Embed Size (px)
Citation preview

Spectrochimica Acta Part B 58(2003) 1109–1124
0584-8547/03/$ - see front matter� 2003 Elsevier Science B.V. All rights reserved.doi:10.1016/S0584-8547Ž03.00067-3
Coherent control of alkali cluster fragmentation dynamics�
Albrecht Lindinger, Cosmin Lupulescu, Andreas Bartelt, Stefan Vajda, Ludger Woste*ˇ ¨
Institut fur Experimentalphysik, Freie Universitat Berlin, Arnimallee 14, D-14195 Berlin, Germany¨ ¨
Received 11 December 2002; accepted 27 February 2003
Abstract
Metal clusters exhibit extraordinary chemical and catalytic properties, which sensitively depend upon their size.This behavior makes them interesting candidates for the real-time analysis of ultrafast photo-induced processes—ultimately leading to coherent control scenarii. We have performed transient multi-photon ionization experiments onsmall alkali clusters of different size in order to probe their wave packet dynamics, structural reorientations, chargetransfers and dissociative events in different vibrationally excited electronic states including their ground state. Theobserved processes were highly dependent on the irradiated pulse parameters, like its phase, amplitude and duration;an emphasis to employ a feedback control system for generating the optimum pulse shapes. Their spectral andtemporal behavior reflects interesting properties about the investigated system and the irradiated photochemicalprocess. We present first the vibrational dynamics of bound, dissociated, and pre-dissociated electronically excitedstates of alkali dimers and trimers. The scheme for observing the wave packet dynamics in the electronic groundstate using stimulated Raman-pumping is shown. Since the employed pulse parameters significantly influence theefficiency of the irradiated dynamic pathways photo-induced fragmentation experiments on bifurcating reactionchannels were carried out. In these experiments different branching ionization and fragmentation pathways ofelectronically excited Na K were investigated. By employing an evolutionary algorithm for optimizing the phase and2
amplitude of the applied laser field, the yield of the resulting parent or fragment ions could significantly be influencedand interesting features could be concluded from the obtained optimum pulse shapes revealing the characteristicmolecular oscillation period. Moreover, the influence on the optimal pulse shape due to fragmentation from largerclusters into NaK is obtained. The substructure of the optimal pulse shape thereby offers new insight into thefragmentation channel during the control process. Characteristic motions of the involved wave packets are proposed,in order to explain the optimized dynamic dissociation pathways.� 2003 Elsevier Science B.V. All rights reserved.
Keywords: Alkali cluster; Fragmentation; Coherent control experiment
� This paper was presented at the International Conference on Laser Probing(LAP-2002), held in Leuven, Flanders, Belgium,July 2002, and is published in the Special Issue ofSpectrochimica Acta Part B, dedicated to that conference.
*Corresponding author. Fax:q49-30-838-55567.E-mail address: [email protected](L. Woste).¨

1110 A. Lindinger et al. / Spectrochimica Acta Part B 58 (2003) 1109–1124
Fig. 1. The principle of pump and probe spectroscopy bymeans of transient two-photon ionization: a first fs-laser pulseelectronically excites the particle into an ensemble of vibra-tional states creating a wave packet. Its temporal evolution isprobed by a second probe pulse, which ionizes the excitedparticle as a function of the time-dependent Franck–Condon-window. Shown is the principle for a bound–bound transition,where the oscillative behavior of the wave packet will appear.
1. Introduction
Alkali metal clusters are excellent model sys-tems for time-resolved investigations in the fem-tosecond (fs)-regime: they exhibit pronouncedabsorption bandsw1x, which are well located withinthe tuning range of the available fs-laser sources;their detachment and photo-ionization energiesw2xare also well within their reach, and the vibrationalfrequencies can well be resolved with the availablefs-pulse lengths. We have developed a scheme forobserving wave packet and reaction dynamics alsoin the electronic ground state by means of stimu-lated Raman-pumpingw3x. As demonstrated forNa , the photo-induced intramolecular dynamics3
of a system critically depends upon the employedpulse parametersw4x. Based on a suggestion ofJudson and Rabitzw5x we have, therefore, carriedout active control experiments, in which we excit-ed differently branching fragmentation and ioni-zation pathways of photo-excited Na K, its2
corresponding fragment NaKw6x and variousorganometallic compoundsw7x. An evolutionaryalgorithm w8x for optimizing the phase and ampli-tude of the applied laser field could significantlyinfluence the yield of the resulting parent andfragment ions. Another major goal of closed loopexperiments is to gain information about the photo-induced control process itselfw6x. The observable,suitable for this inversion, is the acquired optimalpulse shape. Since the obtained pulse shapes areusually complicated and difficult to interpret, it isuseful to start with these simple model systems,where the number of possible pathways is reduced.This will allow an easier access to the interpreta-tion of the optimized process. By increasing thecomplexity of the investigated system, one canprogressively gain further information about theadded parts. The additional complexity can berealized e.g. by attaching auxiliary atoms to themolecules. This can be achieved in a molecularbeam by modifying the stagnation conditions likesource pressure and temperature. In this way,clusters of different size distribution can be pro-duced. These clusters may fragment into smallermolecules during the optimization. Therefore, theacquired pulse shape should also reveal theenhancement of the fragmentation efficiency.
Thereby, the obtained optimal pulse shapes carryfingerprints of the chosen dynamic pathway of theinvolved molecules. The resulting modification ofthe optimal pulse shapes offers new insights intocompeting fragmentation channels during the con-trol process.
2. Experimental set-up of the pump and probeexperiment
The principle of the pump and probe observationscheme is plotted in Fig. 1. The clusters are excitedfrom a low vibrational level of the electronicground state to an excited state. Due to the spectralwidth of the employed fs-pump pulse, a coherentsuperposition of several vibrational states is excit-ed, which leads to the formation of a wave packet.

