Embed Size (px)
Citation preview

Design of an infrared laser pulse to control the multiphoton dissociationof the Fe–CO bond in CO-heme compounds
Sitansh Sharma,1,a� Harjinder Singh,1,b� Jeremy N. Harvey,2,c� and Gabriel G. Balint-Kurti2,d�
1Center for Computational Natural Sciences and Bioinformatics, International institute of InformationTechnology, Hyderabad 500032, India2Center for Computational Chemistry, School of Chemistry, University of Bristol, Bristol BS8 1TS,United Kingdom
�Received 24 June 2010; accepted 9 September 2010; published online 1 November 2010�
Optimal control theory is used to design a laser pulse for the multiphoton dissociation of the Fe–CObond in the CO-heme compounds. The study uses a hexacoordinated iron–porphyrin–imidazole–COcomplex in its ground electronic state as a model for CO liganded to the heme group. The potentialenergy and dipole moment surfaces for the interaction of the CO ligand with the heme group arecalculated using density functional theory. Optimal control theory, combined with a time-dependentquantum dynamical treatment of the laser-molecule interaction, is then used to design a laser pulsecapable of efficiently dissociating the CO-heme complex model. The genetic algorithm method isused within the mathematical framework of optimal control theory to perform the optimizationprocess. This method provides good control over the parameters of the laser pulse, allowingoptimized pulses with simple time and frequency structures to be designed. The dependence ofphotodissociation yield on the choice of initial vibrational state and of initial laser field parametersis also investigated. The current work uses a reduced dimensionality model in which only the Fe–Cand C–O stretching coordinates are explicitly taken into account in the time-dependent quantumdynamical calculations. The limitations arising from this are discussed in Sec. IV. © 2010 AmericanInstitute of Physics. �doi:10.1063/1.3494543�
I. INTRODUCTION
Control of molecular processes at the atomic level is along held scientific dream. Lasers would seem to be wellsuited to the task of manipulating the flow of energy withtime so as to steer a system to a desired quantum state orphysical configuration. In the past two decades, coherentcontrol of molecular quantum dynamics by means of exter-nal laser pulses has experienced significant progress boththeoretically1–10 as well as experimentally.11–16 The applica-tion of lasers to the control of complex photophysical pro-cesses, such as photoisomerization of the retinal chro-mophore in bacteriorhodopshin17 and of energy flow innatural as well as artificial light-harvesting complexmolecules,18–20 has been demonstrated. The developments inlaser pulse shaping techniques21,22 has opened the way tocontrolling molecular processes even in the femtosecond andattosecond time regimes.23 The recent progress in the designof ultrafast and intense midinfrared laser pulses24,25 providesan efficient way of controlling molecular processes such asthe dissociation of molecules in their ground electronic statesby means of vibrational ladder climbing or through othermechanisms. It may also provide the opportunity to monitorand understand the dynamics of large biological systemssuch as proteins.
Hemoglobin and myoglobin are among the most studiedproteins. The structure and binding energies of ligands suchas CO, O2, and NO binding with the heme group withinthese proteins have been extensively investigated.26–32 Vari-ous techniques, such as time-resolved IR spectroscopy,33–35
x-ray crystallography,36–39 and molecular simulation,40–43
have been used to characterize the structure and dynamics ofprotein and ligand before and after dissociation. The CO vi-brational spectra have been used to understand the mecha-nism of the ligand transfer to the docking site after its disso-ciation from heme binding site.44–46 It has also beensuggested that laser induced dissociation of carboxy-hemoglobin47 might be used as a treatment for carbon mon-oxide poisoning. This suggestion has not been tested in anyin vivo experiments, and must therefore, for the present, betreated as speculative.
Various aspects of the laser controlled dynamics of theCO-heme and other compounds exhibiting similar propertieshave been extensively studied. Joffre and co-workers,48 intheir pump-probe studies, demonstrated coherent vibrationalladder climbing up to the v=6 state of the CO vibration incarboxy-heomoglobin by means of midinfrared laser pulses.In contrast to previous studies on internal vibrational redis-tribution �IVR� mediated dissociation of the metal-CO bondupon CO-stretch excitation in a similar metal-carbonyl sys-tem, Cr�CO�6,49 they observed less yield for IVR mediateddissociation of Fe–CO bond. Quantum wave packet studiesperformed by Kühn on CO-heme compounds50 demonstratedthat even if the initial average energy of CO-stretch is abovethe dissociation limit, there is only a small probability of
a�Electronic mail: [email protected]�Author to whom correspondence should be addressed. Electronic mail:
[email protected]�Electronic mail: [email protected]�Electronic mail: [email protected].
THE JOURNAL OF CHEMICAL PHYSICS 133, 174103 �2010�
0021-9606/2010/133�17�/174103/11/$30.00 © 2010 American Institute of Physics133, 174103-1

dissociation of the Fe–CO bond. The possibility of laser con-trol of vibrational excitation in a two dimensional math-ematical model of CO-heme compounds was alsoinvestigated.51,52 Recently, Gollub et al.53 performed theoret-ical studies on the vibrational ladder climbing in the transi-tion metal carbonyl complex, MnBr�CO�5 �having similarcharacteristics to the CO-heme complex�. They observed avery low quantum yield ��2%� after 3 ps for the IVR me-diated dissociation of the Mn–CO bond. This led them toconclude that the C–O mode can be regarded as an isolateddegree of freedom within the polyatomic system on the pi-cosecond to nanosecond time scale. All these studies con-firmed that the mode coupling between the Fe–CO stretchingmode and the C–O stretch is small, and that pumping energyinto the C–O stretch will not constitute an efficient way ofputting energy into the Fe–CO stretching motion. Prelimi-nary studies for the present paper confirm this view and ourattempts to design efficient laser pulses to dissociate theFe–CO bond by pumping energy into the C–O vibrationalmotion have met with failure.
