Embed Size (px)
Citation preview

Desacyl-ghrelin and Synthetic GH-secretagogues Modulate the Productionof Inflammatory Cytokines in MouseMicroglia Cells Stimulated byb-Amyloid Fibrils
Ilaria Bulgarelli,1 Laura Tamiazzo,1 Elena Bresciani,1 Daniela Rapetti,1
Simona Caporali,1 Donatella Lattuada,2 Vittorio Locatelli,1,3
and Antonio Torsello1,3*1Department of Experimental Medicine, University of Milano-Bicocca, Milan, Italy2Department of Pharmacology, University of Milan, Milan, Italy3Interdepartmental Center for Bioinformatics and Proteomics,University of Milano-Bicocca, Milan, Italy
Data from Alzheimer’s disease (AD) patients and ADanimal models demonstrate the accumulation of inflam-matory microglia at sites of insoluble fibrillar b-amyloidprotein (fAb) deposition. It is known that fAb binds toCD36, a type B scavenger receptor also involved ininternalization of oxidized low-density lipoprotein (LDL),and initiate a signaling cascade that regulates microglialrecruitment, activation, and secretion of inflammatorymediators leading to neuronal dysfunction and death.The recent demonstration of a binding site for thegrowth hormone secretagogues (GHS) on CD36prompted us to ascertain whether ghrelin and syntheticGHS could modulate the synthesis of inflammatory cyto-kines in fAb-activated microglia cells. We demonstratethat N9 microglia cells express the CD36 and are a suit-able model to study the activation of inflammatory cyto-kines synthesis. In fact, in N9 cells exposed to fAb25–35
for 24 hr, the expression of interleukin (IL)-1b and IL-6mRNA significantly increased. Interestingly, 1027 Mdesacyl-ghrelin, hexarelin, and EP80317 in the nanomo-lar range effectively counteracted fAb25–35 stimulation ofIL-6 mRNA levels, whereas ghrelin was ineffective. Simi-larly, the effects of fAb25–35 on IL-1b mRNA levels wereattenuated by desacyl-ghrelin, hexarelin, and EP80317,but not ghrelin. Because we have observed that thespecific GHS receptor GHS-R1a is not expressed in N9cells, the actions of GHS should be mediated by differ-ent receptors. Reportedly, hexarelin and EP80317 arecapable of binding the CD36 in mouse macrophagesand reducing atherosclerotic plaque deposition in mice.We conclude that desacyl-ghrelin, hexarelin, andEP80317 might interfere with fAb activation of CD36 inmicroglia cells. VVC 2009 Wiley-Liss, Inc.
Key words: ghrelin; hexarelin; N9; CD36; Alzheimer’sdisease
Among neurodegenerative diseases, Alzheimer’sdisease (AD) is a leading cause of dementia in elderlyindividuals. AD is clinically characterized by loss ofmemory and progressive deficits in other cognitivedomains and intellectual functions. Many efforts havebeen made in understanding the causes and mechanismsof AD, and at least three different primum movens havebeen evoked for AD pathogenesis. Many investigatorshave postulated the cholinergic dysfunction as the pri-mary/direct memory decline causative event in AD, buttherapeutic approaches based on the administration ofacetylcholinesterase inhibitors have led only to transitorysymptomatic palliative treatments. Two other hypotheseshave been proposed that are based on the atypical patho-physiology in AD of b-amyloid neuritic plaques and/orthe tau proteins neurofibrillary tangles (Giordano et al.,2007). Interestingly, not all people who at autopsy havepathologic plaques and/or tangles in brain parenchymawere clinically manifesting AD symptoms (Giordanoet al., 2007). Much attention is now given to cholinergicsynapses loss, which appears much better related to cog-nition decline than the number of plaques and tangles.
Microglia is the cell population primarily involvedin this inflammatory reaction (El Khoury et al., 1998;Meda et al., 1995). Cholinergic synapses loss might
Contract grant sponsor: Fondo di Ateneo per la Ricerca of the University
of Milano-Bicocca (to V.L. and A.T.).
*Correspondence to: Antonio Torsello, DIMS, Univ. of Milano-Bicocca,
via Cadore 48, 20052 Monza (MI), Italy.
E-mail: [email protected]
Received 3 October 2008; Revised 20 February 2009; Accepted 22
February 2009
Published online 20 April 2009 in Wiley InterScience (www.
interscience.wiley.com). DOI: 10.1002/jnr.22088
Journal of Neuroscience Research 87:2718–2727 (2009)
' 2009 Wiley-Liss, Inc.

results from microglia release of oxygen radicals andother inflammatory neurotoxic mediators. It is welldocumented that AD is characterized by the extracellulardeposition of insoluble fibrillar b-amyloid protein (fAb)in the brain (Wisniewski et al., 1989). b-Amyloid (Ab)is a 39– to 43–amino acid peptide encoded within theamyloid precursor protein (Masters et al., 1985), andreportedly its fragment, 25–35 (Ab25–35), contains thefunctional domain of Ab required for both neurotrophicand neurotoxic effects (Ashenafi et al., 2005; Yankneret al., 1990). Cellular reactions to the parenchymal dep-osition of fAb include the production of reactive oxygenspecies and tumor necrosis factor (TNF)-a (El Khouryet al., 2003). These and other microglial proinflamma-tory cytokines might play a relevant role in the progres-sion of AD. Interestingly, the toxicity of fAb is almostabsent in cultures of brain cells from which microgliahad been removed, indicating that microglia is neededto mediate the cytotoxic effects of fAb on primary neu-rons (Giulian et al., 1996). The molecular events thatoccur between the deposition of fAb and the activationof microglia have been characterized only in part, but ithas been demonstrated that binding of fAb to receptorCD36 is needed to stimulate the production of proin-flammatory cytokines from microglia (El Khoury et al.,2003). CD36 is a type B scavenger receptor largelyinvolved in cell interactions with oxidatively modifiedLDL, anionic phospholipids, fAb, fatty acid, and otherfactors (Febbraio et al., 2001). The adhesion of mono-cytes/macrophages to surfaces containing oxidativelymodified LDL induces the activation of CD36, whichsignal the cell to secrete reactive oxygen species andinflammatory cytokines. More recently, it has beenshown that the CD36 receptor also contains a bindingsequence for the growth hormone secretagogues (GHS),a family of endogenous and synthetic compoundsendowed with endocrine and extraendocrine activities(Demers et al., 2004; Marleau et al., 2005, 2006).
Reportedly, the GHS stimulates growth hormone(GH) secretion through their binding to a specific G-protein-coupled receptor (designated GHS-R1a), whoseendogenous ligand was identified as ghrelin in 1999(Kojima et al., 1999). Ghrelin circulates in the acylatedand desacylated forms, but only the acylated protein iscapable of binding the GHS-R1a (Kojima et al., 1999).In the cardiovascular system, hexarelin, a classical GHS-R1a agonist, and EP80317, an hexarelin derivative withno GH-releasing activity, have been shown to inhibitatherosclerotic plaque deposition (Avallone et al., 2006;Marleau et al., 2005) through their interaction with theCD36, thus supporting a GH-independent role forGHS.
On the basis of the foregoing, we wonderedwhether ghrelin, desacyl-ghrelin, hexarelin, andEP80317 could interfere with fAb activation of theCD36. It might be anticipated that drugs capable of in-hibiting activation of brain microglia and release of neu-rotoxic mediators could be considered in the treatmentof AD. We decided to perform our experiments with
the N9 immortalized mouse microglia cells, which havepreviously been demonstrated to be a suitable model forstudies on microglia (Bureau et al., 2008; Corradinet al., 1993; Pirami et al., 1991; Santambrogio et al.,2001; Wang et al., 2008) and might be a valuable alter-native to primary mouse microglia culture for use inpharmacological and toxicological investigations. Theaims of the present study were first, to characterize invitro the capability of fAb to stimulate the production ofproinflammatory cytokines in N9 microglia cells; andsecond, to ascertain whether ghrelin, its desacylated ana-logue, and two synthetic GHS (hexarelin and EP80317)could modulate fAb activation of N9 cells.