1111A. Lindinger et al. / Spectrochimica Acta Part B 58 (2003) 1109–1124
Fig. 2. Experimental set-up of the pump and probe experiment, showing the molecular beam irradiated by laser pulse sequencescoming from a Ti:Sapphire fs-laser system. The resulting photoions are detected by a quadrupole mass spectrometer.
If a bound electronic state is excited(Fig. 1), thiswave packet will oscillate between the inner andouter turning point of the potential energy surface,thereby reflecting the vibrational motion of theexcited molecule. The temporal evolution of thiswave packet can be monitored with the probepulse, which—at a well-defined variable timedelay—ionizes the particle into a size-selectivelydetectable state. This process may occur directlyor via a higher electronic excited state. Since,however, the efficiency of this ionization step
sensitively depends on the position of the wavepacket along the reaction coordinate, the obtainedionization efficiency (time-dependent Franck–Condon factor) changes significantly as a functionof time delay between excitation(pump) andionization (probe). By tuning this time delay, thetemporal evolution of the oscillating wave packetappears as an intensity modulation on the corre-sponding ion channel.
The scheme of the experiment is plotted in Fig.2. The metal clusters are produced in an adiabatic

1112 A. Lindinger et al. / Spectrochimica Acta Part B 58 (2003) 1109–1124
beam expansion consisting of a radiatively heatedoven cartridge, from which the alkali metal vaporis co-expanded with 3 bars of argon carrier gaspressure across a nozzle of 70mm diameter intothe vacuum. For this purpose, the cartridge issurrounded with seven resistively heated hairpin-shaped tungsten filaments and a threefold tantalumradiation shield. During the experiment the ovenis heated to a temperature, at which an alkali vaporpressure of some 10 bars is achieved, wherebyy1
a slightly higher temperature in the nozzle regionis set to prevent clogging. The expansion zone isevacuated by a diffusion pump system of 5000 lys pumping speed, which results in a pressure of3=10 mbar in the oven chamber during they4
experiment. From the expansion a molecular beamis skimmed off, using a skimmer of 1 mm diam-eter. It is placed approximately 10 mm behind theoven orifice. The skimmer leads into the detectionchamber, which is differentially pumped by twoturbomolecular pumps of 2200 and 500 lys. Thisresults in a pressure of approximately 1=10y5
mbar during the experiment. The mass flux of theneutral alkali cluster beam is permanently moni-tored by a Langmuir–Taylor detector, which islocated 50 cm downstream from the skimmer. Ata distance of 10 cm downstream from the skimmer,a quadrupole mass spectrometer(Balzers QMG420) is perpendicularly oriented to the clusterbeam, in order to extract the resulting photoions.Window ports at the detection chamber allow oneto irradiate the interaction zone perpendicular toboth, the neutral and the extracted ion beam.
For the experiment a commercial fs-laser is used(Spectra-Physics 3960 Tsunami), which is pumpedby a Nd:YVO laser(Spectra-Physics Millennia4
X). The laser operates at a repetition rate of 80.6MHz; it is tunable in a wavelength range between730 and 850 nm, producing pulses of 80 fsduration at a total power of 1.6 W. These condi-tions allow one to operate the experiment at a100% duty cycle, since each molecule of thecontinuous molecular beam is irradiated severaltimes by the pulsed laser. Furthermore, the corre-sponding low laser peak power prevents undesiredmulti-photonic transitions, which would camou-flage the sought information. The wavelengthrange of the laser can significantly be extended by
using a second harmonic generator(SHG) or anoptical parametric oscillator. The employed laserpulses are analyzed with a spectrometer, autocor-relator, spectrally-resolved cross-correlation andSHG–FROG(Femtos). In the pump and probeexperiment, the laser pulses were split up andrecombined in a Michelson interferometer system,allowing to generate pump and probe sequencesof a variable delay.
3. Pump and probe spectra of bound electronicstates
Transient two-photon ionization experiments ontrimer systems were motivated by the need for atime-resolved verification of the pseudo-rotationmotion, which can be considered as a superpositionof the asymmetric stretch(q ) and the bendingx
vibration (q ) w9x. In this respect, the situation ofy
a triatomic molecule with its three vibrationalmodes is quite different from an isolated oscillatingdimer, which vibrates in its single mode untileventually it radiates back to the electronic groundstate or predissociates. The coupling of vibrationalmodes in a trimer system can be considered as theonset of internal vibrational redistribution—a cru-cial phenomenon in metal cluster dynamicsw10x.
A typical result, which was obtained for theelectronic Na B§X-transition with transform-lim-3
ited pulses of approximately 100 fs duration(FWHM), is shown in Fig. 3. The progressionshows a pronounced molecular vibration, indicat-ing only one vibrational mode of 320 fs duration,which corresponds to the symmetric stretch(q )smode of Na in the B-state. There is, however, no3
indication for the asymmetric stretch mode, thebending mode or for pseudo-rotation.
4. Spectroscopy of electronic ground states usingchirped pulses
In the time-resolved experiments described sofar, the pump laser simply populates the interme-diate excited state. Therefore, the experimentbecomes a means to study that particular excitedstate. Dynamic processes like catalysis and theor-etical studies, however, are often more concernedabout the electronic ground state than about excited