In this work, we investigated the possibility of directlydissociating the Fe–CO bond in CO-heme compounds intheir ground electronic state using a single laser pulse.54 Atwo dimensional mathematical model of the Fe–C–O portionof the CO-heme complex is used to study the dissociation ofFe–CO bond. Our aim is to design a laser pulse havingsimple time and frequency structures, which can maximizethe outgoing flux in the dissociating channel correspondingto breaking the Fe–CO bond. The genetic algorithm �GA�method is used as the optimization procedure within theframework of optimal control theory �OCT�.55 In this par-ticular approach, each individual of the GA population ischaracterized by a small set of chosen laser pulse parameterssuch as the initial amplitude of the laser field and the timeparameters governing the shape of the laser pulse. The fitnessof each individual of the population is compared using thevalue of a field dependent “cost functional,” which includesa measure of the probability of successfully attaining thedesired objective, as well as other constraint terms, such as apenalty term for the overall laser pulse energy. The penaltyterm is multiplied by a positive factor �0, which determinesthe weight assigned to it. The Franck–Condon overlap be-tween the bound states of the system and the continuum statewave functions is small; therefore, in order to achieve theefficient dissociation of the Fe—CO bond, a single infraredlaser pulse that operates through a multiphoton pathway toexcite the system into its dissociation continuum is designedusing the optimal control theory and the GA optimizationmethod. The effect of the initial vibrational state on the pho-todissociation dynamics of the Fe–CO bond is also investi-gated by considering different vibrational states as startingpoints. Laser pulses for three different choices of �0 valuesare designed for each initial vibrational state.
This paper is organized in the following way: Sec. II Adescribes the active site model of the system and the relatedelectronic structure calculations, Sec. II B discusses the time-dependent quantum dynamics used, Sec. II C outlines theoptimal control theory used, and Sec. II D briefly describesthe implementation of the genetic algorithm methodology
within the framework of the optimal control theory for thelaser pulse shaping. Section III presents the results obtained.Finally, a conclusion is given in Sec. IV.
II. THEORY
A. Active site model and electronic structurecalculations
X-ray studies of carbon monoxide ligated myoglobinshow a nearly perfect square planar symmetry of the planeformed by the four nitrogen atoms of the pyrrole unit. Thesestudies also show an almost linear geometry for the Fe–C–Ogroup.56 In order to model the dynamics of CO bound to theheme group, we choose hexacoordinated iron–porphyrin–imidazole–CO as the active site model. This model is shownin Fig. 1�a�. The imidazole ligand is used to mimic the effectof the proximal histidine residue in the model. The geometryoptimization of the active site model was performed usingdensity functional theory �DFT� in combination with a hy-brid density functional, built up using Becke’s three param-eter exchange and Lee–Yang–Parr’s correlation functional�abbreviated as B3LYP�.57–59 A standard Gaussian 6-311G�
all electron basis set was used to represent Fe, O, N, and Catoms,60–63 while the hydrogen atoms were described using a6-31G basis.64 All electronic structure calculations were car-ried out using the GAUSSIAN 03 suite of quantum chemicalprograms.65 The coordinates of all atoms, except for those ofthe CO ligand, were fixed at the equilibrium geometry of theground electronic state.29 The Fe–C and C–O coordinateswere varied systematically over the ranges of 1.3–5.0 and0.9–1.5 Å, respectively, so as to obtain the computed groundstate energy and dipole moment at 184 nuclear geometries. Atwo dimensional spline interpolation between these pointswas then used to generate a continuous potential and dipolemoment functions. The calculated potential energy and di-pole moment surfaces are shown in Fig. 2.
B. Quantum dynamics
For our optimal control studies, we have assumed thatthe Fe–C–O moiety is linear and also that dissociation occursentirely on the singlet potential energy surface without in-volving any other electronic surfaces.28 The theoretical mod-eling of the Fe–CO bond dissociation is done using the Ja-cobi coordinates R and r, where R is the distance of the ironatom from the center of mass of the C–O bond and r is thelength of the carbon oxygen bond �see Fig. 1�b��. In this set
FIG. 1. �a� Hexacoordinated active site model of CO-heme compounds,where the imidazole ring mimics the effect of the proximal histidine residue.�b� Jacobi coordinate representation of linear triatomic fragment Fe–C–O,where R is the distance of Fe from the center of mass of C–O bond, while ris the length of the C–O bond.
174103-2 Sharma et al. J. Chem. Phys. 133, 174103 �2010�

of coordinates, the kinetic energy operator T�R ,r� for thelinear Fe–C–O system can be written as atomic units �a.u.�are used throughout the paper, ��=1�
T�R,r� = −1
2�1
�2
�r2 −1
2�2
�2
�R2 , �1�
having the following reduced masses:
1
�1=
1
mC+
1
mO,
1
�2=
1
mFe+
1
mC + mO, �2�
where the quantities mC, mO and mFe denote the masses ofthe carbon, oxygen, and iron atoms, respectively.
The vibrational eigenfunctions and eigenvalues of themodel �Fe–C–O� system in its ground electronic state havebeen calculated using a numerical variational method devel-oped previously within our group.66 Since we are interestedin the bond dissociation of the Fe–CO bond, the nuclearwave function is represented on a spatial grid spanning Rcoordinate values from 2.8a0 to 10.40a0 in 256 steps and rcoordinate values from 1.7a0 to 3.2a0 in 128 steps.
Laser induced time evolution of the nuclear wave packet��R ,r ; t� is described by solving the time-dependentSchrödinger equation, within the semiclassical dipole ap-proximation, as
i�
�t��R,r;t� = �H0�R,r� − ��R,r� · ��t����R,r;t� , �3�
where H0�R ,r� is the molecular Hamiltonian operator de-
scribed as a sum of the kinetic energy operator T�R ,r� and
the potential energy operator V�R ,r�. The quantity ��R ,r� isthe dipole moment of the system in its ground electronicstate and ��t� is the electric field component of the laser pulseprojected onto the Fe–C–O direction. The time evolution ofthe initial wave function is performed numerically by solvingthe time-dependent Schrödinger equation using the Split op-erator method.67 In order to avoid the artificial reflection ofthe wave packet at the edges of the grid in the R direction,we have applied the following damping function:68
FIG. 3. Some eigenfunctions for the model system �Fe–C–O� obtained using a variational calculation. Each eigenfunction is labeled according to its quantumnumber �m ,n�, where indices m and n correspond to the number of quanta of excitation in the Fe–C and C–O modes, respectively �see text�.
FIG. 2. Variation in the potential energy and x component �along Fe–C–O� of the dipole moment with a change in bond lengths of Fe–C and C–O bonds,obtained using DFT.
