Embed Size (px)
Citation preview

Molecular Ecology (2003)
12
, 2707–2717 doi: 10.1046/j.1365-294X.2003.01941.x
© 2003 Blackwell Publishing Ltd
Blackwell Publishing Ltd.
Cryptic species and morphological plasticity in long-lived bivalves (Unionoida: Hyriidae) from inland Australia
ANDREW M. BAKER, CHRIS BARTLETT, STUART E . BUNN, KATRINA GOUDKAMP, FRAN SHELDON and JANE M. HUGHES
Cooperative Research Centre for Freshwater Ecology, Australian School of Environmental Studies, Griffith University, Nathan, Queensland, 4111, Australia
Abstract
Molecular (mitochondrial DNA, isozyme) and morphological diversity of freshwater mus-sels (Family Hyriidae) was examined at 21 sites encompassing four large river systems,across southwest Queensland, Australia. Evidence was found for two major morphologicalgroups. One group, which occurred in every river system, closely matched a recognized spe-cies (
Velesunio ambiguus
) both morphologically and in a well-supported lineage within amitochondrial phylogeny generated from partial cytochrome
c
oxidase subunit I (COI)sequences. The second group most closely matched
Velesunio wilsonii
in shell morphologybut formed three deeply divergent mitochondrial DNA lineages. All four lineages occurredsympatrically in some areas and displayed corresponding fixed differences at nuclear allozymeloci, which suggests an absence of recent hybridization and the presence of separatespecies.
Keywords
: cryptic species, diversity, freshwater mussels, isozymes, mitochondrial DNA, morpho-logical plasticity
Received 1 April 2003; revision received 24 June 2003; accepted 24 June 2003
Introduction
The freshwater mussels (Order Unionoidea) include twosuperfamilies, Unionoidea and Etherioidea. Within theUnionoidea there are three families, the Margaritiferidae(Eurasia, North America), Unionidae (Eurasia, Africa, Indiaand North America) and Hyriidae (Australasia, SouthAmerica). North America has the greatest diversity offreshwater mussels, with nearly 300 recognized unionidspecies (Williams
et al
. 1993).In Australia, freshwater mussel diversity is much lower,
with only 17 species currently recognized. However, thediagnosis of species, genera and even subfamilies is madedifficult in hyriids owing to a traditional emphasis on shellcharacters as a taxonomic tool and their plastic reactions inresponse to distinct environmental conditions. In fact, mostAustralian freshwater mussel species exhibit high levels ofphenotypic variation and differences in shell morphology,and may vary in physiology and behaviour (Walker
et al
.
2001). Thus, the Australian freshwater mussel fauna maywell harbour cryptic species, and molecular techniques incombination with morphometric data offer the scope forimproved understanding about the diversity and evolu-tion of freshwater mussels in this country.
Molecular systematics of unionid mussels has beenwell researched (e.g. Lydeard
et al
. 1996, 2000; Roe &Lydeard 1998; King
et al
. 1999; Roe
et al
. 2001; Graf 2002)and an evolutionary origin for the hyriids has been proposed(Graf & Ó Foighil 2000). However, thus far no detailedgenetic analysis has been conducted on the Australianhyriids.
The objective of the present study therefore, was to com-pare and contrast molecular [mitochondrial DNA (mtDNA),isozyme] and morphological variation of the freshwatermussel fauna from four river systems in southwestQueensland, Australia, which form catchments in twomajor drainage basins: the Lake Eyre and the Murray–Darling. Our research represents the first detailed geneticsurvey of hyriid mussel diversity in Queensland, thereforea first-order exploratory sampling regime was used acrossa broad geographical range.
Correspondence: Dr Andrew Baker. Fax: + 61 73875 7615; E-mail:[email protected]

2708
A . M . B A K E R
E T A L .
© 2003 Blackwell Publishing Ltd,
Molecular Ecology
, 12, 2707–2717
Materials and methods
Study area
The Warrego and Condamine are tributary river systemsof the Darling River in the Murray–Darling Basin in south-eastern Australia, which has an area of 1.06 million squarekilometres (Kingsford 2000). The Darling catchment ischaracterized by small tributary systems feeding intoprogressively fewer and larger channels (Williams 1980).
The Bulloo and the Cooper are river systems of the aridendorheic Lake Eyre Basin in central Australia, which hasan area of 1.14 million square kilometres, making it one ofthe largest areas of internal drainage in the world. Thechannels of these rivers are predominantly anastomosing,with the river existing as multiple channels separated byfloodplain islands (Knighton & Nanson 1994). The topo-graphy of all the drainages is predominantly flat and thehydrology is inherently variable, with extensive floodingalternating with long, dry periods of little flow (Puckridge
et al
. 1998; Thoms & Sheldon 2000).
Sampling
Mussels were collected between late 2000 and Septem-ber 2001 from 21 sites in southwest Queensland (Fig. 1and Table 1). Samples were taken from a variety of flowenvironments to maximize morphological variation, giventhat Walker
et al
. (2001) suggested that the morphologicalfeatures in these taxa are likely to vary in response to local
environmental flow conditions. Mussels were collected byhand from waterholes, placed in large cotton bags to keepthem cool, and returned to the laboratory alive. Shell meas-urements were made for each specimen using digitizedcallipers. Shells were then opened by inserting reversepliers between the valves and applying a gentle but con-tinuous pressure, until the mussel relaxed. This minimizeddamage to the hinge. The body of the mussel was removedand a sample of the mantle tissue was taken for molecularanalysis.
