Embed Size (px)
Citation preview

of October 14, 2013.This information is current as
Circulating HistonesHuman CRP Defends against the Toxicity of
Cheng-Hock TohWang, Jecko Thachil, Yunyan Guan, Guozheng Wang and Simon T. Abrams, Nan Zhang, Caroline Dart, Susan Siyu
http://www.jimmunol.org/content/191/5/2495doi: 10.4049/jimmunol.1203181July 2013;
2013; 191:2495-2502; Prepublished online 26J Immunol
MaterialSupplementary
1.DC1.htmlhttp://www.jimmunol.org/content/suppl/2013/07/26/jimmunol.120318
Referenceshttp://www.jimmunol.org/content/191/5/2495.full#ref-list-1
, 16 of which you can access for free at: cites 48 articlesThis article
Subscriptionshttp://jimmunol.org/subscriptions
is online at: The Journal of ImmunologyInformation about subscribing to
Permissionshttp://www.aai.org/ji/copyright.htmlSubmit copyright permission requests at:
Email Alertshttp://jimmunol.org/cgi/alerts/etocReceive free email-alerts when new articles cite this article. Sign up at:
Print ISSN: 0022-1767 Online ISSN: 1550-6606. Immunologists, Inc. All rights reserved.Copyright © 2013 by The American Association of9650 Rockville Pike, Bethesda, MD 20814-3994.The American Association of Immunologists, Inc.,
is published twice each month byThe Journal of Immunology
at Liverpool U
niversity Library on O
ctober 14, 2013http://w
ww
.jimm
unol.org/D
ownloaded from
at L
iverpool University L
ibrary on October 14, 2013
http://ww
w.jim
munol.org/
Dow
nloaded from
at Liverpool U
niversity Library on O
ctober 14, 2013http://w
ww
.jimm
unol.org/D
ownloaded from
at L
iverpool University L
ibrary on October 14, 2013
http://ww
w.jim
munol.org/
Dow
nloaded from
at Liverpool U
niversity Library on O
ctober 14, 2013http://w
ww
.jimm
unol.org/D
ownloaded from
at L
iverpool University L
ibrary on October 14, 2013
http://ww
w.jim
munol.org/
Dow
nloaded from
at Liverpool U
niversity Library on O
ctober 14, 2013http://w
ww
.jimm
unol.org/D
ownloaded from
at L
iverpool University L
ibrary on October 14, 2013
http://ww
w.jim
munol.org/
Dow
nloaded from
at Liverpool U
niversity Library on O
ctober 14, 2013http://w
ww
.jimm
unol.org/D
ownloaded from

The Journal of Immunology
Human CRP Defends against the Toxicity of CirculatingHistones
Simon T. Abrams,*,†,1 Nan Zhang,‡,1 Caroline Dart,x Susan Siyu Wang,{ Jecko Thachil,†
Yunyan Guan,‖ Guozheng Wang,*,† and Cheng-Hock Toh*,†
C-reactive protein (CRP) is an acute-phase protein that plays an important defensive role in innate immunity against bacterial
infection, but it is also upregulated inmany noninfectious diseases. The generic function of this highly conservedmolecule in diseases
that range from infection, inflammation, trauma, and malignancy is not well understood. In this article, we demonstrate that CRP
defends the human body against the toxicity of histones released into the circulation after extensive cell death. In vitro, CRP sig-
nificantly alleviates histone-induced endothelial cell damage, permeability increase, and platelet aggregation. In vivo, CRP rescues
mice challenged with lethal doses of histones by inhibiting endothelial damage, vascular permeability, and coagulation activation, as
reflected by significant reductions in lung edema, hemorrhage, and thrombosis. In patients, elevation of CRP significantly increases
the capacity to neutralize extracellular histones in the circulation. We have also confirmed that CRP interacts with individual his-
tones in vitro and forms CRP–histone complexes in serum from patients with both elevated CRP and histones. CRP is able to
compete with phospholipid-containing liposomes for the binding to histones. This explains how CRP prevents histones from
integrating into cell membranes, which would otherwise induce calcium influx as the major mechanism of cytotoxicity caused by
extracellular histones. Because histone elevation occurs in the acute phase of numerous critical illnesses associated with extensive
cell death, CRP detoxification of circulating histones would be a generic host defense mechanism in humans. The Journal of
Immunology, 2013, 191: 2495–2502.
The pentraxin protein, C-reative protein (CRP), has longbeen recognized as a major acute-phase protein involvedin both innate and adaptive immunity (1, 2). It commonly
exists in pentameric form (3), but denatured forms have also beendescribed with different functions (4). Earlier studies have dem-onstrated that CRP activates complement, opsonizes pathogenssuch as Streptococcus pneumoniae, and facilitates their clearance(5–7). CRP binds FcgRs and FcaRI receptors to activate neu-trophils and phagocytosis (8–11). CRP also enhances uptake andpresentation of bacterial Ags by dendritic cells to stimulateadaptive immunity (12). Overall, CRP plays a crucial role in hostdefense against bacterial infection.
CRP is also significantly upregulated in many clinical scenarioswithout infection, such as trauma, pancreatitis, myocardial in-farction, neoplasia, and inflammatory disorders. However, its use inclinical assessment is often as a nonspecific marker, and its role(s)in these diseases is largely unknown. In addition to opsonizingpathogens, CRP has also been found to opsonize damaged cells,including nuclear breakdown products, such as chromatin andhistones to facilitate their clearance (7, 13–16). Because the pres-ence of circulating nuclear breakdown products has been linked tothe development of autoimmune diseases (17), CRP has beenproposed as a therapeutic reagent in mouse models of these dis-orders (18–20).Recently, histone toxicity has been demonstrated in vitro and in
mice (21–27), which prompted us to investigate the potential ofCRP in neutralizing these toxic effects. In this article, we dem-onstrate that CRP detoxifies histones both in vitro and in vivo withthe major protective mechanism being through CRP–histone com-plex formation to block histone–cell membrane integration and theconsequent increase in intracellular calcium.
Materials and MethodsPatients
Sera from patients on the Intensive Care Unit at the Royal Liverpool Univer-sity Hospital, United Kingdom, with diagnoses of severe trauma, necrotizingpancreatitis, and severe sepsis were collected in accordance with protocolapproved by the Local Research Ethics Committee and the Hospital Trust.
Animals
C57/BL6 male mice of average weight ∼22 g from the Shanghai Labo-ratory Animal Center (Shanghai, China) were housed and used at theResearch Center of Gene Modified Mice, State Education Ministry Lab-oratory of Developmental Genes & Human Diseases, Southeast University,China. All procedures were performed according to state laws and moni-tored by local inspectors. These were also in compliance with BritishHome Office laws. Histones and CRP were injected through the tail veinswith outcome monitored for up to 6 d in survival assays. To compare the
*Department of Blood Sciences, Royal Liverpool and Broadgreen University Hos-pitals National Health Service Trust, Liverpool L7 8XP, United Kingdom;†Institute of Infection and Global Health, University of Liverpool, Liverpool L693GA, United Kingdom; ‡The Medical School, Southeast University, Nanjing 210009,China; xInstitute of Integrative Biology, University of Liverpool, Liverpool L69 7ZB,United Kingdom; {School of Clinical Medicine, University of Cambridge, Cam-bridge CB2 0SP, United Kingdom; and ‖Intensive Care Unit, Wuxi Traditional Chi-nese Medicine Hospital, Wuxi 214001, China
1S.T.A. and N.Z. contributed equally to this study.
Received for publication November 19, 2012. Accepted for publication June 24,2013.
This work was supported by the National Institute of Health Research and the NorthWest Development Agency.
Address correspondence and reprint requests to Dr. Guozheng Wang and Cheng-HockToh, University of Liverpool, 216G Ronald Ross Building, Derby Street, Liver-pool L69 7BE, U.K. E-mail addresses: [email protected] (G.W.) and [email protected] (C.-H.T.)