174103-3 Laser pulse to control dissociation of Fe–CO J. Chem. Phys. 133, 174103 �2010�

f�Ri� = sin��
2
�Rmask + Rmask − Ri�Rmask
, Ri Rmask, �4�
where Rmask=8.503a0 is the point at which the dampingfunction is activated and Rmask=Rmax−Rmask is the distanceover which the damping function decays from 1 to 0, Rmax isthe maximum value of R on the grid. Similarly, for the rcoordinate, we have used the following damping function:
f�ri� = sin��
2
�rmask + rmask − ri�rmask
, ri rmask, �5�
where rmask=3.011a0 is the point at which the damping isstarted and rmask=0.201a0 is the width of the dampingfunction over which it decays from 1 to 0.
C. Optimal control theory
Our aim is to design a laser pulse having a simple timeand frequency structure that can effectively break the Fe–CObond in the CO-heme complex in its ground electronic state.In order to accomplish this, the laser pulse must deposit en-ergy in one or both of the two normal modes of the lineartriatomic system. At first, similar to many others,48,50 wethought that this could be accomplished most efficiently byinitially pumping the radiation into the C–O stretching mode.As with the previous investigations, we found that we couldnot design a laser pulse to perform this task. We thereforeturned our attention to attempting to pump energy directlyinto the Fe–CO stretching motion. As the system absorbs
progressively more energy, the Fe–CO separation, on aver-age, increases and a progressively greater portion of the mol-ecules is dissociated. We choose some large separation R�,beyond which we consider the system to have dissociated.The integral over time of the particle flux crossing a line withR=R� is a measure of the portion of the system that hasundergone dissociation. In these calculations, R� is set at7.53a0. Using optimal control theory, we want to maximizethe time integral of the flux integrated over the duration �T�of the laser pulse. Therefore, following the theoretical frame-work proposed by Rabitz and co-workers,7,8,69,70 the costfunctional for the optimal control of dissociation in theFe–CO part of CO-heme complexes can be defined as
J = 0
T
dt���t��FR���t�� − �00
T
dt���t��2. �6�
The flux operator FR in the first term of the above equation isgiven as
FR =1
2� pR
�2��R − R�� + ��R − R��
pR
�2 , �7�
where pR=−i�� /�R� is the momentum operator conjugate toR and �2 is the corresponding reduced mass. The secondterm in Eq. �6� is a penalty term, included so as to lower thetotal energy of the laser pulse, and �0 is a constant weightingfactor. Using GA we aim to optimize the selected laser pulseparameters so as to maximize the cost functional �see Eq.�6��.
In principle, while we optimize for maximal Fe–CO dis-sociation, we would also wish to minimize the energy beingpumped into the C–O stretching motion. The implementationof such a constraint would necessitate an additional term inEq. �6�. However, as mentioned earlier, the mode couplingbetween motions in the two coordinates R and r is negligible,thus eliminating the need for this constraint. This is con-firmed in the analysis of our results below.
D. Laser pulse optimization methodology
In this section, we give a brief description of the GAoptimization procedure. Further details can be found inRef. 55.
The laser pulse whose parameters are to be adjusted dur-ing the GA optimization procedure is defined to be of theform
TABLE I. Computed energies of the lowest ten vibrational levels of themodel CO-heme compound measured from the lowest point of the potential.The assignments �m ,n� correspond to the R and r motions, respectively.
Assignment��m ,n��
Energy�cm−1�
�00� 1269.83�10� 1724.99�20� 2171.11�30� 2608.52�40� 3037.44�01� 3301.39�50� 3457.73�11� 3757.49�60� 3869.25�21� 4204.81�70� 4271.88
FIG. 4. Electric field of the optimized laser pulse as a function of time �A1�, frequency spectrum of the pulse �A2�, time dependence of the time-integratedflux and norm of the wave function �A3� for �00� as the initial vibrational state; Total time �T�=60 000 a.u. and �0=0.001.
174103-4 Sharma et al. J. Chem. Phys. 133, 174103 �2010�

�GA = �k sin� kt� · s�t� , �8�
where �k is the peak field strength, k is the frequency of thelaser pulse, and s�t� is the pulse shape function or envelope.The pulse envelope is zero at the beginning and end of thepulse and is preserved unaltered throughout the optimization.This guarantees that at least this aspect of the optimized laserpulse will be experimentally feasible. For the smooth switch-ing on and off of the field, s�t� has a sine-squared form in thefirst and last parts of the pulse and a constant plateau value inbetween. In mathematical form, this can be written as
s�t� =�sin2��
2� t − t0
t1 − t0� for t0 � t � t1
1 for t1 � t � t2
sin2��
2� t3 − t
t3 − t2� for t2 � t � t3 = T ,� �9�
where t0, t1, t2, and t3 are four positive time parameters thatobey the relationship t0� t1� t2� t3. In the present optimiza-tion procedure, we set t0 and t3 equal to 0 and T, respectively.Only the t1 and t2 parameters, characterizing the shape of thelaser pulse, are adjusted during the optimization procedure;t1 is allowed to take values between 0.2T and T, and t2 is
allowed to take values from t0=0 to 0.8T, subject to theconstraints of the relationship just mentioned. So, for a laserpulse with a fixed frequency value, the different possiblecombinations of the amplitude ��k� and the two time param-eters �t1 and t2� are searched using GA with the objective ofmaximizing the cost functional �see Eq. �6��.
The GA is a random search algorithm71 that follows theprinciples of natural selection and evolution to find that com-bination of laser pulse parameters that best maximizes thecost functional in Eq. �6�. A starting population of ten indi-viduals, characterized by a specific choice of the above men-tioned laser pulse parameters, is randomly generated. So, ev-ery individual of the GA population corresponds to a laserfield having some random values for these laser pulse param-eters. Each parameter is discretized into 1024 possible valueslying between a predefined minimum and maximum value.The fitness of each individual of the population is judged onthe basis of the corresponding cost functional value. Usingtournament selection, a new generation of fitter individuals isproduced and the algorithm is iterated until it satisfies theconvergence criteria. In order to ensure the reproduction ofthe most highly fit individuals of the present generation tothe future generations, the elitism procedure is used in thepresent GA procedure. The possibility of biological inter-breeding is eliminated by using the highly efficientmicro-GA approach.72 In the micro-GA approach, during theoptimization, when the individuals of a generation become
FIG. 5. Snapshots of the probability density in the ground electronic state for �00� at different times during the laser driven dynamics for a pulse duration of60 000 a.u. and �0=0.001.
