Embed Size (px)
Citation preview

JOURNAL OF CELLULAR PHYSIOLOGY 162:15-25 (1995)
Chloride Is Required for Receptor-Mediated Divalent Cation Entry in Mesangial Cells
SIDNEY C. KREMER, WENJIA ZENC, ROGER HURST, TERRl NING, CATHARINE WHITESIDE, AND KARL 1. SKORECKI*
MRC Croup in Membrane Biology, Hospital for Sick Children and Toronto Hospital Research Institutes, Division of Nephrology, University of Toronto,
Toronto, Canada M5C 2C4
Agonists which stimulate the inositol 1,4,5 trisphosphate ([1,4,5]-IP,)-dependent mobilization of Ca2+ from intracellular stores also stimulate entry of divalent cations across the cell membrane. Under appropriate experimental conditions, divalent cation entry across the cell membrane can be monitored as the rate at which the intracellular fluorescence of divalent cation indicators is quenched by the addition of Mn2+ to the extracellular medium. We report that addition of vasopressin to fura-2-loaded glomerular mesangial cells in culture markedly ac- celerated the rate at which Mn2+ quenched fura-2 fluorescence at its Ca2+- insensitive wavelength in the presence of extracellular NaCI, but that this quench response was attenuated when CI- was removed from the extracellular medium by equimolar substitution with impermeant anions (gluconate, methanesulfonate, acetate, lactate). Similarly, loss of agonist-induced quench also occurred when CI- was substituted with gluconate in K+-containing media. Addition of the CI- channel inhibitor, 5-nitro-2-(3-phenylpropylaminobenzoic acid) (NPPB), also in- hibited Mn2 +-induced quench of fura-2 fluorescence following vasopressin addi- tion. in contrast, in the presence of gramicidin to provide an alternate conduc- tance pathway to accompany divalent cation entry, agonist-dependent Mn2+ quench occurred even in the absence of extracellular CI-, indicating that the requirement for CI- was not the result of cotransport on a common transporter nor the result of CI- serving as a necessary cofactor for divalent cation entry. A similar dependence on extracellular CI- was observed for other Ca'+-mobilizing ago- nists such as endothelin, as well as the intracellular Ca2+ ATPase inhibitor, thapsigargin. Extracellular CI- dependence for agonist-induced divalent cation entry was also reflected in a corresponding extracellular CI- dependence for agonist-induced mesangial cell contraction. it has been previously shown by ourselves (Kremer et al., 1992a, Am. J. Physiol., 262:F668-F678) and others that agonist-stimulated calcium mobilization in mesangial cells i s accompanied by inhibition of K+ conductance and increased CI- conductance. Accordingly, we conclude that the current findings suggest that activation of CI- conductance provides regulated charge compensation for receptor-mediated divalent cation entry in response to Ca2+-mobilizing vasoconstrictor agonists in mesangial cells. @ 1995 Wiley-Liss, Inc
Agonists which stimulate phospholipase C and cause the inositol 1,4,5 trisphosphate ([1,4,5I-IP3)-dependent mobilization of Ca2+ from intracellular stores also stimulate entry of divalent cations across the cell mem- brane. In the case of Ca2+, this contributes to the over- all rise in cytosolic Ca2+ that follows stimulation of cells with such agonists (Putney, 1990; Felder et al., 1992). Numerous studies have sought to identify the signalling mechanism for such receptor-mediated diva- lent cation entry (Irvine, 1992). However, very little attention has been focused on the other ionic events across the cell membrane which accompany such diva- lent cation entry. Indeed, we and others have recently reported that stimulation of mesangial cells with Ca2+- mobilizing agonists such as vasopressin, angiotensin, and endothelin results in an increase in C1- conduc- 0 1995 WILEY-LISS, INC.
tance across the cell membrane which is accompanied by inhibition of potassium conductance (Okuda et al., 1986; Kremer et al., 1989, 1992a,b; Hanrahan et al., 1990). These studies demonstrated that a rise in cyto- solic Ca2+ and activation of protein kinase C were each sufficient to increase C1- conductance, but that in addi- tion a G-protein-mediated pathway independent of a rise in cytosolic Ca2+ and activation of protein kinase C
Received July 19,1993; accepted June 3,1994. *To whom reprint requestskorrespondence should be addressed at Division of Nephrology, Hospital for Sick Children, 555 Univer- sity Avenue, Room 5128, Elm Wing, Toronto, Ontario, Canada M5G 1x8.

KREMER ET AL. 16
could also increase C1- conductance (Kremer et al., 1992a). Accordingly, in the current study, we tested the possibility that C1- is required to support agonist stim- ulation of divalent cation entry as measured by the quench of fluorescence of divalent cation indicators fol- lowing the addition of Mn2+ (Anderson, 1983; Hallam and Rink, 1985; Glennon et al., 1992; Huang et al., 1993).
MATERIALS AND METHODS Materials
Tissue culture media were obtained from Gibco (Bur- lington, Ontario). Fetal bovine serum was obtained from Flo Laboratories (Mississauga, Ontario). Fura-2/ AM, BCECFiAM (2’,7’-bis(carboxyethyl)-5,6-carboxyflu- orescein-acetoxymethyl ester), and Bis-oxonol (bis-( 1’3- diethy1thiobarbiturate)trimethineoxonol) were obtained from Molecular Probes Inc. (Eugene, OR). Thapsigargin was obtained from L-C Services Corp. (Woburn, MA). NPPB (5-nitro-2-(3-phenylpropylaminobenzoic acid) was kindly provided by Dr. John Hanrahan (McGill Univer- sity, Montreal, Canada).
Cell culture Mesangial cells are vascular smooth muscle cells of
the renal glomerular microcirculation (Mene et al., 1989). Homogeneous cultures were derived from colla- genase-treated glomeruli isolated from young Sprague- Dawley rats and characterized as previously described (Kremer et al., 1988a,b, 1992a). Cell cultures were maintained in Corning 75 cm2 dishes in Dulbecco’s Modified Eagle’s medium (DMEM) supplemented with 20% bovine serum and incubated at 37°C with 5% CO,.
