Embed Size (px)
Citation preview

Characterization of the protein ubiquitination responseinduced by DoxorubicinGiorgia Mandili1, Amina Khadjavi1, Valentina Gallo1, Valerio G. Minero2, Luca Bessone1,Franco Carta3, Giuliana Giribaldi1,* and Francesco Turrini1,*
1 Department of Genetics, Biology and Biochemistry, University of Turin, Turin, Italy
2 Department of Experimental Medicine and Oncology, University of Turin, Turin, Italy
3 Nurex SRL, Sassari, Italy
Keywords
cancer; Doxorubicin; neuroblastoma;
proteomics; ubiquitinated proteins
Correspondence
G. Mandili, Department of Genetics, Biology
and Biochemistry, University of Turin, via
Santena 5 bis, 10126 Torino, Italy
Fax: +39 11 6705845
Tel: +39 11 6705850
E-mail: [email protected]
*These authors contributed equally to this
work
(Received 4 January 2012, revised 13 March
2012, accepted 13 April 2012)
doi:10.1111/j.1742-4658.2012.08602.x
Doxorubicin is commonly considered to exert its anti-tumor activity by trig-
gering apoptosis of cancer cells through DNA damage. Recent reports have
shown that Doxorubicin elicits a marked heat shock response, and that either
inhibition or silencing of heat shock proteins enhance the Doxorubicin apop-
totic effect in neuroblastoma cells. In order to investigate whether Doxorubi-
cin may also act through protein modification, we performed a proteomic
analysis of ubiquitinated proteins. Here we show that nanomolar Doxorubicin
treatment of neuroblastoma cells caused: (a) dose-dependent over-ubiquitina-
tion of a specific set of proteins in the absence of measurable inhibition of
proteasome, (b) protein ubiquination patterns similar to those with Bortezo-
mib, a proteasome inhibitor, (c) depletion and loss of activity of ubiquitinated
enzymes such as lactate dehydrogenase and a-enolase, and (d) a decrease
in HSP27 solubility, probably a consequence of its binding to denatured
proteins. These data strongly reinforce the hypothesis that Doxorubicin may
also exert its effect by damaging proteins.
Introduction
Doxorubicin is an anti-tumor agent of the anthracy-
cline family that is widely used in the treatment of
solid tumors, including neuroblastoma (NB). NB is a
neoplasm that originates in the neural crest, and is the
most common extracranial solid tumor in children [1].
Advanced NB is still frequently fatal despite aggressive
management [2]: even though chemosensitive, it usually
recurs and resists further treatment [3]. Doxorubicin is
a well-known topoisomerase II inhibitor and also
inhibits DNA and RNA synthesis and produces single-
stranded DNA breaks [4,5].
Additional evidence suggests extra-nuclear action of
Doxorubicin. It is well-known that Doxorubicin
induces free radical formation [6], lipid peroxidation [7],
membrane damage [8,9], protein carbonylation and
morphological changes in the mitochondria [10]. More-
over, we previously demonstrated that Doxorubicin
treatment elicits a pronounced heat shock response [11].
It is commonly accepted that increased levels of heat
shock proteins HSPs indicate an accumulation of par-
tially denatured proteins [12]. Damaged proteins follow
two possible fates: repair by HSPs, or, if they are irre-
versibly damaged, degradation, mainly through the
ubiquitin–proteasome system [13].
Ubiquitination is a process during which ubiquitin, a
76 amino acid peptide, is covalently conjugated to the
protein substrate [14]. Substrates may be modified by
either a single ubiquitin, multiple single ubiquitins or a
polyubiquitin chain [15]. Ubiquitin conjugation may
occur on multiple lysine residues of the substrate or
Abbreviations
HSP, heat shock protein; LDH, lactate dehydrogenase; NB, neuroblastoma.
2182 FEBS Journal 279 (2012) 2182–2191 ª 2012 The Authors Journal compilation ª 2012 FEBS
ubiquitin itself (K6, K11, K48 and K63). The
polyubiquitin chain formed through the K48 residue of
ubiquitin with four or more ubiquitins is a recognition
signal for the 26S proteasome, and can therefore
mediate the degradation of substrates [14,16,17]. Ubi-
quitination has emerged as a central regulatory mecha-
nism, controlling not only protein stability but also
localization, interactions and functional activity of a
large number of protein substrates [15]. Protein degra-
dation via the ubiquitin–proteasome pathway may play
a critical role in the regulation of cell-cycle progression
and apoptosis [17]; thus, malignant cells, which often
show disruption to cell cycle, have an increased depen-
dency on proteasome-mediated degradation of aberrant
proteins, and consequently are more sensitive to pro-
teasome inhibitors than normal cells [18].
In the present work, we performed a comprehensive
study of the effect of nanomolar concentrations of
Doxorubicin on protein ubiquitination changes. The
protein ubiquitination patterns affected by Doxorubi-
cin displayed a marked resemblance to the patterns
obtained by inhibiting the proteasome using Bortezo-
mib. Mass spectrometry analysis confirmed that the
same proteins were ubiquitinated following Doxorubi-
cin or Bortezomib treatments, leading to the hypothe-
sis that the two drugs affect the same protein targets,
but with different mechanisms. As further evidence
that therapeutic Doxorubicin concentrations cause pro-
found protein modifications and may elicit the
observed heat shock response, the solubility of HSP27
was found to be reduced in a dose-dependent way,
suggesting that it binds to denatured proteins [19].
Results
Effect of Doxorubicin on protein ubiquitination
Given our previous data indicating that Doxorubicin
may induce cellular protein modifications [11], we per-
formed a comprehensive analysis of protein ubiquitina-
tion after treatment of NB cells with Doxorubicin
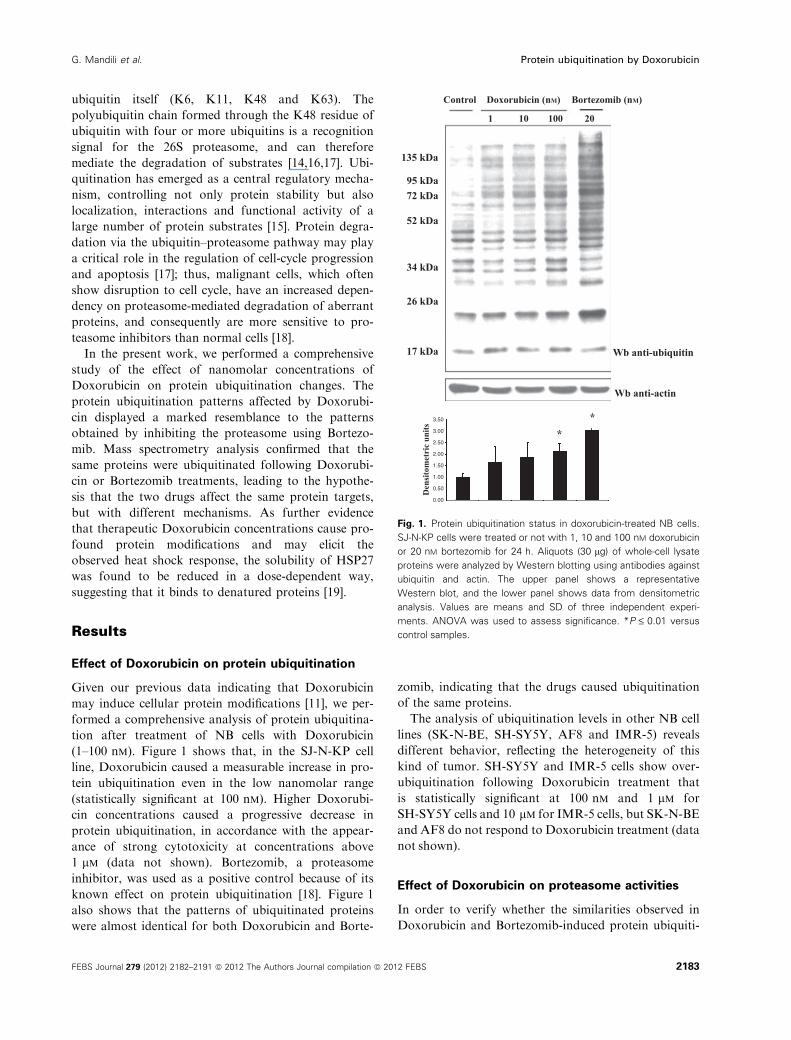
(1–100 nm). Figure 1 shows that, in the SJ-N-KP cell
line, Doxorubicin caused a measurable increase in pro-
tein ubiquitination even in the low nanomolar range
(statistically significant at 100 nm). Higher Doxorubi-
cin concentrations caused a progressive decrease in
protein ubiquitination, in accordance with the appear-
ance of strong cytotoxicity at concentrations above
1 lm (data not shown). Bortezomib, a proteasome
inhibitor, was used as a positive control because of its
known effect on protein ubiquitination [18]. Figure 1
also shows that the patterns of ubiquitinated proteins
were almost identical for both Doxorubicin and Borte-
zomib, indicating that the drugs caused ubiquitination
of the same proteins.
