Embed Size (px)
Citation preview

ARTHRITIS & RHEUMATISMVol. 50, No. 5, May 2004, pp 1624–1635DOI 10.1002/art.20211© 2004, American College of Rheumatology
Defective Costimulatory Function Is a Striking Feature ofAntigen-Presenting Cells in an HLA–B27–Transgenic
Rat Model of Spondylarthropathy
Cecile Hacquard-Bouder,1 Geraldine Falgarone,1 Antoine Bosquet,1 Faıza Smaoui,1
Dominique Monnet,1 Marc Ittah,1 and Maxime Breban2
Objective. A disease resembling the human spon-dylarthropathies develops in HLA–B27–transgenic rats.This disease in rats is mediated by CD4� T cells, butantigen-presenting cells (APCs) may also play a role.Dendritic cells (DCs) have been reported to be defectivein allogeneic mixed lymphocyte culture in this model.Here, we further investigated the functional defect ofAPCs.
Methods. DCs and B cells from nontransgenic,HLA–B27 (33-3)–transgenic, and HLA–B7 (120-4)–transgenic rats were used to stimulate T cells. Surfaceexpression of HLA–B transgene and rat molecules onAPCs and the formation of conjugates between DCs andT cells were monitored by flow cytometry.
Results. We observed a strikingly defective stim-ulation of allogeneic and syngeneic T lymphocytes byAPCs from HLA–B27 but not HLA–B7 rats, even ifstimulation was driven in the presence of anti–T cellreceptor (TCR) antibody. We found no evidence thatHLA–B27 DCs were immature, lacked production of
some diffusible factor, or produced an inhibitory factorfor T cells. When comparing the levels of expression ofclass II major histocompatibility complex, CD2, inter-cellular adhesion molecule 1, lymphocyte function–associated antigen 1, B7, and CD40 molecules at thesurface of DCs from 33-3, 120-4, and nontransgenicrats, we found little difference. However, HLA–B27–transgenic DCs formed fewer conjugates with T cellsthan did nontransgenic DCs. Furthermore, the propor-tion of conjugates formed between DCs and T cells, aswell as the difference between nontransgenic and HLA–B27–transgenic DCs, were in large part reduced byblocking CD86 on DCs.
Conclusion. We confirmed defective stimulationof T cells by APCs in HLA–B27 rats, the mechanism ofwhich appears to implicate APC/T cell contact, indepen-dent of TCR engagement. In addition, decreased use ofthe CD86 costimulatory molecule by B27 DCs wasobserved. Impaired costimulatory function could resultin a loss of tolerance toward microbial flora in thismodel.
The HLA–B27 class I major histocompatibilitycomplex (MHC) allele is strongly associated with spondy-larthropathy (SpA), a group of inflammatory rheumaticdisorders that may also combine with psoriasis andinflammatory bowel disease. Both skeletal and extraar-ticular features of SpA are likely determined by sharedfactors, including HLA–B27 (1).
Several lines of rats transgenic for HLA–B27 andhuman �2-microglobulin (�2m) develop a spontaneousmultisystem inflammatory disease that strikingly resem-bles human SpA, by combining arthritis with colitis andpsoriatic skin lesions (2). The immune system is criticallyinvolved in the pathogenesis of disease in B27-transgenicrats, which is transmittable to nontransgenic rats byimmature hematopoietic cell engraftment and also re-
Supported by grants from Association de Recherche sur laPolyarthrite and Societe Francaise de Rhumatologie (SFR). Dr.Hacquard-Bouder’s work was supported in part by grants from theAssociation de Recherche Clinique en Rhumatologie and SFR. Dr.Monnet’s work was supported by a grant from Fondation pour laRecherche Medicale.
1Cecile Hacquard-Bouder, MD, Geraldine Falgarone, MD,PhD (current address: Hopital Avicenne, Bobigny, France), AntoineBosquet, MD, Faıza Smaoui, MSc, Dominique Monnet, MD, MarcIttah, BSc: Cochin Hospital, INSERM, CNRS, Universite Rene Des-cartes, Paris, France; 2Maxime Breban, MD, PhD: Cochin Hospital,INSERM, CNRS, Universite Rene Descartes, Paris, and AmbroisePare Hospital, AP-HP, Boulogne-Billancourt, France.
Drs. Hacquard-Bouder and Falgarone contributed equally tothis work.
Address correspondence and reprint requests to MaximeBreban, MD, PhD, Service de Rhumatologie, Hopital Ambroise Pare,9 Avenue Charles de Gaulle, Boulogne 92104, France. E-mail:[email protected].
Submitted for publication September 17, 2003; accepted inrevised form January 23, 2004.
1624

quires the presence of thymically derived T cells for itsdevelopment (3,4). This disease appears to be specificfor high expression levels of HLA–B27, and the presenceof resident bacterial flora is required for the develop-ment of gut and joint disease in B27-transgenic rats (5).Based on these findings, it is hypothesized that disease inB27-transgenic rats arises as a consequence of interac-tion between antigen-presenting cells (APCs) expressinghigh levels of HLA–B27 and peripheral T lymphocytes,with the CD4� T cell subset, in contrast to CD8� Tcells, appearing pathogenic (4,6). Accordingly, sponta-neous inflammation could result from a breakdown ofperipheral tolerance, particularly in the gut mucosa,which is a prominent site of inflammation in this model.
Dendritic cells (DCs) are hematopoietically de-rived cells present at a low rate in all lymphoid organsand also in solid organs, where they are capable ofacquiring antigen and thereafter migrating to secondarylymphoid organs. Among APCs, DCs play a key role inthe induction of a primary T cell response, in large partbecause of their capacity to dispense accessory signals,in addition to presenting antigen, acquired by endocyto-sis and loaded on MHC molecules, to the T cell receptor(TCR) (7). Besides promoting a T cell response towardforeign antigen, evidence has accumulated indicatingthat DCs also play a role in establishing tolerance towardself antigens and in the maintenance of peripheraltolerance (8–11). This may notably apply to antigenoriginating from the gut mucosa (12).
In several situations, impaired DC function hasbeen associated with a predisposition to the develop-ment of spontaneous autoimmunity. Such observa-tions were reported in MRL/lpr mice, in biobreedingdiabetes-prone rats, and in humans at risk for insulin-dependent diabetes (13–15). DC function was previouslystudied in 2 disease-prone HLA–B27–transgenic lines ofrats. A decreased capacity of splenic DCs to stimulate aprimary allogeneic T cell response in vitro was describedin both lines (16). In the present study, we confirmedand further expanded this observation. Our results indi-cate that the costimulatory function of APCs must beimpaired in disease-prone HLA–B27–transgenic rats,which may have implications for the spontaneous devel-opment of chronic inflammatory disorders in theseanimals.
MATERIALS AND METHODS
Rats. Inbred nontransgenic Fisher (F344), Lewis(LEW), dark agouti (DA), and brown Norway (BN) rats ages2–3 months were purchased from Iffa Credo (L’Arbresle,
France) or CERJ (Le Genest St. Isle, France). The 2 HLA–B27–transgenic lines 33-3 and 21-4L, and the HLA–B7–transgenic line 120-4 were originally produced at the Univer-sity of Texas Southwestern Medical Center (5). Disease-pronerats hemizygous for the 33-3 transgene locus, bearing 55 copiesof HLA–B*2705 and 66 copies of human �2m on a F344background, were maintained by breeding 33-3 females withnontransgenic F344 males and typing offspring, as previouslydescribed (17). The 33-3 rats homozygous for the rnu (nude)allele, which confers thymic aplasia and protection from HLA–B27–associated inflammatory disease (4), were produced bybackcrossing the 33-3 transgene locus onto nontransgenicF344-rnu/rnu rats, which were kindly provided by Dr. J. D.Taurog (University of Texas Southwestern). The disease-freeDA.33-3 line was produced by backcrossing the 33-3 transgenelocus �4 generations onto a DA background. Disease-freehomozygous 21-4L rats bearing 12 copies each of HLA–B*2705 and human �2m on a LEW background were main-tained by sister-to-brother mating. Disease-free homozygousF344.120-4 rats bearing 52 copies of HLA–B*0702 and 10copies of human �2m on a F344 background were produced bybackcrossing the 120-4 transgene locus originally on a LEWbackground �8 generations onto a F344 background. All ratswere bred and housed under conventional conditions. Ratsages 1–6 months were used and were routinely matched for ageand sex within individual experiments. Study procedures wereapproved by the institutional animal care committee.