Measurements of fura-2 fluorescence quench and cytosolic free calcium
Measurements of intracellular fura-2 fluorescence were conduct,ed using adherent mesangial cells grown to confluency on 25 mm diameter round cover slips. Cells were loaded by incubation with fura-2/AM (2.5 pM) for 40 minutes at 37°C and then the cells were washed free of extracellular dye. In previous studies, we have shown minimal dye leakage for several hours following this loading and washing protocol (Kremer et al., 1988a, 1992a,b). During fluorescence measure- ments, the cells were bathed in a 10 mM 442-hydroxy1)- 1-piperazineethanesulfonic acid (Hepes)-buffered (pH 7.4) physiological saline solution consisting of NaCl 137 mM, KC1 3 mM, glucose 11 mM, Hepes 10 mM, MgC1, 1 mM, and CaC1, 0.5 mM. Where indicated in the Results section, NaCl(137 mM) was replaced with equimolar Na-gluconate (137 mM) or alternatively with combinations of 2/3 Na-gluconate (91 mM) plus one third (46 mM) of either NaC1, NaBr, Na-methane- sulfonate, Na-acetate, or Na-lactate. In the Cl--free media, KCl, MgCl,, and CaC1, were replaced by their corresponding gluconate salts and calcium was ad- justed to achieve equivalent free ionic calcium concen- trations as measured by a calcium-selective electrode. In separate experiments, we also measured the ability of various salts to quench fura-2 fluorescence in cell- free solutions and found no interference with the in- duced Mn2+ quench by the accompanying anion (data not shown).
Fluorescence measurements were conducted in a dual-excitation wavelength SPEX fluorescence spec- trophotometer as previously reported (Kremer et al., 1992a,b). Excitation monochrometers were set a t 340 nm and 360 nm (the Ca2+ isobestic excitation wave- length for fura-2) for measurements of fluorescence quench, or else a t 340 nm and 380 nm for ratiometric determination of changes in cytosolic Ca2+. The emis- sion signal was monitored after passing the fluores- cence output through a 490 nm narrow-bandpass inter- ference filter, and the emitted fluorescence signal collected from a field of cells optically isolated using a Nikon diaphot microscope equipped with a 20 x fluores- cence objective. During fluorescence measurements, the cells were housed in a humidified, thermostatted incubation chamber set to 37°C and equipped with an automated injection system for addition of various re- agents and agonists.
Measurement of intracellular pH Intracellular pH was measured in adherent mesan-
gial cells using the pH-sensitive fluorescent dye BCECF (Grinstein et al., 1989). Fluorescence measure- ments were conducted by setting excitation monochro- mators at 500 nm and 436 nm (the pH isobestic excita- tion wavelength for BCECF) and emission recorded a t 530 nm and the ratio of the 500 nm excitation signal to that a t 436 nm was obtained in order to compensate for photobleaching or drift in the signal due to loss of dye. In order to calibrate the fluorescent signal to pH after each experiment, intra- and extracellular pH were equilibrated using the Kt/H+ exchange ionophore ni- gericin (5 pM) in combination with the Na+/H+ ex- change ionophore monensin (12.5 pM). This sets [K+ + Na+I,/[K+ + Nafl, = [H+],/[H+],,, where i and o denote inside and outside the cell, respectively. Under these conditions, intracellular pH can be determined and altered by measuring and changing extracellular pH. After each experiment, the cells were so treated and pH adjusted for three calibration points. Since flu- orescence of BCECF is linearly related to pH in the physiological range, a linear regression of the three calibration points was conducted and basal cellular pH interpolated for each experiment.
Membrane potential measurements Membrane potential was assessed as previously de-
scribed (Kremer et al., 1989, 1992a,b). Bis-oxonol dis- solved in dimethyl sulfoxide (final concentration, 0.2 pM and 0.2% V/V, respectively) was added to an aliquot of cells and allowed to equilibrate. Excitation and emis- sion wavelengths were 540 nm and 580 nm, respec- tively.
Mesangial cell contraction measurements Mesangial cell contraction was determined a t 30°C as
has been previously described (Kreisberg et al., 1985). The cell planar surface area was measured using an inverted light microscope, a 386SX computer, and JAVA software (Jandel Scientific, San Rafael, CAI. Subconfluent mesangial cells were grown in culture for 24 hours on small (35 mm diameter) Petri dishes with 2 ml of DMEM supplemented with 20% bovine serum. Following the capturing of a zero time point image, the

17 RECEPTOR-MEDIATED DIVALENT CATION ENTRY
medium was gently replaced with a 10 mM Hepes-buff- ered (pH 7.4) physiological saline solution with or with- out C1- ions and vasopressin. Further images were then captured at time points from 5 to 60 minutes. The perimetry of all cells with clearly defined borders (n = 10-15/experiment) was used for planar surface area measurement. Partial retraction of the cytoplas- mic extensions was observed in response to vasoactive agents (in contrast to the rounding up of cells exposed to trypsin, associated with detachment). Mesangial cells without agonist exposure were used as the control for spontaneous contraction in these experiments. The average of individual cell planar area was determined for all the cells at each time point and expressed as the percent of the original cell planar area (100%).
Data presentation All quench data curves (360 nm excitation) were cal-
ibrated by the addition of ionomycin and additional Mn2+ at the end of each run to determine cellular auto- fluorescence. Normalization of the data curves to allow comparison between experiments was done by setting autofluorescence to zero and baseline fluorescence to 1.0 x lo5 counts per second (c.P.s.) and the data repre- sented as normalized C.P.S. Unless indicated otherwise, tracings shown are representative of a t least four ex- periments conducted under the same experimental con- ditions.