174103-5 Laser pulse to control dissociation of Fe–CO J. Chem. Phys. 133, 174103 �2010�

too similar, the optimization procedure is restarted usinghighly fit �elite� individuals, while the rest of the individualsof the populations are randomly selected. Various other GAparameters are fixed as suggested by Carroll andco-workers.73–75
III. RESULTS AND DISCUSSION
In this section, we present and discuss the results ob-tained for the direct photodissociation of the Fe–CO bond inthe CO-heme model compound. The wave functions of thefour selected vibrational states of the Fe–C–O system ob-tained using a variational procedure66 based on a direct prod-uct of one dimensional wave functions obtained using theFourier Grid Hamiltonian �FGH� method76,77 are plotted inFig. 3. From an examination of the nodal structure of thewave functions in the figure, we see that the nodes in thewave functions are parallel to the two axes. This shows thatalthough the Jacobi coordinates R and r have not been de-fined as normal coordinates, they very nearly fill this role.The form of the wave functions shows that the motion in thetwo coordinates is largely uncoupled. The vibrational wavefunctions are labeled as �m ,n�, where the indices m and ncorrespond to the number of quanta of excitation in themodes of motion along the coordinates R and r, respectively.
The motion along the coordinate r corresponds to the C–Ovibration, while that along R is the Fe–CO stretching mode,which eventually leads to the breaking of this bond. Table Igives the computed energies and vibrational quantum num-ber assignments of the lowest ten vibrational levels of thesystem. The dissociation energy from the bottom of the po-tential energy surface to the Fe–CO asymptote is11 237.1 cm−1.
We start with the system in its ground �00� vibrationalstate and choose an initial trial laser field of the form
�GA�t� = �0 sin� 00→10t� · s�t� , �10�
where �0 is the amplitude of the laser pulse and is allowed tovary from 0 to 0.03 a.u., 00→10=455.16 cm−1 is the transi-tion frequency from the ground vibrational state to the firstexcited vibrational state of the Fe–CO stretching mode��10��, and s�t� is the pulse envelope function. As the fre-quency of the laser pulse is fixed, different combinations of�0 and the two time parameters �t1 , t2� characterizing theshape of the laser pulse are searched in the parameter spacein order to optimize �maximize� the cost functional valueusing the GA search procedure. The pulse duration �T� andthe premultiplier �0 of the penalty function are fixed as60 000 a.u. �1.451 ps� and 0.001, respectively.
FIG. 6. Electric field of the optimized laser pulses as a function of time �A1, B1, C1�, frequency spectrum of the laser pulse �A2, B2, C2�, time dependenceof the time-integrated flux and norm of the wave function �A3, B3, C3� for different choices of initial vibrational state; �10� �A1, A2, A3�; �01� �B1,B2,B3�,and �11� �C1,C2,C3� for a pulse of duration 60 000 a.u. and �0=0.001.
174103-6 Sharma et al. J. Chem. Phys. 133, 174103 �2010�

Figure 4 shows the results of the optimization process.The main result is displayed in the rightmost frame of thefigure. This shows the time-integrated flux passing throughthe surface defined by R=R�=7.53a0. The time-integrateflux plotted against time shows the degree of dissociation upto that point in time. We see that the optimized laser pulsecauses the Fe–CO bond in the CO-heme model to start dis-sociating sharply at around t=13 125 a.u. �317.5 fs�. Fol-lowing this sharp rise in the time-integrate flux, the quantitycontinues to increase in a steplike manner until almost com-plete dissociation is achieved �99.8%�. Also shown in thefigure is the norm of the wave packet as a function of time.As the Fe–CO bond dissociates, the wave packet is progres-sively absorbed near the edge of the grid and the norm de-creases. The decrease in the norm mirrors exactly the in-crease in the time-integrate flux.
Frame A1 in Fig. 4 shows the optimized laser field as afunction of time. The maximum amplitude of the optimizedlaser field is 0.0273 a.u. �1.404�1010 V m−1�. The fre-quency spectrum of the optimized laser pulse, obtained bytaking the Fourier transform of the electric field as a functionof time, is shown in frame A2. The smooth variation in thefield amplitude of the laser pulse leads to the simple spec-trum shown in the figure.
Figure 5 shows snapshots of the wave packet, acted onby the optimized laser pulse, at different times superimposedon the potential energy surface. At a time of 7500 a.u., we
see that the probability density begins to move outwardalong the Fe–CO channel. Moreover, at 13 125 a.u. of time,it splits up and a significant part of it leaves the interactionregion. The remaining part of the wave packet oscillatesbackward and forward in the interaction region with somesmall parts dissociating on each occasion when the wavepacket moves toward large R values. At the end of the pulseduration, more than 99.8% of the wave packet is dissociated.This oscillatory dissociation is consistent with the steplikegrowth of the time-integrate flux shown in panel A3 of Fig. 4at longer times, following the steep initial growth of thisquantity. It can also be seen from the figure that the wavepacket remains fairly localized throughout along the C–Ostretching coordinate r, thus confirming the lack of couplingbetween the motions along the R and r coordinates. Thisindicates that the optimized laser pulse dissociates theFe–CO bond without substantially exciting the CO vibra-tional motion. The rapid dissociation of the molecule shownin frame A3 of Fig. 4 suggests that the mechanism of photo-dissociation is not one of stepwise ladder climbing.
A. Effect of initial vibrational state
In this subsection, we examine the effect of the initialvibrational state on the photodissociation dynamics of theFe–CO bond. The results for three different initial vibrationalstates are summarized in Figs. 6–9 and Table II.
FIG. 7. Snapshots of the probability density in the ground electronic state for �10� at different times during the laser driven dynamics for a pulse duration of60 000 a.u. and �0=0.001.
174103-7 Laser pulse to control dissociation of Fe–CO J. Chem. Phys. 133, 174103 �2010�
1. �10‹ as initial vibrational state
For this choice of initial state, the Fe–CO stretchingmode is initially excited with one quantum of vibrationalexcitation. The trial laser field is defined as in Eq. �10�.Frames A1 and A2 in Fig. 6 show the optimized laser fieldand its corresponding frequency spectrum. The maximumamplitude of the optimized laser field is 0.027 65 a.u.�1.422�1010 V m−1�. The variation of time-integrated flux,shown in frame A3, is in general terms similar to that shownin Fig. 4 for photodissociation from the �00� state. The dif-ferences are that the onset of the dissociation is later than forthe �00� state and that the initial growth in the time-integratedflux is less sharp. The stepped nature of the growth after theinitial sharp increase is still present, but to a lesser extentthan in the �00� case. The norm of the wave function de-creases in a “mirror image” way to the increase of the time-integrated flux �see frame A3�. Further insights into the laserdriven dissociation dynamics can be gained with the help ofsnapshots of probability density of the wave function at dif-ferent times during the action of the optimized laser pulse, asshown in Fig. 7. During the first 7500 a.u., there is no sig-nificant change in the probability density, except an increasein the amplitude of the right side peak. At time 15 000 a.u.,the probability density splits up, and by time 22 500 a.u.,more than 83% of the wave packet has crossed the analysisline and has dissociated. The remaining probability densityin the small R region begins to oscillate and dissociatesgradually, a small bit at a time.
