Embed Size (px)
Citation preview

Clemson UniversityTigerPrints
All Dissertations Dissertations
8-2019
Developing Iodoarene Derivatives for UsefulOxidative Transformations and Biodegradable X-ray MaterialsTimothy R. LexClemson University, [email protected]
Follow this and additional works at: https://tigerprints.clemson.edu/all_dissertations
This Dissertation is brought to you for free and open access by the Dissertations at TigerPrints. It has been accepted for inclusion in All Dissertations byan authorized administrator of TigerPrints. For more information, please contact [email protected].
Recommended CitationLex, Timothy R., "Developing Iodoarene Derivatives for Useful Oxidative Transformations and Biodegradable X-ray Materials"(2019). All Dissertations. 2460.https://tigerprints.clemson.edu/all_dissertations/2460

DEVELOPING IODOARENE DERIVATIVES FOR USEFUL OXIDATIVE TRANSFORMATIONS AND BIODEGRADABLE X-RAY MATERIALS
A Dissertation Presented to
the Graduate School of Clemson University
In Partial Fulfillment of the Requirements for the Degree
Doctor of Philosophy Chemistry
by Timothy Robert Lex
August 2019
Accepted by: Dr. Daniel C. Whitehead, Committee Chair
Dr. Brian N. Dominy Dr. Sourav Saha
Dr. Modi Wetzler

ii
ABSTRACT
This thesis discloses two distinct applications of synthesized iodoarene-containing
compounds: as hypervalent iodine (HI) organocatalysts and as biodegradable X-ray
contrasting agents. This thesis also details a short chapter regarding the synthesis of
rarely accessible chemical moieties: diazacyclobutenes and 2-imidothioimidates.
Recently, organohypervalent iodine compounds have prevailed as valuable
synthetic reagents. These reagents have the ability to efficiently perform transition
metal-like transformations while possessing environmentally friendly properties.
However, a persistent obstacle in utilizing hypervalent iodine (HI) reagents has been
promoting synthetic reactions with high reactivities and enantioselectivities. To this fact,
it is necessary to further improve upon the field of hypervalent iodine chemistry,
especially in the context of the development of new, modular, and tunable catalyst
scaffolds.
The first chapter of this thesis reviews the background and history of hypervalent
iodine compounds. The second chapter describes the progress that has been made in the
field of catalysis, with emphasis on asymmetric organocatalysis. The third chapter details
the advances that were made to design, develop, and understand the reactivity of
iodoarene-containing catalysts in hypervalent iodine(III)-mediated transformations.
Furthermore, the third chapter provides insight on merging the fields of hypervalent
iodine chemistry with organocatalysis and asymmetric synthesis to generate chiral
catalysts that can be used in valuable organic transformations. These organocatalysts

iii
have the potential to be broadly applicable and to impact synthetically useful
transformations by imparting high yields and stereoselectivities.
Synthetic materials have proven to have a key role in the medical field. In
particular, polymeric materials have exhibited significance as contrast imaging agents
due to their inherent biodegradable and biocompatible profile. The fourth chapter of this
thesis describes the efficient synthesis, characterization, and X-ray evaluation of several
iodoarene-containing polyesters.
Synthetic chemists are continuously searching for novel and efficient bond-
forming methods to produce new chemical species. The final chapter of this thesis
describes a new strategy to generate unique chemical motifs (diazacyclobutenes and 2-
imidothioimidates) via a formal [2+2] cycloaddition between electron rich alkynes and
azodicarbonyl reagents.

iv
Dedicated to my incredible parents, Kathy “Momma Dukes” and Carmen “Knooch” Lex, for being the best role models I could have ever asked for.

v
ACKNOWLEDGMENTS
I would like to express my sincere gratitude to all who have helped me throughout
my journey in graduate school. Without the help, support and encouragement of several
people, this doctoral thesis would not have been possible.
First, I would like to thank my research advisor, Professor Daniel C. Whitehead. Dan
is one of the most intelligent, supportive, and witty persons I have ever met. I have learned a
great deal from you, and would not be the scientist I am today without your guidance and
advice. I am especially thankful for the intellectual conversations we have had, which always
seemed to spark new ideas, or prompt new avenues to overcome experimental shortcomings.
I will always cherish the group outings and cook-outs, as well as the weekly dose of music
jams we have shared throughout the years. I am also grateful to the members of my
committee: Dr. Brian N. Dominy , Dr. Sourav Saha, and Dr. Modi Wetzler. Thank you for
your dedication and mentorship during my time at Clemson University.
I would like to thank past and present group members of the Whitehead group, as
well as other Clemson graduate students, who have made graduate school the best it could be:
Heeren Gordhan, Beau Brummel, Maria Swasy, Daniel Willett, Timmy Thiounn, McKenzie
Campbell, Brad Stadelman, Kerrick Rees, Anthony Santilli, Chandima Narangoda, Eddie “on
the weekends” Hoegg, Sam Rackley, Alfredo Picado, Soham Panda, Mohamed Fathy Attia,
and Brock Miller. I will always cherish the memories we have made inside and outside of
the lab.
Lastly, I want to thank my incredible family, especially my parents, Kathy and
Carmen, my sister, Steph, my brothers Carmen and Chris, and my grandmother “Grams”,
who have helped me through the tough times. I love you all and can’t wait to see you soon!

vi
TABLE OF CONTENTS
Page
TITLE PAGE ................................................................................................................... i
ABSTRACT ..................................................................................................................... ii
DEDICATION ................................................................................................................ iv
ACKNOWLEDGMENTS ................................................................................................ v
LIST OF TABLES ........................................................................................................ viii
LIST OF FIGURES ........................................................................................................ ix
LIST OF SCHEMES ..................................................................................................... xii
CHAPTER
1 GENERAL ASPECTS OF HYPERVALENT IODINE COMPOUNDS ...................... 1 1.1 HYPERVALENCY ................................................................................. 1 1.2 IODINE AS A HYPERVALENT ELEMENT ........................................ 2 1.3 HYPERVALENT IODINE(III) STRUCTURAL FEATURES ............... 6 1.4 REACTIVITY OF HYPERVALENT IODINE COMPOUNDS ............ 8 1.5 REFERENCES ...................................................................................... 16
2 SYNTHETIC UTILITY OF ASYMMETRIC PEPTIDE-BASED ORGANOCATALYSTS.................................................................................................................................. 21
2.1 INTRODUCTION TO CATALYSIS .................................................... 21 2.2 HOMOGENEOUS AND HETEROGENEOUS CATALYSTS ........... 25 2.3 GENERAL ASPECTS OF ORGANOCATALYSIS ............................ 26 2.4 THE IMPORTANCE OF ASYMMETRIC SYNTHESIS .................... 29 2.5 ASYMMETRIC ORGANOCATALYSTS BASED ON ACTIVATION
MODE .................................................................................................... 30 2.6 ASYMMETRIC ORGANOCATALYSIS FOR 𝛼-CARBONYL
FUNCTIONALIZATIONS .................................................................... 32 2.7 PEPTIDES ............................................................................................. 37 2.8 LICENSE AGREEMENT TO USE FIGURE 2.4 ................................ 45 2.9 REFERENCES ...................................................................................... 46
3 IODOARENE-CONTAINING ORGANOCATALYSTS FOR HYPERVALENT IODINE(III) OXIDATIVE TRANSFORMATIONS .................................................. 55
3.1 HYPERVALENT IODINE ORGANOCATALYSIS ............................ 55

vii
Table of Contents (Continued) Page
3.2 GENERAL REMARKS ON OXIDATIONS ........................................ 56 3.3 CHIRAL HYPERVALENT IODINE(III) REAGENTS FOR
ENANTIOSELECTIVE OXIDATIVE TRANSFORMATIONS ......... 56 3.31 ASYMMETRIC OXIDATION OF SULFIDES TO SULFOXIDES
.................................................................................................... 57 3.32 ASYMMETRIC OXIDATIVE DEAROMATIZATION .......... 59 3.33 ALPHA FUNCTIONALIZATION OF CARBONYLS ............ 65
3.4 IODOARENE-CONTAINING ORGANOCATALYSTS FOR HYPERVALENT IODINE(III) OXIDATIVE TRANSFORMATIONS .. ................................................................................................................ 77
3.5 FUTURE WORK FOR PEPTIDE-BASED HYPERVALENT IODINE(III) CATALYSTS ................................................................. 105
3.6 MACROCYCLES AS CHIRAL HYPERVALENT IODINE(III) CATALYSTS ...................................................................................... 112
3.7 CONCLUSIONS ................................................................................. 117 3.8 EXPERIMENTAL ............................................................................... 119 3.9 REFERENCES .................................................................................... 231
4 IODOARENE-CONTAINING BIODEGRADABLE X-RAY MATERIALS ............ 239 4.1 INTRODUCTION ............................................................................... 239 4.2 MEDICAL IMAGING AND X-RAYCONTRAST AGENTS ........... 240 4.3 DESIGNING BIODEGRADABLE X-RAY CONTRAST AGENT .. 242 4.4 CONCLUSIONS ................................................................................. 250 4.5 FUTURE WORK ................................................................................. 251 4.6 EXPERIMENTAL SECTION ............................................................. 253 4.7 REFERENCES .................................................................................... 268
5 SYNTHESIS OF DIAZACYCLOBUTENES VIA A FORMAL [2 + 2] CYCLOADDITION ................................................................................................ 270
5.1 PREFACE ............................................................................................ 270 5.2 INTRODUCTION ............................................................................... 270 5.3 FUTURE WORK ................................................................................. 278
5.4 REFERENCES .................................................................................... 280 5.5 EXPERIMENTAL ............................................................................... 282

viii
LIST OF TABLES
Table Page
1.1 Oxidative ⍺-oxytosylation of propiophenone at 48 hours and room temperature ............................................................................................ 83
1.2 Oxidative ⍺-oxytosylation of propiophenone at 4 hours and room temperature ............................................................................................ 84
1.3 Oxidative ⍺-oxytosylation of propiophenone at 24 hours and 50 ℃ .... 85
1.4 Oxidative ⍺-oxytosylation of propiophenone at 1 hour and 50 ℃ ........ 86
1.5 Structural analogs of lead Catalyst 11 for the Oxidative ⍺-oxytosylation of propiophenone at 4 hours and room temperature .............................. 88
1.6 Oxidative cyclization of 5-oxo-5-phenylvaleric acid at 24 hours and 50 ℃................................................................................................................ 90
1.7 Oxidative ⍺-oxytosylation of propiophenone derivatives ..................... 91
1.8 Oxidative ⍺-oxytosylation of propiophenone at 24 hours and room temperature catalyzed by Cbz-protected amino methyl ester hypervalent iodine(III) catalysts ................................................................................ 97
1.9 Catalytic ⍺-oxytosylation of propiophenone at 4 hours and room temperature ............................................................................................ 98
1.10 Oxidative ⍺-oxytosylation of propiophenone at 24 hours and room temperature using aniline catalysts ...................................................... 104
1.11 Oxidative ⍺-oxytosylation of propiophenone at 24 hours and 50 ℃ using aniline catalysts .................................................................................... 105

ix
LIST OF FIGURES
Figure Page
1.1 Examples of iodine compounds with different oxidation states .............. 2
1.2 Martin-Arduengo Nomenclature .............................................................. 3
1.3 Notable hypervalent iodine(III) and iodine(V) reagents .......................... 5
1.4 Geometric structure of aryl-l3-iodanes .................................................... 7
1.5 Molecular orbitals to describe the 3c-4e bond in hypervalent iodine(III) compounds ............................................................................................... 7
1.6 Leaving group abilities of various nucleofuges ..................................... 14
2.1 Simplified catalytic cycle ....................................................................... 22
2.2 Reaction coordinate diagram of a catalyzed versus uncatalyzed reaction . ................................................................................................................ 24
2.3 Examples of heterogeneous and homogeneous catalysts ....................... 26
2.4 The amount of publications pertaining to organocatalysts from 1968-2008................................................................................................................ 27
2.5 Select examples demonstrating how chirality affects chemical properties and reactivities ....................................................................................... 29
3.1 Chiral sulfoxide containing drugs .......................................................... 57
3.2 Important pharmaceutical agents that contain an 𝛼-functionalized carbonyl moiety .................................................................................................... 65
3.3 Select examples of chiral hypervalent iodine(I) precatalyst scaffolds ... 74
3.4 Envisioned peptide-based catalytic scaffolds that contain an iodoarene active site at the N-terminus .................................................................. 78

x
List of Figures (Continued)
Figure Page
3.5 N-butyl-iodobenzamide catalysts with corresponding amidation yields ................................................................................................................... 80
3.6 1H relative rate study of the oxidation of Catalysts 11, 8, and 4 to their iodine(III) state ....................................................................................... 89
3.7 Utilizing Fmoc-Solid-Phase-Peptide-Synthesis to incorporate iodoarene active sites into the side chain of peptide scaffolds ............................... 93
3.8 Cbz-protected amino methyl ester catalysts that contain an iodoarene active site ............................................................................................... 95
3.9 Incorporation of Fmoc-Solid-Phase-Peptide-Synthesis compatible catalyst 6l into a peptide sequence ...................................................................... 99
3.10 Hypothetical peptides that contain an iodoarene within the backbone of the scaffold ................................................................................................. 100
3.11 Iodoarene containing aniline derivatives that function as hypervalent iodine(III) catalysts .............................................................................. 101
3.12 Incorporating the iodoarene active site anywhere within the peptide scaffold ................................................................................................. 107
3.13 Illustrating the possible enol geometries of propiophenone in acidic conditions ............................................................................................. 108
3.14 Enol/enolate equivalents of propiophenone as a way to control enol geometry .............................................................................................. 111
3.15 Generic iodoarene-containing macrocycles ......................................... 112
3.16 Structure, yield, and enantioselectivity of cyclic and acyclic catalysts in the oxidative 𝛼-oxytosylation of propiophenone at 24 hours and room temperature .......................................................................................... 116
3.17 Plausible iodoarene-containing macrocyclic derivatives that can be synthesized ........................................................................................... 116

xi
List of Figures (Continued)
Figure Page
4.1 Utilizing iodinated monomers for X-ray enhancements ...................... 240 List of Figures (Continued)
4.2 [A] 1H NMR spectral overlay of (1a) lactide, (1b) aryl-iodo lactide (iLA), (1c) poly(lactic) acid (PLA), and (1d) aryl-iodo poly(lactic) acid (iPLA). [B] FTIR characterization of the aryl-iodo lactide (iLA), poly(lactic) acid(PLA), and the aryl-iodo poly(lactic)acid (iPLA ................................. 245
4.3 (a) Relative X-ray intensity of iodinated polymers compared to non-iodoinated PLA. (b)Relative X-ray intensity of iodinated and non-iodinated PLA copolymer with varying amounts ................................ 247
4.4 Quantitative measurements of relative X-ray intensity. (a) Relative X-ray intensity of iodinated poly(lactic) acid (iPLA) synthesized using various reaction times, in comparison to iodinated polycaprolactone (iPLC). (b) Depiction of polymeric pellets (10 mg) composed of poly(lactic) acid (PLA), iodinated polycaprolactone (iPLC), and iodinated poly(lactic) acid (iPLA) copolymers synthesized using 6, 12, and 24-hour reaction times. (c) In vitro imaging of iPLA powder through different depth of chickentissue .................................................................................................... 249
4.5 Degradation of iPLA pellets over time (days) which was incubated into PBS (pH 7.4) at 37 ℃ .......................................................................... 250
4.6 Biomedical monomers as drug delivery platforms .............................. 252
5.1 Historical examples of diazacyclobutene motifs ................................. 272
5.2 Substrate scope in a [2 + 2] cycloaddition to diazacyclobutene motifs ..... .............................................................................................................. 274
5.3 Substrate scope in a [2 + 2] cycloaddition to 2-imidothioimidate motifs .. .............................................................................................................. 277

xii
LIST OF SCHEMES
Scheme Page
1.1 General structure and oxidative pathways of iodine species ................... 4
1.2 Comparing reactivities of hypervalent iodine species to transition metals .................................................................................................................. 9
1.3 Generic ligand coupling reaction in hypervalent iodine(III) reagents ... 11
1.4 Typical reactivity of electrophilic hypervalent iodine(III) species ........ 13
2.1 Cinchona alkaloids as chiral catalysts from chiral mandelonitriles ....... 32
2.2 A 1960 enantioselective ketene methanolysis by Pracejus .................... 32
2.3 Intramolecular aldol reactions catalyzed by proline .............................. 33
2.4 Intermolecular aldolization using a proline chiral catalyst .................... 34
2.5 Enamine catalysis in natural product synthesis ..................................... 34
2.6 Various asymmetric 𝛼-functionalizations of carbonyls mediated by proline catalysts .................................................................................................. 36
2.7 Early examples of peptide-based asymmetric catalysts in organic synthesis................................................................................................................ 38
2.8 Direct aldolization using peptide-based chiral catalysts ........................ 39
2.9 Kinetic resolution of hydroxyamide ...................................................... 40
2.10 Enantioselective Baylis-Hillman reactions with peptide catalysts ........ 41
2.11 Alcohol cross-coupling for kinetic resolution of diols using peptide catalysts .................................................................................................. 42
2.12 Cinchona alkaloid PTCs for asymmetric ⍺-amino acid derivatives ...... 43

xiii
List of Schemes (Continued)
Scheme Page
3.1 Enantioselective oxidations of sulfides to sulfoxides using hypervalent iodine(III) reagents ................................................................................. 59
3.2 Kita’s first asymmetric dearomatizing spirolactonizations using a hypervalent iodine(III) reagent .............................................................. 60
3.3 Proposed mechanistic pathways for the oxidation of phenols via hypervalent iodine(III)-reagents ............................................................ 61
3.4 Asymmetric dearomatizing spirolactonizations using hypervalent iodine(III) reagents ................................................................................. 63
3.5 Enantioselective hydroxylations using hypervalent iodine(III) reagents ... ................................................................................................................ 64
3.6 Typical reactivity to functionalize ketones via hypervalent iodine(III) “umpolung” reactivity ............................................................................ 66
3.7 Asymmetric ⍺-arylation of indanone derivatives via hypervalent iodine(III) reagents ................................................................................ 68
3.8 Hypervalent iodine(III)-mediated asymmetric oxidative cycloetherification................................................................................................................ 68
3.9 Proposed reaction pathway for in-situ 𝛼-oxytosylation of ketones ....... 69
3.10 Putative mechanistic pathways for the ⍺-oxytosylation of propiopheneone mediated by a hypervalent iodine(III) reagent ....................................... 71
3.11 Asymmetric 𝛼-oxytosylation transformations via hypervalent iodine(III) reagents .................................................................................................. 72
3.12 Legault and Badevant’s utilization of enol acetates for asymmetric catalysis .................................................................................................. 76
3.13 Standard synthesis of the N-butyl-iodobenzamide catalysts ................ 79

xiv
List of Schemes (Continued)
Scheme Page
3.14 Utilizing Fmoc-solid-phase-peptide synthesis to easily generate libraries of peptide-based hypervalent iodine(III) chiral catalysts ....................... 92
3.15 Cbz-protected amino methyl ester catalysts that contain an iodoarene active site ............................................................................................... 94
3.16 Protective group manipulation for the synthesis of Fmoc-Solid-Phase-Peptide synthesis suitable catalysts ........................................................ 98
3.17 Synthetic operations to access the aniline amide Catalyst 4e ............. 102
3.18 Proposed stereoselective effect of enol geometry in the ⍺-oxytosylation of propiophenone mediated by a hypervalent iodine(III) chiral catalyst . 109
3.19 Accessing the E and Z boronate esters of propiophenone in a highly selective fashion ................................................................................... 111
3.20 Synthetic route towards iodoarene macrocycles .................................. 114
4.1 The synthesis of aryl-iodinated biodegradable monomers .................. 243
4.2 Tin-mediated ring opening polymerization ......................................... 244
5.1 General synthetic route toward diazacyclobutenes .............................. 271
5.2 General synthetic route toward N,N-dicarbamoyl 2-iminothioimidates .............................................................................................................. 275
5.3 Synthesis of oxygen and nitrogen derived diazacyclobutenes and 2-imidoimidates. ...................................................................................... 278

1
CHAPTER ONE
GENERAL ASPECTS OF HYPERVALENT IODINE (HI) COMPOUNDS
1.1 HYPERVALENCY
In 1916, Gilbert N. Lewis disclosed a critical scientific concept known as the ‘rule
of eight’ or as later named, the ‘octet rule’.1, 2 This rule states that central atoms are more
stable when their valence shell is filled with eight electrons, thereby attaining a similar
electron configuration as the noble gases. This rule-of-thumb, heuristic device serves to
help predict the position, as well as the bonding, of the atoms and lone pair electrons that
make up a molecule.3 Although this concept has been fundamental in understanding
molecular bonding, it is not directly compatible with molecules that contain atoms with
expanded octets.4-9
In 1969, Jeremy I. Musher coined the term ‘hypervalent’ to describe the ability of
a central, non-metallic atom confined within a molecule to expand its valence shell
beyond the limits of the octet rule.10, 11 Hypervalent molecules are often assembled from
elements within groups 15-18 that can accommodate the expanded octet within its
valence shell.12, 13 One element that has the capacity to formally break the octet rule, and
thereby become hypervalent, is iodine.14, 15

2
1.2 IODINE AS A HYPERVALENT ELEMENT
Iodine, isolated in 1811 by Bernard Courtois, is a chemical element of the
periodic table with an atomic number of 53. Iodine-containing reagents have intrigued
chemists due to their distinct chemical properties. In relation to the other non-radioactive
group 17 elements, iodine is the largest, heaviest, most electropositive, and most
polarizable.
Due to these unique properties, iodine is able to form stable polycoordinate,
multivalent compounds.16-18 Generally, iodine atoms form bonds with elements within
the second period of the periodic table. These kinds of bonds are relatively weak. For
instance, a carbon-iodine bond is the weakest carbon-halogen bond, with a bond
dissociation energy of approximately 57 kcal/mol.19 The formal electron configuration of
iodine is [Kr]d10s2p5, with seven electrons in its valence shell. While iodine prefers to
have an oxidation state of –1, the electronic makeup and large atomic radius permit it to
exist in various oxidation states ranging from –1 to +7 (Figure 1.1).18, 20, 21
Figure 1.1 Examples of iodine compounds with different oxidation states
I
Iodobenzene+1
IF3
Iodine trifluoride+3
OI
O
AcO OAcOAc
Dess-Martin periodinane+5
IOOO
O Na
Sodium periodate+7
HI
Hydroiodic acid-1
1a 1b 1c 1d 1e

3
Polyvalent compounds are classified by using Martin-Arduengo N-X-L
nomenclature, where N represents the number of valence electrons associated with the
central atom, X, and L signifies the number of ligands bound to the central atom (Figure
1.2).22 It should be noted that historically, iodine compounds with a +3 oxidation state
were referred to as iodinanes and iodine compounds with a +5 oxidation state were
referred to as periodinanes. However, IUPAC suggests the use of lambda notation (ln)
when considering compounds with varying valence. This notation specifies an atom with
an abnormal valence state.23, 24 While iodine has a normal valence state of 1, iodine(III)
and iodine(V) compounds contain a non-standard valence, and are respectively named as
l3-iodanes and l5-iodanes.
The large range of possible oxidation states allows for iodine to form up to 7
chemical bonds. These bonds are typically constructed through oxidations and ligand
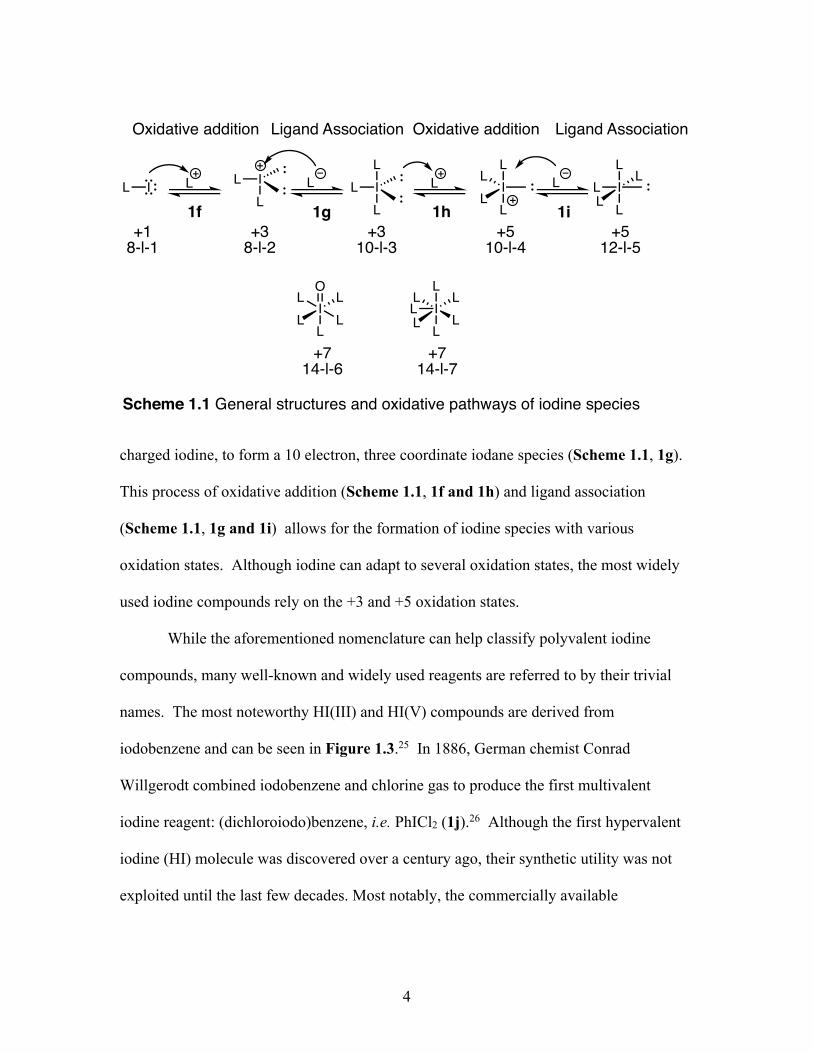
associations (Scheme 1.1).23 In short, the oxidative addition occurs when an iodine(I)
compound utilizes a lone pair of electrons to attack an electrophilic species (L+), which
thereby leaves a positive charge on the central iodine atom (Scheme 1.1, 1f). The ligand
association can then take place, whereby a nucleophilic reagent (L-) attacks a positively
[N-X-L]
Number of valence electrons
Central atom
Number of ligands bound to central atom
Figure 1.2 Martin-Arduengo Nomenclature
4
charged iodine, to form a 10 electron, three coordinate iodane species (Scheme 1.1, 1g).
This process of oxidative addition (Scheme 1.1, 1f and 1h) and ligand association
(Scheme 1.1, 1g and 1i) allows for the formation of iodine species with various
oxidation states. Although iodine can adapt to several oxidation states, the most widely
used iodine compounds rely on the +3 and +5 oxidation states.
While the aforementioned nomenclature can help classify polyvalent iodine
compounds, many well-known and widely used reagents are referred to by their trivial
names. The most noteworthy HI(III) and HI(V) compounds are derived from
iodobenzene and can be seen in Figure 1.3.25 In 1886, German chemist Conrad
Willgerodt combined iodobenzene and chlorine gas to produce the first multivalent
iodine reagent: (dichloroiodo)benzene, i.e. PhICl2 (1j).26 Although the first hypervalent
iodine (HI) molecule was discovered over a century ago, their synthetic utility was not
exploited until the last few decades. Most notably, the commercially available
L I L IL
L L IL
LL
L IL
LLL
L IL
L
LL
L
Scheme 1.1 General structures and oxidative pathways of iodine species
+18-l-1
+38-l-2
+310-l-3
+510-l-4
+512-l-5
Oxidative addition Ligand Association
IL
O
LL
LIL
L
LL
L
+714-l-6
+714-l-7
LL
L
Oxidative addition Ligand Association
1f 1g 1h 1i

5
aryliodine(III) carboxylates, such as phenyliodine diacetate (PIDA) (1k) and
phenyliodine bis(trifluoroacetate) (PIFA) (1l), and organosulfonates, like
[hydroxy(tosyloxy)iodo]benzene (HTIB) (1m), are commonly utilized in synthetic
chemistry as oxidants. The commercially available organoiodine(V) reagent known as
Dess-Martin periodinane (DMP) (1o), a five-membered iodine-containing heterocycle
with acetate ligands, is frequently used to oxidize primary and secondary alcohols to
aldehydes and ketones, respectively.27, 28 Extensive work has been completed by several
laboratories to synthesize these, as well as analogous, hypervalent iodine reagents.12, 16-18,
23, 29-31 While both HI(III) and HI(V) compounds are synthetically useful, the work
described herein focuses on HI(III) compounds due to their more desirable properties. In
comparison to HI(III) species, HI(V) compounds are more shock-sensitive and difficult to
Iodine(III) compounds
ICl
ClIOCOR
OCORIOTs
OH
1jWillgerodt’s reagent
R = CH3 (PIDA) 1kR = CF3 (PIFA) 1l
1mKoser’s reagent
(HTIB)
IO
1niodosobenzene
Iodine(V) compounds
1oDess-Martin periodinane
(DMP)
1p2-iodoxybenzoic acid (IBX)
1qiodoxybenzene
Figure 1.3 Notable hypervalent iodine(III) and iodine(V) reagents
OI
O
OI
O
O OH
IO2
AcO OAcOAc

6
prepare, have lower thermal stabilities, and are less soluble in many common organic
solvents.16, 25
1.3 HYPERVALENT IODINE(III) STRUCTURAL FEATURES
While the traditional definition of a hypervalent state, in which an atom breaks the
octet rule by bearing more than four pairs of electrons in its valence shell, has been
highly debated, the conventional interpretation will be applied in this work.8, 32-42 Akiba
considered the bonding within multivalent iodine species to rationalize the possibility of
expanded octets.13 He reasoned that hypervalent atoms can carry more than eight
electrons in their outermost shell by two plausible modes: (1) utilizing higher energy d-
orbitals to form hybridized dsp3 or d2sp3 orbitals or (2) utilizing orbitals other than d-
orbitals to form highly ionic orbitals. Contemporary computational research has
demonstrated that d-orbitals have little to no contribution in hypervalent bonds.13, 34, 37
The most widely accepted theory to describe hypervalent bonding is the concept of a
highly polarized bond known as a three-center, four-electron (3c-4e) bond. The 3c-4e
bond was independently introduced in 1951 by G.C. Pimentel and R. E. Rundle.38, 39
This concept can be conceptualized by reviewing the structural features of hypervalent
iodine(III) species and how it relates to molecular orbital theory.