RESULTS Effects of extracellular anion on
vasopressin-stimulated entry of Mnz+ into mesangial cells
Figure 1A shows the fluorescence response of fura-2- loaded mesangial cells excited at the Ca2+-insensitive wavelength of 360 nm to the addition of Mn2+ followed by the subsequent addition of vasopressin. In the pres- ence of NaCl in the external medium, addition of Mn2+ alone resulted in a minimal decrease of fluorescence. Subsequent addition of vasopressin resulted in a marked diminution of fluorescence. Replacement of ex- tracellular C1- with the cell impermeant anion glu- conate resulted in a dramatic elimination of the vaso- pressin-stimulated quench (Fig. 1B). Figure 1C shows that quench of fluorescence in NaCl media required the presence of extracellular Mn2+ at the time of agonist addition, indicating that fluorescence quench was in- deed a reflection of entry of Mn2+ across the cell mem- brane via divalent cation entry pathways, rather than a result of movement of Mn2+ from the cytosol into intra- cellular organelles, with quench of compartmentalized fluorescent probes as has been reported in certain other experimental systems (Glennon et al., 1992). Figure 1D shows that readdition of NaCl to cells treated with Mn2+ and stimulated with vasopressin under Cl--free conditions was immediately able to restore fluores- cence quench. This immediate restoration of quench upon replacement of extracellular C1- indicates that attenuation of quench by gluconate substitution was not due to intracellular C1- depletion or due to an effect of the presence of gluconate per se. It should also be noted that the effect of removal of extracellular C1- to attenuate Mn2+-induced quench of fluorescence was
not altered and persisted at varying different loading concentrations of fura-2 (1, 2.5, and 5 p,M; data not shown).
Figure 2 shows that the quench response diminished progressively as the time of successive Mn2+ addition following vasopressin addition was progressively de- layed. This is consistent with the time course for ago- nist-induced increase in C1- conductance in these cells as reported previously by others and ourselves (Okuda et al., 1986; Kremer et al., 1989, 1992a,b; Hanrahan et al., 1990).
In order to further assess the role of C1- channel activation in agonist-induced divalent cation entry, the C1- channel blocker NPPB was used. In preliminary experiments, we have shown that NPPB is the most potent specific inhibitor of C1- channel activation in mesangial cells (Hanrahan et al., unpublished obser- vations). As shown in Figure 3, pretreatment of mesangial cells with NPPB markedly inhibited vaso- pressin-induced Mn2+-dependent quench of fura-2 fluo- rescence.
A similar pattern of C1- dependency for fluorescence quench was observed for the nonmitochondrial Ca2+- ATPase inhibitor, thapsigargin (Fig. 4). We also used thapsigargin to confirm that changes in fluorescence reflect fura-2 in the cytosol rather than fura-2 trapped in intracellular organelles. As shown in the bottom panel, when thapsigargin was added following vaso- pressin in fura-2-loaded mesangial cells, cytosolic Ca2+ increased above baseline as measured by the fluores- cence ratio at excitation wavelengths of 340 nm and 380 nm. Previously it has been reported that a decrease below baseline occurs under those experimental condi- tions in which fura-2 has been compartmentalized into [1,4,51-IP3-sensitive intracellular Ca2+ stores as a re- sult of movement of Ca2+ out of these stores (Glennon et al., 1992). Accordingly, the observed increase in cyto- solic Ca2+ in the current set of experiments provides additional confirmation that changes in fluorescence predominantly reflect fura-2 in the cytosol.
The preceding results suggested the possibility that agonist-stimulated divalent cation entry was depen- dent upon Cl- channel activation and concomitant en- try of only certain anions. Accordingly, experiments were conducted to test the ability of several different anions to support divalent cation entry. Since glu- conate did not support divalent cation entry (Fig. lB), the external medium composition was chosen to con- tain either 213 gluconate in the form of the sodium salt (91 mM) plus one third of either C1-, Br-, methane- sulfonate, acetate, or lactate in the sodium salt (46 mM). As shown in Figure 5A and 5B, both C1- and Br- supported vasopressin-stimulated Mn2+-induced quench of fura-2 fluorescence, whereas neither meth- anesulfonate, acetate, nor lactate was able to restore Mnz+ quench (Fig. 5C-E).
Agonist-induced divalent cation entry persists in KCl but not K-gluconate media
The addition of vasopressin resulted in a marked quench response in mesangial cells incubated in KC1 (140 mM) media (Fig. 6A). However, as shown in Fig- ure 6B, this quench response to vasopressin was mark- edly inhibited in K-gluconate (140 mM) media. These

18
100
80 3 0 %- X
0
Mn+* VP
KREMER ET AL.
VP
NaCl
4
0 I - I X I
- 1 min
1 min \ time -
Fig. 1. Effect of CI- removal on vasopressin-stimulated quench of fluorescence by Mn2+. Mesangial cells were loaded with fura-2 by incubation with its acetomethoxyl ester and cellular fluorescence was measured with excitation at the Ca2 + -insensitive wavelength of 360 nm as described under Materials and Methods. Mn2 + (2 mM), vaso- pressin (VP, 200 nm), and ionomycin (10, 2.5 pM) were added at the times indicated by the arrows. Incubation medium during fluores- cence measurement was as described under Materials and Methods with NaCl medium used in A and C, and Na-gluconate medium used
results indicate that varying extracellular anion can result in loss of quench, even at high extracellular Kt concentrations.