Table 1 Geographic locations of sampled sites and numbers of individuals used in analyses
Catchment Site Site code Geographic coordinates Morphometrics Allozymes mtDNA
Cooper Creek Warrannee Waterhole WN 25°54′30.7′′ S 143°05′23.6′′ E 32 32 5Cooper Creek Pelican Waterhole PC 24°13′35.0′′ S 143°20′06.1′′ E 33 30 4Cooper Creek Tanbar Waterhole TR 25°50′13.3′′ S 141°55′00.3′′ E 6 37 8Cooper Creek Shed Waterhole SH 25°23′41.6′′ S 142°49′39.0′′ E 10 34 10Cooper Creek Glenmurken Waterhole GM 25°26′52.1′′ S 142°40′43.1′′ E — 3 3Cooper Creek Murken Waterhole MU 25°25′46.6′′ S 142°43′57.9′′ E 2 44 8Cooper Creek Waterloo Waterhole WL 24°13′38.2′′ S 143°17′23.3′′ E 3 3 3Cooper Creek Homestead Waterhole HS 25°48′27.8′′ S 143°02′36.2′′ E 3 48 8Cooper Creek Mayfield Waterhole MF 25°26′11.5′′ S 142°43′37.3′′ E 2 42 6Cooper Creek One Mile Waterhole OM 25°50′42.0′′ S 143°03′07.2′′ E 3 25 5Cooper Creek Top Waterhole TH 24°11′02.6′′ S 143°21′05.6′′ E 3 44 11Cooper Creek Wilson WS 27°49′23.1′′ S 142°35′57.6′′ E — 44 4Cooper Creek Yalungah YG 25°51′13.6′′ S 141°58′25.2′′ E — 43 6Bulloo River Bulloo/Thargomindah BT 28°01′01.2′′ S 143°47′04.8′′ E — 44 6Bulloo River Bulloo BU 28°34′00.8′′ S 143°09′60.2′′ E — 44 3Warrego River Sanford Park Lagoon SP 26°55′24.4′′ S 146°02′13.8′′ E 34 33 4Warrego River Tinnenburra TB 28°43′58.5′′ S 145°33′00.7′′ E 3 4 1Warrego River Rocky Waterhole RW 28°17′05.9′′ S 145°38′43.7′′ E 11 11 4Warrego River Thurrulgoonia TG 28°46′10.8′′ S 145°58′36.5′′ E — 1 1Warrego River Key Waterhole KH 28°19′12.6′′ S 145°43′38.7′′ E 5 6 4Condamine River Brigalow BR 26°51′50.4′′ S 150°39′44.4′′ E — — 7
Fig. 1 Sample site localities.

I N L A N D A U S T R A L I A F R E S H W A T E R M U S S E L D I V E R S I T Y 2709
© 2003 Blackwell Publishing Ltd, Molecular Ecology, 12, 2707–2717
Genetic analyses
Enzyme electrophoresis. Twenty enzymes were screenedusing Cellulose Acetate Gel Electrophoresis (Titan III plates,Helena laboratories). Eight enzyme loci were polymorphicand could be scored with confidence: Mdh-1, Mdh-2, α-Est,6-Pgd, Pgm, Pgi, PepC and Mpi. Electrophoresis and stainingprocedures were modified from Richardson et al. (1986)and agar overlays were used to visualize banding patterns.
Allelic designations for each individual at each locuswere scored by eye. The computer program populations(O. Langella, unpublished data) was used to calculate theshared allele distance (DAS, where DAS = 1 − S/2rand the number of shared alleles S is summed over all locir; Jin & Chakraborty 1993) between all pairs of individuals.
Genetic distances were ordinated in two-dimensionalspace by multidimensional scaling (MDS). One of the virtuesof MDS is that it does not assume linearity (a drawbackof both principal component and principal coordinate ana-lyses). Frequency data, such as allele frequencies, willoften violate the assumption of linearity. In addition, MDSallows for the use of a specified distance matrix, such aspairwise distances or similarities, which corrects for the factthat frequencies of alleles for a given locus add up to oneand do not represent independent dimensions (Lessa 1990).
An MDS plot based on the DAS genetic distance matrixwas formulated using the program patn (Belbin 1993). Thealgorithm was invoked using two dimensions, 50 itera-tions and a ratio-ordinal cut value of 0.9. Since the data setwas very large (n = 572), individuals that were identicalacross all loci were deleted prior to MDS analysis.
Mussels were initially separated into putative speciesgroups based on the diagnostic allozyme loci and MDSgroupings. We then sought to determine if the allozyme-based groups would match those generated from mtDNA.
Mitochondrial DNA. For each species group occurring ateach waterhole, individuals were selected randomly formtDNA analysis. All individuals that were sequenced hadpreviously been screened for allozyme variation.
Total genomic DNA was isolated using a modification ofthe cetyltrimethylammonium bromide (CTAB)/phenol–chloroform DNA extraction protocol (Doyle & Doyle1987). A 710-base-pair (bp) fragment of the cytochrome coxidase subunit I (COI) gene was amplified usingpolymerase chain reaction (PCR) in a 25-µL total volumecontaining: 16.9 µL double-distilled H2O, 1 µL each of 10 µmprimer LCO1490 and HCO2198 (Folmer et al. 1994), 0.5 µL10 mm dNTPs, 1 µL 50 mm MgCl2, 2.5 µL 10× ReactionBuffer, 0.1 µL Thermus aquaticus DNA (Taq) polymeraseand 1 µL template DNA. PCR was performed on the Gene-amp PCR System 9700 (PE Applied Biosystems) with aninitial denaturation step of 94 °C for 5 min; 15 cycles of94 °C for 30 s, 40 °C for 30 s, 72 °C for 1 min; 25 cycles of
94 °C for 30 s, 55 °C for 30 s, 72 °C for 1 min and a finalextension step of 68 °C for 5 min. PCR products were puri-fied using the QIAquick PCR purification kit (Qiagen) anddirectly sequenced using the LCO1490 primer, on anApplied Biosystems 377 automated sequencer.