2. �01‹ as initial vibrational state
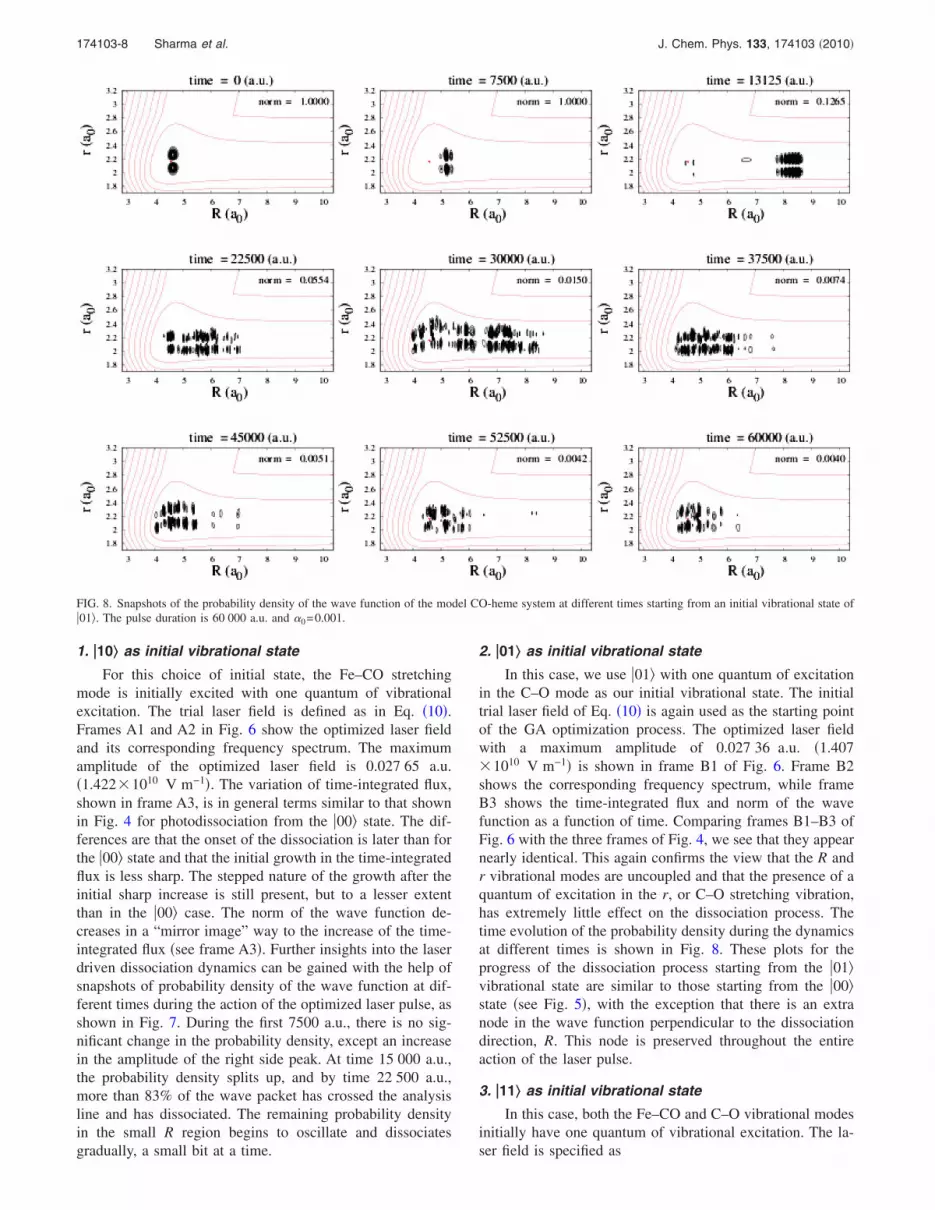
In this case, we use �01� with one quantum of excitationin the C–O mode as our initial vibrational state. The initialtrial laser field of Eq. �10� is again used as the starting pointof the GA optimization process. The optimized laser fieldwith a maximum amplitude of 0.027 36 a.u. �1.407�1010 V m−1� is shown in frame B1 of Fig. 6. Frame B2shows the corresponding frequency spectrum, while frameB3 shows the time-integrated flux and norm of the wavefunction as a function of time. Comparing frames B1–B3 ofFig. 6 with the three frames of Fig. 4, we see that they appearnearly identical. This again confirms the view that the R andr vibrational modes are uncoupled and that the presence of aquantum of excitation in the r, or C–O stretching vibration,has extremely little effect on the dissociation process. Thetime evolution of the probability density during the dynamicsat different times is shown in Fig. 8. These plots for theprogress of the dissociation process starting from the �01�vibrational state are similar to those starting from the �00�state �see Fig. 5�, with the exception that there is an extranode in the wave function perpendicular to the dissociationdirection, R. This node is preserved throughout the entireaction of the laser pulse.
3. �11‹ as initial vibrational state
In this case, both the Fe–CO and C–O vibrational modesinitially have one quantum of vibrational excitation. The la-ser field is specified as
FIG. 8. Snapshots of the probability density of the wave function of the model CO-heme system at different times starting from an initial vibrational state of�01�. The pulse duration is 60 000 a.u. and �0=0.001.