Divalent cation entry occurs in the absence of extracellular Cl-. in cells incubated in the
presence of gramicidin The foregoing results suggested the possibility that
extracellular ClK might be acting as a necessary cofac-
in B. For C, mesangial cells were first preincubated in the presence of 2 mM MnCl, and then washed in Mn2+-free medium prior to fluores- cence measurements. Under these conditions, addition of 200 mM vasopressin failed to induce a quench of fura-2 fluorescence. Subse- quent addition of 2 mM MnCI, as shown by the arrow resulted in a marked quench of the fura-2 fluorescence signal. In D, the cells were incubated in Na-gluconate media initially (1.0 ml) and then NaCl media (0.5 ml) were added at the time indicated by the arrow.
tor for divalent cation entry or might be coupled on a shared cotransporter. In order to test this possibility, conditions were sought in which Mn2+ quench might occur even in the absence of extracellular ClF. We have previously shown that basal membrane potential of mesangial cells is unaffected by anion substitution (Kremer et al., 1989, 1992a). As shown in Figure 7A, when cells were incubated in N-methyl-glucamine (NMG)-gluconate media, the addition of vasopressin re-
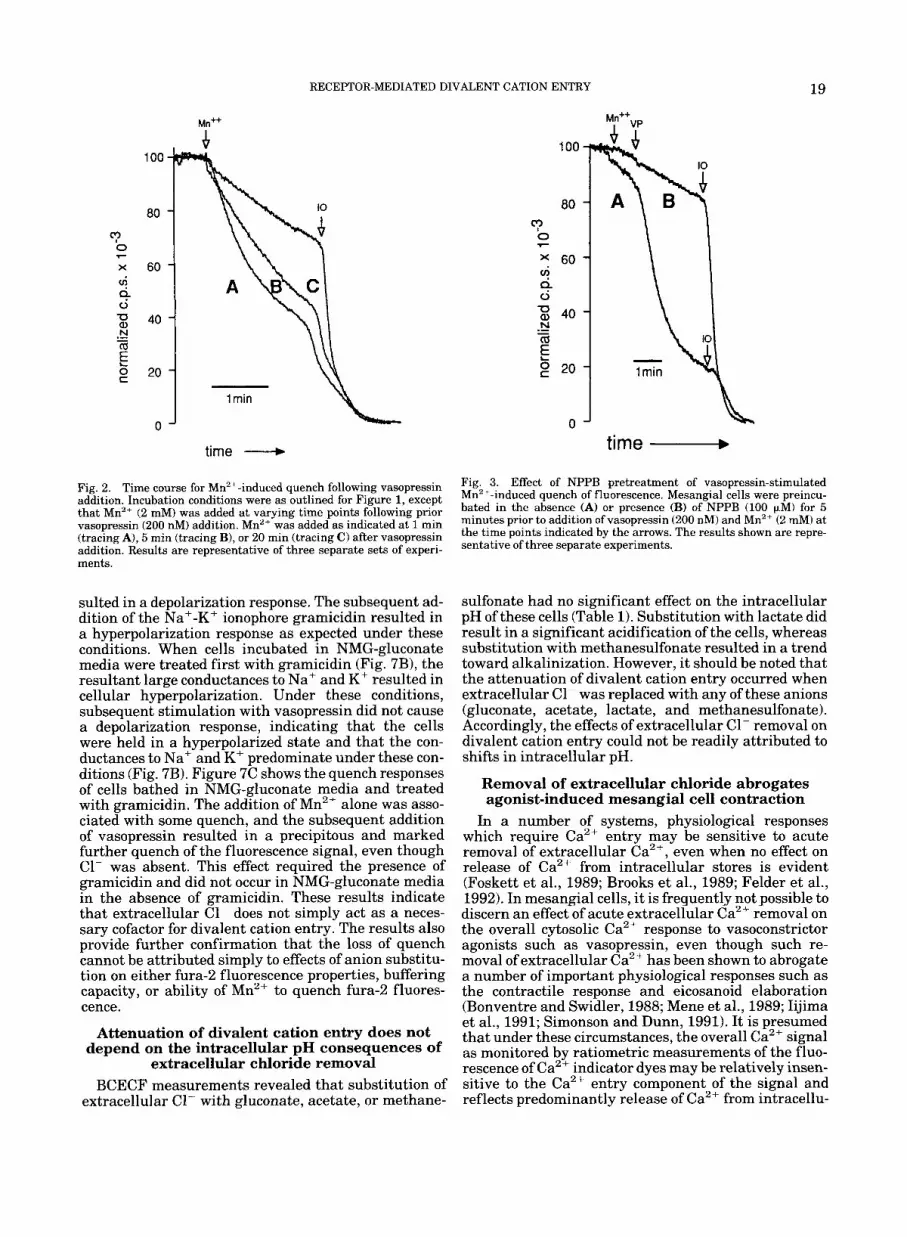
RECEPTOR-MEDIATED DIVALENT CATION ENTRY 19
100
80
@? 0 F
x 60 4 4
40 N
m .- -
E C 20
0
time --+
Fig. 2. Time course for Mn2+-induced quench following vasopressin addition. Incubation conditions were as outlined for Figure 1, except that Mn2' (2 mM) was added at varying time points following prior vasopressin (200 nM) addition. Mn2+ was added as indicated at 1 min (tracing A), 5 min (tracing B), or 20 rnin (tracing C) after vasopressin addition. Results are representative of three separate sets of experi- ments.
sulted in a depolarization response. The subsequent ad- dition of the Na'-K+ ionophore gramicidin resulted in a hyperpolarization response as expected under these conditions. When cells incubated in NMG-gluconate media were treated first with gramicidin (Fig. 7B), the resultant large conductances to Na+ and K+ resulted in cellular hyperpolarization. Under these conditions, subsequent stimulation with vasopressin did not cause a depolarization response, indicating that the cells were held in a hyperpolarized state and that the con- ductances to Na+ and K+ predominate under these con- ditions (Fig. 7B). Figure 7C shows the quench responses of cells bathed in NMG-gluconate media and treated with gramicidin. The addition of Mn2+ alone was asso- ciated with some quench, and the subsequent addition of vasopressin resulted in a precipitous and marked further quench of the fluorescence signal, even though C1- was absent. This effect required the presence of gramicidin and did not occur in NMG-gluconate media in the absence of gramicidin. These results indicate that extracellular C1- does not simply act as a neces- sary cofactor for divalent cation entry. The results also provide further confirmation that the loss of quench cannot be attributed simply to effects of anion substitu- tion on either fura-2 fluorescence properties, buffering capacity, or ability of Mn2+ to quench fura-2 fluores- cence.