1113A. Lindinger et al. / Spectrochimica Acta Part B 58 (2003) 1109–1124
Fig. 3. Transient two-photon ionization spectrum of Na3
recorded with transform-limited 80 fs pulses at a wavelengthof 620 nm. The progression exhibits the symmetric stretchmode of the electronically excited B-state.
Fig. 4. Optical set-up to create linearly chirped laser pulses.
states. For this purpose, it is useful to investigatevibrational wave packet dynamics from thatground state. One possibility to do this is stimu-lated emission Raman-pumping. This requires toexcite the species of interest to an electronicallyexcited state and subsequently to ‘down-pump’ thesystem from there back into an ensemble of highervibrational levels in the electronic ground state,whereby generating an oscillating wave packetthere. This can be achieved by raising the powerlevel of the pump laser pulses. Experiments of thiskind were performed on potassium dimersw3x.Another example obtained on Na will be shown3
below. Besides that, we developed a new methodcalled ‘charge-reversal spectroscopy’ or ‘NeNePo’(negative–positive–neutral). It allowed us to pre-pare vibrational wave packets in very low elec-tronic states or even in the electronic ground stateof size-selected neutral clustersw10x.
The spectrum in Fig. 3 exhibits as mentionedabove the oscillatory features of the symmetricstretch motion of Na in its electronically excited3
B-state, indicating the well-known oscillation timeof 320 fs. The pump and probe spectrum wasobtained with transform-limited pulses of 100 fsduration at a center wavelength of 620 nm. Thenthe experiment was repeated by changing only oneexperimental parameter: the duration of the pumppulse. This was accomplished—as indicated in Fig.4—by passing the pump beam across a set of twoparallel gratings. The assembly creates a linear

1114 A. Lindinger et al. / Spectrochimica Acta Part B 58 (2003) 1109–1124
Fig. 5. Pump and probe spectrum of Na obtained under iden-3
tical experimental conditions, as Fig. 3 with the only exceptionthat the employed pump pulse was linearly downchirped to aduration of 400 fs. The resulting oscillation time of 230 fscorresponds to the symmetric stretch vibration of the electronicground state of Na .3
Fig. 6. Schematic view of the stimulated Raman-pumping pro-cess: the blue section of the chirped pump pulse excites thesystem; the red component stimulated it back down to the elec-tronic ground state, but Stokes-shifted to a higher vibrationallevel. The resulting wave packet is probed by the delayed probepulse, which ionizes the particle from the oscillating groundstate.
frequency down chirp. Its duration and spectralsequence depends only on the incidence anglebetween laser beam and gratings. Fig. 5 shows thecorresponding Na pump and probe spectrum,3
when a 400 fs downchirped pulse(blue precedingred) was applied. Surprisingly, instead of the priorobserved oscillation time of 320 fs(Fig. 3), thisspectrum now shows in the positive time range,where the ‘pump’ pulse occurs prior to the probepulse, an oscillation time of 230 fs, which corre-sponds to the symmetric stretch mode of theelectronic ground state of the sodium trimer. Thisobservation can be explained by the scheme plot-ted in Fig. 6: the frontal frequency components
(shorter wavelengths) create a propagating wavepacket in the B-state of the trimer, which is thendumped down into the ground state(X-state) bythe delayed longer wavelength components of thesame pulse. The resulting vibrational motion ofthe ground state is then monitored by the unchirpedprobe pulse via two-photon ionization(stimulatedRaman-pumping). The length of the employedpulses was determined from wavelength-resolvedcross-correlation measurements. The experimentclearly indicates the very sensitive dependence ofthe occurring intramolecular dynamics with regardto the irradiated pulse shape. The aspect is mostexciting, when competing chemical reaction chan-nels are controled in this way, as shown in thenext chapter.