Sequences contained no indels and were aligned by eyeusing bioedit Version 5.0.9 (Hall 1999). Exploratory dataanalysis of sequences was performed using mega Version2.1 (Kumar et al. 2001). Estimates of mean pairwise sequencediversity ± SE were made using 5000 bootstraps of the data.modeltest Version 3.06 (Posada & Crandall 1998) wasused to determine the DNA substitution model of best fitfor the sequence data.
Avise (1994) recommended that multiple methods ofanalysis involving different philosophical approaches shouldbe undertaken when estimating phylogenies. In the presentstudy therefore, four phylogenetic methods were used toensure concordance among major mitochondrial lineages;Bayesian (Rannala & Yang 1996), Neighbour Joining (Sai-tou & Nei 1987), Maximum Parsimony (Eck & Dayhoff1966) and Maximum Likelihood (Felsenstein & Churchill1996).
A Bayesian phylogeny was inferred from the sequencesusing mrbayes Version 2.01 (Huelsenbeck & Ronquist 2001).mrbayes was invoked using codon partitioning, TrN + GDNA substitution model, empirical base frequencies, among-site rate variation with shape 0.2265, unconstrained branchlength, eight rate categories on the gamma distribution,one million cycles with trees sampled every 100 cycles, andeight chains (seven heated). The program was repeated atthree different chain temperature settings (0.1, 0.2 and 0.5),to ensure topology and posterior probability concordance.Neighbour Joining, Maximum Parsimony and MaximumLikelihood phylogenies were inferred using phylip Version3.6a2 (Felsenstein 1993). The Neighbour Joining methodwas invoked using the F84 model of DNA substitution,gamma-distributed rates, transition/transversion ratio 3.2,coefficient of variation 2.1, 1000 bootstrap pseudoreplic-ates of the data (block size 3) and 10 jumbles. Block size isused to partition the sequence data into codons and thejumble option randomizes the input order of taxa duringtree construction. The Maximum Parsimony method wasinvoked using Search for best tree, most thorough search,1000 bootstrap pseudoreplicates of the data (block size 3)and one jumble. The Maximum Likelihood method wasinvoked using empirical base frequencies, search for besttree, 100 bootstrap pseudoreplicates of the data (block size3), transition/transversion ratio 3.2, gamma-distributedrate variation among sites with shape 0.2265, one jumble ofthe data, coefficient of variation 2.1, three categories in theHidden Markov Model, three categories assigned withrates (1, 1, 4.7). The coefficient of variation (CV) is relatedto the ‘alpha’ (α) shape parameter of the gamma distribu-tion by CV = .
jr∑
112/α

2710 A . M . B A K E R E T A L .
© 2003 Blackwell Publishing Ltd, Molecular Ecology, 12, 2707–2717
A single published freshwater pearly mussel Epioblasmatriquetra (Unionoida: Unionidae) sequence from the USA(GenBank Accession Number AF156528) was used asan outgroup in the phylogeny. The following publishedsequences were used within the ingroup, as a comparisonwith our sampled sequences: one Castalia stevensi (Unionoida:Hyriidae) sequence from South America (GenBank Acces-sion Number AF231736), one Diplodon deceptus (Unionoida:Hyriidae) sequence from South America (GenBank Acces-sion Number AF231744), three Hyridella menziesi (Unionoida:Hyriidae) sequences from New Zealand (GenBank Acces-sion Numbers AF231747; AF305369; AF305370), two Hyridelladepressa (Unionoida: Hyriidae) sequences from New SouthWales, Australia (GenBank Accession Numbers AF156496;AF305368), one Hyridella australis (Unionoida: Hyriidae)sequence from Australia (GenBank Accession NumberAF305367), two Velesunio ambiguus (Unionoida: Hyriidae)sequences from New South Wales, Australia (GenBankAccession Numbers AF305371; AF305372) and one Velesunioangasi (Unionoida: Hyriidae) sequence from Australia (Gen-Bank Accession Number AF231743).
As we were uncertain which of our clades (if any) corre-sponded to Velesunio wilsonii, we attempted to sequencetwo preserved specimens from the Queensland Museum(Southbank Campus, Australia) that had previously beenpositively identified using morphological keys as V. wilsonii(specimens were unregistered; localities — Betoota, southwestQueensland, Australia and Winton, west-central Queensland,Australia). It was intended to use these sequences as a com-parative ingroup in the phylogeny. Two primers [specificfor Velesunio spp. and internal to the COI primers publishedby Folmer et al. (1994)] were designed to amplify two shorterCOI fragments using PCR. Primer A (sequence 5′−3′:GAGTGGWGTTGGRACTGGTTG) in combination withprimer HCO2198 (Folmer et al. 1994) yielded a 425-bp COIfragment. Primer B (sequence 5′−3′: CAGTWCCAACWCC-CCTCTCAAC) in combination with LCO1490 (Folmer et al.1994) yielded a 281-bp COI fragment.
Morphometric analyses
Six shell characters were measured (Fig. 2): total shelllength (TL), beak height (BH), hinge length (HL), maximumheight (MH), long beak length (LBL), and width (W). Topermit comparison with Walker’s (1999) morphological keyto the genera and species of freshwater mussels in Australia,the following six ratios were also calculated: MaximumHeight Index (MHI = MH/TL), Beak Height Index (BHI =BH/MH), Beak Length Index (BLI = LBL/TL), WidthLength Index (WLI = W/TL), Width Height Index (WHI =W/MH) and Hinge Length Index (HLI = HL/TL). Prior toanalysis, measurements for the shell characters were log-transformed, while untransformed values were used forthe ratios.
Principal Components Analysis (PCA) was used to iso-late morphometric characters accounting for the most vari-ation among species groups identified a priori, on the basisof the molecular analyses. PCA was computed in pc-ord(McCune & Mefford 1995) using a double-centreing tech-nique (see Somers 1989; Cadima & Jolliffe 1996). The ‘adjustto mean’ relativization was used with a variance–covariancecross-products matrix. As PCA was used to summarize thesize and shape components for the shells, DiscriminantFunction Analysis (DFA) was then employed to assesshow accurately individual shells had been assigned to thegenetic lineages. DFA was performed using statistica(V.5; Statsoft Inc. 1995) under a standard model.