Tissue culture medium, monoclonal antibodies (mAb),and other reagents. Tissue cultures were performed in RPMI1640 medium with Glutamax I (Life Technologies, Eragny surOise, France) supplemented with 10% fetal calf serum (FCS),penicillin G (100 units/ml), streptomycin (100 �g/ml), 2%sodium pyruvate, 0.05 mM 2-mercaptoethanol, and 5 mMHEPES, unless otherwise stated. Concanavalin A (Con A)–stimulated rat spleen and lymph node cell supernatant plus 50mM �-methyl-mannoside was used as Con A supernatant.
The following mouse IgG1 or IgG2a anti-rat antigenmAb were obtained from the European Collection of CellCulture (Salisbury, Wiltshire, UK) or were purchased from BDPharMingen (Le Pont de Claix, France): R73 (TCR�/�),OX35 (CD4), OX8 (CD8� chain), OX52 (T and natural killer[NK] cell–specific antigen), 3.2.3 (NK cell receptor protein 1A[NKRP1A]), OX62 (an �E2 integrin present on a subset ofDCs), OX33 (a CD45 epitope specific for B cells and a subsetof DCs), OX12 (rat Ig� chain), OX18 (rat class I MHC), OX6(RT1B: class II MHC locus), OX17 (RTID: class II MHClocus), OX41 (signal regulatory protein �/CD172a), OX42(C3bR: macrophages), ED3 (sialoadhesin/CD169: macro-phages), D34-485 (CD32), OX34 (CD2), 1A29 (intracellularadhesion molecule 1 [ICAM-1]), WT.1 (CD11a), WT.3(CD18), 3H5 (B7.1), 24F (B7.2), and A5-7 (interleukin-10[IL-10]).
The following mouse anti-human antigen mAb wereused: W6/32 (IgG2a) and PA2.6 (IgG1); monomorphic anti–HLA–A, anti–HLA–B, and anti–HLA–C; B1.23.2 (IgG2a);monomorphic anti–HLA–B and anti–HLA–C; ME1 (IgG1);anti–HLA–B7 and anti–HLA–B27; HC-10 (IgG2a); anti–HLA–B and anti–HLA–C free heavy chain; BBM.1 (IgG2b);and anti–human �2m. Irrelevant murine mAb includedMPOC-31C (IgG1), MOPC-173 (IgG2a), and MPC-11(IgG2b) (BD PharMingen). Phycoerythrin (PE)-conjugated
FUNCTIONAL IMPAIRMENT OF APCs IN HLA–B27–TRANSGENIC RATS 1625

monoclonal goat anti-mouse (GAM) IgG1 and fluoresceinisothiocyanate (FITC)–conjugated GAM IgG2a were obtainedfrom Caltag (Burlingame, CA), and FITC–GAM IgG wasobtained from Rockland (Gilbertsville, PA). FITC-conjugatedhamster anti-mouse CD40 (cross-reactive with rat CD40) IgM(HM40-3) and irrelevant FITC-conjugated hamster IgM(G235-1) were from BD PharMingen. Recombinant humanIL-2 (rHuIL-2; specific activity 20 IU/ng) was a kind gift fromSanofi (Labege, France). Rat recombinant interferon-�(IFN�) was from PeproTech (Rocky Hill, NJ). NG-methyl-L-arginine �-methyl-mannoside, lipopolysaccharide (LPS; Esch-erichia coli, serotype O111:B4), PKH67 green, and PKH26 redfluorescent cell linker mini kits were obtained from Sigma (St.Quentin Fallavier, France).
Isolation of cells. Splenic DCs were obtained by 2different methods. The first method was adapted from thatdescribed by Knight et al (18), as previously reported (16). Asingle-cell suspension prepared from spleen was culturedovernight in tissue culture flasks (2 � 107 cells/ml), in Dutchmodified RPMI 1640 medium (Life Technologies) supple-mented with 10% FCS, L-glutamine (0.02 mM), penicillin G,streptomycin, sodium pyruvate, and 2-mercaptoethanol, unlessotherwise stated. Recovered nonadherent cells were layeredover 14.5% (weight/volume) metrizamide in culture mediumand centrifuged at 650g for 10 minutes at room temperature.Low-density cells were collected at the interface and consistedmostly of large, dendritic-shaped cells, with few T cells,macrophages, and NK cells (�1% each by flow cytometry) buta significant proportion of B cells (20–40%), which weredepleted as follows: cells were sequentially incubated at 4°Cwith OX12 mAb, followed by GAM IgG–conjugated mi-crobeads (Miltenyi Biotec, Paris, France), and run on aVarioMACS separation unit (Miltenyi Biotec) according to themanufacturer’s recommendations for negative selection. Thefinal yield of DCs represented 0.5–1% of the starting spleno-cytes.
The second method was derived from that described byJosien et al (19,20). Spleen was minced and digested in 2 mg/mlof collagenase D (Roche Diagnostics, Meylan, France) inRPMI supplemented with 1% FCS for 30 minutes at 37°C.EDTA at 10 mM was added during the last 5 minutes, and thecell suspension was then pipetted up and down several timesand filtered. Cells were washed once in phosphate bufferedsaline (PBS)/EDTA, 2 mM/1% FCS, and low-density cellswere obtained after centrifugation on a 14.5% Nycodenzgradient (Nycomed, Oslo, Norway). Cells were then incubatedwith a saturating concentration of OX62 mAb at 4°C for 20minutes, washed twice, and then mixed with GAM IgG–conjugated microbeads. Positive selection was performed onpositive selection columns type MS (Miltenyi Biotec), andyielded a population of OX62� DCs with undetectable T cells,macrophages, or NK cells, and �20% contaminating B cells.This population of DCs was used after overnight incubation incomplete medium containing 4% of the culture supernatantfrom murine hybridoma transfected with rat granulocyte–macrophage colony-stimulating factor.
Resting B and T cells were isolated from lymph nodesingle-cell suspensions by magnetic-activated cell sorting. Neg-ative selection was performed using the combinations ofOX33, OX42, and 3.2.3 to obtain T cells, plus either OX8 orOX35 to purify CD4� or CD8� T cells, respectively. Positiveselection was performed using OX12 to obtain B cells. Incu-
bation with the primary mAb was followed by incubation withGAM IgG–conjugated microbeads. The purity of the selectedpopulations was routinely in the range of 95–100%, byfluorescence-activated cell sorting (FACS) analysis.
Mixed leukocyte culture (MLC). Purified total, CD4�,or CD8� T cells (105 cells/well) were cultured with graded(103–3 � 104 cells/well) or fixed (104 cells/well, unless other-wise stated) doses of irradiated (2,000 rad) DCs or B cells, inround-bottomed 96-well culture dishes, in a final volume of200 �l, at 37°C, 5% CO2. Proliferation of T cells was assessedby measuring the incorporation of 3H-thymidine (3H-TdR)(Amersham, Les Ulis, France), added after 2–4 days of culture(0.5 �Ci/well) 16 hours before harvesting, in a 1450 MicroBetascintillation counter (Wallac, Turku, Finland). Data are ex-pressed as the mean gross counts per minute of triplicates.