The analysis of ubiquitination levels in other NB cell
lines (SK-N-BE, SH-SY5Y, AF8 and IMR-5) reveals
different behavior, reflecting the heterogeneity of this
kind of tumor. SH-SY5Y and IMR-5 cells show over-
ubiquitination following Doxorubicin treatment that
is statistically significant at 100 nm and 1 lm for
SH-SY5Y cells and 10 lm for IMR-5 cells, but SK-N-BE
and AF8 do not respond to Doxorubicin treatment (data
not shown).
Effect of Doxorubicin on proteasome activities
In order to verify whether the similarities observed in
Doxorubicin and Bortezomib-induced protein ubiquiti-
Control
135 kDa
95 kDa72 kDa
52 kDa
34 kDa
26 kDa
17 kDa
Den
sito
met
ric
units
3.50
2.50
1.50
0.50
3.00
2.00
1.00
0.00
Wb anti-ubiquitin
Wb anti-actin
**
1 10 100 20
Doxorubicin (nM) Bortezomib (nM)
Fig. 1. Protein ubiquitination status in doxorubicin-treated NB cells.
SJ-N-KP cells were treated or not with 1, 10 and 100 nM doxorubicin
or 20 nM bortezomib for 24 h. Aliquots (30 lg) of whole-cell lysate
proteins were analyzed by Western blotting using antibodies against
ubiquitin and actin. The upper panel shows a representative
Western blot, and the lower panel shows data from densitometric
analysis. Values are means and SD of three independent experi-
ments. ANOVA was used to assess significance. *P £ 0.01 versus
control samples.
G. Mandili et al. Protein ubiquitination by Doxorubicin
FEBS Journal 279 (2012) 2182–2191 ª 2012 The Authors Journal compilation ª 2012 FEBS 2183
nation patterns were due to proteasome inhibition, we
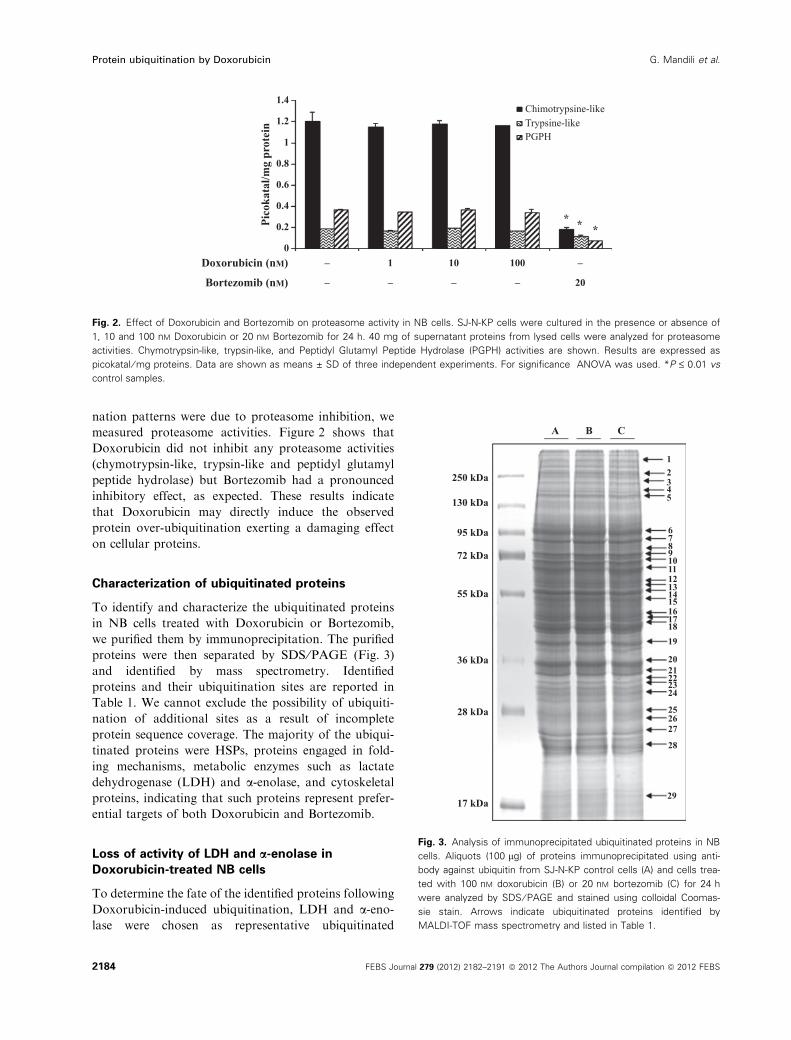
measured proteasome activities. Figure 2 shows that
Doxorubicin did not inhibit any proteasome activities
(chymotrypsin-like, trypsin-like and peptidyl glutamyl
peptide hydrolase) but Bortezomib had a pronounced
inhibitory effect, as expected. These results indicate
that Doxorubicin may directly induce the observed
protein over-ubiquitination exerting a damaging effect
on cellular proteins.