Results
Genetics
Allozymes. Allele frequencies at variable enzyme loci for572 individual mussels are shown in Table 2. A maximumof eight alleles was detected (PepC ).
Several enzyme loci were diagnostic (Table 2), differen-tiating four putative non interbreeding groups (designatedA–D). Group A was distinguished from Groups B, C and Dat Mdh-1-100, Mdh-2-98 and α-Est-98. Group B was distinguishedfrom Group C at Mpi-104 and Group C was distinguishedfrom Group D at Pgi-101. Group B was distinguished fromGroup D at Mdh-1-96 and Pgi-101. The four groups wereclearly separated in the MDS, with greatest separationbetween Group A, Group B and Group C/D (Fig. 3).
After partitioning mussels into Groups (A–D) based ondiagnostic loci and the MDS plot, frequencies of eachgroup at each site were calculated. The corresponding piecharts (Fig. 4) show that all four groups occurred in CooperCreek. Two or more groups were sympatric in 77% (10/13)of sites sampled in Cooper Creek. For example, all four
Fig. 2 Diagrammatic representation of mussel morphometricmeasures.

I N L A N D A U S T R A L I A F R E S H W A T E R M U S S E L D I V E R S I T Y 2711
© 2003 Blackwell Publishing Ltd, Molecular Ecology, 12, 2707–2717
groups were sympatric in the Homestead Waterhole,Top Waterhole contained groups A, B and D, Pelican,Yalungah, Shed, Warrannee and One Mile Waterholes allcontained groups A and D.
All of the sampled Bulloo, Warrego and CondamineRiver mussels exhibited allele frequencies correspondingto group C.
Mitochondrial DNA. Mitochondrial sequences (n = 114) wereobtained using the light strand primer LCO1490 (Folmeret al. 1994), which generated 473 base pairs of unambiguoussequence. A total of 49 unique COI haplotypes wererecovered (sequences are deposited in GenBank, AccessionNumbers AY211550–AY211598). There were no insertionsor deletions detected in the sequences. Of the 473 basepairs sequenced, 137 sites (29%) exhibited variation and106 sites (22%) were phylogenetically informative. Of the106 informative sites, 11 (10%) were at the first position ofthe codon, four (4%) were at the second position and 91 (86%)
Table 2 Allele frequencies at variable enzyme loci for groups A–D
Locus Allele
Group
A B C D
Mdh-1* 94 0 1 0.660 096 0 0 0.340 1
100 1 0 0 0Mdh-2* 98 1 0 0 0
100 0 1 1 1α-Est* 98 1 0 0 0
100 0 1 1 16-Pgd 98 0.005 0 0 0
100 0.995 1 1 1Pgm 96 0.008 0 0.039 0.491
100 0.584 1 0.917 0.496104 0.408 0 0.044 0.013
Pgi* 98 0 0 0.007 099 0.042 0 0 0
100 0.916 1 0.794 0101 0.042 0 0 1104 0 0 0.189 0108 0 0 0.010 0
PepC 97 0 0 0.026 098 0.706 0.011 0.303 0.08699 0.024 0.027 0.541 0
100 0.206 0.881 0.003 0.879101 0 0.061 0.021 0.035102 0.016 0.020 0.098 0103 0 0 0.008 0104 0.048 0 0 0
Mpi* 100 — 0 0 —102 — 0 1 —104 — 1 0 —
Alleles at each locus are designated by relative mobility.*Indicates diagnostic loci among the four lineages; — indicates not scored.
Fig. 3 Multidimensional scaling plot of allozyme shared allelegenetic distances plotted in two dimensions (stress = 0.1781).
Fig. 4 Allozyme-based species-group pie charts of each putativespecies group at each sampled site. The dotted border (top section)delineates sample sites from the Lake Eyre Basin, which isenlarged (bottom section) to show proportions of each putativespecies group (based on the allozyme data) at each site. Allindividuals sampled from the Warrego and Condamine Riversites (top section) were identified as Group C.
2712 A . M . B A K E R E T A L .
© 2003 Blackwell Publishing Ltd, Molecular Ecology, 12, 2707–2717
were at the third. The transition (Ts) to transversion (Tv)substitution ratio was 3.2. Translation of codons toamino acids revealed 27 variable sites. At the 27 variableamino acid sites, there were four first-position codontransitions, six first-position codon transversions, two second-position codon transitions, 11 second-position codon transver-sions and five third-position codon transversions. Of the 27variable amino acid sites, four occurred within lineage
A, 13 within lineage B, six within lineage C and 12 withinlineage D.