7
The most common trivalent iodine species are the aryl-l3-iodanes (ArL1L2). As
shown in Figure 1.4, the atoms that constitute aryl-l3-iodanes share ten valence electrons
(decet structure) to form a distorted trigonal bipyramidal geometry with an overall T-
shape.12, 23 The central iodine atom is bound to two apically positioned ligands (L1 and
L2), one equatorial aryl substituent (Ar), and two equatorial lone pairs of electrons. The
iodine and aryl group overlap orbitals to form a conventional two-center, two-electron
(2c-2e) covalent, carbon-iodine𝜎-bond with 5sp2 hybridization. A highly polarized 3c-
4e bond is created from the interaction of a filled 5p orbital of iodine and half-filled
orbitals of each ligand. This results in three molecular orbitals: a bonding, non-bonding,
and anti-bonding orbital (Figure 1.5). Four valence electrons, two electrons from iodine
and one electron from each ligand, are shared between the three atoms (iodine and two
ligands). Consequently, this electron distribution allows for the two lower molecular
orbitals, the bonding and non-bonding orbitals, to each be filled with two electrons, while
the antibonding orbital is unfilled.
I
L1
L2
Arẟ+
ẟ-
ẟ-
Figure 1.4 Geometric structure of aryl-!3-iodanes

8
The highly polarized nature of the 3c-4e bond is invoked at the highest occupied
molecular orbital (HOMO), in this case, the non-bonding orbital, in which a node is
situated at the central iodine position. This allows for the development of charge
distributions; in particular, there is an intrinsic partial positive charge on the central
iodine atom (+1.0 charge), and a partial negative charge (-0.5 charge) on each apically
positioned ligand.23 Ligands that can better stabilize this partial negative charge (e.g.
halide, tosylate, carboxylate, hydroxyl) tend to reside at the apical positions around the
positively charged central iodine, while the least electronegative ligand, the aryl group,
occupies the equatorial position. Furthermore, the more electropositive the central atom
is the more energetically stable the polycoordinated, multivalent species is, hence aryl-l3-
iodanes are more stable than the related aryl-l3-chloranes and aryl-l3 bromanes.12, 18, 43, 44
1.4 REACTIVITY OF HYPERVALENT IODINE COMPOUNDS
•
IR R
bonding orbital
non-bonding orbital
antibonding orbital
Figure 1.5 Molecular orbitals to describe 3c-4e bond in HI(III) compounds
Ener
gy
Ψ3*
Ψ2
Ψ1

9
Iodine species with expanded octets can result in distinct and interesting chemical
structures, properties, and reactivities. With the ability to possess multiple oxidation
states, it has been suggested that hypervalent iodine species have reactivity patterns that
resemble transition metals. This is apparent when considering the quintessential
reactions involved in both transition metals and polyvalent iodine reagents: oxidative
additions, ligand exchanges, and reductive eliminations (Scheme 1.2).23
The transition metals (1r) and iodine species (in this case, iodoarene 1w) undergo
an increase in coordination number or oxidation state through an oxidative addition. An
exogeneous nucleophile (Nu) can then readily add to the central iodine through an
Oxidative addition
LnM R-L LnM Nu LnM
-L
Ligand Exchange Reductive Elimination
RNu
LnM
General reactivity of transition metal reagents
IL
LPhPh I
L IL
Ph L
Oxidative addition Ligand Association
General reactivity of hypervalent iodine reagentsAssociative pathwayfor ligand exchange
Dissociative pathwayfor ligand exchange
NuIL
LPh
INu
LPh
Nu
L
I
Nu
8-I-22b
12-I-4 (trans)1z
10-I-32c
L
R R
Nu
LINu
LPh L
12-I-4 (cis)2a
Reductive Elimination orLigand Coupling
- PhI
LNu
L
Ph
Scheme 1.2 Comparing reactivities of hypervalent iodine species to transition metals
1w 1x 1y 2d2e
1r1s 1t 1u
1v

10
associative or dissociative ligand exchange process to form the active species (Scheme
1.2, 1t and 2c). Subsequently, a reductive elimination, or as it is analogously called in
hypervalent terminology, ligand coupling, allows for the new bond to form (1v and 2e),
while also liberating the byproducts: the metal reagent (1u) or iodobenzene (2d).
Numerous experimental and computational studies have been conducted to
validate the mechanistic pathway for ligand exchange in hypervalent iodine species.12, 45-
49 The two pathways that have been probed are the associate pathway and the
dissociative pathway. The associative mechanism involves the addition of a nucleophile
to the electropositive central iodine to give a trans 12 electron, 4 ligand (12-I-4) iodate
(1z).12, 49-51 This trans intermediate is in equilibrium with the cis 12-I-4 iodate (2a),
which gives a favorable structural geometry for one of the ligands (L) to depart,
furnishing the desired 10-I-3 hypervalent iodine(III) species (2c). Conversely, the
dissociative pathway commences with the ejection of a ligand (L) to form a cationic 8-I-2
iodonium species (2b). This process typically requires a strong electrophile to assist in
the removal of the ligand.12 This species is then subjected to a nucleophilic addition at
the central iodine atom to afford the trivalent aryl-l3-iodane (2c). Due to the lack of
experimental evidence of the high energy dicoordinated (8-I-2) iodium ion (2b), in the
dissociative pathway, the associative pathway has been the more widely accepted ligand
exchange pathway of l3-iodanes.12, 23
Ligand couplings involved in hypervalent species were presented by Oae to
explain the formation of an intramolecular bond between two ligands that were bound to
the central hypervalent atom.52, 53 As depicted in Scheme 1.3, reductive eliminations, a

11
concept widely invoked in organometallic chemistry, are when the central metal atom
decreases in oxidation state and forms a new bond between the bound ligands. It should
be noted that in non-transition metal chemistry, the term ‘reductive elimination’ has also
been used in several other fashions, for example in the synthesis of alkenes.54, 55 The
aryl-l3-iodanaes can be subjected to a reaction known as ligand coupling (Scheme 1.3).
There is limited experimental evidence of the ligand coupling mechanism, but it has been
proposed that due to the configurational instability of organo-l3-iodanes, rapid
psuedorotation occurs to generate an appropriate coupling configuration (2g). This
psuedorotation is followed by a favorable and concerted coupling of the ligands (Nu and
L) to form the desired product (2i) with retention of configuration of the ligands.13, 52, 56-59
Another detail that illustrates the similarity between hypervalent iodine
complexes and transition metals is the trans influence phenomenon.60-64 This
I
Ph
L
Nu
Nu L
LNu
- PhI
Reductive Elimination
Ligand Coupling
- PhI
psuedorotation
Scheme 1.3 Generic ligand coupling reaction in hypervalent iodine(III) reagents
2g
2h
2i
I
Nu
L
Ph
2f

12
phenomenon states that most electronegative ligands bound to the central atom tend to
configure trans to each other in order to form stable complexes. The mutual influence of
the two ligands occurs when the central atom preserves the ns2 lone pair of electrons.61
In T-shaped hypervalent iodine(III) compounds, such as the generic structure shown in
Figure 1.4, the central iodine atom retains the 5s2 lone pair of electrons, and the I-L1
trans bond becomes influenced by the 𝜎-donating ability of the trans configured ligand
(L2).60 The 3c-4e bond of these hypervalent iodine(III) complexes experience a trans
influence, which is attributed to the inductive effects of the trans positioned ligands, L1
and L2. This phenomenon has been studied and utilized by Ochiai to account for the
stabilities of various HI(III) species.31, 64, 65 In metal complexes, the metal-ligand bond
(M-X) is affected by a trans oriented ligand (L), which weakens the M-X bond.66, 67 In
both metal and hypervalent species, the trans positioned substituents share a central atom,
and can influence and effect each other’s bonding abilities, stabilities, and reactivities.
While multivalent iodine reagents have comparable reactivity profiles as heavy
metal reagents (e.g. Hg(II), Pb(IV), Cr(III), Os(VIII)), the main appeal toward utilizing
hypervalent iodine compounds stems from their environmentally benign character.18, 23, 30,
68 Their less-toxic nature allows hypervalent iodine species to be employed as
sustainable alternatives to the more toxic heavy metal-based reagents, and thus
contributes to ‘greener’ replacement methods in organic synthesis.69
Although organic molecules bearing hypervalent iodine moieties were discovered
over a century ago, they were not employed in synthetically useful transformations until
the last few decades. Due to the partial positive charge around the central iodine atom

13
(cf. Figure 1.4), hypervalent iodine(III) compounds are inherently electrophilic agents.
The distinct 3c-4e bond involved in these species has been investigated, showing that the
iodine to ligand (L) bonds are longer and weaker than analogous covalent bonds, giving
important reactive characteristics to this class of compounds. Allen et al. disclosed a
study concerning the bond length in a hypervalent iodine(III) compound named
diphenyliodonium chloride (Ph2ICl), and showed that the iodine to chlorine bond length
was notably longer (3.06 Å) than the average covalent bond length (2.56 Å).70 These
unique characteristics have contributed to l3-iodane reagents to be routinely utilized as
versatile and selective oxidants and electrophiles. The strong electrophilic nature of l3-
iodanes can be observed as shown in Scheme 1.4.
The predominant reactive property of highly electrophilic HI(III) compounds is
their aptness to readily exchange with nucleophiles by means of a low energy process.
IX
XNu I
Nu
X
X
INu
X
RI
X
Nu R
Scheme 1.4 Typical reactivity of electrophilic hypervalent iodine(III) species
2j 2k
2n
2k 2o
2l
2l
2m

14
Generally, the aryl-λ3-iodane (2j) is susceptible to nucleophilic attack at the positively
charged iodine atom, which affords 2k and the newly displaced ligand 2l. An external
nucleophile (2m) is then added to afford the new Nu-R bond formation (2o).
The functional group known as phenyliodonio (PhIX), highlighted in blue within
structure 2k, is considered to be a potent nucleofuge, or leaving group that retains the
electrons from the broken bond. In fact, Ochiai referred to the phenyliodonio group as a
‘hypernucleofuge’, or an exceptional hypervalent leaving group that has a better leaving
group ability than a “super leaving group”. The phenyliodonio group is estimated to
possess a 1013 times greater leaving group ability than iodine and 106 times higher leaving
ability than triflate (Figure 1.6).12, 13, 71 Furthermore, by using Hammett substituent
constants, Mironova et al. probed the electron nature of phenyl-l3-iodanyl groups, and
Figure 1.6 Leaving group ability of various nucleofuges
Me2S+
Acetoxy (AcO)
F
Cl
CF3CO2
NO3
Br
Nucleofuge krel Nucleofuge krel
I
Mesylate (MsO)
Triflate (TfO)
p-MePh(BF4)I
Ph(BF4)I
p-ClPh(BF4)I
1.4 X 10-6
9.0 X 10-6
5.3 X 10-2
1.0
2.5
7.2
1.4 X 10
9.1 X 10
3.0 X 104
3.7 X 104
1.4 X 108
6.2 X 1013
1.2 X 1014
2.9 X 1014
Tosylate (TsO)

15
showed that they have powerful electron-withdrawing capabilities due to inductive
effects.72
This reaction (Scheme 1.4) proceeds due to the remarkable leaving and electron-
withdrawing ability of the hypervalent iodine group, which can be ascribed from the
entropic favorability of having one molecule (2k) split into three: the anionic
electrophilic ligand (2l), iodoarene (2n), and the intended product (2o). The
thermodynamic driving force of aryl hypervalent iodine(III) reactions is due to the
formation of the highly stable iodoarene by-product (2n).
Chemists have employed l3-iodanes in numerous synthetic transformations due to
their versatility and environmentally benign character. Many aryl-substituted l3-iodanes
are commercially available, and are commonly used as mild and selective reagents in
synthetic chemistry. As described above, these species have a potent electrophilic and
oxidative chemical character, and thus have been particularly impactful in a multitude of
synthetic applications including: oxidations, oxidative rearrangements and
fragmentations, cyclizations, carbon-carbon/carbon-heteroatom/heteroatom-heteroatom
bond forming reactions, and even in total syntheses of natural products.12, 18, 23, 29, 44, 68, 73
The second chapter of this thesis will focus on the utilization of asymmetric
organocatalysts in various synthetic transformations.

16
1.5 REFERENCES
1. Langmuir, I., The Structure of Atoms and the Octet Theory of Valence. Proc. Natl. Acad. Sci. U. S. A. 1919, 5, 252-259.
2 Lewis, G. N., The Atom and the Molecule. J. Am. Chem. Soc. 1916, 38, 762-785. 3. Taber, K. S., College Students' Conceptions of Chemical Stability: The
widespread adoption of a heuristic rule out of context and beyond its range of application. Int. J. Sci. Educ. 2009, 31 (10), 1333-1358.
4. Curnow, O. J., A Simple Qualitative Molecular-orbital/Valence-bond Description of the Bonding in Main Group "Hypervalent" Molecules. J. Chem. Educ. 1998, 75 (7), 910-915.
5. Jensen, W. B., The Origin of the Term "Hypervalent". J. Chem. Educ. 2006, 83 (12), 1751-1752.
6. Galbraith, J. M., On the Role of d Orbital Hybridization in the Chemistry Curriculum. J. Chem. Educ. 2007, 84 (5), 783-787.
7. Suidan, L.; Badenhoop, J. K.; Glendening, E. D.; Weinhold, F., Common Textbook and Teaching Misrepresentations of Lewis Structures. J. Chem. Educ. 1995, 72 (7), 583-586.
8. Noury, S.; Silvi, B.; Gillespie, R. J., Chemical bonding in hypervalent molecules: Is the octet rule relevant? Inorg. Chem. 2002, 41 (8), 2164-2172.
9. Cahill, P. A.; Dykstra, C. E.; Martin, J. C., The Structure and Stability of the 10-F-2 Trifluoride Ion, a Compound of a Hypervalent First Row Element. J. Am. Chem. Soc. 1985, 107 (22), 6359-6362.
10. Musher, J. I., Chemistry of Hypervalent Molecules. Angew. Chem. Int. Ed. 1969, 8 (1), 54.
11. Stirling, A., Assessing Hypervalency in Iodanes. Chem.: Eur. J. 2018, 24 (7), 1709-1713.
12. Ochiai, M., Hypervalent Iodine Chemistry. Modern Developments in Organic Synthesis: Topics in Current Chemistry. Springer: Berlin, 2003; Vol. 224, p Vol. 224 (Ed.: T. Wirth), Springer, Berlin, 2003.
13. Akiba, K., Chemistry of Hypervalent Compounds. Wiley-VCH: New York ; Chichester, 1999; 414 p.
14. Jalalian, N. Development and Applications of Hypervalent Iodine Compounds: Powerful Arylation and Oxidation Reagents. Stockholm University, Stockholm, 2012.
15. Anslyn, E. V.; Dougherty, D. A., Modern Physical Organic Chemistry. Wilsted & Talor Publishing Service: Sausalito, Calif., 2006.
16. Stang, P. J.; Zhdankin, V. V., Organic polyvalent iodine compounds. Chem. Rev. 1996, 96 (3), 1123-1178.
17. Stang, P. J., Polyvalent iodine in organic chemistry. J. Org. Chem. 2003, 68 (8), 2997-3008.
18. Varvoglis, A., Hypervalent Iodine in Organic Synthesis Academic Press: London, 1997.

17
19. Blanksby, S. J.; Ellison, G. B., Bond Dissociation Energies of Organic Molecules. Acc. Chem. Res. 2003, 36 (4), 255-263.
20. Greenwood, N. N.; Earnshaw, A., Chemistry of the Elements. 2nd ed.; Butterworth-Heinemann: Oxford ; Boston, 1997; 1340 p.
21. Falconer, W. E.; Buchler, A.; Stauffer, J. L.; Klemperer, W., Molecular Structure of XeF6 and IF7. J. Chem. Phys. 1968, 48 (1), 312.
22. Perkins, C. W.; Martin, J. C.; Arduengo, A. J.; Lau, W.; Alegria, A.; Kochi, J. K., An electrically neutral .sigma.-sulfuranyl radical from the homolysis of a perester with neighboring sulfenyl sulfur: 9-S-3 species. J. Amer. Chem. Soc. 1980, 102 (26), 7753-7759.
23. Zhdankin, V. V., Hypervalent iodine chemistry : preparation, structure and synthetic applications of polyvalent iodine compounds. John Wiley & Sons, Inc.: Chichester, West Sussex, 2014; p viii, 468 pages.
24. Powell, W. H., Treatment of variable valence in organic nomenclature (lambda convention). Pure Appl. Chem. 1984, 56 (6), 769-778.
25. Dohi, T.; Kita, Y., Hypervalent Iodine Reagents as a New Entrance to Organocatalysts. ChemComm. 2009, (16), 2073-2085.
26. Willgerodt, C., J. Prakt. Chem. 1886, 33, 154. 27. Paquette, L. A.; Burke, S. D.; Edwards, J. P.; Volmer, M.; Ghosez, L. A.,
Encyclopedia of reagents for organic synthesis. Wiley: Chichester, 1995. 28. Dess, D. B.; Martimn, J. C., Readily Accessible 12-I-5 Oxidant for the
Conversion of Primary and Secondary Alcohols to Aldehydes and Ketones. J. Org. Chem. 1983, 48 (22), 4155-4156.
29. Varvoglis, A., Chemical transformations induced by hypervalent iodine reagents. Tetrahedron 1997, 53 (4), 1179-1255.
30. Zhdankin, V. V.; Stang, P. J., Recent developments in the chemistry of polyvalent iodine compounds. Chem. Rev. 2002, 102 (7), 2523-2584.
31. Zhdankin, V. V.; Stang, P. J., Chemistry of Polyvalent Iodine. Chem. Rev. 2008, 108 (12), 5299-5358.
32. Coulson, C. A., Valence. Clarendon Press: Oxford, 1952; p vii, 338 p. 33. Pauling, L., The nature of the chemical bond, and the structure of molecules and
crystals : an introduction to modern structural chemistry. 2nd ed.; Cornell University Press: Ithaca, N.Y., 1948; p xvi, 450 p.
34. Kutzelnigg, W., Chemical Bonding in Higher Main Group Elements. Angew. Chem. Int. Ed. Engl. 1984, 23 (4), 272-295.
35. Magnusson, E., Hypercoordinate molecules of second-row elements: d functions or d orbitals? J. Amer. Chem. Soc. 1990, 112 (22), 7940-7951.
36. Coleman, W. F., Structure and Bonding in Hypervalent Iodine Compounds. J. Chem. Educ. 2010, 87 (9), 999-1000.
37. Reed, A. E.; Schleyer, P. v. R., Chemical bonding in hypervalent molecules. The dominance of ionic bonding and negative hyperconjugation over d-orbital participation. J. Am. Chem. Soc. 1990, 112 (4), 1434-1445.
38. Pimentel, G. C., The Bonding of Trihalide and Bifluoride Ions by the Molecular Orbital Method. J. Chem. Phys. 1951, 19 (4), 446-448.

18
39. Hach, R. J.; Rundle, R. E., The Structure of Tetramethylammonium Pentaiodide. J. Am. Chem. Soc. 1951, 73 (9), 4321-4324.
40. Cioslowski, J.; Mixon, S. T., Rigorous Interpretation of Electronic Wave Functions. 2. Electronic Structures of Selected Phosphorus, Sulfur, and Chlorine Fluorides and Oxides. Inorg. Chem. 1993, 32 (15), 3209-3216.
41. Molina, J. M.; Dobado, J. A., The three-center-four-electron (3c-4e) bond nature revisited. An atoms-in-molecules theory (AIM) and ELF study. Theor. Chem. Acc. 2001, 105 (4-5), 328-337.
42. Cooper, D. L., Spin-coupled Description of the Chemical Bonding to Hypercoordinate Chlorine. Theor. Chem. Acc. 2001, 105 (4-5), 323-327.
43. Amey, R. L.; Martin, J. C., Synthesis and reaction of substituted arylalkoxyiodinanes: formation of stable bromoarylalkoxy and aryldialkoxy heterocyclic derivatives of tricoordinate organoiodine(III). J. Org. Chem. 1979, 44 (11), 1779-1784.
44. Varvoglis, A., The organic chemistry of polycoordinated iodine. VCH Publishers: New York, N.Y., 1992; p xii, 414 p.
45. Moriarty, R. M.; Prakash, O., Hypervalent Iodine in Organic-Synthesis. Acc. Chem. Res. 1986, 19 (8), 244-250.
46. Yusubov, M. S.; Nemykin, V. N.; Zhdankin, V. V., Transition metal-mediated oxidations utilizing monomeric iodosyl- and iodylarene species. Tetrahedron 2010, 66 (31), 5745-5752.
47. Richter, H. W.; Cherry, B. R.; Zook, T. D.; Koser, G. F., Characterization of species present in aqueous solutions of [hydroxy(mesyloxy)iodo]benzene and [hydroxy(tosyloxy)iodo]benzene. J. Am. Chem. Soc. 1997, 119 (41), 9614-9623.
48. Ochiai, M.; Miyamoto, K.; Yokota, Y.; Suefuji, T.; Shiro, M., Synthesis, characterization, and reaction of crown ether complexes of aqua(hydroxy)(aryl)iodonium ions. Angew. Chem. Int. Ed. 2005, 44 (1), 75-78.
49. Edwards, A. J., Crystal Structure of Trichlorosulphonium(IV) Tetrachloroiodate(III). J. Chem. Soc., Dalton Trans. 1978, (12), 1723-1725.
50. Kajigaeshi, S.; Ueda, Y.; Fujisaki, S.; Kakinami, T., Halogenation Using Quaternary Ammonium Polyhalides. XV. An Effective Chlorinating Agent Benzyltrimethylammonium Tetrachloroiodate, Benzylic Chlorination of Alkylaromatic Compounds. Tetrahedron Lett. 1988, 29 (45), 5783-5786.
51. Koser, G. F.; McConville, D. B.; Rabah, G. A.; Youngs, W. J., Crystal and molecular structure of 1-chloro-1,2-benziodoxolin-3(1H)-one center dot tetra-n-butylammonium chloride. J. Chem. Crystallogr. 1995, 25 (12), 857-862.
52. Oae, S.; Uchida, Y., Ligand-coupling reactions of hypervalent species. Acc. Chem. Res. 1991, 24 (7), 202-208.
53. Finet, J.-P., Ligand Coupling Reactions with Heteroatomic Compounds. Pergamon: Oxford, 1998; p xv, 291 p.
54. Kocienski, P. J.; Lythgoe, B.; Waterhouse, I., The Influence of Chain-branching on the Steric Outcome of Some Olefin-forming Reactions. J. Chem. Soc., Perkin Trans. 1 1980, (4), 1045-1050.

19
55. Ono, N.; Tamura, R.; Hayami, J. I.; Kaji, A., Reductive elimination reaction of β-nitrosulfones via one electron transfer process. A new synthetic method for the preparation of α,β-unsaturated nitriles and esters. Tetrahedron Lett. 1978, (8), 763-764.
56. Hoffmann, R.; Howell, J. M.; Muetterties, E. L., Molecular Orbital Theory of Pentacoordinate Phosphorus. J. Am. Chem. Soc. 1972, 94 (9), 3047.
57. Martin-Santamaria, S.; Carroll, M. A.; Carroll, C. M.; Carter, C. D.; Pike, V. W.; Rzepa, H. S.; Widdowson, D. A., Fluoridation of heteroaromatic iodonium salts - experimental evidence supporting theoretical prediction of the selectivity of the process. ChemComm. 2000, (8), 649-650.
58. Ochiai, M.; Shu, T.; Nagaoka, T.; Kitagawa, Y., alpha-Vinylation of 1,3-dicarbonyl compounds with alkenyl(aryl)iodonium tetrafluoroborates: Effects of substituents on the aromatic ring and of radical inhibitors. J. Org. Chem. 1997, 62 (7), 2130-2138.
59. Koser, G. F.; Relenyi, A. G.; Kalos, A. N.; Rebrovic, L.; Wettach, R. H., One-step .alpha.-tosyloxylation of ketones with [hydroxy(tosyloxy)iodo]benzene. J. Org. Chem. 1982, 47 (12), 2487-2489.
60. Sajith, P. K.; Suresh, C. H., Quantification of the Trans Influence in Hypervalent Iodine Complexes. Inorg. Chim. Acta 2012, 51 (2), 967-977.
61. Shustorovich, E. M.; Buslaev, Y. A., Mutual influence of ligands in main group element coordination compounds. Inorg. Chem. 1976, 15 (5), 1142-1147.
62. Landis, C. R.; Firman, T. K.; Cleveland, T.; Root, D. M., What determines the shapes of transition metal hydride and alkyl complexes? Abstracts of Papers of the American Chemical Society 1996, 212, 365.
63. Landis, C. R.; Firman, T. K.; Root, D. M.; Cleveland, T., A valence bond perspective on the molecular shapes of simple metal alkyls and hydrides. J. Am. Chem. Soc. 1998, 120 (8), 1842-1854.
64. Ochiai, M.; Sueda, T.; Miyamoto, K.; Kiprof, P.; Zhdankin, V. V., trans Influences on hypervalent bonding of aryl lambda(3)-iodanes: Their stabilities and isodesmic reactions of benziodoxolones and benziodazolones. Angew. Chem. Int. Ed. 2006, 45 (48), 8203-8206.
65. Kiprof, P., The Nature of Iodine Oxygen Bonds in Hypervalent 10-I-3 Iodine Compounds. Arkivoc 2005, 19-25.
66. Rigamonti, L.; Forni, A.; Manassero, M.; Manassero, C.; Pasini, A., Cooperation between Cis and Trans Influences in cis-Pt-II(PPh3)(2) Complexes: Structural, Spectroscopic, and Computational Studies. Inorg. Chem. 2010, 49 (1), 123-135.
67. Rigamonti, L.; Rusconi, M.; Manassero, C.; Manassero, M.; Pasini, A., Quantification of cis and trans influences in [PtX(PPh3)(3)](+) complexes. A P-31 NMR study. Inorg. Chim. Acta 2010, 363 (13), 3498-3505.
68. Wirth, T.; Antonchick, A. P., Hypervalent iodine chemistry. In Topics in Current Chemistry, [Online] Springer,: Cham, 2016; pp. 1 online resource (viii, 316 pages).

20
69. Arnold, A. M.; Ulmer, A.; Gulder, T., Advances in Iodine(III)-Mediated Halogenations: A Versatile Tool to Explore New Reactivities and Selectivities. Chem. – Eur. J. 2016, 22 (26), 8728-8739.
70. Allen, F. H.; Kennard, O.; Watson, D. G.; Brammer, L.; Orpen, A. G.; Taylor, R., Tables of Bond Lengths Determined by X-Ray and Neutron-diffraction - Bond Lengths in Organic-Compounds. J. Chem. Soc., Perkin Trans. 2 1987, (12), S1-S19.
71. Okuyama, T.; Takino, T.; Sueda, T.; Ochiai, M., Solvolysis of Cyclohexenyliodonium Salt, a New Precursor for the Vinyl Cation: Remarkable Nucleofugality of the Phenyliodonio Group and Evidence for Internal Return from an Intimate Ion-Molecule Pair. J. Amer. Chem. Soc. 1995, 117 (12), 3360-3367.
72. Mironova, A. A.; Soloshonok, I. V.; Maletina, I. I.; Orda, V. V.; Yagupolskii, L. M., Electronic Nature of Substituents, Containing Polyvalent Iodine. Zh. Org. Khim. 1989, 25 (2), 306-311.
73. Silva, L. F.; Olofsson, B., Hypervalent iodine reagents in the total synthesis of natural products. Nat. Prod. Rep. 2011, 28 (10), 1722-1754.

21
CHAPTER TWO
SYNTHETIC UTILITY OF ASYMMETRIC PEPTIDE-BASED ORGANOCATALYSTS
2.1 INTRODUCTION TO CATALYSIS
Chemical synthesis is an essential method used by scientists to construct particular
chemical compounds. From medicines and herbicides to foods and dyes, chemical
synthesis is a fundamental field that impacts several branches of life. Researchers are
constantly searching for new strategies to progress the field of synthesis, and a common
area of exploration involves the use of new reagents to carry out reactions.
Although metal-based reagents continue to demonstrate their importance in the
facilitation of chemical transformations, recent efforts have been made to develop
nontoxic, metal-free synthetic methodologies; it should be noted that while metal
reagents may have drawbacks, they also possess many advantages, especially in the
context of redox chemistry. Small organic molecules, which lack a metallic component,
have become attractive alternatives as they are considered to be “greener” alternatives to
metal reagents.74 In comparison to metal species, metal-free organic molecules are
typically cheaper and more environmentally friendly, with potential to attain highly
efficient synthetic processes.
In 1836, the Swedish chemist J. J. Berzelius introduced the term ‘catalysis’.
According to Berzelius75, catalysis can be conceptualized as a situation whereby:
“…several simple or compound bodies, soluble and insoluble, have the property of exercising on other bodies an action very different from chemical affinity. By means of

22
this action they produce, in these bodies, decompositions of their elements and different recombinations of these same elements to which they remain indifferent.”
In other words, Berzelius hypothesized that chemical reactions can occur by catalytic
contact. During this time in history, the concept of reaction rate was unknown; instead
scientists believed the driving force of a reaction was due to either an ‘affinity’ force, or
as Berzelius stated, a ‘catalytic’ force.75, 76
Catalysts are compounds used in chemical transformations that speed up reactions. In
short, a catalyst forms a bond with a reactant, a chemical reaction transpires to form a
Figure 2.1 Simplified catalytic cycle

23
product, and a subsequent detachment of the unconsumed catalyst from the product
occurs (Figure 2.1).77 When the catalyst is separated from the product, it is available to
be used again in the next reaction sequence, hence the term catalytic cycle. To illustrate a
general catalytic cycle, Figure 2.1 shows a catalyst interacting with compound A and
compound B to form a complex. When bound to the catalyst, A and B react to give a
product, P. The cycle is complete when the catalyst desorbs and departs from the
product, allowing for the cycle to repeat.
The catalyst increases a reaction’s rate by lowering the activation energy that is
needed to get over the high energy activated complex, the transition state. As shown in
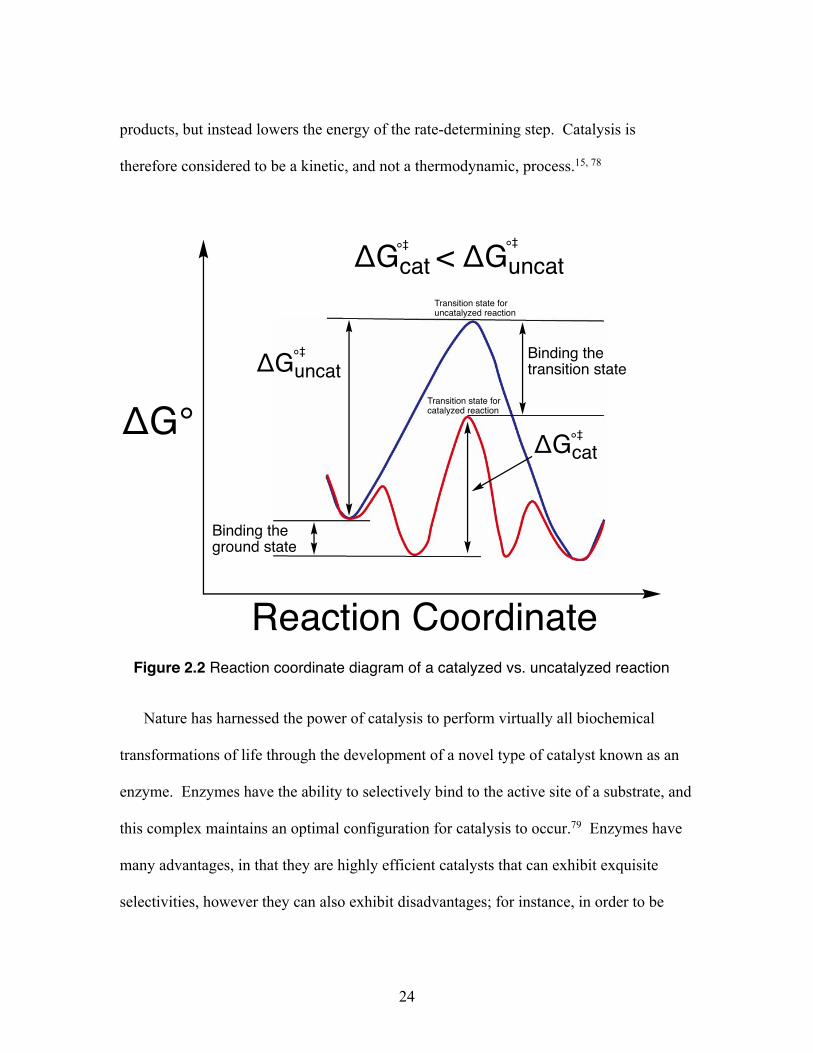
Figure 2.2, a reaction coordinate diagram can emphasize the differences between a
catalyzed (red line) versus an uncatalyzed reaction (blue line).15 In a catalytic reaction,
the energy of the substrate can be lowered when bound to a catalyst, in comparison to the
free, uncatalyzed substrate. Furthermore, the binding between the transition state and the
catalyst can be strong, allowing for a lower activation energy than the uncatalyzed route
(ΔG °‡cat < ΔG °‡
uncat). With a lower activation energy, the rate of the reaction increases.
Therefore, catalysis is accomplished when the catalyst can stabilize the transition state at
a greater extent than it can stabilize the ground state. To put it another way, a catalyst
can react with a starting material to generate a lower energy transition state (in
comparison to the noncatalyzed reaction), and as a result, less energy is needed to
overcome the activation energy barrier; this in turn, accelerates the rate of the reaction.
The catalyst does not change the position of equilibrium between the reactants and
24
products, but instead lowers the energy of the rate-determining step. Catalysis is
therefore considered to be a kinetic, and not a thermodynamic, process.15, 78
Nature has harnessed the power of catalysis to perform virtually all biochemical
transformations of life through the development of a novel type of catalyst known as an
enzyme. Enzymes have the ability to selectively bind to the active site of a substrate, and
this complex maintains an optimal configuration for catalysis to occur.79 Enzymes have
many advantages, in that they are highly efficient catalysts that can exhibit exquisite
selectivities, however they can also exhibit disadvantages; for instance, in order to be
Figure 2.2 Reaction coordinate diagram of a catalyzed vs. uncatalyzed reaction
Binding the transition state
Binding the ground state
ΔG°ΔGuncat
ΔGcat
ΔGcat < ΔGuncat
Reaction Coordinate
Transition state for catalyzed reaction
Transition state for uncatalyzed reaction
°‡
°‡
°‡ °‡

25
effective, they may require specific conditions (temperature, pH, pressure, etc.), and
while they can provide exquisite selectivities, each enzyme has a rather limited substrate
scope. Consequently, leveraging enzymatic catalysis in the laboratory can be tedious,
and it can be difficult to discover/evolve enzymes for broad applicability. To expand the
synthetic chemistry toolbox, scientists have worked toward the development and
application of new catalysts.
2.2 HOMOGENEOUS AND HETEROGENEOUS CATALYSTS
Catalysts can be categorized in many ways, but based on their state of aggregation,
the two main classes are homogeneous or heterogeneous catalysts.77 A homogeneous
catalyst is one that operates in the same phase as the reactant(s) while a heterogeneous
catalyst operates in a separate phase than the reactant(s).77, 80, 81 Ordinarily, for a reaction
to occur with a heterogeneous catalyst, the reactant must first adsorb onto the surface of
the catalyst. On the other hand, homogeneous catalysts merely have to be added to the
reaction mixture. Some of the more notable homogeneous and heterogeneous catalysts
are seen in Figure 2.3.