Characterization of ubiquitinated proteins
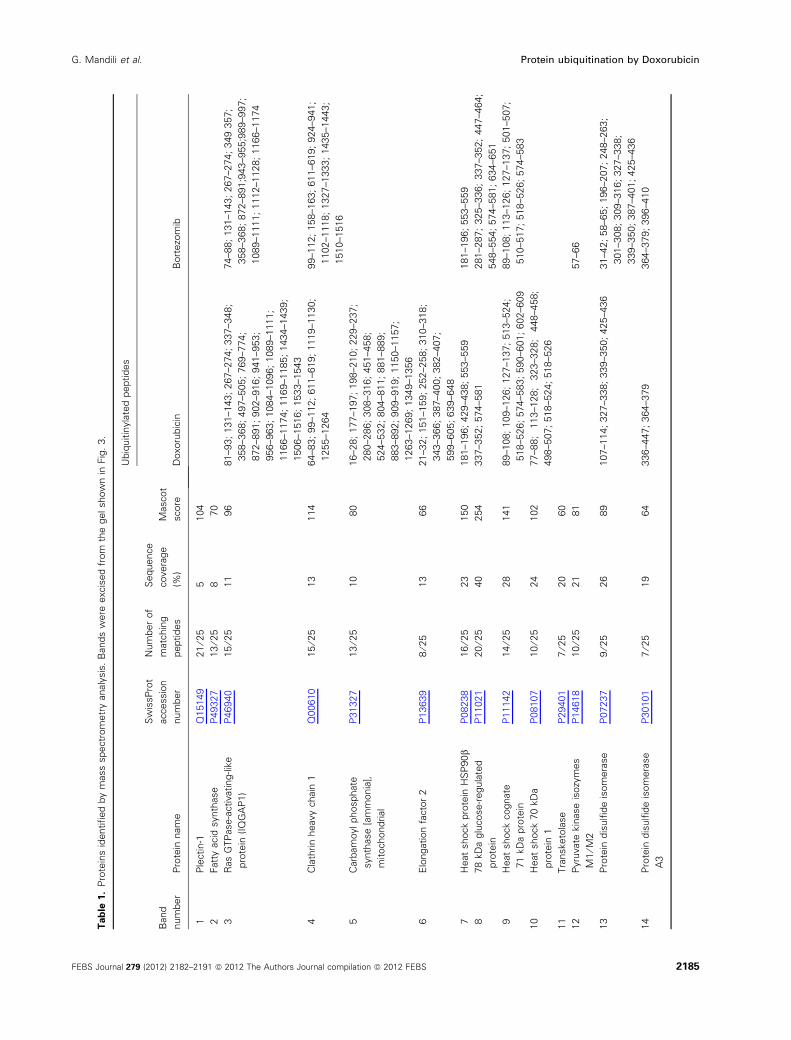
To identify and characterize the ubiquitinated proteins
in NB cells treated with Doxorubicin or Bortezomib,
we purified them by immunoprecipitation. The purified
proteins were then separated by SDS ⁄PAGE (Fig. 3)
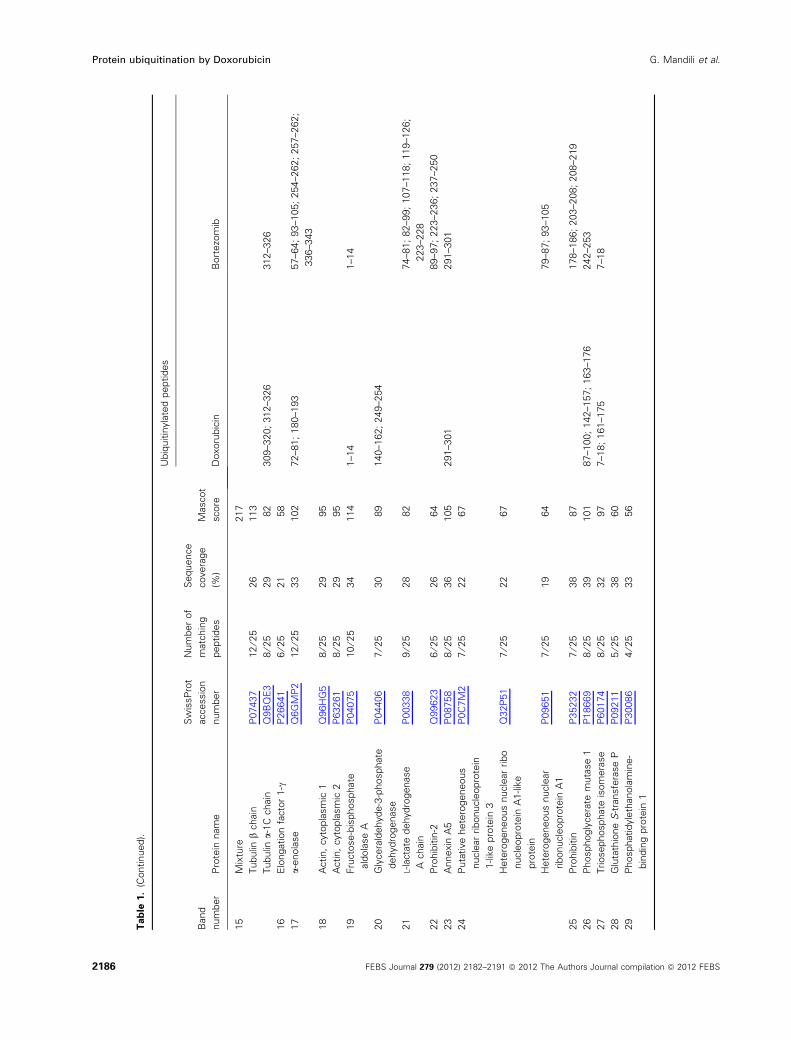
and identified by mass spectrometry. Identified
proteins and their ubiquitination sites are reported in
Table 1. We cannot exclude the possibility of ubiquiti-
nation of additional sites as a result of incomplete
protein sequence coverage. The majority of the ubiqui-
tinated proteins were HSPs, proteins engaged in fold-
ing mechanisms, metabolic enzymes such as lactate
dehydrogenase (LDH) and a-enolase, and cytoskeletal
proteins, indicating that such proteins represent prefer-
ential targets of both Doxorubicin and Bortezomib.
Loss of activity of LDH and a-enolase in
Doxorubicin-treated NB cells
To determine the fate of the identified proteins following
Doxorubicin-induced ubiquitination, LDH and a-eno-lase were chosen as representative ubiquitinated
0
0.2Pico
kata
l/mg
prot
ein
Doxorubicin (nM)
Bortezomib (nM)
–
– – – – 20
1 10 100
* * *
Chimotrypsine-likeTrypsine-likePGPH
–
0.4
0.6
0.8
1
1.2
1.4
Fig. 2. Effect of Doxorubicin and Bortezomib on proteasome activity in NB cells. SJ-N-KP cells were cultured in the presence or absence of
1, 10 and 100 nM Doxorubicin or 20 nM Bortezomib for 24 h. 40 mg of supernatant proteins from lysed cells were analyzed for proteasome
activities. Chymotrypsin-like, trypsin-like, and Peptidyl Glutamyl Peptide Hydrolase (PGPH) activities are shown. Results are expressed as
picokatal ⁄ mg proteins. Data are shown as means ± SD of three independent experiments. For significance ANOVA was used. *P £ 0.01 vs
control samples.
250 kDa
A B C
12345
678910111213141516171819
2021222324
252627
28
29
130 kDa
95 kDa
72 kDa
55 kDa
36 kDa
28 kDa
17 kDa
Fig. 3. Analysis of immunoprecipitated ubiquitinated proteins in NB
cells. Aliquots (100 lg) of proteins immunoprecipitated using anti-
body against ubiquitin from SJ-N-KP control cells (A) and cells trea-
ted with 100 nM doxorubicin (B) or 20 nM bortezomib (C) for 24 h
were analyzed by SDS ⁄ PAGE and stained using colloidal Coomas-
sie stain. Arrows indicate ubiquitinated proteins identified by
MALDI-TOF mass spectrometry and listed in Table 1.
Protein ubiquitination by Doxorubicin G. Mandili et al.
2184 FEBS Journal 279 (2012) 2182–2191 ª 2012 The Authors Journal compilation ª 2012 FEBS
Tab
le1.
Pro
tein
sid
entified
by
mass
spectr
om
etr
yanaly
sis
.B
ands
were
excis
ed
from
the
gelshow
nin
Fig
.3.