Attenuation of divalent cation entry does not depend on the intracellular pH consequences of
extracellular chloride removal BCECF measurements revealed that substitution of
extracellular C1- with gluconate, acetate, or methane-
1 rnin i time -
Fig. 3. Effect of NPPB pretreatment of vasopressin-stimulated Mn2 -induced quench of fluorescence. Mesangial cells were preincu- bated in the absence (A) or presence (B) of NPPB (100 pM) for 5 minutes prior to addition of vasopressin (200 nM) and Mn2+ (2 rnM) at the time points indicated by the arrows. The results shown are repre- sentative of three separate experiments.
sulfonate had no significant effect on the intracellular pH of these cells (Table 1). Substitution with lactate did result in a significant acidification of the cells, whereas substitution with methanesulfonate resulted in a trend toward alkalinization. However, it should be noted that the attenuation of divalent cation entry occurred when extracellular C1- was replaced with any of these anions (gluconate, acetate, lactate, and methanesulfonate). Accordingly, the effects of extracellular C1- removal on divalent cation entry could not be readily attributed to shifts in intracellular pH.
Removal of extracellular chloride abrogates agonist-induced mesangial cell contraction
In a number of systems, physiological responses which require Ca2+ entry may be sensitive to acute removal of extracellular Ca2+, even when no effect on release of Ca2+ from intracellular stores is evident (Foskett et al., 1989; Brooks et al., 1989; Felder et al., 1992). In mesangial cells, it is frequently notpossible to discern an effect of acute extracellular Ca2+ removal on the overall cytosolic Ca2+ response to vasoconstrictor agonists such as vasopressin, even though such re- moval of extracellular Ca2+ has been shown to abrogate a number of important physiological responses such as the contractile response and eicosanoid elaboration (Bonventre and Swidler, 1988; Mene et al., 1989; Iijima et al., 1991; Simonson and Dunn, 1991). It is presumed that under these circumstances, the overall Ca2+ signal as monitored by ratiometric measurements of the fluo- rescence of Ca2+ indicator dyes may be relatively insen- sitive to the Ca2+ entry component of the signal and reflects predominantly release of Ca2+ from intracellu-

20 KREMER ET AL.
,'+ TG
5
4
0 .- c 2 3 al V C al 0 u)
0 3
2 2
ii
1
L
.- f TG
time 7
Fig. 4. Effect of removal of extracellular chloride on thapsigargin- induced manganese quench of fura-2 fluorescence. Top panel: Exper- imental conditions were as outlined for Figure 1. MnCI, ( 2 mM) and thapsigargin (1 p,M) were added a t the times indicated by the arrows. The incubation media during fluorescence measurements contained
137 mM NaCl in tracing A and 137 mM Na-gluconate in tracing B. Bottom panel: Mesangial cells were loaded by incubation with fura- 2/AM as outlined and the ratio of fluorescence emission and excitation wavelengths of 340 nm and 380 nm monitored as described in Materi- als and Methods.
lar stores (Foskett et al., 1989). Therefore, it was not surprising that under the same experimental condi- tions used to measure Mn2+ quench, no readily discern- ible effect of extracellular C1- removal on the overall Ca" signal as measured by the ratio of fura-2 fluores- cence at 340 nm and 380 nm was found except under specialized conditions in which release from stores had been attenuated (data not shown). Accordingly, we monitored the effect of extracellular C1- removal on the
contractile response of mesangial cells since this re- sponse is known to be dependent on Ca2+ entry and is inhibited by removal of extracellular Ca2+ (Mene et al., 1989). As shown in Figure 8 in lVaC1 media, vaso- pressin induced a marked contractile response and this response was eliminated when extracellular C1- was substituted with gluconate. Of note, in two separate experiments Mn2+ itself had no effect on the contractile response (data not shown).

RECEPTOR-MEDIATED DIVALENT CATION ENTRY 21
8 0 - 7
X
4 60-
4
g 2 0 -
m 8 40 - Q .- -
c
0 -
- 1 min
time - Fig. 5. Ability of various anions to restore Mn"+ quench of cellular fura-2 fluorescence by vasopressin. Experimental conditions were as described in Figure 1. The incubation medium contained 91 mM Na- gluconate plus 46 mM of either NaCl (A), NaBr (B), Na-methanesulfonate (C), Na-acetate (D), or Na-lactate (E).
DISCUSSION In the current study, divalent cation entry was mea-
sured as the quench of the fluorescence of intracellular fura-2 at its Ca2+-insensitive isobestic excitation wave- length by Mn2+ added to the extracellular medium. The rate of quench of this fluorescence by Mn2+ was mark- edly accelerated following the addition of vasopressin. Replacement of extracellular C1- by cell-impermeant anions eliminated divalent cation entry. Br- and C1- in the extracellular medium were able to support Mn2+- induced quench, whereas gluconate, methanesulfonate, acetate, and lactate were unable to support Mn2+-in- duced quench. The effect was striking with nearly com- plete loss of Mn2+-induced quench in the absence of extracellular C1-. The ability of another halide (bro- mide) to support Mn2+-induced quench likely reflects
the ability of C1- entry pathways to selectively accom- modate only certain other closely related anions (Wright and Diamond, 1977). The ability of extracellu- lar anions to modulate Mn2'-induced quench of fura-2 fluorescence also speaks to the validity of this quench signal as a measure of Mn2+ entry across the cell mem- brane in the current study, in contrast to quench of compartmentalized fura-2 as occurs in hepatocytes loaded by incubation with fura-2lAM (Glennon et al., 1992). This was further validated by the finding that fluorescence quench in the current study required the presence of Mn2+ in the extracellular medium at the time of agonist addition. Furthermore, the results of experiments using thapsigargin were not consistent with compartmentalization of fura-2 into intracellular organelles, but rather indicated that fluorescence re-

22 KREMER ET AL
Mn++ Mn+VP
1 min
time - Fig. 6. Manganese quench in KC1 and K-gluconate media. Experimental conditions were as outline for Figure 1. The incubation media contained either 140 mM KCl in A or 140 mM K-gluconate in B. MnC1, (2 mM) and vasopressin (200 mM, VP) were added as indicated by the arrows.
sponses were reflecting fura-2 in the cytosol. It is possi- ble that in those cells or experimental systems in which fura-2 is compartmentalized into intracellular stores, and in which Mn2+-induced quench of intracellular flu- orescence does not represent ion movement across the cell membrane, that the effect of extracellular Cl-re- moval to abrogate Mn2+ quench might not be readily evident. Under such circumstances, modified ap- proaches to fura-2 loading of the cytosolic compartment would be called for or alternatively other experimental approaches for assessing divalent cation entry would be required.
