Embed Size (px)
Citation preview

Article No. mb981775 J. Mol. Biol. (1998) 279, 439±447
Conformational Changes of the Tet Repressor Inducedby Tetracycline Trapping
Peter Orth1, Frank Cordes1, Dirk Schnappinger2, Wolfgang Hillen2
Wolfram Saenger1 and Winfried Hinrichs1*
1Institut fuÈ r KristallographieFreie UniversitaÈt BerlinTakustr. 6, D-14195 BerlinGermany2Institut fuÈ r Mikrobiologie undBiochemie, UniversitaÈtErlangen-NuÈrnbergStaudtstr. 5, D-91058 ErlangenGermany
Abbreviations used: TetR, Tet rerepressor of class D; TetR(BD), Tewith class D core sequence and NDNA-reading domain of class B; Tprotein for active ef¯ux; HTH, a-hsequence motif; Tc, collective nountetracyclines; 7ClTc, 7-chlorotetrac7HTc, Tetracycline; r.m.s., root-meTetR(D)/[Mg 7HTc]� complex (thData Bank entry code); 2TCT, TetRcomplex (the Brookhaven Proteincode).
0022±2836/98/220439±09 $25.00/0
The X-ray crystal structure analysis of inducer-free Tet repressor, TetR, at2.4 AÊ resolution identi®es one of two openings of the tunnel-like bindingsite as the entrance for the inducer tetracycline-Mg2�, [Mg Tc]�. Recog-nition and binding of the inducer unleashes conformational changes lead-ing to the induced state of TetR. In the ®rst step, the C-terminal turn ofa-helix 6 unwinds, thereby altering the orientation of a-helix 4. Thisdifferent orientation of a-helix 4 is stabilized by a series of hydrogenbonds mediated through a chain of eight water molecules. The a-helix 4connects the DNA-binding domain (a-helices 1 to 3) to the rigid TetRcore, and thus regulates gene expression through its respective orienta-tions.
# 1998 Academic Press Limited
Keywords: X-ray crystallography; antibiotic resistance; tetracycline; TetRepressor; inducer binding
*Corresponding authorIntroduction
The most frequently observed tetracycline (Tc)resistance mechanism in Gram-negative bacteria isthe active Tc ef¯ux through the cytoplasmic mem-brane mediated by the antiporter protein TetA,which prevents Tc from binding to its ribosomaltarget. TetA is a membrane embedded-pump,which exports a complex of Tc and a divalent cat-ion, e.g. [Mg Tc]� (Figure 1), coupled to equimolaruptake of a proton (Yamaguchi et al., 1990).Expression of the tetA gene is under negative con-trol of the Tet repressor, TetR, which binds to oper-ator DNA preventing transcription of tetR andtetA. [Mg Tc]�-binding induces conformationalchanges in TetR, resulting in its release from theoperator sites, thereby initiating expression.Tc-resistance determinants with these propertiesoccur in at least seven variants (classes A to E, G
pressor; TetR(D), Tett repressor mutant-terminal sequence ofetA, membraneelix-turn-a-helix
for the group ofycline (aureomycin);an-square; 2TRT,
e Brookhaven Protein(D)/[Mg 7ClTc]�
Data Bank entry
and H) on plasmids and transposons (for reviews,see Hillen & Berens, 1994; Schnappinger & Hillen,1996).
Crystal structures of induced TetR of class D,TetR(D), in complex with tetracycline, 7HTc(Hinrichs et al., 1994), and 7-chlorotetracycline,7ClTc (Kisker et al., 1995), as reported previouslyrevealed an all-a-helical TetR-homodimer witha-helix-turn-a-helix motifs located within theN-terminal three-helix bundles of the DNA-bind-ing domains (a1 to a3 and symmetry-related a10 toa30). The DNA-recognition helices (a3 and a30) areseparated by 39 AÊ , which is too wide to allowbinding of TetR to adjacent major grooves of theoperator DNA with 34 AÊ distance. In a ®rst tenta-tive formulation of the induction mechanism, weproposed that [Mg Tc]�-binding in the protein coreinduces a see-saw and lever-like mechanismmediated by helix a4, based on the fact that a4connects the DNA-binding domain to the proteincore (Hinrichs et al., 1994; Kisker et al., 1995). Thelocations and properties of many non-inducibleTetR mutants have, in addition, suggested a role inthe induction process for the loop preceding a6, a6and a9, and the central four-helix bundle (a8, a10,a80 and a100) located in the core of the dimer(MuÈ ller et al., 1995).
Here, we describe the crystal structure analysisof an inducer-free TetR(D)-mutant at 2.4 AÊ resol-ution. Comparison of the structures of induced andinducer-free TetR reveals several conformational
# 1998 Academic Press Limited

Figure 1. Chemical structure of the [Mg Tc]�-complexthat exists under physiological conditions. R7 is H intetracycline (7HTc), and Cl in 7-chlorotetracycline(7ClTc, aureomycin).
440 Tetracycline Trapping by Tet Repressor
changes that occur upon [Mg Tc]�-binding.A detailed description of the entrance, recognitionand binding of [Mg Tc]� to the narrow bindingpocket and a model of the induction mechanismare presented.