Band
num
ber
Pro
tein
nam
e
Sw
issP
rot
accessio
n
num
ber
Num
ber
of
matc
hin
g
peptides
Sequence
covera
ge
(%)
Mascot
score
Ubiq
uitin
yla
ted
peptides
Doxoru
bic
inB
ort
ezo
mib
1P
lectin-1
Q15149
21
⁄25
5104
2Fatt
yacid
synth
ase
P49327
13
⁄25
870
3R
as
GTP
ase-a
ctivating-lik
e
pro
tein
(IQ
GA
P1)
P46940
15
⁄25
11
96
81–93;
131–143;
267–274;
337–348;
358–368;
497–505;
769–774;
872–891;
902–916;
941–953;
956–963;
1084–1096;
1089–1111;
1166–1174;
1169–1185;
1434–1439;
1506–1516;
1533–1543
74–88;
131–143;
267–274;
349
357;
358–368;
872–891;9
43–955;9
89–997;
1089–1111;
1112–1128;
1166–1174
4C
lath
rin
heavy
chain
1Q
00610
15
⁄25
13
114
64–83;
99–112;
611–619;
1119–1130;
1255–1264
99–112;
158–163;
611–619;
924–941;
1102–1118;
1327–1333;
1435–1443;
1510–1516
5C
arb
am
oylphosphate
synth
ase
[am
monia
],
mitochondrial
P31327
13
⁄25
10
80
16–28;
177–197;
198–210;
229–237;
280–286;
308–316;
451–458;
524–532;
804–811;
881–889;
883–892;
909–919;
1150–1157;
1263–1269;
1349–1356
6E
longation
facto
r2
P13639
8⁄2
513
66
21–32;
151–159;
252–258;
310–318;
343–366;
387–400;
382–407;
599–605;
639–648
7H
eat
shock
pro
tein
HS
P90b
P08238
16
⁄25
23
150
181–196;
429–438;
553–559
181–196;
553–559
878
kD
aglu
cose-r
egula
ted
pro
tein
P11021
20
⁄25
40
254
337–352;
574–581
281–287;
325–336;
337–352;
447–464;
548–554;
574–581;
634–651
9H
eat
shock
cognate
71
kD
apro
tein
P11142
14
⁄25
28
141
89–108;
109–126;
127–137;
513–524;
518–526;
574–583;
590–601;
602–609
89–108;
113–126;
127–137;
501–507;
510–517;
518–526;
574–583
10
Heat
shock
70
kD
a
pro
tein
1
P08107
10
⁄25
24
102
77–88;
113–128;
323–328;
448–458;
498–507;
518–524;
518–526
11
Tra
nsketo
lase
P29401
7⁄2
520
60
12
Pyru
vate
kin
ase
isozy
mes
M1
⁄M2
P14618
10
⁄25
21
81
57–66
13
Pro
tein
dis
ulfi
de
isom
era
se
P07237
9⁄2
526
89
107–114;
327–338;
339–350;
425–436
31–42;
58–65;
196–207;
248–263;
301–308;
309–316;
327–338;
339–350;
387–401;
425–436
14
Pro
tein
dis
ulfi
de
isom
era
se
A3
P30101
7⁄2
519
64
336–447;
364–379
364–379;
396–410
G. Mandili et al. Protein ubiquitination by Doxorubicin
FEBS Journal 279 (2012) 2182–2191 ª 2012 The Authors Journal compilation ª 2012 FEBS 2185
Tab
le1.
(Continued).
Band
num
ber
Pro
tein
nam
e
Sw
issP
rot
accessio
n
num
ber
Num
ber
of
matc
hin
g
peptides
Sequence
covera
ge
(%)
Mascot
score
Ubiq
uitin
yla
ted
peptides
Doxoru
bic
inB
ort
ezo
mib
15
Mix
ture
217
Tubulin
bchain
P07437
12
⁄25
26
113
Tubulin
a-1
Cchain
Q9B
QE
38
⁄25
29
82
309–320;
312–326
312–326
16
Elo
ngation
facto
r1-c
P26641
6⁄2
521
58
17
a-enola
se
Q6G
MP
212
⁄25
33
102
72–81;
180–193
57–64;
93–105;
254–262;
257–262;
336–343
18
Actin,
cyto
pla
sm
ic1
Q96H
G5
8⁄2
529
95
Actin,
cyto
pla
sm
ic2
P63261
8⁄2
529
95
19
Fru
cto
se-b
isphosphate
ald
ola
se
A
P04075
10
⁄25
34
114
1–14
1–14
20
Gly
cera
ldehyde-3
-phosphate
dehydro
genase
P04406
7⁄2
530
89
140–162;
249–254
21
L-lacta
tedehydro
genase
Achain
P00338
9⁄2
528
82
74–81;
82–99;
107–118;
119–126;
223–228
22
Pro
hib
itin
-2Q
99623
6⁄2
526
64
89–97;
223–236;
237–250
23
Annexin
A5
P08758
8⁄2
536
105
291–301
291–301
24
Puta
tive
hete
rogeneous
nucle
ar
ribonucle
opro
tein
1-lik
epro
tein
3
P0C
7M
27
⁄25
22
67
Hete
rogeneous
nucle
ar
ribo
nucle
opro
tein
A1-lik
e
pro
tein
Q32P
51
7⁄2
522
67
Hete
rogeneous
nucle
ar
ribonucle
opro
tein
A1
P09651
7⁄2
519
64
79–87;
93–105
25
Pro
hib
itin
P35232
7⁄2
538
87
178–186;
203–208;
208–219
26
Phosphogly
cera
tem
uta
se
1P
18669
8⁄2
539
101
87–100;
142–157;
163–176
242–253
27
Triosephosphate
isom
era
se
P60174
8⁄2
532
97
7–18;
161–175
7–18
28
Glu
tath
ione
S-t
ransfe
rase
PP
09211
5⁄2
538
60
29
Phosphatidyle
thanola
min
e-
bin
din
gpro
tein
1
P30086
4⁄2
533
56
Protein ubiquitination by Doxorubicin G. Mandili et al.
2186 FEBS Journal 279 (2012) 2182–2191 ª 2012 The Authors Journal compilation ª 2012 FEBS
enzymes, and their activities were measured. Figure 4A
shows a loss of intracellular LDH activity following
Doxorubicin treatment, but extracellular LDH activity
was not impaired, demonstrating no evident cytotoxicity
of this Doxorubicin dose (data not shown). Similarly,
Doxorubicin caused a loss of a-enolase activity
(Fig. 4B). These data confirm that ubiquitination of the
two enzymes induced by Doxorubicin leads to their
impairment, suggesting possible protein damage.