Several possible mechanisms can be forwarded to ex- plain the striking dependence of Mn2+-induced quench on extracellular anions observed in the current study. In the first place, an effect mediated through shifts in intracellular pH was ruled out. Also, the effect of extra- cellular C1- removal could not be attributed to rapid depletion of intracellular C1- inasmuch as Mn2+-in- duced quench was restored immediately upon readdi- tion of extracellular C1-. It should also be noted that measurements of extracellular C1- using the fluores- cence C1- indicator, SPQ, revealed that mesangial cells could be maintained in Cl--free media for prolonged periods without depletion of intracellular C1- (data not shown). Furthermore, the loss of Mn2+-induced quench was observed in both Na+- and Kt-containing media. Also, no effect on quench of fura-2 salt fluorescence in cell-free solution was observed with gluconate or the other C1 --substituting anion. Additionally, in the cell fluorescence experiments, the loss of fluorescence in nonhalide-containing media was observed a t varying fura-2-loading concentrations. Finally, Mn2+-induced quench of fluorescence could be restored in the presence
of gramicidin, even in gluconate-containing media. Taken together, these latter sets of results suggest that loss of Mn"-induced quench of fluorescence is not me- diated through effects of anion substitution on fura-2 fluorescence properties, buffering capacity, or ability of Mn2+ to quench fura-2 fluorescence, and truly reflects a C1F dependence for agonist-induced divalent cation entry.
In a recent study, it was reported that volume-regu- lated nonspecific monovalent cation entry in epithelial cells was dependent on the presence of extracellular C1- (Chan and Nelson, 1992). It wias postulated that this effect was due to the role of C1- as a cofactor or as a cotransported ion for monovalent cation entry. How- ever, no direct experimental proof for this formulation was provided. In the current study, the ability to obvi- ate the C1- requirement by providing an alternate con- ductance pathway for charge compensation using gramicidin also ruled out a role for C1- simply as a necessary cofactor or a cotransportedl ion.
A more likely explanation for the finding in the cur- rent study is that the effect of extracellular C1- re- moval results from modulation of th'e electrical compo- nent of the electrochemical gradient for divalent cation entry. In particular, we and others have previously shown that vasopressin and other Ca.2f-mobilizing ago- nists induce an increase in C1-conductance in mesan- gial cells (Okuda et al., 1986; Kremer et al., 1989, 1992a,b). Accordingly, the effect of extracellular C1- removal could be attributed to a diminution in the elec- tromotive driving force for divalent cation entry as a result of enhanced depolarization in Cl--free media. Indeed, such a dependence of Ca2+ entry on the electro- motive driving force has been reported in other systems

RECEPTOR-MEDIATED DIVALENT CATION ENTRY 23
75 1 gram
50
25
- 1 min
0’
25
O J VP
gram
100
80
60
40
20
0
1
J
1 min \
time - Fig. 7. Effect of gramicidin on membrane potential and Mn2+ quench. Cells were incubated in a medium containing as the major salt NMG. Vasopressin (200 nM, VP) and gramicidin (0.2 pM, gram) were added as indicated. A,B: Membrane potential measurements were as outlined under Materials and Methods. An increase in the fluorescence intensity indicates cellular depolarization, whereas a de-
crease in fluorescence intensity indicates hyperpolarization as previ- ously described (Kremer et al., 1989, 1992a). C: Mn2+ quench was measured as outlined for Figure 1, except that cells were bathed in a NMG-gluconate medium and gramicidin was added prior to Mn2 + and vasopressin.
TABLE 1. Effect of anion substitution on intracellular PH of mesangial cells -~ - ~ ~~
NaCl Na-gluconate Na-acetate Na-lactate Na-methanesulfonate
PH 7.18 7.26 7.20 7.00 7.29 S.E. 0.04 0.04 0.05 0.06 0.02 n = 6 5 5 5 5 P-value vs. NaCl - N.S. N.S. P < 0.05 N.S.
(Penner et al., 1988; Hoth and Penner, 1992). It would erned to a great extent by C1- conductance pathways, be of particular interest that in the case of mesangial inasmuch as the signalling pathways for increased C1- cells, the electromotive driving force regulating diva- conductance have been worked out and appear to over- lent cation entry following agonist addition was gov- lap those involved in phospholipase C activation

24 KREMER ET AL.
1 ----t- C17AVP I --1- Gluconate/AVP
- T T
\ T
1 0 2 0 30 4 0 5 0 6 0 7 0 Time (min)
Fig. 8. Effect of extracellular chloride removal on vasopressin-in- duced mesangial cell contraction. Incubation conditions for mesangial cell contraction were as outlined in Materials and Methods. Open circles and open squares indicate the time control in the absence of vasopressin in C1 --containing and gluconate-containing media, re- spectively. Solid circles and solid squares indicate the contractile re- sponse at varying times following addition of vasopressin (0 time, 1 (IM) in C 1 ~ -containing and gluconate-containing media, respectively.
