Embed Size (px)
Citation preview

MINI-REVIEW
Cephalosporin C acylase: dream and(/or) reality
Loredano Pollegioni & Elena Rosini & Gianluca Molla
Received: 6 December 2012 /Revised: 24 January 2013 /Accepted: 24 January 2013 /Published online: 16 February 2013# Springer-Verlag Berlin Heidelberg 2013
Abstract Cephalosporins currently constitute the mostwidely prescribed class of antibiotics and are used to treatdiseases caused by both Gram-positive and Gram-negativebacteria. Cephalosporins contain a 7-aminocephalosporanicacid (7-ACA) nucleus which is derived from cephalosporinC (CephC). The 7-ACA nucleus is not sufficiently potent forclinical use; however, a series of highly effective antibioticagents could be produced by modifying the side chainslinked to the 7-ACA nucleus. The industrial production ofhigher-generation semi-synthetic cephalosporins starts from7-ACA, which is obtained by deacylation of the naturallyoccurring antibiotic CephC. CephC can be converted to 7-ACA either chemically or enzymatically using D-amino acidoxidase and glutaryl-7-aminocephalosporanic acid acylase.Both these methods show limitation, including the produc-tion of toxic waste products (chemical process) and theexpense (the enzymatic one). In order to circumvent theseproblems, attempts have been undertaken to design a single-step means of enzymatically converting CephC to 7-ACA inthe course of the past 10 years. The most suitable approachis represented by engineering the activity of a knownglutaryl-7-aminocephalosporanic acid acylase such that itwill bind and deacylate CephC more preferentially overglutaryl-7-aminocephalosporanic acid. Here, we describethe state of the art in the production of an effective andspecific CephC acylase.
Keywords Cephalosporin C . Protein engineering .
Biocatalysis . Acylase
Introduction
Cephalosporin C (CephC—the C stands for chromatogra-phy) was the second β-lactam antibiotic to be discoveredafter penicillin was established as a clinical drug. Threemilestones characterize the history of CephC. Firstly, in1945, the medical doctor Giuseppe Brotzu isolated a fungusfrom seawater near a sewage outlet in Cagliari city (Italy).The new fungus—initially identified as a strain ofCephalosporium acremonium and reclassified later asAcremonium chrysogenum—showed high antagonistic po-tency against Staphylococcus aureus, Vibrio cholera, andBacillus anthracis. Secondly, in 1953 in Oxford, GuyNewton and Edward Abraham isolated and purified theantibacterial substance CephC, which was initially namedpenicillin N. CephC showed a broad spectrum of action, butonly moderate antibacterial activity: minimum inhibitoryconcentration values were in the range of 25–100 and 12–25 μg/mL for Gram-positive and Gram-negative bacteria,respectively. Thirdly, in 1962, Robert Morin, a researcher atthe Lilly Company, discovered a process by which the D-α-amino adipoyl side chain of CephC could be removed,generating 7-aminocephalosporanic acid (7-ACA). Thisprocess paved the way for being able to chemically modifyCephC at position C7 and for discovering new and moreeffective cephalosporins. As compared to penicillin, whichpossesses a single site for derivatization, the cephalosporinmolecule offers two sites: the 7-amino group and the C3acetoxy group (see Scheme 1). Thus, a larger number ofsemi-synthetic derivatives can be synthesized from CephCthan from the penicillin-based counterpart. Nonetheless, itwas not as easy to synthesize the new cephalosporins as was
L. Pollegioni : E. Rosini :G. MollaDipartimento di Biotecnologie e Scienze della Vita,Università degli studi dell’Insubria, Varese, Italy
L. Pollegioni (*) : E. Rosini :G. MollaCentro Interuniversitario di Ricerca in Biotecnologie Proteiche“The Protein Factory”, Politecnico di Milano, ICRM CNR Milanoand Università degli studi dell’Insubria, Italy, via J.H. Dunant 3,21100 Varese, Italye-mail: [email protected]
Appl Microbiol Biotechnol (2013) 97:2341–2355DOI 10.1007/s00253-013-4741-0

the case for penicillins because of the restricted availabilityof the key intermediate 7-ACA.
The first practical method for removing the D-α-aminoadipoyl side chain of CephC was a chemical deacylationprocedure which requires harsh reaction conditions andreleases toxic waste (Fechtig et al. 1968; Morin et al. 1969;Sonawane 2006). Importantly, residual dichloromethane andphosphates must be eliminated from the mother liquor beforetreating the wastewater; indeed, the final 7-ACA preparationcontains 2–3% oligomers, dichloromethane, and dimethylani-line, which are almost impossible to get rid of. For details onthe chemical methods, see (Cabri et al. 1999).
Enzymatic production of 7-ACA
There are several advantages to producing 7-ACA fromCephC by using an enzymatic process as compared to achemical one (Cabri et al. 1999). These include: (1) safetyand environmental contamination: dangerous and toxicreagents (i.e., trimethylchlorosilane, phosphorus pentachlor-ide, dichloromethane, dimethylaniline, etc.) are absent in theenzymatic transformation; (2) selectivity: owing to enzymeselectivity, the use of permanent/temporarily protectivegroups is avoided; (3) energy consumption: the chemicalprocess uses low temperatures and exothermic steps, whilethe enzymatic approach is carried out at a fixed temperature of20–30 °C; (4) equipment: owing to the use of aggressivereagents, the chemical process requires glass-lined vesselsand dedicated and expensive equipment to transport andcharge these materials, to trap spilling or vapors, etc.; and(5) quality: in the enzymatic 7-ACA preparations, the levels ofoligomers, 7-desacetoxycephalosporanic acid, and desacetyl-7-ACA (or corresponding lacton) are considerably lower—the
typical assay is 3–6 % higher than for the chemical process.The purity of the final 7-ACA preparation depends by andlarge on the quality of the enzyme preparations used: theseenzymes should be free of anyβ-lactamase, esterase, protease,and lipase activity.
The most widely used enzymatic approach is representedby a two-step pathway that employs D-amino acid oxidase(DAAO, EC 1.4.3.3) and glutaryl-7-aminocephalosporanicacid (Gl-7-ACA) acylase (GA, EC 3.5.1.93; Pilone et al.1995; Pilone and Pollegioni 2002, 2010; Volpato et al. 2010;see Scheme 2). For the production of 7-ACA using enzymaticcatalysis, today, multi-enzymatic sequential reactions are per-formed in the absence of purification steps, whereby enzymespecificity prevents the formation of by-products. The fla-voenzyme DAAO oxidizes the D-α-amino adipyl moiety ofCephC to give α-ketoadipyl-7-ACA, which is converted toGl-7-ACA non-enzymatically; this latter compound is suscep-tible to enzymatic attack by various acylases (see Scheme 2).For a review on the structural and functional properties ofDAAO from different sources, see Harris et al. (2001) andPollegioni et al. (2007, 2008a). GA activity has been reportedin a limited number of genera, e.g., in bacteria fromPseudomonas, Arthrobacter, Bacillus, Aeromonas, andFlavobacterium and from the fungus Paceilomyces (Aramoriet al. 1991a; Sonawane 2006; see below). The two-step pro-cess developed by Boehringer Mannheim (Tischer et al. 1992)used DAAO immobilized on an organic polymer (25 kU/kg)and GA immobilized on an inorganic polymer (110 kU/kg)and yielded 94 % conversion of 75 mM CephC into Gl-7-ACA in 30min at pH 8.0 and 20 °C and 96% of conversion ofGl-7-ACA into 7-ACA in 30 min at pH 8.0 and 28 °C. Thesimplicity of this enzymatic alternative as compared to thechemical routes even compensates for the complex conditionsof using two enzymes (and separate reactors due to differentoperational conditions and stability).
One main drawback of the two-step enzymatic process isthat DAAO produces hydrogen peroxide as a by-product.Hydrogen peroxide negatively affects flavoenzyme stability(as well as the stability of GA when the two enzymes areused in a single reactor), although most of it is consumed toconvert the α-ketoadipyl-7-ACA intermediate into Gl-7-ACA (see Scheme 2). The addition of catalase eliminatesthe hydrogen peroxide and prevents enzyme inactivationduring the two-step catalysis (Lopez-Gallego et al. 2005).For a recent review concerning the multistep catalysis ofCephC, see Pollegioni and Molla (2011). Otherwise, D-amino acid transaminase from Bacillus licheniformisATCC 9945 was used instead of DAAO. It employsCephC as the amino donor and α-ketoglutarate as an accep-tor, thus yielding α-keto adipyl aminocephalosporanic acid,a substrate of GA (Aretz and Sauber 1988).