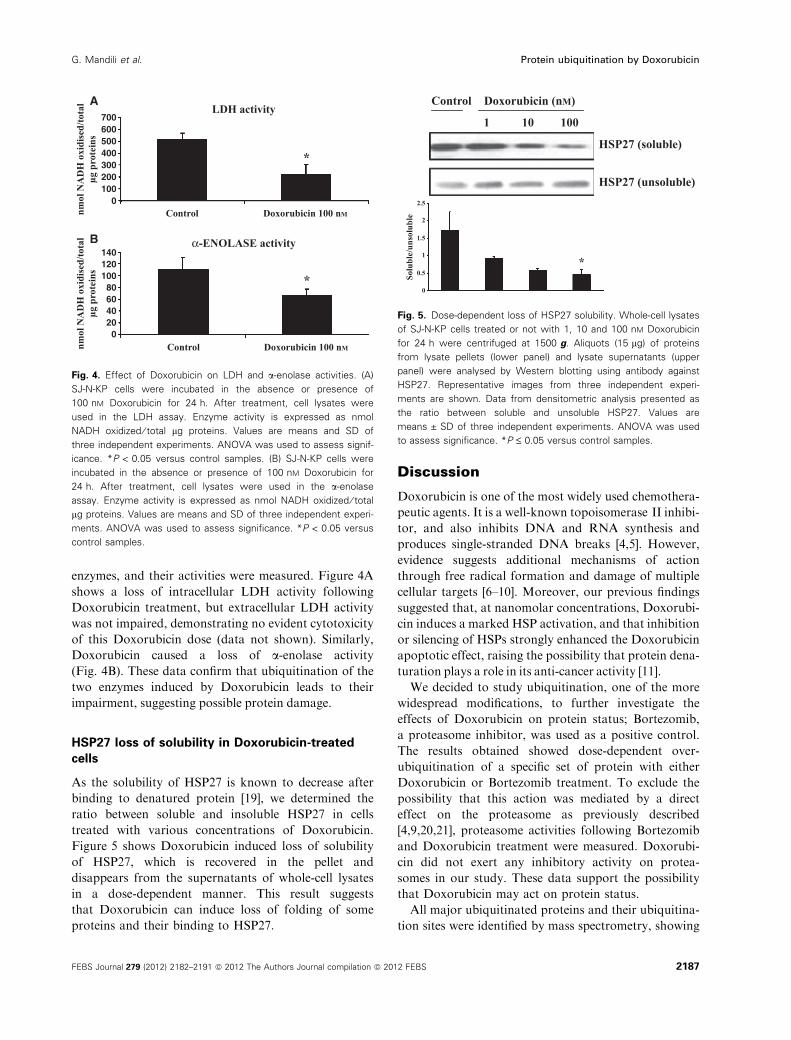
HSP27 loss of solubility in Doxorubicin-treated
cells
As the solubility of HSP27 is known to decrease after
binding to denatured protein [19], we determined the
ratio between soluble and insoluble HSP27 in cells
treated with various concentrations of Doxorubicin.
Figure 5 shows Doxorubicin induced loss of solubility
of HSP27, which is recovered in the pellet and
disappears from the supernatants of whole-cell lysates
in a dose-dependent manner. This result suggests
that Doxorubicin can induce loss of folding of some
proteins and their binding to HSP27.
Discussion
Doxorubicin is one of the most widely used chemothera-
peutic agents. It is a well-known topoisomerase II inhibi-
tor, and also inhibits DNA and RNA synthesis and
produces single-stranded DNA breaks [4,5]. However,
evidence suggests additional mechanisms of action
through free radical formation and damage of multiple
cellular targets [6–10]. Moreover, our previous findings
suggested that, at nanomolar concentrations, Doxorubi-
cin induces a marked HSP activation, and that inhibition
or silencing of HSPs strongly enhanced the Doxorubicin
apoptotic effect, raising the possibility that protein dena-
turation plays a role in its anti-cancer activity [11].
We decided to study ubiquitination, one of the more
widespread modifications, to further investigate the
effects of Doxorubicin on protein status; Bortezomib,
a proteasome inhibitor, was used as a positive control.
The results obtained showed dose-dependent over-
ubiquitination of a specific set of protein with either
Doxorubicin or Bortezomib treatment. To exclude the
possibility that this action was mediated by a direct
effect on the proteasome as previously described
[4,9,20,21], proteasome activities following Bortezomib
and Doxorubicin treatment were measured. Doxorubi-
cin did not exert any inhibitory activity on protea-
somes in our study. These data support the possibility
that Doxorubicin may act on protein status.
All major ubiquitinated proteins and their ubiquitina-
tion sites were identified by mass spectrometry, showing
Control
*
*
LDH activity
α-ENOLASE activity
Doxorubicin 100 nM
Control Doxorubicin 100 nM
nmol
NA
DH
oxi
dise
d/to
tal
μg p
rote
ins
nmol
NA
DH
oxi
dise
d/to
tal
μg p
rote
ins
020406080
100120140
100200300400500600
0
700
A
B
Fig. 4. Effect of Doxorubicin on LDH and a-enolase activities. (A)
SJ-N-KP cells were incubated in the absence or presence of
100 nM Doxorubicin for 24 h. After treatment, cell lysates were
used in the LDH assay. Enzyme activity is expressed as nmol
NADH oxidized ⁄ total lg proteins. Values are means and SD of
three independent experiments. ANOVA was used to assess signif-
icance. *P < 0.05 versus control samples. (B) SJ-N-KP cells were
incubated in the absence or presence of 100 nM Doxorubicin for
24 h. After treatment, cell lysates were used in the a-enolase
assay. Enzyme activity is expressed as nmol NADH oxidized ⁄ total
lg proteins. Values are means and SD of three independent experi-
ments. ANOVA was used to assess significance. *P < 0.05 versus
control samples.
Control Doxorubicin (nM)
1 10
0
0.5Solu
ble/
unso
lubl
e
1
2
2.5
1.5
*
100
HSP27 (soluble)
HSP27 (unsoluble)
Fig. 5. Dose-dependent loss of HSP27 solubility. Whole-cell lysates
of SJ-N-KP cells treated or not with 1, 10 and 100 nM Doxorubicin
for 24 h were centrifuged at 1500 g. Aliquots (15 lg) of proteins
from lysate pellets (lower panel) and lysate supernatants (upper
panel) were analysed by Western blotting using antibody against
HSP27. Representative images from three independent experi-
ments are shown. Data from densitometric analysis presented as
the ratio between soluble and unsoluble HSP27. Values are
means ± SD of three independent experiments. ANOVA was used
to assess significance. *P £ 0.05 versus control samples.
G. Mandili et al. Protein ubiquitination by Doxorubicin
FEBS Journal 279 (2012) 2182–2191 ª 2012 The Authors Journal compilation ª 2012 FEBS 2187

a remarkable correspondence between over-ubiquitinat-
ed proteins after Doxorubicin or Bortezomib treat-
ments. The finding that both Bortezomib and
Doxorubicin treatments also induce over-expression of
HSP27 is in accordance with the accumulation of similar
patterns of modified proteins [11,22], and demonstrates
that Bortezomib and Doxorubicin act on the same tar-
gets, although through different mechanisms. Moreover,
this result suggests that these protein targets are more
likely to be ubiquitinated than other proteins. Most of
the identified proteins were metabolic enzymes, HSPs
and various proteins engaged in the folding of client
proteins. In addition to chaperone activity, HSPs have
been shown to facilitate the degradation of highly mis-
folded proteins by transferring them to the ubiquitin–
proteasome degradation system [23]. In particular,
HSP90b, the heat shock cognate 71 kDa protein and
heat shock 70 kDa protein are known to interact with
the ubiquitin ligase CHIP (carboxy terminus of HSP70-
interacting protein). Following ubiquitination by CHIP,
they cooperate in substrate targeting to the proteasome
[23–26]. A link between GRP78 (78 kDa glucose-regu-
lated protein) and the ubiquitination system has also
been proposed recently [27]. Among the identified pro-
teins, we also found actin and glyceraldehyde-3-phos-
phate dehydrogenase, which are degraded in a heat
shock cognate 71 kDa protein-dependent way [28].
To determine the effect of proteasome-independent
ubiquitination induced by Doxorubicin on protein
functionality, the fate of two of the identified ubiquiti-
nated proteins was monitored: LDH and a-enolase.Interestingly, consistent loss of activity of both
enzymes was observed following Doxorubicin treat-
ment, and this observation reinforces the idea of a
damaging effect of Doxorubicin even though we can-
not exclude a different significance for ubiquitination
induced by Doxorubicin in other proteins.
Finally, as HSP27 was one of most interesting HSPs
in our cellular system [11], and is known to link mis-
folded proteins [19], its solubility following Doxorubi-
cin treatment was investigated. The increased loss of
solubility with increasing Doxorubicin dose supports
the possibility that Doxorubicin may act on protein
folding, and that HSP27 may precipitate as a conse-
quence of binding to unfolded proteins.