Uncorrected pairwise intralineage sequence divergencesranged from 0.2 to 3.8% (lineage A, 0.2–0.8%; lineage B,0.2–3.8%; lineage C, 0.2–2.5%; lineage D, 0.2–2.7). Uncorrectedpairwise interlineage sequence divergences ranged from9.3 to 15.5%. Mean sequence divergence among lineages was9.2% ± 0.8. The two recognized Velesunio species (V. angasi and
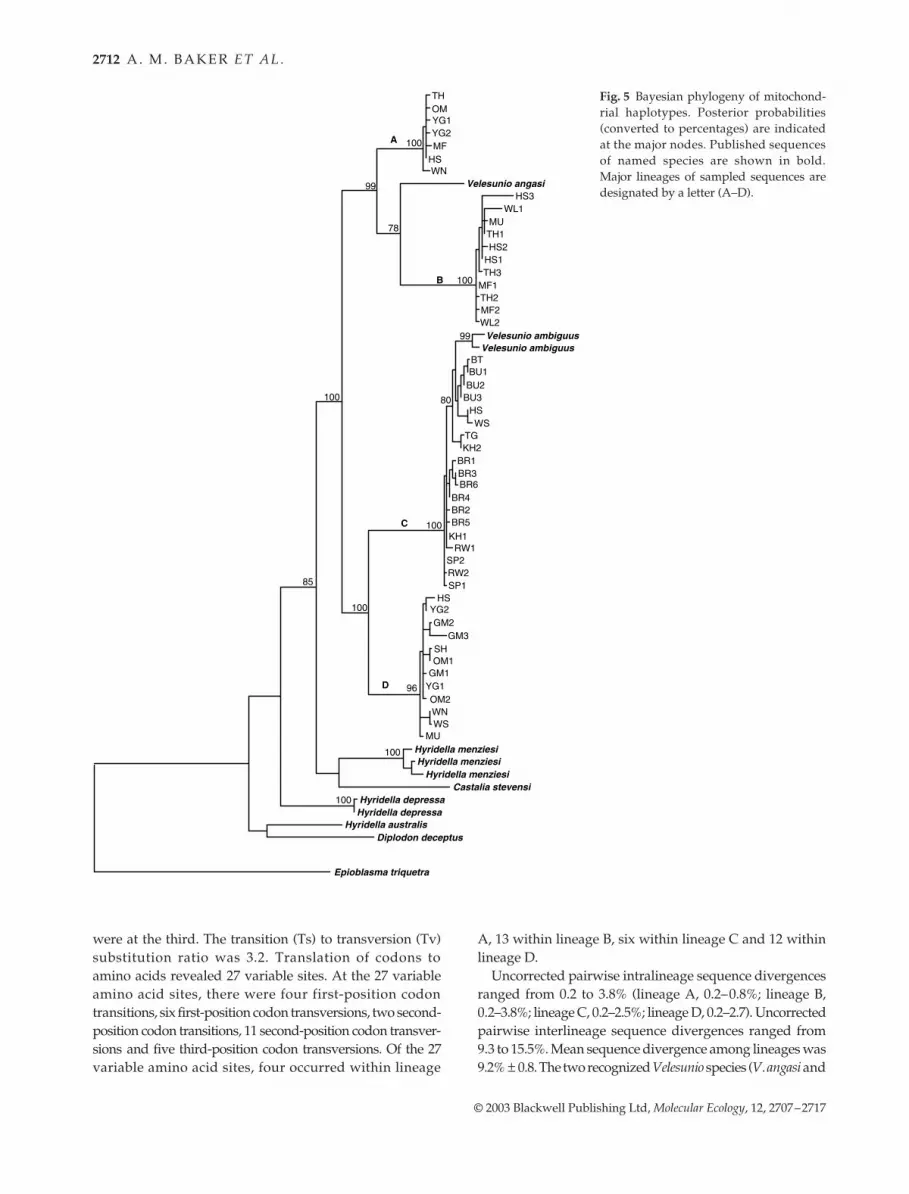
Fig. 5 Bayesian phylogeny of mitochond-rial haplotypes. Posterior probabilities(converted to percentages) are indicatedat the major nodes. Published sequencesof named species are shown in bold.Major lineages of sampled sequences aredesignated by a letter (A–D).

I N L A N D A U S T R A L I A F R E S H W A T E R M U S S E L D I V E R S I T Y 2713
© 2003 Blackwell Publishing Ltd, Molecular Ecology, 12, 2707–2717
V. ambiguus) included in the analysis were 9% ± 0.01 diver-gent, which was very similar to mean sequence divergencebetween all pairs of lineages A–D (range 10.4% ± 1.3 to12.9% ± 1.4).
All attempts at PCR on the preserved Velesunio wilsoniimuseum specimens proved unsuccessful, despite using arange of DNA extraction procedures (CTAB, Chelex, Dneasytissue kit: QIAGEN). However, amplification was successfulusing both fresh mussel material and a preserved museumspecimen of V. ambiguus collected in 1995 (QueenslandMuseum Number MO55581). Presumably the considerablyolder fixed V. wilsonii DNA material (collected in 1978 and1983) was too degraded to be amplified using PCR.
The Bayesian phylogeny (Fig. 5) separated the 49 mito-chondrial haplotypes into four deep, well-supported line-ages (these lineages A–D correspond to the allozyme-basedgroupings A–D). All individuals fell into the same mito-chondrial and allozyme species grouping.
The four mitochondrial lineages were also well supported(90–100% bootstrap values) in the neighbour joining, max-imum parsimony and maximum likelihood trees (not shown).The sampled mussel sequences and the three Velesunio sp.sequences formed a strongly supported monophyletic group,with respect to the Hyridella sp., Castalia stevensi and Diplodondeceptus ingroup sequences. Lineage B formed a sister groupto the single Velesunio angasi sequence. The two Velesunioambiguus sequences formed a strongly supported subcladeembedded within lineage C.
Morphology
Evidence was found for two major morphological groupsin our sample of mussels. The first group keyed out mostclosely to Velesunio ambiguus, although the Maximum HeightIndex was often less than 0.60 in our sample (Fig. 6). Thesecond group keyed out most closely to Velesunio wilsonii,
Fig. 6 Quantile box plots showing range[90th, 75th, 50th (median), 25th and 10th]percentiles in each lineage for calculatedratios used in the morphological key(Walker 1999). The sample mean for eachplot is represented as a solid line.

2714 A . M . B A K E R E T A L .
© 2003 Blackwell Publishing Ltd, Molecular Ecology, 12, 2707–2717
although the Maximum Height Index was sometimesabove 0.55 in our sample (Fig. 6). No Alathyria specieswere found.
In the PCA of all shells using log-transformed values forshell measurements and calculated values for ratios (Fig. 7),more than half of the variation (61%) was explained byPCI, with 30% explained by PCII and only 4% by PCIII. Thefirst component correlated with aspects of width, clearlyseparating the much wider lineage C shells from the rest(Table 3). Although there was considerable overlap amonglineages A, B and D, there was a tighter grouping of lineageD shells compared with A and B, which were widely dis-tributed along both PCI and PCII (Fig. 7).