174103-8 Sharma et al. J. Chem. Phys. 133, 174103 �2010�

�GA�t� = �0 sin� 01→11t� · s�t� �11�
where 01→11=456.09 cm−1 is the frequency correspondingto the transition �01�→ �11�. Table II shows that an optimizedlaser field with a maximum amplitude of 0.027 51 a.u.�1.415�1010 V m−1� is able to dissociate more than 99.7%of the CO-heme model system by breaking its Fe–CO bond.Frame C1 in Fig. 6 shows the variation of electric field of theoptimized laser field as a function of time. The frequencyspectrum of the optimized laser field is plotted in frame C2and shows a simple frequency structure. The variation of thetime-integrated flux and the norm of the wave function areshown in frame C3. As in the previous cases, we see a rela-tively sharp rise in the dissociation probability as a function
of time, followed by a stepwise increase to nearly completedissociation. Note that frames A1–A3 in Fig. 6 are very simi-lar to frames C1–C3. The main difference between thesegroups of figures is that the first corresponds to using thestate �10� as the initial state, while the second group uses �11�as the initial state. Their great similarity again demonstratesthe fact that excitation of the C–O stretching motion does noteffect the photodissociation dynamics. Figure 9 shows snap-shots of the probability density at different times during thedissociation process. This figure is again very similar to Fig.7, except for the fact that the wave function has an extranode along the r direction.
B. Effect of penalty function and �0
The penalty function −�0�0Tdt���t��2 in the cost functional
of Eq. �6� provides a means of limiting the strength andoverall energy of the laser pulse. In order to examine itseffect on the dissociation process and on the resulting opti-mized laser pulse, we have performed a set of calculationsusing larger values of the �0 parameters equal to 0.01 and0.1. The overall length of the pulse is maintained at60 000 a.u.
The results for all the initial vibrational states studied aretabulated in Table III. The table shows that for a particularchoice of the initial vibrational state, the dissociation yield,the cost functional value, and the maximum amplitude of theoptimized laser field ��peak� all increase with a decrease in the
FIG. 9. Snapshots of the probability density of the wave function of the model CO-heme system at different times starting from an initial vibrational state of�11�. The pulse duration is 60 000 a.u. and �0=0.001.
TABLE II. Results of the direct photodissociation of the Fe–CO bond in theCO-heme compound starting from different initial vibrational states ��m ,n��using a pulse duration of 60 000 a.u. �0 is set at 0.001. “J” refers to thevalue of the cost functional and “�peak” refers to the value of amplitude ofthe electric field of the optimized laser pulse at its peak. All quantities are ina.u.
Initial state��m ,n�� Dissociation J �peak
�00� 0.997 627 0.986 167 2.730�10−2
�10� 0.997 690 0.982 535 2.765�10−2
�01� 0.995 957 0.986 647 2.736�10−2
�11� 0.997 453 0.982 766 2.751�10−2
174103-9 Laser pulse to control dissociation of Fe–CO J. Chem. Phys. 133, 174103 �2010�

�0 value. The higher the value of �0, the lower is that of �peak
in the optimized laser pulse and vice versa. The dissociationyield for the lower values of �0 is almost independent of thechoice of the initial vibrational state.
IV. CONCLUSION
We have used the optimal control theory combined witha genetic algorithm method of optimization to design infra-red laser pulses to dissociate the Fe–CO bond in a model ofthe active site of the CO-heme complex. Our model systemconsists of 48 atoms and is shown in Fig. 1�a�. The potentialenergy and dipole moment surfaces for the variation of theFe–C and C–O separations in the Fe–C–O moiety were com-puted using an all-electron ab initio �DFT� approach and alarge �6-311G�� atomic orbital basis set. The system wastaken to be in its ground electronic state and all the otheratoms were maintained in their equilibrium geometry.
We have successfully designed infrared laser pulses withduration of 1.451 ps, which have a very simple frequencyand amplitude structure and which will completely �99.7%�dissociate the Fe–CO bond in the CO-heme system withoutexciting the vibrational motion in the fragment of the COmolecule.
We show �see Fig. 3� that the Fe–CO and C–O stretchingmotions are very nearly normal modes of the Fe–C–O moi-ety, so we can validly speak of these stretching motions as ifthey were normal mode motions. Our calculations show, inkeeping with the findings of other groups,48,50,53 that there isalmost no dynamical coupling between the modes. If, in theinitial state of the system, we excite the C–O stretching mo-tion, the dissociation proceeds in a manner identical in allobservable aspects to that in which it would proceed if wehad not excited this vibrational motion. The C–O vibrationalexcitation acts purely as a spectator mode. It follows alsothat it is not possible to efficiently dissociate the Fe–CObond in the CO–heme complexes by pumping energy intothe C–O stretching motion.
The maximum amplitudes of the electric field of the la-ser pulses we have designed are modest, of the order of1.4�1010 V m−1. If desired, these fields may easily be re-duced. This may be done either by increasing the premulti-plier of the penalty function �0 in the cost functional �see Eq.�6� and Table III� or by increasing the length of the laserpulse.78,79 If the former approach is taken, a penalty must bepaid in that the total dissociation probability will decreasefrom its near 100% value.
Finally, it is necessary to conclude with some cautionarynotes. The current calculations are of a “reduced dimension-ality” nature. That is, the dynamics takes account of only thethree atoms most directly involved in the light absorptionand dissociation process. In a more complete calculation, itwould be desirable to take account of the other atoms in thesystem, especially in regard to their role in IVR, which hasthe potential to divert the absorbed photon energy away fromthe Fe–CO dissociation mode. In a protein environment, wewould expect to encounter many other absorption features inthe vicinity of the frequencies of the laser pulses��450 cm−1� used in the current work. This might hinderthe effective use of the pulses in an in vivo environment. Apossible way of circumventing this problem is to utilize astrong absorption band of the CO-heme group, such as theSoret band,80 in the ultraviolet �UV� region together with atwo photon stimulated emission pumping procedure.81,82 Amore detailed consideration also needs to be given to theorientation dependence of the light absorption; in the currentwork, we have assumed that the laser is polarized along theFe–C–O bond direction. The time-dependent quantum dy-namics are performed using quantum mechanics in thepresent work. It is extremely difficult to extend the dimen-sionality of such calculations as they scale exponentiallywith an increase of dimensionality. The inclusion of furtherdegrees of freedom would therefore require the use of ap-proximations in the dynamics. Two possible approacheswould be the multiconfiguration time-dependent Hartree83
and the semiclassical frozen Gaussian approach.84
ACKNOWLEDGMENTS
We thank Professor S. Mahapatra for the useful discus-sions. S.S. thanks CSIR, New Delhi, for a senior researchfellowship. We thank the Royal Society for an exchange fel-lowship.