(Kremer et al., 1992a). Further evidence relating to activation of C1- conductance pathways with divalent cation entry was provided by the effect of the C1- chan- nel inhibitor NPPB to also attenuate Mn2+-induced quench of fura-2 fluorescence. However, it should be noted that the results in the current study do not distin- guish an effect of extracellular C1- removal mediated through a gating phenomenon (i.e., membrane poten- tial-dependent closure of divalent cation entry path- ways) from an effect mediated through altered electro- motive driving forces for divalent cation entry. Although both effects are electrical in nature, a clear distinction between these two possible mechanisms would require single channel recordings of divalent cat- ion entry using patch clamping under varying condi- tions of membrane potential. To date, single channel recordings of receptor-operated divalent cation entry pathways in mesangial cells have not been published to our knowledge.
It has been pointed out that use of Mn2+ quench to monitor divalent cation entry may not be informative with respect to the properties of heterogeneous popula- tions of nonspecific and Ca2+-specific divalent cation channels in all systems (Fasolato et al., 1993). Mn2+ entry pathways may represent only a minor subset of the divalent cation entry pathways responsible for re- ceptor-mediated Ca2+ entry. Indeed in other systems, a nonspecific 50 pS divalent cation entry pathway which permits Mn2+ entry may account for only a small pro- portion of total Ca2+ entry (Hoth and Penner, 1992).
While multiple different conductance pathways may contribute to overall receptor-mediated Ca2+ entry, the effect of electrochemical driving forces (exclusive of gating effects) regulating the actual movement of diva- lent cations should be similar for Ca'2+ and Mn2+ entry (even if they are occurring through different cation en- try pathways) and have been studied using Mn2+
uench in the current study. Isotopic measurements of 'Ca2+ transport are more specific for Ca2+ movement per se, but may not resolve the relative contributions of influx vs. efflux to overall 45Ca2+ movement. In the mesangial cell system, Mn2+ quench appears to be a useful experimental approach for measuring the effect on divalent cation entry of changes in other ionic con- ductance (Simonson and Dunn, 1991 ). This approach is much more sensitive than direct measurements of changes in cytosolic Ca2+ using ratiometric measure- ments of fura-2 fluorescence since the latter reflects poorly the contribution of Ca2+ entry across the cell membrane (Bonventre et al., 1986). Even though alter- ations in Ca2+ entry across the cell membrane are not readily discernible through measurements of overall cytosolic Ca2+, they are of crucial importance to Ca2+- dependent cell physiological responses including con- traction and prostanoid elaboration among others (Bon- ventre and Swidler, 1988; Mene et al., 1989; Iijima et al., 1991; Simonson and Dunn, 1991). Therefore, it was reassuring to find that the effect of removal of extracel- lular C1- to abrogate Mn2+ quench was matched by a corresponding effect of removal of extracellular C1- to abrogate agonist-stimulated mesaingial cell contrac- tion.
It is now widely appreciated that sustained contrac- tion of mesangial and vascular smooth muscle cells is dependent on the continued presence and influx of ex- tracellular Ca2+ (Iijima et al., 1991; Simonson and Dunn, 1991). Numerous studies have sought to identify the signalling mechanism for receptor-mediated Ca2+ entry (Irvine, 1992; Randriamampiita and Tsien, 1993; Parekh et al., 1993). However, relatively less attention has been focused on the other ionic events across the cell membrane which accompanies such divalent cation entry. In particular, i t is clear that by whatever signal- ling mechanisms divalent cation conductance path- ways are opened, it is necessary to also understand the concomitant regulation of those conductance pathways and ion movements which serve to maintain electro- neutrality and without which divalent cation entry would not proceed even in the face of a favorable chem- ical gradient. The results of previous studies (Okuda et al., 1986; Kremer et al., l989,1992a,b) and the findings in the current study indicate that following the addi- tion of Ca2 '-mobilizing vasoconsitrictor agonists to mesangial cells in culture, C1- channels are activated. Concomitant with this response, divalent cation entry becomes sensitive to extracellular C1- removal. This finding does not necessarily predict the effect of C1- channel inhibition on agonist-induced divalent cation entry. The latter would depend on the prevailing resid- ual potassium conductance following agonist addition. Recent preliminary electrophysiological studies in mes- angial cells have shown that concomitant with C1- channel activation following agonist addition, K + con- ductance is reduced (Hanrahan et al., 1990; Hanrahan

25 RECEPTOR-MEDIATED DIVALENT CATION ENTRY
Grinstein, Sy, Coh&, S., Goeti-Smith, J.D., and Dixon, S.J. (1989) c i systems'. Physiol. Rev.,'57:109-157. -
et al, personal communication). Indeed in the current study, the potent C1- channel inhibitor NPPB also re- sulted in nearly complete attenuation of divalent cation entry, corroborating an important role for C1- channel activation in modulating this response. However, since NPPB as well as other currently available classes of C1- channel inhibitory compounds are all relatively nonspecific, definitive determination of the role of C1- channel activation will probably await isolation and characterization of the specific class of Ca2+-activated C1- channels that mediate this response in mesangial cells.
ACKNOWLEDGMENTS These studies were supported by grants to C.W. and
K.L.S. from the Medical Research Council of Canada and to K.L.S. from the Kidney Foundation of Canada. The financial support of Bristol-Myers Squibb Canada is also gratefully acknowledged. The secretarial assis- tance of Mrs. Vally Brown is gratefully acknowledged.
LITERATURE CITED Anderson, M. (1983) Mn2+ ions pass through Caz+ channels. A possi-
ble explanation. J . Gen. Physiol., 81t805-827. Bonventre, J.V., and Swidler, M. (1988) Calcium dependency of pros-
taglandin E, production in rat glomerular mesangial cells. J . Clin. Invest. 82:68-76.
Bonventre, J.V., Skorecki. K.L.. Kreisbere, J.I., and Cheune, J.Y. (1986) Vasopressin increases cytosolic free Ca2 ' concentration in glomerular mesangial cells. Am. J. Physiol., 251 tF9PF102.
