda1-Rpd3L-NuA4 relationship, now to include H2A.Z as part of the hypothesis that H2A.Z is a critical substrate in this interaction network.
Epl1
Esa1
NuA4
Yng2
Sin3
Rpd3L Ume1 Rpd3 Sds3
Hda1 Hda2
Hda3 Hda1
Acetylation Deacetylation
Non-histone substrates
H2A.Z
Histones

135
and under different growth conditions than the bypass strain growth. It remains
possible that a more parallel experimental set-up would be more revealing. In
either case, the apparent lack of overlap between Htz1 binding and differential
expression does not rule out the possibility that H2A.Z is misincorporated in
epl1∆ sds3∆. To address H2A.Z misincorporation, Htz1 and acetylated-Htz1
binding could be mapped by ChIP-seq in epl1∆ sds3∆ and in epl1∆ sds3∆
hda1∆ to better understand how bypass of NuA4 and the rescue of the bypass
strains affect H2A.Z regulation.
Given that Htz1 is important for the formation of boundaries between
silent and active chromatin, the aberrant spreading of silent chromatin is
another hypothesis that may be tested in epl1∆ sds3∆ for critical rescue by
hda1∆. Deletion of HDA1 rescued the rDNA silencing defect of both esa1∆
sds3∆ (Torres-Machorro and Pillus 2014) and epl1∆ sds3∆ (Figure 3-8).
Additionally, ultrastructural studies demonstrating widespread distribution of
electron dense material in the yeast nucleus upon mutation of ESA1 (Clarke et
al. 1999) may be suggestive of widespread aberrant chromatin silencing that
may be resolved upon deletion of HDA1. There are many ways to test for
aberrant genomic silencing, but distinguishing between silencing defects at
individual loci and abnormal global silencing will be key to characterizing the
role for Hda1 in rescuing the NuA4 bypass strains defects. Additional
integrated silencing reporters, such as was used for rDNA silencing (Figure 3-
8a) may be used to assess silencing at the mating type loci and telomeres

136
[reviewed in (Grunstein and Gasser 2013)]. Mapping the regions of silent
chromatin in WT, epl1∆ sds3∆ (or esa1∆ sds3∆), and the hda1∆ triple mutant
may more broadly address the question of the involvement of silencing in the
hda1∆-mediated rescue. The spread of silent chromatin relies on the SIR-
silencing complex composed of Sir2, Sir3, and Sir4 that bind and spread
throughout the silent domain. Using ChIP-seq to map either Sir3 or Sir4 as
proxies for regions of silent chromatin (Ellahi et al. 2015) would begin to
address aberrant silencing upon NuA4 bypass and the subsequent rescue
upon deletion of HDA1.
Esa1 is critical for proper regulation of ribosome biogenesis. In
Chapter 4, high throughput RNA sequencing coupled with differential
expression analysis and polysome profiling defined Esa1 as an important
regulator of ribosome biogenesis through a mechanism involving regulating
levels of gene expression. It will be important to more fully establish the
precise details of this mechanism.
An interesting unresolved aspect of the involvement of Esa1 in
regulating ribosome biogenesis is related to the apparent amorphous
nucleolus in esa1 (Clarke et al. 1999), the dedicated site of ribosome
biogenesis. The particular significance of nucleolar morphology in normal
conditions, where it exhibits a crescent-like structure, and in the esa1 mutant,
where the nucleolus appears to be unstructured throughout the nuclear
compartment remains to be determined [reviewed in (Taddei et al. 2010)].

137
Broadly, RNA pol I is important for the crescent-like morphology of the
nucleolus. When rDNA transcription was driven either by RNA pol II or by a
mutated RNA pol I with impaired function, it exhibited a more circular
nucleolus that was not tightly associated with the nuclear envelope,
underscoring the importance of RNA pol I-driven rDNA transcription for
nucleolar morphology (Oakes et al. 1993; Oakes et al. 1998). In contrast, in
cells treated with rapamycin, resulting in the downregulation of ribosome
biogenesis, the nucleolus remained tethered to the nuclear envelope, but the
size was minimized, and the nucleolus occupied a much smaller circular area
at the periphery of the nucleus. This shift in nucleolar morphology was
accompanied by a reduction in rDNA transcription, and the release of RNA pol
I from the nucleolus. This same phenotype was, interestingly, also observed
with H4 hypoacetylation (Tsang et al. 2003).
Understanding the role of Esa1 in the regulation of nucleolar
morphology may lead to further insight into how Esa1 regulates ribosome
biogenesis. Whereas esa1 results in H4 hypoacetylation, mutation of ESA1
results in a larger, amorphous nucleolus rather than a diminished nucleolus
(Clarke et al. 1999). However, the upregulation of ribosome biogenesis, in
direct opposition to that caused by rapamycin, has the opposite affect on
nucleolar morphology as well. It will be important to better understand how
complete bypass of ESA1 affects nucleolar morphology since previous
characterization relied on ESA1 hypomorphic alleles. Additionally, it will be