MATERIALS AND METHODS
Chemicals
Human ghrelin, desacyl-ghrelin, hexarelin, EP80317 andhuman Ab25–35 (Gly-Ser-Asn-Lys-Gly-Ala-Ile-Ile-Gly-Leu-Met) have been synthesized by conventional solid-phase syn-thesis (Neosystem, Strasburg, France).
To allow formation of Ab fibrils, Ab25–35 was resus-pended in phosphate-buffered saline (PBS) at 1 mg/ml andincubated at 378C for 72 hr as previously described (Coraciet al., 2002). Unless otherwise specified, all other reagentswere purchased from Sigma-Aldrich (St. Louis, MO).
Cell Cultures
The murine microglial cell line N9 was the gift of Dr.Paola Ricciardi-Castagnoli (University of Milano–Bicocca,Milan, Italy). N9 cells were cultured in Iscove’s modifiedDulbecco’s medium (EuroClone, Pero, Italy) with 5% heat-inactivated fetal bovine serum (EuroClone), 100 IU/ml peni-cillin, 100 lg/ml streptomycin, 2 mM L-glutamine, and50 lM b-mercaptoethanol. Cells were plated at a density of0.6 3 106 cells per well in six-well plates and incubated at378C for 72 hr.
Cell Viability Assay
To study the toxicity of fAb cells were cultured for 24hr with increasing concentrations of fAb25–35, and cell viabil-ity was determined by the MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) protocol as previouslydescribed (Pozzi et al., 2004). Similarly, the effects on cell via-bility of 24-hr incubation with hexarelin and EP80317 at the1027 M concentration was also determined. The concentra-tion of 1027 M of the peptides was chosen because it wasshown to be active in previous reports (Avallone et al., 2006).In dose–response experiments, cells were incubated withincreasing concentrations of fAb25–35 (1–60 lM) for 24 hr.The time-course effects of fAb were characterized by incubat-ing the N9 cells with fAb (30 lM) for 12, 24, and 48 hr.
Semiquantitative Reverse Transcriptase–PolymeraseChain Reaction (RT-PCR) Assay
To study the effects of fAb alone or in association withthe GHS, in the following experiments, cells were incubatedfor 24 hr with 30 lM fAb, 30 lM fAb 1 1027 M ghrelin,30 lM fAb 1 1027 M desacyl-ghrelin, 30 lM fAb 1 1027
GHS Inhibit IL-1b and IL-6 in Microglia 2719
Journal of Neuroscience Research

M hexarelin, 30 lM fAb 1 1027 M EP80317, or culture me-dium alone. Each experiment was independently repeated atleast three times on three different days.
At the end of incubation, culture medium was removed,and total RNA was extracted from cells with TriZol-like rea-gent as previously described (Torsello et al., 2003) and quanti-fied with a NanoDrop spectrophotometer (Thermo Scientific,Wilmington, DE). One thousand nanograms of total RNAwas incubated with rDNase I (Ambion, Austin, TX) for 20minutes at 378C to digest contaminating genomic DNA.
Four hundred nanograms total RNA of each samplewas subjected to reverse transcription with Moloney MurineLeukemia Virus (MMLV) (Invitrogen, Carlsbad, CA) followedby amplification with specific primers based on the publishedsequence of murine GHS-R1a (forward: 50-CCATCAACCCCATTCTGTA-30; reverse: 50-GTTGATGCTCGACTTTGT-30), CD36 (forward: 50-AGACAATCAAAAGGGAAGTT-30;reverse: 50-GAAGGCTCAAAGATGGCTC-30), TNF-a (for-ward: 50-CCAAAGGGATGAGAAGTT-30; reverse: 50-TGACGGCAGAGAGGAGGT-30), peroxisome proliferator acti-vated receptor-g (PPAR-g) (forward: 50-ACTCGCATTCCTTTGACATC-30; reverse: 50-CGCACTTTGGTATTCTTGG-30), COX-1 (forward: 50-GCCCTCACC AGTCAATCCCT-30; reverse: 50-TACACCTCTCCGTCC AGCAC-30), and COX-2 (forward: 50-ATGGGTGTGAAGGGAAATA-30; reverse: 50-GGATGTGAGGAGGGTAGA-30).PCR analysis of total RNA yielded a DNA fragment of theexpected length for all specific mRNAs. To normalize resultsfor differences in RNA sampling, an aliquot of the RT reac-tion was used to amplify a glyceraldehyde-6-phosphate dehy-drogenase (GAPDH) 600-bp fragment (forward: 50-GCCAT-CAACGACCCCTTCATTG-30; reverse: 50-TCTGTCAT-GAGGTTGGCTTTCAG-30). Negative controls of the PCRreaction were made omitting the specific primers from thereaction mixture.
To assure that PCR was performed in the linear ampli-fication range, samples were initially analyzed after 15, 17, 20,25, 27, 30, 35, and 40 cycles (data not shown). For eachamplicon, we chose the cycle number that gave half of themaximal amplification.
Real-Time Quantitative PCR Assay (Q-PCR)
Because results from semiquantitative RT-PCR indi-cated that IL-1b and IL-6 mRNA levels were altered bytreatments, we decided to perform quantitative determinationsof these two mRNAs by real-time quantitative PCR assay.
Four hundred nanograms of total digested RNA of eachsample was subjected to reverse transcription with revertAidH Minus First Strand cDNA Synthesis Kit (Fermentas,Canada).
Q-PCR for IL-1b and IL-6 was performed with Assay-on-Demand technology (Applied Biosystems, Warrington,UK), with a Brilliant QPCR Master Mix (Stratagene, La Jolla,CA,USA). All Q-PCR reactions were performed on the ABI7900 HT system (Applied Biosystems). Samples were normal-ized to GaPDH (Assay-on-Demand, Applied Biosystems) and2DDCt analysis supplied. The RT-PCR reactions wererepeated in triplicate for each sample.
Fluorescence-activated Cell Sorter (FACS) Analysis
N9 cells were suspended at 5 3 105 cells/ml in PBScontaining 1% fetal calf serum (FCS) (PBS-FCS) and FcRswere blocked by incubating with FCS for 30 min at roomtemperature. After washing, each sample was resuspendedwith 100 l1 PBS-FCS (unstained control) or 100 l1 of PBScontaining a specific monoclonal antibody against mouseCD36 (Tebu-Bio, France) and incubated for 30 min at 48C.After 30 min of dark incubation at 48C, cells were washedthree times with PBS-FCS. After a final wash, the cells wereresuspended in 0.5 ml PBS-FCS. For the surface marker,20,000 gated cells were analyzed with a FACSCalibur flowcytometer (Becton Dickinson, San Jose, CA) equipped withan argon ion laser exciting fluorescein isothiocyanate at 488nm. The data are reported as percentage of label-positive cells.The analysis was performed by a Cell Quest software package(Becton Dickinson, San Jose, CA).