The previously published structures of TetR(D)/[Mg 7HTc]� and TetR(D)/[Mg 7ClTc]� will bedenoted by their Brookhaven Protein Data Bankcodes 2TRT and 2TCT, respectively.
Figure 2. Amino acid sequences of wild-type TetR(D), chimminus of wild-type TetR(D) is framed (residues 209 to 218and 2TCT greater than 1 AÊ are shaded. The a-helices areinduced upon [Mg Tc]�-binding. LOOP denotes the ¯exibTetR(BD)).
Results and Discussion
The TetR-variant
For the structure determination of a Tc-free TetRwe crystallized a TetR(BD) chimera, which consistsof the TetR(D) protein core carrying the TetR(B)sequence in the N-terminal DNA-binding domainand differs from TetR(D) by only ten amino acidresidues, see Figure 2. The stop codon was placedat position 209 to truncate the last ten residues,since their cleavage from the wild-type protein isessential for crystal growth (Kisker et al., 1995).The inducer-free TetR(BD) crystallizes isomor-phously to TetR(D) and to corresponding Tccomplexes, with a remarkable improvement in theX-ray diffraction pattern extending the resolutionto 2.4 AÊ resolution, as opposed to inducer-freeTetR(D) or TetR(B) crystals, which diffract to about2.8 AÊ . The in vivo repression and inductionef®ciences of TetR(BD) are identical with TetR(B)and even enhanced compared to TetR(D)(Schnappinger et al., 1998).
Crystal structure
The asymmetric unit contains one monomer ofTetR(BD), because the dyad of the biologicallyactive TetR homodimer coincides with a crystallo-graphic 2-fold axis (Figure 3). The 207 residuespolypeptide chain (residues 2 to 208) is folded intoten a-helices, denoted a1 to a10. The N-terminalthree-helix-bundles a1 to a3 and the symmetry-
eric TetR(BD) and wild-type TetR(B). The truncated C ter-). Main-chain r.m.s. deviations between Tc-free TetR(BD)
marked in blue, red parts indicate structural changesle loop connecting a8 and a9 (residues 152 to 165 in

Figure 3. Overall structure of theinduced form of TetR(D), 2TCT.The two monomers are colouredyellow and orange. Major struc-tural differences of the superim-posed TetR(BD) are indicated inblue. These include the loop con-necting a6 and a7, the N-terminalpart of a7, and the entire lengthof a9. The C-terminal turn of a8is unwound in the inducer-freeTetR(BD) (not marked). The adja-cent disordered loop connection(residues 152 to 165) to a9 iscoloured pink. The 7ClTc is shownin red with a blue Mg2�-sphere.
Figures 3, 6, 7 and 8 were cre-ated with SETOR (Evans, 1993).
Tetracycline Trapping by Tet Repressor 441
related a10 to a30, together constitute the DNA-binding domains. a2, a3 form the a-helix-turn-a-helix motif, HTH, of the DNA-binding domain,which is connected to the core by a4. The core ofthe TetR homodimer is formed by a5 to a10 anda50 to a100, and harbours two symmetry-relatedTc-binding sites, each formed by both polypeptidechains. The monomers are clearly separated,merely a disordered loop and the adjacent a9 wraparound the other half of the core domain in ahand-shake fashion. The central part of the core isformed by the antiparallel a8 and a10 in combi-nation with their symmetry-related counterpartsa80 and a100. These pairs of long a-helices arepositioned at an angle of �50�, typical of theglobin fold (BraÈnden & Tooze, 1991). Each of thetwo [Mg Tc]�-binding sites forms a continuoustunnel buried entirely within the TetR core; the®rst is de®ned by a-helices 4 to 8, 80 and 90, thesecond by 40 to 80, 8 and 9. Each tunnel has twoopenings to the bulk solvent. One is C-terminal toa4, at a position where the Tc-ring A forms hydro-gen bonds to His64; the other is next to the C ter-minus of a90, where the Tc-ring D is in van derWaals contact with side-chains of Leu1740 andMet1770 of a90.
Comparison of the induced andinducer-free TetR
Superposition of the Tc-free TetR(BD) structurewith the induced form of TetR(D), 2TCT, showssigni®cant structural differences; the overall r.m.s.deviations for main-chain atoms amounting to1.13 AÊ . Induction causes large displacements of up
to 4 AÊ including the C-terminal part of a6, theadjacent loop connecting a6 to a7, as well as theN-terminal part of a7; furthermore, a9 is displacedby about 2.4 AÊ as a unit, see Figure 4(a). TheC-terminal turn of a6 is converted into a type IIb-turn following Mg2� coordination, while a8 isextended C-terminally by one turn and the N-term-inal turn of a7 adopts 310-helical conformation oninduction. Small displacements of the DNA-bind-ing domain and a4 are also observed. The mostrigid part of TetR(BD) is represented by a5 and thecore helices a8 and a10. The observed differencesbetween the two structures are accompanied bygreater ¯exibility of the inducer-free TetR(BD), asindicated by increased thermal parameters(Figure 4(b)), which are generally larger in Tc-freeTetR(BD) than in 2TCT.