26
2.3 GENERAL APSECTS OF ORGANOCATALYSIS
A pivotal advancement in the field of synthetic chemistry has been the
implementation of organocatalysts, or small organic molecules that serve as catalysts.
Organocatalysis has been known and described for over a century, yet this field has only
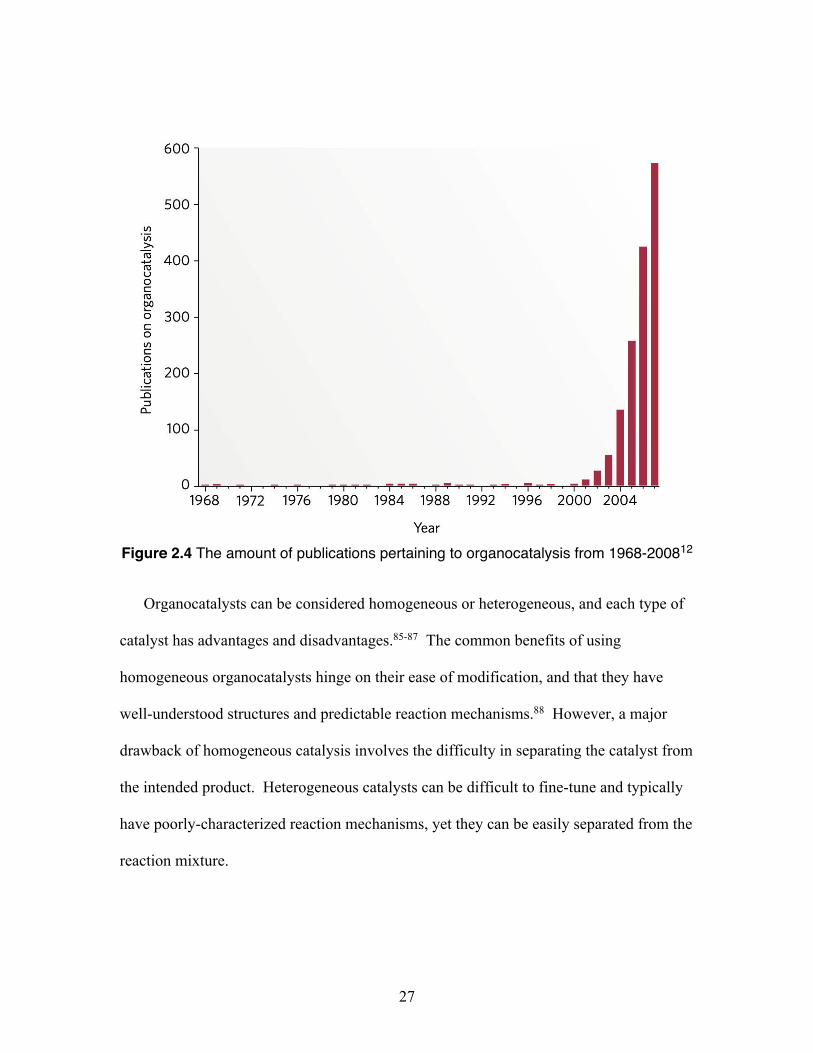
recently emerged as a prominent concept.82, 83 As reported by MacMillan, a staggering
statistic shows the growing interest of utilizing organic catalysts (Figure 2.4).84 From
1968-1997, there were no review articles, and only a limited number of manuscripts
disclosing the use of organocatalysts, however from 1998-2008, more than 1,500
publications reported the use of organic molecules to catalyze over 130 distinct reaction
types.
Figure 2.3 Examples of heterogeneous and homogeneous catalysts
Heterogeneous catalyst
Type of CatalystCatalystProcess
Contact process
Haber-Bosch process
Ostwald process
Product Synthesized
Sulfuric acid (H2SO4)
Ammonia (NH3)
Nitric acid (HNO3)
Vanadium oxides (V2O5)
Iron oxides on alumina (Al2O3)
Unsupported Pt-Rh
Andrussov oxidation Hydrogen cyanide (HCN)
Ziegler-Natta polymerization Polymerized olefins TiCl3 or MgCl2
Heterogeneous catalyst
Heterogeneous catalyst
Heterogeneous catalyst
Heterogeneous catalyst
Pt-Rh
Homogeneous catalystHydrolysis of esters
Hydrogenation of olefins
Enzymatic catalysis
Carboxylic acids
Alkanes
Acid
Wilkinson’s or Crabtree’s catalyst
Carbonic anhydrases
Cativa process Acetic acid (CH3CO2H)
Wacker process Acetaldehyde (CH3CHO)
Homogeneous catalyst
Homogeneous catalyst
Homogeneous catalyst
Homogeneous catalyst
Iridium complex
CO2
Pd(II)
27
Organocatalysts can be considered homogeneous or heterogeneous, and each type of
catalyst has advantages and disadvantages.85-87 The common benefits of using
homogeneous organocatalysts hinge on their ease of modification, and that they have
well-understood structures and predictable reaction mechanisms.88 However, a major
drawback of homogeneous catalysis involves the difficulty in separating the catalyst from
the intended product. Heterogeneous catalysts can be difficult to fine-tune and typically
have poorly-characterized reaction mechanisms, yet they can be easily separated from the
reaction mixture.
Figure 2.4 The amount of publications pertaining to organocatalysis from 1968-200812

28
While biocatalysis78, 87, 89-91 and metal catalysis78, 92-94 continue to be valuable within
the realm of synthesis, organocatalysis has become a complementary, and often superior
strategy to effectively catalyze chemical transformations.15, 95-101 The energy-efficient
nature of organocatalysis enable chemists to obtain desired products in a timely and
practical fashion. Organocatalysts are typically more stable to air and moisture, and are
readily accessible and operationally simple to implement. Furthermore, because organic
catalysts are metal-free and are used in substoichiometric amounts without being
consumed, they are considered to be more atom-economical and sustainable alternatives,
with the potential to be recovered, recycled, and reused.88 It should be noted that the
metal-free aspect of organocatalysts is particularly crucial within the pharmaceutical and
food industries, owing to the fact that the FDA has placed strict regulations on metal
contamination, even at trace amounts, of final products, due to possible adverse
toxicological effects.88
Organocatalysis has become an attractive area of research over the last decade.95, 97, 98
Scientists have applied organocatalysts in a vast array of important synthetic
transformations.74, 95-98 Some of the more well-known reactions that have been impacted
by organocatalysis include: Hajos–Parrish reactions102, Knoevenagel condensations103, 104,
esterifications105, Baylis-Hillman reactions106, Stetter reactions107, Aldol reactions108, 109,
Diels-Alder cycloadditions110, 111, Michael reactions112, Mannich transformations113-115,
and natural product syntheses116, 117. While organic catalysts have been successfully
utilized in various synthetic transformations, one of the most pivotal aspects has been
their capacity to act as enantioselective catalysts.

29
2.4 THE IMPORTANCE OF ASYMMETRIC SYNTHESIS
Many scientific areas depend on the chirality of organic molecules. Depending on the
chemical connectivity, or simply put, how atoms are arranged within a compound, can
affect a compound’s properties and reactivity. When enantiomers are in an achiral
environment, they maintain the same chemical and physical properties (other than the
direction in which they rotate plane-polarized light). However, in chiral conditions, the
spatial arrangement of the atoms can have drastic effects. As an example, the difference
in smell between oranges and lemons can be attributed to the existence of the
enantiomeric forms of limonene: (R)-limonene and (L)-limonene (Figure 2.5).118
Similarly, the enantiomeric forms of carvone can give discrete smells of either spearmint
or caraway seed. Another, more extreme example, involves the formerly prescribed drug
named thalidomide. In the 1960s, many West European countries advocated pregnant
women to use thalidomide to alleviate the symptoms of morning sickness.119 Although
Figure 2.5 Select examples demonstrating how chirality affects chemical properties and reactivities
O
H
O
H
(R)-carvoneSpearment oil smell
(S)-carvoneCaraway oil smell
N
O
O
NHO
O N
O
O
NHO
O
(R)-thalidomideEffective sedative
(S)-thalidomideTeratogenic
H H
(R)-limoneneFresh citrus, orange smell
(S)-limoneneSharp, lemon smell
H H

30
some women responded well to the thalidomide medication, other women witnessed
severe birth defects in their newborn babies. The drug was administered as a mixture of
enantiomers, and doctors soon realized that one of the isomers, the (S)-enantiomer of
thalidomide, was causing birth defects.
Since scientists have recognized the extent in which enantiomers can impact science
and life, there has been an increase of interest in the field of enantioselective synthesis.
One of the more elegant and eco-friendly strategies that can install chirality into a
molecule is to apply a substoichiometric amount of a metal-free chiral catalyst; this is the
basis of asymmetric organocatalysis.95, 97, 98, 100 Asymmetric organocatalysis can be
broadly defined as an enantioselective process that is accelerated through the use of a
catalytic amount of organic-based chiral reagent. In contrast to transition metal and
enzymatic catalysis, the key detail to recognize is that asymmetric organocatalysis
employs small organic molecules to carry out the chemical reaction. These chiral
organocatalysts are typically more stable, more affordable, less toxic, and easier to
synthesize and apply to various chemical transformations.88, 100
2.5 ASYMMETRIC ORGANOCATALYSTS BASED ON ACTIVATION MODE
In general, organocatalytic transformations can be classified based on how they
interact with a substrate, which is referred to as their mode of action. The
organocatalysts react mainly as Lewis bases, Lewis acids, Brønsted bases, and Brønsted
acids, but these can be simplified into two main modes of action: covalent and non-
covalent organocatalysts.99, 101, 120 Covalent organocatalysts involve a strong interaction

31
between a substrate and the catalyst in which a new covalent bond is formed. The
covalent modes of action commonly encompass enamine, iminium, singly occupied
molecular orbital (SOMO), carbene, and Lewis-base catalysts. Non-covalent
organocatalysts depend on weak bonds between the substrate and organocatalyst. The
non-covalent modes of action predominantly involve hydrogen bonding, Brønsted base,
Brønsted acid, bifunctional, and phase transfer catalysts. While publications related to
asymmetric organocatalysis continue to flourish, in the interest of brevity, the following
content will feature a select few chiral organic frameworks that illustrate the innovations
that have been made in asymmetric organocatalysis. Particular emphasis will be placed
on asymmetric organocatalysts that closely relate to the work displayed in this thesis: 𝛼-
functionalizations of carbonyl compounds, oxidations, and peptide related
organocatalysts. This review is intended to only cover specific topics, and therefore
many pioneering works will not be represented. However, for a more thorough and all-
inclusive understanding of asymmetric organocatalysis, the reader is directed to the
remarkable multitude of books87, 99, 101, 121 and reviews74, 84, 96, 120, 122-138 that have been
published. Some of the more influential works that pertain to asymmetric
organocatalysis will not be discussed but that are worth mentioning include: Fu’s planar-
chiral catalysts139, MacMillan chiral imidazolidinones for Diels-Alder, Friedel-Craft
alkylations, direct alkylations of furans and indoles, and Mukaiyama−Michael
additions110, 140-143,and Shi144, Yang145, and Denmark146 catalytic scaffolds for
asymmetric epoxidations.
32
2.6 ASYMMETRIC ORGANOCATALYSIS FOR 𝛼-CARBONYL
FUNCTIONALIZATIONS
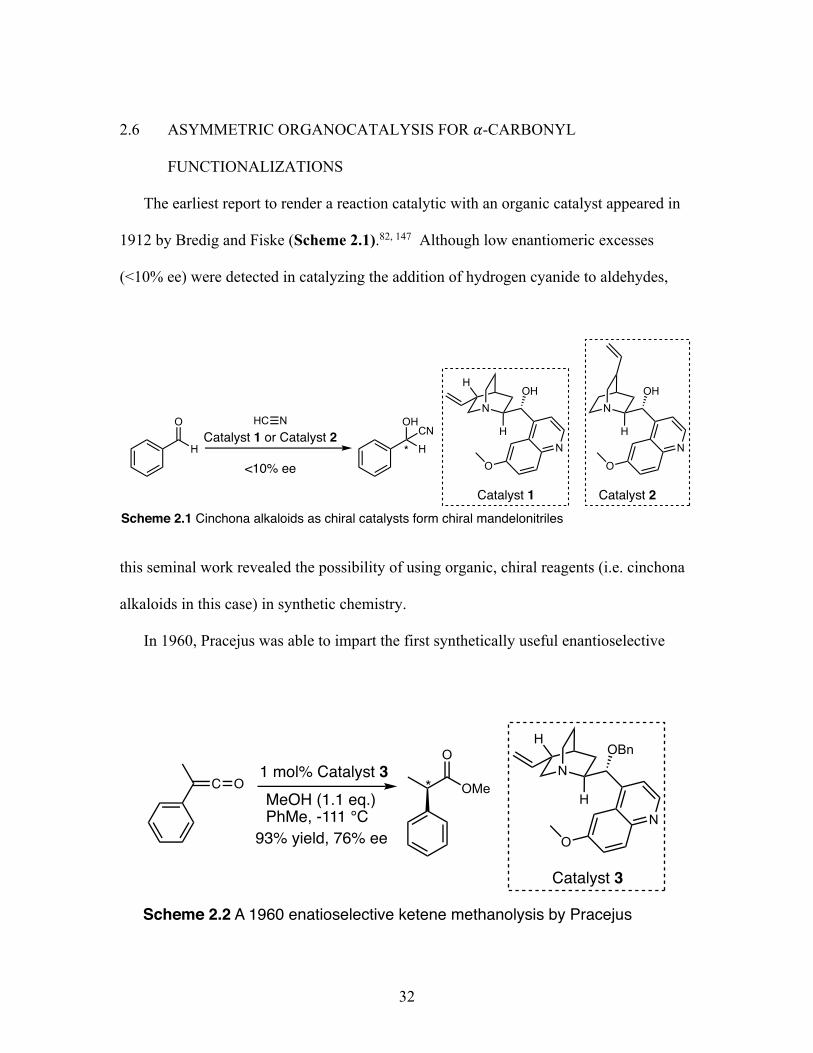
The earliest report to render a reaction catalytic with an organic catalyst appeared in
1912 by Bredig and Fiske (Scheme 2.1).82, 147 Although low enantiomeric excesses
(<10% ee) were detected in catalyzing the addition of hydrogen cyanide to aldehydes,
this seminal work revealed the possibility of using organic, chiral reagents (i.e. cinchona
alkaloids in this case) in synthetic chemistry.
In 1960, Pracejus was able to impart the first synthetically useful enantioselective
H
O HC N
H
OHCN
*
Scheme 2.1 Cinchona alkaloids as chiral catalysts form chiral mandelonitriles
Catalyst 1 or Catalyst 2
<10% ee
N
N
O
H
OHH
Catalyst 1
N
N
O
H
OH
Catalyst 2
Scheme 2.2 A 1960 enatioselective ketene methanolysis by Pracejus
C O1 mol% Catalyst 3MeOH (1.1 eq.)PhMe, -111 °C
93% yield, 76% ee
OMe
ON
N
O
H
OBnH
Catalyst 3
*
33
reaction through the methanolysis of phenylmethylketene by utilizing an O-
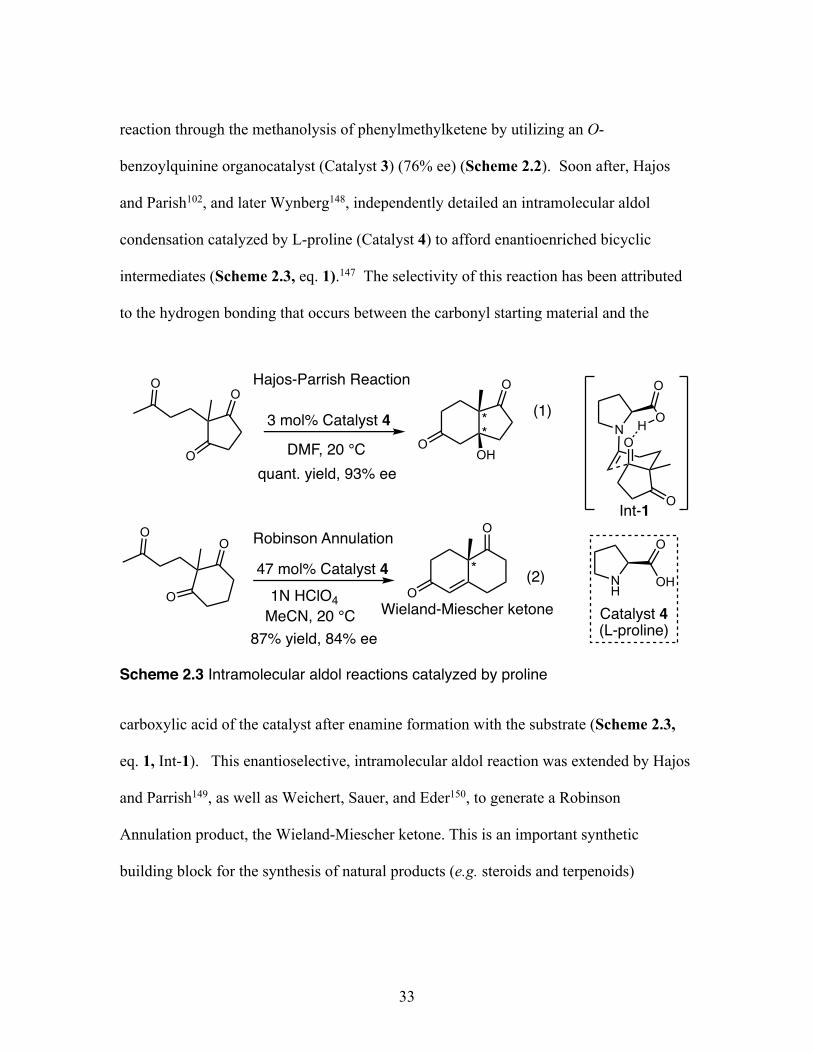
benzoylquinine organocatalyst (Catalyst 3) (76% ee) (Scheme 2.2). Soon after, Hajos
and Parish102, and later Wynberg148, independently detailed an intramolecular aldol
condensation catalyzed by L-proline (Catalyst 4) to afford enantioenriched bicyclic
intermediates (Scheme 2.3, eq. 1).147 The selectivity of this reaction has been attributed
to the hydrogen bonding that occurs between the carbonyl starting material and the
carboxylic acid of the catalyst after enamine formation with the substrate (Scheme 2.3,
eq. 1, Int-1). This enantioselective, intramolecular aldol reaction was extended by Hajos
and Parrish149, as well as Weichert, Sauer, and Eder150, to generate a Robinson
Annulation product, the Wieland-Miescher ketone. This is an important synthetic
building block for the synthesis of natural products (e.g. steroids and terpenoids)
O
O
O
3 mol% Catalyst 4
NH
O
OH
DMF, 20 °C
Catalyst 4(L-proline)
O
OOH
Scheme 2.3 Intramolecular aldol reactions catalyzed by proline
quant. yield, 93% ee
**
O
47 mol% Catalyst 41N HClO4 O
87% yield, 84% ee
*O
O
O
Hajos-Parrish Reaction
Wieland-Miescher ketone
(1)
(2)
Robinson Annulation
MeCN, 20 °C
N
O
O
OH O
Int-1

34
(Scheme 2.3, eq. 2).125, 148, 151, 152 These catalytic scaffolds have helped solve major
synthetic challenges and have demonstrated their applicability in organic synthesis.
List and Barbas reported a carbon-carbon bond forming reaction by means of a
direct asymmetric intermolecular aldol condensation.37 Proline was used as the amine-
based catalyst, and similar to the Hajos-Parrish reaction (Scheme 2.3, eq. 1), it has been
suggested that the enantiofacial selectivity arises from the stereodirecting carboxylate of
proline (Scheme 2.4, Int-2).108, 125, 153 Further research from Woodward154 and
Danishefsky155 exhibited the utilization of enamine catalysts in application to natural
products such as erythromycin (Scheme 2.5, eq. 1) and estrone (Scheme 2.5, eq. 2).
Me Me
O
H
O 30 mol% Catalyst 4
DMSO, rtMe
O
97% yield96% ee
Intermolecular aldol reactionH OH
Scheme 2.4 Intermolecular aldolization using a proline chiral catalyst
N
O
MeOH O
Int-2
H
N O O
O
N
O
O
200 mol% Catalyst 5MeCN, HClO4
80 °C82% yield, 86% ee
HO
O
NH2
Catalyst 5((S)-phenylalanine)
S
O
S
O
O
S
O
S
OH
OH
H
70% yield, 36% eeCatalyst 4
Erythromycin
Scheme 2.5 Enamine catalysis in natural product synthesis
Estrone precursor
(1)
(2)
7ak

35
The ability to use small, organic compounds, such as the natural amino acid
proline, to catalyze chemical reactions has allowed for an expanded repertoire in the field
of organocatalysis. As shown in Scheme 2.6, many carbonyl moieties have been
functionalized at the ⍺-position with use of L-proline as the organocatalyst. The
asymmetric Mannich reaction (Scheme 2.6, eq. 1) can generate syn β-amino ketones and
aldehydes.113, 153, 156, 157 This has become an attractive enantioselective strategy for
constructing decorated 𝛼-amino acids and alcohols.153, 157 MacMillan158, Hayashi159, and
Zhong160 independently published enantioselective methods to assemble new carbon-
oxygen bonds at the alpha position of aldehydes and ketones (Scheme 2.6, eq. 2). The
proposed transition state (Scheme 2.6, eq. 2, int-3) involves hydrogen bonding to the
nitrogen atom of the nitroso species, which has an enhanced Brønsted basic character,
and allows for O-regioselectivity.161 These procedures have been exploited to produce
optically active 𝛼-oxy-functionalized carbonyl compounds, which can be transformed
into biologically relevant chiral compounds.161 Strategies that utilize asymmetric
organocatalysis to form carbonyl compounds equipped with nitrogen substituents at the
𝛼-position have been disclosed by Jørgensen 162 and List163 (Scheme 2.6, eq. 3). These
approaches can accompany the enantioselective Strecker and Mannich reactions toward
𝛼-amino carbonyl compounds, however, List and Jørgensen illustrate a direct 𝛼-
amination by reacting enamines with alkyl diazodicarboxylates.162 Proline has also been
utilized to afford a direct asymmetric catalytic dihydroxylation (Scheme 2.6, eq. 4). This
process is the first to exercise small organic catalysts en route to enantiopure anti-1,2-
diols, and has been used as a complementary protocol to the Sharpless dihydoxylation.164
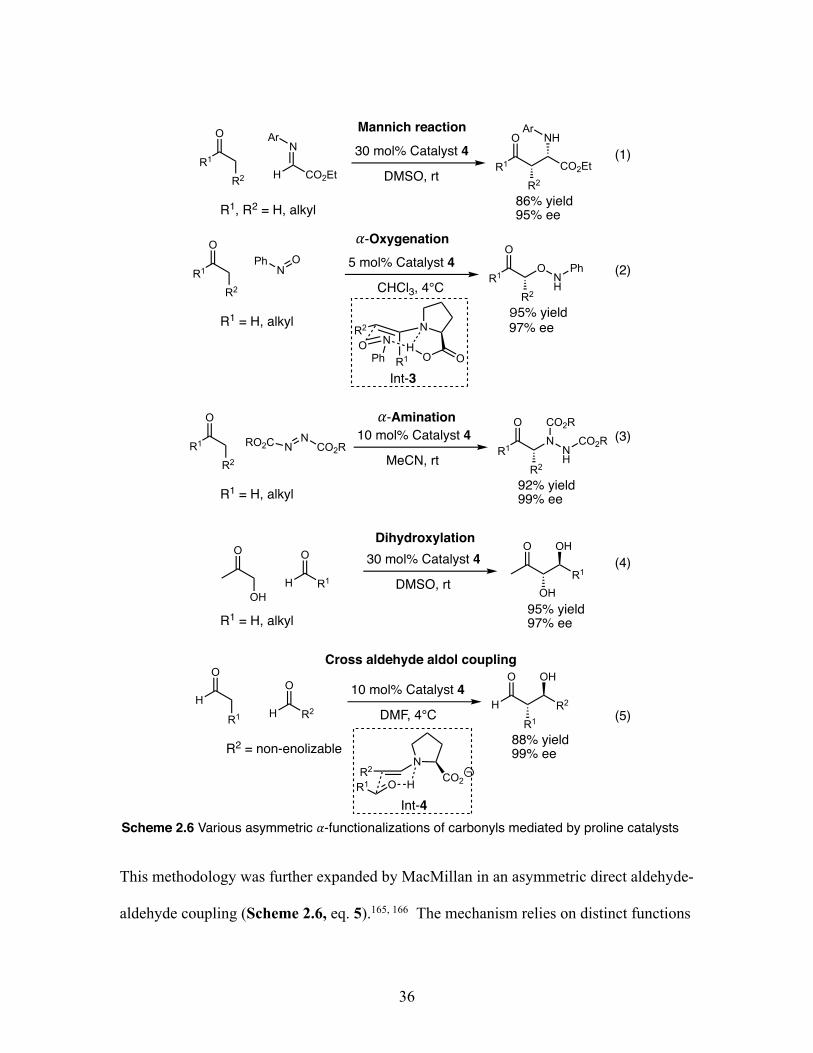
36
This methodology was further expanded by MacMillan in an asymmetric direct aldehyde-
aldehyde coupling (Scheme 2.6, eq. 5).165, 166 The mechanism relies on distinct functions
Scheme 2.6 Various asymmetric !-functionalizations of carbonyls mediated by proline catalysts
R1
O
R2 H CO2Et
NAr
30 mol% Catalyst 4
DMSO, rt
R1, R2 = H, alkyl
R1
O
R2CO2Et
NHAr
86% yield95% ee
Mannich reaction
H
O
R1 H R2
O 10 mol% Catalyst 4
DMF, 4℃
R2 = non-enolizable
H
O
R1R2
OH
88% yield99% ee
Cross aldehyde aldol coupling
R1
O
R2
5 mol% Catalyst 4
CHCl3, 4℃
R1 = H, alkyl
R1
O
R2
ONH
95% yield97% ee
!-Oxygenation
NOPh Ph
R1
O
R2
10 mol% Catalyst 4
MeCN, rt
R1 = H, alkyl
R1
O
R2
NNH
92% yield99% ee
!-AminationCO2R
(1)
(2)
(3)
O
H R1
O 30 mol% Catalyst 4
DMSO, rt
O
OHR1
OH
95% yield97% ee
Dihydroxylation
OH
(4)
CO2R
NNCO2RRO2C
R1 = H, alkyl
N
HO O
O NR2
Ph R1
Int-3
H
N
O CO2R1R2
Int-4
(5)

37
of the two aldehydes, one aldehyde must function as a nucleophilic donor while the other
operates as an electrophilic acceptor. The donor aldehyde transposes to an enamine form,
and permits the enantioinduction to ensue to form the desired product, a β-hydroxy
aldehyde, which is an important building block for the synthesis of natural products; the
intermediate is shown in Scheme 2.6, eq. 5, Int-4. The aforementioned reactions have
made it possible for small organic catalysts to be applied in various organic reactions, and
thereby gives additional evidence of the prominent role that organocatalysts play in chiral
catalytic transformations.74
2.7 PEPTIDES
One innovative strategy that has spawned from proline-type catalysts has been the
utilization of short peptide-based chiral organocatalysts.128, 167-169 Peptide scaffolds are
highly modular and easy to fine-tune due to the immense “chiral space” provided by the
various commercially available and structurally diverse amino-acid residues. The easily
iterative and facile assembly of peptides can be attributed to the development of Fmoc
solid-phase peptide synthesis (Fmoc SPPS).170-172 Peptides are unique in that they can
have small molecule characteristics (e.g. relatively low molecular weight, broad substrate
scope), yet can mimic many sophisticated features of enzymes (e.g. site-selectivity,
chemoselectivity).173, 174 The synthetic accessibility of peptides allows for large catalyst
libraries to be generated, and these libraries can be rapidly tested using combinational
optimization/selection and high-throughput methods.167, 175-177 Asymmetric catalysis
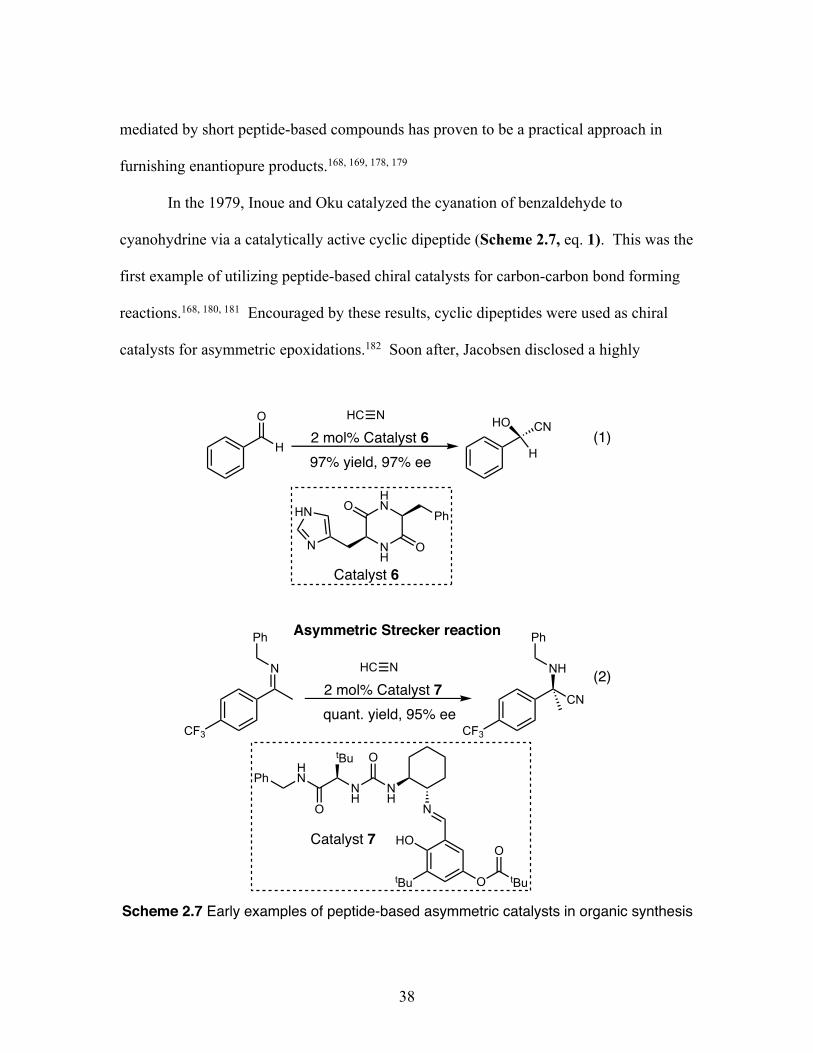
38
mediated by short peptide-based compounds has proven to be a practical approach in
furnishing enantiopure products.168, 169, 178, 179
In the 1979, Inoue and Oku catalyzed the cyanation of benzaldehyde to
cyanohydrine via a catalytically active cyclic dipeptide (Scheme 2.7, eq. 1). This was the
first example of utilizing peptide-based chiral catalysts for carbon-carbon bond forming
reactions.168, 180, 181 Encouraged by these results, cyclic dipeptides were used as chiral
catalysts for asymmetric epoxidations.182 Soon after, Jacobsen disclosed a highly
H
O HC N HO2 mol% Catalyst 697% yield, 97% ee
CN
NH
HNO
O
PhHN
N
Catalyst 6
O tBu
O
tBu
HO
N
NH
NH
OHN
O
Ph
tBu
Catalyst 7
N HC N NH
2 mol% Catalyst 7quant. yield, 95% ee
CN
Ph Ph
Scheme 2.7 Early examples of peptide-based asymmetric catalysts in organic synthesis
(1)H
CF3 CF3
Asymmetric Strecker reaction
(2)