The online version of this article contains supplemental material.
Abbreviations used in this article: ahscFv, antihistone single-chain variable fragment;CRP, C-reactive protein; dCRP, denatured C-reactive protein; PC, phosphatidylcho-line; PE, phosphatidylethanolamine; PI, propidium iodide; PS, phosphatidylserine;sTM, soluble thrombomodulin; TAT, thrombin–antithrombin.
Copyright� 2013 by The American Association of Immunologists, Inc. 0022-1767/13/$16.00
www.jimmunol.org/cgi/doi/10.4049/jimmunol.1203181
pathological changes of different treatments, we euthanized mice at 1 or4 h after injection. Blood was collected and serum isolated within 2 h andstored at 280˚C. Organs were fixed in 4% (w/v) paraformaldehyde for24 h and then stored in 70% (v/v) ethanol until embedded and sectioned.
Tissue culture
EAhy926 (human endothelial) cell line was cultured in DMEM (Sigma)supplemented with 20% (v/v) FBS (Sigma). HUVECs were isolated asdescribed previously (28) and cultured in DMEM supplemented with 20%(v/v) FBS and 5 ng/ml epidermal growth factor (Invitrogen) for use withinthree passages.
Reagents
Human CRP (Merck) was extensively dialyzed and concentrated. Afterconfirmation of the pentamer form of each protein by HPLC gel filtration,CRP at final concentrations of 1, 2, or 4 mg/ml was stored at 280˚C.Antihistone single-chain variable fragment (ahscFv) was expressed inE. coli and purified, as previously described, with demonstration of anti-histone specificity (27). LPS contamination was monitored using E-Toxate
reagents (Sigma). Recombinant histones (New England Biolabs) and calfthymus histones (Roche) were also monitored using E-Toxate.
Histone cytotoxicity assay
A propidium iodide (PI) method was used, as previously described (29). Inbrief, EAhy926 cells were grown to 70–90% confluence and treated withhistones or histones preincubated with CRP or activated protein C for 30min in DMEM supplemented with 2% (v/v) FBS. After 1h of incubation,the medium was removed to a fresh tube, and the remaining cells weredetached using Versene and transferred to the same tube. The cells werepelleted, washed, and fixed in 70% (v/v) ethanol for 30 min at 220˚Cbefore staining with 20 mg/ml PI. Flow cytometric analysis of PI-staineddamaged nuclei results in a broad peak of hypodiploid particles, clearlyseparated from the distinct diploid DNA peak of viable cells.
Quantification of circulating histones, histone–CRP complexes,CRP, soluble thrombomodulin, and thrombin–antithrombin
A Cell Death Detection ELISAPLUS kit (Roche Diagnostics) was modifiedfor measuring histone–CRP complexes in serum by replacing anti-DNA
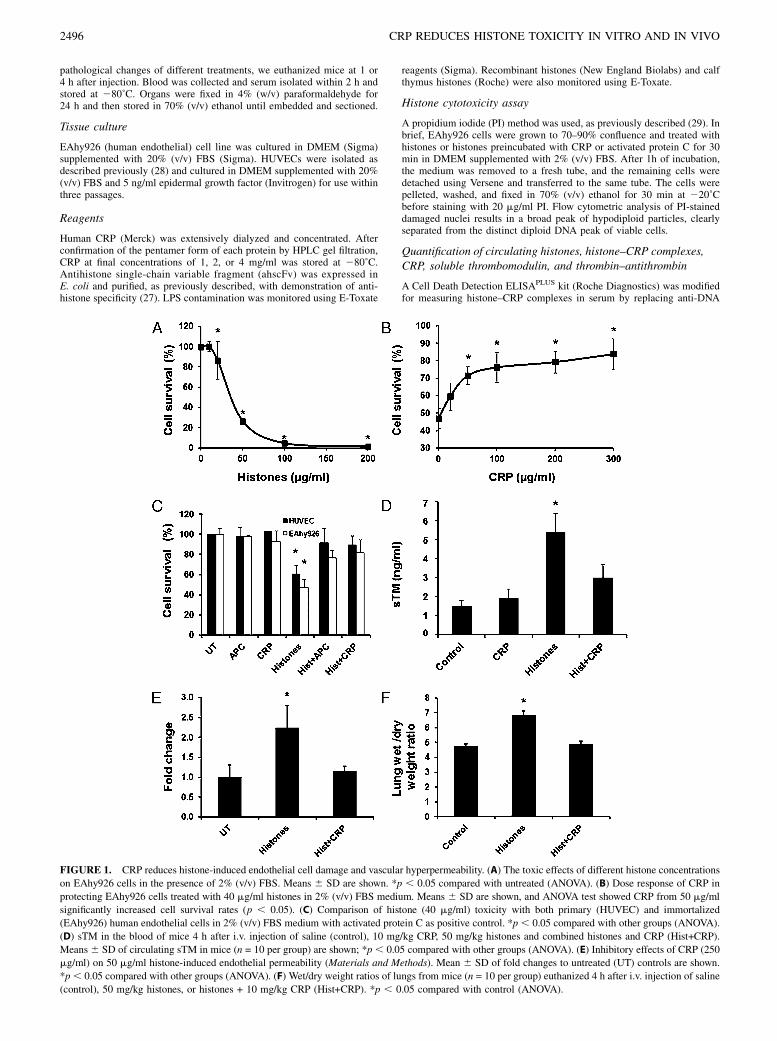
FIGURE 1. CRP reduces histone-induced endothelial cell damage and vascular hyperpermeability. (A) The toxic effects of different histone concentrations
on EAhy926 cells in the presence of 2% (v/v) FBS. Means 6 SD are shown. *p , 0.05 compared with untreated (ANOVA). (B) Dose response of CRP in
protecting EAhy926 cells treated with 40 mg/ml histones in 2% (v/v) FBS medium. Means 6 SD are shown, and ANOVA test showed CRP from 50 mg/ml
significantly increased cell survival rates (p , 0.05). (C) Comparison of histone (40 mg/ml) toxicity with both primary (HUVEC) and immortalized
(EAhy926) human endothelial cells in 2% (v/v) FBS medium with activated protein C as positive control. *p , 0.05 compared with other groups (ANOVA).
(D) sTM in the blood of mice 4 h after i.v. injection of saline (control), 10 mg/kg CRP, 50 mg/kg histones and combined histones and CRP (Hist+CRP).
Means6 SD of circulating sTM in mice (n = 10 per group) are shown; *p , 0.05 compared with other groups (ANOVA). (E) Inhibitory effects of CRP (250
mg/ml) on 50 mg/ml histone-induced endothelial permeability (Materials and Methods). Mean 6 SD of fold changes to untreated (UT) controls are shown.
*p, 0.05 compared with other groups (ANOVA). (F) Wet/dry weight ratios of lungs from mice (n = 10 per group) euthanized 4 h after i.v. injection of saline
(control), 50 mg/kg histones, or histones + 10 mg/kg CRP (Hist+CRP). *p , 0.05 compared with control (ANOVA).
2496 CRP REDUCES HISTONE TOXICITY IN VITRO AND IN VIVO
Ab with HRP-conjugated anti-CRP mAb (Abcam). ELISA kits were alsoused to determine CRP (Diamed), soluble thrombomodulin (sTM), andthrombin–antithrombin (TAT; Cusabio Biotech) levels in serum. Eachsample was performed in duplicate. Histone H3 in plasma were detectedby Western blotting using anti-histone H3 Ab (Abcam) and calculatedusing human recombinant histone H3 protein as standard.