Identification of the entrance of the[Mg Tc]�-binding tunnel
Under physiological conditions, the inducerexists and binds to TetR as a [Mg Tc]�-complex(Takahashi et al., 1986; Jogun & Stezowski, 1976).The narrow openings of the binding tunnel necessi-tate an initial directed insertion of the inducer witheither Tc-ring A or D. For a comparison of theopenings, see Figure 5. In Tc-free TetR(BD), theopening next to the C-terminus of a90 is twice aslarge as that C-terminal to a4. Whereas the latterremains almost unchanged upon [Mg Tc]�-bind-ing, the opening at a90 narrows signi®cantly as aresult of the movement of a90. The docking of [MgTc]� induces a lateral motion of a90 comparable toa ``sliding door'' (Figure 3). The r.m.s. positional
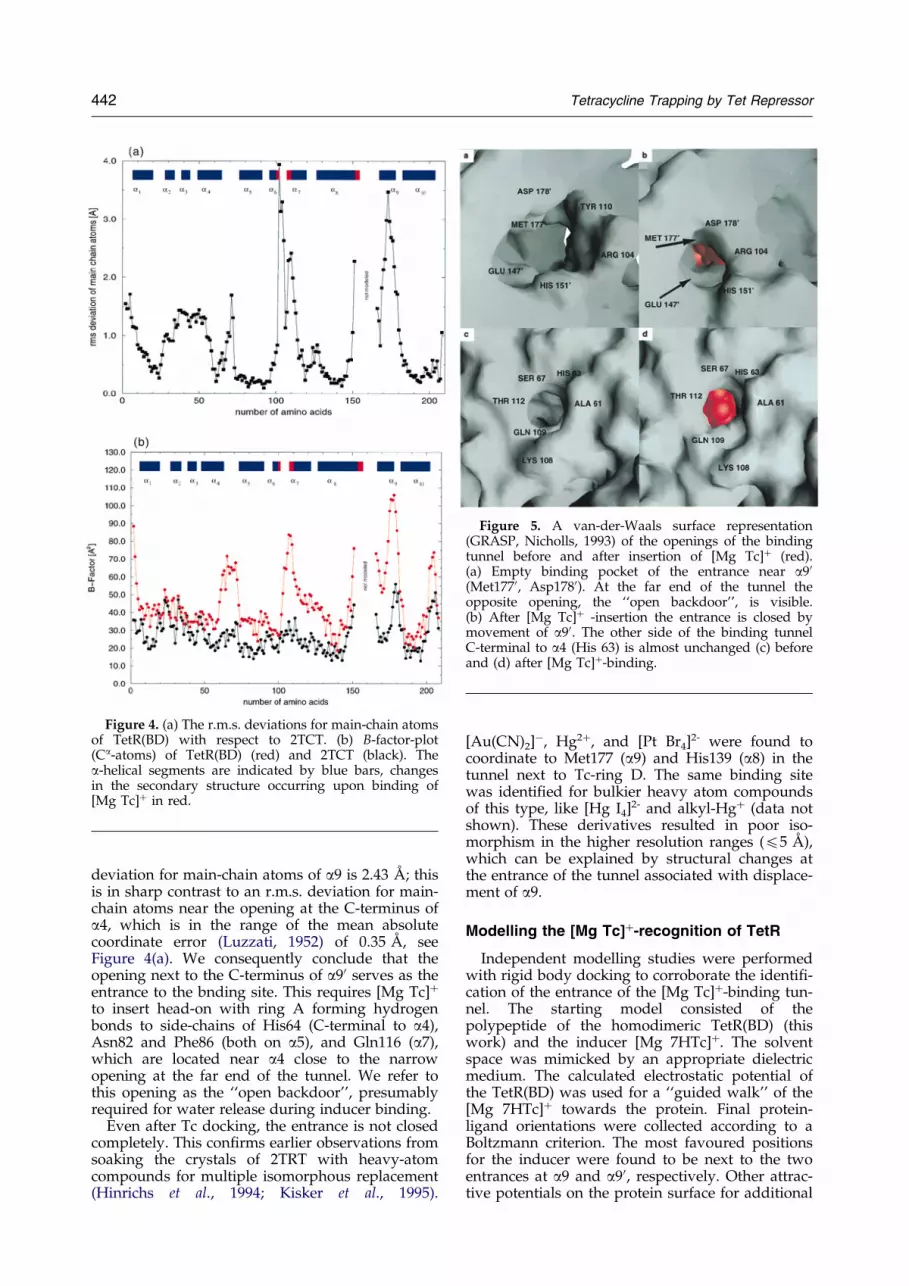
Figure 4. (a) The r.m.s. deviations for main-chain atomsof TetR(BD) with respect to 2TCT. (b) B-factor-plot(Ca-atoms) of TetR(BD) (red) and 2TCT (black). Thea-helical segments are indicated by blue bars, changesin the secondary structure occurring upon binding of[Mg Tc]� in red.
Figure 5. A van-der-Waals surface representation(GRASP, Nicholls, 1993) of the openings of the bindingtunnel before and after insertion of [Mg Tc]� (red).(a) Empty binding pocket of the entrance near a90(Met1770, Asp1780). At the far end of the tunnel theopposite opening, the ``open backdoor'', is visible.(b) After [Mg Tc]� -insertion the entrance is closed bymovement of a90. The other side of the binding tunnelC-terminal to a4 (His 63) is almost unchanged (c) beforeand (d) after [Mg Tc]�-binding.