39
enantioselective Strecker-type reaction by using a rigidified peptide-urea catalyst
(Scheme 2.7, eq. 2) .183-186 The direct asymmetric aldolization of acetone with aldehydes
was established by utilizing tripeptides that incorporate a secondary amine moiety
(proline) (Scheme 2.8).187 The difference in reactivity and selectivity when using
catalyst 8 versus catalyst 9 has been ascribed to the secondary structure of the peptide
chiral catalyst. In fact, the two catalysts give products with opposite absolute
configurations.
Remarkable advances in the domain of peptide-based asymmetric organocatalysis
have been accomplished by the Miller laboratory.128, 167, 169, 173, 178, 179, 188 With short
peptide catalysts, Miller’s group successfully performed kinetic resolutions to produce
enantiopure alcohols (Scheme 2.9).189 The enantiodiscrimination of the reaction is
enabled by a well-documented β-turn secondary conformation190-193 within catalyst 10
Scheme 2.8 Direct aldolization using peptide-based chiral catalysts
NH O
N
ONH
NH2
O
CO2H
NH
O
HN HN
ONH2
O
CO2HCatalyst 8 Catalyst 9
H
OCatalyst 8 (1 mol%)
or
Catalyst 9 (10 mol%)
OH O
Acetone, rtWith Catalyst 8: 69% yield, 78% ee (S)With Catalyst 9: 58% yield, 66% ee (R)
*
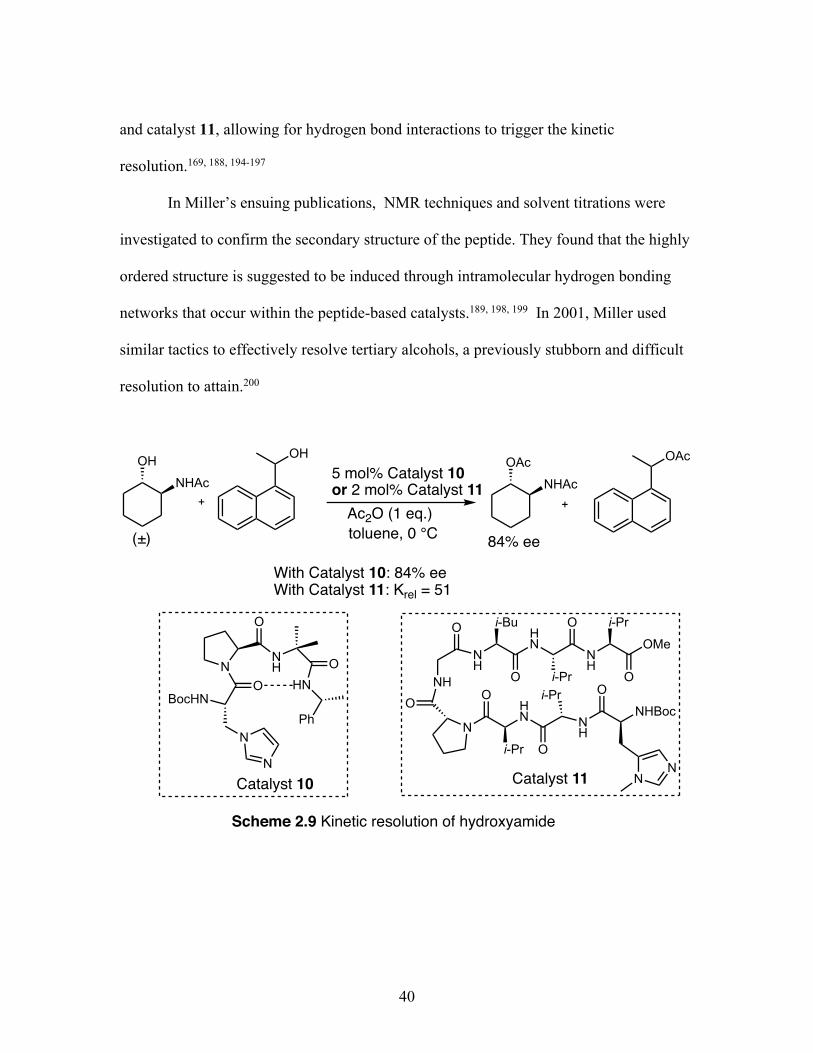
40
and catalyst 11, allowing for hydrogen bond interactions to trigger the kinetic
resolution.169, 188, 194-197
In Miller’s ensuing publications, NMR techniques and solvent titrations were
investigated to confirm the secondary structure of the peptide. They found that the highly
ordered structure is suggested to be induced through intramolecular hydrogen bonding
networks that occur within the peptide-based catalysts.189, 198, 199 In 2001, Miller used
similar tactics to effectively resolve tertiary alcohols, a previously stubborn and difficult
resolution to attain.200
NHAc
OH
(±)
OH5 mol% Catalyst 10or 2 mol% Catalyst 11
Ac2O (1 eq.)toluene, 0 ℃
NHAc
OAc OAc
NO
BocHN
N
O
NH O
HN
Ph
NCatalyst 10
84% ee
NN
NHBocNH
Oi-PrHN
Oi-Pr
N
OONH
O
NH
O
HN
i-Bu O
NH
i-Pr
OMe
O
i-Pr
Catalyst 11
With Catalyst 10: 84% eeWith Catalyst 11: Krel = 51
Scheme 2.9 Kinetic resolution of hydroxyamide
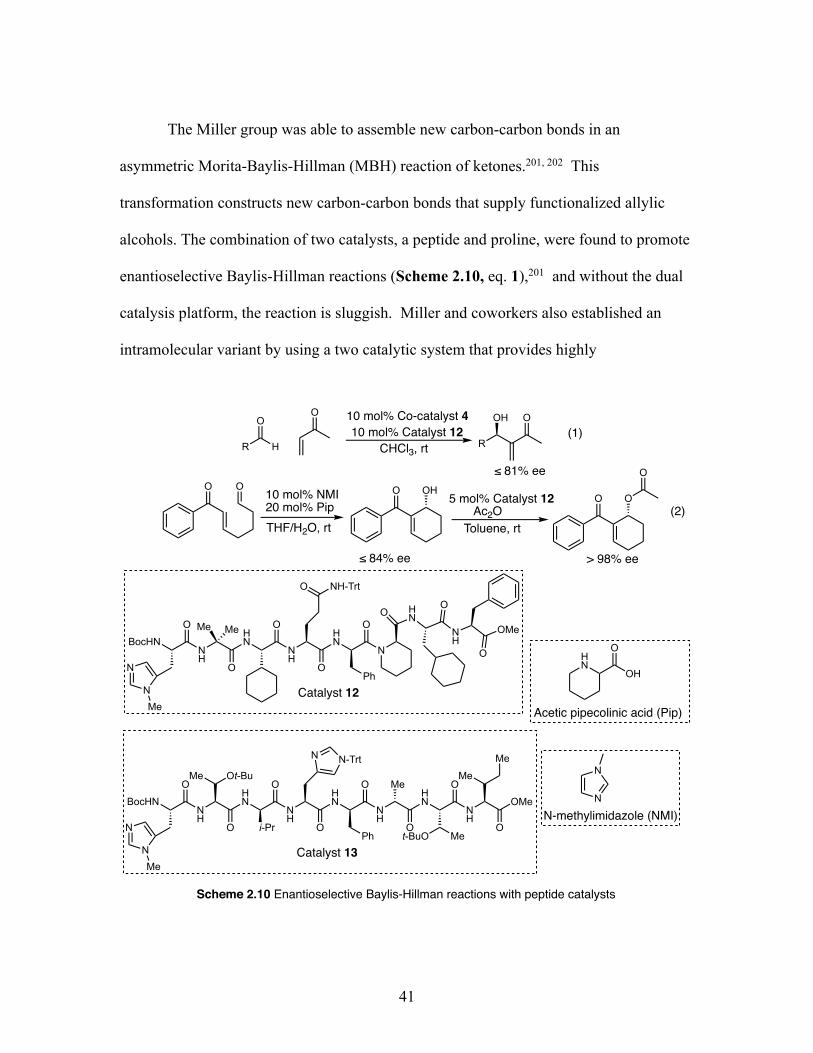
41
The Miller group was able to assemble new carbon-carbon bonds in an
asymmetric Morita-Baylis-Hillman (MBH) reaction of ketones.201, 202 This
transformation constructs new carbon-carbon bonds that supply functionalized allylic
alcohols. The combination of two catalysts, a peptide and proline, were found to promote
enantioselective Baylis-Hillman reactions (Scheme 2.10, eq. 1),201 and without the dual
catalysis platform, the reaction is sluggish. Miller and coworkers also established an
intramolecular variant by using a two catalytic system that provides highly
O O O OH
≤ 84% ee
Ac2OO O
> 98% ee
O
R H
O O
R
OH
Scheme 2.10 Enantioselective Baylis-Hillman reactions with peptide catalysts
≤ 81% ee
N
NMe
BocHNNH
OHN
O
NH
OHN
O
MeMe
O NH-Trt
Ph
N
OO H
NNH
O
OMe
O
Catalyst 12
10 mol% Co-catalyst 410 mol% Catalyst 12
CHCl3, rt
N
NMe
BocHNNH
OHN
O
NH
OHN
Oi-PrPh
NH
O
Catalyst 13
Me Ot-BuN-TrtN
HN
O
Me
Met-BuO
NH
O
OMe
O
Me
Me
10 mol% NMI20 mol% Pip
HN
OH
O
Acetic pipecolinic acid (Pip)
Toluene, rt
O
THF/H2O, rt
(1)
(2)
N
N
N-methylimidazole (NMI)
5 mol% Catalyst 12

42
enantioselective transformations (Scheme 2.10, eq. 2).202 The products obtained contain
a new substituent at the carbonyl ⍺-position, as well as a newly set stereocenter at the β-
position.
The concept of utilizing a peptide-based catalyst that contains a conformational
rigid scaffold has been implemented in various other selective reactions.203-208 In 2015,
Schreiner kinetically resolved racemic diols, in conjunction with a catalyzed alcohol
cross coupling, to form enantioenriched esters (Scheme 2.11).209 This enantioselective
reaction was catalyzed by a peptidic catalyst that incorporates a sterically bulk adamantyl
group, a moiety that causes structural rigidification.210, 211
N
HN
OO
NH
OHN
ONH
OHN O
ON
NCatalyst 14
1.) 2.5 mol% Catalyst 14m-CPBA, toluene, rt
OH
OH
R1
OH O
OH
O
R1
⩽97% ee
2.) DIC (2 eq.), rt
3.)
Scheme 2.11 Alcohol cross-coupling for kinetic resolution of diols using peptide catalysts
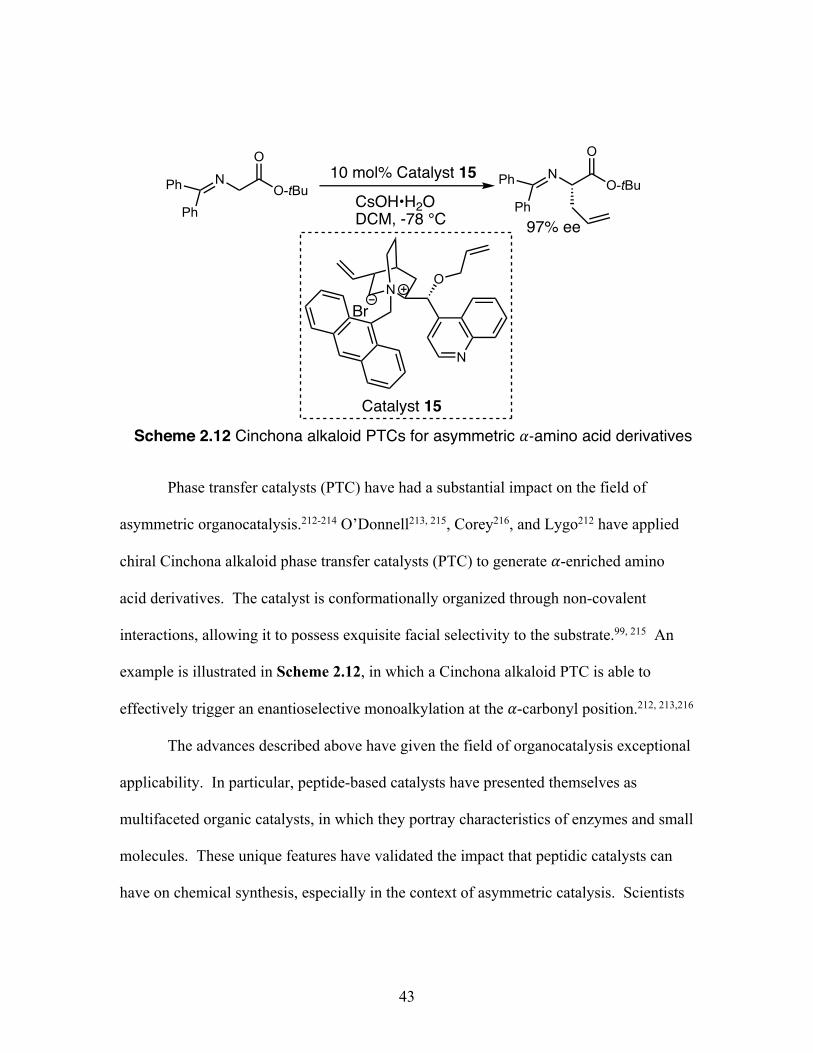
43
Phase transfer catalysts (PTC) have had a substantial impact on the field of
asymmetric organocatalysis.212-214 O’Donnell213, 215, Corey216, and Lygo212 have applied
chiral Cinchona alkaloid phase transfer catalysts (PTC) to generate 𝛼-enriched amino
acid derivatives. The catalyst is conformationally organized through non-covalent
interactions, allowing it to possess exquisite facial selectivity to the substrate.99, 215 An
example is illustrated in Scheme 2.12, in which a Cinchona alkaloid PTC is able to
effectively trigger an enantioselective monoalkylation at the 𝛼-carbonyl position.212, 213,216
The advances described above have given the field of organocatalysis exceptional
applicability. In particular, peptide-based catalysts have presented themselves as
multifaceted organic catalysts, in which they portray characteristics of enzymes and small
molecules. These unique features have validated the impact that peptidic catalysts can
have on chemical synthesis, especially in the context of asymmetric catalysis. Scientists
Catalyst 15
N
N
O
Br
Ph
Ph
NO-tBu
O10 mol% Catalyst 15
CsOH•H2ODCM, -78 ℃
Ph
Ph
NO-tBu
O
97% ee
Scheme 2.12 Cinchona alkaloid PTCs for asymmetric !-amino acid derivatives

44
have geared toward using “greener” and more environmentally benign catalysts, and
organocatalysts meet these desires. One area of research that has also gained attraction as
a versatile, yet eco-friendly strategy has been the implementation of hypervalent iodine
organic catalysts. While many remarkable achievements have been made, the integration
of hypervalent iodine chemistry with peptide-based organocatalysts has remained widely
unexplored. Developing methodologies that can exercise the unique aspects of chiral
peptide-based catalysts and hypervalent iodine species offers a potential new paradigm to
construct complex molecular skeletons in a highly reactive and enantioselective fashion.
The next chapter of this thesis will describe the efforts that have been made in utilizing
chiral hypervalent iodine(III) catalysts as versatile and “greener” alternatives to toxic
heavy metal chiral catalysts.

45
2.8 LICENSE AGREEMENT TO USE FIGURE 2.4

46
2.9 REFERENCES
74. Hernandez, J. G.; Juaristi, E., Recent efforts directed to the development of more sustainable asymmetric organocatalysis. ChemComm. 2012, 48 (44), 5396-5409.
75. Berzelius, J. J., Annls. Chim. Phys. 1836, 61, 146. 76. Moulijn, J. A.; Leeuwen, P. W. N. M. v.; Santen, R. A. v., Catalysis : an
integrated approach to homogeneous, heterogeneous and industrial catalysis. Elsevier: Amsterdam ; New York, 1993; p xviii, 460 p.
77. Chorkendorff, I.; Niemantsverdriet, J. W., Concepts of modern catalysis and kinetics. Third edition. ed.; Wiley-VCH,: Weinheim, 2017; p. 1 online resource.
78. Hanefeld, U.; Lefferts, L., Catalysis : an integrated textbook. 1st ed.; Wiley-VCH,: Weinheim, 2017; p. 1 online resource.
79. Drauz, K.; Gröger, H.; May, O.; Waldmann, H., Enzyme catalysis in organic synthesis. 3rd, completely rev. and enl. ed.; Wiley-VCH: Weinheim, 2012.
80. Coperet, C.; Chabanas, M.; Saint-Arroman, R. P.; Basset, J. M., Homogeneous and heterogeneous catalysis: Bridging the gap through surface organometallic chemistry. Angew. Chem. Int. Ed. 2003, 42 (2), 156-181.
81. Carey, F. A.; Sundberg, R. J., Advanced organic chemistry. Part B, Reactions and synthesis. 5th ed.; Springer: New York, 2007; p 1 online resource.
82. Bredig, G.; Fiske, P. S., Biochem. Z. 1912, 46, 7-23. 83. von Liebig, J., Ueber die Bildung des Oxamids aus Cyan. Annalen der Chemie
und Pharmacie 1860, 113 (2), 246-247. 84. MacMillan, D. W. C., The advent and development of organocatalysis. Nature
2008, 455 (7211), 304-308. 85. Ertl, G.; Knözinger, H.; Weitkamp, J., Handbook of heterogeneous catalysis.
VCH: Weinheim, 1997. 86. Sheldon, R. A.; Downing, R. S., Heterogeneous catalytic transformations for
environmentally friendly production. Appl. Catal., A 1999, 189 (2), 163-183. 87. Zecchina, A.; Califano, S., The development of catalysis : a history of key
processes and personas in catalytic science and technology. p 1 online resource. 88. Shaikh, I. R., Organocatalysis: Key Trends in Green Synthetic Chemistry,
Challenges, Scope towards Heterogenization, and Importance from Research and Industrial Point of View. J. Catal. 2014, 402860, 1-35.
89. Goswami, A.; Stewart, J. D., Organic synthesis using biocatalysis. Academic Press,: Amsterdam, 2015; p. 1 online resource.
90. Patel, R. N., Green biocatalysis. Wiley,: Place of publication not identified, 2016; p. 1 online resource (792 pages).
91. Williams, G.; Hall, M., Modern biocatalysis : advances towards synthetic biological systems. In Catalysis series 32 [Online] 1st ed.; Royal Society of Chemistry,: Cambridge, 2018; p. 1 online resource.
92. Chiusoli, G. P.; Maitlis, P. M., Metal-catalysis in Industrial Organic Processes. 1st ed.; Royal Society of Chemistry,: 2019; p. 1 online resource (328 pages).
93. Tsuji, J., Transition metal reagents and catalysts : innovations in organic synthesis. Wiley: Chichester, 2000; p xv, 477 p.

47
94. Hartwig, J. F., Organotransition metal chemistry : from bonding to catalysis. University Science: Sausalito, Calif., 2010; p xxx, 1127 p.
95. Banik, B. K., Advances in Organocatalysis. Curr. Organocatal. 2018, 5 (3), 164-164.
96. Bertelsen, S.; Jorgensen, K. A., Organocatalysis-after the gold rush. Chem. Soc. Rev. 2009, 38 (8), 2178-2189.
97. Brahmachari, G., Recent Advances in Organocatalysis. Curr. Organocatal. 2016, 3 (2), 92-92.
98. Csaky, A. G., Metal-Free Organocatalysis. Catalysts 2018, 8 (5). 99. Berkessel, A.; Gröger, H., Asymmetric organocatalysis : from biomimetic
concepts to applications in asymmetric synthesis. Wiley-VCH: Weinheim, 2005; p xiv, 440 p.
100. Dalko, P. I., Comprehensive enantioselective organocatalysis : catalysts, reactions, and applications. New edition. ed.; Wiley-VCH: Weinheim, Germany, 2013; p 3 volumes (xlv, 1438 pages).
101. List, B.; Arseniyadis, S., Asymmetric organocatalysis. Springer: Berlin, 2010; p xi, 460 p.
102. Hajos, Z. G.; Parrish, D. R., Asymmetric synthesis of bicyclic intermediates of natural product chemistry. J. Org. Chem. 1974, 39 (12), 1615-1621.
103. List, B., Emil Knoevenagel and the Roots of Aminocatalysis. Angew. Chem. Int. Ed. 2010, 49 (10), 1730-1734.
104. Dakin, H. D., The catalytic action of amino-acids, peptones and proteins in effecting certain syntheses. J. Biol. Chem. 1909, 7 (1), 49-55.
105. Neises, B.; Steglich, W., 4-Dialkylaminopyridines as Acylation Catalysts. Angew. Chem. Int. Ed. Engl. 1978, 17 (7), 522-524.
106. Basavaiah, D.; Veeraraghavaiah, G., The Baylis-Hillman reaction: a novel concept for creativity in chemistry. Chem. Soc. Rev. 2012, 41 (1), 68-78.
107. Stetter, H., Catalyzed Addition of Aldehydes to Activated Double Bonds—A New Synthetic Approach. Angew. Chem. Int. Ed. Engl. 1976, 15 (11), 639-647.
108. List, B.; Lerner, R. A.; Barbas, C. F., Proline-catalyzed direct asymmetric aldol reactions. J. Am. Chem. Soc. 2000, 122 (10), 2395-2396.
109. An, Y. J.; Zhang, Y. X.; Wu, Y.; Liu, Z. M.; Pi, C.; Tao, J. C., Simple amphiphilic isosteviol-proline conjugates as chiral catalysts for the direct asymmetric aldol reaction in the presence of water. Tetrahedron: Asymmetry 2010, 21 (6), 688-694.
110. Ahrendt, K. A.; Borths, C. J.; MacMillan, D. W. C., New strategies for organic catalysis: The first highly enantioselective organocatalytic Diels-Alder reaction. J. Amer. Chem. Soc. 2000, 122 (17), 4243-4244.
111. Rideout, D. C.; Breslow, R., Hydrophobic acceleration of Diels-Alder reactions. J. Am. Chem. Soc. 1980, 102 (26), 7816-7817.
112. Betancort, J. M.; Sakthivel, K.; Thayumanavan, R.; Barbas, C. F., Catalytic enantioselective direct Michael additions of ketones to alkylidene malonates. Tetrahedron Lett. 2001, 42 (27), 4441-4444.

48
113. Notz, W.; Sakthivel, K.; Bui, T.; Zhong, G. F.; Barbas, C. F., Amine-catalyzed direct asymmetric Mannich-type reactions. Tetrahedron Lett. 2001, 42 (2), 199-201.
114. Sakthivel, K.; Notz, W.; Bui, T.; Barbas, C. F., Amino acid catalyzed direct asymmetric aldol reactions: A bioorganic approach to catalytic asymmetric carbon-carbon bond-forming reactions. J. Am. Chem. Soc. 2001, 123 (22), 5260-5267.
115. An, Y. J.; Wang, C. C.; Liu, Z. P.; Tao, J. C., Isosteviol?Proline Conjugates as Highly Efficient Amphiphilic Organocatalysts for Asymmetric Three-Component Mannich Reactions in the Presence of Water. Helv. Chim. Acta 2012, 95 (1), 43-51.
116. Wegler, R., Uber die mit verschiedener Reaktionsgeschwindigkeit erfolgende Veresterung der optischen Antipoden eines Racemates durch opt. akt.Katalysatoren. Justus Liebigs Annalen der Chemie 1932, 498, 62-73.
117. Vavon, M. M.; Peignier, P., L’application des alcalo¨ıdes dans la synth`ese organique. Bulletin de la Soci´et´eChimique deFrance 1929, 45, 293.
118. Clayden, J.; Greeves, N.; Warren, S. G., Organic chemistry. 2nd ed.; Oxford University Press: Oxford, 2012; p xxv, 1234 p.
119. Tokunaga, E.; Yamamoto, T.; Ito, E.; Shibata, N., Understanding the Thalidomide Chirality in Biological Processes by the Self-disproportionation of Enantiomers. Sci. Rep. 2018, 8.
120. Seayad, J.; List, B., Asymmetric organocatalysis. Org. Biomol. Chem. 2005, 3 (5), 719-724.
121. Pellissier, H., Recent developments in asymmetric organocatalysis. Royal Society of Chemistry: Cambridge, 2010; p xv, 241 p.
122. List, B., Asymmetric aminocatalysis. Synlett 2001, (11), 1675-1686. 123. Melchiorre, P.; Marigo, M.; Carlone, A.; Bartoli, G., Asymmetric aminocatalysis
- Gold rush in organic chemistry. Angew. Chem. Int. Ed. 2008, 47 (33), 6138-6171.
124. List, B., The ying and yang of asymmetric aminocatalysis. ChemCommun. 2006, (8), 819-824.
125. Mukherjee, S.; Yang, J. W.; Hoffmann, S.; List, B., Asymmetric enamine catalysis. Chem. Rev. 2007, 107 (12), 5471-5569.
126. Lelais, G.; MacMillan, D. W. C., Modern strategies in organic catalysis: The advent and development of iminium activation. Aldrichimica Acta 2006, 39 (3), 79-87.
127. France, S.; Guerin, D. J.; Miller, S. J.; Lectka, T., Nucleophilic chiral amines as catalysts in asymmetric synthesis. Chem. Rev. 2003, 103 (8), 2985-3012.
128. Jarvo, E. R.; Miller, S. J., Amino acids and peptides as asymmetric organocatalysts. Tetrahedron 2002, 58 (13), 2481-2495.
129. Doyle, A. G.; Jacobsen, E. N., Small-molecule H-bond donors in asymmetric catalysis. Chem. Rev. 2007, 107 (12), 5713-5743.
130. Dalko, P. I.; Moisan, L., Enantioselective organocatalysis. Angew. Chem. Int. Ed 2001, 40 (20), 3726-3748.

49
131. Dalko, P. I.; Moisan, L., In the golden age of organocatalysis. Angew. Chem. Int. Ed 2004, 43 (39), 5138-5175.
132. Guillena, G.; Najera, C.; Ramon, D. J., Enantioselective direct aldol reaction: the blossoming of modern organocatalysis. Tetrahedron: Asymmetry 2007, 18 (19), 2249-2293.
133. Guillena, G.; Ramon, D. J., Enantioselective alpha-heterofunctionalisation of carbonyl compounds: organocatalysis is the simplest approach. Tetrahedron: Asymmetry 2006, 17 (10), 1465-1492.
134. Kacprzak, K.; Gawronski, J., Cinchona alkaloids and their derivatives: Versatile catalysts and ligands in asymmetric synthesis. Synthesis-Stuttgart 2001, (7), 961-998.
135. Houk, K. N.; List, B., Asymmetric organocatalysis. Acc. Chem. Res. 2004, 37 (8), 487-487.
136. Ventura, M. R., Asymmetric Syntheses Based on Organocatalysis. Mini-Rev. Org. Chem. 2014, 11 (2), 154-163.
137. Abbasov, M. E.; Romo, D., The ever-expanding role of asymmetric covalent organocatalysis in scalable, natural product synthesis. Nat. Prod. Rep. 2014, 31 (10), 1318-1327.
138. Dibello, E.; Gamenara, D.; Seoane, G., Organocatalysis in the Synthesis of Natural Products: Recent Developments in Aldol and Mannich Reactions, and 1,4-Conjugated Additions. Curr. Organocatal. 2015, 2 (2), 124-149.
139. Fu, G. C., Asymmetric catalysis with "planar-chiral" derivatives of 4-(dimethylamino)pyridine. Acc. Chem. Res. 2004, 37 (8), 542-547.
140. Northrup, A. B.; MacMillan, D. W. C., The first general enantioselective catalytic Diels-Alder reaction with simple alpha,beta-unsaturated ketones. J. Am. Chem. Soc. 2002, 124 (11), 2458-2460.
141. Jen, W. S.; Wiener, J. J. M.; MacMillan, D. W. C., New strategies for organic catalysis: The first enantioselective organocatalytic 1,3-dipolar cycloaddition. J. Am. Chem. Soc. 2000, 122 (40), 9874-9875.
142. Paras, N. A.; MacMillan, D. W. C., New strategies in organic catalysis: The first enantioselective organocatalytic Friedel-Crafts alkylation. J. Am. Chem. Soc. 2001, 123 (18), 4370-4371.
143. Brown, S. P.; Goodwin, N. C.; MacMillan, D. W. C., The first enantioselective organocatalytic Mukaiyama- Michael reaction: A direct method for the synthesis of enantioenriched gamma-butenolide architecture. J. Am. Chem. Soc. 2003, 125 (5), 1192-1194.
144. Shi, Y., Organocatalytic asymmetric epoxidation of olefins by chiral ketones. Acc. Chem. Res. 2004, 37 (8), 488-496.
145. Yang, D., Ketone-catalyzed asymmetric epoxidation reactions. Acc. Chem. Res. 2004, 37 (8), 497-505.
146. Denmark, S. E.; Wu, Z. C., The development of chiral, nonracemic dioxiranes for the catalytic, enantioselective epoxidation of alkenes. Synlett 1999, 847-859.
147. Lelais, G.; MacMillan, D. W. C., New Frontiers in Asymmetric Catalysis: History and Perspective of Chiral Organic Catalysts. Wiley&Sons 2007; pp 313-358.

50
148. Eder, U.; Sauer; G.; Wiechert, R., Angew. Chem. Int. Ed. 1971, 10, 496-497. 149. Hajos, Z. G.; Parrish, D. R. Asymmetric synthesis of optically active polycyclic
organic compounds. 1971. 150. Eder, U.; Sauer, G. R.; Wiechert, R. Optically active 1,5-indanone and 1,6-
napthalenedione derivatives. 1971. 151. Corey, E. J.; Ohno, M.; Mitra, R. B.; Vatakencherry, P. A., Total Synthesis of
D,L-Longifolene. J. Am. Chem. Soc. 1961, 83 (5), 1251. 152. Corey, E. J.; Ohno, M.; Mitra, R. B.; Vatakencherry, P. A., Total Synthesis of
Longifolene. J. Am. Chem. Soc, 1964, 86 (3), 478. 153. Gaunt, M. J.; Johansson, C. C. C.; McNally, A.; Vo, N. T., Enantioselective
organocatalysis. Drug Discovery Today 2007, 12 (1-2), 8-27. 154. Woodward, R. B.; Logusch, E.; Nambiar, K. P.; Sakan, K.; Ward, D. E.; Au-
Yeung, B. W.; Balaram, P.; Browne, L. J.; Card, P. J.; Chen, C. H., Asymmetric total synthesis of erythromcin. 1. Synthesis of an erythronolide A secoacid derivative via asymmetric induction. J. Am. Chem. Soc. 1981, 103 (11), 3210-3213.
155. Danishefsky, S.; Cain, P., Optically specific synthesis of estrone and 19-norsteroids from 2,6-lutidine. J. Am. Chem. Soc. 1976, 98 (16), 4975-4983.
156. Notz, W.; Tanaka, F.; Barbas, C. F., Enamine-based organocatalysis with proline and diamines: The development of direct catalytic asymmetric Aldol, Mannich, Michael, and Diels-Alder reactions. Acc. Chem. Res. 2004, 37 (8), 580-591.
157. Cordova, A.; Notz, W.; Zhong, G. F.; Betancort, J. M.; Barbas, C. F., A highly enantioselective amino acid-catalyzed route to functionalized alpha-amino acids. J. Am. Chem. Soc. 2002, 124 (9), 1842-1843.
158. Brown, S. P.; Brochu, M. P.; Sinz, C. J.; MacMillan, D. W. C., The direct and enantioselective organocatalytic alpha-oxidation of aldehydes. J. Am. Chem. Soc. 2003, 125 (36), 10808-10809.
159. Hayashi, Y.; Yamaguchi, J.; Hibino, K.; Shoji, M., Direct proline catalyzed asymmetric alpha-aminooxylation of aldehydes. Tetrahedron Lett. 2003, 44 (45), 8293-8296.
160. Zhong, G. F., A facile and rapid route to highly enantiopure 1,2-diols by novel catalytic asymmetric alpha-aminoxylation of aldehydes. Angew. Chem. Int. Ed. 2003, 42 (35), 4247-4250.
161. Merino, P.; Tejero, T., Organocatalyzed asymmetric alpha-aminoxylation of aldehydes and ketones - An efficient access to enantiomerically pure alpha-hydroxycarbonyl compounds, diols, and even amino alcohols. Angew. Chem. Int. Ed. 2004, 43 (23), 2995-2997.
162. Kumaragurubaran, N.; Juhl, K.; Zhuang, W.; Bogevig, A.; Jorgensen, K. A., Direct L-proline-catalyzed asymmetric alpha-amination of ketones. J. Am. Chem. Soc. 2002, 124 (22), 6254-6255.
163. List, B., Direct catalytic asymmetric alpha-amination of aldehydes. J. Am. Chem. Soc. 2002, 124 (20), 5656-5657.
164. Notz, W.; List, B., Catalytic asymmetric synthesis of anti-1,2-diols. J. Am. Chem. Soc. 2000, 122 (30), 7386-7387.

51
165. Northrup, A. B.; MacMillan, D. W. C., The first direct and enantioselective cross-aldol reaction of aldehydes. J. Am. Chem. Soc. 2002, 124 (24), 6798-6799.
166. Mangion, I. K.; Northrup, A. B.; MacMillan, D. W. C., The importance of iminium geometry control in enamine catalysis: Identification of a new catalyst architecture for aldehyde-aldehyde couplings. Angew. Chem. Int. Ed. 2004, 43 (48), 6722-6724.
167. Berkessel, A., The discovery of catalytically active peptides through combinatorial chemistry. Current Opinion in Chemical Biology 2003, 7 (3), 409-419.
168. Davie, E. A. C.; Mennen, S. M.; Xu, Y. J.; Miller, S. J., Asymmetric catalysis mediated by synthetic peptides. Chem. Rev. 2007, 107 (12), 5759-5812.
169. Miller, S. J., In search of peptide-based catalysts for asymmetric organic synthesis. Acc. Chem. Res. 2004, 37 (8), 601-610.
170. Chan, W. C.; White, P. D., Fmoc solid phase peptide synthesis : a practical approach. Oxford University Press: Oxford, 2000; p xv, 346 p.
171. Kent, S. B. H., Solid-phase peptide synthesis. In Chemical biology : the role of chemistry in our fundamental understanding of biology [Online] Henry Stewart Talks,: London, 2008; p. 1 online resource.
172. Albericio, F., Solid-Phase Synthesis : a Practical Guide. CRC Press,: Boca Raton, FL, 2000; p. 1 online resource.
173. Blank, J. T.; Miller, S. J., Studies of folded peptide-based catalysts for asymmetric organic synthesis. Biopolymers 2006, 84 (1), 38-47.
174. Breslow, R., Biomimetic chemistry: a frontier at the chemistry/biology interface. Chem. Bio. 1998, 5 (2), R27-R28.
175. Revell, J. D.; Wennemers, H., Peptidic catalysts developed by combinatorial screening methods. Curr. Op. Chem. Bio. 2007, 11 (3), 269-278.
176. Furka, A.; Sebestyen, F.; Asgedom, M.; Dibo, G., General-method for Rapid Synthesis of Multicomponent Peptide Mixtures. Int. J. Pep. Protein Res. 1991, 37 (6), 487-493.
177. Houghten, R. A.; Pinilla, C.; Blondelle, S. E.; Appel, J. R.; Dooley, C. T.; Cuervo, J. H., Generation and use of synthetic peptide combinatorial libraries for basic research and drug discovery. Nature 1991, 354 (6348), 84-86.
178. Wennemers, H., Asymmetric catalysis with peptides. ChemComm. 2011, 47 (44), 12036-12041.
179. Lewandowski, B.; Wennemers, H., Asymmetric catalysis with short-chain peptides. Curr. Op. Chem. Bio. 2014, 22, 40-46.
180. Oku, J. I.; Inoue, S., Asymmetric cyanohydrin synthesis catalysed by a synthetic cyclic dipeptide. J. Chem. Soc. Chem. Comm. 1981, (5), 229-230.
181. Oku, J. I.; Ito, N.; Inoue, S., Asymmetric cyanohydrin synthesis catalyzed by synthetic cyclic dipeptide. Makromol. Chem. 1979, 180 (4), 1089-1091.
182. Juliá, S.; Guixer, J.; Masana, J.; Rocas, J.; Colonna, S.; Annuziata, R.; Molinari, H., Synthetic enzymes. Part 2. Catalytic asymmetric epoxidation by means of polyamino-acids in a triphase system. J. Chem. Soc., Perkin Trans. 1, 1982, 1317 1982, (6), 1317-1324.