1115A. Lindinger et al. / Spectrochimica Acta Part B 58 (2003) 1109–1124
Fig. 7. Schematic view of the investigated three-photonic frag-mentation and ionization process. Shown is the channel for thebound–bound transition leading to the mother ion formationand the fragmentative process across a pre-dissociated state,where the wave packet progressively leaks into a fragmentationchannel.
5. Reaction control with shaped pulses
Pulse-shaping techniques, allowing amplitudeand phase modulation of fs-laser pulses, haveopened fascinating perspectives for driving themolecular reaction dynamics in real-time. Thesubject becomes most exciting, when—as pro-posed in 1992 by Judson and Rabitzw5x—feed-back-loop algorithms are employed. In this way,the optimum laser pulse shapes are generated,which drive the reaction to a maximum yield on adesired path. Thermally, these reactions may verywell be inaccessible. An important first experimen-tal step in this regard was performed by Gerber etal. w11x, who employed a genetic algorithmw8x tooptimize a fs-laser pulse profile, which then chan-neled the highly fragmentative multi-photon ioni-zation process of metal carbonyl compounds eitherto maximize the mother ion or a fragment ionyield. As emphasized by Rabitz, the experimentcan be viewed as an analog computer, which solvesthe Schroedinger equation in real-time and drivesthe reaction into a desired direction. An importantissue in this regard remains the question about theinformation content in the optimized laser pulseshape. The extraction of information from theacquired laser field is the most attractive aspect,since it may lead to a conceptionally new approachto the investigation of molecular dynamics.Employing evolutionary strategies—nature’s con-cept itself—the experiment does not only learn tocontrol the dynamics of a molecular system byoptimizing a problem of high dimensionality, butalso acquires valuable information about themolecular potential energy surface along the reac-tion path. To extract this information from theoptimized laser field, fundamental questions like,for example, the energy dissipation and the loss ofcoherence in a system of increasing complexitymust be understood. In this regard it is veryimportant to investigate systems which are simple.Small clusters are therefore, good models. Thesolution then reflects their unique dynamical fea-tures. Based on these findings, related systems ofslowly increased degrees of freedom may thenbecome controllable and understandable, as well.
A schematic view of the involved transientmulti-photon ionization process is shown in Fig.7. In the pump–probe experiment the first fs-pumppulse excites Na K in a one-photonic transition2
from its ground state to an electronically excitedstate, thereby creating a coherent vibrational wavepacket. According to ab initio calculations ofBonacic-Koutecky(private communication), the´excitation wavelength corresponds to a transitioninto the bound excited electronic state 3 A . This2
1
state is crossed by a second electronic state 1 A22
above its vibrational dissociation limit. As a result,the oscillating wave packet leaks into a fragmen-tation channel, forming NaK* and Na. The tem-poral evolution of the oscillating mother moleculeand its fragment are then monitored by the probepulse, which ionizes the electronically excitedspecies in a two-photon process, leading either toNa K or NaK . The systematically performedq q
2

1116 A. Lindinger et al. / Spectrochimica Acta Part B 58 (2003) 1109–1124
Fig. 8. Temporal evolution of a transient three-photon ionization signal of decaying Na K(a) and its emerging fragment NaK(b).2
The decay time of the excited trimer corresponds well to the rise time of its diatomic fragment. The superposed oscillations indicatewave packet oscillations in the coherently excited system.
pump and probe spectroscopy on alkali clustersprovided a good indication about suited candidatesfor a coherent control experiment. Among these,the fragmentation dynamics of the heteronucleartrimer Na K appeared to us the most appropriate.2
The corresponding pump–probe spectrum isshown in Fig. 8a. It clearly exhibits—superim-posed on an exponential decay with a time constantof 3.28 ps—an oscillatory behavior with a periodof roughly 500 fs. The Fourier-transform of thisoscillation w12x shows two peaks at 18 and 67.4ycm, which reflect two of the three vibrationalmodes of the photo-excited trimer. Fig. 8b showsthe transient of the fragment NaK, where theexponential rise time of 3.25 ps indicates withinthe experimental error its origin from the equallyfast decaying Na K. The observed period of oscil-2
lation of 440 fs corresponds to the well-knowneigenfrequency√s75ycm of this electronicallyexcited dimerw13x. It should be pointed out, thatin this one-color experiment the pump and theprobe pulse originate both from the same laser
source. This allows a decisive modification, inwhich the pump–probe scheme is replaced bytailor-made pulse shapes, generated from single fs-pulses w14x. In order to optimize the describedthree-photon ionization process to maximum yieldsof either mother ion or fragment signal channel,we can now establish a self-learning feedback-loop w5x between ion signal detector and pulseshaper. With this experiment the question will beanswered, if the obtained pulse shapes reflect theintrinsic properties of the product ions.
6. Set-up of the coherent control experiment
In the experimental set-up basic components ofthe already described pump and probe experimentlike the supersonic molecular beam apparatus,quadrupole mass spectrometer, and the fs-laser arere-used (Fig. 2). As shown in Fig. 9 in theoptimization experiment, the laser beam is passedacross a pulse shaper system, which allows tomodulate simultaneously the phase and amplitude

1117A
.L
indingeret
al./
Spectrochimica
Acta
Part
B58
(2003)1109–1124
Fig. 9. Schematic view of the feedback-loop for optimizing the ion yields of particular reactive channels by employing a self-learning algorithm.