138
interesting to understand if Hda1 is at all involved in restoration of nucleolar
organization. Florescence microscopy of nucleolar proteins that are not
differentially expressed upon loss of ESA1, such as Sir2 and one of several
RNA pol I subunits, such as A190, will help characterize the distribution of
nucleolar proteins to help assess overall nucleolar morphology.
Whereas it is evident that Esa1 is critical for regulation of ribosome
biogenesis, it is not yet apparent how the pieces of such a mechanism fit
together to encompass transcription, rDNA silencing, and nucleolar
morphology. Future studies, such as those proposed here, would allow for
better resolution of how Esa1 is specifically involved in the critical process of
ribosome biogenesis.
Summary. The powerful tool of bypass suppression facilitated the
expanded characterization of two critical chromatin regulators in the NuA4
HAT complex, Epl1 and Esa1. Bypass suppression can introduce added
challenges in interpretation of results, since an additional mutation
accompanies the deletion of an essential gene. Here, bypass suppression not
only allowed for comprehensive study of essential genes in vivo, but
simultaneously highlighted the importance of the network of interactions
between HAT and HDAC mutlimeric complexes, specifically Rpd3L, Hda1, and
NuA4. The studies presented and discussed in this thesis define new roles for
Epl1, Esa1, and for how these critical subunits interact with opposing HDAC
enzymes. These studies set the fundamental groundwork for future analyses,

139
where the focus can now shift from initial characterization toward increasingly
deeper understanding of the mechanisms behind many of the new phenotypes
and functions discussed. Given that the tight regulation of chromatin is critical
for basic cellular functioning and is frequently dysregulated in human disease,
it is particularly important to understand how the key chromatin modifiers
discussed here are specifically controlled, and why they are explicitly
necessary for biological function.

140
Acknowledgements This chapter, in part, contains material in-press for publication as it
appears in Searle NE, Pillus L. 2017. Critical genomic regulation mediated by
Enhancer of Polycomb. Current Genetics. In-press. The dissertation author
was the primary investigator and first author of this paper.

141
References Attwooll C, Oddi S, Cartwright P, Prosperini E, Agger K, Steensgaard P,
Wagener C, Sardet C, Moroni MC, Helin K. 2005. A novel repressive E2F6 complex containing the polycomb group protein, EPC1, that interacts with EZH2 in a proliferation-specific manner. J Biol Chem 280: 1199-1208.
Avvakumov N, Côté J. 2007. The MYST family of histone acetyltransferases and their intimate links to cancer. Oncogene 26: 5395-5407.
Clarke AS, Lowell JE, Jacobson SJ, Pillus L. 1999. Esa1p is an essential histone acetyltransferase required for cell cycle progression. Molecular and Cellular Biology 19: 2515-2526.
Dong Y, Isono KI, Ohbo K, Endo TA, Ohara O, Maekawa M, Toyama Y, Ito C, Toshimori K, Helin K, Ogonuki N, Inoue K, Ogura A, Yamagata K, Kitabayashi I, Koseki H. 2017. EPC1/TIP60-mediated histone acetylation facilitates spermiogenesis in mice. Mol Cell Biol.
Downey M, Johnson JR, Davey NE, Newton BW, Johnson TL, Galaang S, Seller CA, Krogan N, Toczyski DP. 2015. Acetylome profiling reveals overlap in the regulation of diverse processes by sirtuins, gcn5, and esa1. Mol Cell Proteomics 14: 162-176.
Ellahi A, Thurtle DM, Rine J. 2015. The Chromatin and Transcriptional Landscape of Native Saccharomyces cerevisiae Telomeres and Subtelomeric Domains. Genetics 200: 505-521.
Grunstein M, Gasser SM. 2013. Epigenetics in Saccharomyces cerevisiae. Cold Spring Harb Perspect Biol 5.
Guillemette B, Bataille AR, Gevry N, Adam M, Blanchette M, Robert F, Gaudreau L. 2005. Variant histone H2A.Z is globally localized to the promoters of inactive yeast genes and regulates nucleosome positioning. PLoS Biol 3: e384.
Hamza A, Tammpere E, Kofoed M, Keong C, Chiang J, Giaever G, Nislow C, Hieter P. 2015. Complementation of Yeast Genes with Human Genes as an Experimental Platform for Functional Testing of Human Genetic Variants. Genetics 201: 1263-1274.
Huang HT, Kathrein KL, Barton A, Gitlin Z, Huang YH, Ward TP, Hofmann O, Dibiase A, Song A, Tyekucheva S, Hide W, Zhou Y, Zon LI. 2013. A