52
183. Iyer, M. S.; Gigstad, K. M.; Namdev, N. D.; Lipton, M., Asymmetric catalysis of the Strecker amino acid synthesis by a cyclic dipeptide. J. Am. Chem. Soc. 1996, 118 (20), 4910-4911.
184. Sigman, M. S.; Vachal, P.; Jacobsen, E. N., A general catalyst for the asymmetric Strecker reaction. Angew. Chem. Int. Ed. 2000, 39 (7), 1279.
185. Yet, L., Recent developments in catalytic asymmetric Strecker-type reactions. Angew. Chem. Int. Ed. 2001, 40 (5), 875-877.
186. Vachal, P.; Jacobsen, E. N., Enantioselective catalytic addition of HCN to ketoimines. Catalytic synthesis of quaternary amino acids. Org. Lett. 2000, 2 (6), 867-870.
187. Krattiger, P.; Kovasy, R.; Revell, J. D.; Ivan, S.; Wennemers, H., Increased structural complexity leads to higher activity: Peptides as efficient and versatile catalysts for asymmetric aldol reactions. Org. Lett. 2005, 7 (6), 1101-1103.
188. Freundand, M. T., S. B., Peptides for asymmetric catalysis in Catalytic methods in asymmetric synthesis : advanced materials, techniques, and applications. Wiley: Hoboken, N.J., 2011; p xviii, 702 p.
189. Miller, S. J.; Copeland, G. T.; Papaioannou, N.; Horstmann, T. E.; Ruel, E. M., Kinetic resolution of alcohols catalyzed by tripeptides containing the N-alkylimidazole substructure. J. Amer. Chem. Soc. 1998, 120 (7), 1629-1630.
190. Imperiali, B.; Fisher, S. L.; Moats, R. A.; Prins, T. J., A conformational study of peptides with the general structure AC-l-XAA-PRO-d-XAA-l-XAA-NH2– spectroscopic evidence for a peptide with significant beta turn character in water and in dimethyl-sulfoxide. J. Am. Chem. Soc. 1992, 114 (9), 3182-3188.
191. Raghothama, S. R.; Awasthi, S. K.; Balaram, P., beta-hairpin nucleation by Pro-Gly beta-turns. Comparison of D-Pro-Gly and L-Pro-Gly sequences in an apolar octapeptide. J. Chem. Soc.-Perkin Trans 2 1998, (1), 137-143.
192. Chung, Y. J.; Christianson, L. A.; Stanger, H. E.; Powell, D. R.; Gellman, S. H., A beta-peptide reverse turn that promotes hairpin formation. J. Am. Chem. Soc. 1998, 120 (40), 10555-10556.
193. Stanger, H. E.; Gellman, S. H., Rules for antiparallel beta-sheet design: D-Pro-Gly is superior to L-Asn-Gly for beta-hairpin nucleation. J. Am. Chem. Soc. 1998, 120 (17), 4236-4237.
194. Copeland, G. T.; Jarvo, E. R.; Miller, S. J., Minimal acylase-like peptides. Conformational control of absolute stereospecificity. J. Org. Chem. 1998, 63 (20), 6784-6785.
195. Fierman, M. B.; O'Leary, D. J.; Steinmetz, W. E.; Miller, S. J., Structure-selectivity relationships and structure for a peptide-based enantioselective acylation catalyst. J. Am. Chem. Soc. 2004, 126 (22), 6967-6971.
196. Jarvo, E. R.; Vasbinder, M. M.; Miller, S. J., Asymmetric acylation reactions catalyzed by conformationally biased octapeptides. Tetrahedron 2000, 56 (50), 9773-9779.
197. Miller, S. J., Peptide-based catalysts for asymmetric reactions. Biopolymers 2005, 80 (4), 488-488.

53
198. Vasbinder, M. M.; Jarvo, E. R.; Miller, S. J., Incorporation of peptide isosteres into enantioselective peptide-based catalysts as mechanistic probes. Angew. Chem. Int. Ed. 2001, 40 (15), 2824-2827.
199. Jarvo, E. R.; Copeland, G. T.; Papaioannou, N.; Bonitatebus, P. J.; Miller, S. J., A biomimetic approach to asymmetric acyl transfer catalysis. J. Am. Chem. Soc. 1999, 121 (50), 11638-11643.
200. Jarvo, E. R.; Evans, C. A.; Copeland, G. T.; Miller, S. J., Fluorescence-based screening of asymmetric acylation catalysts through parallel enantiomer analysis. Identification of a catalyst for tertiary alcohol resolution. J. Org. Chem. 2001, 66 (16), 5522-5527.
201. Imbriglio, J. E.; Vasbinder, M. M.; Miller, S. J., Dual catalyst control in the amino acid-peptide-catalyzed enantioselective Baylis-Hillman reaction. Organic Lett. 2003, 5 (20), 3741-3743.
202. Aroyan, C. E.; Vasbinder, M. M.; Miller, S. J., Dual catalyst control in the enantioselective intramolecular Morita-Baylis-Hillman reaction. Organic Lett. 2005, 7 (18), 3849-3851.
203. Formaggio, F.; Barazza, A.; Bertocco, A.; Toniolo, C.; Broxterman, Q. B.; Kaptein, B.; Brasola, E.; Pengo, P.; Pasquato, L.; Scrimin, P., Role of secondary structure in the asymmetric acylation reaction catalyzed by peptides based on chiral C-alpha-tetrasubstituted alpha-amino acids. J. Org. Chem. 2004, 69 (11), 3849-3856.
204. Licini, G.; Bonchio, M.; Broxterman, Q. B.; Kaptein, B.; Moretto, A.; Toniolo, C.; Scrimin, P., C-alpha-tetrasubstituted amino acid based peptides in asymmetric catalysis. Biopolymers 2006, 84 (1), 97-104.
205. Papaioannou, N.; Evans, C. A.; Blank, J. T.; Miller, S. J., Enantioselective synthesis of a mitosane core assisted by diversity-based catalyst discovery. Org. Lett. 2001, 3 (18), 2879-2882.
206. Papaioannou, N.; Blank, J. T.; Miller, S. J., Enantioselective synthesis of an aziridinomitosane and selective functionalizations of a key intermediate. J. Org. Chem. 2003, 68 (7), 2728-2734.
207. Copeland, G. T.; Miller, S. J., Selection of enantioselective acyl transfer catalysts from a pooled peptide library through a fluorescence-based activity assay: An approach to kinetic resolution of secondary alcohols of broad structural scope. J. Am. Chem. Soc. 2001, 123 (27), 6496-6502.
208. Evans, C. A.; Miller, S. J., Proton-activated fluorescence as a tool for simultaneous screening of combinatorial chemical reactions. Curr. Opin. Chem. Bio. 2002, 6 (3), 333-338.
209. Hofmann, C.; Schumann, J. M.; Schreiner, P. R., Alcohol Cross-Coupling for the Kinetic Resolution of Diols via Oxidative Esterification. J. Org. Chem. 2015, 80 (3), 1972-1978.
210. Muller, C. E.; Wanka, L.; Jewell, K.; Schreiner, P. R., Enantioselective kinetic resolution of trans-cycloalkane-1,2-diols. Angew. Chem. Int. Ed. 2008, 47 (33), 6180-6183.

54
211. Muller, C. E.; Zell, D.; Hrdina, R.; Wende, R. C.; Wanka, L.; Schuler, S. M. M.; Schreiner, P. R., Lipophilic Oligopeptides for Chemo- and Enantioselective Acyl Transfer Reactions onto Alcohols. J. Org. Chem. 2013, 78 (17), 8465-8484.
212. Lygo, B.; Andrews, B. I., Asymmetric phase-transfer catalysis utilizing chiral quaternary ammonium salts: Asymmetric alkylation of glycine imines. Acc. Chem. Res. 2004, 37 (8), 518-525.
213. O'Donnell, M. J., The enantioselective synthesis of alpha-amino acids by phase-transfer catalysis with achiral Schiff base esters. Acc. Chem. Res. 2004, 37 (8), 506-517.
214. Maruoka, K.; Ooi, T., Enantioselective amino acid synthesis by chiral phase-transfer catalysis. Chem. Rev. 2003, 103 (8), 3013-3028.
215. Ojima, I., Catalytic asymmetric synthesis. 2nd ed.; Wiley-VCH: New York ; Chichester, 2000; p xiv, 864 p.
216. Corey, E. J.; Xu, F.; Noe, M. C., A rational approach to catalytic enantioselective enolate alkylation using a structurally rigidified and defined chiral quaternary ammonium salt under phase transfer conditions. J. Am. Chem. Soc. 1997, 119 (50), 12414-12415.

55
CHAPTER THREE
IODOARENE-CONTAINING ORGANOCATALYSTS FOR HYPERVALENT IODINE(III)
OXIDATIVE TRANSFORMATIONS
3.1 HYPERVALENT IODINE ORGANOCATALYSIS
To facilitate asymmetric transformations, the scientific community has turned a great
deal of attention toward the use of chiral organocatalysts. Recently, one area that has
witnessed tremendous growth has been the employment of chiral, hypervalent iodine
organocatalysts. As discussed in Chapter 1, hypervalent iodine species can act as highly
efficient and selective electrophiles and/or oxidants, and are appealing due to their mild
and environmentally friendly nature.18, 30, 44, 68 Although organic molecules bearing
hypervalent iodine moieties were discovered over a century ago, chiral hypervalent
iodine reagents were not utilized as asymmetric organocatalysts until the last few
decades. The following chapter will highlight the work that has helped advance
asymmetric chemistry in the context to hypervalent iodine-mediated catalysis. Particular
focus will be placed on chiral hypervalent iodine(III) catalysts that assist in oxidative
transformations and/or that furnish alpha-functionalized carbonyl products. For a more
broad overview of the contributions that have been made toward asymmetric
transformations mediated by hypervalent iodine compounds, the reader should survey the
exceptionally thorough books13, 23, 44, 68 and reviews217-226 that have recently appeared.
Significant hypervalent iodine-mediated developments that will be featured at a minimum
but that warrant a mention include: hypervalent iodine(V) reagents68, 217, 225-227,

56
asymmetric rearrangements217, 223, 228-230 alkene functionalizations217, 228, 231-233, and
alcohol oxidations234-236.
3.2 GENERAL REMARKS ON OXIDATIONS
At the most basic level, an oxidation/reduction (redox) reaction is a process that
involves the transfer of an electron from one species to another. Oxidation refers to a
loss of electron(s), and reduction indicates a gain in electron(s). Oxidative reactions are
central for life and are commonly performed in nature as well as in the laboratory. In
fact, approximately 30% of total chemical production involves oxidation, which
represents the second largest chemical process utilized in industry.237 A functional group
is commonly converted to a more highly oxidized form by the addition of oxygen and/or
removal of hydrogen.81 The currently available oxidation methods allow for scientists to
synthesize many key chemicals and intermediates, including aldehydes, ketones,
alcohols, epoxides, esters, and carboxylic acids. Despite the advances that have been
made toward oxidative transformations, oxidation methods have been traditionally
dominated by the use of toxic and heavy metal reagents or catalysts. To further improve
and expand upon the synthetic chemist’s arsenal of asymmetric oxidation methods, the
following review will familiarize the reader with the progress that has been made in
utilizing chiral hypervalent iodine(III) reagents to effectively carry out asymmetric
oxidative transformations.
3.3 CHIRAL HYPERVALENT IODINE(III) REAGENTS FOR
ENANTIOSELECTIVE OXIDATIVE TRANSFORMATIONS

57
3.31 ASYMMETRIC OXIDATION OF SULFIDES TO SULFOXIDES
Chiral sulfoxides have dominated as premier chiral organosulfur compounds and
have displayed great importance in many chemical applications. Two of the more
impactful areas in which enantiopure sulfoxides have been used are: as chiral auxiliaries
and chiral ligands in asymmetric synthesis238, 239 and as pharmaceutically active
compounds240 such as Nexium®, Nuvigil® and Clinoril® (Figure 3.1). The preparation of
optically active chiral sulfoxides has been a challenging area of organic chemistry, yet
there is a growing interest in seeking synthetic routes in order to obtain this valuable class
of chiral compounds.
The introduction of chiral hypervalent iodine reagents can be attributed to Pribam,
Amey and Martin. In 1906, Pribam synthesized diphenyliodonium tartrate to use as the
first chiral hypervalent iodine reagent.241 In the 1970s, Amey and Martin described the
synthesis of cyclic, chiral iodanes.242 While these were essential discoveries, chiral
hypervalent iodine compounds were not synthetically utilized until decades later. In
1986, Imamoto and Koto oxidized prochiral sulfides (3d) to enantio-enriched sulfoxides
N
O
S
N
N
ONa
Nexium® (Esomeprazole sodium)Proton pump inhibitor
Nuvigil® (Armodafinil)Wakefulness stimulant
Figure 3.1 Chiral sulfoxide containing drugs
SO
FCOOH
Clinoril® (Sulindac)Antiarthritic
OS
NH2
OO

58
(3e) with the use of aryl-l3-iodane reagents (3c) (Scheme 3.1, eq. 1).68, 243 Interestingly,
the chiral oxidizing agent (3c), which was generated in situ from reacting an L-tartaric
acid anhydride derivative (3a) with iodosylbenzene (3b), was only able to induce high
enantioselectivities when a C2-symmetric shape was incorporated into the proposed
seven-membered ring (3c). When using chiral acetyl-L-lactic acids, the authors reported
racemic mixtures of sulfoxides. Koser also created an in situ active chiral I(III) agent by
adding either (diacetoxyiodo)benzene (3f, PIDA) or iodosylbenzene (3b) to tartaric
anhydride analogs (3a).244 In contrast to Imamoto’s work, Koser suggested that the active
chiral iodine(III) benzoyl tartrate reagent was a polymer (3g) rather than a seven-
membered ring as previously proposed (i.e. 3c).
Varvoglis and co-workers245 generated novel (+)-camphorsulfonic acid
derivatives (3i) to act as chiral hypervalent iodine(III) reagents, and Chen246 utilized a
similar methodology for the preparation of organosulfonyloxy analogs (3j) in good
yields, but poor enantioselectivies (Scheme 3.1, eq. 2). In 1990, another asymmetric
strategy toward the synthesis of optically active sulfoxides was detailed by Koser and
Ray (Scheme 1.3, eq. 3).247 Through ligand exchange, (+)- or (–)-menthol (3k) could be
attached to an iodine(III) moiety, thereby creating an efficient enantioselective iodine(III)
reagent (3l). Following recrystallization and base-mediated hydrolysis, this method
furnished chiral sulfoxides (3m) in up to 99% ee.
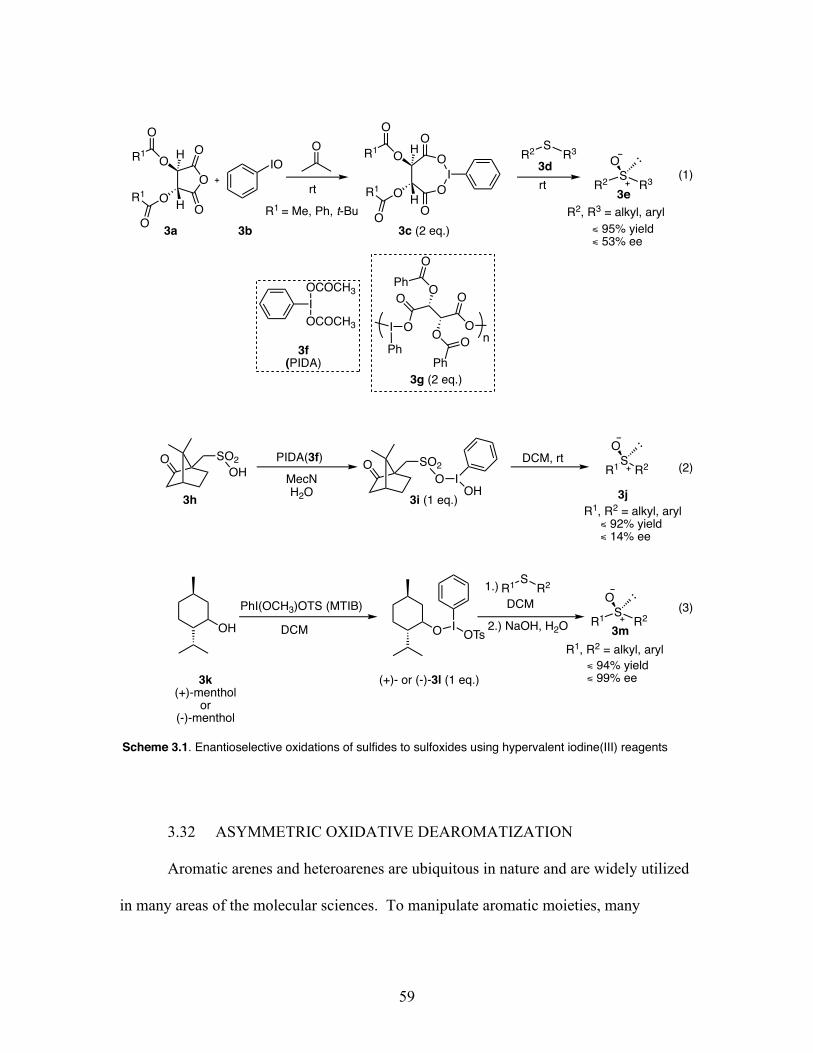
59
3.32 ASYMMETRIC OXIDATIVE DEAROMATIZATION
Aromatic arenes and heteroarenes are ubiquitous in nature and are widely utilized
in many areas of the molecular sciences. To manipulate aromatic moieties, many
O
O
O
O
O
H
H
IOO
3a 3b 3c (2 eq.)R1 = Me, Ph, t-Bu
(1)
(2)
R1 SR2
rt R2 SR3
O
R2, R3 = alkyl, aryl
rt3e
SO2O I
OH
O
⩽ 95% yield⩽ 53% ee
SO2OH
O PIDA(3f)MecNH2O3h 3i (1 eq.)
⩽ 92% yield⩽ 14% ee
R1SR2
O
3jR1, R2 = alkyl, aryl
O I OTsOH
(+)- or (-)-3l (1 eq.)3k(+)-menthol
or(-)-menthol
DCM
PhI(OCH3)OTS (MTIB)R1 S
R2
O
R1, R2 = alkyl, aryl3m
⩽ 94% yield⩽ 99% ee
1.)
2.) NaOH, H2O
DCM (3)
R2 SR3
DCM, rt
Scheme 3.1. Enantioselective oxidations of sulfides to sulfoxides using hypervalent iodine(III) reagents
O
R1
O
R1
O
O
H
H
O
R1
O
R1 OI
OO
O
3g (2 eq.)
Ph O
O
OO
O
PhO
O
OI
Phn
3d
IOCOCH3
OCOCH3
3f (PIDA)

60
chemists rely on well-established transformations, such as Friedel-Crafts and Sandmeyer
reactions. One specific transformation, dearomatization, has perpetually sparked interest
in the synthetic community. Dearomatization reactions have the potential to transform
aromatic substrates into more complex ring systems or into nonaromatic products that can
act as valuable synthetic building blocks.248-251
Siegel and Antony252 were the first to utilize hypervalent iodine reagents for
dearomatizations, however Kita253 further developed this chemistry by disclosing the first
enantioselective dearomatization strategy. Kita’s asymmetric phenolic oxidation was
carried out with a novel l3-iodane reagent that contained a conformationally rigid
spirocyclic backbone (Scheme 3.2, 7n).
OH
R
OO
O
RCHCl3, -50 ℃
⩽ 86% yield⩽ 86% ee
OH
O
Scheme 3.2 Kita’s first asymmetric dearomatizing spirolactonizations using a hypervalent iodine(III) reagent
3n0.55 eq
I
IO
OAc
OAc
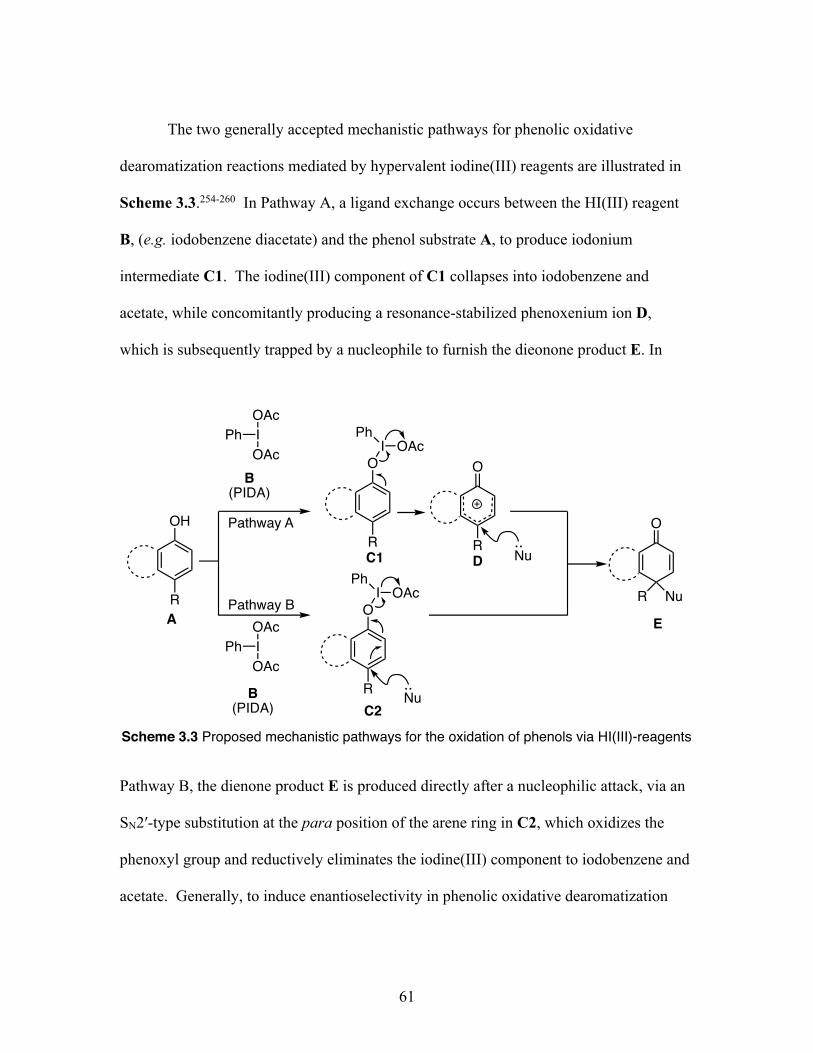
61
The two generally accepted mechanistic pathways for phenolic oxidative
dearomatization reactions mediated by hypervalent iodine(III) reagents are illustrated in
Scheme 3.3.254-260 In Pathway A, a ligand exchange occurs between the HI(III) reagent
B, (e.g. iodobenzene diacetate) and the phenol substrate A, to produce iodonium
intermediate C1. The iodine(III) component of C1 collapses into iodobenzene and
acetate, while concomitantly producing a resonance-stabilized phenoxenium ion D,
which is subsequently trapped by a nucleophile to furnish the dieonone product E. In
Pathway B, the dienone product E is produced directly after a nucleophilic attack, via an
SN2′-type substitution at the para position of the arene ring in C2, which oxidizes the
phenoxyl group and reductively eliminates the iodine(III) component to iodobenzene and
acetate. Generally, to induce enantioselectivity in phenolic oxidative dearomatization
OH
RO
R
IPh
OAc
O
R
IPh
OAc
Nu
O
RNu
O
R Nu
Ph IOAc
OAcB
(PIDA)
Ph IOAc
OAc
B (PIDA)
Pathway A
Pathway B
Scheme 3.3 Proposed mechanistic pathways for the oxidation of phenols via HI(III)-reagents
A
C1 D
C2
E
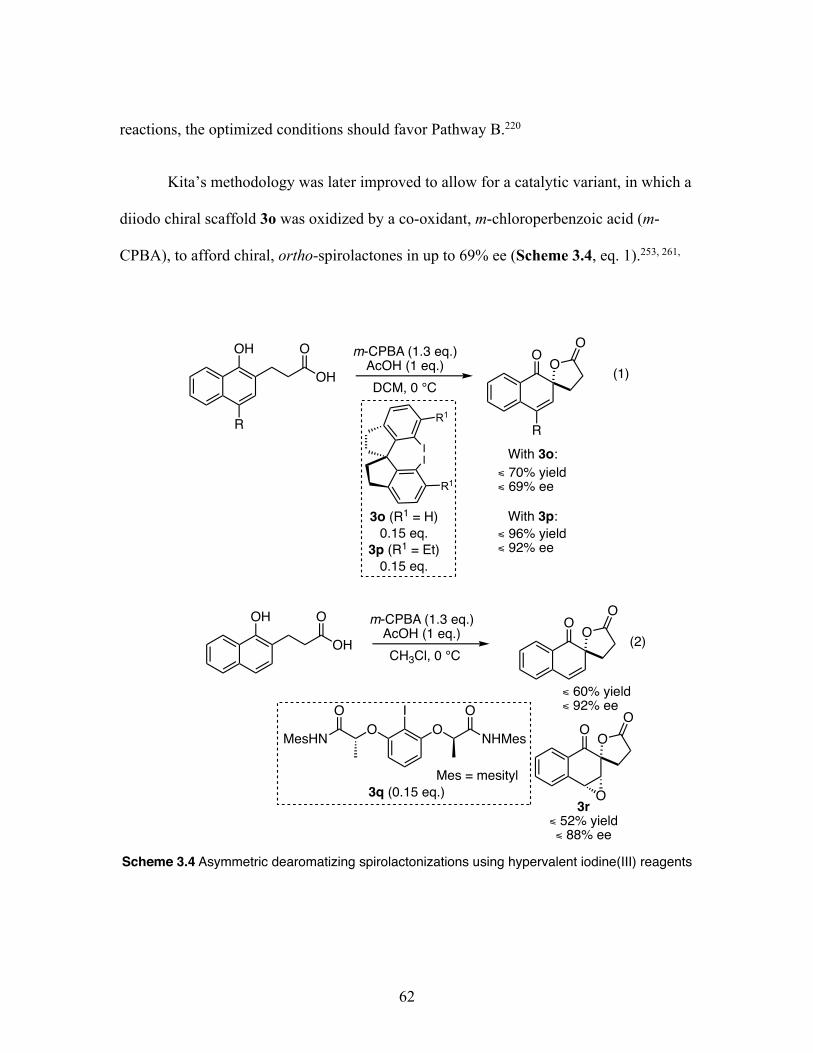
62
reactions, the optimized conditions should favor Pathway B.220
Kita’s methodology was later improved to allow for a catalytic variant, in which a
diiodo chiral scaffold 3o was oxidized by a co-oxidant, m-chloroperbenzoic acid (m-
CPBA), to afford chiral, ortho-spirolactones in up to 69% ee (Scheme 3.4, eq. 1).253, 261,
OH
R
OO
O
R
DCM, 0 ℃
m-CPBA (1.3 eq.)AcOH (1 eq.)
IOO NHMes
O
MesHN
O
(1)
3o (R1 = H)0.15 eq.
3p (R1 = Et)0.15 eq.
⩽ 70% yield⩽ 69% ee
OH
O
With 3o:
With 3p:⩽ 96% yield⩽ 92% ee
OHO
OO
CH3Cl, 0 ℃
m-CPBA (1.3 eq.)AcOH (1 eq.) (2)OH
O
3q (0.15 eq.)Mes = mesityl
⩽ 60% yield⩽ 92% ee
Scheme 3.4 Asymmetric dearomatizing spirolactonizations using hypervalent iodine(III) reagents
3r⩽ 52% yield⩽ 88% ee
OO
O
O
II
R1
R1

63
262 To understand the structural influence the catalyst had on the chiral induction, Kita et
al. investigated the ortho positioned R1 groups relative to the aryl iodine.263 By
synthesizing various spirobiindane-containing chiral reagents, they found an ethyl
substituent in the ortho-position (3p) could increase the enantioselectivity of the
dearomatizing spirocyclization of napthols up to 92% ee. With the optimized conditions,
Kita subjected Ishihara and co-worker’s C2-symmetrical chiral hypervalent iodine(III)
scaffold (3q)264, 265 to the oxidative dearomatization of naphthols, which returned an
unsubstituted spirocyclic product in a similar 92% ee (Scheme 3.4, eq. 2).263 It should be
noted that while this reaction is successful with lower equivalents of the co-oxidant, if
five or more equivalents of m-CPBA are added, the olefin-containing product is
selectively oxidized to an epoxide (Scheme 3.4, eq. 2, 3r).264
This work was extended by Quideau to allow for an iodoarene-mediated
asymmetric hydroxylative phenol dearomatization, which can provide chiral o-quinols
(Scheme 3.5, eq. 1, 3u).266 The active catalyst was generated in situ with use of a co-
oxidant, m-CPBA, but as similarly described by Kita and Ishihara, if the co-oxidant is in
excess, the resulting product undergoes subsequent epoxidation (3v). Harned et. al.
applied DFT calculations to design tricyclic chiral catalyst 3x, a dimethyl tartrate
derivative.260, 267 This chiral aryl iodide catalyst was employed in an asymmetric
oxidative dearomatization reaction to yield enantioenriched p-quinols (Scheme 3.5, eq. 2,
3y). Quideau also performed an intramolecular hypervalent iodine-induced oxidative
dearomatization to generate a 2,5-cyclohexadienone derivative 3aa in 40% ee (Scheme
3.5, eq. 3).260
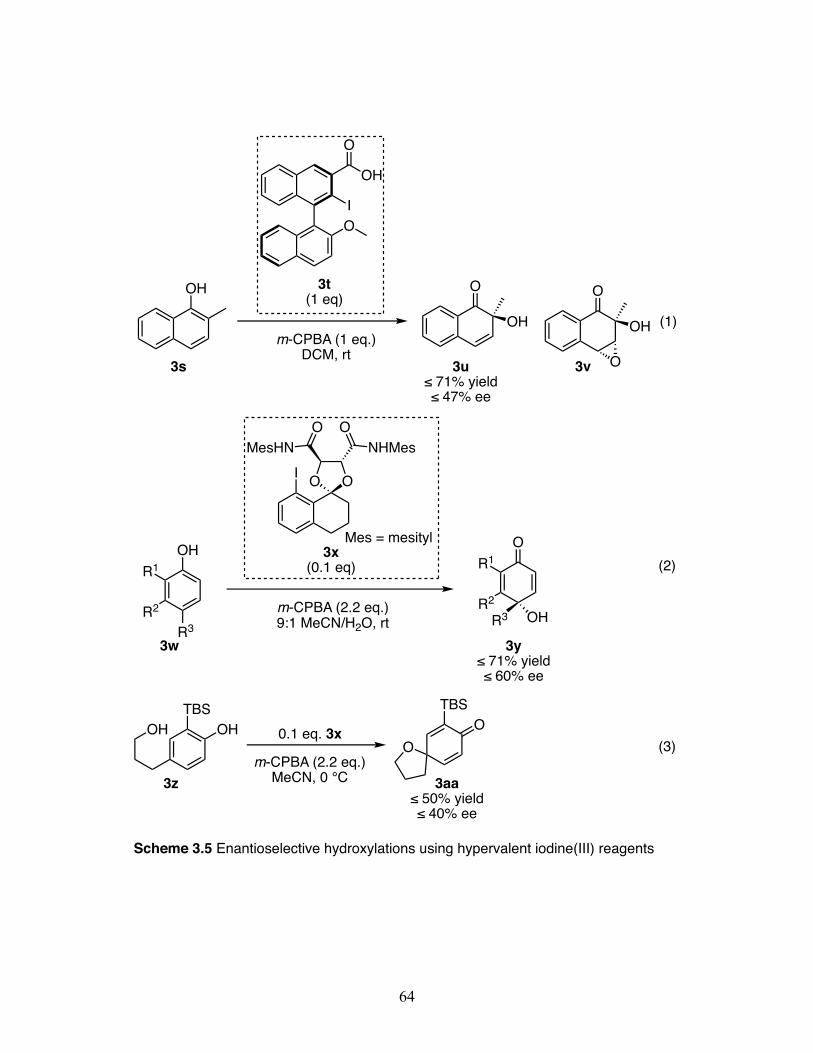
64
I
OH
O
O
OH O
O
OH
O
OH
OO
OMesHN
ONHMes
I
OHR1
R2
R3
O
R3 OH
R1
R2
3t (1 eq)
m-CPBA (1 eq.)DCM, rt
3u≤ 71% yield≤ 47% ee
3v3s
(1)
Scheme 3.5 Enantioselective hydroxylations using hypervalent iodine(III) reagents
3x(0.1 eq)
m-CPBA (2.2 eq.)9:1 MeCN/H2O, rt
3y≤ 71% yield≤ 60% ee
3w
(2)
Mes = mesityl
OH OHTBS
m-CPBA (2.2 eq.)MeCN, 0 ℃
0.1 eq. 3x OTBS
O
3aa≤ 50% yield≤ 40% ee
3z
(3)

65
3.33 ALPHA FUNCTIONALIZATION OF CARBONYLS
Enantiopure 𝛼-functionalized carbonyl compounds are valuable and versatile
chiral synthons that enable rapid access to synthetic targets.96, 133, 224, 226 They can be
used for the construction of complex natural products, pharmaceutical agents, or key
synthetic intermediates. For example, Figure 3.2 highlights several pharmaceutically
active molecules that contain an α-functionalized carbonyl moiety. A significant amount
of progress toward the α-functionalization of carbonyl compounds through the use of
selective hypervalent iodine(III) reagents is presented below.
The general reactivity profile of carbonyl compounds in the context of
hypervalent iodine(III) reagents is illustrated in Scheme 3.6.268 Under acidic or basic
conditions, ketones (Scheme 3.6, eq. 1, 3ab) can equilibrate to their enol/enolate isomers
(3ac). A lone pair of electrons from the oxygen can swing down to produce a C=O π-
bond, which breaks the C=C π-bond, and subsequently permits the α-carbon to trap an
electrophile. This overall process generates an 𝛼-substituted carbonyl compound (3ad),
in which the electrophile is trapped at the α-position. Similarly, the same enolizable
Figure 3.2 Important pharmaceutical agents that contain an !-functionalized carbonyl
ON
Amfepramone (diethylpropion)Appetite suppressant
Cl
N
S
O
H3CO
Plavix® (Clopidogrel)Antiplatelet agent
H3CO
O HN
Ritalin® (Methylphenidate)Central nervous system stimulant
N
S
O
AcO
F
Effient® (Prasugrel)Antiplatelet agent