Intracellular Ca21 Mobilization Assay
N9 cells were plated at 20,000 cells per well into black-walled, clear-bottomed 96-well plates (Corning, Germany)and cultured for 2 days to up to 80–90% of confluence.Before assay, cells were incubated in dark conditions with 100 llof Hanks’ balanced salt solution containing HEPES 20 mM,probenecid 2.5 mM, and FLUO-4 NW 4.5 lM (MolecularProbes, Eugene, OR) at 378C and 5% CO2 for 45 minutes.Fluorescence emissions from 96 wells were measured with themultilabel spectrophotometer Victor3 (Perkin Elmer) at 485/535 nm (excitation/emission filters) for the 60 sec after injec-tion of the stimuli. Ghrelin (1027 M), hexarelin (1027 M),des-acyl-ghrelin (1027 M), EP80317 (1027 M), and adenosinetriphosphate (ATP) (1024 M) were diluted in Hanks’ balancedsalt solution and injected into the well by an automated injec-tor system. As positive control for our experimental settings,we tested the ability of ghrelin, des-acyl-ghrelin, and EP80317to stimulate intracellular calcium in a line of Chinese hamsterovary (CHO) cells stably transfected with a plasmid containingthe human GHS-R1a (CHO-GHS-R1a). All the experimentswere performed at 378C.
Statistical Analysis
Values are expressed as mean 6 standard error of themean (SEM). The statistical significance of differencesbetween groups was evaluated with Tukey-Kramer’s t-test formultiple comparisons, preceded by analysis of variance. A Pvalue of less than 0.05 was considered significant.
RESULTS
Characterization of the Expression of CD36 andGHS-R1a in N9 Microglia Cells
In the first experiment, we ascertained whethermurine N9 microglia cells express or do not express themRNA for GHS-R1a and CD36. RT-PCR analysisdemonstrated that GHS-R1a mRNA was undetectablein N9 cells, whereas the same cells expressed detectableamounts of CD36 mRNA (Fig. 1A).
To test whether CD36 mRNA was effectivelytranslated and the protein expressed on the cell surface,
2720 Bulgarelli et al.
Journal of Neuroscience Research
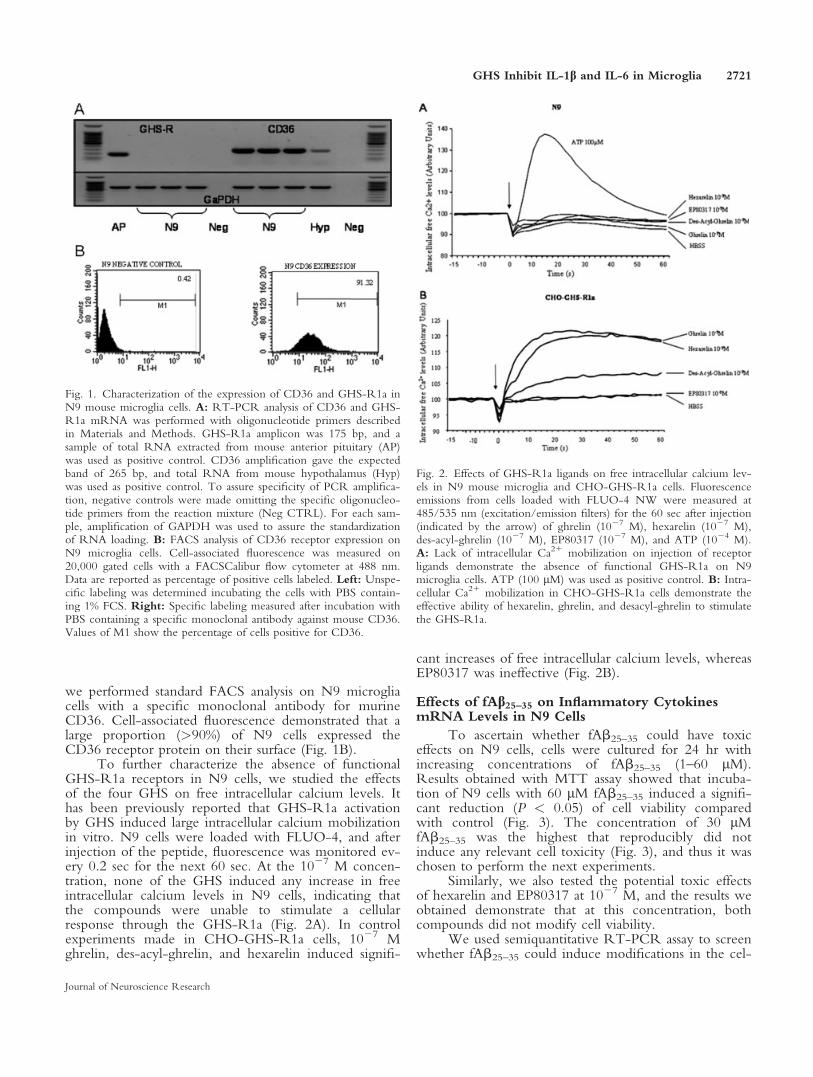
we performed standard FACS analysis on N9 microgliacells with a specific monoclonal antibody for murineCD36. Cell-associated fluorescence demonstrated that alarge proportion (>90%) of N9 cells expressed theCD36 receptor protein on their surface (Fig. 1B).
To further characterize the absence of functionalGHS-R1a receptors in N9 cells, we studied the effectsof the four GHS on free intracellular calcium levels. Ithas been previously reported that GHS-R1a activationby GHS induced large intracellular calcium mobilizationin vitro. N9 cells were loaded with FLUO-4, and afterinjection of the peptide, fluorescence was monitored ev-ery 0.2 sec for the next 60 sec. At the 1027 M concen-tration, none of the GHS induced any increase in freeintracellular calcium levels in N9 cells, indicating thatthe compounds were unable to stimulate a cellularresponse through the GHS-R1a (Fig. 2A). In controlexperiments made in CHO-GHS-R1a cells, 1027 Mghrelin, des-acyl-ghrelin, and hexarelin induced signifi-
cant increases of free intracellular calcium levels, whereasEP80317 was ineffective (Fig. 2B).
Effects of fAb25–35 on Inflammatory CytokinesmRNA Levels in N9 Cells
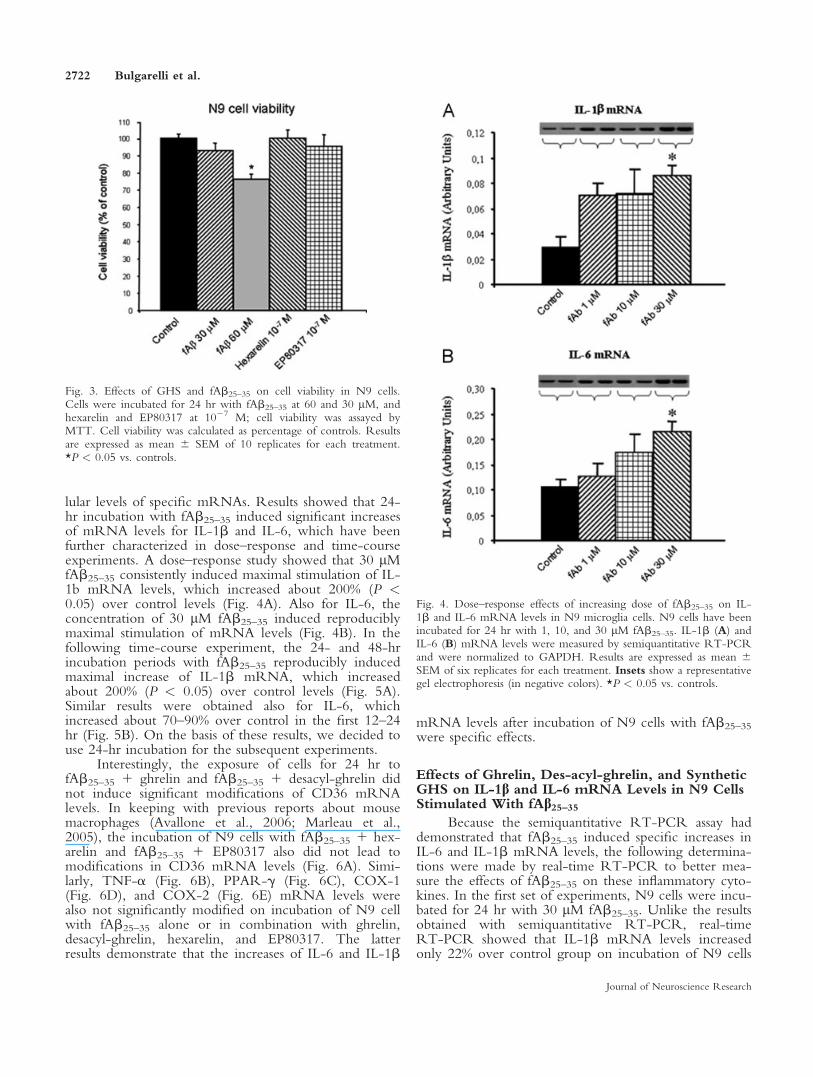
To ascertain whether fAb25–35 could have toxiceffects on N9 cells, cells were cultured for 24 hr withincreasing concentrations of fAb25–35 (1–60 lM).Results obtained with MTT assay showed that incuba-tion of N9 cells with 60 lM fAb25–35 induced a signifi-cant reduction (P < 0.05) of cell viability comparedwith control (Fig. 3). The concentration of 30 lMfAb25–35 was the highest that reproducibly did notinduce any relevant cell toxicity (Fig. 3), and thus it waschosen to perform the next experiments.