In conclusion, our results provide the first evidence
that Doxorubicin, in the low nanomolar range, induces
a complex protein ubiquitination response without
inhibition of proteasome activities. They support the
hypothesis that Doxorubicin, in addition to its action
on nucleic acids, may exert its effect through intracel-
lular accumulation of damaged proteins, even though
the mechanism is not yet fully understood.
A very recent study [29] demonstrating that inducers
of protein misfolding such as hypertemia or Puromycin,
in combination with the proteasome inhibitor Bortezo-
mib, have a proteotoxic effect that is correlated with
increased cytotoxicity may provide support for a clinical
application of our data. Indeed, it has been demon-
strated that combined treatment of normal and chemo-
resistant NB cell lines using Bortezomib and other
chemotherapeutic agents, including Doxorubicin, has a
synergistic effect on the cells, considerably decreasing
the half-maximal effective concentration of Doxorubi-
cin, and combined treatment appears to be a promising
tool for a clinical trial [30]. Moreover, it was also dem-
onstrated by Zanini et al. [11] that combined treatment
with Quercetin and Doxorubicin causes sensitization of
NB cells at nanomolar concentrations of Doxorubicin,
suggesting a clinical interest. A better understanding of
the mechanism underlying the observed synergy may
improve NB treatment strategies in the future by provid-
ing a rationale for the design of novel therapeutic com-
binations with the aim of decreasing the effective dosage
of anticancer drugs and preventing ⁄overcoming the
development of drug resistance.
Experimental procedures
Materials
All materials were obtained from Sigma-Aldrich (St Louis,
MO, USA) unless otherwise stated. Doxorubicin was
obtained from Pharmacia (Milan, Italy); synthetic fluorogenic
peptide substrate Bz-VGR-AMC and monoclonal antibody
against ubiquitin were obtained from Biomol (Plymouth
Meeting, PA, USA); the DC protein assay kit, acrylamide and
enhanced chemiluminescence kit were obtained from Bio-Rad
(Hercules, CA, USA); fetal bovine serum (USA origin) was
obtained from EuroClone (Paignton, UK); the poly(vinyli-
dene fluoride) membrane Immobilon-P was obtained from
Millipore (Milan, Italy); antibodies against HSP27 and ubi-
quitin (polyclonal antibody) were obtained from Stressgen
(Ann Arbor, MI, USA); antibody against actin was obtained
from Santa Cruz Biotechnology (Santa Cruz, CA, USA);
horseradish-peroxidase labeled antibodies were obtained
fromGEHealthcare Bio-Sciences (Milan, Italy).
Cell cultures and treatments
The established NB cell line SJ-N-KP [31] was cultured at
37 �C, 5% CO2 in RPMI-1640 supplemented with 10%
heat-inactivated fetal bovine serum (USA origin),
100 lgÆmL)1 Streptomycin and 100 UÆmL)1 Penicillin. All
experiments were performed using sub-confluent cells. For
Western blotting experiments and measurement of protea-
some activities, cells were cultured in the presence or
Protein ubiquitination by Doxorubicin G. Mandili et al.
2188 FEBS Journal 279 (2012) 2182–2191 ª 2012 The Authors Journal compilation ª 2012 FEBS

absence of 1, 10 or 100 nm Doxorubicin or 20 nm Bortezo-
mib (a generous gift from Molinette Hospital, Turin, Italy)
for 24 h. For immunoprecipitation experiments, cells were
cultured in the presence or absence of 100 nm Doxorubicin
or 20 nm Bortezomib for 24 h. For measurement of lactate
dehydrogenase and a-enolase activities, cells were cultured
in the presence or absence of 100 nm Doxorubicin for 24 h.
Western blotting assays
SJ-N-KP cells were washed twice for few seconds in NaCl ⁄Pi
and solubilized in Laemmli buffer [32] containing protease
inhibitors and nuclease. After centrifugation at 17 000 g for
15 min at 4 �C, proteins were quantified using a DC protein
assay kit and supplemented with 60 mm dithiothreitol and
0.01% bromophenol blue. Lysates containing equal amounts
of proteins (30 lg) were subjected to SDS ⁄PAGE (10% gel)
using the Mini PROTEAN system (Bio-Rad).
The separated proteins were transferred to an Immo-bilon-
P membrane using a Bio-Rad transfer cell unit according to
the manufacturer’s instructions. The blot was blocked using
5% w ⁄ v BSA in NaCl ⁄Pi containing 0.1% Tween-20, and
probed using polyclonal rabbit antibody against HSP27
(diluted 1 : 5000), polyclonal rabbit antibody against ubiqui-
tin (diluted 1 : 5000) or polyclonal goat antibody against
actin (diluted 1 : 100) overnight at 4 �C. After washing using
NaCl/P containing 0.1% Tween-20 for 35 min, the blot was
incubated for 1 h with horseradish-peroxidase labeled anti-
bodies against rabbit or goat IgG (diluted 1 : 5000 and
1 : 20 000, respectively), and immunoreactivity was detected
using an enhanced chemilumine-scence kit (Bio-Rad).
Measurement of proteasome activities
Proteasome activities in SJ-N-KP cells were determined by
cleavage of specific fluorogenic substrates as previously
described [33]. Briefly, SJ-N-KP cells were washed twice with
NaCl ⁄Pi, solubilized in lysis buffer (20 mm Tris ⁄HCl, pH
7.2, containing 0.1 mm EDTA, 1 mm 2-mercaptoethanol,
5 mm ATP, 20% glycerol and 0.04% v ⁄ v Nonidet P-40) and
centrifuged at 17 000 g for 15 min at 4 �C, and the superna-
tant was collected. Protein concentration was determined
using a DC protein assay kit. Aliquots corresponding to
40 lg of proteins were incubated for 1 h at 37 �C in reaction
buffer (50 mm HEPES, pH 8.0, 5 mm EGTA) with the syn-
thetic fluorogenic peptide substrates Suc-LLVY-AMC, Bz-
VGR-AMC and Z-LLE-AMC at 40 lm concentrations, to
measure chymotrypsin-like, trypsin-like and peptidyl glutam-
yl peptide hydrolase activities, respectively. Following incu-
bation, the reaction was blocked using 0.01% ice-cold
trichloroacetic acid, and fluorescence was measured at an
excitation wavelength of 380 nm and an emission wavelength
of 460 nm, and read using an LS 55 spectrofluorometer (Per-
kin-Elmer, Waltham, MA, USA). Free aminomethyl couma-
rin (AMC) was used as a working standard.
Immunoprecipitation assays
To immunoprecipitate ubiquitinated proteins, cells were
washed twice with NaCl ⁄Pi and lysed in modified radioim-
munoprecipitation buffer (154 mm NaCl, 65 mm Tris ⁄HCl,
pH 7.4, 1% w ⁄ v Nonidet P-40, 0.25% w ⁄ v sodium deoxy-
cholate, 1 mm EDTA) containing protease inhibitors and
nuclease. Aliquots (10 mg) of whole-cell lysate were pre-
cleared using Protein A–agarose beads (3 mgÆmL)1) for 1 h
at 4 �C with rotation, and incubated with monoclonal
mouse antibody against ubiquitin (100 lg per sample) over-
night at 4 �C with rotation. Protein A–agarose beads were
then added for 3 h at 4 �C with rotation. Pellets were
washed three times with ice-cold radioimmunoprecipitation
buffer, solubilized in Laemmli buffer, and protein concen-
tration quantified using a DC protein assay kit. For protein
identification by mass spectrometry, 100 lg of immunopre-
cipitated proteins were run in a 10% polyacrylamide SDS
gel (big format gel, 18 · 20 · 0.1 cm) using a Bio-Rad XI
cell, and stained using colloidal Coomassie stain (18% v ⁄ vethanol, 15% w ⁄ v ammonium sulfate, 2% v ⁄ v phosphoric
acid, 0.2% w ⁄ v Coomassie G-250) for 48 h.