66
carbonyl compound (Scheme 3.6, eq. 2, 3ae) can trap an electrophilic species, in this
case a hypervalent iodine(III) species, to produce the α-substituted intermediate 3af.
However, due to the highly electrophilic nature of the iodine(III) species, this unstable
intermediate is susceptible to a subsequent nucleophilic attack. Upon nucleophilic attack,
the iodine(III) collapses to a univalent iodine by-product (iodobenzene) via reductive
elimination, to yield an α-substituted carbonyl product that formally contains a
nucleophile in the α-position (3ag). It can be reasoned that this reaction efficiently
proceeds to 3ag, owing to some key factors: (1) The iodine(III) is highly electrophilic,
O
RZ
O
RZ
R
EO
RZ
E
Typical reactivity of enolates with electrophiles
“Umpolung” reactivity of enolates with nucleophiles
O
RZ
R
IIIIO
RZ
IIIINu O
RZ
Nu
Scheme 3.6 Typical reactivity to functionalize ketones via HI(III) “umpolung” reactivity
3ab 3ac 3ad
3ae 3af 3ag
(1)
(2)
R, Z = substituentNu = Generic nucleophileE = Generic electrophileIIII = Generic iodine(III) reagent

67
and is considered a hypernucleofuge, allowing for it to act as an excellent leaving group,
(2) The iodine(III) leaving process is entropically favorable, in which one molecule (3af)
is split into three molecules (the product 3ag, iodobenzene, and an anionic ligand that
was supplied by the iodine(III) reagent) (cf. chapter 1, Scheme 1.4), (3) The high stability
of the iodobenzene by-product thermodynamically drives the reaction forward.
Therefore, hypervalent iodine(III) compounds have the unique ability to alter the typical
reactivity profile of enolizable carbonyl compounds by trapping a nucleophile, rather than
an electrophile, in the α-position; this inversion of polarity is known as an umpolung
strategy. The umpolung nature of hypervalent iodine(III) species has been exploited in
various chemical transformations.268, 269
Whilethe synthesis of α-functionalized chiral carbonyl compounds has
immensely advanced over the last decade, the introduction of α-carbonyl substituents in
an asymmetric fashion mediated by hypervalent iodine compounds has only recently
emerged.223, 224, 226 In 1990, Ochaia and co-workers synthesized chiral diaryliodonium
salts (Scheme 3.7, eq. 1, 3ai) to induce the first hypervalent iodine-mediated direct 𝛼-
phenylation of cyclic β-ketoesters (3aj).270 The chiral l3-organoiodane gave a moderate
enantioinduction (up to 53% ee), but prevailed as a highly regioselective reagent (i.e. no
O-phenyl congener was isolated). Metal-free enantioselective arylations were further
developed by Olofsson and Wirth.14, 230, 271, 272
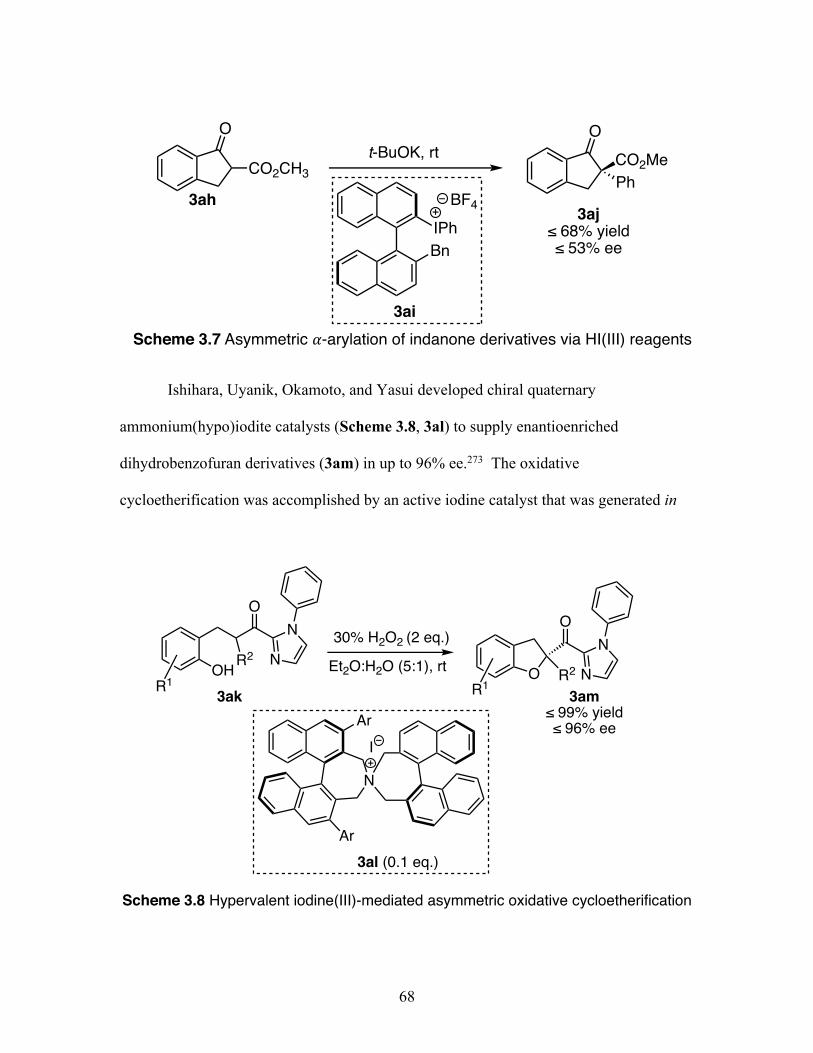
68
Ishihara, Uyanik, Okamoto, and Yasui developed chiral quaternary
ammonium(hypo)iodite catalysts (Scheme 3.8, 3al) to supply enantioenriched
dihydrobenzofuran derivatives (3am) in up to 96% ee.273 The oxidative
cycloetherification was accomplished by an active iodine catalyst that was generated in
IPhBn
Scheme 3.7 Asymmetric !-arylation of indanone derivatives via HI(III) reagents
O
CO2CH3
OCO2MePh
t-BuOK, rt
3aj≤ 68% yield≤ 53% ee
BF4
3ai
3ah
OH
O
N
N
R1
R2
O
N
N
R1R2Et2O:H2O (5:1), rt
30% H2O2 (2 eq.)
O
N
Ar
ArI
3al (0.1 eq.)
3ak 3am≤ 99% yield≤ 96% ee
Scheme 3.8 Hypervalent iodine(III)-mediated asymmetric oxidative cycloetherification
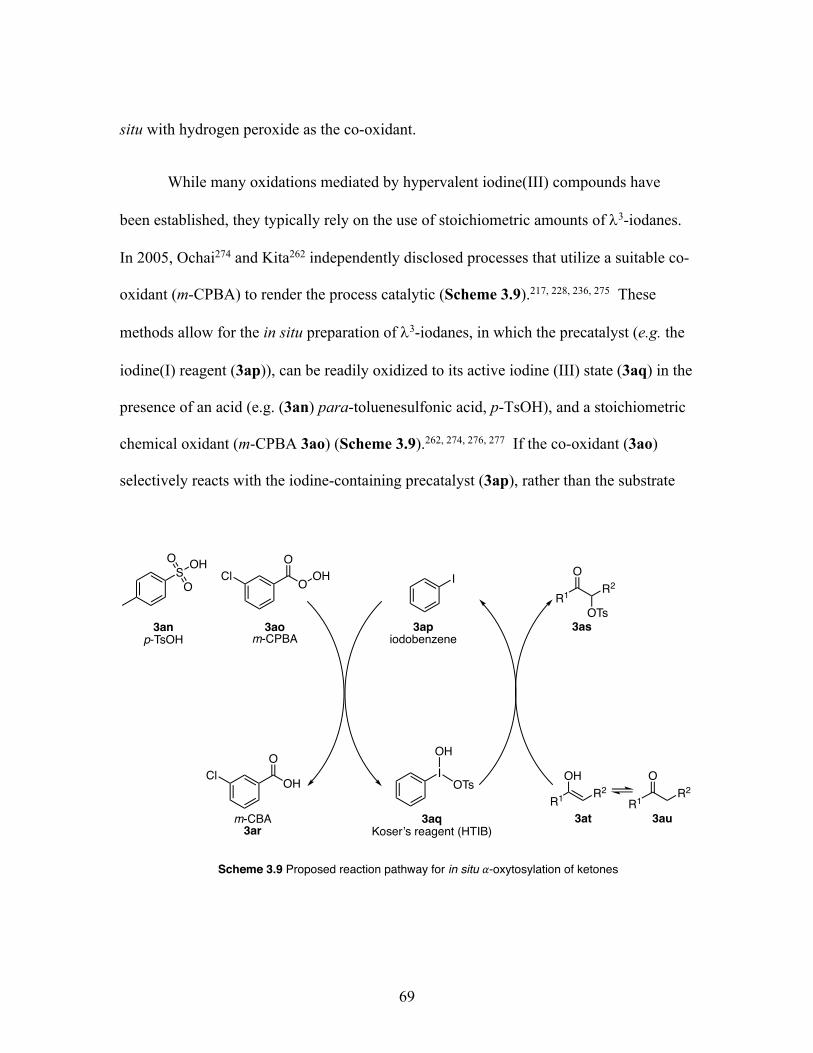
69
situ with hydrogen peroxide as the co-oxidant.
While many oxidations mediated by hypervalent iodine(III) compounds have
been established, they typically rely on the use of stoichiometric amounts of l3-iodanes.
In 2005, Ochai274 and Kita262 independently disclosed processes that utilize a suitable co-
oxidant (m-CPBA) to render the process catalytic (Scheme 3.9).217, 228, 236, 275 These
methods allow for the in situ preparation of l3-iodanes, in which the precatalyst (e.g. the
iodine(I) reagent (3ap)), can be readily oxidized to its active iodine (III) state (3aq) in the
presence of an acid (e.g. (3an) para-toluenesulfonic acid, p-TsOH), and a stoichiometric
chemical oxidant (m-CPBA 3ao) (Scheme 3.9).262, 274, 276, 277 If the co-oxidant (3ao)
selectively reacts with the iodine-containing precatalyst (3ap), rather than the substrate
Scheme 3.9 Proposed reaction pathway for in situ !-oxytosylation of ketones
I
IOTs
OH
Cl O
OOH
3aom-CPBA
SO
O
OH
Cl OH
O
3anp-TsOH
m-CBA3ar
R1 R2O
OTs
R1R2
O
R1 R2OH
3aqKoser’s reagent (HTIB)
3apiodobenzene
3as
3au3at

70
(3at), the catalytic cycle can continue by a re-oxidation of the reduced iodine(I) (3ap) by-
product to regenerate the active HI(III) catalyst (3aq).
The enantioselective α-functionalization of carbonyl compounds has been an
attractive approach toward the production of valuable chiral carbonyl synthons.224, 226
Hypervalent iodine(III) compounds in conjunction with acidic conditions (e.g. p-TsOH)
can allow for ketones, such as propiophenone, to become susceptible to an
enantioselective α-oxysulfonylation process, which delivers an enantioenriched 𝛼-
substituted carbonyl compound that is equipped with an excellent leaving group at the α-
carbon stereocenter. The putative mechanistic pathways of this reaction are shown in
Scheme 3.10. Theoretical and experimental evidence has been reported in the literature
to determine which pathway is operative, yet the reports seem to dispute one another and
the true course of transformation remains unclear.59, 68, 278, 279 The two competing
mechanistic proposals for this transformation that have emerged, invoke either an SN2’ or
an SN2 displacement (Scheme 3.10 , Pathway 1 and Pathway 2 , respectively).277, 280-282
Both mechanistic pathways commence with the ketone (4a) existing in an acid-catalyzed
equilibrium with its (E) and (Z) enol tautomers (4c and 4d, respectively). Mechanistic
Pathway 1 (shown using red arrows) involves the hypervalent I(III) species (4e, Koser’s
reagent) undergoing a nucleophilic attack from the enol oxygen atom, resulting in an O-
bonded iodono intermediate (4f). This intermediate expels water to form the O-bonded
iodonium ion (4g). The iodonium intermediate subsequently undergoes an SN2’
displacement by the tosylate ion to create the observed product (4h). In this scenario, the
SN2’ nucleophilic attack is the stereo-defining step.281 Mechanistic Pathway 2 (shown
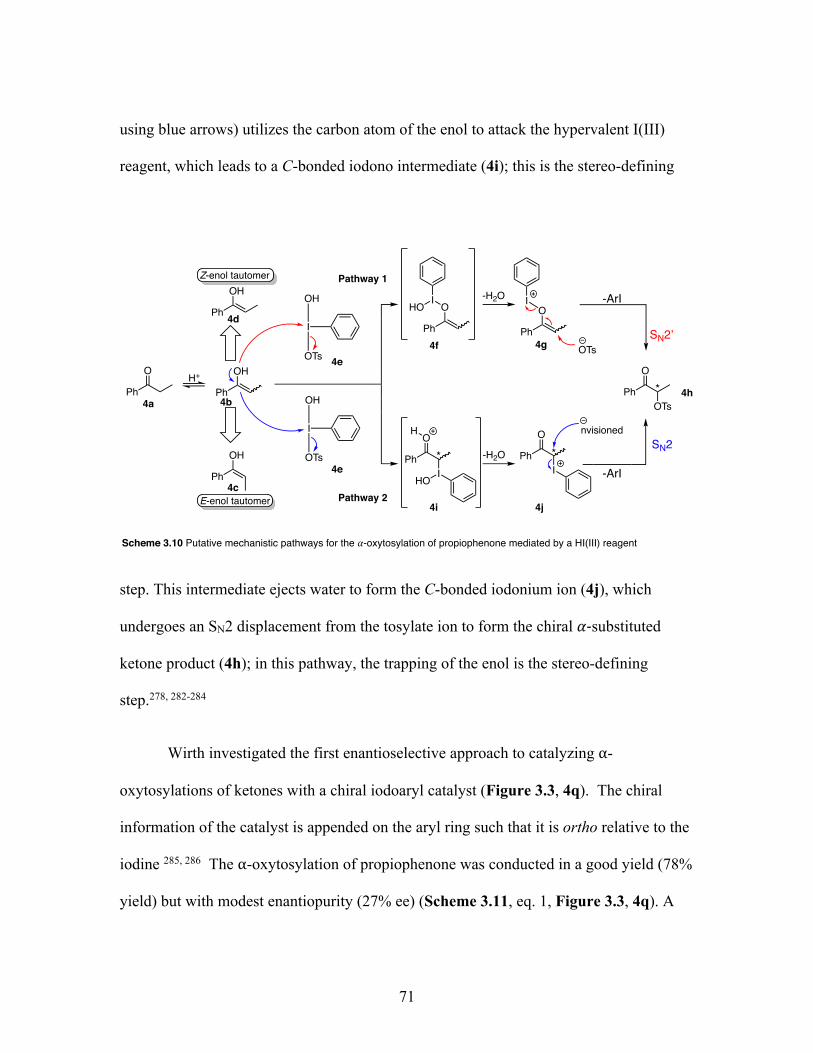
71
using blue arrows) utilizes the carbon atom of the enol to attack the hypervalent I(III)
reagent, which leads to a C-bonded iodono intermediate (4i); this is the stereo-defining
step. This intermediate ejects water to form the C-bonded iodonium ion (4j), which
undergoes an SN2 displacement from the tosylate ion to form the chiral 𝛼-substituted
ketone product (4h); in this pathway, the trapping of the enol is the stereo-defining
step.278, 282-284
Wirth investigated the first enantioselective approach to catalyzing ⍺-
oxytosylations of ketones with a chiral iodoaryl catalyst (Figure 3.3, 4q). The chiral
information of the catalyst is appended on the aryl ring such that it is ortho relative to the
iodine 285, 286 The ⍺-oxytosylation of propiophenone was conducted in a good yield (78%
yield) but with modest enantiopurity (27% ee) (Scheme 3.11, eq. 1, Figure 3.3, 4q). A
Ph
OH+
Ph
OH
I
OH
OTs
I
OH
OTs
Ph
O
Ph
O
I
IHO
HO
H
*
-H2O
-H2O
Ph
OI
Ph
O
I*
Ph
O
OTs
OTs
nvisioned
-ArI
-ArI
*
Ph
OH
Ph
OH
Z-enol tautomer
E-enol tautomer
SN2
SN2’
4a 4b
4c
4d
4e
4e4f
4i
4g
4j
4h
Pathway 1
Pathway 2
Scheme 3.10 Putative mechanistic pathways for the !-oxytosylation of propiophenone mediated by a HI(III) reagent
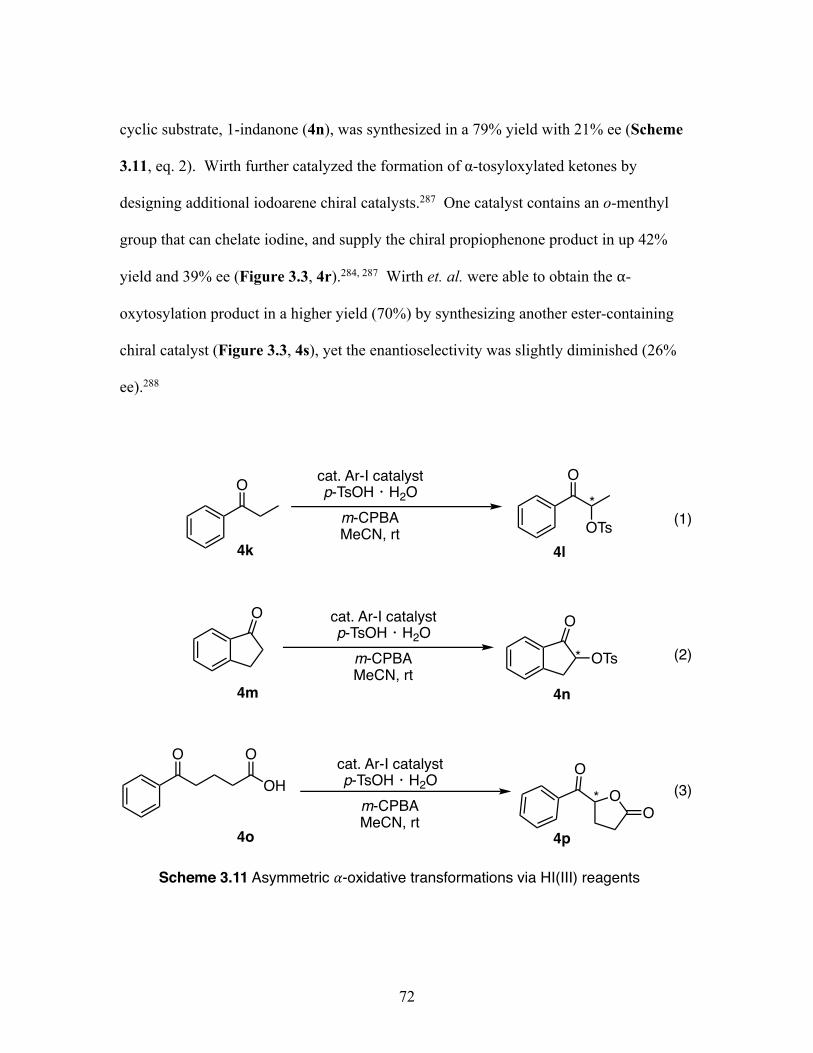
72
cyclic substrate, 1-indanone (4n), was synthesized in a 79% yield with 21% ee (Scheme
3.11, eq. 2). Wirth further catalyzed the formation of α-tosyloxylated ketones by
designing additional iodoarene chiral catalysts.287 One catalyst contains an o-menthyl
group that can chelate iodine, and supply the chiral propiophenone product in up 42%
yield and 39% ee (Figure 3.3, 4r).284, 287 Wirth et. al. were able to obtain the ⍺-
oxytosylation product in a higher yield (70%) by synthesizing another ester-containing
chiral catalyst (Figure 3.3, 4s), yet the enantioselectivity was slightly diminished (26%
ee).288
Scheme 3.11 Asymmetric !-oxidative transformations via HI(III) reagents
O O
OTsm-CPBAMeCN, rt
p-TsOH独H2Ocat. Ar-I catalyst
O
OH
O
OO
O
O O
OTs
4l
4n
(1)
(2)
(3)
m-CPBAMeCN, rt
p-TsOH独H2Ocat. Ar-I catalyst
m-CPBAMeCN, rt
p-TsOH独H2Ocat. Ar-I catalyst
4p
*
*
*
4k
4m
4o

73
Moran and Rodríguez synthesized and assessed chiral aryl iodides in an
intermolecular as well as an intramolecular asymmetric oxidation. (Scheme 3.11, eq. 1
and eq. 3).289 The most interesting catalyst was a pseudoephedrine derivative (Figure
3.3, 4t) that oriented the iodine ortho in relation to the chiral information. In the a-
oxytosylation of propiophenone, the catalyst supplied the intended product (4l) in a
moderate yield (59% yield), but with a negligible enantioselectivity (3% ee).
Nonetheless, catalyst 4t was able to selectively induce the asymmetric oxylactonization
of 5-oxo-5-phenylvaleric acid in 51% ee (Scheme 3.10, eq. 3), which was a significant
increase in selectivity over Wirth’s reagent (Figure 3.3, 4s, ~3% ee).288
Legault’s laboratory was able to apply iodoaryloxazoline-based chiral catalysts
for ⍺-functionalizations (Figure 3.3, 4u).277, 278 The most effective catalyst was
comprised of an aryl ring that accommodates an o-methyl and p-chlorine substituent
(relative to iodine) (4u). With slow addition of m-CPBA to the reaction mixture,
propiophenone was converted to ⍺-oxytosylpropiophenone in an 80% yield with a
stereoinduction of 48% ee (Scheme 3.10, eq. 1, Figure 3.3, 4u). It should be noted that
without the ortho methyl substituent, the reaction did not proceed.278
Through a seven-step synthesis, Zhang was able to synthesize spirobiindane-
based chiral iodoarenes, which he utilized as catalysts in the enantioselective ⍺-
oxytosylation of propiophenone (Scheme 3.11, eq. 1, Figure 3.3, 4v). With ethyl acetate
as the optimal solvent, Zhang’s catalyst gave an enantio-enriched product in 53% ee.290
Brenet, Berthiol, and Einhorn suggested that sterics around the iodine center may play an
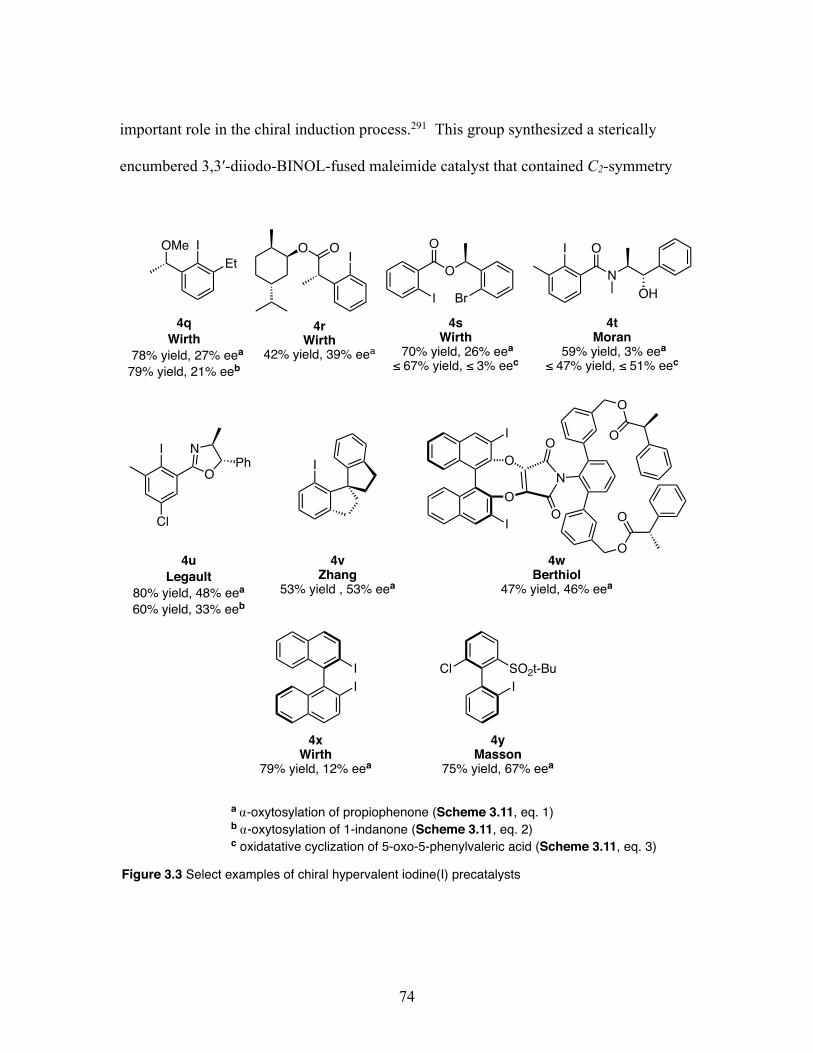
74
important role in the chiral induction process.291 This group synthesized a sterically
encumbered 3,3′-diiodo-BINOL-fused maleimide catalyst that contained C2-symmetry
Cl SO2t-BuI
4qWirth
78% yield, 27% eea
79% yield, 21% eeb
IOMe
I
Cl
I
O
NPh
Et
I
O
O
Br
4sWirth
70% yield, 26% eea
≤ 67% yield, ≤ 3% eec
I
N
O
OH
4uLegault
80% yield, 48% eea
60% yield, 33% eeb
4vZhang
53% yield , 53% eea
4rWirth
42% yield, 39% eea
II
4xWirth
79% yield, 12% eea
4yMasson
75% yield, 67% eea
IOO
4wBerthiol
47% yield, 46% eea
O
ON
O
O
O
O
O
OI
I
4tMoran
59% yield, 3% eea
≤ 47% yield, ≤ 51% eec
a ⍺-oxytosylation of propiophenone (Scheme 3.11, eq. 1)b ⍺-oxytosylation of 1-indanone (Scheme 3.11, eq. 2) c oxidatative cyclization of 5-oxo-5-phenylvaleric acid (Scheme 3.11, eq. 3)
Figure 3.3 Select examples of chiral hypervalent iodine(I) precatalysts

75
(Figure 3.3, 4w). In the ⍺-oxytosylation of propiophenone, catalyst 4w imparted an
enantioselective outcome with up to 46% ee.
Richardson and Wirth also implemented axial chirality by synthesizing a
binaphthyl-based chiral diiodo catalyst (Figure 3.3, 4x).286 This catalyst gave poor
enantioselectivities (£12% ee) but moderate yields (79% yield) in the oxytosylation of
propiophenone (Scheme 3.11, eq. 1). Wirth and Mizar further developed protocols that
employ silyl enol ethers and nucleophiles other than tosylates to broaden the scope of the
α-functionalization of carbonyl compounds.268, 286, 287 In 2017, Masson disclosed the use
of chiral, non C2-symmetric iodoarene catalysts for the direct α‑oxysulfonylation of
ketones (4y).220 This unique catalyst can offer respectable yields (75% yield) and
enantioselectivities up to 67% ee in the α-oxytosylation of propiophenone (Scheme 3.11,
eq. 1).
Legault and Basdevant made great strides in the area of α-oxytosylated ketones.
By coupling the use of Ishihara’s bis-lactate-derived, C2-symmetric iodoarene catalyst
scaffold (Scheme 3.12, 4y) and preformed enol acetate substrates (4z), enantio-enriched
α-oxytosylated ketones (5b) were accessible in synthetically useful yields and
enantioselectivities (up to 70% yield and 90% ee).292 While this is an impressive feat, the
requirement to use preformed enol acetates is cumbersome, entailing the in situ
preparation of lithium diisopropylamide (LDA), a very strong and moisture/air sensitive
base, as well as a tedious dropwise addition protocol over 13 h. Furthermore, when
utilizing these conditions in the direct α-oxytosylation of ketones, this method produces

76
low enantioselectivities. Finally, under catalytic conditions, the only substrate that gave a
high steroinduction was propiophenone. While a broader scope of ketones underwent the
reaction in synthetically useful enantioselectivities, they were only evaluated using a
stoichiometric amount of the chiral catalyst 5a.292
The previously described studies include some of the pioneering breakthroughs
that have greatly advanced methodologies designed for catalytic asymmetric oxidative
transformations. Nevertheless, a persistent obstacle has been the inability to promote
hypervalent iodine-mediated organic reactions in an a direct, catalytic, and
enantioselective manner. Particularly problematic in this regard are two of the most
studied hypervalent asymmetric iodine-mediated reactions: the α-oxytosylation of
propiophenone and the oxidative cyclization of 5-oxo-5-phenylvaleric acid. These
particular reactions have been studied for decades, however efficiently promoting the
formation of products with synthetically useful enantioselectivity and high yields has
O O
OTsm-CPBA (1.1 eq.)MeCN, rt, 13 h
p-TsOH独H2O (1.1 eq)0.2 eq. Ar-I catalyst
O
IOO N
O
N
O
Me Me
5a
Me Me
Scheme 3.12 Legault and Basdevant’s utilization of enol acetates for asymmetric catalysis
5b70% yield90% ee
4z

77
proven to be a formidable challenge which warrants further investigation. Furthermore,
most of the hypervalent I(III) asymmetric catalysts described in this chapter require
lengthy and sometimes tedious protocols for their preparation, which limits the ability to
make iterative changes to the catalyst scaffold for further optimization. The efforts
described below aim to resolve the aforementioned obstacles associated with HI(III)-
mediated oxidative transformations by synthesizing and implementing modular and
tunable iodoarene-containing catalysts that are capable of providing high yielding,
enantiopure products.
3.4 IODOARENE-CONTAINING ORGANOCATALYSTS FOR HI(III)
OXIDATIVE TRANSFORMATIONS
Our group has investigated the incorporation of an iodoarene functional group
into a chiral framework, such that the chiral information can be transferred from the
HI(III) reagent to the substrate in order to generate high yielding, enantioenriched
products. We envisioned the use of peptide-based scaffolds, that contain an active
iodoarene component, to operate as the chiral inducing agent. As discussed in Chapter 2,
peptides have the ability to induce high enantioselectivities in organic transformations
due to the immense chiral space they provide, and can thereby create a chiral
environment for selective substrate recognition.128, 168, 169 Fmoc Solid-Phase Peptide
Synthesis (SPPS) techniques allow for the rapid access and evaluation of catalytic
libraries.293 Figure 3.4 illustrates our envisioned generic peptide-based scaffolds that
incorporate iodoarene moieties (highlighted in blue) at the N-terminus of a linear (6a) or
β-turn (6b) scaffold, which has been documented in literature to be nucleated from a D-

78
Ala-Pro sequence.128, 169, 173, 190, 294, 295 The overall goal of this research is to unite the
fields of hypervalent iodine(III) catalysis with peptide-mediated asymmetric synthesis to
generate chiral HI(III) peptide-based catalysts that can effectively induce enantioselective
and high yielding outcomes in a range of synthetically useful oxidative transformations.
Initial investigations were focused on the installation of the catalytically active
iodoarene moiety into a highly modular chiral peptide framework. We considered that
this concept can be achieved through a peptide bond, or more specifically a benzamide
linkage, as portrayed in blue in Figure 3.4. The amide linkage presents itself as a facile
method for installing aryl iodo active sites onto a variety of amino acid side chains,
thereby allowing for iterative modifications. Therefore, in the context of our strategy,
probing the influence of an amide bond on the catalysis of the desired transformations
was critical in order to ensure that this strategy for incorporation of the catalytic site into
the peptide framework could be done while maintaining acceptable activity. To reveal
NH2
HN
NH
O
IR
O
OR2
R1 n
6a
Figure 3.4 Envisioned peptide-based catalytic scaffolds that contain an iodoarene active site at the N-terminus
6bLinear peptide-scaffold
H2N
NH
HNO
O
OR1
R2
NH
CH3O
NO
NH
R3
I
O
Rβ-turn peptide-scaffold