Similarly, we also tested the potential toxic effectsof hexarelin and EP80317 at 1027 M, and the results weobtained demonstrate that at this concentration, bothcompounds did not modify cell viability.
We used semiquantitative RT-PCR assay to screenwhether fAb25–35 could induce modifications in the cel-
Fig. 2. Effects of GHS-R1a ligands on free intracellular calcium lev-els in N9 mouse microglia and CHO-GHS-R1a cells. Fluorescenceemissions from cells loaded with FLUO-4 NW were measured at485/535 nm (excitation/emission filters) for the 60 sec after injection(indicated by the arrow) of ghrelin (1027 M), hexarelin (1027 M),des-acyl-ghrelin (1027 M), EP80317 (1027 M), and ATP (1024 M).A: Lack of intracellular Ca21 mobilization on injection of receptorligands demonstrate the absence of functional GHS-R1a on N9microglia cells. ATP (100 lM) was used as positive control. B: Intra-cellular Ca21 mobilization in CHO-GHS-R1a cells demonstrate theeffective ability of hexarelin, ghrelin, and desacyl-ghrelin to stimulatethe GHS-R1a.
Fig. 1. Characterization of the expression of CD36 and GHS-R1a inN9 mouse microglia cells. A: RT-PCR analysis of CD36 and GHS-R1a mRNA was performed with oligonucleotide primers describedin Materials and Methods. GHS-R1a amplicon was 175 bp, and asample of total RNA extracted from mouse anterior pituitary (AP)was used as positive control. CD36 amplification gave the expectedband of 265 bp, and total RNA from mouse hypothalamus (Hyp)was used as positive control. To assure specificity of PCR amplifica-tion, negative controls were made omitting the specific oligonucleo-tide primers from the reaction mixture (Neg CTRL). For each sam-ple, amplification of GAPDH was used to assure the standardizationof RNA loading. B: FACS analysis of CD36 receptor expression onN9 microglia cells. Cell-associated fluorescence was measured on20,000 gated cells with a FACSCalibur flow cytometer at 488 nm.Data are reported as percentage of positive cells labeled. Left: Unspe-cific labeling was determined incubating the cells with PBS contain-ing 1% FCS. Right: Specific labeling measured after incubation withPBS containing a specific monoclonal antibody against mouse CD36.Values of M1 show the percentage of cells positive for CD36.
GHS Inhibit IL-1b and IL-6 in Microglia 2721
Journal of Neuroscience Research
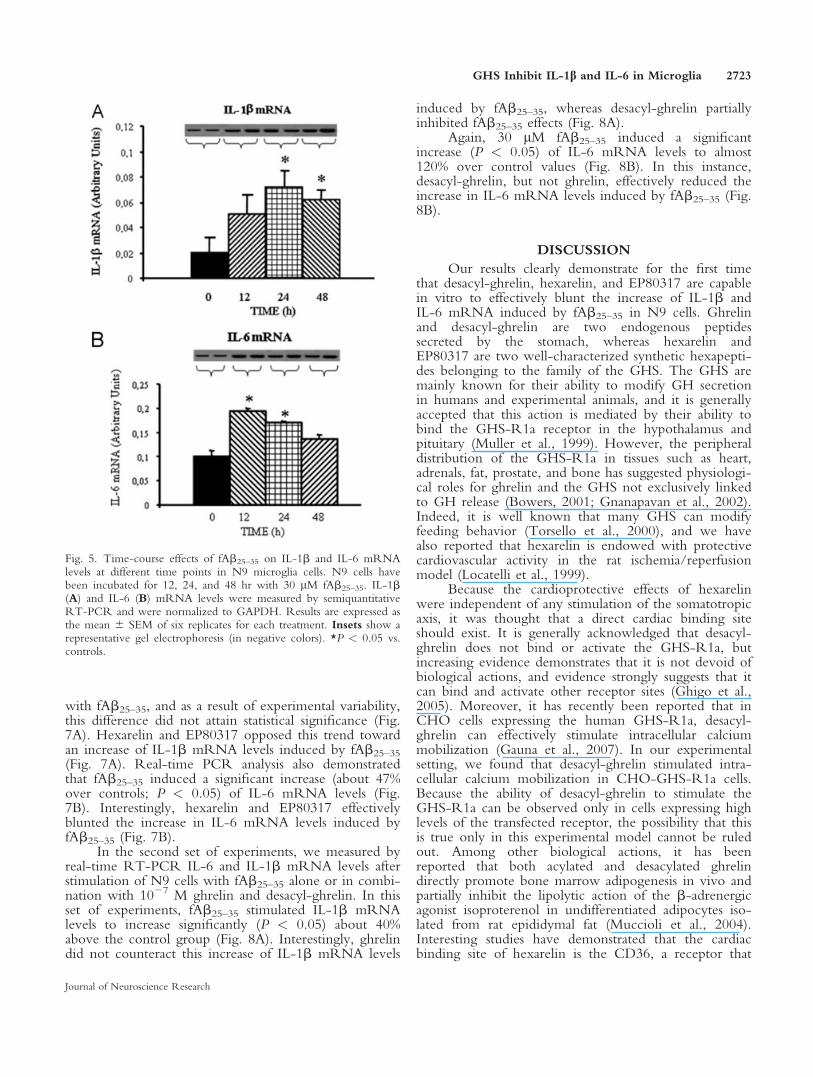
lular levels of specific mRNAs. Results showed that 24-hr incubation with fAb25–35 induced significant increasesof mRNA levels for IL-1b and IL-6, which have beenfurther characterized in dose–response and time-courseexperiments. A dose–response study showed that 30 lMfAb25–35 consistently induced maximal stimulation of IL-1b mRNA levels, which increased about 200% (P <0.05) over control levels (Fig. 4A). Also for IL-6, theconcentration of 30 lM fAb25–35 induced reproduciblymaximal stimulation of mRNA levels (Fig. 4B). In thefollowing time-course experiment, the 24- and 48-hrincubation periods with fAb25–35 reproducibly inducedmaximal increase of IL-1b mRNA, which increasedabout 200% (P < 0.05) over control levels (Fig. 5A).Similar results were obtained also for IL-6, whichincreased about 70–90% over control in the first 12–24hr (Fig. 5B). On the basis of these results, we decided touse 24-hr incubation for the subsequent experiments.