The alternative enzymatic approach is a one-step processin which CephC is converted directly into 7-ACA by a true
Scheme 1 Cephalosporin molecule, its different constituent moieties,and sites amenable to enzymatic modification. The glutaryl side chainin N7 is present in Gl-7-ACA only
2342 Appl Microbiol Biotechnol (2013) 97:2341–2355

CephC acylase (CA; Scheme 2). This approach is veryattractive at the industrial level because of the prospects ofsimplifying the process and reducing costs. As shown inScheme 1, acylases can cleave the amide bond throughwhich the cephem nucleus is linked to the acyl side chain.Following the original classification based on substratespecificity (Aramori et al. 1991b), acylases have been clas-sified into five types on the basis of their gene structures,molecular masses, and enzyme properties (Table 1 andFig. 1). The deduced amino acid sequence of the mem-bers of each class is at least 90 % homologous to each
other, and their functional properties are also similar (Liet al. 1999; Oh et al. 2003). Members of the differentclasses can be distinguished in terms of substrate spec-ificity: while all GAs are active on Gl-7-ACA, onlymembers of classes I and III show an appreciable activity onCephC (Fig. 1). Various GA enzymes also active on CephChave been identified, e.g., in Pseudomonas sp. strain N176(Aramori et al. 1991a, b), Pseudomonas sp. V22 (Aramori etal. 1991b), and Pseudomonas sp. strain SE83 (Matsuda etal. 1987a, b). GA from Pseudomonas diminuta N176showed the highest activity on both Gl-7-ACA (Vmax=
Scheme 2 Bioconversion ofcephalosporin C. Left Two-stepbioconversion of CephC to 7-amino cephalosporanic acid byD-amino acid oxidase andglutaryl-7-ACA acylase. RightOne-step conversion bycephalosporin C acylase
Table 1 Structural properties of acylases
Class Precursor (kDa) Signal peptide (AA) α-subunitmolecularmass (kDa)
Spacerpeptide (AA)
β-subunitmolecularmass (kDa)
Source Reference
I 70 29 16 10 54 Pseudomonas sp. 130 Li et al. (1999)
II 89 29 28 n.d. 61 Pseudomonas sp. A14 Aramori et al. (1991b)
III 80 – 22 10 58 Pseudomonas diminuta N176 Aramori et al. (1991b)
IV 64 – 40 n.d. 22 Pseudomonas sp. SE83 [Acy I] Matsuda et al. (1987a, b)
V 70 27 – 70 – Bacillus laterosporus J1 Aramori et al. (1991c)
AA amino acids, n.d. not determined, – no spacer or signal
Appl Microbiol Biotechnol (2013) 97:2341–2355 2343

100 U/mg protein) and CephC (Vmax=3.1 U/mg protein;Aramori et al. 1992).
The genes for GAs have been cloned and the encodedproteins frequently produced in large quantities and character-ized. For example, enzymes active on CephC were reported byMerck & Co. in the 1980s (Lein 1988, 1989; Crawford 1991):they identified a CA activity in Bacillus megaterium ATCC53667 and also isolated, cloned, and purified a CephC amidasefrom the same source. This latter enzyme (constituted by twosubunits of 45 and 37 kDa) used desacetyl CephC as the bestsubstrate and showed a specific activity of 0.02–0.05 μmolmin−1mg−1 protein and a Km of 1.3 mM for CephC. Amongthe known acylases, the most promising enzyme was a CephCamidohydrolase (formerly an acylase) that was identified andcloned from Pseudomonas vesicularis B965 at Glaxo labora-tories (Burr et al. 1999). On CephC, the recombinant enzymeexpressed in Escherichia coli showed 8 % of the activitydetermined on Gl-7-ACA and a specific activity of0.14 U/mg protein. In 1996, an enzyme active on CephC andfully inactive toward Gl-7-ACAwas also reported (Deshpandeet al. 1996). Up to 1.1 U/L of this enzyme was produced fromAeromonas sp. ACY95, representing the only known true CA(Fig. 1). Of note is that in this review, we use the term CA toindicate all GA members that show a significant activity onCephC, even if it is not the preferred substrate.
During the years, 3D structural analyses and site-directedmutagenesis studies have increased our understanding of thestructure–function relationship of GAs, as detailed in thefollowing paragraph(s). This information was used by anumber of groups for developing GA variants with higherhydrolytic activity on the natural antibiotic CephC.
Structure–function relationships in GAs
General fold
From a structural point of view, acylases belong to the N-terminal nucleophile aminohydrolase (Ntn hydrolase) super-family (according to the SCOP classification; Murzin et al.1995). All members of this protein superfamily have severalpeculiar features in common: (1) the core region of the proteinis formed by four layers of α-helices and β-sheets arranged inan αββα fold. (2) Ntn hydrolases are produced as an inactiveprecursor. As a matter of fact, the gene structure of the openreading frame of the acylases differs within each enzyme, butgenerally consists of a signal peptide followed by the α-subunit, a spacer sequence (9–11 residues), and the β-subunit. For maturation, the protein requires two autoproteo-lytic cleavages, resulting in an active αβ heterodimer (Kim etal. 2000) or an (αβ)2 heterotetramer (Oh et al. 2003). (3) TheN-terminal residue of theβ-chain provides the two fundamen-tal catalytic functions for hydrolysis of the amide bond of thesubstrate, i.e., the nucleophile and the proton donor.
Currently, the 3D structure of a few members of class Iacylase is known: CA from P. diminuta (CAD, pdb code1fm2; Kim et al. 2000), Pseudomonas sp. 130 (P130, pdbcode 1gk0; Fritz-Wolf et al. 2002), and Pseudomonas sp.strain GK16 (pdb code 1or0; Kim et al. 2003; sequenceidentity of 94.6–97.6 %). In the following paragraphs,CAD will be used as the reference since the structure ofthe mature form of this protein is available both in theabsence of ligands (1fm2) and in complex with the substrateGl-7-ACA or the product glutarate (1jvz and 1jw0, respec-tively; Kim and Hol 2001). In addition, the structure of theinactive precursor was also solved (1keh; Kim et al. 2002).
Although the structure of the mature form of CAD isformed by a small α-subunit and a large β-subunit, they donot form two discrete domains, but rather the two subunitsare deeply engaged with each other and form a single-domain heterodimer (Fig. 2a; Kim et al. 2000). This struc-tural aspect is common in multimeric proteins derived fromthe cleavage of a single-chain precursor. The overall shapeof mature CAD resembles a saddle formed by a large glob-ular region surmounted by two apical “knobs.” The centralregion possesses the characteristic four-layer (αββα) foldof Ntn hydrolase family members with the essential active-site residue Ser1β located at the end of the first β-sheet ofthis region. The Ntn fold is flanked by the α-subunit (mainlyα-helices), which has an important role in forming part ofthe active site and is surmounted by the two “knob”domains. These latter domains have a similar dimensionand shape, but completely different secondary structurecompositions: the “A knob” contains mostly α-helices,while the “B knob” is formed almost exclusively byβ-strands (Fig. 2a).