1 M. Shapiro and P. Brumer, Principles of the Quantum Control of Molecu-lar Processes �Wiley, Canada, 2003�.
2 S. A. Rice and M. Zhao, Optical Control of Molecular Dynamics �WileyInterscience, New York, 2000�.
3 G. G. Balint-Kurti, S. Zou, and A. Brown, Adv. Chem. Phys. 138, 43�2008�.
4 J. Werschnik and E. K. U. Gross, J. Phys. B 40, R175 �2007�.5 P. Gross, H. Singh, H. Rabitz, K. Mease, and G. M. Huang, Phys. Rev. A
47, 4593 �1993�.6 D. J. Tannor and S. A. Rice, J. Chem. Phys. 83, 5013 �1985�.7 A. P. Peirce, M. A. Dahleh, and H. Rabitz, Phys. Rev. A 37, 4950 �1988�.8 M. A. Dahleh, A. P. Peirce, and H. Rabitz, Phys. Rev. A 42, 1065 �1990�.9 J. Combariza, B. Just, J. Manz, and G. Paramonov, J. Phys. Chem. 95,10351 �1991�.
10 S. Sharma, P. Sharma, H. Singh, and G. G. Balint-Kurti, J. Mol. Model.15, 623 �2009�.
11 P. Nuernberger, G. Vogt, T. Brixner, and G. Gerber, Phys. Chem. Chem.
TABLE III. Results for direct photodissociation of Fe–CO bond in CO-heme compounds from different initial vibrational states ��m ,n�� using apulse duration of 60 000 a.u. “J” refers to the value of the cost functional;“�peak” refers to the value of amplitude of the electric field of the optimizedlaser pulse at its peak. All quantities are in a.u.
Initial state��m ,n�� �0 Dissociation J �peak
�00� 0.1 0.780 421 0.474 709 1.422�10−2
0.01 0.965 850 0.893 654 2.495�10−2
0.001 0.997 627 0.986 167 2.730�10−2
�10� 0.1 0.717 224 0.482 905 1.411�10−2
0.01 0.961 805 0.881 369 2.261�10−2
0.001 0.997 690 0.982 535 2.765�10−2
�01� 0.1 0.751 181 0.489 159 1.469�10−2
0.01 0.979 079 0.881 184 2.704�10−2
0.001 0.995 957 0.986 647 2.736�10−2
�11� 0.1 0.636 237 0.385 028 1.428�10−2
0.01 0.977 531 0.897 016 2.670�10−2
0.001 0.997 453 0.982 766 2.751�10−2
174103-10 Sharma et al. J. Chem. Phys. 133, 174103 �2010�

Phys. 9, 2470 �2007�.12 C. Winterfeldt, C. Spielmann, and G. Gerber, Rev. Mod. Phys. 80, 117
�2008�.13 T. Hornung, R. Meier, and M. Motzkus, Chem. Phys. Lett. 326, 445
�2000�.14 T. Hornung, R. Meier, D. Zeidler, K. Kompa, D. Proch, and M. Motzkus,
Appl. Phys. B: Lasers Opt. 71, 277 �2000�.15 C. Daniel, J. Full, L. González, C. Lupulescu, J. Manz, A. Merli, T.
Vajda, and L. Wöste, Science 299, 536 �2003�.16 M. Dantus and V. V. Lozovoy, Chem. Rev. �Washington, D.C.� 104, 1813
�2004�.17 V. I. Prokhorenko, A. M. Nagy, S. A. Waschuk, L. S. Brown, R. R. Birge,
and R. J. D. Miller, Science 313, 1257 �2006�.18 J. Herek, W. Wohllehen, R. Cogdell, D. Zeidler, and M. Motzkus, Nature
�London� 417, 533 �2002�.19 J. Savolainen, R. Fanciulli, N. Dijkhuizen, A. L. Moore, J. Hauer, T.
Buckup, M. Motzkus, and J. L. Herek, Proc. Natl. Acad. Sci. U.S.A. 105,7641 �2008�.
20 D. G. Kuroda, C. P. Singh, Z. Peng, and V. D. Kleiman, Science 326, 263�2009�.
21 D. Goswami, Phys. Rep. 374, 385 �2003�.22 A. M. Weiner, Rev. Sci. Instrum. 71, 1929 �2000�.23 I. Znakovskaya, P. von den Hoff, S. Zherebtsov, A. Wirth, O. Herrwerth,
M. J. J. Vrakking, R. de Vivie-Riedle, and M. F. Kling, Phys. Rev. Lett.103, 103002 �2009�.
24 D. B. Strasfeld, S.-H. Shim, and M. T. Zanni, Adv. Chem. Phys. 141, 1�2009�.
25 C. Middleton, D. Strasfeld, and M. Zanni, Opt. Express 17, 14526�2009�.
26 F. G. Parak and G. U. Nienhaus, ChemPhysChem 3, 249 �2002�.27 A. Dreuw, B. D. Dunietz, and M. Head-Gordon, J. Am. Chem. Soc. 124,
12070 �2002�.28 J. N. Harvey, J. Am. Chem. Soc. 122, 12401 �2000�.29 N. Strickland, A. J. Mulholland, and J. N. Harvey, Biophys. J. 90, L27
�2006�.30 N. Strickland and J. N. Harvey, J. Phys. Chem. B 111, 841 �2007�.31 A. Sato, Y. Gao, T. Kitagawa, and Y. Mizutani, Proc. Natl. Acad. Sci.
U.S.A. 104, 9627 �2007�.32 The Smallest Biomolecules: Diatomics and Their Interactions with Heme
Proteins, edited by A. Ghosh �Elsevier B.V., Italy, 2008�.33 L. Rothberg, T. M. Jedju, and R. H. Austint, Biophys. J. 57, 369 �1990�.34 D. E. Sagnella, J. E. Straub, T. A. Jackson, M. Lim, and P. A. Annrud,
Proc. Natl. Acad. Sci. U.S.A. 96, 14324 �1999�.35 J. Helbing, L. Bonacina, R. Pietri, J. Bredenbeck, P. Hamm, F. van
Mourik, F. Chaussard, A. Gonzalez-Gonzalez, M. Chergui, and C.Ramos-Alvarez, Biophys. J. 87, 1881 �2004�.
36 J. C. Kendrew and M. F. Perutz, Annu. Rev. Biochem. 26, 327 �1957�.37 T.-Y. Teng, V. Srajer, and K. Moffat, Nat. Struct. Biol. 1, 701 �1994�.38 V. Šrajer, Z. Ren, T. Y. Teng, M. Schmidt, T. Ursby, D. Bourgeois, C.
Pradervand, W. Schildkamp, M. Wulff, and K. Moffat, Biochemistry 40,13802 �2001�.