1118 A. Lindinger et al. / Spectrochimica Acta Part B 58 (2003) 1109–1124
of the laser pulses by applying voltages to a doublemask liquid crystal spatial light modulator(SLM)consisting of 2=128 pixels w14x. The SLM isplaced in the Fourier plane of a zero dispersioncompressor, which is a linear set-up of two gratings(1200 lymm) and two plano-convex lenses(fs200 mm) in a 4f-arrangement. Voltages can inde-pendently be applied on each pixel of themodulator, allowing the modification of the ampli-tude and phase of the transmitted light. By com-puter control, pulses of arbitrary form can thus begenerated. These shaped pulses are then focusedinto the detection chamber and the current of thedesired mass-selected ion is taken as a feedbacksignal.
The optimization algorithm iteratively drives thepulse shaper in order to maximize the ion signal.A schematic view of this procedure is given inFig. 9. We apply an algorithm based on evolution-ary strategiesw15x in order to find the pulse shapein the 128-dimensional searching space, whichyields to the highest feedback signal. We performexclusively phase optimizations in order to keepthe pulse intensity constant. Evolutionary strategiesare based on the concept of evolution, applyingschemes like ‘cross-over’, ‘mutation’ and ‘survivalof the fittest’. In the beginning of the cycle 10parent arrays of 256 random numbers are created(parent individuals). The first 128 elements specifythe spectral phase shift, the others define thespectral attenuation of the modulator pixels. Whensent to the modulator, a certain pulse shape iscreated from a bandwidth-limited pulse. In thecrossover stage, 15 pairs of new individuals arecreated from pairs of parent individuals by ran-domly distributing copies of the respective parentarray elements between the new pair. Thereby, apopulation of 30 individuals is formed. Each arrayelement is then altered by adding a random numberfrom a Gaussian probability distribution(muta-tion). The whole population is subsequently testedby recording the ion yield for each modulatorsetting. Only those pulse shapes, which producethe highest ion yield are selected(survival of thefittest) and taken as parents for the next generation.The optimization proceeds until a convergence ofthe ion yield is achieved.
The produced optimal pulse shapes are analyzedby SHG–FROG, intensity auto-correlation andintensity cross-correlation technique. When theauto-correlation trace suggests a pulse train, theanalysis of the auto-correlation trace allows one toretrieve the pulse form by a downhill simplex fit,starting with a function consisting of up to fiveinput Gaussian peaks. In the next step an auto-correlation trace is generated. This calculated auto-correlation trace is compared with the experimentaltrace. The width, height and position of the indi-vidual peaks are then searched iteratively withinthe downhill simplex method. When testing thisprocedure against the SHG–FROG—as will bediscussed below—data consistent for NaK wereobtained. Since neither the analysis of both SHG–FROG traces nor SHG auto-correlation data definethe real-time ordering of the pulses contained insuch pulse trains, intensity cross-correlation usingan unchirped fs-laser pulse as a reference pulsewas applied. The intensity cross-correlation tran-sients provide an intuitive and direct informationabout the time ordering of the pulse elements. Inthe case of the Na K optimization experiment2
discussed in this paper, the full FROG trace of theemerging pulse form could not be recorded withour present set-up. This is due to the long temporalstructure of the wave form consisting of a leadingintense double-pulse sequence followed by twolow-intensity pulses occurring at times 3.7 and 8.0ps(observed via auto-correlation) and thus exceed-ing the time-window of our SHG–FROG config-uration (part of the FROG trace was cut off). Inthis case, the pulse from was obtained from inten-sity auto-correlation measurements and the timesequence of the leading pulses was determined byintensity cross-correlation. The two weak pulsesappearing at later times contribute to the overallintensity by;10%. Taking into account the frag-mentation time of Na K* and that under given2
experimental conditions no larger aggregates werepresent in the molecular beam, the occurrence ofthe two delayed weak peaks can be explained onlyby a failure of the optimization algorithm todiminish them.
In order to investigate the more complex pulsestructures, we applied the spectrally and temporallyresolved up-conversion technique(STRUT). The

1119A. Lindinger et al. / Spectrochimica Acta Part B 58 (2003) 1109–1124
Fig. 10. Evolution of the ion yield during the optimization experiment plotted for the Na K-signal(a) and the NaK -signal(b).q2
reference pulse, directly taken from the Ti:Sapphirelaser, is characterized by SHG–FROG. In STRUT,the cross-correlation signal of the shaped test pulseand the reference pulse is frequency-resolved. Thisis achieved by recording the cross-correlation trac-es for different frequency components of the spec-trum. When operating in the weak field regime,STRUT offers the advantage of providing anintuitive viewgraph, indicating the time–frequencycharacter of the pulse form. In order to fullycharacterize the pulse form, the amplitude andphase can be calculated from the STRUT spectro-gram by an iterative phase-retrieval algorithm.
7. Results and discussion
Fig. 10 shows a typical evolution of the ionyield during the optimization procedure for thetriatomic mother ion Na K (a) and the diatomicq
2
fragment ion NaK (b). In the first generation, allq
pulses are random; hence, the resulting ion yieldis small. As the iteration proceeds, the ion yieldincreases and reaches convergence roughly after150 and 70 generations for Na K and NaK ,q q
2
respectively. We have to point out, that under thegiven experimental conditions the overall NaKq
ion signal consists not only of photo-fragments.There is still another contribution from directlyphoto-ionized NaK dimers, which are also presentin the molecular beam. As estimated from thepump–probe spectrum of NaK, the contribution ofionic fragments to the overall signal is approxi-mately 20%. It should be noted, that under certainexperimental conditions also an Ar -mass peakq
emerges between the two K -isotopes, which isq
due to multi-photon ionization of the largely abun-dant carrier gas.
The evolution of the ion yields during theoptimization procedure(as shown in Fig. 10a andb) clearly indicate, that we have successfullyperformed a control experiment, in which theefficiency of competing reactive pathways wasoptimized by applying feedback looped, phase-shaped fs-laser pulses. The most important aspectin this regard is the information content, which isacquired during the optimization procedure. In thecase of the mother trimer Na K, a double-pulse2
sequence(as shown in Fig. 11) was retrieved from