Fig. 1 Substrate specificity profiles of glutaryl acylases: while all GAsare active on Gl-7-ACA (black bars), only members of classes I and IIIshow a significant activity on CephC (gray bars) and only Aeromonas sp.ACY95 is absolutely specific for CephC. Specific activity values set as100 %: Aeromonas sp. ACY95 on CephC: not reported (Deshpande et al.1996); P130 (class I) on Gl-7-ACA: not reported (Li et al. 1999); A14(class II) on Gl-7-ACA: 7.1 U/mg protein (Aramori et al. 1991b); N176(class III) on Gl-7-ACA: 100 U/mg protein (Aramori et al. 1991b); SE83acyI (class IV) on Gl-7-ACA: not reported (Matsuda et al. 1987a); J1(class V) on Gl-7-ACA: 5.3 U/mg protein (Aramori et al. 1991b). Thespecific activity values were determined on purified proteins
2344 Appl Microbiol Biotechnol (2013) 97:2341–2355

Active site and substrate binding
The active site is positioned in a cleft in the center of theprotein molecule between the two knobs (Fig. 2a). Thecomplex between CAD and Gl-7-ACA (or glutarate) is anexample of the “lock-and-key”mode of substrate interactionsince binding of the substrate to the active site causes noevident conformational changes: the root-mean-square de-viation between the free enzyme and the enzyme in complexwith Gl-7-ACA is 0.23 Å for all 627 Cα atoms (Kim andHol 2001). CAD represents one of the few cases where theexperimental structure of the Michaelis complex is avail-able, allowing the direct assignment of the role of the active-site residues for substrate binding. The molecule of the
substrate Gl-7-ACA is constituted by three parts: (1) theglutaryl chain (i.e., the substituent at position 7); (2) thecephem nucleus formed by a β-lactam ring fused to a six-membered dihydrothiazine ring; and (3) the two branchedchains (the carboxylate and acetoxymethyl substituents, po-sition 3) attached to the six-membered ring (see Scheme 1for structural details). According to this subdivision, active-site residues can be clustered into three groups depending onwhich part of substrate they interact with. The most exten-sive interactions between Gl-7-ACA and CAD active siteinvolve the glutaryl side chain of the substrate that is buriedinside a deep pocket in the innermost part of the active-sitecleft (substrate specificity pocket; Fig. 2). Although theoverall geometry of this region is conserved between class
Fig. 2 Three dimensional structure of CAD (class I acylase, PDB code1jvz; Kim and Hol 2001). a Cartoon representation of the overallstructure of mature CAD. The α-subunit is represented in green andthe β-subunit in different tones of blue. The substrate Gl-7-ACA andSer1β are depicted showing the van der Waals radii of their atoms ingray and purple, respectively. b Close-up of the glutaryl side chainspecificity pocket at the active site of CAD (α-subunit is represented ingreen and the β-subunit in blue) in complex with Gl-7-ACA (yellow). cSchematic representation of the main interactions between CAD and
Gl-7-ACA (brown). Residues of CAD are indicated by black labels,while corresponding residues of class III N176 acylase are indicated byblue labels. Residues placed above the plane (on the top) of thesubstrate are depicted in gray. Non-covalent interactions are indicatedby dotted lines: H-bonds are in blue, interactions of the oxyanion holein purple, and contacts between the catalytic triad residues in green.Hydrophobic interactions are shown in orange (Fritz-Wolf et al. 2002).Tyr178α and Arg184α as in Fig. 3 correspond to Tyr149α andArg155α, respectively, in Fritz-Wolf et al. (2002)
Appl Microbiol Biotechnol (2013) 97:2341–2355 2345

I acylases and penicillin G acylase (PGA, EC 3.5.1.11), thespecificity pocket in the latter is lined by exclusively hydro-phobic residues, while in CAD several polar residues arepresent. The most important residues are Arg57β (number-ing as reported in Fig. 3), which electrostatically interactswith the negatively charged carboxylate group of glutaryl,and two tyrosines (Tyr178α and Tyr33β) whose phenolhydroxyl groups form hydrogen bonds with the oxygenatoms of the glutaryl carboxylate. In addition to these polarinteractions, affinity for Gl-7-ACA is enhanced by hydro-phobic interactions between the aliphatic portion of theglutaryl moiety and neighboring hydrophobic residues:Leu24β, Val70β, and, at a larger distance, Gln50β andPhe177β. Interestingly, although the substituted cephemring possesses several functional groups potentially able toestablish polar and hydrogen bond interactions, in CAD itforms only few hydrophobic interactions (with Tyr178α,Leu24β, and Val70β) and only one single H-bond (betweenthe acetoxymethyl group at C3 and Arg184α). It is remark-able that each of the two most important residues for sub-strate binding and recognition (the arginines 184α and 57β,which anchor the two opposite ends of Gl-7-ACA) belongsto a different subunit (Kim and Hol 2001).
Specificity
Although class I acylases show a residual activity on CephC(≈40-fold lower in comparison to Gl-7-ACA; Aramori et al.1992), there is no detectable CAD activity on this substrate.The strict specificity of this enzyme for Gl-7-ACA residesalmost completely in the numerous and precise interactionsbetween the glutaryl moiety of the substrate and the sidechains that line the active site specificity pocket. Modelingof CephC in the CAD active site revealed the rationale forthe absence of activity on the natural antibiotic. Since theD-α-amino adipyl moiety is ≈41 Å3 larger than the glutarylgroup, the distal (terminal) amino acidic portion of CephCwould collide with critical active-site residues, mainlyTyr178α, Tyr33β, and Arg57β. In order to improve activityvs. CephC, Kim and co-workers suggested increasing theroom available at the active site of the enzyme to host thelarger chain of this substrate (Kim and Hol 2001) and/orincluding a histidine or glutamate residue to specificallybind the positively charged α-amino group of this com-pound. The following mutations have been suggested:Leu24βArg, Gln50βArg, Thr176βAsp, and Phe177βHis(see below; Fritz-Wolf et al. 2002).
Catalysis
The catalytic mechanism of Ntn hydrolases is very similar tothat of serine hydrolases with a critical nucleophile (sidechain hydroxyl or thiol group of Ser, Thr, or Cys) which
attacks the carbonyl carbon of the scissile amide bond. Apeculiar feature is that the nucleophile is the first residue ofthe β-subunit of the enzyme (Ser1β in CAD) and that thegeneral base, which extracts the proton from the nucleo-phile, is the α-amino group of the same residue. The N-terminal nucleophile is engaged in a tight hydrogen bondnetwork and is part of an arrangement reminiscent of thecatalytic triad of serine proteases. The catalytic mechanismof CAD was inferred from the data available on PGAbecause of the close similarity between the two enzymes(Duggleby et al. 1995). Although different hypotheses wereformulated concerning the precise role of the catalytic res-idues —hydrolysis has been proposed to be mediated via asingle catalytic residue (Kim et al. 2000), a double dyad(Fritz-Wolf et al. 2002), or a catalytic triad (Kim et al.2003)— a general picture of the catalytic mechanismemerges: the substrate (Gl-7-ACA) is bound at the activesite with the carbonyl carbon of the amide bond close (at3.4 Å) to the nucleophile Oγ of Ser1β. In analogy topenicillin acylase, catalysis is initiated by the nucleophilicattack of Ser1β Oγ to the carbonyl carbon of the amidebond of the substrate. Interestingly, in contrast to classicserine proteases, His23β (the central member of the catalytictriad) does not interact directly with Ser1β Oγ, but rather isconnected to this atom through the α-amino group of Ser1β(at 2.8 Å distance). This group, which is uncharged at the pHoptimum of the enzyme (i.e., 7.8), works as a general base thatabstracts the hydroxyl proton of the serine, thus enhancing itsnucleophilicity. The role of His23β and Glu455β, which areunprotonated at the physiological pH, is to stabilize the tran-sient positive charge that is formed during catalysis on the α-amino group of Ser1β (Fig. 2; Fritz-Wolf et al. 2002; Mao etal. 2004). During catalysis, the variation in the geometry of thecarbonyl group from planar to tetrahedral results in a 1 Ådecrease in the distance between the negatively charged oxy-gen and the oxyanion hole (formed by the main chain NH ofVal70β, the side chain ND2 of Asn244β, and, possibly, themain chain NH of His23β). As a result of this movement, twoadditional hydrogen bonds are produced while the transitionstate is forming (Duggleby et al. 1995; Kim and Hol 2001).Collapse of this intermediate leads to the release of 7-ACA,
Fig. 3 Amino acid alignment of glutaryl acylases used to evolve theactivity on CephC. N176: acylase from Pseudomonas sp. N176 (codeCAA01318.1); CAD: from Pseudomonas sp. KAC-1, also namedPseudomonas sp. 130 acylase (code AAC34685.2); SE83: acyII fromPseudomonas sp. SE83 (code CAP12264.1); SY-77: from Pseudomo-nas SY-77 (code AAN39264.1). Multiple alignment was performedusing ClustalW (http://npsa-pbil.ibcp.fr/cgi-bin/npsa_automat.pl?page=npsa_clustalw.html) at the Pole Bioinformatique Lyonnais andoptimized based on structural and functional information. Identical andsimilar residues are shaded in black and gray, respectively. Residuesmaking contacts with Gl-7-ACA in CAD are indicated by black tri-angles and catalytic residues (Ser1β, His23β, Glu455β for CAD andSer1β, Asp21β, His23β for N176) are indicated by black circles.Signal and spacer peptide of CAD are indicated by gray lines
�
2346 Appl Microbiol Biotechnol (2013) 97:2341–2355

Appl Microbiol Biotechnol (2013) 97:2341–2355 2347

while the second product (i.e., glutarate) remains bound to theactive site in a conformation similar to that of the transitionstate intermediate (Kim and Hol 2001). Finally, the nucleo-philic attack of an active-site water molecule on the boundadduct leads to the release of glutarate (Fritz-Wolf et al. 2002).