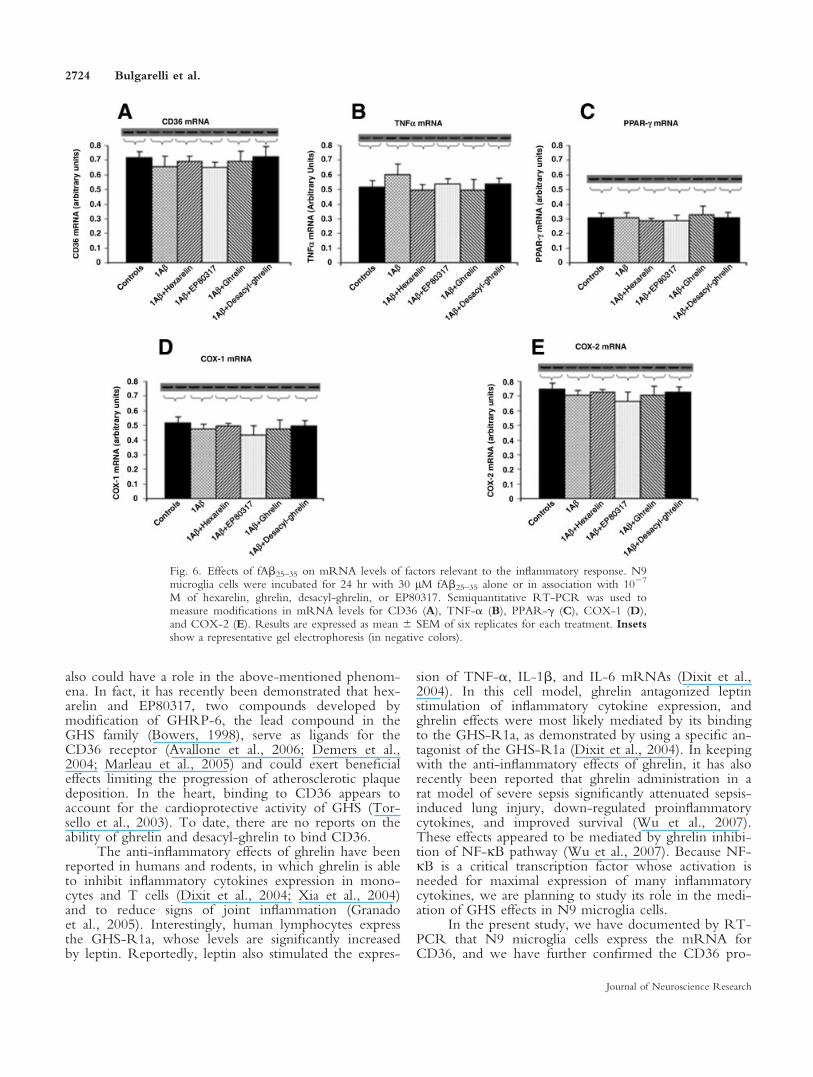
Interestingly, the exposure of cells for 24 hr tofAb25–35 1 ghrelin and fAb25–35 1 desacyl-ghrelin didnot induce significant modifications of CD36 mRNAlevels. In keeping with previous reports about mousemacrophages (Avallone et al., 2006; Marleau et al.,2005), the incubation of N9 cells with fAb25–35 1 hex-arelin and fAb25–35 1 EP80317 also did not lead tomodifications in CD36 mRNA levels (Fig. 6A). Simi-larly, TNF-a (Fig. 6B), PPAR-g (Fig. 6C), COX-1(Fig. 6D), and COX-2 (Fig. 6E) mRNA levels werealso not significantly modified on incubation of N9 cellwith fAb25–35 alone or in combination with ghrelin,desacyl-ghrelin, hexarelin, and EP80317. The latterresults demonstrate that the increases of IL-6 and IL-1b
mRNA levels after incubation of N9 cells with fAb25–35
were specific effects.
Effects of Ghrelin, Des-acyl-ghrelin, and SyntheticGHS on IL-1b and IL-6 mRNA Levels in N9 CellsStimulated With fAb25–35
Because the semiquantitative RT-PCR assay haddemonstrated that fAb25–35 induced specific increases inIL-6 and IL-1b mRNA levels, the following determina-tions were made by real-time RT-PCR to better mea-sure the effects of fAb25–35 on these inflammatory cyto-kines. In the first set of experiments, N9 cells were incu-bated for 24 hr with 30 lM fAb25–35. Unlike the resultsobtained with semiquantitative RT-PCR, real-timeRT-PCR showed that IL-1b mRNA levels increasedonly 22% over control group on incubation of N9 cells
Fig. 4. Dose–response effects of increasing dose of fAb25–35 on IL-1b and IL-6 mRNA levels in N9 microglia cells. N9 cells have beenincubated for 24 hr with 1, 10, and 30 lM fAb25–35. IL-1b (A) andIL-6 (B) mRNA levels were measured by semiquantitative RT-PCRand were normalized to GAPDH. Results are expressed as mean 6SEM of six replicates for each treatment. Insets show a representativegel electrophoresis (in negative colors). *P < 0.05 vs. controls.
Fig. 3. Effects of GHS and fAb25–35 on cell viability in N9 cells.Cells were incubated for 24 hr with fAb25–35 at 60 and 30 lM, andhexarelin and EP80317 at 1027 M; cell viability was assayed byMTT. Cell viability was calculated as percentage of controls. Resultsare expressed as mean 6 SEM of 10 replicates for each treatment.*P < 0.05 vs. controls.
2722 Bulgarelli et al.
Journal of Neuroscience Research
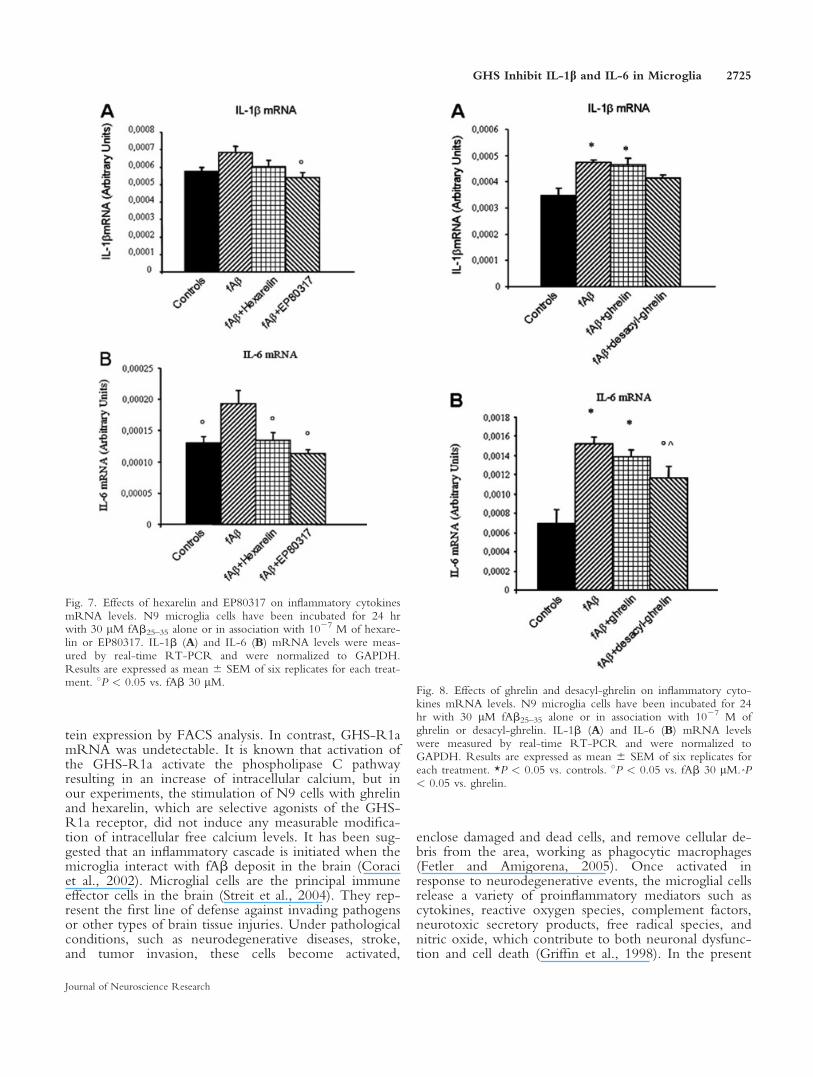
with fAb25–35, and as a result of experimental variability,this difference did not attain statistical significance (Fig.7A). Hexarelin and EP80317 opposed this trend towardan increase of IL-1b mRNA levels induced by fAb25–35
(Fig. 7A). Real-time PCR analysis also demonstratedthat fAb25–35 induced a significant increase (about 47%over controls; P < 0.05) of IL-6 mRNA levels (Fig.7B). Interestingly, hexarelin and EP80317 effectivelyblunted the increase in IL-6 mRNA levels induced byfAb25–35 (Fig. 7B).