1120 A. Lindinger et al. / Spectrochimica Acta Part B 58 (2003) 1109–1124
Fig. 11. Oscillatory component of the transient pump and probeNa K -signal compared with the acquired pulse shape for pro-q
2
ducing a maximum yield of Na K .q2
the analysis of the auto-correlation trace. The timedifference between the first and second pulse isapproximately 1240 fs. This corresponds exactlyto 2.5 oscillation periods of the electronicallyexcited trimer. The intensity of the second pulseis approximately 80% higher than the first pulse.This can be understood by the fact, that the firstpulse excites the system in a one-photonic resonanttransition, whereas the following ionization steprequires two-photons associated with a lower tran-sition probability. It is known from pump andprobe spectroscopyw12x, that the photo-ionizationof the electronically excited Na K molecule occurs2
in the Franck–Condon-window at the outer turningpoint of the propagating wave packet. The obser-vation, that the time delay for the feedback-loopoptimized ion yield only occurs 2.5 oscillationperiods after excitation, is less evident. It mayresult from the fact, that the procedure is startedat random phases, which corresponds to rather
long pulses, so earlier passages at the open FC-window are missed. It may also be due to compli-cated non-adiabatic dynamics involving severalelectronically excited states, which have to befurther investigated.
The optimized NaK fragment channel is alsoobtained in the presence of Na K in the cluster2
beam. For these experimental conditions, theoptimization factorI yI amounts to 1.8(com-opt tl
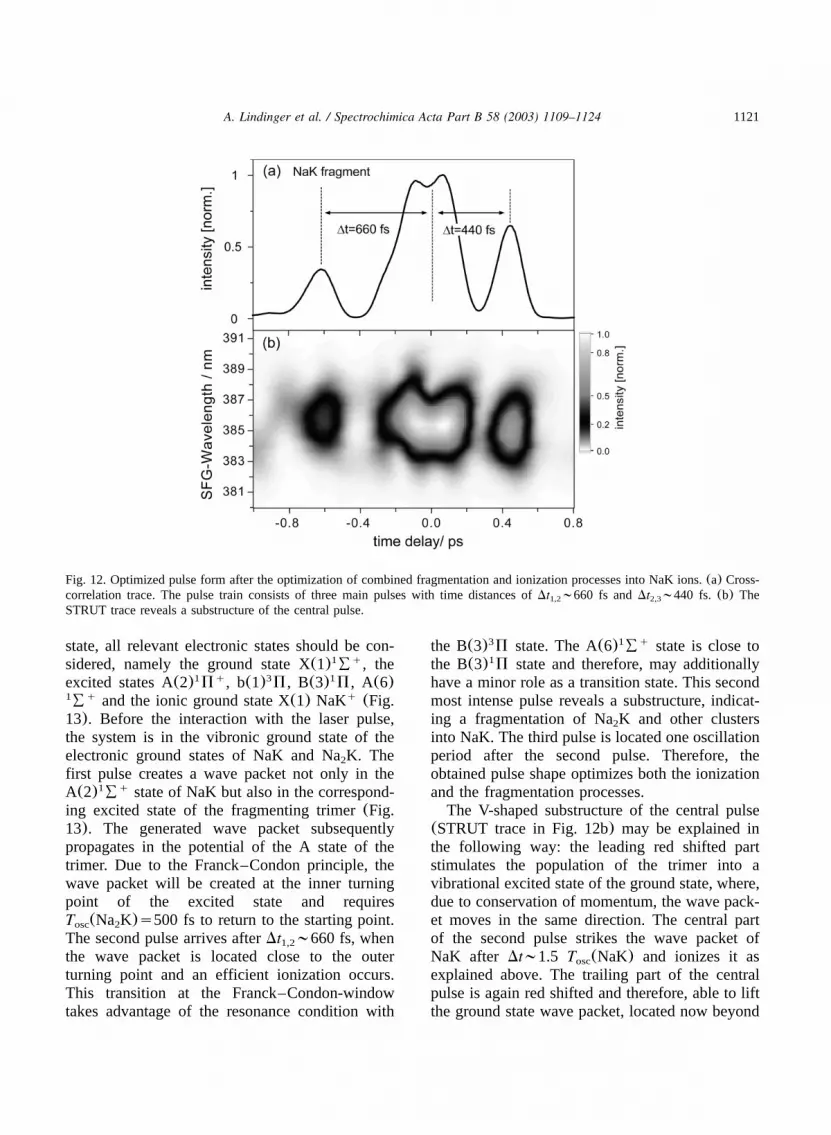
pared with the ion signal of a transform-limitedpulse). In Fig. 12 the optimized pulse for NaK isshown in its cross-correlation(a) and STRUTrepresentation(b). The pulse structure now is apulse train consisting of three main pulses with amost intense middle pulse. The time delaysbetween the first and second pulse and betweenthe second and third amount toDt ;660 and1,2
Dt ;440 fs, respectively. The intensity of the2,3
central pulse is almost three times higher than thatof the first pulse and twice as high as the thirdone, leading to a subpulse intensity ratio of1:2.9:1.8. The STRUT trace in Fig. 12b displaysthe time behavior of the frequency components ofthe pulses. The central pulse reveals a pronouncedchirped substructure. On the red side of the spec-trum the pulse is divided into two subpulsesDt s150 fs, whose intensity maxima converge2a,2b
in a V-form going to shorter wavelengths. Thus,an up chirp is followed by a down chirp. Themain intensity on the short wavelength side of themiddle pulse marks the center of the pulse. Theabove mentioned pulse distances refer to thismaximum.
In the following section an explanation of theoptimized pulse shape will be given, based on thetime-dependent analysis including the involvedpotential curves and transition dipole moments.The applied laser pulses are chosen with a suffi-ciently low peak intensity in order to be in theweak field limit. This aids the interpretation of theobtained results, since saturation effects can beneglected and perturbation theory can be applied.The discussion starts with transient three-photonionization with fragmentation from the Na K. Later2
the additional contribution of fragmentation fromlarger clusters will be explained.
For the analysis of optimal photo-induced tran-sitions from the ground state of NaK into the ion
1121A. Lindinger et al. / Spectrochimica Acta Part B 58 (2003) 1109–1124
Fig. 12. Optimized pulse form after the optimization of combined fragmentation and ionization processes into NaK ions.(a) Cross-correlation trace. The pulse train consists of three main pulses with time distances ofDt ;660 fs andDt ;440 fs. (b) The1,2 2,3
STRUT trace reveals a substructure of the central pulse.
state, all relevant electronic states should be con-sidered, namely the ground state X(1) 8 , the1 q
excited states A(2) P , b(1) P, B(3) P, A(6)1 q 3 1
8 and the ionic ground state X(1) NaK (Fig.1 q q
13). Before the interaction with the laser pulse,the system is in the vibronic ground state of theelectronic ground states of NaK and Na K. The2
first pulse creates a wave packet not only in theA(2) 8 state of NaK but also in the correspond-1 q
ing excited state of the fragmenting trimer(Fig.13). The generated wave packet subsequentlypropagates in the potential of the A state of thetrimer. Due to the Franck–Condon principle, thewave packet will be created at the inner turningpoint of the excited state and requiresT (Na K)s500 fs to return to the starting point.osc 2
The second pulse arrives afterDt ;660 fs, when1,2
the wave packet is located close to the outerturning point and an efficient ionization occurs.This transition at the Franck–Condon-windowtakes advantage of the resonance condition with
the B(3) P state. The A(6) 8 state is close to3 1 q
the B(3) P state and therefore, may additionally1
have a minor role as a transition state. This secondmost intense pulse reveals a substructure, indicat-ing a fragmentation of Na K and other clusters2
into NaK. The third pulse is located one oscillationperiod after the second pulse. Therefore, theobtained pulse shape optimizes both the ionizationand the fragmentation processes.
The V-shaped substructure of the central pulse(STRUT trace in Fig. 12b) may be explained inthe following way: the leading red shifted partstimulates the population of the trimer into avibrational excited state of the ground state, where,due to conservation of momentum, the wave pack-et moves in the same direction. The central partof the second pulse strikes the wave packet ofNaK after Dt;1.5 T (NaK) and ionizes it asosc
explained above. The trailing part of the centralpulse is again red shifted and therefore, able to liftthe ground state wave packet, located now beyond