142
network of epigenetic regulators guides developmental haematopoiesis in vivo. Nat Cell Biol 15: 1516-1525.
Keogh MC, Mennella TA, Sawa C, Berthelet S, Krogan NJ, Wolek A, Podolny V, Carpenter LR, Greenblatt JF, Baetz K, Buratowski S. 2006. The Saccharomyces cerevisiae histone H2A variant Htz1 is acetylated by NuA4. Genes Dev 20: 660-665.
Kim JR, Kee HJ, Kim JY, Joung H, Nam KI, Eom GH, Choe N, Kim HS, Kim JC, Kook H, Seo SB, Kook H. 2009. Enhancer of polycomb1 acts on serum response factor to regulate skeletal muscle differentiation. J Biol Chem 284: 16308-16316.
Kim Y, Sun H. 2007. Functional genomic approach to identify novel genes involved in the regulation of oxidative stress resistance and animal lifespan. Aging Cell 6: 489-503.
Lee KK, Workman JL. 2007. Histone acetyltransferase complexes: one size doesn't fit all. Nat Rev Mol Cell Bio 8: 284-295.
Lin YY, Lu JY, Zhang J, Walter W, Dang W, Wan J, Tao SC, Qian J, Zhao Y, Boeke JD, Berger SL, Zhu H. 2009. Protein acetylation microarray reveals that NuA4 controls key metabolic target regulating gluconeogenesis. Cell 136: 1073-1084.
Lin YY, Qi Y, Lu JY, Pan X, Yuan DS, Zhao Y, Bader JS, Boeke JD. 2008. A comprehensive synthetic genetic interaction network governing yeast histone acetylation and deacetylation. Genes Dev 22: 2062-2074.
Mehta M, Braberg H, Wang S, Lozsa A, Shales M, Solache A, Krogan NJ, Keogh MC. 2010. Individual lysine acetylations on the N terminus of Saccharomyces cerevisiae H2A.Z are highly but not differentially regulated. J Biol Chem 285: 39855-39865.
Meneghini MD, Wu M, Madhani HD. 2003. Conserved histone variant H2A.Z protects euchromatin from the ectopic spread of silent heterochromatin. Cell 112: 725-736.
Millar CB, Xu F, Zhang K, Grunstein M. 2006. Acetylation of H2AZ Lys 14 is associated with genome-wide gene activity in yeast. Genes Dev 20: 711-722.
Mitchell L, Huard S, Cotrut M, Pourhanifeh-Lemeri R, Steunou AL, Hamza A, Lambert JP, Zhou H, Ning Z, Basu A, Côté J, Figeys DA, Baetz K. 2013. mChIP-KAT-MS, a method to map protein interactions and

143
acetylation sites for lysine acetyltransferases. Proc Natl Acad Sci U S A 110: E1641-1650.
Neves LT, Douglass S, Spreafico R, Venkataramanan S, Kress TL, Johnson TL. 2017. The histone variant H2A.Z promotes efficient cotranscriptional splicing in S. cerevisiae. Genes Dev 31: 702-717.
Oakes M, Aris JP, Brockenbrough JS, Wai H, Vu L, Nomura M. 1998. Mutational analysis of the structure and localization of the nucleolus in the yeast Saccharomyces cerevisiae. J Cell Biol 143: 23-34.
Oakes M, Nogi Y, Clark MW, Nomura M. 1993. Structural alterations of the nucleolus in mutants of Saccharomyces cerevisiae defective in RNA polymerase I. Mol Cell Biol 13: 2441-2455.
Perry J. 2006. The Epc-N domain: a predicted protein-protein interaction domain found in select chromatin associated proteins. BMC Genomics 7: 6.
Prasad P, Ronnerblad M, Arner E, Itoh M, Kawaji H, Lassmann T, Daub CO, Forrest AR, Lennartsson A, Ekwall K, consortium F. 2014. High-throughput transcription profiling identifies putative epigenetic regulators of hematopoiesis. Blood 123: e46-57.
Squatrito M, Gorrini C, Amati B. 2006. Tip60 in DNA damage response and growth control: many tricks in one HAT. Trends Cell Biol 16: 433-442.
Taddei A, Schober H, Gasser SM. 2010. The budding yeast nucleus. Cold Spring Harb Perspect Biol 2: a000612.
Torres-Machorro AL, Pillus L. 2014. Bypassing the requirement for an essential MYST acetyltransferase. Genetics 197: 851-863.
Tsang CK, Bertram PG, Ai W, Drenan R, Zheng XF. 2003. Chromatin-mediated regulation of nucleolar structure and RNA Pol I localization by TOR. EMBO J 22: 6045-6056.
Yi C, Ma M, Ran L, Zheng J, Tong J, Zhu J, Ma C, Sun Y, Zhang S, Feng W, Zhu L, Le Y, Gong X, Yan X, Hong B, Jiang FJ, Xie Z, Miao D, Deng H, Yu L. 2012. Function and molecular mechanism of acetylation in autophagy regulation. Science 336: 474-477.
Zeng X, Han L, Singh SR, Liu H, Neumuller RA, Yan D, Hu Y, Liu Y, Liu W, Lin X, Hou SX. 2015. Genome-wide RNAi screen identifies networks involved in intestinal stem cell regulation in Drosophila. Cell Rep 10: 1226-1238.