Maturation
Maturation of acylases requires two sequential autoproteolyticsteps that excise the short spacer sequence connecting the α-andβ-subunits. After protein folding, class I acylases undergoprimary proteolysis involving a N→O acyl shift. This firstreaction is triggered by a conformational flip of the spacerpeptide that is in a high-energy state due to conformationalconstraints (Kim et al. 2006). Subsequently, the peptide bondbetween Gly198α (Gly169) and Ser1β (Ser170) of the pre-cursor is attacked by the Ser1β-OH group, whose nucleophi-licity is enhanced by a nearby active-site water molecule—thenumbering in parentheses refers to the polypeptide chainprecursor of CAD without the signal peptide of 29 aminoacids, according to Kim et al. (2003, 2006). The reaction iscompleted by the deacylation step catalyzed by a secondactive-site water molecule forming a catalytic triad withHis23β (His192) and Glu455β (Glu624) side chains (Fig. 4,reaction A; Kim et al. 2003, 2006). This first cleavage pro-motes a conformational change of the space peptide thattriggers the second proteolytic cleavage. The nature of theresidues involved in the second reaction is still debated: fromanalysis of the 3D structure and maturation kinetics of severalvariants of GA from the Pseudomonas sp. strain GK16, it wasproposed that a water molecule, activated by Glu188α(Glu159), could attack the peptide bond between Gly189α(Gly160) and Asp190α (Asp161), this reaction resulting inthe dissociation of the spacer (Fig. 4, reaction B; Kim et al.2006). Based on the biochemical characterization ofPseudomonas sp. 130 acylase variants possessing shortenedspacer sequences, on the other hand, it was recently proposedthat even the second cleavage could be catalyzed by Ser1β(Ser170, Fig. 4, reaction C; Yin et al. 2011). According to theauthors, the fundamental role of Glu188α (Glu159) is tomimic the side chain of substrate Gl-7-ACA, leading thescissile bond (between Glu188α and Gly189α) into the cor-rect position (next to Ser1β) at the active site for cleavage. InP130 acylase (PDB code 1ghd), there is a 22 Å gap betweenthe two cleavage sites; therefore, a large-scale conformationalchange would be required between the first and second cleav-age in order to place the scissile bond close to the catalyticSer1β (Ser170). This conformational change could be favoredby the peculiar amino acidic sequence of the spacer, whichcontains three regularly spaced aspartate residues Asp190α,Asp193α, and Asp196α (xDxxDxxDx motif in CAD).Following the first cleavage, these repeated residues couldallow the spacer to fold into a compact structure (e.g., an α-
helix) that could insert easily into the catalytic pocket andtrigger the second cleavage (Yin et al. 2011).
Class III acylases
Class I P130 and class III N176 acylases share about 23 %of sequence identity (50 % of sequence similarity).Sequence alignment between these two acylases suggeststhat the two enzymes adopt the same catalytic mechanism:the active site nucleophile Ser1β is part of a catalytic triad(the sole difference is the substitution of glutamate 455β inP130 with aspartate 21β in N176) and the oxyanion hole isformed by three NH groups (Fritz-Wolf et al. 2002). In theabsence of the 3D experimental structure of class III acy-lases —which should be published shortly (Anandan et al.2010)— investigation of the mode of Gl-7-ACA (andCephC) binding at the active site of N176 acylase was basedon 3D models built by homology modeling using class ICAD structure as the template (Pollegioni et al. 2005). Closeinspection of the model confirmed that the β-subunit (thathosts the catalytic triad and a large part of the substraterecognition pocket) is largely conserved. The few but sig-nificant differences in the residue composition of this regionprobably account for the different activity on CephC be-tween the two enzymes (Fig. 1). In particular, the mostimportant substitutions are represented by Leu24β andArg57β of CAD that, in N176 acylase, are replaced by anarginine and a histidine residue, respectively (Fritz-Wolf etal. 2002; Pollegioni et al. 2005).
Evolution of CA from GAs
N176 acylase
In the pioneering paper of Aramori et al. (1992), GA fromPseudomonas sp. N176 was demonstrated to act on CephC(≈4 % of the activity on Gl-7-ACA). N176 acylase possessesinteresting kinetic properties on the natural antibiotic (Table 2),although the product 7-ACA acts as a strong competitiveinhibitor (Ki=1.4 mM). Subsequently, Fujisawa Pharm. dem-onstrated by site-directed mutagenesis that the substitution ofM31β (numbering as in Fig. 3 where the protein sequence ofN176 acylase is reported in comparison to the sequence of theother cited acylases) with an aromatic amino acid (F or Y)increases the activity on CephC (up to 1.8-fold) and suggestedfurther substitutions at positions A33β (in Y) and C67β (in S;Niwa et al. 1994; Ishii et al. 1995).
Starting from the previous information, site-directed mu-tagenesis was carried out next at positions A50α, M165α,S167α, M175α, D117β, M227β, M268β, and M512β(Niwa et al. 1998). Relative to the activity of wild-typeN176 acylase on CephC, the M165αL/M175αA/M31βY
2348 Appl Microbiol Biotechnol (2013) 97:2341–2355

and the A50αL/M165αL/M175αA/M31βY variantsshowed an over twofold increase in specific activity(Table 2). Concerning single point substitutions, theS167αA and the M512βA most significantly affected theactivity on CephC, while the A50αL positively affected theamount of expressed protein. A 20 % increase in activity onCephC was also reported for the M165αL and the M175αAvariants of N176 acylase (Saito et al. 1996). Simultaneously,the systematic substitution of every cysteine with serine
demonstrated that the C67βS substitution increases proteinexpression threefold (Yamada et al. 1996). Furthermore, theM31βY/C67βS double variant showed a 1.6-fold increasein activity on CephC as compared to wild-type N176 acy-lase (Table 2). These studies demonstrated that differentsubstitutions can affect to a similar extent the activity ofN176 acylase on CephC and that a rationale of such alter-ations was not elucidated to allow an additive combinationof the identified substitutions.
Fig. 4 Autoproteolyticmaturation of class I acylases.The first autoproteolyticcleavage is catalyzed by Ser1β(Ser170, reaction A; Kim et al.2003, 2006). Two alternativemechanisms are shown for thesecond autoproteolyticcleavage: it is catalyzed byresidue Glu188α (Glu159,reaction B; Kim et al. 2006) orby Ser1β (Ser170, reaction C;Yin et al. 2011). Numbering isreferred to mature CAD proteinas reported in Fig. 3; numbersin parentheses refer tonumeration of the precursorwithout the signal peptide (29amino acids), as stated in Kimet al. (2003, 2006) and Yin et al.(2011)
Appl Microbiol Biotechnol (2013) 97:2341–2355 2349