1122 A. Lindinger et al. / Spectrochimica Acta Part B 58 (2003) 1109–1124
Fig. 13. Optimal multi-photon induced ionization and fragmentation pathways into NaK.(a) Multi-photon Ionization scheme usingthree pulses.(b) Multi-photon fragmentation scheme which circumvents the predissociative curve crossing.
the curve crossing, onto the repulsive potentialcurve of the trimer. Next, the third pulse ionizesboth, the residual wave packet of the NaKA(2) 8 state and the NaK fragments coming1 q
from this dissociative channel.If one modifies the stagnation conditions in such
a way that larger clusters are present in the beam,the NaK ion yield rises to a factor ofI yI s1.9.opt tl
For producing the NaK fragments numerousmother molecules are available now. Besides thetrimers Na K and K Na, also larger clusters such2 2
as Na K(108 amu), Na K (124 amu), Na K (1313 2 2 4
amu), K Na (140 amu), Na K (147 amu),3 3 2
Na K (163 amu) and K Na(179 amu) are present2 3 4
in the cluster beam. The intensity reductionRsI yI is dependent on the particular cluster ion.opt tl
For trimers one getsR(Na K)s0.4, R(K Na)s2 2
0.34 andR(Na K)s0.31, whereas the ion intensity2
for larger clusters is reduced on average by a factorof 0.5. It has to be considered that the reduction
of ion peaks is due to both, enhanced fragmenta-tion and diminished ionization probabilities.
The optimized pulse shape is a complex pulsetrain containing several sub pulses of differentintensities at various spacings of 250–440 fs(Fig.13). The second pulse is again divided in twosubpulses with a spacing ofDt ;180 fs. The2a,2b
distance between the first pulse and the center ofthe second pulse amounts toDt ;440 fs, whereas1,2
for the other spacings values ofDt ;420,2,3
Dt ;330 and Dt ;250 fs are obtained. The3,4 4,5
third pulse has a positive and the fourth pulse anegative chirp(Fig. 13b).
No simple relation of the pulse timings to thedynamics of NaK is possible. Thus, the pulse traincan only be explained as inducing fragmentationprocesses in clusters of different sizes down toNaK, rather than optimizing the ionization effi-ciency of NaK itself. Due to the high number ofpulses a sequential fragmentation Na K™n m