79
the steric and electronic properties that affect catalytic performance, a series of twelve
iodoarene containing amides (Figure 3.5, Catalysts 1-12) were synthesized (Scheme
3.13, yields shown in parentheses) and then utilized in the direct α-oxytosylation of
propiophenone.296 A generic amidation reaction, which was used to synthesize the N-
butyl-iodobenzamide organocatalysts, is shown in Scheme 3.13. This conventional
amidation reaction allows for an iodoarene-containing carboxylic acid substrate (6c) to
react with thionyl chloride to form an acyl chloride (6d), which can be displaced by N-
butyl amine to form the desired amide (e.g. Catalyst 3) (Scheme 3.13).
The suite of iodoarene containing catalysts shown in Figure 3.5 were chosen due
to their varying electronic and steric substituents. The influence of the electron-
withdrawing amide motif was probed in the α-oxytosylation of propiophenone (Table
1.1). The α-oxytosylation reaction was conducted at room temperature on a 0.1 mmol
scale with acetonitrile as the solvent, para-toluenesulfonic acid monohydrate (p-TsOH・
H2O) as the acid and nucleophile, iodo-benzamide as the catalyst (Catalysts 1-12), and
OH
O 1.) Thionyl Chloride2.) Butylamine
Dry DCM0-25 °C
NH
O
Catalyst 3 (93%)6c
Scheme 3.13 Standard synthesis of the N-butyl-iodobenzamide catalysts
Cl
O
6d
I
I
I
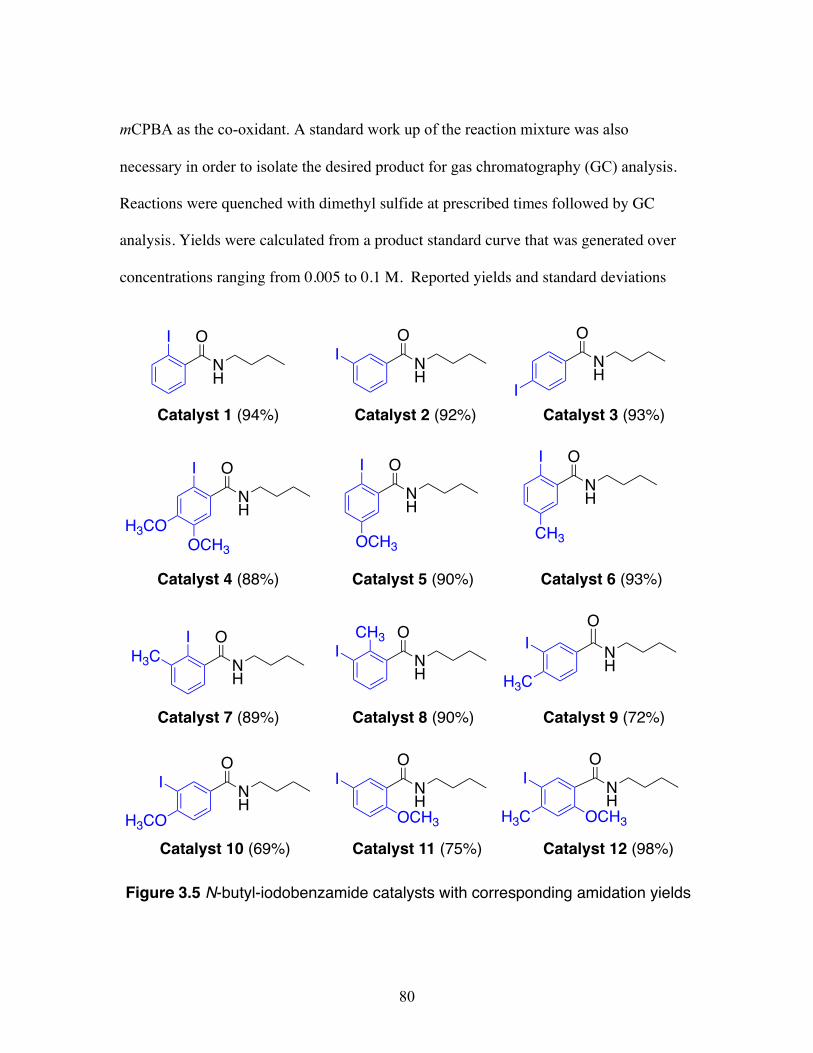
80
mCPBA as the co-oxidant. A standard work up of the reaction mixture was also
necessary in order to isolate the desired product for gas chromatography (GC) analysis.
Reactions were quenched with dimethyl sulfide at prescribed times followed by GC
analysis. Yields were calculated from a product standard curve that was generated over
concentrations ranging from 0.005 to 0.1 M. Reported yields and standard deviations
ONH
I ONH
IO
NH
I
ONH
H3CO
I
OCH3
ONH
I
OCH3
ONH
I
CH3
ONH
IH3C
ONH
ICH3
O
NH
I
H3C
ONH
H3CO
ONH
I
OCH3
ONH
H3C
Catalyst 2 (92%) Catalyst 3 (93%)Catalyst 1 (94%)
Catalyst 4 (88%) Catalyst 5 (90%) Catalyst 6 (93%)
Catalyst 7 (89%) Catalyst 8 (90%) Catalyst 9 (72%)
Catalyst 10 (69%) Catalyst 11 (75%) Catalyst 12 (98%)
I
OCH3
I
Figure 3.5 N-butyl-iodobenzamide catalysts with corresponding amidation yields

81
were collected from averaged triplicate runs with tetraglyme being utilized as the internal
standard.
Conducting 48-hour reactions revealed the relative rates of the differently
substituted iodoarene catalysts (Entries 1-12, Table 1.1). Relative to the iodine, meta-
and para-positioned amides were more effective as catalysts than ortho-substituted
amides (Entries 1-3, Table 1.1). This suggests that the sluggish behavior observed with
using Catalyst 1 may be due to steric hindrance as well as the inductive electron
withdrawing nature of the ortho positioned amide. Catalysts 4-7 were synthesized to
investigate how electronics and sterics can affect the rate in relation to the parent ortho-
substituted iodoarene (Catalyst 1). With the aim to diminish the electron-withdrawing
nature of the amide, an electron donating group (i.e., methoxy) was positioned para to the
amide substituent (Catalyst 4). An additional electron donating group, such as a
methoxy or methyl group, was situated para to the iodine with the intent to generate a
more electron rich iodine center (Catalysts 4-6). These analogs were inspired by the
work done by Wirth284 and Legault278, who reported slight increases in catalytic activity
when iodine was situated para to an electron-releasing group. With little to no rate
enhancement detected, these studies indicate that electronics may play a rather minor role
in the catalytic activity of the iodobenzamides (Table 1.1, entries 4-6). Catalyst 7 was
created to induce the “hypervalent twist” phenomenon, which in essence, states that
catalysts can achieve higher relative rates if sterically bulky substituents are located ortho
to the iodine.278, 284, 297 With an approximate 10 fold increase in rate, the “hypervalent
twist” phenomenon appears to be validated in our system (Table 1.1, entry 1 vs. entry 7).

82
Legault’s computational research suggests that if a Lewis base (e.g. an amide) is
positioned ortho to the aryl iodine(I) precatalyst, when activated to iodine(III), the
catalyst is stabilized, which slows down the ensuing displacement, and therefore
decreases catalytic reactivity.278 Invoking this reasoning, Catalyst 1 underperforms in
the oxidative 𝛼-oxytosylation of propiophenone, when compared to Catalyst 2 and
Catalyst 3 (Table 1.1, entry 1 vs. entries 2 and 3). This same concept becomes apparent
when comparing two “hypervalent twist” congeners, Catalyst 7 and Catalyst 8, in which
a meta positioned amide relative to the iodine, performs at a much higher rate than an
ortho positioned amide (Table 1.1 entry 7 versus entry 8). Finally, the “hypervalent
twist” was probed between meta positioned amide catalysts, Catalyst 8 and Catalyst 9.
Evidently, the “hypervalent twist” phenomenon is inoperable when the Lewis base and
the iodine are distal to each other, and instead ortho-positioned steric effects play a large
role in influencing the rate of catalysis (Table 1.1, entry 8 and 9). Finally, electron
donating groups were situated around a meta positioned amide (relative to iodine), which
gave the α-oxytosylated product in relatively good to quantitative yields (Table 1.1
entries 10-12).

83
The highest relative rates obtained after 48 h were quantitative yields (Table 1.1
entries 2, 11, and 12), and as a result these, as well as the other top four catalysts, were
subjected to a four-hour reaction time in order compare the catalysts prior to completion
Entry Catalyst Yield (%)a,b
1 1 3 ± 0
2 2 quant.
3 3 82 ± 6
4 4 2 ± 0
5 5 2 ± 0
6 6 3± 0
7 7 29 ± 4
8 8 91 ± 3
9 9 76 ± 3
10 10 67 ± 4
11 11 quant.
12 12 quant.
Table 1.1 Oxidative ⍺-Oxytosylation of Propiophenone (48 h, rt)
a Average yields of triplicate runs employing tetraglyme as the ISTD.b 0.1 mmol scale.
O O
OTs
mCPBA (3 equiv)pTSA•H2O (3 equiv)Catalyst (0.1 equiv)
MeCN (1 mL)48 hrs, r.t.
*
4k 4l

84
(Table 1.2, entries 1-7). The catalyst that gave the most significant rate acceleration was
observed to be Catalyst 11, thereby supporting Wirth284 and Legualt’s277 findings that
electron donating substituents in the para position (relative to I) can promote increases in
reaction rates (Entry 6, Table 1.2). These rate studies provide the idea that attaching the
amide functionality onto the iodoarene can potentially have tremendous effects on
catalytic activity.
Entry Catalyst Yield (%)a,b
1 2 39 ± 3
2 3 19 ± 3
3 8 28 ± 2
4 9 24 ± 3
5 10 36 ± 1
6 11 44 ± 2
7 12 40 ± 1
Table 1.2 Oxidative α-Oxytosylation of Propiophenone (4 h, rt)
a Average yields of triplicate runs employing tetraglyme as the ISTD.b 0.1 mmol scale.
O O
OTs
mCPBA (3 equiv)pTSA•H2O (3 equiv)Catalyst (0.1 equiv) *
MeCN (1 mL)4 h, r.t.4k 4l

85
All 12 catalysts were evaluated in the oxytosylation of propiophenone over a 24 h
time period at 50 ℃, and the top seven catalysts were further screened at 50 ℃ for 1 hour
(Table 1.3 and Table 1.4, respectively). Similar trends were witnessed in the 50 ℃
Entry Catalyst Yield (%)a,b
1 1 16 ± 4
2 2 96 ± 2
3 3 96 ± 3
4 4 11 ± 3
5 5 12 ± 2
6 6 16 ± 3
7 7 65 ± 1
8 8 92 ± 4
9 9 quant.
10 10 quant.
11 11 97 ± 2
12 12 60 ± 5
Table 1.3 Oxidative α-Oxytosylation of Propiophenone (24 h/50 ºC)
a Average yields of triplicate runs employing tetraglyme as the ISTD.b 0.1 mmol scale.
O O
OTs
mCPBA (3 equiv)pTSA•H2O (3 equiv)Catalyst (0.1 equiv)
MeCN (1 mL)24 hr, 50 ℃
*
4k 4l

86
trials, as those observed in the room temperature reactions. The catalyst that repeatedly
furnished the oxytosylated ketone (4l) in the highest yield was Catalyst 11. This catalyst
therefore became our lead catalyst, and provided the oxytosylated ketone in high yields.
To further investigate how the amide moiety may affect the overall catalytic
activity of iodoarenes, two significant studies were completed. The first study entailed
subjecting a series of structural analogs of lead Catalyst 11 to four-hour reaction
conditions (Table 1.5, entries 1-7). The second study involved kinetic 1H NMR rate
studies involving the best, an intermediate, and an inefficient catalyst (Catalyst 11,
Entry Catalyst Yield (%)a,b
1 2 63 ± 2
2 3 57 ± 2
3 8 43 ± 1
4 9 52 ± 3
5 10 63 ± 1
6 11 68 ± 1
7 12 48 ± 1
Table 1.4. Oxidative α-Oxytosylation of Propiophenone (1 h/50 ºC)
a Average yields of triplicate runs employing tetraglyme as the ISTD.b 0.1 mmol scale.
O O
OTs
mCPBA (3 equiv)pTSA•H2O (3 equiv)Catalyst (0.1 equiv)
MeCN (1 mL)1 hr, 50 ℃
*
4k 4l

87
Catalyst 8, and Catalyst 4, respectively) in order to examine the relative rates of
oxidation from their iodine(I) to iodine(III) active state (Figure 3.6).
The structural analog study revealed that an amide bond, which will link the
active iodoarene containing catalyst and the peptide, is a valid strategy since there is no
appreciable effect on the catalytic activity when incorporating an amide functionality
(Table 1.5, entry 2 versus entries 1, 3-7). It seems that by positioning a methoxy group
para to the iodine, and placing the iodine meta to the deactivating amide, an effective
catalyst template is established (i.e. Catalyst 11). This presents a distinct catalytic
platform that differs from many other HI(III) scaffolds, in which an o-iodo scaffold is
required for an effective catalytic, chiral outcome (see Figure 3.3).
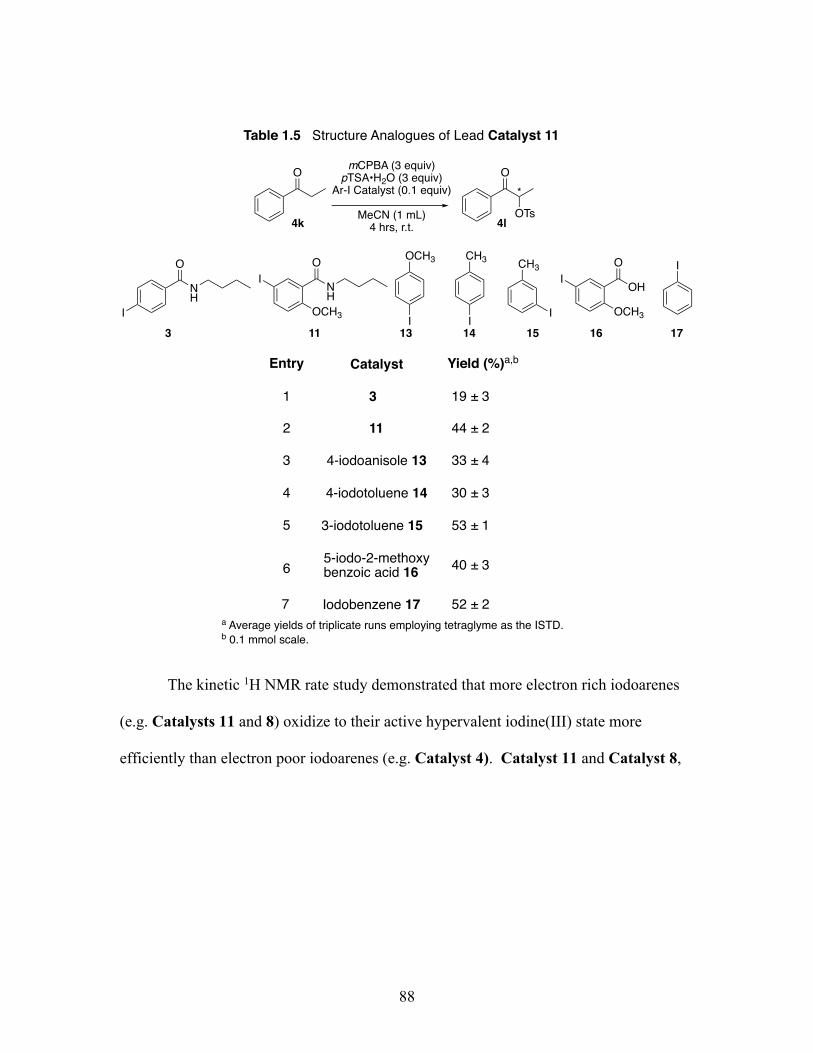
88
The kinetic 1H NMR rate study demonstrated that more electron rich iodoarenes
(e.g. Catalysts 11 and 8) oxidize to their active hypervalent iodine(III) state more
efficiently than electron poor iodoarenes (e.g. Catalyst 4). Catalyst 11 and Catalyst 8,
Entry Catalyst Yield (%)a,b
1 3 19 ± 3
2 11 44 ± 2
3 4-iodoanisole 13 33 ± 4
4 4-iodotoluene 14 30 ± 3
5 3-iodotoluene 15 53 ± 1
65-iodo-2-methoxy benzoic acid 16 40 ± 3
7 Iodobenzene 17 52 ± 2a Average yields of triplicate runs employing tetraglyme as the ISTD.b 0.1 mmol scale.
OCH3
I
CH3
I
O
OH
OCH3
I
CH3
I13 14 15 16
O O
OTs
mCPBA (3 equiv)pTSA•H2O (3 equiv)
Ar-I Catalyst (0.1 equiv)
MeCN (1 mL)4 hrs, r.t.
*
O
NH
3
I
O
NH
I
OCH3
11
Table 1.5 Structure Analogues of Lead Catalyst 11
I
17
4k 4l
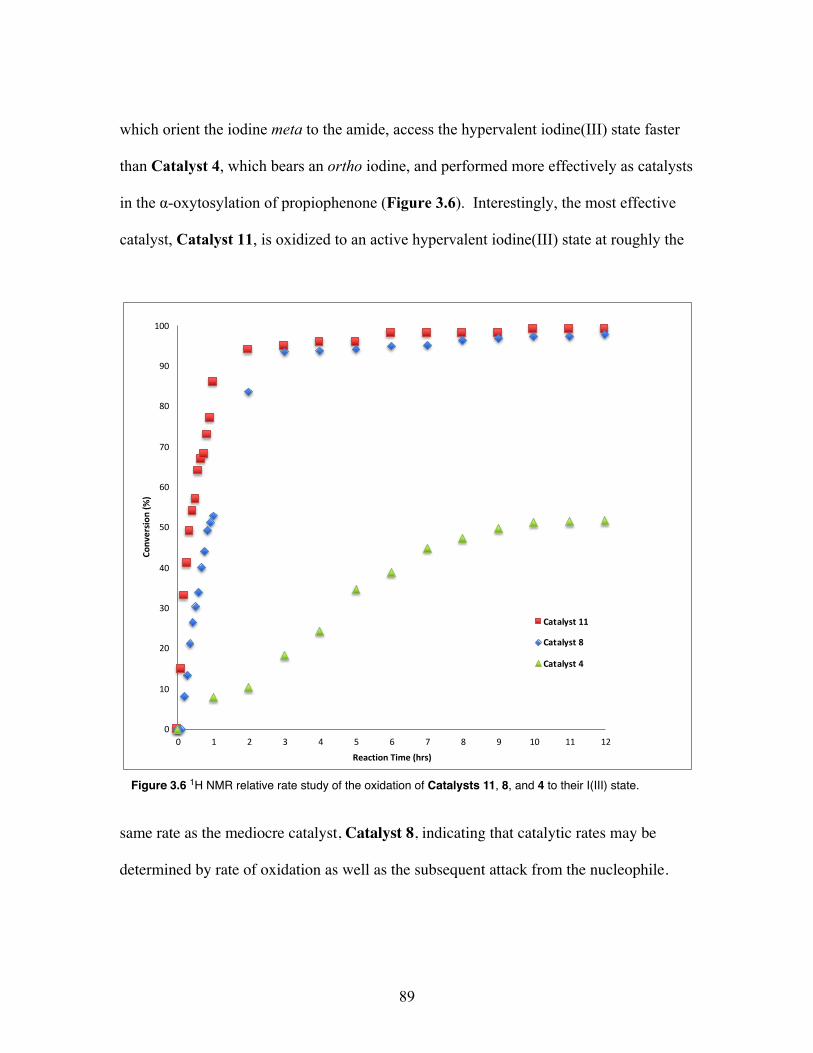
89
which orient the iodine meta to the amide, access the hypervalent iodine(III) state faster
than Catalyst 4, which bears an ortho iodine, and performed more effectively as catalysts
in the α-oxytosylation of propiophenone (Figure 3.6). Interestingly, the most effective
catalyst, Catalyst 11, is oxidized to an active hypervalent iodine(III) state at roughly the
same rate as the mediocre catalyst, Catalyst 8, indicating that catalytic rates may be
determined by rate of oxidation as well as the subsequent attack from the nucleophile.
Figure 3.6 1H NMR relative rate study of the oxidation of Catalysts 11, 8, and 4 to their I(III) state.
0
10
20
30
40
50
60
70
80
90
100
0 1 2 3 4 5 6 7 8 9 10 11 12
Conv
ersio
n (%
)
Reaction Time (hrs)
Catalyst 11
Catalyst 8
Catalyst 4

90
The suite of iodo-arene containing catalysts (Catalysts 1-12, Figure 3.5) were
also applied to an intramolecular transformation, in which a ketocarboxylic acid is
converted to a ketolactone via oxidative cyclization mediated by hypervalent iodine(III)
Entry Catalyst Yield (%)a,b
1 2 12 ± 1
2 3 54 ± 3
3 7 24 ± 2
4 8 58 ± 4
5 9 52 ± 4
6 10 64 ± 3
7 11 71 ± 3
8 12 61 ± 3
Table 1.6 Oxidative Cyclization of 5-oxo-5-phenylvaleric acid (24 h/50 ºC)
PhO
OHO
OO
Ph O
mCPBA (3 equiv)pTSA•H2O (3 equiv)Catalyst (0.1 equiv)
a Average yields of triplicate runs employing tetraglyme as the ISTD.b 0.1 mmol scale.c Isolated yields after silica gel column chromatography.d 1 mmol scale.e pTSA•H2O (1.5 eq.), Catalyst (0.2 eq.)f 1.3 mmol scale.
9c,d
10c,e,f
11
11
72
89
MeCN (1 mL)24 hrs, 50 ◦C
*
4o 4p

91
catalysts (Table 1.6). This is a prominent transformation because not only are lactones
prevalent in nature, they have found great importance in the synthetic and medicinal
community.298-300 The catalytic scaffold that achieved the highest yielding product (11b)
at 50 ℃ was again Catalyst 11, which includes a meta positioned iodine and para
positioned methoxy group (Table 1.6, entry 7). The oxidative cyclization of 5-oxo-5-
phenylvaleric acid (4o) was also carried at 1 and 1.3 mmol scales in the presence of either
10-20 mol % Catalyst 11, to afford the oxylactone (4p) in decent (72%, entry 9) to good
yields (89 %, entry 10).
Entry R Product n Time (hr) Temperature (ºC) Yield (%)a,b
1 CH3 18 1 36 50 87
2 OCH3 19 1 40 50 72
3 F 20 1 24 50 94
4 Cl 21 1 24 50 75
5 Br 22 1 24 50 85
6 H 23 0 24 50 94
7 H 4l 1 24 50 99
8 H 4l 1 48 r.t. 94
O
Table 1.7. Oxidative α-Oxytosylation of Propiophenone Derivatives
R
n
O
R
nOTs
mCPBA (3 equiv)pTSA•H2O (3 equiv)
Ar-I Catalyst 11 (0.1 equiv)
MeCN (1 mL)
a Isolated yields after silica gel column chromatography.b 1 mmol scale.

92
To exemplify and assess the value of our most reactive catalyst, Catalyst 11, a
small substrate scope was evaluated (Table 1.7, entries 1-8). Propiophenone,
acetophenone, and propiophenone derivatives with various para-substituents on the
phenyl ring were successfully synthesized in good isolated yields in up to a 1 mmol scale.
The previously mentioned studies have allowed our group to understand how an
amide linkage, which appends the iodine-containing active site to the peptide scaffold,
influences the catalytic reactivity, and possibly selectivity, in hypervalent iodine(III)-
mediated oxidations. In the context of the well-known reactivity-selectivity principle
(RSP)301, which states that there tends to be an inverse relationship between the reactivity
and selectivity of a reagent, it appears evident that being able to access a tunable scaffold
may greatly impact the efficiency of the developed asymmetric catalytic system. With
the use of Fmoc solid-phase peptide synthesis protocols, we have the capability of
O
OHI
6eOCH3
OCH3
In
O
NH
R2 O
R1O
HN
Fmoc Solid Phase Peptide Synthesis
NH2
Scheme 3.14 Utilizing Fmoc-SPPS to easily generate libraries of peptide-based HI(III) chiral catalysts
6f
6g
H2N
NH
HNO
O
OR1
R2
NH
CH3O
NO
NHR3
O
I
OCH3

93
installing the most active catalysts as caps on the N-terminus of the peptide (Figure 3.4
and Scheme 3.14). For example, we can mimic the most effective iodoarene benzamide
(Catalyst 11), by utilizing the carboxylic acid version (6e), and through well-established
Fmoc solid-phase peptide synthesis, we can cap the N-terminus of peptide-based
scaffolds with active iodoarenes to quickly build up peptide-based catalyst libraries (e.g.
Scheme 3.14, 6f and 6g). The peptide-based scaffolds are amendable to easy iterations
through the use of various commercially available amino acids that contain diverse side
chains.
To diversify the concept further, we next devised strategies that allow for the
active site to be incorporated anywhere in the peptide sequence. We generated chiral
amino acids that allow for the incorporation of the iodine active site into the amino acid
side-chain, thereby allowing us to assimilate the active site in a new, variable location
(Figure 3.7).302
This study was designed around the synthesis of Fmoc-protected amino acids that
integrate the aryl-iodo active site onto the amino acid side chain through an amide
H2NHN
NH
O
IR3
O
O R2
R1 n
H2NHN
O
O
R1 nNH
IR3
HNO
O HN
R2
Figue 1. Strategies for incorporating an aryl-iodo active site into a chiral peptide paradigmPrevious work: Reactive iodoarene-containing n-butyl amide catalysts to be used as caps in peptide scaffolds
New Strategy: Incorporating aryl-iodo active sites into the side chain of peptide scaffolds
O
NH
IO
NH
I
OCH3
O
NH
I
OCH3
Figure 3.7 Utilizing Fmoc-SPPS to incorporate iodoarene active sites into the side chain of peptide scaffolds

94
linkage. The initial route involved the synthesis of Nα-protected carboxybenzyl (i.e. Cbz)
diamino methyl esters that contain an iodoarene active site (Scheme 3.15). This was
accomplished by first converting commercially available Cbz-protected diamino acids
(6h) into their corresponding methyl esters (6i) via thionyl chloride and methanol.
Through a conventional amidation coupling reaction, involving thionyl chloride and
triethyl amine, the aryl iodo moiety (6j) was installed onto the side-chain of the diamino
acid (6k). This synthetic route is attractive for multiple reasons, chiefly in the fact that it
is high yielding, and based on an X-Ray crystal structure, little to no racemization occurs
(see experimental section for X-ray crystallography information).
This synthetic route successfully provides a suite of Cbz-protected methyl ester
catalysts (Figure 3.8), which vary in electronic and structural makeup based on their aryl
iodo active site and the length between the active site and the amino acid scaffold (carbon
tether length i.e. n =1-4). Catalysts 1a-1d feature an iodoarene active site with the iodine
situated in the meta position relative to the amide, with one to four carbons tethered
between the active site and the amino acid chain. The most effective iodoarene moiety
that was uncovered in our previous studies contained a meta positioned iodine, with a
6iQuantatative yields
Scheme 3.15 Cbz-protected amino methyl ester catalysts that contain an iodoarene active site
6h
HN
O
ONH
n
O
O
O
I
R1 R2
H2N nHN
O
OH
O
OH2N n
HN
O
O
O
O1.) SOCl2, 0°C2.) MeOH Et2O
OH
OI
R1 R2R1 = H, CH3R2 = H, OCH3
NEt3, Dry DCMSOCl2, 0°C
6k73-96 % yields over 2 steps
n = 1, 2, 3, 4
6j

95
methoxy group para to that element as well (refer back to Figure 3.5, Catalyst 11); this
functionality was incorporated into Catalysts 2a-2d. The final catalyst scaffold
encompassed a methoxy and methyl substituent on the arene ring coinciding with a meta
positioned iodine (Catalysts 3a-3d). These catalysts were synthesized in moderate to
excellent yields (73-96% yields), and were investigated in the direct, catalytic α-
oxytosylation of propiophenone (Table 1.8). The relative rates for the formation of the
desired product (4l) were acquired by means of gas chromatography, and the
enantioselectivities (%ee) were obtained via chiral stationary phase HPLC analysis.
The direct α-oxytosylation of propiophenone was initially conducted at 24 h at
room temperature in the presence of 10 mol % catalyst (Table 1.8). These data suggest
that an iodoarene that situates the iodine meta to the amide, and methoxy group(s) ortho
and/or para to the iodine will have a relatively high reaction rate (Table 1.8, entries 5-
12); this recapitulates the conclusions that were found when using the N-butyl-
iodobenzamides as catalysts (refer back to Figure 3.5). Furthermore, the optimal carbon
tether length appears to be two or three methylenes away from the amino ⍺-carbon
HN
OON
Hn
CH3
O
OO
IHN
OON
Hn
CH3
O
OO
I
OCH3HN
OON
Hn
CH3
O
OO
I
H3C OCH3
Catalyst 1a (n = 1), 84 % yieldCatalyst 1b (n = 2), 79 % yieldCatalyst 1c (n = 3), 81 % yieldCatalyst 1d (n = 4), 86 % yield
Catalyst 2a (n = 1), 95 % yieldCatalyst 2b (n = 2), 73 % yieldCatalyst 2c (n = 3), 74 % yieldCatalyst 2d (n = 4), 77 % yield
Catalyst 3a (n = 1), 83 % yieldCatalyst 3b (n = 2), 82 % yieldCatalyst 3c (n = 3), 87 % yieldCatalyst 3d (n = 4), 96 % yield
Figure 3.8 Cbz-protected amino methyl ester catalysts that contain an iodoarene active site

96
(Table 1.8, entries 2, 3, 6, 7, 10, and 11). The Cbz-protected amino methyl ester
precatalysts that include a 5-iodo-2-methoxy moiety and that contain a two to three
carbon spacer (Figure 3.8, Catalysts 2b and 2c) were shown to afford the product 4l in
quantitative yields (Table 1.8, entries 6 and 7). Two additional precatalysts granted
quantitative yields (Table 1.8, entries 10 and 11), and those rely on a 5-iodo-2-methoxy-
4-methyl iodoarene scaffold (Figure 3.8, Catalysts 3b and 3c). The top four catalysts
were screened in the same reaction conditions, but were quenched with dimethylsulfide
after four hours; the results are shown in Table 1.9. While all four catalysts oxidized
propiophenone (4k) into oxytosylated propiophenone (4l) at about the same relative rate,
the catalyst that produced the highest yield was Catalyst 3c (49%, Table 1.9, entry 4).
The structure of Catalyst 3c is derived from ornithine, and the active arene ring bears a
meta iodine and two electron donating groups (methoxy and methyl). With no surprise,
in all cases, Catalysts 1a-3d returned the desired product 4l with very low enantiomeric
excess (%ee).
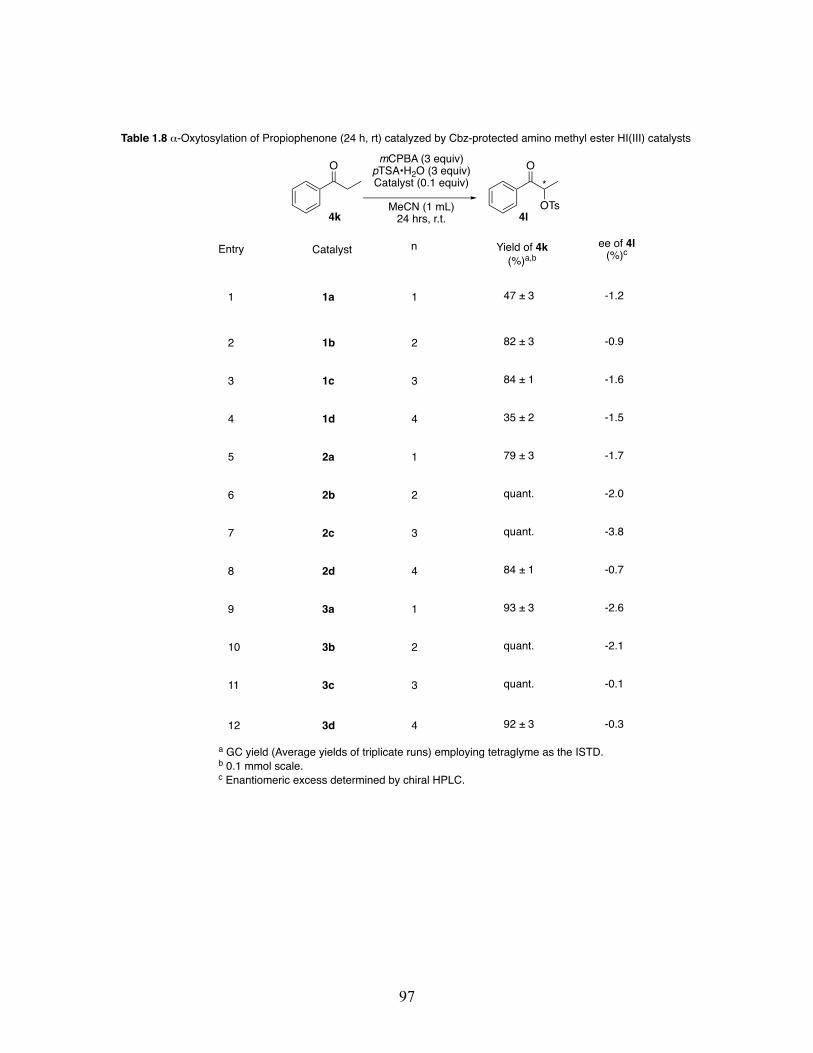
97
Table 1.8 α-Oxytosylation of Propiophenone (24 h, rt) catalyzed by Cbz-protected amino methyl ester HI(III) catalysts
a GC yield (Average yields of triplicate runs) employing tetraglyme as the ISTD.b 0.1 mmol scale.c Enantiomeric excess determined by chiral HPLC.
O O
OTs
mCPBA (3 equiv)pTSA•H2O (3 equiv)Catalyst (0.1 equiv)
MeCN (1 mL)24 hrs, r.t.
*
4k 4l
Catalyst Yield of 4k (%)a,b
Entry
1
2
3
4
5
6
7
8
9
10
11
12
1a
1b
1c
1d
2a
2b
2c
2d
3a
3b
3c
3d
1
2
3
4
1
2
3
4
1
2
3
4
n ee of 4l(%)c
47 ± 3 -1.2
82 ± 3 -0.9
84 ± 1 -1.6
35 ± 2 -1.5
-1.779 ± 3
quant. -2.0
-3.8quant.
84 ± 1 -0.7
-2.693 ± 3
quant. -2.1
-0.1quant.
92 ± 3 -0.3
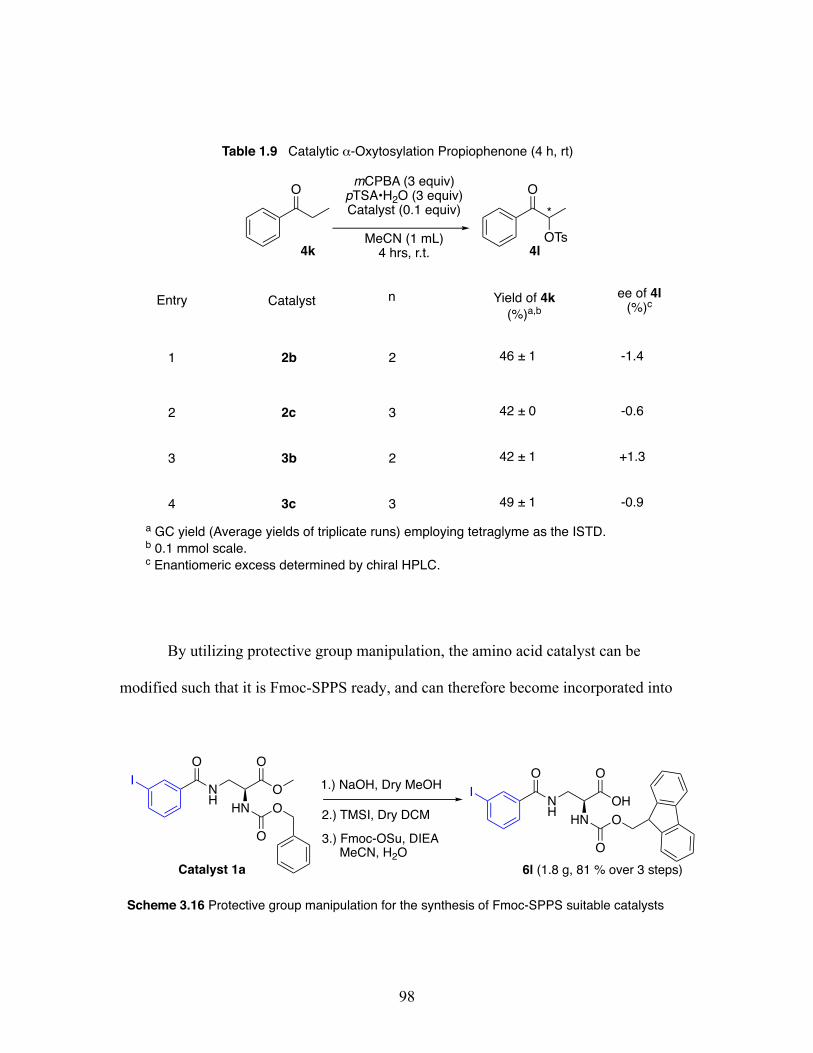
98
By utilizing protective group manipulation, the amino acid catalyst can be
modified such that it is Fmoc-SPPS ready, and can therefore become incorporated into
Scheme 3.16 Protective group manipulation for the synthesis of Fmoc-SPPS suitable catalysts
HN
O
ONH
O
O
O
I
Catalyst 1a
2.) TMSI, Dry DCM
1.) NaOH, Dry MeOH
3.) Fmoc-OSu, DIEA MeCN, H2O
HN
O
OHNH
OI
6l (1.8 g, 81 % over 3 steps)O
O
Table 1.9 Catalytic α-Oxytosylation Propiophenone (4 h, rt)
a GC yield (Average yields of triplicate runs) employing tetraglyme as the ISTD.b 0.1 mmol scale.c Enantiomeric excess determined by chiral HPLC.
O O
OTs
mCPBA (3 equiv)pTSA•H2O (3 equiv)Catalyst (0.1 equiv)
MeCN (1 mL)4 hrs, r.t.
*
4k 4l
Catalyst Yield of 4k (%)a,b
Entry
1
2
3
4
2b
2c
3b
3c
2
3
2
3
n ee of 4l(%)c
46 ± 1 -1.4
42 ± 0 -0.6
42 ± 1 +1.3
49 ± 1 -0.9