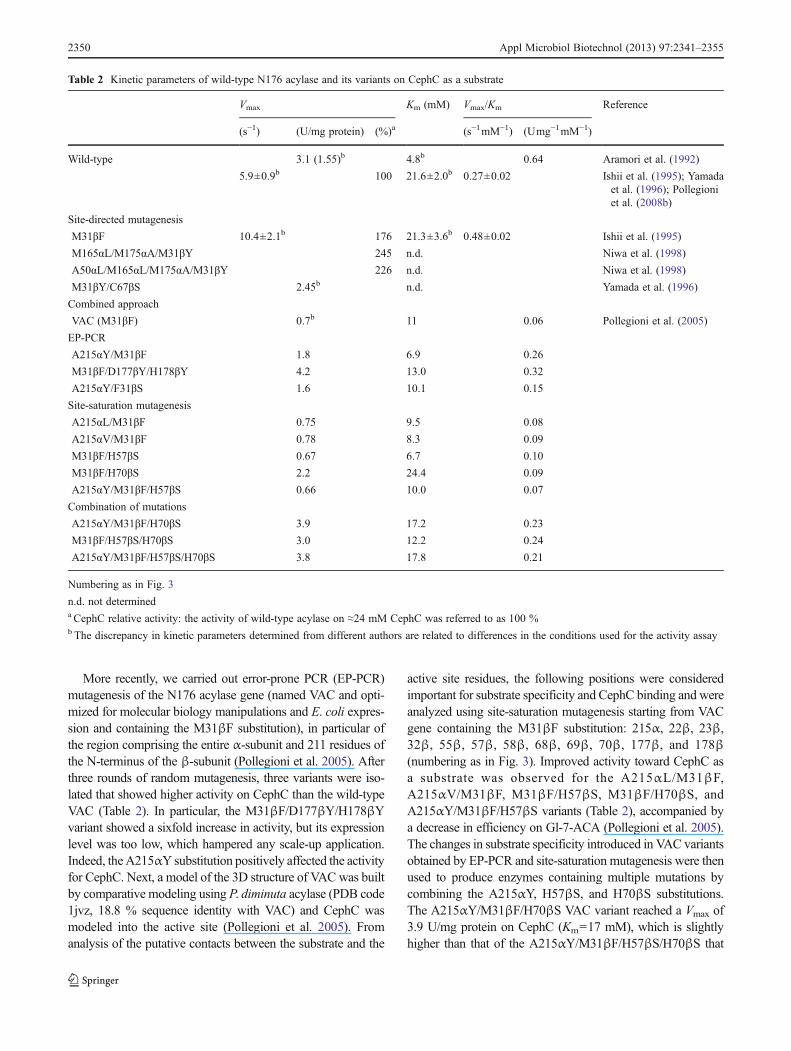
More recently, we carried out error-prone PCR (EP-PCR)mutagenesis of the N176 acylase gene (named VAC and opti-mized for molecular biology manipulations and E. coli expres-sion and containing the M31βF substitution), in particular ofthe region comprising the entire α-subunit and 211 residues ofthe N-terminus of the β-subunit (Pollegioni et al. 2005). Afterthree rounds of random mutagenesis, three variants were iso-lated that showed higher activity on CephC than the wild-typeVAC (Table 2). In particular, the M31βF/D177βY/H178βYvariant showed a sixfold increase in activity, but its expressionlevel was too low, which hampered any scale-up application.Indeed, theA215αY substitution positively affected the activityfor CephC. Next, a model of the 3D structure of VACwas builtby comparative modeling using P. diminuta acylase (PDB code1jvz, 18.8 % sequence identity with VAC) and CephC wasmodeled into the active site (Pollegioni et al. 2005). Fromanalysis of the putative contacts between the substrate and the
active site residues, the following positions were consideredimportant for substrate specificity and CephC binding and wereanalyzed using site-saturation mutagenesis starting from VACgene containing the M31βF substitution: 215α, 22β, 23β,32β, 55β, 57β, 58β, 68β, 69β, 70β, 177β, and 178β(numbering as in Fig. 3). Improved activity toward CephC asa substrate was observed for the A215αL/M31βF,A215αV/M31βF, M31βF/H57βS, M31βF/H70βS, andA215αY/M31βF/H57βS variants (Table 2), accompanied bya decrease in efficiency on Gl-7-ACA (Pollegioni et al. 2005).The changes in substrate specificity introduced in VAC variantsobtained by EP-PCR and site-saturation mutagenesis were thenused to produce enzymes containing multiple mutations bycombining the A215αY, H57βS, and H70βS substitutions.The A215αY/M31βF/H70βS VAC variant reached a Vmax of3.9 U/mg protein on CephC (Km=17 mM), which is slightlyhigher than that of the A215αY/M31βF/H57βS/H70βS that
Table 2 Kinetic parameters of wild-type N176 acylase and its variants on CephC as a substrate
Vmax Km (mM) Vmax/Km Reference
(s−1) (U/mg protein) (%)a (s−1mM−1) (Umg−1mM−1)
Wild-type 3.1 (1.55)b 4.8b 0.64 Aramori et al. (1992)
5.9±0.9b 100 21.6±2.0b 0.27±0.02 Ishii et al. (1995); Yamadaet al. (1996); Pollegioniet al. (2008b)
Site-directed mutagenesis
M31βF 10.4±2.1b 176 21.3±3.6b 0.48±0.02 Ishii et al. (1995)
M165αL/M175αA/M31βY 245 n.d. Niwa et al. (1998)
A50αL/M165αL/M175αA/M31βY 226 n.d. Niwa et al. (1998)
M31βY/C67βS 2.45b n.d. Yamada et al. (1996)
Combined approach
VAC (M31βF) 0.7b 11 0.06 Pollegioni et al. (2005)
EP-PCR
A215αY/M31βF 1.8 6.9 0.26
M31βF/D177βY/H178βY 4.2 13.0 0.32
A215αY/F31βS 1.6 10.1 0.15
Site-saturation mutagenesis
A215αL/M31βF 0.75 9.5 0.08
A215αV/M31βF 0.78 8.3 0.09
M31βF/H57βS 0.67 6.7 0.10
M31βF/H70βS 2.2 24.4 0.09
A215αY/M31βF/H57βS 0.66 10.0 0.07
Combination of mutations
A215αY/M31βF/H70βS 3.9 17.2 0.23
M31βF/H57βS/H70βS 3.0 12.2 0.24
A215αY/M31βF/H57βS/H70βS 3.8 17.8 0.21
Numbering as in Fig. 3
n.d. not determineda CephC relative activity: the activity of wild-type acylase on ≈24 mM CephC was referred to as 100 %b The discrepancy in kinetic parameters determined from different authors are related to differences in the conditions used for the activity assay
2350 Appl Microbiol Biotechnol (2013) 97:2341–2355

was more active on CephC than on Gl-7-ACA. Theevolved variants exhibited a >100-fold increase in thespecificity constant (the Vmax/Km ratio between CephCand Gl-7ACA) compared to the wild-type VAC: theA215αY/M31βF/H57βS/H70βS variant is a “true” CA sinceit exhibits a higher Vmax on CephC than on Gl-7ACA.
A 90 % production of 7-ACA from 50 mM CephC, atpH 8.5 and 25 °C, was reached in approx. 3 h using 5 U/mLof the M31βF/H57βS/H70βS VAC variant (Pollegioni et al.2005, 2008b). The latter VAC variant is not inhibited by thereaction product 7-ACA; interestingly, the substitutions ofsuch active site residues in N176 acylase favor the binding(and orientation) of CephC for hydrolysis and do not stabi-lize product binding. Anyway, the increase in product inhi-bition is frequently observed in evolved acylases since largepart of interactions in the enzyme–substrate complex arealso maintained in the ensuing product (see that observedfor SE83 acylase variants, below).
N176 acylase seems a suitable choice for the industrialproduction of cephalosporin derivatives since it was alsoefficiently overexpressed in BL21(DE3)pLysS E. coli cells(Volontè et al. 2008). By alternating screenings of mediumcomponents and factorial experimental designs, ≈1,400 U/gcell and 16,100 U/L of fermentation broth (on Gl-7-ACA asa substrate)—corresponding to ≈10.5 mg/g cell and120 mg/L, respectively—were produced in shake flasks ora 2-L fermentor. The very high expression level and thesingle-step chromatographic purification of the His-taggedrecombinant enzyme make N176 acylase an economicaltool for producing 7-ACA (Volontè et al. 2008).
Very recently, an in-depth investigation of the substratepreference of M31βF and M31βF/H57βS/H70βS VAC var-iants on a number of C7 derivatives of 7-ACA/ADCA (7-desacetoxycephalosporanic acid) or C3 derivatives of Gl-7-ACA/ADCAwas reported (Rosini et al. 2012). These achieve-ments will help us choose the best enzyme for the hydrolysisof different cephalosporin derivatives of industrial interest.
CAD acylase
A model of CephC binding mode to the active site of CADidentified ten residues that were involved in the substratebinding: these positions were the object of mutagenesis (Ohet al. 2003). The Y33βS and Q50βM CAD variants —residues that are located in the side chain pocket— showedimproved activity on CephC (Table 3, numbering as inFig. 3). The latter CAD variant was used as the startingpoint for saturation mutagenesis at positions Y178α, Y33β,and F58β. Y178αK/Q50βM, Y33βD/Q50βM, andQ50βM/F58βG showed the greatest improvement in deacy-lation activity with regard to CephC. A third mutation on theY178αK/Q50βM DNA template was carried out acting onthe M174α, Y33β, and F177β positions: the triple
Y178αK/Q50βM/F177βS, Y178αK/Y33βS/Q50βM, andM174αL/Y178αK//Q50βM CAD variants showed a ≥6-fold increase in activity on CephC as compared to thewild-type (Table 3). Improved deacylation activity on thenatural antibiotic was related to the most favorable accom-modation of the D-α-amino adipyl portion of CephC in thenewly generated binding pocket mainly because of morespace, as in the Y178αK/Q50βM/F177βS variant (Oh etal. 2003). A patent claiming that a novel, improved CADcan be produced based on a combination of substitutions atposition L24β(→R), R85α(→E), Q50β(→A), Q50β(→R),R57β(→H), Y153β(→L), T176β(→D), F177β(→H),Y178α(→A) was filed, but details about the increased prop-erties were lacking (Koller et al. 2002).