The preactivated allogeneic T cells used in someexperiments were prepared as follows: DA T cells (105 cells/well) were submitted to a first round of allogeneic stimulation,by culture in the presence of irradiated F344 or 33-3 DCs (3 �104 cells/well), as described above. After 5 days, T cells wereharvested and centrifuged over Lymphoprep (Nycomed) toremove dead cells. These preactivated T cells (2 � 104
cells/well) were further cultured in the presence of APCs for 2days, before measuring 3H-TdR incorporation.
In experiments using a Transwell apparatus (CorningCostar, Cambridge, MA), DA T cells (5 � 105 cells/well) werestimulated with irradiated F344 or 33-3 DCs (5 � 104 cells/well) in the lower compartments of 24-well culture dishes, in a1-ml volume, in the presence of a Transwell insert (6.5-mmdiameter, 0.4-�m pore size) containing either an MLC of DAT cells (2 � 105 cells/insert) and irradiated F344 or 33-3 DCs(5 � 104 cells/well), identical numbers of DA T cells, DCsalone, or no cells, in 350 �l of medium. After 3 days of MLC,cells from the lower compartments were split into triplicatesand transferred to 96-well culture dishes with 200 �l ofmedium, before adding 3H-TdR and measuring its incorpora-tion.
In vivo priming. Splenic DCs were obtained by themethod adapted from that described by Knight et al (16,18), asdescribed above, with a minor modification as follows: FCSwas replaced by 1% homologous rat serum in the overnightculture medium. Naive rats were primed by injecting each footpad with 5 � 105 DCs resuspended in 100 �l of PBS (2 � 106
DCs/recipient). Afferent popliteal and epitrochlear lymphnode cells were harvested 6 days later, resuspended, and testedfor their capacity to proliferate in vitro (3 � 105 cells/well) toallogeneic (F344), syngeneic (DA), and third-party (LEW)irradiated splenic DCs (103 cells/well), as described above(MLC). Incorporation of 3H-TdR was performed after 1–2days.
Cytofluorometry and cell sorting. For FACS of surfaceantigen, cells (2–5 � 105) were incubated with saturatingconcentrations of the appropriate primary mAb (mouse IgG1,IgG2a, IgG2b, or FITC-conjugated hamster IgM) for 30minutes, washed, then incubated with secondary PE-conjugated GAM IgG1, FITC-conjugated GAM IgG, or FITC-conjugated IgG2a as needed, for 30 minutes. The cells wereanalyzed using an Epics XL cytometer (Beckman Coulter,Villepinte, France), and Expo32 Analysis software (BeckmanCoulter). For analysis of conjugate formation between T cellsand DCs, both types of cells were labeled with membranousfluorescent dye (PKH67 green and PKH26 red, respectively),
1626 HACQUARD-BOUDER ET AL
according to the manufacturer’s recommendations. Then, Tcells and DCs were resuspended in culture medium, mixedtogether at a ratio varying from 1:1 to 5:1, and forced toestablish contact by centrifugation (50g for 2 minutes). After aperiod of incubation at 37°C, varying from 5 minutes to 18hours, the proportion of conjugates formed between cells wasanalyzed with an Epics cytometer, by setting the number ofevents acquired in the DC gate (i.e. upper quadrants) to10,000. The proportion of conjugates was expressed as apercentage of the number of events in the DC gate. T cells andDCs incubated alone and mixed together immediately beforeFACS served as the control.
Experiments using mAb to block specific moleculesexpressed at the surface of DCs were performed as describedabove, with the following changes: PKH26-labeled DCs weresequentially preincubated with anti-CD32 mAb D34-485 (10�g/ml) for 15 minutes at room temperature to block Fcreceptors, then with tested mAb at a saturating concentrationor isotype-matched control for 15 minutes at room tempera-ture, before being allowed to form conjugates with T cells inthe continuous presence of a saturating concentration of mAb.The blocking effect was expressed as a percentage of thereduction of conjugates formed in the presence of tested mAb,as compared with those formed with isotype-matched control.
For the conjugate-sorting experiment, single positive Tcells and DC–T cell conjugates were sorted after 2 hours ofcontact, using an Epics Elite cytometer (Beckman Coulter),and cultured for 1–3 days. Apoptosis of T cells was measuredby FACS analysis, using annexin V–PE (BD PharMingen),according to the manufacturer’s recommendations.
Statistical analysis. Wilcoxon’s matched pairs test wasused to compare the levels of reduction of conjugate formationbetween F344 or 33-3 DCs and T cells achieved with anti-CD86 mAb. P values less than 0.05 were considered significant.
RESULTS
Specific defective T cell stimulation by APCsexpressing a disease-prone HLA–B27–transgenic locus.Splenic DCs from the disease-prone 33-3 rats bearing ahigh copy number of HLA–B27–transgenic locus exhib-ited a dramatically decreased capacity to stimulate allo-geneic T cells from DA rats, as compared with DCs fromnontransgenic rats. In contrast, the stimulatory capacityof DCs from rats homozygous for either a low copynumber of HLA–B27–transgenic locus (21-4L; data not
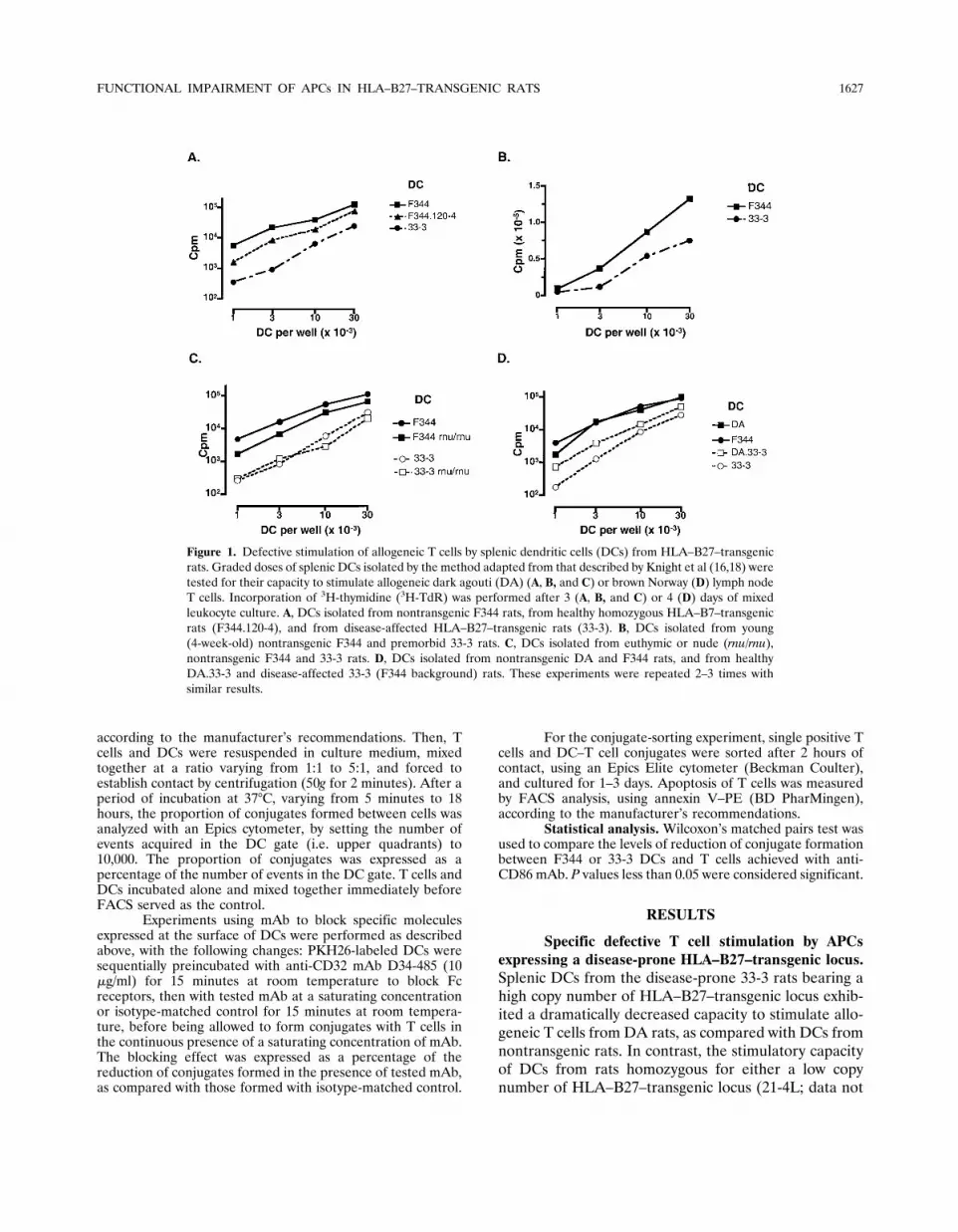
Figure 1. Defective stimulation of allogeneic T cells by splenic dendritic cells (DCs) from HLA–B27–transgenicrats. Graded doses of splenic DCs isolated by the method adapted from that described by Knight et al (16,18) weretested for their capacity to stimulate allogeneic dark agouti (DA) (A, B, and C) or brown Norway (D) lymph nodeT cells. Incorporation of 3H-thymidine (3H-TdR) was performed after 3 (A, B, and C) or 4 (D) days of mixedleukocyte culture. A, DCs isolated from nontransgenic F344 rats, from healthy homozygous HLA–B7–transgenicrats (F344.120-4), and from disease-affected HLA–B27–transgenic rats (33-3). B, DCs isolated from young(4-week-old) nontransgenic F344 and premorbid 33-3 rats. C, DCs isolated from euthymic or nude (rnu/rnu),nontransgenic F344 and 33-3 rats. D, DCs isolated from nontransgenic DA and F344 rats, and from healthyDA.33-3 and disease-affected 33-3 (F344 background) rats. These experiments were repeated 2–3 times withsimilar results.