442 Tetracycline Trapping by Tet Repressor
deviation for main-chain atoms of a9 is 2.43 AÊ ; thisis in sharp contrast to an r.m.s. deviation for main-chain atoms near the opening at the C-terminus ofa4, which is in the range of the mean absolutecoordinate error (Luzzati, 1952) of 0.35 AÊ , seeFigure 4(a). We consequently conclude that theopening next to the C-terminus of a90 serves as theentrance to the bnding site. This requires [Mg Tc]�
to insert head-on with ring A forming hydrogenbonds to side-chains of His64 (C-terminal to a4),Asn82 and Phe86 (both on a5), and Gln116 (a7),which are located near a4 close to the narrowopening at the far end of the tunnel. We refer tothis opening as the ``open backdoor'', presumablyrequired for water release during inducer binding.
Even after Tc docking, the entrance is not closedcompletely. This con®rms earlier observations fromsoaking the crystals of 2TRT with heavy-atomcompounds for multiple isomorphous replacement(Hinrichs et al., 1994; Kisker et al., 1995).
[Au(CN)2]ÿ, Hg2�, and [Pt Br4]2- were found to
coordinate to Met177 (a9) and His139 (a8) in thetunnel next to Tc-ring D. The same binding sitewas identi®ed for bulkier heavy atom compoundsof this type, like [Hg I4]
2- and alkyl-Hg� (data notshown). These derivatives resulted in poor iso-morphism in the higher resolution ranges (45 AÊ ),which can be explained by structural changes atthe entrance of the tunnel associated with displace-ment of a9.
Modelling the [Mg Tc]�-recognition of TetR
Independent modelling studies were performedwith rigid body docking to corroborate the identi®-cation of the entrance of the [Mg Tc]�-binding tun-nel. The starting model consisted of thepolypeptide of the homodimeric TetR(BD) (thiswork) and the inducer [Mg 7HTc]�. The solventspace was mimicked by an appropriate dielectricmedium. The calculated electrostatic potential ofthe TetR(BD) was used for a ``guided walk'' of the[Mg 7HTc]� towards the protein. Final protein-ligand orientations were collected according to aBoltzmann criterion. The most favoured positionsfor the inducer were found to be next to the twoentrances at a9 and a90, respectively. Other attrac-tive potentials on the protein surface for additional

Tetracycline Trapping by Tet Repressor 443
[Mg 7HTc]� were found near the C terminus andbetween the HTH motifs, but the backdoor next toa4 does not show such a potential and therefore isnot recognized by the inducer molecules. Thissimulation clearly supports the identi®cation of theopening C-terminal to a9 as the entrance of thebinding tunnel for the antibiotic.
Flexibility of the loop connecting aaa8 and aaa9
The mobility of a9 is supported by the ¯exibilityof the loop connecting a8 and a9. This oligopeptidesegment is disordered in all known TetR crystalstructures, and was not modelled because the cor-responding regions of the electron density mapswere poorly de®ned. In 2TRT and 2TCT the loopcomprises residues 156 to 164, but in the inducer-free TetR(BD) this disordered segment is extendedand covers residues 152 to 165. One C-terminalturn of a8 and residue 165 at the end of the loop,N-terminal to a9, became more ¯exible, asindicated by weak and uninterpretable portions ofthe electron density map in TetR(BD).
Alignment of the seven known TetR sequencesindicates that, apart from C-terminal sequencevariations, this loop is the least conserved regionand the only part where deletions or insertions arefound. The amino acid composition of this loopgives rise to a net negative charge in all TetRsequences, indicating a supporting role in [MgTc]�-capture. Deletions in this loop lead to reduced[Mg Tc]� af®nity of TetR mutants, correlating withlack of inducibility. In contrast, sequence variationsby substitutions to alanine show minor effects only(Berens et al., 1997). A suf®cient length of this loopis important for the transition of the open TetR
Figure 6. Stereoview of the superposition of the [Mg 7TetR(BD) (blue), Tc(red). 7ClTc is shown in red with a bl(red spheres). The induced stepwise conformational changesbinding tunnel through the entrance next to (9 in a directGln116 (event a). After Mg2�-coordination (event b) to Hinitiates reorientation of the adjacent loop connection to a7hydrogen bonds to two water ligands of Mg2� (event d).after a90 has closed the entrance. Hydrogen bonds and Mg2�
structure to the closed [Mg Tc]�-induced confor-mation.
[Mg Tc]�-recognition and changes leadingto induction
After passing the entrance of the binding tunnel,speci®c [Mg Tc]�-recognition triggers a sequence ofevents (a to e, see Figure 6), including confor-mational changes that result in the narrowing ofthe binding pocket and the reorientation of a4,required for a changed af®nity of TetR for theoperator-DNA.
Event a
The functional groups amide O21, enolate O3and dimethylammonium N4 of Tc-ring A formtight hydrogen bonds to His64 (a4), Asn82(a5) andGln116 (a7). These residues are identical in pos-ition and orientation in the Tc-bound and Tc-freeTetR, and may be assumed to constitute the ®rstspeci®c recognition targets of TetR.