1123A. Lindinger et al. / Spectrochimica Acta Part B 58 (2003) 1109–1124
Fig. 14. Optimized pulse shape for fragmentation from larger clusters. The obtained pulse structure reveals a complex pulse trainconsisting of five separated pulses.
Na K ™NaK cannot be excluded. Here, firstnyk myl
the larger clusters form smaller clusters beforethey fragment further in a second stepw16–18x. Inorder to control the process of stepwise fragmen-tation at maximizing a single product, the popula-tion of the intermediates must be regarded.Therefore, a complex sequence of pulses is neededin order to excite and break the right fragment atthe exact time.
8. Summary
In conclusion, we have performed systematicpump and probe measurements on small alkaliclusters and analyzed the temporal behavior of thesystem after bound–bound and bound-free excita-tions including pre-dissociated states. The resultscould strongly be influenced by the shapes of theirradiated pulses. This observation prepared theway to perform a feedback control experiment onthe photo-fragmenting system Na K, which shows2
pronounced wave packet dynamics on the signalchannels of the mother ion and the NaK fragment.
As a result of the achieved coherent control pro-cess, it was possible to extract from the optimizedlaser field specific information about molecularvibration of the investigated particles. Additionally,we have investigated the adaptive feedback controlof multi-photon dissociation and ionization pro-cesses in the NaK dimer fragmentation channel,whereby optimization was achieved by maximizingthe NaK ion yield. Information about wave packetpropagation, oscillation periods and the involvedenergy curves could be gained from the shape ofthe pulse forms, which optimize the multi-photonionization. In the presence of Na K clusters the2
effect of fragmentation was examined, which gavenew insights into the photo-induced controled frag-mentation processes. A model for the avoidanceof the predissociation curve crossing in Na K was2
proposed. According to this concept, the wavepacket is dumped in a stimulated Raman scatteringprocess on the ground state potential, and subse-quently lifted back on the repulsive branch of theNa K potential, thereby avoiding the predissocia-2
tion curve crossing. In the case of enhanced

1124 A. Lindinger et al. / Spectrochimica Acta Part B 58 (2003) 1109–1124
fragmentation, the more complex pulse sequencesreflect the sequential fragmentation processes oflarger alkali clusters. The presented method oflearning about the involved molecular system fromthe obtained pulse shapes is a promising approachfor future control experiments(Fig. 14).
Acknowledgments
The authors thank Dr Porfirio Rosendo-Francis-co for his participation in the earlier experiments.In addition we thank Prof. Herschel Rabitz, Prof.Vlasta Bonacic-Koutecky and Prof. Jorn Manz for¨´stimulating discussions. Dr Thomas Feurer collab-orated with us in developing the evolutionaryalgorithm. The generous support of this researchproject by the Deutsche Forschungsgemeinschaftin the frame of the SFB 450 research project isgratefully acknowledged.
References
w1x S. Vajda, T. Leisner, S. Wolf, L. Woste, Philos. Mag. Bˇ ¨79 (1999) 1353–1366.
w2x A. Herrmann, S. Leutwyler, E. Schumacher, L. Woste,¨Chem. Phys. Lett. 52(1977) 418–425.
w3x R. de Vivie-Riedle, K. Kobe, J. Manz, W. Meyer, B.Reischl, S. Rutz, E. Schreiber, L. Woeste, J. Phys.Chem. 100(1996) 7789–7796.
w4x R. de Vivie-Riedle, J. Gaus, V. Bonacic-Koutecky, J.´Manz, B. Reischl, S. Rutz, E. Schreiber, L. Woeste,Femtochemistry, In: M. Chergui(Ed.), World Scientific,
Lausanne, Singapore, New Jersey, London, Hong Kong,September 4–8, 1995, pp. 319–403.
w5x R.S. Judson, H. Rabitz, Phys. Rev. Lett. 68(1992)1500–1503.
w6x S. Vajda, A. Bartelt, E.-C. Kaposta, T. Leisner, C.ˇ
Lupulescu, S. Minemoto, P. Rosendo-Francisco, L.Woeste, Chem. Phys. 267(2001) 231–239.
w7x C. Daniel, J. Full, L. Gonzalez, C. Kaposta, M. Krenz,C. Lupulescu, J. Manz, S. Minemoto, M. Oppel, S.Vajda, L. Woeste, Chem. Phys. 267(2001) 247–260.
w8x I. Rechenberg, Evolutionstrategie, Frommann-HozboogStuttgart, 1994.
w9x G. Delacretaz, E. Grant, R. Whetten, L. Woeste, J.´Zwanziger, Phys. Rev. Lett. 56(1986) 2598–2601.
w10x S. Wolf, G. Sommerer, S. Rutz, E. Schreiber, T. Leisner,L. Woste, R.S. Berry, Phys. Rev. Lett. 74(1995)¨4177–4180.
w11x A. Assion, T. Baumert, M. Bergt, T. Brixtner, B. Kiefer,V. Seyfried, M. Strehle, G. Gerber, Science 282(1998)919–922.
w12x S. Vajda, S. Rutz, J. Heufelder, P. Rosendo, H. Ruppe,ˇ
P. Wetzel, L. Woste, J. Chem. Phys. A 102(1998)¨4066–4068.
w13x S. Rutz, E. Schreiber, L. Woeste, in: O. Svelto, S.De Silvestri, G. Denardo(Eds.), Ultrafast Processes inSpectroscopy, Plenum Publ, New York, 1996.
w14x A. Weiner, D.E. Leaird, J.S. Patel, J.R. Wullert II, IEEE.J. Quant. Electron. 28(1992) 908–919.
w15x H.-P. Schwefel, Evolution and Optimum Seeking, Wiley,New York, 1995.
w16x H. Kuhling, S. Rutz, K. Kobe, E. Schreiber, L. Woste,¨¨J. Phys. Chem. 97(1993) 12500–12503.
w17x A. Ruff, S. Rutz, E. Schreiber, L. Woste, Z. Phys. D 37¨(1996) 175–180.
w18x S. Rutz, K. Kobe, H. Kuhling, A. Ruff, E. Schreiber,¨G. Sommerer, L. Woste, Surf. Rev. Lett. 3(1996)¨583–589.





