FUNCTIONAL IMPAIRMENT OF APCs IN HLA–B27–TRANSGENIC RATS 1627
shown) or a high copy number of HLA–B7–transgeniclocus (120-4) was weakly decreased, as compared withthat observed with DCs from nontransgenic rats (Figure1A).
Allogeneic T cell stimulation by DCs from the33-3 line could theoretically be impaired as a conse-quence of the chronic inflammatory process in theserats. However, this possibility was ruled out becauseDCs from young premorbid 33-3 rats exhibited a weakercapacity to stimulate allogeneic T cells than did thenontransgenic DCs (Figure 1B). Similarly, DCs fromnude 33-3 rat (Figure 1C) and DCs from DA.33-3 rats(Figure 1D), both of which are protected from diseasedevelopment, displayed an impaired allogeneic T cellstimulatory capacity.
T cells from various rat backgrounds (DA, BN,
and F344) were used, all of which demonstrated im-paired stimulation by splenic DCs expressing the trans-genic locus from the 33-3 line on an F344 or DAbackground (Figure 1 and 2, and data not shown). Thedefect concerned both purified CD4� and CD8� T cellsand was consistently observed in the stimulation ofallogeneic and syngeneic T cells (Figure 2A).
The foregoing results were obtained with a popula-tion of splenic DCs isolated by the method adapted fromthat described by Knight et al, as previously reported(16,18). This population displays a cytologic aspect, antigensurface expression, and functional properties characteristicof DCs (16). Nevertheless, only a minority of those cells(�10%) express the �E2 integrin (CD103) identified bythe mAb OX62, which is characteristic of a population ofDCs isolated from the spleen by a different method
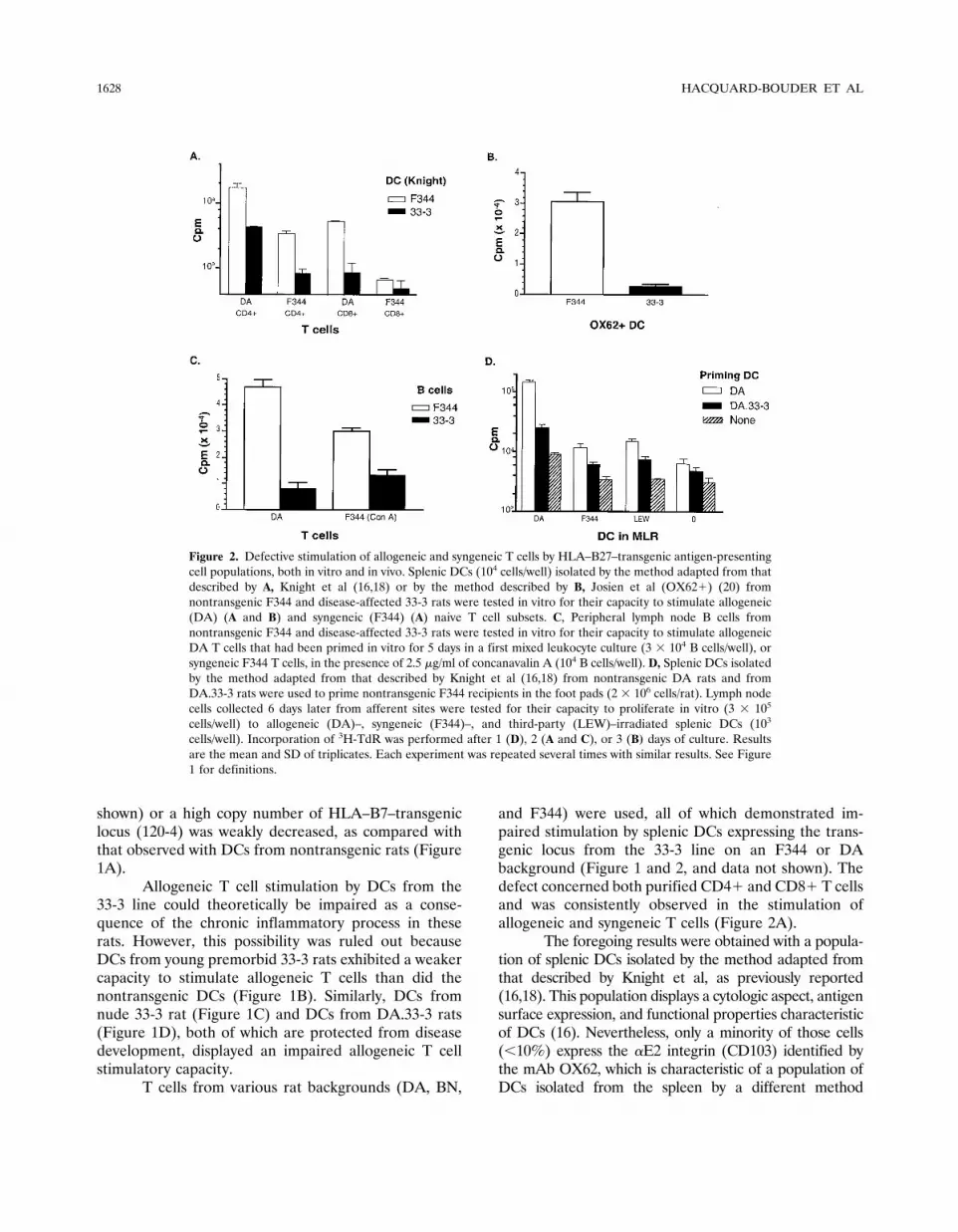
Figure 2. Defective stimulation of allogeneic and syngeneic T cells by HLA–B27–transgenic antigen-presentingcell populations, both in vitro and in vivo. Splenic DCs (104 cells/well) isolated by the method adapted from thatdescribed by A, Knight et al (16,18) or by the method described by B, Josien et al (OX62�) (20) fromnontransgenic F344 and disease-affected 33-3 rats were tested in vitro for their capacity to stimulate allogeneic(DA) (A and B) and syngeneic (F344) (A) naive T cell subsets. C, Peripheral lymph node B cells fromnontransgenic F344 and disease-affected 33-3 rats were tested in vitro for their capacity to stimulate allogeneicDA T cells that had been primed in vitro for 5 days in a first mixed leukocyte culture (3 � 104 B cells/well), orsyngeneic F344 T cells, in the presence of 2.5 �g/ml of concanavalin A (104 B cells/well). D, Splenic DCs isolatedby the method adapted from that described by Knight et al (16,18) from nontransgenic DA rats and fromDA.33-3 rats were used to prime nontransgenic F344 recipients in the foot pads (2 � 106 cells/rat). Lymph nodecells collected 6 days later from afferent sites were tested for their capacity to proliferate in vitro (3 � 105
cells/well) to allogeneic (DA)–, syngeneic (F344)–, and third-party (LEW)–irradiated splenic DCs (103
cells/well). Incorporation of 3H-TdR was performed after 1 (D), 2 (A and C), or 3 (B) days of culture. Resultsare the mean and SD of triplicates. Each experiment was repeated several times with similar results. See Figure1 for definitions.