Event b
In the inducer-free TetR(BD), a6 comprisesresidues 96 to 102; this is shortened to residues 96to 100 in the induced structure. Mg2�-binding tothe imidazole group of His100 triggers the for-mation of a type II b-turn, His100 to Thr103, at theexpense of the C-terminal turn of a6. His100remains in position, but the Ca positions of Gly102and Thr103 are displaced by 3.92 and 3.12 AÊ ,respectively. Hydrogen bonding of Thr103 Og to aMg2�-coordinated water molecule supports for-mation of the b-turn. This induced ®t identi®es the
ClTc]�-binding sites of 2TCT (yellow) and inducer-freeue Mg2�-sphere and three coordinating water molecules
are marked a to e (see the text). [Mg 7ClTc]� enters thefashion. Tc-ring A hydrogen bonds to His64, Asn82 andis100, the formation of the b-turn (residues 100 to 103)
(event c). The carboxylate group of Glu1470 (a80) formsFinally (event e), Arg104 forms a salt-bridge to Asp1780-coordination are indicated by broken lines.

Figure 7. View along a4 showing the DNA-bindingdomain relative to the [Mg 7ClTc]�-binding site. Theinducer-free TetR(BD) (blue) is superimposed upon2TCT (yellow). The DNA-binding domain a1 to a3 anda4 are represented by tubes. Red arrows indicateinduced structural movements. [Mg 7ClTc]� (red, blueMg2�-sphere) triggers the transformation of the C-term-inal turn of a6 into a b-turn (residues 100 to 103). Thehydrogen bond of this induced b-turn (O100 . . . NH103)is formed at the expense of a hydrogen bond in a6(O98 . . . NH102; broken lines). Helix a4 is in contactwith the unwinding part of a6 and the resulting void is®lled by a4 moving into the same direction. This move-ment is associated with a displacement of the DNA-binding domain a1 to a3 into a position where thehomodimeric TetR no longer recognizes the operatorDNA.
444 Tetracycline Trapping by Tet Repressor
binding site of Tc-chelated Mg2� as the initiationpoint for conformational rearrangements associ-ated with induction.
Event c
Adjacent to this b-turn, the loop with residues104 to 106 is reoriented to form a part of the hydro-phobic pocket by methylene side-chains of Arg104and Pro105 for the Tc-ring D.
Event d
After a rotation by �90�, the carboxylate groupof Glu1470 (a80) forms hydrogen bonds to Gly102Nof the new b-turn and to two Mg2�-coordinatingwater molecules.
Event e
The hydrophobic pocket is completed by resi-dues Leu1700, Leu1740 and Met1770 of a90, whichcloses the entrance of the binding tunnel like asliding door and ®nally places the C-terminalAsp1780 in an appropriate position to form a salt-bridge to Arg104.
Induction
As mentioned above, binding of [Mg Tc]� toTetR triggers the unwinding of the C-terminal turnof a6, which is in contact with the central part ofa4. These two a-helices form angles of about 60�and 53� in 2TCT and TetR(BD), respectively. Thepositioning of a4 (residues 48 to 63) is determinedby a hydrophobic contact region (Leu52, Ala56) tothe C-terminal helical turn of a6 (Val99, Thr103),see Figure 7. This region was proposed to rep-resent a pivot for a4, in a see-saw- and lever-likemechanism for induction (Hinrichs et al., 1994),which we can now analyse in more detail. TheMg2�-induced transformation of the C-terminalturn of a6 into a b-turn produces free space at thecontact surface to a4, which now moves to ®ll thevoid. Compared to 2TCT, the main-chain r.m.s.deviations of a4 increase from the C terminus inthe direction of the N-terminal DNA-bindingdomain a1 to a3, see Figure 4(a). The position ofthe C terminus of a4 remains essentially unalteredwith the pivoting of Tc-binding His64, and theadjustment of the DNA-binding domains forinduction or operator recognition can be describedas a hinge-like movement of a4. In the inducedTc-complexes, 2TRT and 2TCT, the distancebetween the recognition helices a3 and a30 is main-tained at about 39 AÊ and in the inducer-freeTetR(BD) crystal structure their distance isincreased slightly by �1 AÊ . In the operator/TetRcomplex, the recognition helices a3 and a30 of theHTH motifs have to insert into adjacent majorgrooves in an antiparallel orientation at �34 AÊ sep-aration, assuming canonical B-DNA. The inducer-free TetR(BD) de®ned as the pre-induced state, has
the DNA-binding domains suf®ciently mobile toallow docking to the operator DNA. In the crystallattice, neighbouring molecules are arranged head-to-head (Kisker et al., 1995) and it remains possiblethat intermolecular contacts play a role in prevent-ing the recognition helices from moving towardseach other. Another reason why the recognitionhelices are not in a position compatible with DNAbinding could be the existence of equivalentcharged regions on each DNA-binding domain,which would interact unfavourably in the absenceof DNA.