The DFA of eigenvalues from the PCA for each shell forall lineages (Fig. 8) confirmed the distinctiveness of the lin-eage C shells (tolerance 0.008). The DFA suggested that66% (10/15) of lineage A, 20% (2/10) of lineage B, 100%(54/54) of lineage C and 100% (71/71) of lineage D indi-viduals could be correctly assigned by morphology. In theDFA, the first function (Root 1) gave the best overall dis-crimination among the lineages (Wilks’ Lambda = 0.082;approximate F9,350 = 69.45; P < 0.001). The DFA suggests aclear separation between lineages A, B and D comparedwith lineage C, but also suggests a degree of morphologicaldistinctiveness between lineages A, B and D since there wasonly partial overlap between their 95% confidence limitellipses (Fig. 8).
Discussion
The mitochondrial phylogeny revealed four distinct, highlydivergent lineages among the mussels sampled from thefour southwest Queensland river drainages. Mussel group-ings derived from diagnostic enzyme loci and MDS analysisalways matched the mitochondrial lineages. Where thegroups are sympatric, no heterozygotes were detected atdiagnostic allozyme loci. Taken together therefore, thegenetic data strongly suggests that the four mussel groupsidentified in the present study have differentiated genepools and should be considered separate species.
Given the distribution of Australian freshwater musselsrecognized by Walker (1999), it could have been expectedto find Velesunio ambiguus, V. wilsonii, Alathyria jacksoni andA. pertexta in the present survey. Morphological and mito-chondrial data suggest that lineage C represents a variantof the mussel Velesunio ambiguus. One of the mitochondriallineages A, B, or D may indeed represent Velesunio wilsonii,
Fig. 7 Principal components (PC) analysis plot of shell shapes forPCI vs. PCII.
Table 3 Eigenvalues, variance explained and correlations of shellmeasurements and ratios with the first three principal com-ponents axes (PCI, PCII and PCIII) for the principal componentsanalysis (PCA) on morphological data
PCI PCII PCIII
Eigenvalue 7.046 6.453 0.459% variance explained 61.35 30.07 3.99Total length (TL) −0.1346 −0.3969 0.0816Beak height (BH) −0.2462 −0.2507 0.2772Hinge length (HL) −0.0676 −0.5272 −0.4134Maximum height (MH) −0.2291 −0.2746 0.2746Beak length (LBL) −0.1469 −0.422 0.1054Width (W) −0.5922 −0.0833 0.0497IndicesMHI −0.122 0.1505 0.2419BHI −0.0374 0.0512 0.0081BLI −0.0203 −0.0429 0.0427WLI −0.3938 0.281 −0.0686WHI −0.559 0.3206 −0.3945HLI 0.0875 −0.1799 −0.6602
Fig. 8 Relationships between scores on Root 1 and Root 2 ofdiscriminant function analyses of log-transformed shell measure-ments for all lineages. Ellipses encompass 95% confidence limitsfor each lineage.

I N L A N D A U S T R A L I A F R E S H W A T E R M U S S E L D I V E R S I T Y 2715
© 2003 Blackwell Publishing Ltd, Molecular Ecology, 12, 2707–2717
but since we were unable to obtain genetic information frommuseum specimens of this species, this remains uncertain.Mean COI sequence divergences among pairs of lineagesA, B and D (range 11.3% ± 1.3 to 12.9% ± 1.4) were the sameorder of magnitude as those among COI sequences of recog-nized freshwater mussel species used in the current study(Velesunio ambiguus/V. angasi 9.0% ± 0.01; Hyridella depressa/H. australis 8.5% ± 0.01) and elsewhere (1.2–14.5% amongPotamilus species, Roe & Lydeard 1998; 3.8–15.0% amongfour Lasmigona species, King et al. 1999).
The PCA based on shell morphology did not success-fully distinguish between lineages A, B and D. The PCAand DFA both revealed more differentiation within thanbetween lineages A and B, suggesting high levels of mor-phological variation within species in this group. In theDFA however, lineage D showed a more distinctive tightclustering of all shells.
Variation in morphology (and possibly physiology) ofthese mussels may have evolved in response to variablelocal environmental flow conditions (Walker et al. 2001).Plastic reactions allowing the mussels to mature andreproduce under different environmental conditions couldbe selectively favoured, provided genetic variation in reac-tions is available and the benefits of the response are notoutweighed by the costs (Smekens & van Tienderen 2001).Future taxonomic investigations may more clearly resolvethe apparent morphological differences between lineagesand facilitate field identification of ‘live’ shells.
Graf & Ó Foighil (2000) proposed that since the Austral-asian Hyridellinae were paraphyletic with respect to theSouth American Hyriinae in a 28S rDNA phylogeny, theHyriidae probably arose in Australia and spread via Ant-arctica to South America, prior to the break up of Gondwa-naland. The topology of the published Hyridellinae andHyriinae COI sequences in our Bayesian phylogeny sup-ports this hypothesis, since the South American Castaliastevensi is nested within the New Zealand and AustralianHyridella clades.
In addition to the large mitochondrial diversity amonglineages, notable diversity within lineages was found, aswell as a large number of intralineage amino acid changes,particularly considering that COI is a relatively conservedcoding mtDNA gene in terms of its amino acid evolution.Taken together, these data suggest that these lineages havebeen reproductively isolated for some time. Imposing amolecular clock on mitochondrial sequence divergence amongthe lineages presented here, and assuming a COI sequencedivergence rate of 0.6% per million years (Baldwin et al.1996; Renard et al. 2000), it can be estimated that a commonancestor existed 14–17 million years ago, during the Miocene.The Early Tertiary climate in Australia was warm and wet,but towards the end of the Miocene Epoch the continent hadprobably started to become cooler and drier (Christophel& Greenwood 1989; Pole et al. 1993). It is conceivable that
our mussel lineages became separated during this drier period,evolved in isolation for a time, and have since recontacted asa result of episodic flooding.