1628 HACQUARD-BOUDER ET AL

Figure 3. Exposure of HLA–B27 DCs to lipopolysaccharide (LPS) and/or interferon-� (IFN�) does not restore their capacityto stimulate allogeneic T cells. Graded doses of splenic DCs (isolated by the method adapted from that described by Knightet al [16,18]) from nontransgenic F344 and disease-affected 33-3 rats and cultured for 24 hours in the presence of LPS (5�g/ml), rat recombinant IFN� (100 IU/ml), or both, were tested for their capacity to stimulate allogeneic DA T cells.Incorporation of 3H-TdR was performed after 3 days of mixed leukocyte culture. This experiment was repeated twice withsimilar results. See Figure 1 for other definitions.
Figure 4. Antigen expression on splenic DCs from F344, HLA–B27–transgenic, and HLA–B7–transgenic rats. Antigenexpression was analyzed by fluorescence-activated cell sorting at the surface of splenic DCs (isolated by the method adaptedfrom that described by Knight et al [16,18]) from age-matched nontransgenic F344, homozygous HLA–B7–transgenicF344.120-4, and disease-affected HLA–B27–transgenic 33-3 rats. The following monoclonal antibodies (mAb) were used:PA2.6, B1.23.2, and ME1, which recognize the HLA–B molecule complexed with human �2-microglobulin (�2m); HC-10, whichrecognizes HLA–B free heavy chain; BBM.1 (anti-human �2m); OX18 (anti-rat class I major histocompatibility complex[MHC]; OX6, anti-RT1-B, and OX17 anti-RT1D rat class II loci; WT.1 (anti-rat CD11a/lymphocyte function–associatedantigen 1� [LFA-1�] chain); WT.3 (anti-rat CD18/LFA-1� chain); 1A29 (anti-rat CD54/intercellular adhesion molecule 1[ICAM-1]); HM40-3 (anti-rat CD40); 3H5 (anti-rat B7.1/CD80); and 24F (anti-rat B7.2/CD86). Staining of DCs withisotype-matched control mAb is shown for each histogram. The results are representative of at least 2 different experimentswith each mAb tested. See Figure 1 for other definitions.
FUNCTIONAL IMPAIRMENT OF APCs IN HLA–B27–TRANSGENIC RATS 1629

(19,20). A defect similar to the one described with DCsisolated by the method described by Knight et al was alsoobserved with OX62� splenic DCs (Figure 2B).
We also used B cells, either to restimulate invitro–primed allogeneic T cells or to stimulate syngeneicT cells in the presence of Con A. In both conditions, weobserved a defective stimulatory function of peripherallymph node B cells from sick (Figure 2C), premorbid, ornude 33-3 rats (data not shown). Finally, in vivo induc-tion of a primary allogeneic response by splenic DCsexpressing the HLA–B27–transgenic locus from the 33-3line was also impaired (Figure 2D).
Phenotype analysis of APCs from HLA–B27– andHLA–B7–transgenic rats. It has been reported thatimmature DCs are poor stimulators of T cells. Hence, a
defective stimulatory capacity of 33-3 DCs might beconsecutive to an underactivated state of those DCs. Toaddress this possibility, we studied the stimulatory func-tion of DCs on allogeneic DA T cells after culture in thepresence of stimulatory agents. Overnight incubation of33-3 DCs with LPS, IFN�, or both failed to enhance thecapacity of those cells to stimulate allogeneic DA T cells(Figure 3).
Cell surface expression of several antigens wasstudied by FACS analysis. The HLA–B27– and HLA–B7–transgenic molecules, as recognized by mAb PA2.6,B1.23.2, ME1, HC-10 (Figure 4), and W6/32 (data notshown), were expressed at similar levels on DCs from33-3 and homozygous F344.120-4 rats. The level ofexpression of human �2m, as recognized by mAb
Figure 5. Defective stimulation of allogeneic T cells by HLA–B27 splenic dendritic cells (DCs) does not involvean inhibitory mechanism, a diffusible factor, or induction of T cell anergy. Splenic DCs were isolated fromnontransgenic F344 and disease-affected 33-3 rats by the method adapted from that described by Knight et al(16,18). A, Mixtures of F344 and 33-3 DCs at graded doses were tested for their capacity to stimulate allogeneicdark agouti (DA) T cells. Incorporation of 3H-thymidine (3H-TdR) was performed after 4 days of mixedleukocyte culture (MLC). B, MLCs of DA T cells (5 � 105 cells/well) and 33-3 or F344 DCs (5 � 104 cells/well)were set up in 24-well culture dishes (lower wells), in the presence of a Transwell insert containing an MLC ofDA T cells (2 � 105 cells/insert) and F344 DCs (5 � 104 cells/insert), the same numbers of DA T cells, F344 DCsalone, or no cells. After 3 days of MLC, triplicates of the cells contained in the lower wells were transferred to96-well culture dishes and tested for incorporation of 3H-TdR. C, 33-3 and F344 DCs were tested for theircapacity to stimulate DA T cells, with or without addition of human recombinant interleukin 2 (IL-2; 50 ng/ml).Incorporation of 3H-TdR was performed after 4 days of MLC. D, Graded doses of 33-3 and F344 DCs were testedfor their capacity to restimulate in a secondary MLC, allogeneic DA T cells that had been primed in vitro byculture for 5 days with F344 or 33-3 DC (first MLC, 105 T cells � 3 � 104 DCs/well). Incorporation of 3H-TdRwas performed after 2 days of the second MLC. Experiments shown were performed at least twice with similarresults. Cpm � counts per minute.
1630 HACQUARD-BOUDER ET AL

BBM.1, was also equivalent between both transgeniclines (Figure 4). The proportion of positive cells and thelevel of expression were comparable between splenicDCs from 33-3 and nontransgenic or homozygousF344.120-4 rats for most rat antigen studied, such asRT1B (class II MHC locus), LFA-1 (CD11a/CD18),ICAM-1 (CD54), CD40, B7.1 (CD80), B7.2 (CD86)(Figure 4), CD45R, CD32, NKRP1A, CD4, CD11b/c,CD103, CD169, and CD172a (data not shown). Similarresults were observed with DCs isolated from euthymic(Figure 4) and nude rats (data not shown). In contrast,the expression of endogenous rat class I MHC andRT1D (class II MHC locus) was decreased at the surfaceof DCs from 33-3 rats, as compared with DCs fromnontransgenic and to a lesser extent from homozygousF344.120-4 transgenic rats (Figure 4).