In the second set of experiments, we measured byreal-time RT-PCR IL-6 and IL-1b mRNA levels afterstimulation of N9 cells with fAb25–35 alone or in combi-nation with 1027 M ghrelin and desacyl-ghrelin. In thisset of experiments, fAb25–35 stimulated IL-1b mRNAlevels to increase significantly (P < 0.05) about 40%above the control group (Fig. 8A). Interestingly, ghrelindid not counteract this increase of IL-1b mRNA levels
induced by fAb25–35, whereas desacyl-ghrelin partiallyinhibited fAb25–35 effects (Fig. 8A).
Again, 30 lM fAb25–35 induced a significantincrease (P < 0.05) of IL-6 mRNA levels to almost120% over control values (Fig. 8B). In this instance,desacyl-ghrelin, but not ghrelin, effectively reduced theincrease in IL-6 mRNA levels induced by fAb25–35 (Fig.8B).
DISCUSSION
Our results clearly demonstrate for the first timethat desacyl-ghrelin, hexarelin, and EP80317 are capablein vitro to effectively blunt the increase of IL-1b andIL-6 mRNA induced by fAb25–35 in N9 cells. Ghrelinand desacyl-ghrelin are two endogenous peptidessecreted by the stomach, whereas hexarelin andEP80317 are two well-characterized synthetic hexapepti-des belonging to the family of the GHS. The GHS aremainly known for their ability to modify GH secretionin humans and experimental animals, and it is generallyaccepted that this action is mediated by their ability tobind the GHS-R1a receptor in the hypothalamus andpituitary (Muller et al., 1999). However, the peripheraldistribution of the GHS-R1a in tissues such as heart,adrenals, fat, prostate, and bone has suggested physiologi-cal roles for ghrelin and the GHS not exclusively linkedto GH release (Bowers, 2001; Gnanapavan et al., 2002).Indeed, it is well known that many GHS can modifyfeeding behavior (Torsello et al., 2000), and we havealso reported that hexarelin is endowed with protectivecardiovascular activity in the rat ischemia/reperfusionmodel (Locatelli et al., 1999).
Because the cardioprotective effects of hexarelinwere independent of any stimulation of the somatotropicaxis, it was thought that a direct cardiac binding siteshould exist. It is generally acknowledged that desacyl-ghrelin does not bind or activate the GHS-R1a, butincreasing evidence demonstrates that it is not devoid ofbiological actions, and evidence strongly suggests that itcan bind and activate other receptor sites (Ghigo et al.,2005). Moreover, it has recently been reported that inCHO cells expressing the human GHS-R1a, desacyl-ghrelin can effectively stimulate intracellular calciummobilization (Gauna et al., 2007). In our experimentalsetting, we found that desacyl-ghrelin stimulated intra-cellular calcium mobilization in CHO-GHS-R1a cells.Because the ability of desacyl-ghrelin to stimulate theGHS-R1a can be observed only in cells expressing highlevels of the transfected receptor, the possibility that thisis true only in this experimental model cannot be ruledout. Among other biological actions, it has beenreported that both acylated and desacylated ghrelindirectly promote bone marrow adipogenesis in vivo andpartially inhibit the lipolytic action of the b-adrenergicagonist isoproterenol in undifferentiated adipocytes iso-lated from rat epididymal fat (Muccioli et al., 2004).Interesting studies have demonstrated that the cardiacbinding site of hexarelin is the CD36, a receptor that
Fig. 5. Time-course effects of fAb25–35 on IL-1b and IL-6 mRNAlevels at different time points in N9 microglia cells. N9 cells havebeen incubated for 12, 24, and 48 hr with 30 lM fAb25–35. IL-1b(A) and IL-6 (B) mRNA levels were measured by semiquantitativeRT-PCR and were normalized to GAPDH. Results are expressed asthe mean 6 SEM of six replicates for each treatment. Insets show arepresentative gel electrophoresis (in negative colors). *P < 0.05 vs.controls.
GHS Inhibit IL-1b and IL-6 in Microglia 2723
Journal of Neuroscience Research
also could have a role in the above-mentioned phenom-ena. In fact, it has recently been demonstrated that hex-arelin and EP80317, two compounds developed bymodification of GHRP-6, the lead compound in theGHS family (Bowers, 1998), serve as ligands for theCD36 receptor (Avallone et al., 2006; Demers et al.,2004; Marleau et al., 2005) and could exert beneficialeffects limiting the progression of atherosclerotic plaquedeposition. In the heart, binding to CD36 appears toaccount for the cardioprotective activity of GHS (Tor-sello et al., 2003). To date, there are no reports on theability of ghrelin and desacyl-ghrelin to bind CD36.
The anti-inflammatory effects of ghrelin have beenreported in humans and rodents, in which ghrelin is ableto inhibit inflammatory cytokines expression in mono-cytes and T cells (Dixit et al., 2004; Xia et al., 2004)and to reduce signs of joint inflammation (Granadoet al., 2005). Interestingly, human lymphocytes expressthe GHS-R1a, whose levels are significantly increasedby leptin. Reportedly, leptin also stimulated the expres-
sion of TNF-a, IL-1b, and IL-6 mRNAs (Dixit et al.,2004). In this cell model, ghrelin antagonized leptinstimulation of inflammatory cytokine expression, andghrelin effects were most likely mediated by its bindingto the GHS-R1a, as demonstrated by using a specific an-tagonist of the GHS-R1a (Dixit et al., 2004). In keepingwith the anti-inflammatory effects of ghrelin, it has alsorecently been reported that ghrelin administration in arat model of severe sepsis significantly attenuated sepsis-induced lung injury, down-regulated proinflammatorycytokines, and improved survival (Wu et al., 2007).These effects appeared to be mediated by ghrelin inhibi-tion of NF-jB pathway (Wu et al., 2007). Because NF-jB is a critical transcription factor whose activation isneeded for maximal expression of many inflammatorycytokines, we are planning to study its role in the medi-ation of GHS effects in N9 microglia cells.