Lineage C is widespread, occurring in all four of theriver systems surveyed, a (straight line) distance of morethan 800 kilometres. Notwithstanding the diversity acrossthe sampled range, smaller-scale gene flow within thislineage may occur, since a number of haplotypes werecommon to populations separated by substantial geo-graphical distances (unpublished data). Spatial geneticstructure within lineage C and lineage D (which waswidespread in Cooper Creek) is currently being examinedin detail.
There is considerable potential for dispersal in fresh-water mussels, since the glochidial larvae are obligateparasites on one or more fish species for a period of daysto several weeks, after which the juveniles settle andbecome predominantly sessile as adults (Walker 1981).Provided some mussel larvae are parasitizing fish duringthe flooding period, patterns of gene flow among water-holes of the mussels and their teleost host(s), are likely tobe closely linked. It is not known which fish species areparasitized by our mussel lineages, but this is currentlybeing investigated.
Areas of the extensive Lake Eyre Basin are recognizedto have flourishing communities of molluscs (Sheldon &Puckridge 1998; Sheldon et al. 2002). In the present study,a rich diversity of freshwater mussel fauna has beendemonstrated in Cooper Creek, within the Lake EyreBasin. It is plausible that the lineages identified here werehistorically more widespread, have since been extirpatedfrom areas of the much disturbed Murray–Darling Basin,and are now restricted to catchments of the Lake EyreBasin.
The sampling strategy employed in the present studydid not permit a formal analysis of the relationshipbetween environment and mussel diversity. However, weare currently conducting more detailed sampling to deter-mine if the distribution and diversity of mussel lineagesobserved in the present study is representative of other west-ern Queensland drainages within the Murray–Darlingand Lake Eyre Basins. This should allow us to investigatecausal relationships between environmental change andgenetic diversity in Queensland’s freshwater musselfauna.
Acknowledgements
We thank Darryl Potter and John Stanisic of the QueenslandMuseum for access to Velesunio wilsonii specimens, and RachelKing for help with patn. We also thank Angus Emmott, SandyKidd and Bob Morrish for granting us access to the study sites andfor their hospitality. This study forms part of a large project ondryland river refugia, funded by the Cooperative Research Centrefor Freshwater Ecology.

2716 A . M . B A K E R E T A L .
© 2003 Blackwell Publishing Ltd, Molecular Ecology, 12, 2707–2717
References
Avise JC (1994) Molecular markers. Natural History and Evolution.Chapman & Hall, New York.
Baldwin BS, Black M, Sanjur O, Gustafson R, Lutz RA, VrijenhoekRC (1996) A diagnostic molecular marker for zebra mussels(Dreissena polymorpha) and potentially co-occuring bivalves:mitochondrial COI. Molecular Marine Biology and Biotechnology,5, 9–14.
Belbin L (1993) PATN Technical Reference Manual. CSIRO Divisionof Wildlife and Ecology, Canberra.
Cadima JFCL, Jolliffe IT (1996) Size- and shape-related PrincipalComponent Analysis. Biometrics, 52, 710–716.
Christophel DC, Greenwood DR (1989) Changes in climate andvegetation in Australia during the Tertiary. Review of Palaeobotanyand Palynology, 58, 95–109.
Doyle JJ, Doyle JL (1987) A rapid DNA isolation procedure forsmall quantities of leaf tissue. Phytochemistry Bulletin, 19, 11–15.
Eck RV, Dayhoff MO (1966) Atlas of Protein Sequence and Structure1966. National Biomedical Research Foundation, Silver SpringMD.
Felsenstein J (1993) phylip — Phylogeny Inference Package(Version 3.2). Cladistics, 5, 164–166.
Felsenstein J, Churchill GA (1996) A hidden Markov modelapproach to variation among sites in rate of evolution. MolecularBiology and Evolution, 13, 93–104.
Folmer O, Black M, Hoeh W, Lutz R, Vrijenhoek R (1994) DNAprimers for amplification of mitochondrial cytochrome c oxi-dase subunit I from diverse metazoan invertebrates. MolecularMarine Biology and Biotechnology, 3, 294–299.
Graf DL (2002) Molecular phylogenetic analysis of two problem-atic freshwater mussel genera (Unio and Gonidea) and a re-evaluation of the classification of nearctic Unionidae (Bivalvia:Palaeoheterodonta: Unionoida). Journal of Molluscan Studies, 68,65–71.
Graf DL, Ó Foighil D (2000) Molecular phylogenetic analysis of28S rDNA supports a Gondwanan origin for AustralasianHyriidae (Mollusca: Bivalvia: Unionoida). Vie et Milieu, 50, 245–254.
Hall TA (1999) BioEdit: a user-friendly biological sequence align-ment editor and analysis program for Windows 95/98/NT.Nucleic Acids Symposium Series, 41, 95–98.
Huelsenbeck JP, Ronquist F (2001) mrbayes: Bayesian inference ofphylogenetic trees. Biometrics, 17, 754–755.
Jin L, Chakraborty R (1993) Estimation of genetic distance andcoefficient of gene diversity from single-probe multilocusDNA fingerprinting data. Molecular Biology and Evolution, 11,120–127.
King TL, Eackles MS, Gjetvaj B, Hoeh WR (1999) Intraspecificphylogeography of Lasmigona subviridis (Bivalvia: Unionidae):conservation implications of range discontinuity. Molecular Ecology,8, S65–S78.
Kingsford RT (2000) Protecting rivers in arid regions or pumpingthem dry? Hydrobiologia, 427, 1–11.