Evidence for impaired accessory molecule func-tion from HLA–B27 DCs. Allogeneic MLC performedusing a mixture of nontransgenic F344 and 33-3 DCs atvarious ratios resulted in a purely additive proliferativeeffect upon DA T cells (Figure 5A). Furthermore, usingTranswell chambers, we observed no evidence that adiffusible factor produced during allogeneic MLC per-formed with 33-3 DCs (data not shown) or F344 DCs(Figure 5B) in the upper compartments could, respec-tively, inhibit MLC performed with nontransgenic F344DCs (data not shown) or enhance MLC performed with33-3 DCs in the lower compartments (Figure 5B). Theseresults ruled out the possibility that 33-3 DCs could exertan inhibitory effect on the stimulation of allogeneic Tcells by nontransgenic DCs, and presumably also indi-cated that nontransgenic DCs cannot compensate forthe 33-3 DC defect. The impaired stimulatory functionof 33-3 DCs on allogeneic DA T cells could not berestored by the addition of exogenous Con A superna-tant (10%; data not shown), rHuIL-2 (50 ng/ml) (Figure5C), anti-rat IL-10 mAb (5 �g/ml) (data not shown), orNG-methyl-L-arginine to block nitric oxide productionduring the culture (5 �M) (data not shown). We foundno evidence for either increased apoptosis of T cellsinduced by DCs from 33-3 rats, as detected by annexin Vstaining (data not shown), or for the induction of ananergic state in T cells (Figure 5D).
Altogether, it could be inferred from the forego-ing results that the defective stimulation of T cells by33-3 APCs likely resulted from impaired activation atthe level of direct cell contact, which in turn couldinvolve decreased signaling through the TCR or de-creased accessory molecule function. To examine thelatter possibility, we performed syngeneic T cell stimu-lation by DCs in the presence of soluble anti-TCR�/�
mAb R73, which allows direct crosslinking of the TCR.In this assay, a defective APC function of 33-3 DCs wasconsistently observed (Figure 6). Because a defectivestimulatory function of 33-3 DCs existed independentlyof TCR engagement, this indicated that the function ofsome accessory molecule from 33-3 DCs was impaired.
Decreased formation of conjugates betweenHLA–B27 DCs and T cells. Defective accessory cellfunction could result from impaired contact between33-3 APCs and T cells. To test this hypothesis, allogeneicor nontransgenic syngeneic T cells and DCs weremarked with vital membranous fluorescent dyes andallowed to form contact. The formation of conjugatesbetween T cells and DCs during the first hours of theMLC was monitored by FACS. The proportion ofconjugates formed between 33-3 DCs and allogeneic orsyngeneic resting T cells was decreased as comparedwith the proportion of those formed between F344 DCsand T cells (Figure 7). A decrease of a magnitudevarying from 1.2-fold to 5.3-fold (median 2.3-fold) wasconsistently observed in 19 independent consecutiveexperiments, by using DCs isolated by the methoddescribed by Knight et al (18) (Figure 7) or by usingOX62�-purified DCs (data not shown). The decreasewas detected as early as 5 minutes after the onset ofcontact (Figure 7B); it was also observed with peripherallymph node B cells that had been preactivated with LPS.Interestingly, the rate of conjugates formed between
Figure 6. Defective stimulation of syngeneic T cells by soluble anti–Tcell receptor (anti-TCR), in the presence of HLA–B27 DCs. SplenicDCs were isolated by the method adapted from that described byKnight et al (16,18) from nontransgenic F344 and disease-affected 33-3rats, and were used to stimulate syngeneic (F344) lymph node CD4�T cells with or without soluble anti-TCR�/� monoclonal antibody R73(1 �g/ml), or control murine IgG1 (1 �g/ml). Incorporation of 3H-TdRwas performed after 2 days of MLC. Results are the mean and SD oftriplicates. This experiment was repeated 5 times with similar results.See Figure 5 for other definitions.
FUNCTIONAL IMPAIRMENT OF APCs IN HLA–B27–TRANSGENIC RATS 1631

DCs and allogeneic or syngeneic T cells was very similar,regardless of which DC (F344 or 33-3) was used (Figure7A). The formation of conjugates between F344 or 33-3DCs and T cells was proportionally increased by Con A,leaving the difference unchanged (data not shown). Incontrast, the rates of conjugates formed between alloge-neic or syngeneic T cells and DCs from F344.120-4homozygous HLA–B7 or from nontransgenic F344 ratswere comparable (Figure 7A).
Sorting experiments demonstrated that the Tcells engaged in conjugates were those that proliferate(data not shown). Conjugate formation between DCsand CD4� T cells was performed in the presence ofblocking mAb to examine the role played by severalmolecules in the decreased formation of conjugatesbetween 33-3 DCs and T cells. In these experiments, thecontribution of rat MHC molecules to the formation ofconjugates was undetectable (mAb OX18 and OX17) orweak (OX6) and could not explain the difference be-tween F344 and 33-3 DCs (data not shown). In contrast,blocking the CD86 costimulatory molecule on F344 DCsresulted in a strong reduction of the proportion ofconjugates formed, which was significantly greater thanthat with 33-3 DCs (mean � SD percent reduction in 25independent experiments, 46 � 12 versus 31 � 12,respectively; P � 0.0001 by Wilcoxon’s matched pairstest). Hence, the difference between F344 and 33-3 DCsin the proportion of conjugates formed with CD4� Tcells was strikingly reduced in the presence of anti-CD86(Figure 7B).
DISCUSSION
Impaired stimulation of allogeneic DA T cells bysplenic DCs from the 2 disease-prone HLA–B27–transgenic rat lines, 33-3 on a F344 background and21-4H on a LEW background, was previously describedby comparison with DCs from nontransgenic rats (16).In this study we have confirmed that splenic DCs fromthe disease-prone 33-3 line have a striking defect in theircapacity to stimulate the allogeneic T cell proliferativeresponse in vitro. We have also established the relativespecificity of this finding for a disease-prone HLA–B27–transgenic locus, because the stimulatory capacity ofDCs from healthy homozygous F344.120-4 HLA–B7–transgenic rats was only weakly decreased but stillremained much closer to that of the nontransgenic thanto that of the 33-3 DCs. These B7 rats representadequate control for the HLA–B27 disease-prone trans-genic locus effect, because their copy number and levelof expression of HLA–B transgene in hematopoietic
Figure 7. Decreased formation of conjugates between HLA–B27DCs and naive T cells. Splenic DCs isolated by the method adaptedfrom that described by Knight et al (16,18) from nontransgenicF344, healthy HLA–B7–transgenic F344.120-4, and disease-affectedHLA–B27–transgenic 33-3 rats, were labeled with PKH26 red.Allogeneic DA or nontransgenic syngeneic F344 lymph node CD4�T cells were labeled with PKH67 green. DCs and T cells at a 1:1ratio were incubated at 37°C. The formation of conjugates betweenT cells and DCs was monitored by fluorescence-activated cellsorting, by acquiring 10,000 events in the DC gate (i.e., upperquadrants), and expressed as the proportion of double-positive cellsin the DC gate. A, Dot plots showing the proportion of conjugates(upper right quadrants) formed between allogeneic dark agouti(DA) or syngeneic F344 CD4� T cells and F344, F344.120-4, or33-3 DCs, after 120 minutes of incubation. B, Kinetics of formationof conjugates between syngeneic F344 CD4� T cells and F344 or33-3 DCs (mean and SD of 3 independent experiments differentfrom those described in A) during the first 2 hours of culture, in thepresence of isotype-matched irrelevant monoclonal antibody (mAb;control IgG1) or 24F anti-CD86 mAb (20 �g/ml). See Figure 5 forother definitions.