Permeability assay
The in vitro permeability was analyzed in a dual-chamber system usingEvans blue–labeled BSA, as described previously (30). In brief, EAhy926cells were grown on the upper chamber of Transwell polycarbonatemembranes (Corning) to a confluent monolayer and treated with histonesin DMEM with 2% (v/v) FBS for 1 h. Permeability was then assessed byreplacing the media in the upper chamber with 100 ml Evans blue–BSAand in the lower chamber with 500 ml DMEM with 4% (w/v) BSA. After10 min, a 100 ml aliquot was taken from the lower chamber, and absor-bance was measured at 650 nm using a spectrometer. In vivo permeabilityassay was carried out as follows: wild type C57BL/6 male mice of 22 60.5 g body weight were challenged with 50 mg/kg histones (nonlethaldose; i.v.) or histones + 10 mg/kg CRP for 4 h. Pulmonary edema wasquantified by measuring the wet/dry weight ratio of the right lung. Wetweight was obtained immediately after euthanasia and dry weight after4 d of drying at 60˚C.
Electrophysiology
Whole-cell currents were recorded using the perforated patch configura-tion from single EAhy926 cells using an Axopatch 200B amplifier (AxonInstruments) as previously described (31). Recorded membrane currentswere filtered at 5 kHz, digitized using a Digidata 1320A interface (AxonInstruments), and analyzed using pCLAMP software. Histones (20 mg/ml),CRP (250 mg/ml), or histones preincubated with CRP for 30 min wereadded to the extracellular solution and applied to the cell by bath super-fusion. All experiments were performed at room temperature (18–22˚C).
Measurement of intracellular calcium
Intracellular Ca2+ concentration was determined by measuring fluores-cence emission at 510 nm during excitation at 340 and 380 nm accordingto published protocols (32) with fura 2-AM as the fluorescent probe.Fluorescence was monitored continuously using a Hitachi F-7000 fluo-rescence spectrometer, and intracellular Ca2+ concentration was calculatedusing the software provided. Ca2+ influx was stimulated by adding calfthymus histones to the fura 2–loaded EAhy926 cells.
Fluorescent staining
ahscFv and histones were labeled using a FluoroTag FITC conjugation kit(Sigma), and CRP was conjugated with Cy5 (GE Healthcare) separately.Free FITC or Cy5 was removed using a Sephadex G25 column. EAhy926cells were fixed with 4% (w/v) paraformaldehyde, permeabilized with 1%(v/v) Triton X-100, and blocked with 5% (w/v) defatted milk before thecostaining with 10 mg/ml FITC-ahscFv and Cy5-CRP for 1 h. Images weretaken using LSM 710 confocal microscope. For histone–plasma membraneinteractions, EAhy926 cells were seeded in glass-bottom dishes for 24 hand incubated with FITC-histones (10 mg/ml) alone or with CRP, dena-tured CRP (dCRP) (250 mg/ml), and liposomes (100 mM) made of phos-phatidylcholine (PC), phosphatidylethanolamine (PE), and phosphatidylserine(PS). The images were taken after 10-min incubation using LSM 710 con-focal microscope.
Binding assays
Gel overlay was used to determine which histones bind CRP. In brief, equalamounts of proteins were subjected to SDS-PAGE. One gel was stained withCoomassie brilliant blue to ensure equal loading and the other electroblottedonto polyvinylidence difluoride membranes, which were incubated at 4˚Covernight with 3 mg/ml CRP. Bound CRP was detected using anti–CRP-HRP (Sigma). In addition, the competitive binding of histones to CRP or
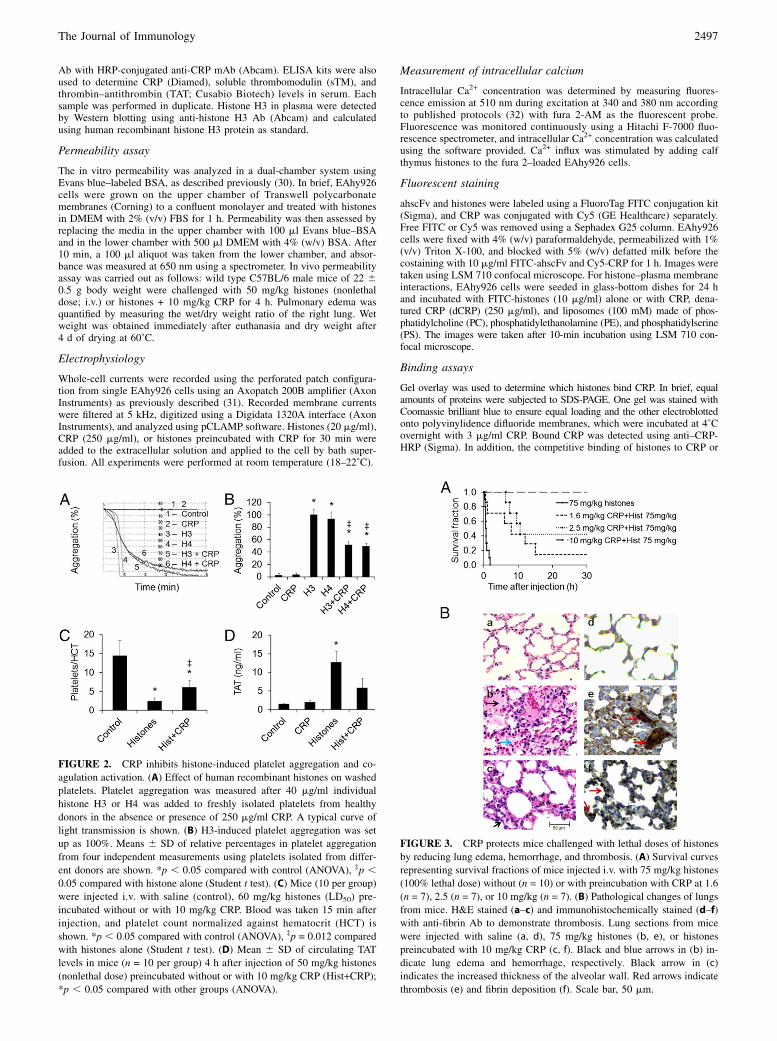
FIGURE 2. CRP inhibits histone-induced platelet aggregation and co-
agulation activation. (A) Effect of human recombinant histones on washed
platelets. Platelet aggregation was measured after 40 mg/ml individual
histone H3 or H4 was added to freshly isolated platelets from healthy
donors in the absence or presence of 250 mg/ml CRP. A typical curve of
light transmission is shown. (B) H3-induced platelet aggregation was set
up as 100%. Means 6 SD of relative percentages in platelet aggregation
from four independent measurements using platelets isolated from differ-
ent donors are shown. *p , 0.05 compared with control (ANOVA), ‡p ,0.05 compared with histone alone (Student t test). (C) Mice (10 per group)
were injected i.v. with saline (control), 60 mg/kg histones (LD50) pre-
incubated without or with 10 mg/kg CRP. Blood was taken 15 min after
injection, and platelet count normalized against hematocrit (HCT) is
shown. *p , 0.05 compared with control (ANOVA), ‡p = 0.012 compared
with histones alone (Student t test). (D) Mean 6 SD of circulating TAT
levels in mice (n = 10 per group) 4 h after injection of 50 mg/kg histones
(nonlethal dose) preincubated without or with 10 mg/kg CRP (Hist+CRP);
*p , 0.05 compared with other groups (ANOVA).
FIGURE 3. CRP protects mice challenged with lethal doses of histones
by reducing lung edema, hemorrhage, and thrombosis. (A) Survival curves
representing survival fractions of mice injected i.v. with 75 mg/kg histones
(100% lethal dose) without (n = 10) or with preincubation with CRP at 1.6
(n = 7), 2.5 (n = 7), or 10 mg/kg (n = 7). (B) Pathological changes of lungs
from mice. H&E stained (a–c) and immunohistochemically stained (d–f)
with anti-fibrin Ab to demonstrate thrombosis. Lung sections from mice
were injected with saline (a, d), 75 mg/kg histones (b, e), or histones
preincubated with 10 mg/kg CRP (c, f). Black and blue arrows in (b) in-
dicate lung edema and hemorrhage, respectively. Black arrow in (c)
indicates the increased thickness of the alveolar wall. Red arrows indicate
thrombosis (e) and fibrin deposition (f). Scale bar, 50 mm.