In the present study, we have documented by RT-PCR that N9 microglia cells express the mRNA forCD36, and we have further confirmed the CD36 pro-
Fig. 6. Effects of fAb25–35 on mRNA levels of factors relevant to the inflammatory response. N9microglia cells were incubated for 24 hr with 30 lM fAb25–35 alone or in association with 1027
M of hexarelin, ghrelin, desacyl-ghrelin, or EP80317. Semiquantitative RT-PCR was used tomeasure modifications in mRNA levels for CD36 (A), TNF-a (B), PPAR-g (C), COX-1 (D),and COX-2 (E). Results are expressed as mean 6 SEM of six replicates for each treatment. Insetsshow a representative gel electrophoresis (in negative colors).
2724 Bulgarelli et al.
Journal of Neuroscience Research
tein expression by FACS analysis. In contrast, GHS-R1amRNA was undetectable. It is known that activation ofthe GHS-R1a activate the phospholipase C pathwayresulting in an increase of intracellular calcium, but inour experiments, the stimulation of N9 cells with ghrelinand hexarelin, which are selective agonists of the GHS-R1a receptor, did not induce any measurable modifica-tion of intracellular free calcium levels. It has been sug-gested that an inflammatory cascade is initiated when themicroglia interact with fAb deposit in the brain (Coraciet al., 2002). Microglial cells are the principal immuneeffector cells in the brain (Streit et al., 2004). They rep-resent the first line of defense against invading pathogensor other types of brain tissue injuries. Under pathologicalconditions, such as neurodegenerative diseases, stroke,and tumor invasion, these cells become activated,
enclose damaged and dead cells, and remove cellular de-bris from the area, working as phagocytic macrophages(Fetler and Amigorena, 2005). Once activated inresponse to neurodegenerative events, the microglial cellsrelease a variety of proinflammatory mediators such ascytokines, reactive oxygen species, complement factors,neurotoxic secretory products, free radical species, andnitric oxide, which contribute to both neuronal dysfunc-tion and cell death (Griffin et al., 1998). In the present
Fig. 8. Effects of ghrelin and desacyl-ghrelin on inflammatory cyto-kines mRNA levels. N9 microglia cells have been incubated for 24hr with 30 lM fAb25–35 alone or in association with 1027 M ofghrelin or desacyl-ghrelin. IL-1b (A) and IL-6 (B) mRNA levelswere measured by real-time RT-PCR and were normalized toGAPDH. Results are expressed as mean 6 SEM of six replicates foreach treatment. *P < 0.05 vs. controls. 8P < 0.05 vs. fAb 30 lM.ˆP< 0.05 vs. ghrelin.
Fig. 7. Effects of hexarelin and EP80317 on inflammatory cytokinesmRNA levels. N9 microglia cells have been incubated for 24 hrwith 30 lM fAb25–35 alone or in association with 1027 M of hexare-lin or EP80317. IL-1b (A) and IL-6 (B) mRNA levels were meas-ured by real-time RT-PCR and were normalized to GAPDH.Results are expressed as mean 6 SEM of six replicates for each treat-ment. 8P < 0.05 vs. fAb 30 lM.
GHS Inhibit IL-1b and IL-6 in Microglia 2725
Journal of Neuroscience Research

study, ghrelin had no significant effects on IL-1b andIL-6 mRNA levels in fAb-stimulated N9 cells. Interest-ingly, it has been reported that both ghrelin and hexare-lin stimulated the proliferation of adult rat hippocampalprogenitor cells, but only hexarelin protected these cellsfrom growth factor deprivation–induced apoptosis andnecrosis (Johansson et al., 2008). Similar to results thatwe obtained with N9 cells, adult rat hippocampal pro-genitor cells also do not express the GHS-R1a but arecapable of specifically binding both hexarelin and ghre-lin, suggesting that alternative GHS-R subtypes or dif-ferent receptors are involved (Johansson et al., 2008).Recently it has also been reported that circulating ghre-lin enters the hippocampus and stimulates dendritic spinesynapses formation and the generation of long-termpotentiation phenomena in neurons of the hippocampalregion (Diano et al., 2006).
Our data clearly demonstrate that hexarelin andEP80317 blunt the inflammatory process induced byfAb in N9 microglial cells, as evaluated in terms of IL-6and IL-1b mRNA levels. Further studies are needed inanimal models to ascertain whether the GHS might havea therapeutic potential in the treatment of AD.
REFERENCES
Ashenafi S, Fuente A, Criado JM, Riolobos AS, Heredia M, Yajeya J.
2005. Beta-amyloid peptide25–35 depresses excitatory synaptic trans-
mission in the rat basolateral amygdala ‘‘in vitro.’’ Neurobiol Aging
26:419–428.
Avallone R, Demers A, Rodrigue-Way A, Bujold K, Harb D, Anghel S,
Wahli W, Marleau S, Ong H, Tremblay A. 2006. A growth hormone–
releasing peptide that binds scavenger receptor CD36 and ghrelin
receptor up-regulates sterol transporters and cholesterol efflux in macro-
phages through a peroxisome proliferator–activated receptor gamma-
dependent pathway. Mol Endocrinol 20:3165–3178.
Bowers CY. 1998. Growth hormone-releasing peptide (GHRP). Cell
Mol Life Sci 54:1316–1329.
Bowers CY. 2001. Unnatural growth hormone-releasing peptide begets
natural ghrelin. J Clin Endocrinol Metab 86:1464–1469.
Bureau G, Longpre F, Martinoli MG. 2008. Resveratrol and quercetin,
two natural polyphenols, reduce apoptotic neuronal cell death induced
by neuroinflammation. J Neurosci Res 86:403–410.
Coraci IS, Husemann J, Berman JW, Hulette C, Dufour JH, Campanella
GK, Luster AD, Silverstein SC, El-Khoury JB. 2002. CD36, a class B
scavenger receptor, is expressed on microglia in Alzheimer’s disease
brains and can mediate production of reactive oxygen species in
response to beta-amyloid fibrils. Am J Pathol 160:101–112.
Corradin SB, Mauel J, Donini SD, Quattrocchi E, Ricciardi-Castagnoli
P. 1993. Inducible nitric oxide synthase activity of cloned murine
microglial cells. Glia 7:255–262.
Demers A, McNicoll N, Febbraio M, Servant M, Marleau S, Silverstein
R, Ong H. 2004. Identification of the growth hormone-releasing pep-
tide binding site in CD36: a photoaffinity cross-linking study. Biochem
J 382(Pt 2):417–424.
Diano S, Farr SA, Benoit SC, McNay EC, da Silva I, Horvath B, Gaskin
FS, Nonaka N, Jaeger LB, Banks WA, Morley JE, Pinto S, Sherwin
RS, Xu L, Yamada KA, Sleeman MW, Tschop MH, Horvath TL.
2006. Ghrelin controls hippocampal spine synapse density and memory
performance. Nat Neurosci 9:381–388.
Dixit VD, Schaffer EM, Pyle RS, Collins GD, Sakthivel SK, Palaniappan
R, Lillard JW Jr, Taub DD. 2004. Ghrelin inhibits leptin- and activa-
tion-induced proinflammatory cytokine expression by human mono-
cytes and T cells. J Clin Invest 114:57–66.
El Khoury J, Hickman SE, Thomas CA, Loike JD, Silverstein SC. 1998.
Microglia, scavenger receptors, and the pathogenesis of Alzheimer’s dis-
ease. Neurobiol Aging 19(1 Suppl):S81–S84.
El Khoury JB, Moore KJ, Means TK, Leung J, Terada K, Toft M, Free-
man MW, Luster AD. 2003. CD36 mediates the innate host response
to beta-amyloid. J Exp Med 197:1657–1666.
Febbraio M, Hajjar DP, Silverstein RL. 2001. CD36: a class B scavenger
receptor involved in angiogenesis, atherosclerosis, inflammation, and
lipid metabolism. J Clin Invest 108:785–791.
Fetler L, Amigorena S. 2005. Neuroscience. Brain under surveillance: the
microglia patrol. Science 309(5733):392–393.