SE83 acylase
Two different acylase-encoding genes have been identifiedin Pseudomonas sp. SE83 (Matsuda et al. 1987a, b): acyI
Table 3 Mutagenesis of CAD acylase and effect on CephC acylaseactivity (Oh et al. 2003)
CephC relativeactivity (%)
Wild-type 100
1-point substitution
Y33βS 120
Q50βM 180
2-point substitutions starting from Q50βM
Y178αK/Q50βM 600
Q50βM/Y33βN 350
Q50βM/Y33βD 450
Q50βM/Y33βL 202
Q50βM/F58βS 260
Q50βM/F58βT 230
Q50βM/F58βP 240
Q50βM/F58βG 290
3-point substitutions starting from Y178αK/Q50βM
Y178αK/Q50βM/M174αT 433
Q50βM/Y149αK/M145αP 442
Q50βM/Y149αK/M145αL 704
Y178αK/Q50βM/Y33βT 424
Q50βM/Y149αK/Y33βN 460
Q50βM/Y149αK/Y33βS 568
Y178αK/Q50βM/F177βG 456
Q50βM/Y149αK/F177βS 787
The specific activity of wild-type CAD on CephC as substrate is 2.3 U/mg protein (Li et al. 1999). Here, 100 % is referred to as the activitydetermined at pH 7.0 on ≈24 mM CephC (assay temperature was notstated; Oh et al. 2003)
Numbering as in Fig. 3
Appl Microbiol Biotechnol (2013) 97:2341–2355 2351

and acyII, which do not share sequence similarity with eachother. Both enzymes were reported to be active on Gl-7-ACA, but only AcyII deacylated CephC (≈4 % of activityon the reference substrate Gl-7-ACA).
Site-directed mutagenesis at positions F170α, M31β,F58β, H70β, and I176β (compared to the sequence reportedin Fig. 3; the numbering in this case does not take into accountMet1α) was performed on the acyII gene—encoding for aprotein constituted by an α-subunit of 230 residues (plus aspacer of nine amino acids) and a 535-residue-long β-subunit(Shin et al. 2007). The M31βL/F58βM double variant wasidentified because of ≈20 % increase in activity on CephC:enzymes with three, four, or five substitutions were subse-quently prepared whose specific activity on CephC was in-creased over the previous AcyII generation (Table 4). EP-PCRexperiments further increased the activity on CephC as dem-onstrated for the TnS5β (having six point substitutions) or theS12 variant (with five point substitutions and a specificactivity on CephC of 5.8 U/mg protein; see Table 4). Asignificant decrease in Km for CephC was apparent forF170αY/M31βL/F58βM/H70βS/I176βV: 8 vs. 50 mM forwild-type SE83 acylase.
The 7-ACA can strongly inhibit this acylase (Ki for wild-type SE83 is ≈0.4 mM, a value increased up to 1.8 mM inthe S12 variant). Interestingly, reversion of S70β into theoriginal histidine residue resulted in a lower end-productinhibition.
The S12 variant of SE83 acylase was overexpressed in E.coli BL21(DE3) cells using the pET29 plasmid, reaching1,207 U/L of fermentation broth (activity on CephC wasassayed at pH 8.0 and 37 °C). Bioconversion of 50 mMCephC with 5 U/mL enzyme at 25 °C produced after 1 h60 % of 7-ACA using wild-type SE83 acylase, and 80 and90 % conversion to 7-ACA with the TnS5β and S12 var-iants, respectively (Shin et al. 2007).
Recently, the gene encoding for a SE83 acylase contain-ing six point mutations (Whang et al. 2012), named sAcy(see Table 4), was redesigned and optimized for E. coliexpression (pET28 expression plasmid). A model of thisacylase in complex with CephC suggested that (1) residuesR24β, Y32β, V49β, H57β, V68β, and H70β directlyinteract with CephC by means of hydrogen bonds; (2)residues V49β and V68β form hydrophobic weak interac-tions with atoms C9 and C5 of CephC; (3) residues P22β,H23β, M31β, and P56β should be located close to thestretched D-α-amino adipyl moiety of CephC and mightaffect essential residues, such as Ser1β; and (4) residuesL427β, W434β, A435β, A436β, L437β, and L438β arelocated in the substrate transport tunnel and might influencethe binding of CephC by steric hindrance. Based on thisprediction, residues R24β, M31β, H57β, H70β, P56β,L427β, A436β, and L438β were selected for mutation.The purified SE83 variant containing the additionalA436βG substitution showed increased activity —up to2.6 U/mg protein on ≈24 mM CephC and a Vmax of 12.3±2.2 U/mg protein— but lower efficiency because of a sig-nificant increase in Km for CephC, 105 vs. 50 mM for thewild-type SE83 acylase. Most importantly, optimization ofculture conditions in JM109 E. coli strain (grown at 28 °Cusing a semi-defined medium in which the corn steep liquoris the key nutrient and lactose replaced IPTG as an inducer)made it possible to significantly increase productivity:5,350 U/L (about 450 mg/L) vs. 3,900 U/L with sAcy(Whang et al. 2012). A436βG sAcy variant immobilizedon an epoxy support (LX-1000EP) was used in a single-stepconversion of CephC, giving a 96.7 % yield in terms of the7-ACA produced (30 mM CephC, 5 U/mL, ≈1 h conver-sion; Whang et al. 2012).
Very recently, a recombinant, engineered SE83 acylaseexpressed in E. coli BL21(DE3) using a pET28 plasmid —and characterized by a specific activity on CephC of≈10 U/mg protein and a volumetric fermentation yield of3,000 U/L— was immobilized on the epoxy support LX-1000EP with a ≈34 % recovery in terms of enzyme activity(Zhu et al. 2011). Immobilized SE83 CA showed a good
Table 4 Relative activity of SE83 acyI acylase variants on CephC(Shin et al. 2007; Whang et al. 2012)
CephC relativeactivity (%)
Wild-type 100
Site-directed mutagenesis
M31βL/F58βM 120
M31βL/F58βM/H70βS 520
F170αY/M31βL/F58βM/H70βS 730
F170αY/M31βL/F58βM/H70βS/I176βV 1120
EP-PCR1
G140αS/F170αY/M31βL/F58βM/I176βV 800
V122αA/F170αY/M31βL/F58βM/I176βV 800
V122αA/G140αS/F170αY/M31βL/F58βM/I176βV (TnS5β)
950
EP-PCR2
V122αA/G140αS/F58βN/I176βV 1,900
V122αA/G140αS/F58βN/L175βT/I176βV 1,900
V122αA/G140αS/F58βN/I176βV/S471βC (S12) 1,900
V122αA/G140αS/F58βN/I75βT/I176βV/S471βC 1,900
Site-directed mutagenesis of sAcy
V122αA/G140αS/F58βR/L75βV/I176βV/S471βC (sAcy)
395
V122αA/G140αS/F58βR/I75βV/I176βV/A436βG/S471βC
395
The specific activity determined for the wild-type enzyme at pH 8.0 or8.5 and 37 °C on ≈24 mM CephC was fixed to 100 % (thecorresponding specific activity is 0.65 U/mg protein)
Numbering as in Fig. 3
2352 Appl Microbiol Biotechnol (2013) 97:2341–2355

stability at a pH of between 8.0 and 9.5 and at a temperatureup to 40 °C. At 25 °C and pH 8.5, 96.7 % of 7-ACA wasproduced from 70 mM CephC in 60 min using 5 U/mL ofthe immobilized enzyme: interestingly, no 7-ACA inhibitionwas observed. Indeed, 50 % of the initial activity wasmaintained after 24 cycles.
SY77 acylase
A region and saturation mutagenesis approach performed onGA from Pseudomonas sp. SY-77 identified variants withincreased activity on adipyl-7-ADCA: concerning the kinet-ic efficiency for adipyl-7-ADCA, the F177βC variantshowed a sixfold increase (mainly because of a decreasedKm) and the F177βH variant a 2.4-fold increase (because ofan increase in kcat) with respect to the wild-type enzyme (Sioet al. 2002, 2003). Additional studies from the same groupfurther increased the performance on adipyl-7-ADCA,reaching up to a tenfold improved kinetic efficiency forthe N68βH variant —arising from both a double kcat and asixfold tighter substrate binding (Otten et al. 2002)— and15-fold increase for the N68βM variant, for which kcat wasas high as on Gl-7-ACA (Otten et al. 2004). Although theactivity on CephC was slightly higher for the N68βQ var-iant, and even more so in the Y178αH (2.8-fold) andY178αH/N68βH variants (2.5-fold) than for the wild-type(Sio et al. 2006; Otten et al. 2007), its value was still too lowto be practically employed (i.e., the activity assay required>75 μg of pure protein).
Additional acylases
An alternative CephC acylase was recently obtained bya rational design approach (based on the CAD 3Dstructure and modeling studies) of an acylase fromPseudomonas FERM BO-817 strain (Yoon and Choi2010). A variant containing six point substitutions (i.e.,A129αV/F147αY/L37βM/F55βN/L154βI/L175βV) wasoverexpressed in BL21(DE3) E. coli strain using thepET24 expression plasmid; the recombinant enzyme(used at 19 U/mL) converted 96.5 % of a 50-mMCephC solution at 25 °C and pH 8.5. Unfortunately,no details on the kinetic properties of the wild-typeand evolved enzyme were reported.