The Journal of Immunology 2497
phospholipids was examined by a competitive ELISA. In brief, histoneswere coated on ELISA plates and incubated with different doses of lip-osomes mixed with 50 mg/ml CRP. The bound CRP was detected withanti–CRP-HRP, and the absorbance at 450 nM was used to represent therelative amount of CRP that bound to coated histones. The liposomes wereprepared as described previously (33) using PS and PE at a ratio 1:2 toreflect the ratio in plasma membrane. PC, another major phospholipid incell membranes, was omitted to avoid the possibility of its interaction withCRP (34).
Immunohistochemical staining
Paraffin-embedded lung sections were dewaxed and rehydrated followedby Ag retrieval using DAKO PT-Pre-Treatment link system. Anti-fibrin Ab(Abcam) and EnVision+ kit (DAKO) was used to probe fibrin. Images weretaken using Olympus Microscopy and Nikon ACT-1 software.
Platelet aggregation assay
Blood was withdrawn from healthy donors, and freshly isolated washedplatelets (35) were treated with recombinant human histone H3 and H4(New England Biolabs), preincubated with or without CRP. Platelet ag-gregation was conducted using a Born Aggregometer (pap 8, Bio-DATA).
Statistical analysis
Intergroup differences were analyzed using ANOVA followed by theStudent–Newman–Keuls test. Two-group comparison before and aftertreatment used Student t test with animal survival time analyzed using alog-rank test. Association analysis used simple linear correlation. Mean 6SD are from at least three independent experiments.
ResultsCRP reduces histone-induced endothelial cell damage andpermeability increase
Extracellular histones have been demonstrated to be toxic to en-dothelial cells (24). Fig. 1A showed that calf thymus histones inculture medium with 2% (v/v) FBS are significantly toxic tocultured EAhy926 cells, a human endothelial cell line. The tox-icity of 40 mg/ml histones to EAhy926 cells can be significantlyreduced when CRP was .50 mg/ml (Fig. 1B). BSA, as control,was unable to block histone toxicity to these cells (data notshown). Primary HUVECs isolated from cords were also used andshowed similar responses to indicate that histone toxicity and CRP
protection are not cell-line–specific effects (Fig. 1C). Activatedprotein C, which can cleave histones (24), was also used as pos-itive control and could reduce histone toxicity in this assay.In vivo translation of these data are shown in Fig. 1D–F. sTM,
a circulating marker of endothelial damage, increased significantlyin mice injected with a nonlethal infusion of histones at 50 mg/kg.When this was coinjected with 10 mg/kg CRP, sTM levels de-creased significantly (Fig. 1D). Because the immediate consequenceof endothelial damage is an increase in vascular permeability, pre-incubation with CRP was able to reduce significantly histone-induced permeability in vitro (Fig. 1E) and in vivo (Fig. 1F). Col-lectively, these data indicate that CRP can inhibit extracellularhistone-induced endothelial damage and its consequences.
CRP inhibits histone-induced platelet aggregation andcoagulation activation
CRP significantly inhibited histone-induced platelet aggregationby up to 50% but did not block it completely even at a very highconcentration of 250 mg/ml (Fig. 2A, 2B). In mice, coinfusion ofCRP (10 mg/kg) partially alleviated thrombocytopenia induced byinjection of 60 mg/kg histones (Fig. 2C). Activation of coagula-tion, as measured by TAT complexes, was also significantly re-duced in mice injected with 50 mg/kg histones + 10 mg/kg CRPcompared with histone treatment alone (Fig. 2D). These datastrongly indicate that CRP is able to attenuate histone-inducedplatelet and thrombin activation. The significant but incompleteblockage of these effects by CRP might serve to retain vital he-mostatic processes whereas limiting excessive histone-induceddamage at sites of vascular injury.
CRP protects mice challenged with lethal doses of histones byreducing lung edema, hemorrhage, and thrombosis
A total of 75 mg/kg histones killed all mice within an hour. Survivaltimes were prolonged with one in seven mice surviving for .6 dwhen low-dose CRP (1.6 mg/kg) was coinfused. When CRP levelwas increased to 5 mg/kg, 3 of 7 mice survived for .6 d and 4 of 7mice died between 7 and 11 h. At 10 mg/kg CRP, all seven micesurvived .6 d (Fig. 3A). A log-rank test showed significant differ-
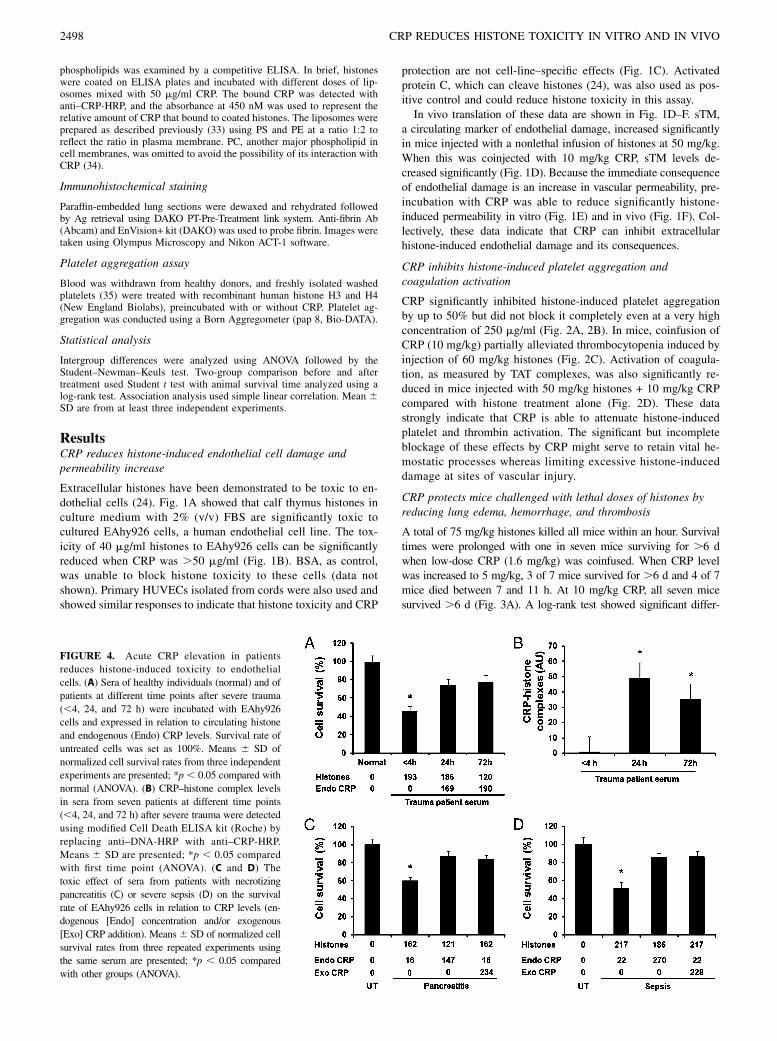
FIGURE 4. Acute CRP elevation in patients
reduces histone-induced toxicity to endothelial
cells. (A) Sera of healthy individuals (normal) and of
patients at different time points after severe trauma
(,4, 24, and 72 h) were incubated with EAhy926
cells and expressed in relation to circulating histone
and endogenous (Endo) CRP levels. Survival rate of
untreated cells was set as 100%. Means 6 SD of
normalized cell survival rates from three independent
experiments are presented; *p , 0.05 compared with
normal (ANOVA). (B) CRP–histone complex levels
in sera from seven patients at different time points
(,4, 24, and 72 h) after severe trauma were detected
using modified Cell Death ELISA kit (Roche) by
replacing anti–DNA-HRP with anti–CRP-HRP.