Knighton AD, Nanson GC (1994) Waterholes and their signi-ficance in the anastomosing channel system of Cooper Creek,Australia. Geomorphology, 9, 311–324.
Kumar S, Tamura T, Jakobsen IB, Nei M (2001) mega2: molecularevolutionary genetics analysis software. Bioinformatics, 17,1244–1245.
Lessa EP (1990) Multidimensional analysis of geographic geneticstructure. Systematic Zoology, 39, 242–252.
Lydeard C, Mulvey M, Davis GM (1996) Molecular systematicsand evolution of reproductive traits of North American fresh-water unionacean mussels (Mollusca: Bivalvia) as inferred from16S rRNA DNA sequences. Proceedings of the Royal Society ofLondon Series B, 351, 1593–1603.
Lydeard C, Minton RL, Williams JD (2000) Prodigious polyphylyin imperiled freshwater pearly-mussels (Bivalvia: Unionidae): aphylogenetic test of species and generic designations. In: TheEvolutionary Biology of the Bivalvia (eds Harper EM, Taylor JD,Crame JA), pp. 145–158, Special Publications 177. GeologicalSociety, London.
McCune B, Mefford MJ (1995) Multivariate Analysis of EcologicalData, PC-ORD, Version 3.0. MjM Sofware, Gleneden Beach, OR.
Pole MS, Hill RS, Green N, Macphail MK (1993) The Miocene Ber-wick Quarry — rain-forest in a drying environment. AustralianSystematic Botany, 6, 399–427.
Posada D, Crandall KA (1998) Modeltest: testing the model ofDNA substitution. Bioinformatics, 14, 817–818.
Puckridge JT, Sheldon F, Walker KF, Boulton AJ (1998) Flow vari-ability and the ecology of large rivers. Marine and FreshwaterResearch, 49, 55–72.
Rannala B, Yang ZH (1996) Probability distribution of molecularevolutionary trees: a new method of phylogenetic inference.Journal of Molecular Evolution, 43, 304–311.
Renard E, Bachmann V, Cariou ML, Moreteau C (2000) Mor-phological and molecular differentiation of invasive freshwaterspecies of the genus Corbicula (Bivalvia, Corbiculidea) suggestthe presence of three taxa in French rivers. Molecular Ecology, 9,2009–2016.
Richardson BJ, Baverstock PR, Adams M (1986) Allozyme Electro-phoresis. Academic Press, Australia.
Roe K, Lydeard C (1998) Molecular systematics of the freshwatermussel genus Potamilus (Bivalvia: Unionidae). Malacologia, 39,195–205.
Roe K, Hartfield PD, Lydeard C (2001) Phylogeographic analysisof the threatened and endangered superconglutinate-producingmussels of the genus Lampsilus (Bivalvia: Unionidae). MolecularEcology, 10, 2225–2234.
Saitou N, Nei M (1987) The neighbor-joining method: a newmethod for reconstructing phylogenetic trees. Molecular Biologyand Evolution, 4, 406–425.
Sheldon F, Puckridge JT (1998) Macroinvertebrate assemblagesof Goyder Lagoon, Diamantina River, South Australia. Transac-tions of the Royal Society of South Australia, 122, 17–31.
Sheldon F, Boulton AJ, Puckridge JT (2002) Conservation valueof variable connectivity: aquatic invertebrate assemblagesof channel and floodplain habitats of a central Australianarid-zone river, Cooper Creek. Biological Conservation, 103, 13–31.
Smekens MJ, van Tienderen PH (2001) Genetic variation andplasticity of Plantago coronopus under saline conditions. ActaOecologica, 22, 187–200.
Somers KM (1989) Allometry, isometry and shape in PrincipalComponent Analysis. Systematic Zoology, 38, 169–173.
Statsoft Inc. (1995) Statistica Software. Thompson Learning, Tulsa,OK.
Thoms MC, Sheldon F (2000) Water resource development andhydrological change in a large dryland river: The Barwon-Darling River, Australia. Journal of Hydrology, 228, 10–21.
Walker KF (1981) Ecology of freshwater mussels in the River Murray.Australian Water Resources Council Technical Paper, 63, 1–119.
Walker KF (1999) A provisional guide to the freshwater mussels

I N L A N D A U S T R A L I A F R E S H W A T E R M U S S E L D I V E R S I T Y 2717
© 2003 Blackwell Publishing Ltd, Molecular Ecology, 12, 2707–2717
(Hyriidae, Unionidae) of Australasia. In: Interactive Guide toAustralian Aquatic Invertebrates 2nd Edition (eds Gunn B, CranstonPS, Dimitriadis S, Trueman JWH). CSIRO Division of Entomology,Melbourne.
Walker KF, Byrne M, Hickey CW, Roper DS (2001) Freshwatermussels (Hyriidae) of Australasia. In: Ecology and Evolution of theFreshwater Mussels Unionoida (eds Bauer G, Wächtler K), pp. 5–31. Springer Verlag, Berlin.
Williams WD (1980) Australian Freshwater Life. Macmillan Pub-lishing, Melbourne.
Williams JD, Warren ML, JrCummings KS, Harris JL, Neves RJ(1993) Conservation status of freshwater mussels of the UnitedStates and Canada. Fisheries, 18, 6–22.
A.M.B. is a postdoctoral researcher investigating spatial geneticpatterns in Australian freshwater macroinvertebrates, ranging indiversity from caddis flies to spiny crayfish. F.S. is a postdoctoralresearch fellow in the Australian School of Environmental Studies.C.B. and K.G. were research assistants on the dryland river refugiaproject. J.M.H. is Professor in the Australian School of Envir-onmental Studies and her research focuses on using moleculartechniques to answer ecological and evolutionary questions ona range of animal and plant species. The work presented hereforms part of a large cooperative research project on the biodiversityof dryland river refugia run by S.E.B.





