A chain of water molecules along aaa4 linksthe inducer-binding site to theDNA-binding domain
The N-terminal helical turn (residues 107 to 109)of a7 (107 to 121) of Tc-free TetR(BD) is distortedinto a 310-helical conformation after inducer bind-ing (2TCT, 2TRT). This is due to the formation ofan extended water-mediated hydrogen bonding

Figure 8. In the induced structure, the DNA-bindingdomains are tightly locked in a conformation unable tobind to the operator DNA. This rigidity is supported byan extended network of hydrogen bonds (broken lines)that stabilize the positions of a4 to a6 after induction.The network becomes possible only after the loop seg-ment connecting a6 and a7 moves into position follow-ing [Mg 7ClTc]�-recognition and induction, which startsclosing the indicated ``water zipper'' with b-turn for-mation of residues His100 to Thr103. A zigzag-chain ofwater molecules (red spheres) forms on one side hydro-gen bonds to carbonyl oxygen atoms (53, 56, 57, 60 and64) of a4. On the opposite side, this water chain formshydrogen bonds to the amide group of 7ClTc at ring Avia water 7, peptide NH104 (water 2) and O104 (water4). The side-chain of Gln109 has a central position inthis network through Gln109 Ne, bridging the watermolecules 5 and 6. Gln109 Oe forms hydrogen bonds toNH106 and water 9, which is in turn hydrogen bondedto a water ligand of Mg2� (not shown).
Tetracycline Trapping by Tet Repressor 445
network, which links the [Mg Tc]�-binding site viathe amide side-chain of Gln109 to a4, see Figure 8.Gln109 Oe forms hydrogen bonds both to the pep-tide backbone NH106 and, via a water molecule, toone aqua-ligand of the Mg2�-coordination sphere.Gln109 Ne is part of a zigzag-chain with eightwater molecules hydrogen bonded to peptide car-bonyl oxygen atoms 53, 56, 57, 63 and 64 of a4.This hydrogen bonding network ®xes the [MgTc]�-binding site to a4 carrying the N-terminalDNA-binding domain. In the inducer-free TetR,this water network is not observed, because severalanchor points associated with [Mg Tc]� are miss-ing. The inducer-free structure is more open/loosewith greater ¯exibility around the empty bindingpocket.
The rigid core
Non-inducible TetR mutants resulting frommutations at the dimer interface have suggested arole for the core helices a8 and a10, and their sym-metry-related mates, in the mechanism of induc-tion (MuÈ ller et al., 1995). A shear motion of the twocore subunits (each encompassing two of the fourcentral helices) was proposed to increase the dis-tance between the DNA-binding domains by�5 AÊ . This now appears unlikely, especially as theorientation of a8 and a10 with respect to their sym-metry-related counterparts is observed to representthe most rigid part of known structures of TetR.We therefore conclude that the mobility of theDNA-binding domains, supported by rearrange-ments of helices a4 and a6 is suf®cient to ensureoperator binding or release after Tc recognition.The reason for the above-mentioned group ofmutants interfering with induction is presumablyrelated to the reduced stability of the central core,which is required as a compact unit for Tc anchor-ing.
Materials and Methods
Mutagenesis, expression and purification
Escherichia coli RB791 was used for cloning andoverexpression (Brent & Ptashne, 1981). Isolation,manipulation and sequencing of DNA was performedaccording to Maniatis et al. (1982). For constructionof pWH610(B/D)51-208 (Schnappinger et al., 1998),which was used for overexpression of TetR(BD),pWH853(B/D)51-208 was digested with XbaI and NcoI,and the tetR (BD) fragment was cloned into similarlydigested pWH1950 (Ettner et al., 1996). The presence oftetR (BD) was veri®ed by sequencing the entire gene.Overproduction and puri®cation of TetR was performedas described for TetR(B) (Ettner et al., 1996).
Crystallization and Data collection
TetR(BD)-protein solution (10 ml of 10 mg/mlTetR(BD), 200 mM NaCl, 50 mM Tris-HCl, pH 8.0)mixed with 5 ml of reservoir (0.8 M potassium phosphatebuffer and 50 mM Tris-HCl, pH 8.0) were equilibrated tothe reservoir buffer in a vapour diffusion assay (hangingdrop) at 18�C. Crystals with a maximum size of1.5 mm � 1.5 mm � 0.5 mm grew within two weeks.TetR(BD) crystals were found to be isomorphous tothose of 2TRT and 2TCT, see Table 1.
X-ray diffraction data of the TetR(BD) mutant werecollected at 4�C with a single crystal on an MARResearch image plate detector, using synchrotronradiation at LURE, Paris-Orsay (beamline DW32). Thedataset was processed with MOSFLM (Leslie, 1992) andother programs of the CCP4 (1994) suite. Statistics of thedata collection are summarized in Table 1.