Brooks, R.C., McCarthy, K.D., Lapetina, E.G., and Morell, P. (1989) Receptor-stimulated phospholipase A, activation is coupled to in- flux of external Ca2+ and not to mobilization ofintracellular Ca2+ in C62B glioma cells. J . Biol. Chem., 264t20147-20153.
Chan, H.C., and Nelson, D.J. (1992) Chloride-dependent cation con- ductance activated during cellular shrinkage. Science, 257t669- 671 - . _.
Duddy, S.K., Kass, G.E., and Orrenius, S. (1989) Ca2+-mobilizing hor- mones stimulate Ca2+ efflux from hepatocytes. J . Biol. Chem., 264: 20863-20866.
Fasolato, C., Hoth, M., Matthews, G., and Penner, R. (1993) Ca2' and Mn2 ' influx through receptor mediated activation of nonspecific cation channels in mass cells. Proc. Natl. Acad. Sci. USA, 90t3068- 3072.
Felder, C.C., Poulter, M.O., and Wess, J. (1992) Muscarinic receptor- operated Ca2+ influx in transfected fibroblast cells is independent of inositol uhosuhates and release of intracellular Ca2+. Proc. Natl. Acad. Scfi. USA, 89509-513.
Foskett, J.K., Gunther-Smith, P.J., Melvin, J.E., and Turner, R.J. (1989) Physiological localization of an agonist-sensitive pool of Ca2+ in parotic acinar cells. Proc. Natl. Acad. Sci. USA, 86t167-171.
Glennon, M.C., Bird, G.S., Kwan, C.Y., and Putney, J.W., J r . (1992) Actions of vasopressin and the Ca2 ' -ATPase inhibitor, thapsigargin on Ca2 sienallinz in heuatocvtes. J. Biol. Chem.. 267:8230-8233.
Measurements of cytoplasmic pH and cellular volume for detection of Na+/H+ exchange in lymphocytes. Methods Enzymol., 173t777- 790.
Hallam, T.J., and Rink, T.J. (1985) Agonists stimulate divalent cation channels in the plasma membrane of human platelets. FEBS Lett., 186t175-179.
Hanrahan, J.W., Kremer, S.G., and Skorecki, K.L. (1990) Whole cell recording of vasopressin activated chloride conductance in cultured rat mesangial cells. Biophys. J . , 57t309.
Hoth, M., and Penner, R. (1992) Depletion of intracellular Ca2+ stores activates a Ca2+ current in mesangial cells. Nature, 355t353-355.
Huang, S., Simonson, M.S., and Dunn, M.J. (1993) Manidipine inhib- its endothelin-1-induced [Ca2+]i signalling but potentiates endothe- lin's effect on c-fos and c-jun induction in vascular smooth muscle and glomerular mesangial cells. Am. Heart J., 125589-597.
Iijima, K., Lin, L., Nasjletti, A., and Goligorsky, M. (1991) Intracellu- lar ramification of endothelin signal. Am. J . Physiol., 260:C982- C992.
Irvine, R.F. (1992) Inositol phosphates and Ca2+ entry: Toward a pro- liferation or a simplication. FASEB J. 6:3085-3091.
Kremer, S., Harper, P., Hegele, R., and Skorecki, K. (1988a) Bradyki- nin stimulates a rise in cytosolic Ca2+ in renal glomerular mesan- gial cells via a pertussis toxin insensitive pathway. Can. J . Physiol. Pharmacol.. 66t43-48.
Kremer, S., Troyer, D., Kreisberg, J., and Skorecki, K. (1988b) Inter- action of atrial natriuretic peptide stimulated guanylate cyclase and vasopressin-stimulated Ca'+signalling pathways fn the glomerular mesangial cell. Arch. Biochem. Biophys., 260t763-770.
Kremer, S.G., Breuer, W.V., and Skorecki, K.L. (1989) Vasoconstric- tor hormones depolarize renal glomerular mesangial cells by acti- vating chloride channels. J . Cell. Physiol., 138t97-105.
Kremer, S.G., Zeng, W., Sridhara, S., and Skorecki, K.L. (1992a) Mul- tiple routes of chloride dependent depolarization in glomerular mes- angial cells. Am. J . Physiol., 262tF668-F678.
Kremer, S.G., Zeng, W., and Skorecki, K.L. (199213) Simultaneous fluorescence measurement of Ca2+ and membrane potential re- sponses to vasopressin and endothelin in glomerular mesangial cells. Am. J. Physiol., 263:C1302-C1309.
Kreisberg, J.I., Venkatachalam, M., and Troyer, D. (1983) Contractile Properties of Cultured Mesangial Cells. Am. J. Physiol. 249:F457- 463.
Mene, P., Simonson, M.S., and Dunn, M.J. (1989). Physiology of the mesangial cell. Physiol. Rev., 69t1347-1424.
Okuda, T., Yamashita, N., and Kurokawa, K. (1986) Angiotensin I1 and vasopressin stimulate Ca2+-activated chloride conductance in rat mesangial cells. J. Clin. Invest., 78:1443-1448.
Parekh, A.B., Terlau, H., and Stuhmer, W. (1993) Depletion of IP, stores activates a Ca2' and K + current by means of a phosphatase and a diffusible messenger. Nature, 364t81P818.
Penner, R., Matthews, G., and Neher, E. (1988) Regulation of Ca2+ influx by second messengers in mast cells. Nature, 334t499-504.
Putney, J.W., Jr . (1990) Capacitative Ca" entry revisited. Cell Cal- cium, 11t611-624.
Randriamamptia, C., and Tsien, R.Y. (1993) Emptying of intracellular CaZ ' stores releases a novel small messenger that stimulates Ca2+ influx. Nature, 364t809-814.
Simonson, M.S., and Dunn, M.J. (1991) Calcium signaling by distinct endothelin peptide in glomerular mesangial cells. Exp. Cell Res., 192t148-156.
Wright. E.M., and Diamond. J.M. (1977) Anion selectivitv in bioloei-





