Means 6 SD are presented; *p , 0.05 compared
with first time point (ANOVA). (C and D) The
toxic effect of sera from patients with necrotizing
pancreatitis (C) or severe sepsis (D) on the survival
rate of EAhy926 cells in relation to CRP levels (en-
dogenous [Endo] concentration and/or exogenous
[Exo] CRP addition). Means 6 SD of normalized cell
survival rates from three repeated experiments using
the same serum are presented; *p , 0.05 compared
with other groups (ANOVA).
2498 CRP REDUCES HISTONE TOXICITY IN VITRO AND IN VIVO
ences in survival times among the four groups (p , 0.001). His-tological examination showed that mice that died at 1 h after in-jection of 75 mg/kg histones alone had obvious lung edema andhemorrhage (Fig. 3Bb), but mice euthanized 1 h after injection of75 mg/kg histones + 10 mg/kg CRP had less edema and hemor-rhage, although congestion and increased alveolar wall thicknesswere observed (Fig. 3Bc). Immunohistochemical staining for fibrinshowed obvious thrombosis in lungs from mice injected with 75mg/kg histones alone (Fig. 3Be), but not in mice injected withhistones preincubated with 10 mg/kg CRP. However, fibrin depo-sition was clearly observed in lungs (Fig. 2Bf). These pathologicalchanges support the finding that CRP reduces histone-inducedendothelial damage and coagulation activation.
Acute CRP elevation in patients reduces histone toxicity toendothelial cells
When serum samples from patients with severe trauma were in-cubated with endothelial cells, those collected within 4 h of injurycould induce significant cell death (Fig. 4A). However, this effectwas not observed at later time points of 24 and 72 h despite his-tone levels remaining elevated. The functional difference could bebecause of endogenous CRP levels, which were not raised within4 h but were .150 mg/ml at 24 and 72 h after trauma. Indeed, thiscorresponded to significant elevation of CRP–histone complexlevels, but only at 24 and 72 h (Fig. 4B). These data suggest thatCRP is a major factor in combating histone-induced toxicity inpatients.To confirm this, we collected sera from patients with necrotiz-
ing pancreatitis (Fig. 4C) and severe sepsis (Fig. 4D), which areconditions also associated with extensive cell damage. Sera thatcontained high histone levels but low CRP concentrations weretoxic to endothelial cells, whereas sera that were high in both CRPand histones were much less toxic. If exogenous CRP was addedto sera with high histone levels, their toxic effects could be at-tenuated. These data indicate that elevated CRP is able to reduceclinically toxicity of circulating histones.
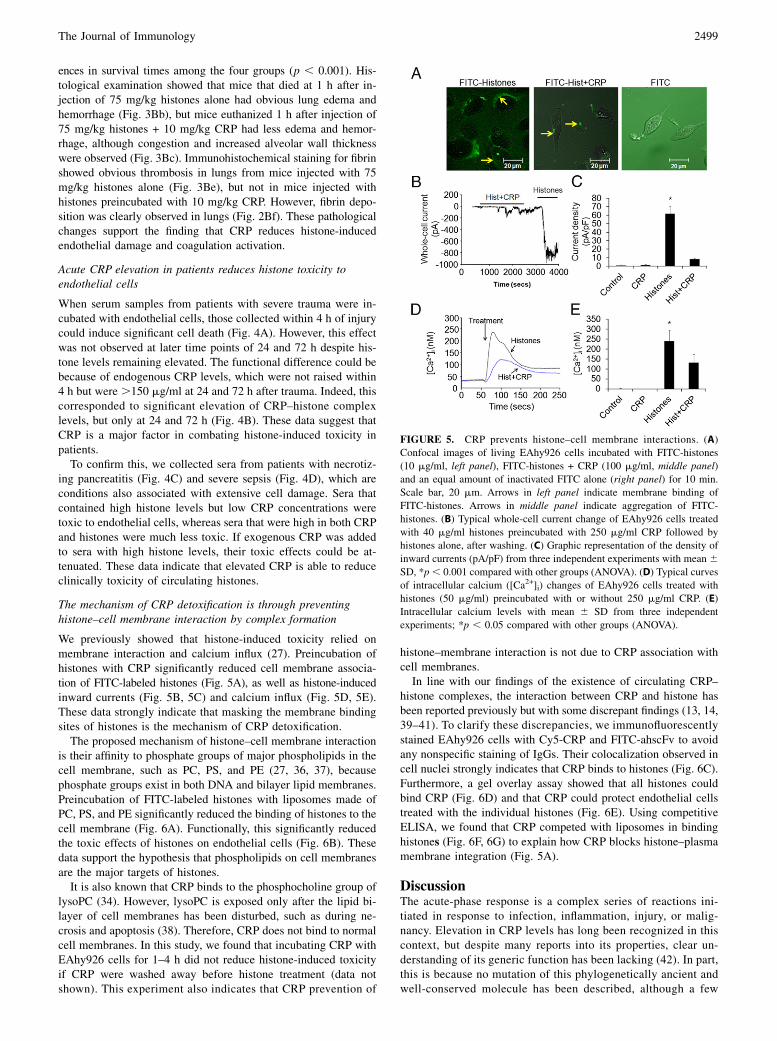
The mechanism of CRP detoxification is through preventinghistone–cell membrane interaction by complex formation
We previously showed that histone-induced toxicity relied onmembrane interaction and calcium influx (27). Preincubation ofhistones with CRP significantly reduced cell membrane associa-tion of FITC-labeled histones (Fig. 5A), as well as histone-inducedinward currents (Fig. 5B, 5C) and calcium influx (Fig. 5D, 5E).These data strongly indicate that masking the membrane bindingsites of histones is the mechanism of CRP detoxification.The proposed mechanism of histone–cell membrane interaction
is their affinity to phosphate groups of major phospholipids in thecell membrane, such as PC, PS, and PE (27, 36, 37), becausephosphate groups exist in both DNA and bilayer lipid membranes.Preincubation of FITC-labeled histones with liposomes made ofPC, PS, and PE significantly reduced the binding of histones to thecell membrane (Fig. 6A). Functionally, this significantly reducedthe toxic effects of histones on endothelial cells (Fig. 6B). Thesedata support the hypothesis that phospholipids on cell membranesare the major targets of histones.It is also known that CRP binds to the phosphocholine group of
lysoPC (34). However, lysoPC is exposed only after the lipid bi-layer of cell membranes has been disturbed, such as during ne-crosis and apoptosis (38). Therefore, CRP does not bind to normalcell membranes. In this study, we found that incubating CRP withEAhy926 cells for 1–4 h did not reduce histone-induced toxicityif CRP were washed away before histone treatment (data notshown). This experiment also indicates that CRP prevention of
histone–membrane interaction is not due to CRP association withcell membranes.In line with our findings of the existence of circulating CRP–
histone complexes, the interaction between CRP and histone hasbeen reported previously but with some discrepant findings (13, 14,39–41). To clarify these discrepancies, we immunofluorescentlystained EAhy926 cells with Cy5-CRP and FITC-ahscFv to avoidany nonspecific staining of IgGs. Their colocalization observed incell nuclei strongly indicates that CRP binds to histones (Fig. 6C).Furthermore, a gel overlay assay showed that all histones couldbind CRP (Fig. 6D) and that CRP could protect endothelial cellstreated with the individual histones (Fig. 6E). Using competitiveELISA, we found that CRP competed with liposomes in bindinghistones (Fig. 6F, 6G) to explain how CRP blocks histone–plasmamembrane integration (Fig. 5A).