Phase determination and refinement
The present model was re®ned against all data to amaximum resolution of 2.4 AÊ using X-PLOR (BruÈ nger,1996), with reference parameters for stereochemicalrestraints derived by Engh & Huber (1991). The free

Table 1. Data collection and re®nement statistics
Source LURE, beamline DW32Wavelength (AÊ ) 0.94Space group I4122Cell dimensions at 4�Ca�b (AÊ ) 69.81 (5)c (AÊ ) 184.95 (45)Resolution range (AÊ ) 18.5±2.4Number of unique reflections 9076Redundancya 4.8
All data 2.47±2.40 AÊ shellCompleteness (%) 97.8 98.6Rsym
b (%) 7.3 24.8Ratio I/s(I) 6.13 2.9RefinementR-factorc (%) 21.4Free R-factord (%) 27.5Number of protein atoms 1541Number of solvent molecules 28r.m.s. deviations from ideality:
Bond lengths (AÊ ) 0.007Bond angles (�) 1.20Dihedrals (�) 19.9Improper dihedrals (�) 1.18
Average overall isotropic B-factors (AÊ 2)Main-chain atoms 45.6Side-chain atoms 49.5Solvent molecules 48.9
Ramachandran parameterse
Most favoured region (%) 93.6Additionally allowed region (%) 5.8Mean absolute error (AÊ ), (Luzzati, 1952) 0.35
PDB entry code 1a6i, data available within one year
a Redundancy � number observations/number unique re¯ections.b Rsym � �i �hkljIi,hkl ÿ hIhklij/�i�hklhIhkli, where Ii,hkl is the intensity of symmetry redundant re¯ections and hIhkli is the mean
intensityc R-factor � �jFobs ÿ Fcalcj/�Fobsd Free R-factor � R-factor for the test set of re¯ections (10% of the total) omitted in the re®nement process for cross-validation.e According to PROCHECK (Laskowski et al., 1993).
446 Tetracycline Trapping by Tet Repressor
R-value was used to monitor the quality of all re®ne-ment and model building steps (BruÈ nger, 1992). Relevantre®nement statistics are given in Table 1. The crystalstructure of TetR(BD) was suf®ciently isomorphous to2TRT and 2TCT for initial phasing. The polypetide chainof the 2TCT complex (excluding [Mg 7ClTc]� and allwater molecules) was used as a starting model for rigidbody and simulated annealing re®nement using molecu-lar dynamics with a slow-cooling protocol (BruÈ nger,1996) from 2000 to 300 K and an overall B-factor of30.0 AÊ 2. Manual model building was supported byFRODO (Jones, 1978). Water molecules were identi®edin difference electron density maps employing athreshold of three r.m.s. deviations above mean densityin (Fo ÿ Fc) maps, and 1 r.m.s. in (2Fo ÿ Fc) maps. Watermolecules located at a distance of 2.0 to 4.5 AÊ of poten-tial hydrogen bond donors or acceptors were added tothe existing model. They were rejected if their B-factorsexceeded 75 AÊ 2 during re®nement. The quality of themodel was controlled by 2Fo ÿ Fc simulating annealingomit maps (BruÈ nger, 1996), calculated in the vicinity offunctionally important polypeptide segments, e.g. resi-dues 100 to 104. Following model completion, bulk sol-vent correction was applied for ®nal re®nement ofpositional and thermal parameters. The loop segment ofresidues 152 to 165 in TetR(BD) was not included in themodel, because the corresponding electron densitieswere not clearly interpretable. The Ramachandran plotshowed main-chain torsion angles in the commonlyexpected regions. Only Leu204 lies in the ``generously
allowed'' region; this amino acid residue is part of theC-terminal loop and located in well-de®ned electrondensity.
Docking
The geometry of the [Mg Tc]�-complex was optimizedwith the program SPARTAN (Hehre et al., 1993) and thesemi-empirical PM3 method. The magnesium ion is octa-hedrally coordinated by the chelating ketoenolate groupO-11/O-12 of the antibiotic, three water ligands and afourth water molecule, replacing the His100 Ne2 coordi-nation. Atomic charges of the [Mg Tc]� complex were®tted against the quantum chemically calculated electro-static potential. Subsequently, we used the programEPOS written by our group to perform a ®nite-differencesolution of the linearized Poisson-Boltzmann-equation(Gilson et al., 1987) for a given charge distribution of theprotein, based on the GROMOS force-®eld (vanGunsteren et al., 1996), and the quantum chemically cal-culated charges for the ligand. The electrostatic calcu-lation was done on a grid with 1.0 AÊ spacing, a minimaldistance between protein surface and grid edges of 15 AÊ ,0.1 molar ionic strength, and dielectric constants of 1.0and 80.0 for the protein interior and the solvent area,respectively. The program EPOS was also used to obtaintrajectories for the [Mg Tc]� ligand within the previouslycalculated electrostatic potential of the repressor. Theseelectrostatically ``guided walks'' started from positions atleast 15 AÊ away from the protein, chosen from a spheri-

Tetracycline Trapping by Tet Repressor 447
cal distribution of ligands around the target. New orien-tations for the ligand were generated by random trans-lations and rotations, and accepted by minimizing theenergy. The ®nal positions of all trajectories wereweighted against each other according to a Boltzmanndistribution, in order to describe statistically the ensem-ble of inducer positions on the TetR surface.
Acknowledgements
We thank H. Roscher for help with protein prep-aration and crystallization, and Drs G. Bains, P. Jekowand W.-D. Schubert for critical reading of the manu-script. Preliminary X-ray data were collected at the SRSDaresbury, UK. Financial support by the DeutscheForschungsgemeinschaft (SfB 344) and the Fonds derChemischen Industrie is gratefully acknowledged.
References
Berens, Ch. , Schnappinger, D. & Hillen, W. (1997). Therole of the variable region in Tet repressor forinducibility by tetracycline. J. Biol. Chem. 272, 6936±6942.
BraÈndeÂn, C.-I. & Tooze, J. (1991). Introduction to ProteinStructure, Garland Publishing, Inc., New York.