Gauna C, van de Zande B, van Kerkwijk A, Themmen AP, van der Lely
AJ, Delhanty PJ. 2007. Unacylated ghrelin is not a functional antagonist
but a full agonist of the type 1a growth hormone secretagogue receptor
(GHS-R). Mol Cell Endocrinol 274:30–34.
Ghigo E, Broglio F, Arvat E, Maccario M, Papotti M, Muccioli G.
2005. Ghrelin: more than a natural GH secretagogue and/or an orexi-
genic factor. Clin Endocrinol (Oxf) 62:1–17.
Giordano V, Peluso G, Iannuccelli M, Benatti P, Nicolai R, Calvani M.
2007. Systemic and brain metabolic dysfunction as a new paradigm for
approaching Alzheimer’s dementia. Neurochem Res 32:555–567.
Giulian D, Haverkamp LJ, Yu JH, Karshin W, Tom D, Li J, Kirkpatrick
J, Kuo LM, Roher AE. 1996. Specific domains of beta-amyloid from
Alzheimer plaque elicit neuron killing in human microglia. J Neurosci
16:6021–6037.
Gnanapavan S, Kola B, Bustin SA, Morris DG, McGee P, Fairclough P,
Bhattacharya S, Carpenter R, Grossman AB, Korbonits M. 2002. The
tissue distribution of the mRNA of ghrelin and subtypes of its receptor,
GHS-R, in humans. J Clin Endocrinol Metab 87:2988.
Granado M, Priego T, Martin AI, Villanua MA, Lopez-Calderon A.
2005. Anti-inflammatory effect of the ghrelin agonist growth hormone-
releasing peptide-2 (GHRP-2) in arthritic rats. Am J Physiol Endocri-
nol Metab 288:E486–E492.
Griffin WS, Sheng JG, Royston MC, Gentleman SM, McKenzie JE,
Graham DI, Roberts GW, Mrak RE. 1998. Glial-neuronal interactions
in Alzheimer’s disease: the potential role of a ‘‘cytokine cycle’’ in dis-
ease progression. Brain Pathol 8:65–72.
Johansson I, Destefanis S, Aberg ND, Aberg MA, Blomgren K, Zhu C,
Ghe C, Granata R, Ghigo E, Muccioli G, Eriksson PS, Isgaard J. 2008.
Proliferative and protective effects of growth hormone secretagogues on
adult rat hippocampal progenitor cells. Endocrinology 149:2191–2199.
Kojima M, Hosoda H, Date Y, Nakazato M, Matsuo H, Kangawa K.
1999. Ghrelin is a growth-hormone-releasing acylated peptide from
stomach. Nature 402(6762):656–660.
Locatelli V, Rossoni G, Schweiger F, Torsello A, De Gennaro Colonna
V, Bernareggi M, Deghenghi R, Muller EE, Berti F. 1999. Growth
hormone–independent cardioprotective effects of hexarelin in the rat.
Endocrinology 140:4024–4031.
Marleau S, Harb D, Bujold K, Avallone R, Iken K, Wang Y, Demers A,
Sirois MG, Febbraio M, Silverstein RL, Tremblay A, Ong H. 2005.
EP 80317, a ligand of the CD36 scavenger receptor, protects apolipo-
protein E-deficient mice from developing atherosclerotic lesions.
FASEB J 19:1869–1871.
Marleau S, Mulumba M, Lamontagne D, Ong H. 2006. Cardiac and pe-
ripheral actions of growth hormone and its releasing peptides: relevance
for the treatment of cardiomyopathies. Cardiovasc Res 69:26–35.
Masters CL, Simms G, Weinman NA, Multhaup G, McDonald BL,
Beyreuther K. 1985. Amyloid plaque core protein in Alzheimer disease
and Down syndrome. Proc Natl Acad Sci USA 82:4245–4249.
Meda L, Cassatella MA, Szendrei GI, Otvos L, Jr., Baron P, Villalba M,
Ferrari D, Rossi F. 1995. Activation of microglial cells by beta-amyloid
protein and interferon-gamma. Nature 374(6523):647–650.
2726 Bulgarelli et al.
Journal of Neuroscience Research

Muccioli G, Pons N, Ghe C, Catapano F, Granata R, Ghigo E. 2004.
Ghrelin and des-acyl ghrelin both inhibit isoproterenol-induced lipolysis
in rat adipocytes via a non-type 1a growth hormone secretagogue re-
ceptor. Eur J Pharmacol 498:27–35.
Muller EE, Locatelli V, Cocchi D. 1999. Neuroendocrine control of
growth hormone secretion. Physiol Rev 79:511–607.
Pirami L, Stockinger B, Corradin SB, Sironi M, Sassano M, Valsasnini P,
Righi M, Ricciardi-Castagnoli P. 1991. Mouse macrophage clones
immortalized by retroviruses are functionally heterogeneous. Proc Natl
Acad Sci USA 88:7543–7547.
Pozzi G, Guidi M, Laudicina F, Marazzi M, Falcone L, Betti R, Crosti
C, Muller EE, DiMattia GE, Locatelli V, Torsello A. 2004. IGF-I stim-
ulates proliferation of spontaneously immortalized human keratinocytes
(HACAT) by autocrine/paracrine mechanisms. J Endocrinol Invest
27:142–149.
Santambrogio L, Belyanskaya SL, Fischer FR, Cipriani B, Brosnan CF,
Ricciardi-Castagnoli P, Stern LJ, Strominger JL, Riese R. 2001. Devel-
opmental plasticity of CNS microglia. Proc Natl Acad Sci USA
98:6295–6300.
Streit WJ, Mrak RE, Griffin WS. 2004. Microglia and neuroinflamma-
tion: a pathological perspective. J Neuroinflammation 1:14.
Torsello A, Locatelli V, Melis MR, Succu S, Spano MS, Deghenghi R,
Muller EE, Argiolas A. 2000. Differential orexigenic effects of hexarelin
and its analogs in the rat hypothalamus: indication for multiple growth
hormone secretagogue receptor subtypes. Neuroendocrinology 72:327–
332.
Torsello A, Bresciani E, Rossoni G, Avallone R, Tulipano G, Cocchi D,
Bulgarelli I, Deghenghi R, Berti F, Locatelli V. 2003. Ghrelin plays a
minor role in the physiological control of cardiac function in the rat.
Endocrinology 144:1787–1792.
Wang X, Li C, Chen Y, Hao Y, Zhou W, Chen C, Yu Z. 2008. Hy-
poxia enhances CXCR4 expression favoring microglia migration via
HIF-1alpha activation. Biochem Biophys Res Commun 371:283–288.
Wisniewski HM, Robe A, Zigman W, Silverman W. 1989. Neuropatho-
logical diagnosis of Alzheimer disease. J Neuropathol Exp Neurol
48:606–609.
Wu R, Dong W, Zhou M, Zhang F, Marini CP, Ravikumar TS, Wang
P. 2007. Ghrelin attenuates sepsis-induced acute lung injury and mor-
tality in rats. Am J Respir Crit Care Med 176:805–813.
Xia Q, Pang W, Pan H, Zheng Y, Kang JS, Zhu SG. 2004. Effects of
ghrelin on the proliferation and secretion of splenic T lymphocytes in
mice. Regul Pept 122:173–178.
Yankner BA, Duffy LK, Kirschner DA. 1990. Neurotrophic and neuro-
toxic effects of amyloid beta protein: reversal by tachykinin neuropepti-
des. Science 250:279–282.
GHS Inhibit IL-1b and IL-6 in Microglia 2727
Journal of Neuroscience Research





