1632 HACQUARD-BOUDER ET AL

cells exceed the threshold required for disease expres-sion in HLA–B27–transgenic lines (5)
The defective function of DCs precedes diseaseonset in 33-3 rats. Moreover, the defect affects DCsfrom nude 33-3 and DA.33-3 rats, which are protectedfrom disease (4,5), indicating that it is not secondary tochronic inflammation but rather happens as a directconsequence of high levels of HLA–B27 expression.Therefore, the defective function could be critical fordisease pathogenesis.
DCs are heterogeneous cell populations thathave been split into subsets with distinct phenotypes andfunctional capacities, including stimulation of allogeneicT cell proliferation (21,22). The majority of rat splenicDCs isolated by the method initially described by Knightet al (18) lacked expression of specific antigen such asthe �E2 integrin (CD103) recognized by OX62 mAb,although their morphologic aspect and their potentstimulatory activity in primary allogeneic MLC werecharacteristic of DCs. Importantly, however, we demon-strated a similar defective stimulatory function by usingOX62� DCs and even peripheral lymph node B cells.The stimulatory function of B cells was tested usingpreactivated allogeneic T cells or Con A–stimulatedsyngeneic T cells, because B cells did not support aprimary T cell response in vitro. Taken together, ourresults indicate that the defective function was notlimited to a particular subset of DCs and was notattributable to another contaminating population oflymphoid cells but rather was a general feature of APCsin 33-3 rats.
Impaired stimulation by 33-3 APCs applied to avariety of T cell populations, i.e. allogeneic and synge-neic T cells from different strains of rats, CD4� andCD8� subsets, naive or preactivated T cells. Those Tcells have distinctive requirements for their stimulation.It is therefore unlikely that involvement of the mostspecific ones, such as a selective competition betweenHLA–B27 and particular rat class I or class II MHCmolecules, could account for a phenomenon that is sobroad. If antigen presentation to T cells was affected byHLA–B27 overexpression in APCs, it could rather resultfrom some general antigen-processing disruption. How-ever, 33-3 DCs were also defective in supporting naivesyngeneic T cell stimulation by Con A, which is largelyindependent of engagement with MHC molecules (datanot shown) or by soluble R73 mAb, which directlycrosslinks the TCR�/�. These results strongly argue foran antigen-independent mechanism of APC impairmentin 33-3 rats, although it cannot entirely be ruled out that
engagement of the TCR by MHC molecules couldadditionally be involved.
Results from several experiments (competition,Transwell, addition of exogenous cytokines or blockingagents, monitoring of T cell apoptosis) indicated thatimpaired accessory molecule function taking place at thelevel of direct contact between 33-3 APCs and T cellswas most likely responsible for the defective stimulationof T cells. Accordingly, the proportion of conjugatesformed between 33-3 DCs and allogeneic or syngeneic Tcells was similarly decreased, as early as 5 minutes aftercontact. Antigen-independent accessory molecules,which are recognized as essential for the adhesion ofDCs to T cells and/or costimulation of T cells, are mostlikely involved in the decreased formation of conjugatesand presumably also in the decreased proliferation of Tcells. Those that appear to be restricted to DCs are lesslikely to be important than are molecules shared byother APCs such as B cells.
We found no difference in the surface expressionof major class II MHC locus RT1B, LFA-1 (CD11a/CD18), ICAM-1 (CD54), CD2, B7.1 (CD80), B7.2(CD86), or CD40, between 33-3 and control DCs.However, this finding does not entirely rule out thepossibility that the function of some of these moleculescould be impaired in 33-3 DCs. Such could notably bethe case for LFA-1 integrin, the functional activation ofwhich involves conformational changes that are unde-tectable with the mAb used in the present study (23,24).A number of other molecules, such as DC-SIGN, B7-DC, OX40-L or TRANCE-R, the expression of which atthe surface of APCs participates in T cell activation,were not examined in the present study but couldpresumably be involved in the defective function of 33-3APCs (25,26). Interestingly, however, the proportion ofconjugates formed between DCs and CD4� T cells wasstrongly decreased by blocking CD86 on DCs, and thelevel of reduction achieved with F344 was 1.5-foldgreater than that with 33-3 DCs, indicating that thiscostimulatory molecule was less used during interactionbetween 33-3 DCs and T cells.
It has been shown that the CD4 molecule isinvolved in the regulation of human T cell adhesion toAPCs in an antigen-independent manner, and that li-gands binding to CD4 could down-regulate early adhe-sion of CD4� T cells to APCs (27–29). Interestingly, itwas previously reported that the HLA–B27 moleculewas recognized by human CD4� T lymphocytes, and itwas suggested that binding of the CD4 molecule to ahomodimeric form of B27 could be responsible for thisobservation (30). Such an interpretation is consistent
FUNCTIONAL IMPAIRMENT OF APCs IN HLA–B27–TRANSGENIC RATS 1633

with the detection of high levels of homodimeric formsof B27 on DCs from 33-3 rats (Bird L, et al: unpublishedobservations) and could potentially account for thedecreased formation of conjugates between CD4� Tcells and 33-3 DCs. If this interpretation is true, how-ever, a different mechanism should explain the de-creased stimulation of CD8� T cells by 33-3 APCs.Furthermore, results from preliminary experiments us-ing several mAb to block the putative interaction be-tween different forms of HLA–B27 molecules on DCsand CD4 on T cells failed to support this hypothesis.
DCs in the immature state have relatively poorefficacy in stimulating T cells (31). In biobreeding rats,immaturity of splenic DCs was found to explain theimpaired activation of syngeneic and allogeneic T cells invitro (14). In our study, however, 33-3 rat DCs wereunlikely to be immature, because they displayed highlevels of MHC and costimulatory molecules on theirsurfaces. A weak decrease in rat class I MHC and inRT1D class II MHC expression levels was observed on33-3 DCs, and to a lesser extent on F344.120-4 cells. It ismore likely that this decrease resulted directly fromHLA–B transgene expression than from DC immaturity,because the decrease was also present on resting B cells(data not shown). Furthermore, pretreatment of 33-3DCs with LPS and/or INF� failed to enhance the level ofT cell stimulation.
Previously, we speculated that in the HLA–B27–transgenic rat model of SpA, CD4� T cell–mediateddisease might arise from a failure of tolerance, related atleast in part to high-level B27 expression in APCs and tothe immune response to gut bacteria (2,4). The defectiveAPC function that is specific to disease-prone HLA–B27–transgenic lines is all the more consistent with thishypothesis, because we have now shown that the in vitrodefect translates in vivo, as a failure of 33-3 DCs, toprime rats for an allogeneic response or an ovalbumin-specific response (Monnet D, Breban M: unpublishedobservations). Accumulating evidence suggests that DCsare important in maintaining tolerance, especially to-ward gut antigens (8,12). Of note, it was recently shownthat costimulation is essential to the expansion of regu-latory T cells in vivo (32). Hence, it can be speculatedthat defective activation of regulatory T cells couldresult in a loss of tolerance toward self antigen and,presumably, also toward bacterial flora. Alternatively (oradditionally), impaired T cell stimulation could result inaltered control of gut bacteria, as was suggested by theresults of other experiments (33,34), thereby sustainingstimulation of other immune defense arms such asmacrophages. Accordingly, arthritis could develop as a
secondary consequence of sustained gut inflammation,which is a hallmark of disease in both HLA–B27–transgenic rats and human SpA (2,35,36).
ACKNOWLEDGMENT
We gratefully acknowledge the technical assistance ofEliette Lallemand.
REFERENCES
1. Breban M, Said-Nahal R, Hugot JP, Miceli-Richard C. Familialand genetic aspects of spondyloarthropathy. Rheum Dis ClinNorth Am 2003;29:575–94.
2. Breban M, Hacquard-Bouder C, Falgarone G. Animal models ofHLA-B27-associated diseases. Curr Top Med Chem 2004;4:31–40.
3. Breban M, Hammer RE, Richardson JA, Taurog JD. Transfer ofthe inflammatory disease of HLA-B27 transgenic rats by bonemarrow engraftment. J Exp Med 1993;178:1607–16.