Indeed, optimization of the growth conditions wasreported to reach a significant production of a novel CAfrom Micrococcus luteus (Gaurav et al. 2010): approx.10 U/mL of fermentation broth. The kinetic properties ofthis αβ heterodimeric, 80 kDa enzyme on CephC seempromising (Vmax=4.1 U/mg protein, Km,CephC=22 mM), al-though the time of assay employed was unusual for CAs(180 vs. 10 min), suggesting that this report should beconsidered carefully.
Evolution of CA from alternative scaffolds
As an alternative strategy, some authors proposed the engi-neering of PGA, which is also a member of the Ntn hydrolasefamily and consists of two different chains generated byposttranslational processing from an inactive precursor.Indeed, starting from the structure of acylase sp. 130 in com-plex with phosphate, ethylene glycol, and glycerol, a reactionmechanism similar to that of PGAwas proposed (Fritz-Wolf etal. 2002). A thorough site-directed mutagenesis study of PGAidentified a variant containing seven point substitutions(F146αY/F24βL/T32βY/P49βQ/V56βR/W154βY/I177β-Y) and showing 2.1 % of deacylation activity on Gl-7-ACArelatively to wild-type CAD. Unfortunately, the combinationof these mutations did not result in a PGAvariant with CephCdeacetylation activity (Oh et al. 2004).
S im i l a r l y, t h e r e p l a c emen t o f D333 o f γ -glutamyltranspeptidase of E. coli K12 (EC 2.3.2.2) by anasparagine produced an enzyme that deacylated Gl-7-ACAat a rate of 0.12 U/mg protein, but that did not cleave CephC(Suzuki et al. 2004).
Conclusions
β-Lactam antibiotics are still the most widely employedantibiotics in clinical practice, mainly because a large vari-ety of semi-synthetic derivatives of penicillins and cepha-losporins can be synthesized using their starting scaffolds 6-APA and 7-ACA, respectively. Cephalosporin offers moreopportunity to generate new derivatives as it possesses twomajor sites for enzymatic modifications. Furthermore, third-generation, semi-synthetic cephalosporins have remarkablepotency against a wide range of bacteria, includingPseudomonas aeruginosa, a pathogen that does not respondto several antibiotics.
The two-step enzymatic production of 7-ACA fromCephC is a well-established industrial process that has anumber of advantages over the chemical method. It can befurther improved by establishing the single-step enzymaticprocess based on CA. In recent years, protein engineeringstudies have made it possible to substantially improve theefficiency of known GAs on CephC (up to 20-fold for SE83acyI), reaching a significant activity on the natural antibiotic(≈4 and 10 U/mg protein for variants of N176 and SE83acylase, respectively). Nonetheless, these properties are notsufficient to encourage 7-ACA manufacturers to shift to thesingle-step enzymatic conversion of CephC into 7-ACA atthe industrial level. In order to make that a reality in the nearfuture, an integrated approach is required based on micro-biology and bioinformatics (to increase production and toidentify new CAs using sequence homology analyses), pro-tein engineering (to improve their properties vs. CephC and
Appl Microbiol Biotechnol (2013) 97:2341–2355 2353

simultaneously avoid 7-ACA inhibition), immobilization (toproduce reusable and stable enzyme preparations), and re-actor design (to facilitate CephC conversion as well as insitu extraction of the product).
Indeed, fermentative production of cephem nucleus witheasily cleavable adipyl side chains can represent an attrac-tive alternative for the production of 7-ACA.
Acknowledgments We thank the support of Fondo di Ateneo per laRicerca, Centro Grandi Attrezzature (Università dell’Insubria), and Con-sorzio Interuniversitario per le Biotecnologie. The authors are grateful toall members of their laboratory and particularly to Mirella Pilone.
References
Anandan A, Vallet C, Coyle T, Moustafa IM, Vrielink A (2010)Crystallization and preliminary diffraction analysis of an engi-neered cephalosporin acylase. Acta Crystallogr Sect F Struct BiolCryst Commun 66:808–810
Aramori I, Fukagawa M, Tsumura M, Iwami M, Yogota Y, Kojo H,Kohsaka M, Ueda Y, Imanaka H (1991a) Isolation of soil strainsproducing new cephalosporin acylases. J Ferment Bioeng 72:227–231
Aramori I, Fukagawa M, Tsumura M, Iwami M, Isogai T, Ono H,Ishitani Y, Kojo H, Kohsaka M, Ueda Y, Imanaka H (1991b)Cloning and nucleotide sequencing of new glutaryl 7-ACA andcephalosporin-C acylase genes from Pseudomonas strains. JFerment Bioeng 72:232–243
Aramori I, Fukagawa M, Tsumura M, Iwami M, Ono H, Kojo H,Kohsaka M, Ueda Y, Imanaka H (1991c) Cloning and nucleotidesequencing of a novel 7 beta-(4-carboxybutanamido)cephalospor-anic acid acylase gene of Bacillus laterosporus and its expression inEscherichia coli and Bacillus subtilis. J Bacteriol 173:7848–7855
Aramori I, Fukagawa M, Tsumura M, Iwami M, Ono H, Ishitani Y,Kojo H, Kohsaka M, Ueda Y, Imanaka H (1992) Comparativecharacterization of new glutaryl-7-ACA and cephalosporin Cacylases. J Ferment Bioeng 73:185–192
Aretz W, Sauber K (1988) Novel D-amino acid transaminase. Ann NYAcad Sci 542:366–370
Burr KW, Ramsden M, Illing G, Harrison LA, Maishman NJ, SpenceDW (1999) Industrial enzymes. US Patent 5,919,648
Cabri W, Verga R, Cambiaghi S, Bernasconi E (1999) Environmentallyfriendly production of 7-ACA. Chimica e Industria 81:461–464
Crawford MS (1991) One-step cephalosporin C amidase enzyme, agene encoding the same, and expression thereof in a suitable host.EP Patent 0,405,846
Deshpande BS, Ambedkar SS, Shewale JG (1996) Cephalosporin Cacylase and penicillin V acylase formation by Aeromonas sp.ACY 95. World J Microbiol Biotechnol 12:373–378
Duggleby HJ, Tolley SP, Hill CP, Dodson EJ, Dodson G, Moody PC(1995) Penicillin acylase has a single-amino-acid catalytic centre.Nature 373:264–268
Fechtig B, Peter H, Bickel H, Vischer E (1968) Concerning the prep-aration of 7-amino-cephalosporanic acid. Helv Chim Acta51:1108–1119
Fritz-Wolf K, Koller KP, Lange G, Liesum A, Sauber K, Schreuder H,Aretz W, Kabsch W (2002) Structure-based prediction of modifi-cations in glutarylamidase to allow single-step enzymatic produc-tion of 7-aminocephalosporanic acid from cephalosporin C.Protein Sci 11:92–103
Gaurav K, Kundu K, Kundu S (2010) Biosynthesis of cephalosporin-Cacylase enzyme: optimal media design, purification, and
characterization. Artif Cells Blood Substit Immobil Biotechnol38:277–283
Harris CM, Ghisla S, Pollegioni L (2001) pH and kinetic effects in D-amino acid oxidase catalysis. Evidence for a concerted mecha-nism in substrate dehydrogenation via hydride transfer. Eur JBiochem 268:5504–5520
Ishii Y, Saito Y, Fujimura T, Sasaki H, Noguchi Y, Yamada H, Niwa M,Shimomura K (1995) High-level production, chemical modifica-tion and site-directed mutagenesis of a cephalosporin C acylasefrom Pseudomonas strain N176. Eur J Biochem 230:773–778
Kim Y, Hol WG (2001) Structure of cephalosporin acylase in complexwith glutaryl-7-aminocephalosporanic acid and glutarate: insightinto the basis of its substrate specificity. Chem Biol 8:1253–1264
Kim Y, Yoon K, Khang Y, Turley S, Hol WG (2000) The 2.0 A crystalstructure of cephalosporin acylase. Structure 8:1059–1068
Kim Y, Kim S, Earnest TN, Hol WG (2002) Precursor structure ofcephalosporin acylase. Insights into autoproteolytic activation in anew N-terminal hydrolase family. J Biol Chem 277:2823–2829