MS analysis and peptide mass fingerprinting
Gel slices from Coomassie-stained gels were excised and
destained by several washes in 50% v ⁄ v acetonitrile in 5 mm
NH4HCO3, and successively dried using pure acetonitrile.
The gel slices were rehydrated for 45 min at 4 �C in 20 lL of
5 mm NH4HCO3 digestion buffer containing 10 ngÆlL)1
trypsin. Excess protease solution was then removed, and the
volume was adjusted using 5 mm NH4HCO3 to cover the gel
slices. Digestion was allowed to proceed overnight at 37 �C[34]. MS analysis of peptides was performed using a
MALDI-TOF spectrometer (MALDI micro MX; Waters,
Milford, MA, USA) equipped with a delayed extraction unit,
according to the tuning procedures suggested by the manu-
facturer, operating on reflectron mode. Samples were loaded
onto the MALDI target using 1.5 lL of the tryptic digest
mixed 1 : 1 with a solution of a-cyanohydroxycinnamic acid
(10 mgÆmL)1) in 40% v ⁄ v acetonitrile, 60% v ⁄ v trifluoro-
acetic acid 0.1%. Peak lists were generated by ProteinLynx
(Waters, Milford, MA, USA) data preparation using the fol-
lowing parameters: external calibration with lock mass using
a mass of 2465.1989 Da for ACTH (adrenocorticotropic hor-
mone), background subtract type adaptive combining all
scans, and de-isotoping with a threshold of 1%. The 25 most
intense masses were used for database searches against the
SWISSPROT database using the free search program MAS-
COT (http://www.matrixscience.com). Peak lists created as
above described were used for a ProteinLynx Global Server
2.2.5 search using the SWISSPROT database (downloaded
from ftp://ftp.ncbi.nlm.nih.gov/blast/db/FASTA/; release
2011_08, 27 July 2011) and used for identification of ubiquiti-
nated sequences. In all cases, the search settings allowed one
G. Mandili et al. Protein ubiquitination by Doxorubicin
FEBS Journal 279 (2012) 2182–2191 ª 2012 The Authors Journal compilation ª 2012 FEBS 2189

missed cleavage with the trypsin enzyme selected, oxidation
of methionine as a potential variable modification, a peptide
tolerance of 100 ppm, taxon human. In the case of Protein-
Lynx Global Server searches, ubiquitination as potential var-
iable modification was added: peptides containing an
ubiquitinated site show an increase in molecular mass of
114 Da for each targeted lysine residue as trypsin digestion
cannot occur at the modified lysine and tandem glycines are
conjugated to the target lysine residue [14].
Enzymatic assays
Cell culture lysates were used in the LDH (lactate deydrogen-
ase) assay, as previously described [35], and in the
a-enolase assay (cells were lysed in 1 m Tris ⁄HCl, pH 8, and
mixed with 10 mm MgCl2, 1 m KCl, 100 lm 2-phosphogly-
ceric acid, 4 mm ADP, 6.8 UÆmL)1 pyruvate kinase,
9.9 UÆmL)1 LDH, 200 lm NADH). Activities for both
enzymes, measured spectrophotometrically using a Packard
EL340 microplate reader (Bio-Tek Instruments, Winooski,
VT, USA) as absorbance variation at 340 nm at 37 �C, wereexpressed as nmol NADH oxidized per total lg proteins.
Measurement of HSP27 solubility
SJ-N-KP cells were washed twice in NaCl ⁄Pi and solubi-
lized in radioimmunoprecipitation buffer containing prote-
ase inhibitors and nuclease. Whole-cell lysates were then
centrifuged at 1500 g for 10 min, and the protein content
of obtained pellets and supernatants was quantified using a
DC protein assay kit. Proteins (15 lg) from the pellet or
supernatant were then added to Laemmli buffer supple-
mented with 60 mm dithiothreitol and 0.01% w ⁄ v brom-
ophenol blue and subjected to SDS ⁄PAGE (12% gel) using
the Mini PROTEAN system (Bio-Rad) and Western blot-
ting with antibody against HSP27 (see above).
Statistical analysis
Experimental data are presented as means ± standard devi-
ation (SD) of three independent experiments. Data signi
ficance was determined by one-way analysis of variance
(ANOVA).
Acknowledgements
This work was supported by the ‘Oncology Special
Project’, Compagnia di San Paolo ⁄FIRMS (Fondazione
Internazionale di Ricerca inMedicina Sperimentale), and
by the ‘Ricerca Sanitaria Finalizzata’, Piedmont Region
(Italy). The authors thank Dr Mauro Prato (Department
of Genetics, Biology and Biochemistry, University of
Turin, Turin, Italy) andDrAndrea Bonetto (Department
of Cancer and Biology, Kimmel Cancer Center, Thomas
Jefferson University, Philadelphia, PA, USA) for critical
reading of themanuscript.
References
1 Brignole C, Marimpietri D, Pastorino F, Nico B,
Di Paolo D, Cioni M, Piccardi F, Cilli M, Pezzolo A,
Corrias M et al. (2006) Effect of bortezomib on human
neuroblastoma cell growth, apoptosis, and angiogenesis.
J Natl Cancer Inst 98, 1142–1157.
2 Blanc E, Goldschneider D, Ferrandis E, Barrois M, Le
Roux G, Leonce S, Douc-Rasy S, Benard J & Raguenez
G (2003) MYCN enhances P-gp ⁄MDR1 gene expression
in the human metastatic neuroblastoma IGR-N-91
model. Am J Pathol 163, 321–331.
3 Ho R, Eggert A, Hishiki T, Minturn J, Ikegaki N,
Foster P, Camoratto A, Evans A & Brodeur G (2002)
Resistance to chemotherapy mediated by TrkB in
neuroblastomas. Cancer Res 62, 6462–6466.
4 LoConte N, Thomas J, Alberti D, Heideman J, Binger
K, Marnocha R, Utecht K, Geiger P, Eickhoff J,
Wilding G et al. (2008) A phase I pharmacodynamic
trial of bortezomib in combination with doxorubicin in
patients with advanced cancer. Cancer Chemother
Pharmacol 63, 109–115.
5 Dees E, O’Neil B, Lindley C, Collichio F, Carey L,
Collins J, Riordan W, Ivanova A, Esseltine D &
Orlowski R (2008) A phase I and pharmacologic
study of the combination of bortezomib and
pegylated liposomal doxorubicin in patients with
refractory solid tumors. Cancer Chemother Pharmacol
63, 99–107.
6 Cummings J, Willmott N, Hoey B, Marley E & Smyth
J (1992) The consequences of doxorubicin quinone
reduction in vivo in tumour tissue. Biochem Pharmacol
44, 2165–2174.
7 Benchekroun M, Pourquier P, Schott B & Robert J
(1993) doxorubicin-induced lipid peroxidation and gluta-
thione peroxidase activity in tumor cell lines selected for
resistance to doxorubicin. Eur J Biochem 211, 141–146.
8 Huang R, Kowalski D, Minderman H, Gandhi N &
Johnson E (2007) Small ubiquitin-related modifier
pathway is a major determinant of doxorubicin
cytotoxicity in Saccharomyces cerevisiae. Cancer Res 67,
765–772.
9 Yamamoto Y, Hoshino Y, Ito T, Nariai T, Mohri T,
Obana M, Hayata N, Uozumi Y, Maeda M, Fujio Y
et al. (2008) Atrogin-1 ubiquitin ligase is upregulated by
doxorubicin via p38-MAP kinase in cardiac myocytes.