4. Breban M, Fernandez-Sueiro JL, Richardson JA, Hadavand RR,Maika SD, Hammer RE, et al. T cells, but not thymic exposure toHLA-B27, are required for the inflammatory disease of HLA-B27transgenic rats. J Immunol 1996;156:794–803.
5. Taurog JD, Maika S, Satumtira N, Dorris ML, McLean IL,Yanagisawa H, et al. Inflammatory disease in HLA-B27 transgenicrats. Immunol Rev 1999;169:209–23.
6. May E, Dorris ML, Satumtira N, Iqbal I, Rehman MI, Lightfoot E,et al. CD8 � � T cells are not essential to the pathogenesis ofarthritis or colitis in HLA-B27 transgenic rats. J Immunol 2003;170:1099–105.
7. Banchereau J, Briere F, Caux C, Davoust J, Lebecque S, Liu YJ,et al. Immunobiology of dendritic cells. Annu Rev Immunol2000;18:767–811.
8. Steinman RM, Nussenzweig MC. Avoiding horror autotoxicus: theimportance of dendritic cells in peripheral tolerance. Proc NatlAcad Sci U S A 2002;99:351–8.
9. Menges M, Rossner S, Voigtlander C, Schindler H, Kukutsch NA,Bogdan C, et al. Repetitive injections of dendritic cells maturedwith tumor necrosis factor � induce antigen-specific protection ofmice from autoimmunity. J Exp Med 2002;195:15–21.
10. Chang CC, Ciubotariu R, Manavalan JS, Yuan J, Colovai AI,Piazza F, et al. Tolerization of dendritic cells by T(S) cells: thecrucial role of inhibitory receptors ILT3 and ILT4. Nat Immunol2002;3:237–43.
11. Legge KL, Gregg RK, Maldonado-Lopez R, Lequn L, Caprio JC,Moser M, et al. On the role of dendritic cells in peripheraltolerance and modulation of autoimmunity. J Exp Med 2002;196:217–27.
12. Huang FP, Platt N, Wykes M, Major JR, Powell TJ, Jenkins CD,et al. A discrete subpopulation of dendritic cells transports apo-ptotic intestinal epithelial cells to T cell areas of mesenteric lymphnodes. J Exp Med 2000;191:435–43.
13. Ito A, Woo HJ, Imai Y, Osawa T. Functional deficiencies of spleendendritic cells in autoimmune MRL/lpr mice. Immunol Lett1988;17:223–8.
14. Delemarre FG, Simons PJ, de Heer HJ, Drexhage HA. Signs ofimmaturity of splenic dendritic cells from the autoimmune pronebiobreeding rat: consequences for the in vitro expansion ofregulator and effector T cells. J Immunol 1999;162:1795–801.
15. Takahashi K, Honeyman MC, Harrison LC. Impaired yield,phenotype, and function of monocyte-derived dendritic cells inhumans at risk for insulin-dependent diabetes. J Immunol 1998;161:2629–35.
16. Stagg AJ, Breban M, Hammer RE, Knight SC, Taurog JD.
1634 HACQUARD-BOUDER ET AL

Defective dendritic cell function in a HLA-B27 transgenic ratmodel of spondyloarthropathy (SpA). Adv Exp Med Biol 1995;378:557–9.
17. Blanchard HS, Dernis-Labous E, Lamarque D, Nhieu JT, SzepesZ, Flejou JF, et al. Inducible nitric oxide synthase attenuateschronic colitis in HLA-B27/human �2 microglobulin transgenicrats. Eur Cytokine Netw 2001;12:111–8.
18. Knight SC, Mertin J, Stackpoole A, Clark J. Induction of immuneresponses in vivo with small numbers of veiled (dendritic) cells.Proc Natl Acad Sci U S A 1983;80:6032–5.
19. Josien R, Heslan M, Soulillou JP, Cuturi MC. Rat spleen dendriticcells express natural killer cell receptor protein 1 (NKR-P1) andhave cytotoxic activity to select targets via a Ca2�-dependantmechanism. J Exp Med 1997;186:467–72.
20. Trinite B, Voisine C, Yagita H, Josien R. A subset of cytolyticdendritic cells in rat. J Immunol 2000;165:4202–8.
21. Liu L, Zhang M, Jenkins C, MacPherson GG. Dendritic cellheterogeneity in vivo: two functionally different cell populations inrat intestinal lymph can be distinguished by CD4 expression.J Immunol 1998;161:1146–55.
22. Voisine C, Hubert FX, Trinite B, Heslan M, Josien R. Twophenotypically distinct subsets of spleen dendritic cells in ratsexhibit different cytokine production and T cell stimulatory activ-ity. J Immunol 2002;169:2284–91.
23. Van Kooyk Y, Figdor CG. Avidity regulation of integrins: thedriving force in leukocyte adhesion. Curr Opin Cell Biol 2000;12:542–7.
24. Tamatani T, Kotani M, Miyasaka M. Characterization of the ratleukocyte integrin, CD11/CD18, by the use of LFA-1 subunit-specific monoclonal antibodies. Eur J Immunol 1991;21:627–33.
25. Steinman RM. DC-SIGN: a guide to some mysteries of dendriticcells. Cell 2000;100:491–4.
26. Tseng SJ, Otsuji M, Gorski G, Huang X, Slansky JE, Pai SI, et al.B7-DC, a new dendritic cell molecule with potent costimulatoryproperties for T cells. J Exp Med 2001;193:839–46.
27. Mazerolles F, Amblard F, Lumbroso C, Lecomte O, Van deMoortele PF, Barbat C, et al. Regulation of T helper-B lympho-cyte adhesion through CD4-HLA class II interaction. Eur J Im-munol 1990;20:637–44.
28. Mazerolles F, Hauss P, Barbat C, Figdor CG, Fischer A. Regula-tion of LFA-1-mediated T cell adhesion by CD4. Eur J Immunol1991;21:887–94.
29. Hauss P, Selz F, Cavazzana-Calvo M, Fischer A. Characteristics ofantigen-independent and antigen-dependent interaction of den-dritic cells with CD4� T cells. Eur J Immunol 1995;25:2285–94.
30. Boyle LH, Goodall JC, Opat SS, Gaston JS. The recognition ofHLA-B27 by human CD4� T lymphocytes. J Immunol 2001;167:2619–24.
31. Banchereau J, Steinman RM. Dendritic cells and the control ofimmunity. Nature 1998;392:245–52.
32. Lin CH, Hunig T. Efficient expansion of regulatory T cells in vitroand in vivo with a CD28 superagonist. Eur J Immunol 2003;33:626–38.
33. Warner TF, Madsen J, Starling J, Wagner RD, Taurog JD, BalishE. Human HLA-B27 gene enhances susceptibility of rats to oralinfection by Listeria monocytogenes. Am J Pathol 1996;149:1737–43.
34. Falgarone G, Blanchard HS, Riot B, Simonet M, Breban M. Thecytotoxic T cell-mediated response against Yersinia pseudo-tuberculosis in HLA-B27 transgenic rat. Infect Immun 1999;67:3773–9.
35. De Keyser F, Elewaut D, De Vos M, De Vlam K, Cuvelier C,Mielants H, et al. Bowel inflammation and the spondyloarthropa-thies. Rheum Dis Clin North Am 1998;24:785–813.
36. Lamarque D, Nhieu JT, Breban M, Bernardeau C, Martin-GarciaN, Szepes Z, et al. Lymphocytic expression and expression ofinducible nitric oxide synthase in human duodenal and colonicmucosa is a characteristic feature of ankylosing spondylitis.J Rheumatol 2003;30:2428–36.
FUNCTIONAL IMPAIRMENT OF APCs IN HLA–B27–TRANSGENIC RATS 1635





