39 D. Bourgeois, B. Vallone, F. Schotte, A. Arcovito, A. E. Miele, G. Sciara,M. Wulff, P. Anfinrud, and M. Brunori, Proc. Natl. Acad. Sci. U.S.A.100, 8704 �2003�.
40 J. E. Straub and M. Karplus, Chem. Phys. 158, 221 �1991�.41 J. Meller and R. Elber, Biophys. J. 74, 789 �1998�.42 C. Bossa, A. Amadei, I. Daidone, M. Anselmi, B. Vallone, M. Brunori,
and A. D. Nola, Biophys. J. 89, 465 �2005�.43 M. Devereux and M. Meuwly, J. Phys. Chem. B 113, 13061 �2009�.44 T. Polack, J. P. Ogilvie, S. Franzen, M. H. Vos, M. Joffre, J.-L. Martin,
and A. Alexandrou, Phys. Rev. Lett. 93, 018102 �2004�.45 T. G. Spiro and I. H. Wasbotten, J. Inorg. Biochem. 99, 34 �2005�.46 M. Anselmi, M. Aschi, A. D. Nola, and A. Amadeiz, Biophys. J. 92,
3442 �2007�.
47 M. M. Asimov, R. M. Asimov, and A. N. Rubinov, J. Appl. Spectrosc.72, 454 �2005�.
48 C. Ventalon, J. M. Fraser, M. H. Vos, A. Alexandrou, J.-L. Martin, andM. Joffre, Proc. Natl. Acad. Sci. U.S.A. 101, 13216 �2004�.
49 T. Witte, T. Hornung, L. Windhorn, D. Proch, R. de Vivie-Riedle, and M.Motzkus, J. Chem. Phys. 118, 2021 �2003�.
50 O. Kühn, Chem. Phys. Lett. 402, 48 �2005�.51 C. Meier and M. C. Heitz, J. Chem. Phys. 123, 044504 �2005�.52 H. Singh, S. Sharma, P. Kumar, J. Harvey, and G. G. Balint-Kurti, Lect.
Notes Comput. Sci. 5102, 387 �2008�.53 C. Gollub, B. M. R. Korff, K. L. Kompa, and R. de Vivie-Riedle, Phys.
Chem. Chem. Phys. 9, 369 �2007�.54 Y. Zhao and O. Kühn, Chem. Phys. Lett. 302, 7 �1999�.55 S. Sharma, H. Singh, and G. G. Balint-Kurti, J. Chem. Phys. 132,
064108 �2010�.56 G. S. Kachalova, A. N. Popov, and H. D. Bartunik, Science 284, 473
�1999�.57 A. D. Becke, Phys. Rev. A 38, 3098 �1988�.58 A. D. Becke, J. Chem. Phys. 98, 5648 �1993�.59 W. Kohn, A. D. Becke, and R. G. Parr, J. Phys. Chem. 100, 12974
�1996�.60 A. J. H. Wachters, J. Chem. Phys. 52, 1033 �1970�.61 P. J. Hay, J. Chem. Phys. 66, 4377 �1977�.62 K. Raghavachari and G. W. Trucks, J. Chem. Phys. 91, 1062 �1989�.63 M. J. Frisch, J. A. Pople, and J. S. Binkley, J. Chem. Phys. 80, 3265
�1984�.64 R. Ditchfield, W. J. Hehre, and J. A. Pople, J. Chem. Phys. 54, 724
�1971�.65 M. J. Frisch, G. W. Trucks, H. B. Schlegel et al., GAUSSIAN 03, Revision
C.02, Gaussian, Inc., Wallingford, CT, 2004.66 K. Ahmed, G. G. Balint-Kurti, and C. M. Western, J. Chem. Phys. 121,
10041 �2004�.67 M. Feit and J. Fleck, Jr., J. Chem. Phys. 78, 301 �1983�.68 S. Mahapatra and N. Sathyamurthy, J. Chem. Soc., Faraday Trans. 93,
773 �1997�.69 P. Gross, D. Neuhauser, and H. Rabitz, J. Chem. Phys. 96, 2834 �1992�.70 Q. Ren, G. G. Balint-Kurti, F. Manby, M. Artamonov, T. Ho, and H.
Rabitz, J. Chem. Phys. 124, 014111 �2006�.71 D. E. Goldberg, Genetic Algorithm in Search, Optimization and Machine
Learning �Addison-Wesley, Reading, MA, 1989�.72 K. Krishnakumar, in Intelligent Control and Adaptive Systems, edited by
G. Rodriguez �SPIE, Bellingham, WA, 1989�, Vol. 1196, pp. 289–296.73 D. L. Carroll, Developments in Theoretical and Applied Mechanics �Uni-
versity of Alabama, Tuscaloosa, AL, 1996�, p. 411.74 G. Yang, L. Reinstein, S. Pai, Z. Xu, and D. L. Carroll, Med. Phys. 25,
2308 �1998�.75 D. L. Carroll, AIAA J. 34, 338 �1996�.76 C. C. Marston and G. G. Balint-Kurti, J. Chem. Phys. 91, 3571 �1989�.77 C. L. W. G. G. Balint-Kurti and C. C. Marston, Comput. Phys. Commun.
67, 285 �1991�.78 S. Zou, Q. Ren, G. G. Balint-Kurti, and F. Manby, Phys. Rev. Lett. 96,
243003 �2006�.79 T. Cheng and A. Brown, J. Chem. Phys. 124, 034111 �2006�.80 A. D. Pace, A. Cupane, M. Leone, E. Vitrano, and L. Cordone, Biophys.
J. 63, 475 �1992�.81 Molecular Dynamics and Spectroscopy by Stimulated Emission Pumping,
Advanced Series in Physical Chemistry Vol. 4, edited by H.-L. Dai and R.W. Field �World Scientific, Hackensack, NJ, 1995�.
82 C. Kittrell, E. Abramson, J. L. Kinsey, S. A. McDonald, D. E. Reisner, R.W. Field, and D. H. Katayama, J. Chem. Phys. 75, 2056 �1981�.
83 M. H. Beck, A. Jäckle, G. A. Worth, and H. D. Meyer, Phys. Rep. 324,1 �2000�.
84 A. Kondorskiy and H. Nakamura, Phys. Rev. A 77, 043407 �2008�.
174103-11 Laser pulse to control dissociation of Fe–CO J. Chem. Phys. 133, 174103 �2010�





