DiscussionThe acute-phase response is a complex series of reactions ini-tiated in response to infection, inflammation, injury, or malig-nancy. Elevation in CRP levels has long been recognized in thiscontext, but despite many reports into its properties, clear un-derstanding of its generic function has been lacking (42). In part,this is because no mutation of this phylogenetically ancient andwell-conserved molecule has been described, although a few
FIGURE 5. CRP prevents histone–cell membrane interactions. (A)
Confocal images of living EAhy926 cells incubated with FITC-histones
(10 mg/ml, left panel), FITC-histones + CRP (100 mg/ml, middle panel)
and an equal amount of inactivated FITC alone (right panel) for 10 min.
Scale bar, 20 mm. Arrows in left panel indicate membrane binding of
FITC-histones. Arrows in middle panel indicate aggregation of FITC-
histones. (B) Typical whole-cell current change of EAhy926 cells treated
with 40 mg/ml histones preincubated with 250 mg/ml CRP followed by
histones alone, after washing. (C) Graphic representation of the density of
inward currents (pA/pF) from three independent experiments with mean 6SD, *p, 0.001 compared with other groups (ANOVA). (D) Typical curves
of intracellular calcium ([Ca2+]i) changes of EAhy926 cells treated with
histones (50 mg/ml) preincubated with or without 250 mg/ml CRP. (E)
Intracellular calcium levels with mean 6 SD from three independent
experiments; *p , 0.05 compared with other groups (ANOVA).
The Journal of Immunology 2499
nucleotide polymorphisms that do not alter the protein sequencehave been identified (43). This would suggest a critical functionalrole for CRP. Although the concept of CRP acting to detoxify andclear damaging products of cell destruction was hypothesized
back in 1981 (44), this study demonstrates for the first time, toour knowledge, that high levels of CRP in the acute clinicalphase plays a major role in combating the toxicity of circulatinghistones.
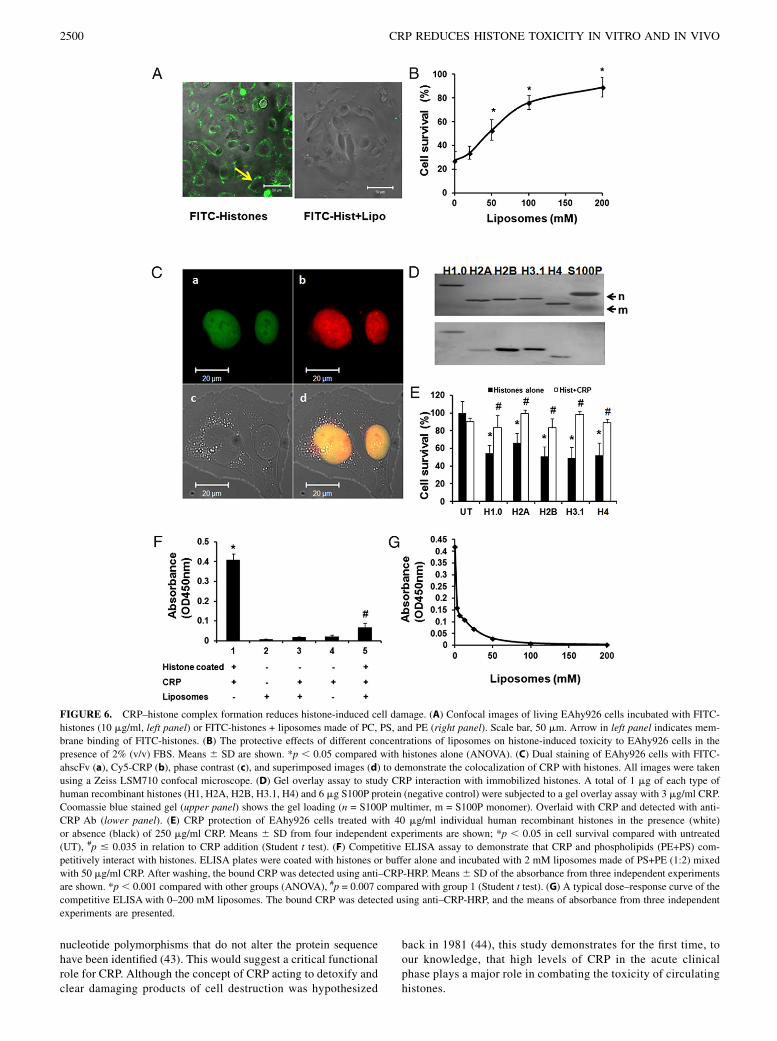
FIGURE 6. CRP–histone complex formation reduces histone-induced cell damage. (A) Confocal images of living EAhy926 cells incubated with FITC-
histones (10 mg/ml, left panel) or FITC-histones + liposomes made of PC, PS, and PE (right panel). Scale bar, 50 mm. Arrow in left panel indicates mem-
brane binding of FITC-histones. (B) The protective effects of different concentrations of liposomes on histone-induced toxicity to EAhy926 cells in the
presence of 2% (v/v) FBS. Means 6 SD are shown. *p , 0.05 compared with histones alone (ANOVA). (C) Dual staining of EAhy926 cells with FITC-
ahscFv (a), Cy5-CRP (b), phase contrast (c), and superimposed images (d) to demonstrate the colocalization of CRP with histones. All images were taken
using a Zeiss LSM710 confocal microscope. (D) Gel overlay assay to study CRP interaction with immobilized histones. A total of 1 mg of each type of
human recombinant histones (H1, H2A, H2B, H3.1, H4) and 6 mg S100P protein (negative control) were subjected to a gel overlay assay with 3 mg/ml CRP.
Coomassie blue stained gel (upper panel) shows the gel loading (n = S100P multimer, m = S100P monomer). Overlaid with CRP and detected with anti-
CRP Ab (lower panel). (E) CRP protection of EAhy926 cells treated with 40 mg/ml individual human recombinant histones in the presence (white)
or absence (black) of 250 mg/ml CRP. Means 6 SD from four independent experiments are shown; *p , 0.05 in cell survival compared with untreated
(UT), #p # 0.035 in relation to CRP addition (Student t test). (F) Competitive ELISA assay to demonstrate that CRP and phospholipids (PE+PS) com-
petitively interact with histones. ELISA plates were coated with histones or buffer alone and incubated with 2 mM liposomes made of PS+PE (1:2) mixed
with 50 mg/ml CRP. After washing, the bound CRP was detected using anti–CRP-HRP. Means6 SD of the absorbance from three independent experiments
are shown. *p , 0.001 compared with other groups (ANOVA), #p = 0.007 compared with group 1 (Student t test). (G) A typical dose–response curve of the
competitive ELISA with 0–200 mM liposomes. The bound CRP was detected using anti–CRP-HRP, and the means of absorbance from three independent
experiments are presented.