Cardiovasc Res 79, 89–96.
10 Liu L, Zhang X, Qian B,Min X&Cheng Y (2008) Heat
shock protein 27 attenuated doxorubicin-induced
myocardial damage by reducing cardiomyocyte apoptosis,
mitochondria damage and protein carbonylation.
Protein ubiquitination by Doxorubicin G. Mandili et al.
2190 FEBS Journal 279 (2012) 2182–2191 ª 2012 The Authors Journal compilation ª 2012 FEBS

Zhonghua Xin Xue Guan Bing Za Zhi 36, 1021–1026 (in
Chinese).
11 Zanini C, Giribaldi G, Mandili G, Carta F, Crescenzio
N, Bisaro B, Doria A, Foglia L, di Montezemolo L,
Timeus F et al. (2007) Inhibition of heat shock proteins
(HSP) expression by quercetin and differential doxoru-
bicin sensitization in neuroblastoma and Ewing’s
sarcoma cell lines. J Neurochem 103, 1344–1354.
12 Mosser D & Morimoto R (2004) Molecular chaperones
and the stress of oncogenesis. Oncogene 23, 2907–2918.
13 Murata S, Chiba T & Tanaka K (2003) CHIP: a
quality-control E3 ligase collaborating with molecular
chaperones. Int J Biochem Cell Biol 35, 572–578.
14 Tan F, Lu L, Cai Y, Wang J, Xie Y, Wang L, Gong Y,
Xu B, Wu J, Luo Y et al. (2008) Proteomic analysis of
ubiquitinated proteins in normal hepatocyte cell line
Chang liver cells. Proteomics 8, 2885–2896.
15 Kirkpatrick DS, Denison C & Gygi SP (2005) Weighing
in on ubiquitin: the expanding role of mass-spectrome-
try-based proteomics. Nat Cell Biol 7, 750–757.
16 LayfieldR, ToothD, LandonM,Dawson S,Mayer J&
AlbanA (2001) Purification of poly-ubiquitinated
proteins by S5a-affinity chromatography.Proteomics 1,
773–777.
17 Khan T, Stauffer J, Williams R, Hixon J, Salcedo R,
Lincoln E, Back T, Powell D, Lockett S, Arnold A
et al. (2006) Proteasome inhibition to maximize the
apoptotic potential of cytokine therapy for murine
neuroblastoma tumors. J Immunol 176, 6302–6312.
18 Bersani F, Taulli R,AccorneroP,MorottiA,Miretti S,
Crepaldi T&PonzettoC (2008)Bortezomib-mediated pro-
teasome inhibition as a potential strategy for the treatment
of rhabdomyosarcoma.Eur JCancer 44, 876–884.
19 Rogalla T, Ehrnsperger M, Preville X, Kotlyarov A,
Lutsch G, Ducasse C, Paul C, Wieske M, Arrigo A,
Buchner J et al. (1999) Regulation of Hsp27 oligomeri-
zation, chaperone function, and protective activity
against oxidative stress ⁄ tumor necrosis factor a by
phosphorylation. J Biol Chem 274, 18947–18956.
20 Bar-On O, Shapira M & Hershko DD (2007) Differen-
tial effects of doxorubicin treatment on cell cycle arrest
and Skp2 expression in breast cancer cells. Anticancer
Drugs 18, 1113–1121.
21 Fekete MR, McBride WH & Pajonk F (2005) Anthra-
cyclines, proteasome activity and multi-drug-resistance.
BMC Cancer 5, 114.
22 Mitsiades N, Mitsiades CS, Poulaki V, Chauhan D,
Fanourakis G, Gu X, Bailey C, Joseph M, Libermann
TA, Treon SP et al. (2002) Molecular sequelae of
proteasome inhibition in human multiple myeloma cells.
Proc Natl Acad Sci USA 99, 14374–14379.
23 Shin Y, Klucken J, Patterson C, Hyman B & McLean
P (2005) The co-chaperone carboxyl terminus of Hsp70-
interacting protein (CHIP) mediates a-synuclein degra-
dation decisions between proteasomal and lysosomal
pathways. J Biol Chem 280, 23727–23734.
24 McDonough H & Patterson C (2003) CHIP: a link
between the chaperone and proteasome systems. Cell
Stress Chaperones 8, 303–308.
25 Qian S, McDonough H, Boellmann F, Cyr D & Patter-
son C (2006) CHIP-mediated stress recovery by sequen-
tial ubiquitination of substrates and Hsp70. Nature 440,
551–555.
26 Kumar P, Ambasta R, Veereshwarayya V, Rosen K,
Kosik K, Band H, Mestril R, Patterson C & Querfurth
H (2007) CHIP and HSPs interact with b-APP in a
proteasome-dependent manner and influence Abmetabolism. Hum Mol Genet 16, 848–864.
27 Wang M, Ye R, Barron E, Baumeister P, Mao C, Luo
S, Fu Y, Luo B, Dubeau L, Hinton D et al. (2010)
Essential role of the unfolded protein response regulator
GRP78 ⁄BiP in protection from neuronal apoptosis. Cell
Death Differ 17, 488–498.
28 Garrido C, Brunet M, Didelot C, Zermati Y, Schmitt E
& Kroemer G (2006) Heat shock proteins 27 and 70:
anti-apoptotic proteins with tumorigenic properties. Cell
Cycle 5, 2592–2601.
29 Neznanov N, Komarov AP, Neznanova L, Stanhope-
Baker P & Gudkov AV (2011) Proteotoxic stress tar-
geted therapy (PSTT): induction of protein misfolding
enhances the antitumor effect of the proteasome inhibi-
tor bortezomib. Oncotarget 2, 209–221.
30 Armstrong M, Schumacher K, Mody R, Yanik G,
Opipari AJ & Castle V (2008) Bortezomib as a thera-
peutic candidate for neuroblastoma. J Exp Ther Oncol
7, 135–145.
31 Te Kronnie G, Timeus F, Rinaldi A, Crescenzio N,
Spinelli M, Rosolen A, Ricotti E & Basso G (2004)
Imatinib mesylate (STI571) interference with growth of
neuroectodermal tumour cell lines does not critically
involve c-Kit inhibition. Int J Mol Med 14, 373–382.
32 Laemmli UK (1970) Cleavage of structural proteins
during the assembly of the head of bacteriophage T4.
Nature 227, 680–685.
33 Mastrocola R, Reffo P, Penna F, Tomasinelli C,
Boccuzzi G, Baccino F, Aragno M & Costelli P (2008)
Muscle wasting in diabetic and in tumor-bearing rats:
role of oxidative stress. Free Radic Biol Med 44, 584–593.
34 Barbero G, Carta F, Giribaldi G, Mandili G, Crobu S,
Ceruti C, Fontana D, Destefanis P & Turrini F (2006)
Protein ⁄RNA coextraction and small two-dimensional
polyacrylamide gel electrophoresis for proteomic ⁄ geneexpression analysis of renal cancer biopsies. Anal Bio-
chem 349, 62–71.
35 Aina V, Perardi A, Bergandi L, Malavasi G, Menabue
L, Morterra C & Ghigo D (2007) Cytotoxicity of
zinc-containing bioactive glasses in contact with human
osteoblasts. Chem Biol Interact 167, 207–218.
G. Mandili et al. Protein ubiquitination by Doxorubicin
FEBS Journal 279 (2012) 2182–2191 ª 2012 The Authors Journal compilation ª 2012 FEBS 2191





