144
Appendix A.
Experimental Methods

145
Experimental Methods
Unless otherwise noted, all experiments were performed independently two or
more times. The following materials and methods for Chapters 2, 3, and 4 are
detailed within the “Materials and Methods” section of Chapter 2:
Yeast strains and plasmids
Growth assays
Flow cytometry
Lysate preparations
Immunoblots
RNA-seq sample preparation and analysis
Two additional methods used in Chapters 3 and 4 were not detailed in
Chapter 2, and are as follows:
Halo assays. Cells were grown at 24˚ to saturation in SC medium. Final
concentrations of 0.1 A600 of cells in 200ul were spread onto SC plates with sterile
glass beads and allowed to dry. A small circle of sterile Whatman paper was added to
the center of the plate center. 20 µl of either H2O or a solution of 3% H2O2 was added
dropwide to the Whatman paper. Plates were incubated at 24˚ for 3-4 days, until a
lawn of cells was sufficiently grown. No sensitivity was observed upon H2O treatment,
and the size of the resulting Halo was insignificant. The H2O2-treated plates were
imaged and the size of the Halo was quantified measuring the diameter of the halo in
technical triplicate. The three measurements were averaged, and the area of the Halo

146
was calculated, and normalized to the Halo area in WT cells. Error bars were
calculated using standard deviation of the mean of 3 independent experiments.
Polysome profiling. Polysome profiling was performed according to (Ingolia
et al. 2009) with minor adaptations. In brief, starter cultures of cells were grown in SC
at 24˚, expanded in 20ml, grown overnight, and then expanded again in 100 ml and
grown to mid-log phase. Cell cultures were treated with 100µg/ml cycloheximide for
30 seconds, stirring continuously. Cells were collected by vacuum filtration, flash-
frozen in conical tubes in a bath of liquid N2, and suspended in 1.1ml of chilled
polysome lysis buffer (20mM Tris 8.0, 140mM KCl, 1.5mM MgCl2, 100µg/ml
cycloheximide, 1% Triton), adding the chilled buffer dropwise to the cells. Frozen cells
were lysed with a ball mill, for 2 rounds or 3 minutes at 400rpm in 1 minute intervals.
Lysed cells were collected from the mixer mill chambers, and flash frozen. Lysates
were thawed in a water bath and spun for 5 minutes at 3000g at 4˚. The supernatant
was recovered and spun again for 10 minutes at 20,000g at 4˚, and the supernatant
between the top layer of debris and the pellet was collected and the A260 was
quantified. Aliquots of approximately 100 A260 units were used for the sucrose
gradient. Sucrose gradients were prepared using 10% and 50% weight/volume
sucrose prepared in polysome gradient buffer (20mM Tris 8.0, 140mM KCl, 5mM
MgCl2, 100µg/ml cycloheximide, 0.5mM DTT, 20 U/ml SUPERase-In (Invitrogen).
Using a gradient-maker, ultracentrifuge tubes were filled using a syringe, halfway with
10% sucrose and then underlaid with 50% sucrose, until the tube was completely
filled, avoiding air bubbles. The Gradient Master was used for form the sucrose
gradients by tilted tube rotation with the following settings: 158 seconds, at a tilt of

147
12˚, at 16rpm. Gradient tubes were placed in SW-41 rotor buckets, and prepared cell
extract was added to the top of the gradient. Gradients were spun in a pre-chilled 4˚
ultracentrifuge (Beckman Coulter Optima LE-80K) for 3 hours at 35,000rpm. A254
measurements were read and recorded, using a solution of 50% sucrose to feed the
polysome gradient into the spectrophotometer.
References
Ingolia NT, Ghaemmaghami S, Newman JR, Weissman JS. 2009. Genome-wide analysis in vivo of translation with nucleotide resolution using ribosome profiling. Science 324: 218-223.

![[Coronary Bypass Surgery in Acute Myocardial Infarction]. Akut Miyokard İnfarktüsünde Koroner Bypass Cerrahisi](https://img.pdfslide.net/doc/110x75/63444ad1596bdb97a9086499/coronary-bypass-surgery-in-acute-myocardial-infarction-akut-miyokard-infarktuesuende.jpg)



