2500 CRP REDUCES HISTONE TOXICITY IN VITRO AND IN VIVO

In this study, we demonstrate that CRP could significantly al-leviate histone-induced endothelial damage and coagulation acti-vation, which are the major toxic effects of extracellular histones.CRP protective effects are due to its interaction with histones, andthis has been further confirmed by coimmunofluorescent stainingand gel overlay assay. How CRP binds histones is not clear, butpositively charged histones are more likely to bind the predomi-nantly negatively charged central pore of the CRP pentamer ina similar way to that of complement C1q. Because complementfactor H has been reported to bind dCRP (nonnative pentamericCRP), we also denatured CRP by heating at 70˚C for 1 h as de-scribed in a previous report (4). The dCRP strongly aggregatedwith FITC-labeled histones, and greatly reduced the binding ofhistones to the cell membrane and reduced histone toxicity toendothelial cells (Supplemental Fig. 1) These data indicate thatboth native and nonnative pentameric CRP are able to bind his-tones and reduce their toxic effects.Because histones released after acute cellular and nuclear
damage may also be present in the form of histone–DNA com-plexes, we have additionally checked their toxicity profile and anyblocking effect by CRP. Using isolated nucleosomes (Supple-mental Fig. 2A–C) and histones from swine liver, prepared asdescribed previously (45), we demonstrate that swine histoneshave similar toxicity to cultured endothelial cells as with re-combinant or calf thymus histones (data not shown). However,intact nucleosomes are not toxic (Supplemental Fig. 2D), butdegraded nucleosomes, either from brief sonication or incubationwith serum, showed toxicity to endothelial cells. Significantly, thiscould be reduced by CRP and dCRP (Supplemental Fig. 2D). Toconfirm this finding, genomic DNA fragments were mixed withisolated histones to create artificial histone–DNA complexes, asdescribed previously (25). Again, we found that the complexesshowed similar toxicity to histones alone, but that DNA alone wasnot toxic (Supplementary Fig. 3). These data suggest that thetoxicity of histone–DNA complexes to endothelial cells is pre-dominantly caused by histones, although histone–DNA complexesare stronger activators of TLR2 and TLR4 receptors and subse-quent release of cytokines than histones alone (25). Similarly,CRP or dCRP can significantly reduce this toxicity (Supplemen-tary Fig 3). As to the clinical implications, our previous study hasshown that circulating histone–DNA complexes were detectableonly within 24 h after severe trauma, and that free histones remaindetectable after 72 h (27). This suggests that histone–DNA com-plexes have been quickly degraded into histones. Collectively,these data would indicate that in vivo toxicity is caused by bothhistone–DNA complexes and histones alone in the circulation, andthat CRP can block both forms, as confirmed by adding CRP toserum from patients in the immediate phases after severe trauma(Fig. 4A).As to the mechanism of histone toxicity, a previous report
showed that histones increased cell membrane permeability (46),but a recent report showed that anti-TLR4 or anti-TLR2 Ab couldrescue mice from fatal liver injury to suggest that TLR4 and TLR2may be involved in histone toxicity (25). We demonstrate thathistones integrate onto the endothelial cell membrane both in vitroand in vivo to induce calcium influx, which is essential for histonetoxicity, at least to endothelial cells (27) and platelets (22). CRPprotection relies on the formation of CRP–histone complexes, whichprevents the integration of histones into the cell membrane andconsequently reduced histone-induced calcium influx and celldamage. We also found the aggregation of FITC-labeled histonesafter incubation with CRP both in tissue culture medium and insidecells. Whether CRP enhances the clearance of circulating histonesstill requires further investigation.
Histones can bind different phospholipids (36, 37, 47, 48), andas these are major components of the cell membrane, we speculatethat histones bind to the phospholipid bilayer in the plasma mem-brane in a manner similar to the interaction between the DNAphosphodiester backbone and the histone cores (49). In this study,we demonstrate that artificial liposomes made of phospholipidscompete with the cell membranes for histone binding and signifi-cantly reduce histone-induced toxicity. CRP is also able to competewith phospholipids for binding to histones, and this indicates thatthe major mechanism underlying the protective effects of CRPin vitro and in vivo is in its interference with the histone associ-ation to cell membranes. Therefore, CRP binds histones, as in itsrole in opsonizing pathogens, to block their toxic effect on cellsand to possibly facilitate their clearance. This action defends thehuman body from the toxicity of circulating histones, especially inthe acute phase of many critical illnesses.
AcknowledgmentsWe are indebted to participating patients and their attending staff. We thank
Prof. Mark Pepys for constructive discussion and advice.
DisclosuresThe authors have no financial conflicts of interest.
References1. Peisajovich, A., L. Marnell, C. Mold, and T. W. Du Clos. 2008. C-reactive
protein at the interface between innate immunity and inflammation. ExpertRev. Clin. Immunol. 4: 379–390.
2. Marnell, L., C. Mold, and T. W. Du Clos. 2005. C-reactive protein: ligands,receptors and role in inflammation. Clin. Immunol. 117: 104–111.
3. Thompson, D., M. B. Pepys, and S. P. Wood. 1999. The physiological structureof human C-reactive protein and its complex with phosphocholine. Structure 7:169–177.
4. Hakobyan, S., C. L. Harris, C. W. van den Berg, M. C. Fernandez-Alonso,E. G. de Jorge, S. R. de Cordoba, G. Rivas, P. Mangione, M. B. Pepys, andB. P. Morgan. 2008. Complement factor H binds to denatured rather than tonative pentameric C-reactive protein. J. Biol. Chem. 283: 30451–30460.
5. Szalai, A. J., D. E. Briles, and J. E. Volanakis. 1995. Human C-reactive protein isprotective against fatal Streptococcus pneumoniae infection in transgenic mice.J. Immunol. 155: 2557–2563.
6. Mold, C., S. Nakayama, T. J. Holzer, H. Gewurz, and T. W. Du Clos. 1981. C-reactive protein is protective against Streptococcus pneumoniae infection inmice. J. Exp. Med. 154: 1703–1708.
7. Du Clos, T. W., and C. Mold. 2011. Pentraxins (CRP, SAP) in the process ofcomplement activation and clearance of apoptotic bodies through Fcgammareceptors. Curr. Opin. Organ Transplant. 16: 15–20.
8. Lu, J., L. L. Marnell, K. D. Marjon, C. Mold, T. W. Du Clos, and P. D. Sun. 2008.Structural recognition and functional activation of FcgammaR by innate pen-traxins. Nature 456: 989–992.
9. Lu, J., K. D. Marjon, L. L. Marnell, R. Wang, C. Mold, T. W. Du Clos, andP. Sun. 2011. Recognition and functional activation of the human IgA receptor(FcalphaRI) by C-reactive protein. Proc. Natl. Acad. Sci. USA 108: 4974–4979.
10. Mold, C., H. D. Gresham, and T. W. Du Clos. 2001. Serum amyloid P com-ponent and C-reactive protein mediate phagocytosis through murine Fc gammaRs. J. Immunol. 166: 1200–1205.
11. Marjon, K. D., L. L. Marnell, C. Mold, and T. W. Du Clos. 2009. Macrophagesactivated by C-reactive protein through Fc gamma RI transfer suppression ofimmune thrombocytopenia. J. Immunol. 182: 1397–1403.
12. Thomas-Rudolph, D., T. W. Du Clos, C. M. Snapper, and C. Mold. 2007. C-reactive protein enhances immunity to Streptococcus pneumoniae by targetinguptake to Fc gamma R on dendritic cells. J. Immunol. 178: 7283–7291.
13. Shephard, E. G., P. D. van Helden, M. Strauss, L. Bohm, and F. C. De Beer.1986. Functional effects of CRP binding to nuclei. Immunology 58: 489–494.
14. Du Clos, T. W., L. T. Zlock, and R. L. Rubin. 1988. Analysis of the binding of C-reactive protein to histones and chromatin. J. Immunol. 141: 4266–4270.
15. Pepys, M. B., and P. J. Butler. 1987. Serum amyloid P component is the majorcalcium-dependent specific DNA binding protein of the serum. Biochem. Bio-phys. Res. Commun. 148: 308–313.
16. Poon, I. K., M. D. Hulett, and C. R. Parish. 2010. Molecular mechanisms of lateapoptotic/necrotic cell clearance. Cell Death Differ. 17: 381–397.
17. Szalai, A. J. 2004. C-reactive protein (CRP) and autoimmune disease: facts andconjectures. Clin. Dev. Immunol. 11: 221–226.
18. Du Clos, T. W., L. T. Zlock, P. S. Hicks, and C. Mold. 1994. Decreased auto-antibody levels and enhanced survival of (NZB x NZW) F1 mice treated with C-reactive protein. Clin. Immunol. Immunopathol. 70: 22–27.
19. Kravitz, M. S., M. Pitashny, and Y. Shoenfeld. 2005. Protective molecules—C-reactive protein (CRP), serum amyloid P (SAP), pentraxin3 (PTX3), mannose-
The Journal of Immunology 2501

binding lectin (MBL), and apolipoprotein A1 (Apo A1), and their autoanti-bodies: prevalence and clinical significance in autoimmunity. J. Clin. Immunol.25: 582–591.