Brent, R. & Ptashne, M. (1981). Mechanism of action ofthe lexA gene product. Proc. Natl Acad. Sci. USA,78, 4204±4208.
BruÈ nger, A. T. (1992). The free R-value: a novel statisti-cal quantity for assessing the accuracy of crystalstructures. Nature, 355, 472±475.
BruÈ nger, A. T. (1996). X-PLOR Manual, Version 3.8: ASystem for X-ray Crystallography and NMR, Yale Uni-versity Press, New Haven, CT.
CCP4, (1994). Collaborative computational project num-ber 4. The CCP4 suite: programms for protein crys-tallography. Acta Crystallog. sect D, 50, 760±763.
Engh, R. A. & Huber, R. (1991). Accurate Bond andAngle Parameters for X-ray Protein-StructureRe®nement. Acta Crystallog. sect A, 47, 392±400.
Ettner, N., MuÈ ller, G., Berens, Ch. , Backes, H.,Schnappinger, D., Schreppel, T., P¯eiderer, K. &Hillen, W. (1996). Fast large scale puri®cation oftetracycline repressor variants from overproducingEscherichia coli strains. J. Chromatog. sect. A, 742, 95±105.
Evans, S. V. (1993). SETOR: Hardware-lighted Three-dimensional Solid Model Representations of Macromol-ecules, University of British Columbia, Vancouver,Canada.
Gilson, M. K., Sharp, K. A. & Honig, B. H. (1987). Calcu-lating the electrostatic potential of molecules in sol-ution: method and error accessment. J. Comput.Chem. 9, 327±335.
Hehre, W. J., Burke, L. D. & Shusterman, A. J. (1993). ASPARTAN Tutorial Version 3. 0, Wavefunction, Inc.,Irvine, CA.
Hillen, W. & Berens, Ch. (1994). Mechanisms under-lying expression of Tn10 encoded tetracycline resist-ance. Annu. Rev. Microbiol. 48, 345±369.
Hinrichs, W., Kisker, C., DuÈ vel, M., MuÈ ller, A., Tovar,K., Hillen, W. & Saenger, W. (1994). Antibioticresistance: structure of the Tet repressor-tetracyclinecomplex and mechanism of induction. Science, 264,418±420.
Jogun, K. H. & Stezowski, J. J. (1976). Coordination andconformational aspects of oxytetracycline metal ioncomplexation. J. Am. Chem. Soc. 98, 6018±6026.
Jones, T. A. (1978). A graphics model building andre®nement system for macromolecules. J. Appl.Crystallog. 11, 268±272.
Kisker, C., Hinrichs, W., Tovar, K., Hillen, W. &Saenger, W. (1995). The complex formed betweenTet repressor and tetracycline-Mg2� reveals mechan-ism of antibiotic resistance. J. Mol. Biol. 247, 260±280.
Laskowski, R. A., MacArthur, M. W., Moss, D. S. &Thornton, J. M. (1993). PROCHECK: a program tocheck the stereochemical quality of protein struc-tures. J. Appl. Crystallog. 26, 283±291.
Leslie, A. G. W. (1992). MOSFLM. In Joint CCP4 andESF-EACMB Newsletter on Protein CrystallographyNo. 26, Daresbury Laboratory, Warrington, UK.
Luzzati, V. (1952). Traitement statistique des erreursdans la deÂtermination des structures cristallines.Acta Crystallog. 5, 802±810.
Maniatis, T., Fritsch, E. F. & Sambrook, J. (1982). Molecu-lar Cloning: A Laboratory Manual, Cold Spring Har-bor Laboratory Press, Cold Spring Harbor, NY.
MuÈ ller, G., Hecht, B., Helbl, V., Hinrichs, W., Saenger,W. & Hillen, W. (1995). Characterisation of non-inducible Tet repressor mutants suggests confor-mational changes necessary for induction. NatureStruct. Biol. 2, 693±703.
Nicholls, A. J. (1993). GRASP Manual, Columbia Univer-sity, New York.
Schnappinger, D. & Hillen, W. (1996). Tetracyclines:antibiotic action, uptake, and resistance mechan-isms. Arch. Microbiol. 165, 359±369.
Schnappinger, D., Schubert, P., P¯eiderer, K. & Hillen,W. (1998). Determinants of protein-protein recog-nition by four helix bundles: changing the dimeriza-tion speci®ty of Tet repressor. EMBO J. 17, 535±543.
Takahashi, M., Altschmied, L. & Hillen, W. (1986). Kin-etic and equilibrium characterization of the Tet-repressor-tetracycline complex by ¯uorescencemeasurements; evidence for divalent ion require-ments and energy transfer. J. Mol. Biol. 187, 341±348.
van Gunsteren, W. F., et al. (1996). Biomolecular Simu-lation: The GROMOS Manual and User Guide, vdfHochschulverlag AG, ETH ZuÈ rich.
Yamaguchi, A., Udagawa, T. & Sawai, T. (1990). Trans-port of divalent cations with tetracycline asmediated by the transposon Tn10-encoded tetra-cycline resistance protein. J. Biol. Chem. 265, 4089±4813.
Edited by R. Huber
(Received 24 November 1997; received in revised form 5 February 1998; accepted 5 February 1998)





