Kim JK, Yang IS, Rhee S, Dauter Z, Lee YS, Park SS, Kim KH (2003)Crystal structures of glutaryl 7-aminocephalosporanic acid acy-lase: insight into autoproteolytic activation. Biochemistry42:4084–4093
Kim JK, Yang IS, Shin HJ, Cho KJ, Ryu EK, Kim SH, Park SS, KimKH (2006) Insight into autoproteolytic activation from the struc-ture of cephalosporin acylase: a protein with two proteolyticchemistries. Proc Natl Acad Sci U S A 103:1732–1737
Koller KP, Lange G, Sauber K (2002) Mutant glutaryl amidase anduses thereof. WO Patent 02/072806
Lein J (1988) One-step enzymatic conversion of cephalosporin C andderivatives to 7-aminocephalosporanic acid and derivatives. EPPatent 0,283,218
Lein J (1989) One-step enzymatic conversion of cephalosporin C andderivatives to 7-aminocephalosporanic acid and derivatives. EPPatent 0,322,032
Li Y, Chen J, Jiang W, Mao X, Zhao G, Wang E (1999) In vivo post-translational processing and subunit reconstitution of cephalosporinacylase from Pseudomonas sp. 130. Eur J Biochem 262:713–719
Lopez-Gallego F, Batencor L, Hidalgo A, Mateo C, Fernandez-Lafuente R, Guisan JM (2005) One-pot conversion of cephalo-sporin C to 7-aminocephalosporanic acid in the absence of hy-drogen peroxide. Adv Synth Catal 347:1804–1810
Mao X, Wang W, Jiang W, Zhao GP (2004) His23beta and Glu455betaof the Pseudomonas sp. 130 glutaryl-7-amino cephalosporanicacid acylase are crucially important for efficient autoproteolysisand enzymatic catalysis. Protein J 23:197–204
Matsuda A, Matsuyama K, Yamamoto K, Ichikawa S, Komatsu KI(1987a) Cloning and characterization of the genes for two distinctcephalosporin acylases from a Pseudomonas strain. J Bacteriol169:5815–5820
Matsuda A, Toma K, Komatsu KI (1987b) Nucleotide sequences of thegenes for two distinct cephalosporin acylases from aPseudomonas strain. J Bacteriol 169:5821–5829
Morin RB, Jackson BG, Flynn EH, Roseske RW, Andrews SL (1969)Chemistry of cephalosporin antibiotics XIV: reaction of cephalo-sporin C with nitrosyl chloride. J Am Chem Soc 91:1396–1400
Murzin AG, Brenner SE, Hubbard T, Chothia C (1995) SCOP: astructural classification of proteins database for the investigationof sequences and structures. J Mol Biol 247:536–540
Niwa M, Saito Y, Sasaki H, Ishii Y (1994) Cephalosporin C acylase.US Patent 5,336,613
Niwa M, Fujimura T, Ishii Y, Noguchi Y (1998) Cephalosporin Cacylase. US Patent 5,804,429
Oh B, Kim M, Yoon J, Chung K, Shin Y, Lee D, Kim Y (2003)Deacylation activity of cephalosporin acylase to cephalosporinC is improved by changing the side-chain conformations ofactive-site residues. Biochem Biophys Res Commun 310:19–27
2354 Appl Microbiol Biotechnol (2013) 97:2341–2355

Oh B, Kim K, Park J, Yoon J, Han D, Kim Y (2004) Modifying thesubstrate specificity of penicillin G acylase to cephalosporin acy-lase by mutating active-site residues. Biochem Biophys ResCommun 319:486–492
Otten LG, Sio CF, Vrielink J, Cool RH, Quax WJ (2002) Altering thesubstrate specificity of cephalosporin acylase by directed evolu-tion of the beta-subunit. J Biol Chem 277:42121–42127
Otten LG, Sio CF, van der Sloot AM, Cool RH, Quax WJ (2004)Mutational analysis of a key residue in the substrate specificity ofa cephalosporin acylase. ChemBioChem 5:820–825
Otten LG, Sio CF, Reis CR, Koch G, Cool RH, Quax WJ (2007) Ahighly active adipyl-cephalosporin acylase obtained via rationalrandomization. FEBS J 274:5600–5610
Pilone MS, Pollegioni L (2002) D-amino acid oxidase as an industrialbiocatalyst. Biocatal Biotrans 20:145–159
Pilone MS, Pollegioni L (2010) In: Flickinger MC (ed) Encyclopediaof industrial biotechnology: bioprocess, bioseparation and celltechnology. Wiley, New York, pp 1–11
Pilone MS, Buto S, Pollegioni L (1995) A process for bioconversion ofcephalosporin C by Rhodotorula gracilis D-amino acid oxidase.Biotechnol Lett 17:199–204
Pollegioni L, Molla G (2011) New biotech applications from evolvedD-amino acid oxidases. Trends Biotechnol 29:276–283
Pollegioni L, Lorenzi S, Rosini E, Marcone GL, Molla G, Verga R,Cabri W, Pilone MS (2005) Evolution of an acylase active oncephalosporin C. Protein Sci 14:3064–3076
Pollegioni L, Piubelli L, Sacchi S, Pilone MS, Molla G (2007)Physiological functions of D-amino acid oxidases: from yeast tohumans. Cell Mol Life Sci 64:1373–1394
Pollegioni L, Molla G, Sacchi S, Rosini E, Verga R, Pilone MS (2008a)Properties and applications of microbial D-amino acid oxidases:current state and perspectives. Appl Microbiol Biotechnol 78:1–16
Pollegioni L, Pilone M, Molla G, Cucchetti E, Verga R, Cabri W(2008b) Cephalosporin C acylases. US Patent 7,399,624
Rosini E, Monelli CS, Pollegioni L, Riva S, Monti D (2012) On thesubstrate preference of glutaryl acylases. J Mol Catal B: Enzym76:52–58
Saito Y, Fujimura T, Ishii Y, Noguchi Y, Miura T, Niwa M, ShimomuraK (1996) Oxidative modification of a cephalosporin C acylasefrom Pseudomonas strain N176 and site-directed mutagenesis ofthe gene. Appl Environ Microbiol 62:2919–2925
Shin YC, Jeon JYJ, Jung KH, Park MR, Kim Y (2007) CephalosporinC acylase mutant and method for preparing 7-ACA using same.US Patent 0,207,519
Sio CF, Riemens AM, van der Laan JM, Verhaert RM, Quax WJ(2002) Directed evolution of a glutaryl acylase into an adipylacylase. Eur J Biochem 269:4495–4504
Sio CF, Otten LG, Cool RH, Quax WJ (2003) Analysis of a substratespecificity switch residue of cephalosporin acylase. BiochemBiophys Res Commun 312:755–760
Sio CF, Otten LG, Cool RH, Quax WJ (2006) Glutaryl amidases andtheir uses. US Patent 0,292,665
Sonawane VC (2006) Enzymatic modifications of cephalosporins by ceph-alosporin acylase and other enzymes. Crit Rev Biotechnol 26:95–120
Suzuki H, Miwa C, Ishihara S, Kumagai H (2004) A single amino acidsubstitution converts gamma-glutamyltranspeptidase to a class IVcephalosporin acylase (glutaryl-7-aminocephalosporanic acidacylase). Appl Environ Microbiol 70:6324–6328
Tischer W, Giesecke U, Lang G, Röder A, Wedekind F (1992)Biocatalytic 7-aminocephaIosporanic acid production. Ann NYAcad Sci 672:502–509
Volontè F, Marinelli F, Gastaldo L, Sacchi S, Pilone MS, Pollegioni L,Molla G (2008) Optimization of glutaryl-7-aminocephalosporanicacid acylase expression in E. coli. Protein Expr Purif 61:131–137
Volpato G, Rodrigues RC, Fernandez-Lafuente R (2010) Use of enzymesin the production of semi-synthetic penicillins and cephalosporins:drawbacks and perspectives. Curr Med Chem 17:3855–3873
Whang Y, Yu H, Song W, An M, Zhang J, Luo H, Shen Z (2012)Overexpression of synthesized cephalosporin C acylase containingmutations in the substrate transport tunnel. J Biosci Bioeng 113:36–41
Yamada H, Ishii Y, Noguchi Y, Miura T, Mori T, Saito Y (1996) Proteinengineering of a cephalosporin C acylase. Ann NY Acad Sci799:74–81
Yin J, Deng Z, Zhao G, Huang X (2011) The N-terminal nucleophile serineof cephalosporin acylase executes the second autoproteolytic cleavageand acylpeptide hydrolysis. J Biol Chem 286:24476–24486
Yoon JC, Choi SY (2010) Mutation cephalosporin C acylase. KoreanPatent 10-2010-0056123
ZhuX, LuoH, ChangY, SuH, Li Q, YuH, Shen Z (2011) Characteristic ofimmobilizer cephalosporin C acylase and its application in one-stepenzymatic conversion of cephalosporin C to 7-aminocephalosporanicacid. World J Microbial Biotechnol 27:823–829
Appl Microbiol Biotechnol (2013) 97:2341–2355 2355





