20. Rodriguez, W., C. Mold, M. Kataranovski, J. Hutt, L. L. Marnell, and T. W. DuClos. 2005. Reversal of ongoing proteinuria in autoimmune mice by treatmentwith C-reactive protein. Arthritis Rheum. 52: 642–650.
21. Semeraro, F., C. T. Ammollo, J. H. Morrissey, G. L. Dale, P. Friese, N. L. Esmon,and C. T. Esmon. 2011. Extracellular histones promote thrombin generationthrough platelet-dependent mechanisms: involvement of platelet TLR2 andTLR4. Blood 118: 1952–1961.
22. Fuchs, T. A., A. A. Bhandari, and D. D. Wagner. 2011. Histones induce rapid andprofound thrombocytopenia in mice. Blood 118: 3708–3714.
23. Ammollo, C. T., F. Semeraro, J. Xu, N. L. Esmon, and C. T. Esmon. 2011. Extra-cellular histones increase plasma thrombin generation by impairing thrombomodulin-dependent protein C activation. J. Thromb. Haemost. 9: 1795–1803.
24. Xu, J., X. Zhang, R. Pelayo, M. Monestier, C. T. Ammollo, F. Semeraro,F. B. Taylor, N. L. Esmon, F. Lupu, and C. T. Esmon. 2009. Extracellular his-tones are major mediators of death in sepsis. Nat. Med. 15: 1318–1321.
25. Xu, J., X. Zhang, M. Monestier, N. L. Esmon, and C. T. Esmon. 2011. Extra-cellular histones are mediators of death through TLR2 and TLR4 in mouse fatalliver injury. J. Immunol. 187: 2626–2631.
26. Huang, H., J. Evankovich, W. Yan, G. Nace, L. Zhang, M. Ross, X. Liao,T. Billiar, J. Xu, C. T. Esmon, and A. Tsung. 2011. Endogenous histones functionas alarmins in sterile inflammatory liver injury through Toll-like receptor 9 inmice. Hepatology 54: 999–1008.
27. Abrams, S. T., N. Zhang, J. Manson, T. Liu, C. Dart, F. Baluwa, S. S. Wang,K. Brohi, A. Kipar, W. Yu, et al. 2013. Circulating histones are mediators oftrauma-associated lung injury. Am. J. Respir. Crit. Care Med. 187: 160–169.
28. Jaffe, E. A., R. L. Nachman, C. G. Becker, and C. R. Minick. 1973. Culture ofhuman endothelial cells derived from umbilical veins. Identification by mor-phologic and immunologic criteria. J. Clin. Invest. 52: 2745–2756.
29. Riccardi, C., and I. Nicoletti. 2006. Analysis of apoptosis by propidium iodidestaining and flow cytometry. Nat. Protoc. 1: 1458–1461.
30. Perez-Casal, M., C. Downey, B. Cutillas-Moreno, M. Zuzel, K. Fukudome, andC. H. Toh. 2009. Microparticle-associated endothelial protein C receptor and theinduction of cytoprotective and anti-inflammatory effects. Haematologica 94:387–394.
31. Barrett-Jolley, R., C. Dart, and N. B. Standen. 1999. Direct block of native andcloned (Kir2.1) inward rectifier K+ channels by chloroethylclonidine. Br.J. Pharmacol. 128: 760–766.
32. Grynkiewicz, G., M. Poenie, and R. Y. Tsien. 1985. A new generation of Ca2+indicators with greatly improved fluorescence properties. J. Biol. Chem. 260:3440–3450.
33. Smirnov, M. D., and C. T. Esmon. 1994. Phosphatidylethanolamine incorpora-tion into vesicles selectively enhances factor Va inactivation by activated proteinC. J. Biol. Chem. 269: 816–819.
34. Volanakis, J. E., and K. W. Wirtz. 1979. Interaction of C-reactive protein withartificial phosphatidylcholine bilayers. Nature 281: 155–157.
35. Alberio, L., O. Safa, K. J. Clemetson, C. T. Esmon, and G. L. Dale. 2000.Surface expression and functional characterization of alpha-granule factor V inhuman platelets: effects of ionophore A23187, thrombin, collagen, and con-vulxin. Blood 95: 1694–1702.
36. Furnrohr, B. G., G. J. Groer, B. Sehnert, M. Herrmann, and R. E. Voll. 2007.Interaction of histones with phospholipids—implications for the exposure ofhistones on apoptotic cells. Autoimmunity 40: 322–326.
37. Pereira, L. F., F. M. Marco, R. Boimorto, A. Caturla, A. Bustos, E. G. De laConcha, and J. L. Subiza. 1994. Histones interact with anionic phospholipidswith high avidity; its relevance for the binding of histone-antihistone immunecomplexes. Clin. Exp. Immunol. 97: 175–180.
38. Gershov, D., S. Kim, N. Brot, and K. B. Elkon. 2000. C-Reactive protein binds toapoptotic cells, protects the cells from assembly of the terminal complementcomponents, and sustains an antiinflammatory innate immune response: impli-cations for systemic autoimmunity. J. Exp. Med. 192: 1353–1364.
39. Minota, S., N. Morino, H. Sakurai, A. Yamada, and Y. Yazaki. 1993. Interrela-tionship between autoepitope, DNA-binding domain, and CRP-binding domainon a histone H1 molecule. Clin. Immunol. Immunopathol. 66: 269–271.
40. Shephard, E. G., P. J. Smith, S. Coetzee, A. F. Strachan, and F. C. de Beer. 1991.Pentraxin binding to isolated rat liver nuclei. Biochem. J. 279: 257–262.
41. Pepys, M. B., S. E. Booth, G. A. Tennent, P. J. Butler, and D. G. Williams. 1994.Binding of pentraxins to different nuclear structures: C-reactive protein binds tosmall nuclear ribonucleoprotein particles, serum amyloid P component binds tochromatin and nucleoli. Clin. Exp. Immunol. 97: 152–157.
42. Pepys, M. B., and G. M. Hirschfield. 2003. C-reactive protein: a critical update.J. Clin. Invest. 111: 1805–1812.
43. Hage, F. G., and A. J. Szalai. 2007. C-reactive protein gene polymorphisms, C-reactive protein blood levels, and cardiovascular disease risk. J. Am. Coll.Cardiol. 50: 1115–1122.
44. Pepys, M. B. 1981. C-reactive protein fifty years on. Lancet 1: 653–657.45. Schnitzler, G. R. 2001. Isolation of histones and nucleosome cores from mam-
malian cells. Curr. Protoc. Mol. Biol. Chapter 21: Unit 21.5.46. Abakushin, D. N., I. A. Zamulaeva, and A. M. Poverenny. 1999. Histones evoke
thymocyte death in vitro; histone-binding immunoglobulins decrease their cy-totoxicity. Biochemistry Mosc. 64: 693–698.
47. Zhao, H., S. Bose, E. K. Tuominen, and P. K. Kinnunen. 2004. Interactions ofhistone H1 with phospholipids and comparison of its binding to giant liposomesand human leukemic T cells. Biochemistry 43: 10192–10202.
48. Gavilanes, J. G., M. A. Lizarbe, A. M. Munico, and M. Onaderra. 1985. Inter-action of dipalmitoyl-phosphatidylcholine with calf thymus histone H1. Int.J. Pept. Protein Res. 26: 187–194.
49. Luger, K., A. W. Mader, R. K. Richmond, D. F. Sargent, and T. J. Richmond.1997. Crystal structure of the nucleosome core particle at 2.8 A resolution.Nature 389: 251–260.
2502 CRP REDUCES HISTONE TOXICITY IN VITRO AND IN VIVO





