99
the side chain of a peptide sequence (Scheme 3.16). The methyl ester can be hydrolyzed
to a free carboxylic acid in the presence of sodium hydroxide (NaOH). Subsequently, the
Cbz protecting group can be removed with the addition of trimethylsilyl iodide (TMSI),
leaving a free amine.303 To render this molecule as a suitable catalyst for solid-phase
peptide synthesis, Fmoc-succinimide (Fmoc-OSu) was added for the reprotection of the
free amine.304, 305 This synthetic process was applied to Catalyst 1a and produced Fmoc-
SPPS compatible compound 6l in 81% over the three steps. By means of standard Fmoc-
SPPS, 6l was incorporated into a small peptide sequence (Ac-Ile-3-I-Dap-Pro-D-Ala-Ala-
Ala) (Figure 3.9). This proof of concept demonstrates the feasibility to synthesize
peptide-based scaffolds that incorporate iodoarene active sites into the side chains of the
amino acids. Moreover, this gives another valuable synthetic approach to varying where
the active site can be situated on the peptide scaffold, and thereby allows for an even
more modular catalytic system.
NHN
NH
H2N
O
O
CH3
O
O
NH
O
O
HN
H3CO
NH
O
I
Figure 3.9 Incorporation of Fmoc SPPS compatible catalyst 6l into a peptide sequence

100
To further progress this strategy in generating novel peptide-based iodoarene
catalysts, we investigated a strategy that can integrate the critical iodoarene active site
into the backbone of the peptide framework (Figure 3.10, 6m and 6n). Achieving this
goal was anticipated through the synthesis of iodoarene containing aniline acetamides
(Figure 3.11, Catalysts 4a-4h).
H2N
NH
HNO
O
OR1
R2
NH
CH3O
NO
HN
O R1
OR2
NH
O
n
IHN
O
R3
NH
OHN
R4 nONH2
I
NHO
HN R3
O
Figure 3.10 Hypothetical peptides that contain an iodoarene within the backbone
6m 6n
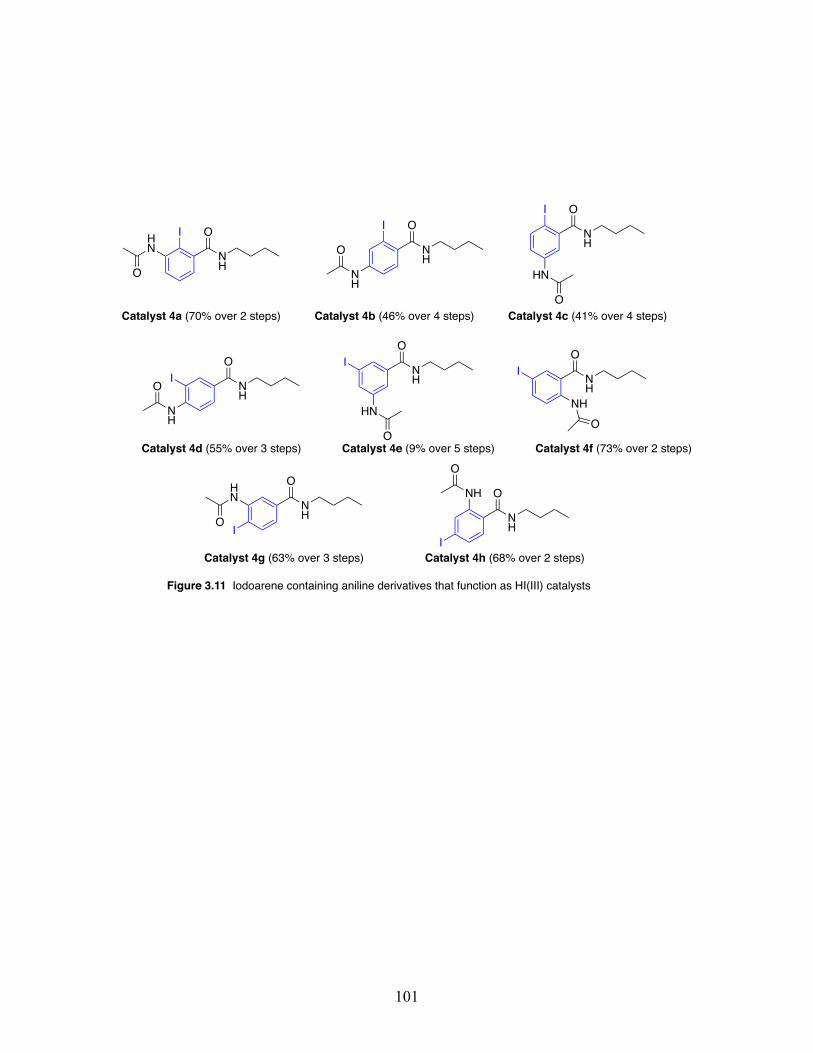
101
I
NH
O I
NH
OHN
O NH
O
NH
OI
NH
O
I
NH
O
HN
O
NH
OI
NH
OI
NH
OHN
O INH
O
I
HN
O
NHO
NH
O
Catalyst 4a (70% over 2 steps) Catalyst 4b (46% over 4 steps) Catalyst 4c (41% over 4 steps)
Catalyst 4d (55% over 3 steps) Catalyst 4e (9% over 5 steps) Catalyst 4f (73% over 2 steps)
Catalyst 4g (63% over 3 steps) Catalyst 4h (68% over 2 steps)
Figure 3.11 Iodoarene containing aniline derivatives that function as HI(III) catalysts

102
The aniline acetamide catalysts were accessed through the chemical manipulation
of commercially available nitro, amino, iodo, and/or carboxylic acid containing arene
rings. As an example, the synthesis of Catalyst 4e is illustrated in Scheme 3.17. Briefly,
commercially available 3,5-dinitrobenzoic acid (6o) was partially reduced by iron powder
in glacial acetic acid to afford nitroaniline 6p.306-308 A diazotization/iodination sequence
transformed the nitroarene into iodoarene derivative 6q.306, 308-311 The second nitro group
was reduced in the presence of Mohr’s salt and concentrated ammonium hydroxide to
give the iodo aniline carboxylic acid 6r in a 68% yield.306, 312 Next, a traditional
amidation employing thionyl chloride and butyl amine was carries out. Subsequently, the
aniline nitrogen was acylated with acetic anhydride to produce the aniline acetamide
Catalyst 4e.
Scheme 3.17 Synthetic operations to access the aniline amide Catalyst 4e
6o
COOH
NO2O2N
COOH
NH2O2N
Fe PowderGlacial acetic acid (120 ºC)
COOH
IO2N
conc. HClNaNO2, H2O, 0 ºCKI (90 ºC)
6p45%
6q59%
conc. NH4OHMohr’s salt, H2O
COOH
IH2N
1.) NEt3, Dry DCMSOCl2, 0 ºC
2.) NEt3, Dry DCM
NH2
IH2N
OHN
6r68%
pyridine, r.t. INH
OHN
O
6s73%
Catalyst 4e64%
O
O O

103
These catalysts incorporate an aryl iodo active site that is flanked by an electron
donating substituent, an aniline, and an electron withdrawing substituent, an amide. The
electron withdrawing amide functionality is necessary to mimic the peptide linkage that
we utilize in our strategy for the incorporation of the active site into the peptide scaffold.
The “push/pull” electronic nature of the compounds, due to the electron-donating aniline
acetamide and the electron-withdrawing N-butyl amide, respectively, could have
immense effects on the catalytic performance.. Therefore, these catalysts were subjected
to the ⍺-oxytosylation of propiophenone in a 24 h reaction at room temperature, as well
as reaction at 50 ˚C (Table 1.10 and Table 1.11). Interestingly, only one catalyst was
relatively effective in the oxytosylation of propiophenone. Catalyst 4f was drastically
superior as a catalyst in comparison to the other aniline acetamide derivatives, and
supplied the oxytosylated propiophenone product 4l in a moderate 73% yield at room
temperature and 80% yield at 50 ℃ (Table 1.10, entry 6 and Table 1.11, entry 6).
Catalyst 4f positions the electron withdrawing N-butyl amide moiety in the meta position
relative iodine while the aniline acetamide is oriented in the para position. The other
catalysts showed comparatively poorer catalytic rates. Although Catalyst 4f and
Catalyst 4d orient the iodine meta to the amide, Catalyst 4d is more sterically
incumbered by the ortho aniline. Comparing the reactivity of these catalysts, Catalyst 4f
supplies the product in a much higher yield (Table 1.10, 73% vs 17% yield), suggesting
that sterics may be an important factor when designing aniline acetamide HI(III)
catalysts. As our previous studies have shown, evidently, an effective catalyst relies on
positioning arene substituents in such a way that the iodine is more electron rich. A

104
catalyst configuration that fits this description involves orienting the iodine meta to the
electron withdrawing amide, and aligning an electron donating group (i.e. aniline or
methoxy) para to the iodine. This study illustrates that drastic effects on catalytic
activity can be present depending on the positioning of the arene substituents, and gives
another variance in which the iodoarene active site can be placed in the backbone of the
peptide scaffold.
a Average yields of triplicate runs employing tetraglyme as the ISTD.b 0.1 mmol scale.
O O
OTs
mCPBA (3 equiv)pTSA•H2O (3 equiv)Catalyst (0.1 equiv) *
MeCN (1 mL)24 h, rt
8
13 ± 27
6c
5c
4
3
2
73 ± 3
17 ± 1
16 ± 1
15 ± 2
11 ± 21
Entry Catalyst Yielda,b
9 ± 2
9 ± 1
Table 1.10 Oxidative α-Oxytosylation of Propiophenone Using Aniline Catalysts (24 h/rt)
4h
4g
4f
4e
4d
4c
4b
4a
4k 4l

105
3.5 FUTURE WORK FOR PEPTIDE-BASED HI(III) CATALYSTS
The preceding studies were directed toward oxidations by means of hypervalent
iodine(III) catalysis, and emphasized how sterics and electronics can affect the catalytic
performance. In particular, we explored how an amide linkage, which joins the active
site to the peptide, influenced the catalytic reactivity. While the majority of known
HI(III) catalysts scaffolds orient the necessary chiral information in the ortho position of
the arene relative to I, in our hands, it appears that orienting the chiral information in the
a Average yields of triplicate runs employing tetraglyme as the ISTD.b 0.1 mmol scale.
O O
OTs
mCPBA (3 equiv)pTSA•H2O (3 equiv)Catalyst (0.1 equiv) *
MeCN (1 mL)24 h, 50 °C
8
25 ± 17
6c
5c
4
3
2
28 ± 3
26 ± 1
28 ± 2
24 ± 11
4h
4g
4f
4e
4d
4c
4b
4a
Entry Catalyst Yielda,b
15 ± 2
17 ± 2
80 ± 3
Table 1.11 Oxidative α-Oxytosylation of Propiophenone Using Aniline Catalysts (24 h/50 ℃)
4k 4l

106
meta position can exhibit a higher active catalyst. Of course, a balance must be struck
between achieving optimal catalytic activity to ensure high yields while also maintaining
optimal positioning of the chiral information for productive enantioinduction in the
transformation. Furthermore, as demonstrated previously by Legault277 and Wirth229,
positioning a suitable electron donating group (i.e. methoxy or aniline) para to the iodine,
can also promote reasonable catalytic activity. These findings provide a scaffolding
template that can allow for the construction of highly reactive hypervalent iodine
catalysts. The developed methodology is compatible with Fmoc-SPPS, and has been
validated as a way to generate functional peptides that can be employed as catalysts in a
variety of synthetic transformations mediated by hypervalent iodine catalysis. Current
efforts are focused on installing and evaluating our most reactive iodoarene-containing
Fmoc-protected amino acids and anilines into peptide scaffolds. We have demonstrated
that highly reactive iodoarenes (i.e. Catalyst 11 Figure 3.5, Catalyst 3c Figure 3.8, and
Catalyst 4f Figure 3.11) can be easily installed anywhere along the peptide scaffold,
including at the N-terminus, on the side-chain of the amino acid, or within the peptide
backbone (Figure 3.12). Libraries of modified peptide scaffolds can be generated in a
facile manner through the use of well-established Fmoc-SPPS protocols, and these
compounds have the capability to be utilized as chiral organocatalysts in various HI-
mediated organic transformations, including, but not limited to, the ⍺-oxytoslyation of
propiophenone, the ⍺-oxytoslyation of 1-indanone, and the oxidative lactonization of 5-
oxo-5-phenylpentanoic acid (Scheme 3.11, eq. 1-3).
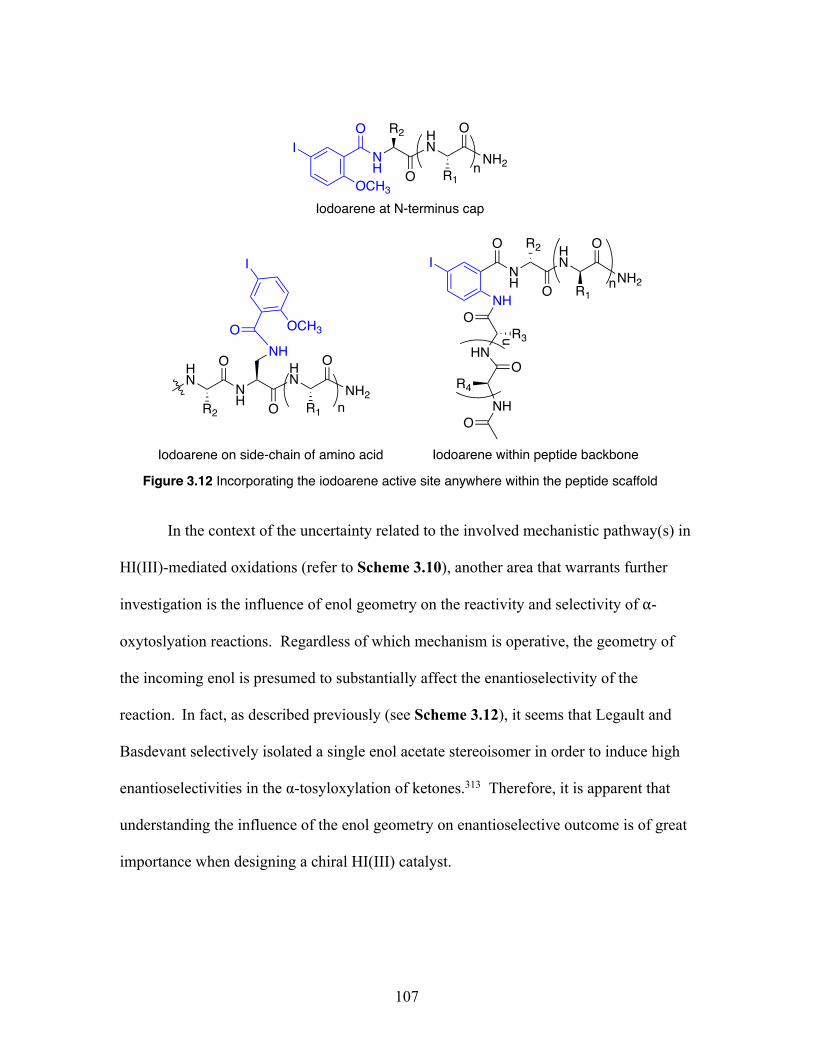
107
In the context of the uncertainty related to the involved mechanistic pathway(s) in
HI(III)-mediated oxidations (refer to Scheme 3.10), another area that warrants further
investigation is the influence of enol geometry on the reactivity and selectivity of ⍺-
oxytoslyation reactions. Regardless of which mechanism is operative, the geometry of
the incoming enol is presumed to substantially affect the enantioselectivity of the
reaction. In fact, as described previously (see Scheme 3.12), it seems that Legault and
Basdevant selectively isolated a single enol acetate stereoisomer in order to induce high
enantioselectivities in the α-tosyloxylation of ketones.313 Therefore, it is apparent that
understanding the influence of the enol geometry on enantioselective outcome is of great
importance when designing a chiral HI(III) catalyst.
nR1
R2 O
O
O
NH
HN
NH2I
R2
HN
O
ONH
NH R1
O
O
HN
NH2
I
OCH3
HN
O R1
OR2
NH
O
nNH2
I
NHO
HNR3
NH
OR4
O
OCH3
n
Iodoarene at N-terminus cap
Iodoarene on side-chain of amino acid Iodoarene within peptide backbone
Figure 3.12 Incorporating the iodoarene active site anywhere within the peptide scaffold
n
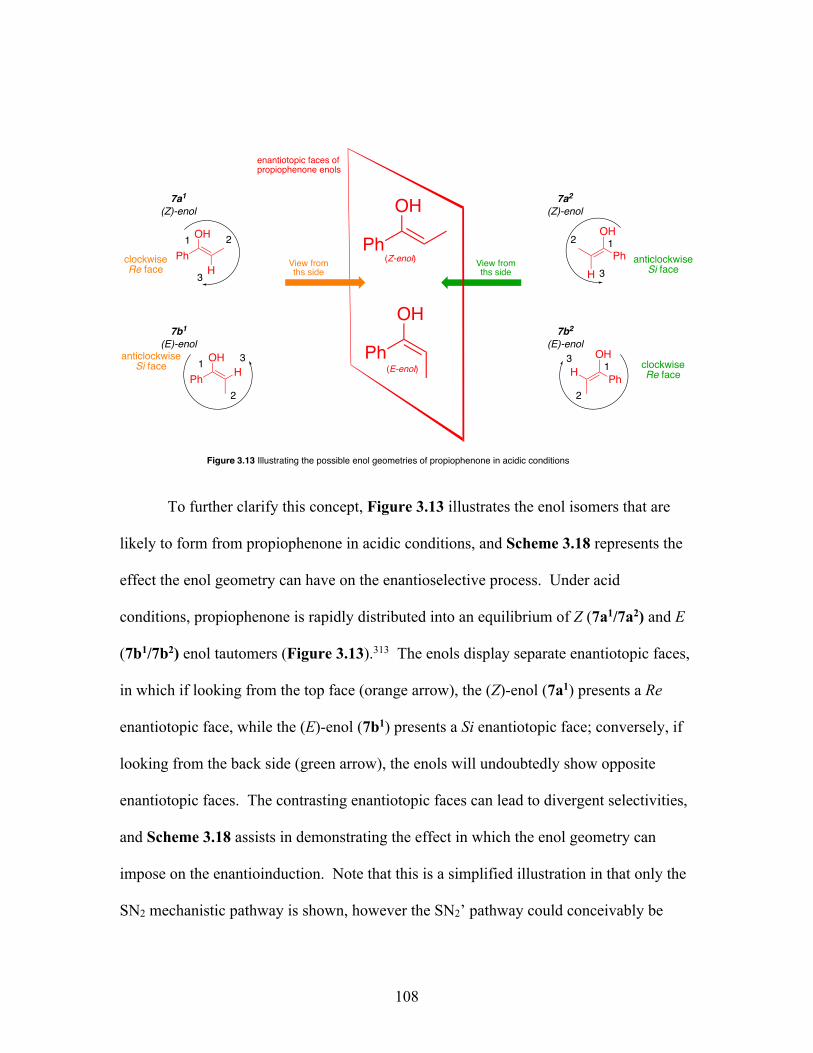
108
To further clarify this concept, Figure 3.13 illustrates the enol isomers that are
likely to form from propiophenone in acidic conditions, and Scheme 3.18 represents the
effect the enol geometry can have on the enantioselective process. Under acid
conditions, propiophenone is rapidly distributed into an equilibrium of Z (7a1/7a2) and E
(7b1/7b2) enol tautomers (Figure 3.13).313 The enols display separate enantiotopic faces,
in which if looking from the top face (orange arrow), the (Z)-enol (7a1) presents a Re
enantiotopic face, while the (E)-enol (7b1) presents a Si enantiotopic face; conversely, if
looking from the back side (green arrow), the enols will undoubtedly show opposite
enantiotopic faces. The contrasting enantiotopic faces can lead to divergent selectivities,
and Scheme 3.18 assists in demonstrating the effect in which the enol geometry can
impose on the enantioinduction. Note that this is a simplified illustration in that only the
SN2 mechanistic pathway is shown, however the SN2’ pathway could conceivably be
Ph
OH
Ph
OH
View fromths side
View fromths side
enantiotopic faces of propiophenone enols
Ph
OH
H
1 2
3clockwiseRe face
2
31H
OHclockwiseRe face
7a1(Z)-enol
7b2(E)-enol
anticlockwiseSi face
Ph
OHH
1 3
2
7b1(E)-enol
3
2 1
H
OH
anticlockwiseSi face
7a2(Z)-enol
Ph
Ph
Figure 3.13 Illustrating the possible enol geometries of propiophenone in acidic conditions
(Z-enol)
(E-enol)

109
affected by the E/Z enol population as well. In short, with the SN2 pathway being
operative, one could reasonably expect that the (Z)-enol would produce the S-enantiomer
(7e), while the (E)-enol would likely generate the R-enantiomer (7i) even if the catalyst
promotes complete facial selectivity during the course of the reaction (Scheme 3.18).
The geometry of the enol can thus lead to different enantiomeric products, and if a
mixture of (E)- and (Z)-enols are present in the reaction mixture, the product will reflect
this ratio and will be generated as a mixture of enantiomers. Therefore, even if a chiral
catalyst was able to exclusively deliver the iodine(III) to one face of the substrate, the
enantioselective outcome could only be as selective as the population of (E)- and (Z)-
tautomers present at equilibrium. On that account, in order to develop a highly
enantioselective chiral catalyst, considerable attention must be dedicated to controlling,
or at least understanding, the enol geometry in HI(III)-mediated oxidative reactions.
One method to control the enol geometry is to selectively synthesize the (E)- and
(Z)-enol, such that they are stereo-defined, and can be subjected to HI(III)-oxidative
conditions, such as the ⍺-oxytoslyation of propiophenone. The relative rates and
OHH CH3 PhOH
CH3
H
(Z)-enol IOH
ArOTs Ph
OPh CH3
IPh
OH
OTsPh
OCH3
OTs*
S-enantiomer
PhOH
HCH3
(E)-enol IOH
ArOTs Ph
OH
IPh
OHPh
OCH3
OTs*
R-enantiomer
CH3
H
PhO
CH3
IPh
OH
*OTs
HPhH CH3
HO
Scheme 3.18 Proposed stereoselective effect of enol geometry in the ⍺-oxytosylation of propiophenone mediated by a HI(III) chiral catalyst
7c
7d7e
7f
7g 7h
7i

110
enantioselectivities could be evaluated, and could ultimately provide evidence on the
influence of the enol geometry. Our group, as well as others, have been unsuccessful in
utilizing silyl enol ethers for the catalytic ⍺-oxytoslyation of propiophenone, as the silyl
group quickly decomposes in acidic conditions (i.e. pTsOH).313, 314 One could envision
the use of enol/enolate equivalents, such as those shown in Figure 3.14, to selectively
trap desirable enol confirmations. In particular, one convenient method to selectively
control the conversion of ketones to preferred geometries is through the use of boronate
esters. Historically, boron reagents have been used to selectively convert ketones into
enol boronates for directed stereoselective aldol reactions.315, 316 By using tertiary amines
in the presence of dialkylboron reagents, ketones can be converted into boronate ester
analogs with very high selectivities and yields (Scheme 3.19). Brown et. al. utilized
diisoproylethylamine (DIPEA) as a tertiary amine, and 9-BBN triflate (7k) to generate
the (Z)-enol boronate of propiophenone (7l) in a 80% yield with a significant selectivity
of 99:1 (Z:E) (Scheme 3.19, eq. 1).316 By using triethylamine and
chlorodicyclohexylborane (7m), they were able to afford the (E)-boronate ester (7n) in a
96% yield with a very high selectivity of 1:99 (Z)/(E) (Scheme 3.19, eq. 2). With this
methodology, we could preferentially form the (E)- and/or (Z)-boronate esters of ketones,
and thereby assess the influence of enol geometry in order to achieve broadly applicable,
highly enantioselective catalysts in ketone α-oxytosylation reactions.
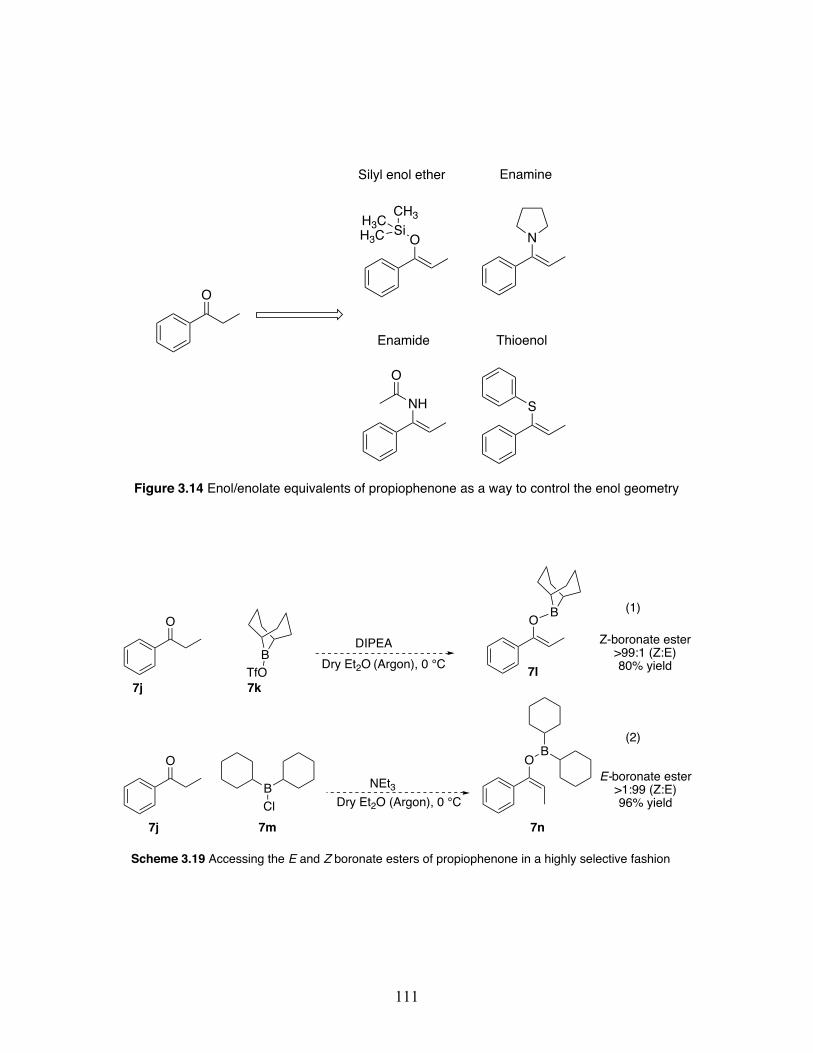
111
O O
Dry Et2O (Argon), 0 °C
O OB
E-boronate ester>1:99 (Z:E)96% yieldDry Et2O (Argon), 0 °C
BTfO
DIPEA
B
Z-boronate ester>99:1 (Z:E)80% yield
BCl
NEt3
Scheme 3.19 Accessing the E and Z boronate esters of propiophenone in a highly selective fashion
7j 7k
7j
7l
7m 7n
(1)
(2)
O
N
NH
O
S
OSiCH3H3C
H3C
Enamine
Enamide
Silyl enol ether
Thioenol
Figure 3.14 Enol/enolate equivalents of propiophenone as a way to control the enol geometry

112
3.6 MACROCYCLES AS CHIRAL HI(III) CATALYSTS
Recently, our group has explored the implementation of macrocycles as an
alternative strategy toward achieving high yields and enantioselectivities in HI-mediated
transformations. In general, a macrocycle is a molecule that contains at least one cyclic
framework of eight or more atoms.317, 318 In comparison to their acyclic analogs,
macrocycles are conformationally restricted, possessing a lower number of rotatable
bonds, and are thus capable of having powerful stereoselective abilities.317, 319
Macrocycles are unique in that they combine beneficial features of small molecules,
including relative ease of synthesis and small molecular weights, and complex
biomolecules (i.e. enzymes and proteins), such as highly selective target engagement.320
Macrocycles have been successfully applied as chiral reagents in various asymmetric
organic reactions.317, 319-326
We envisioned the use of iodoarene-containing chiral macrocycles to be used as
chiral catalysts in hypervalent iodine-mediated oxidative transformations (Figure 3.15,
7o and 7p). As frequently reported in the literature, structural rigidity of a chiral agent
N
O
RR
NNN
O
HN
I
Figure 3.15 Generic iodoarene-containing macrocycles
N
O
R
R
NNN
O
HN
I7o 7p

113
can play a significant role in enantioselective recognition.168, 327, 328 This approach has
the possibility of providing a novel and robust method to generate enantioenriched
species and reinforce the relationship between structural rigidity and enantioselective
control. The routes toward synthesizing iodoarene-containing macrocycles, such as 7o
and 7p (Figure 3.15), are illustrated in Scheme 3.20.329 The synthesis of Macrocycle A
began with the conversion of commercially available L-phenylalanine (7q) to ⍺-azido
acid 7r via a copper(II)-catalyzed diazo transfer; 7r was used at a later synthetic stage.329,
330 Commercially available propargylamine (7s) was transformed into N-(2,4-
dimethoxybenzyl)propargylamine hydrochloride (7u) by means of a reductive
elimination with sodium borohydride. The propargyl hydrochloride 7u was coupled to
commercially available Fmoc-4-iodo-L-phenylalaine (7v) with the use of an amide
coupling agent known as hexafluorophosphate azabenzotriazole tetramethyl uronium
(HATU); this produced the Fmoc-protected, iodoarene containing alkyne 7w in an 88%
yield. 7w was converted into the azido terminal alkyne 7x in a 69% yield over the
following two steps: (1) Fmoc deprotection in the presence of diethylamine, (2) HATU -
coupling with the ⍺-azido acid 7r. Lastly, a copper catalyzed alkyne-azide cycloaddition
was accomplished with use of sodium ascorbate and a dilute sample of 7x to generate
Macrocycle A.331-333 Similar synthetic steps were utilized to produce macrocyclic
derivatives that contained a phenyl (Macrocycle B) or tert-butyl (Macrocycle C)
substituent, rather than a benzyl group (Macrocycle A) (Figure 3.16). The macrocycles
(Macrocycles A-C) and their acyclic precursors (7x-7z) were evaluated in the ⍺-
oxytosylation of propiophenone at room temperature for 24 hours (Figure 3.16). Under
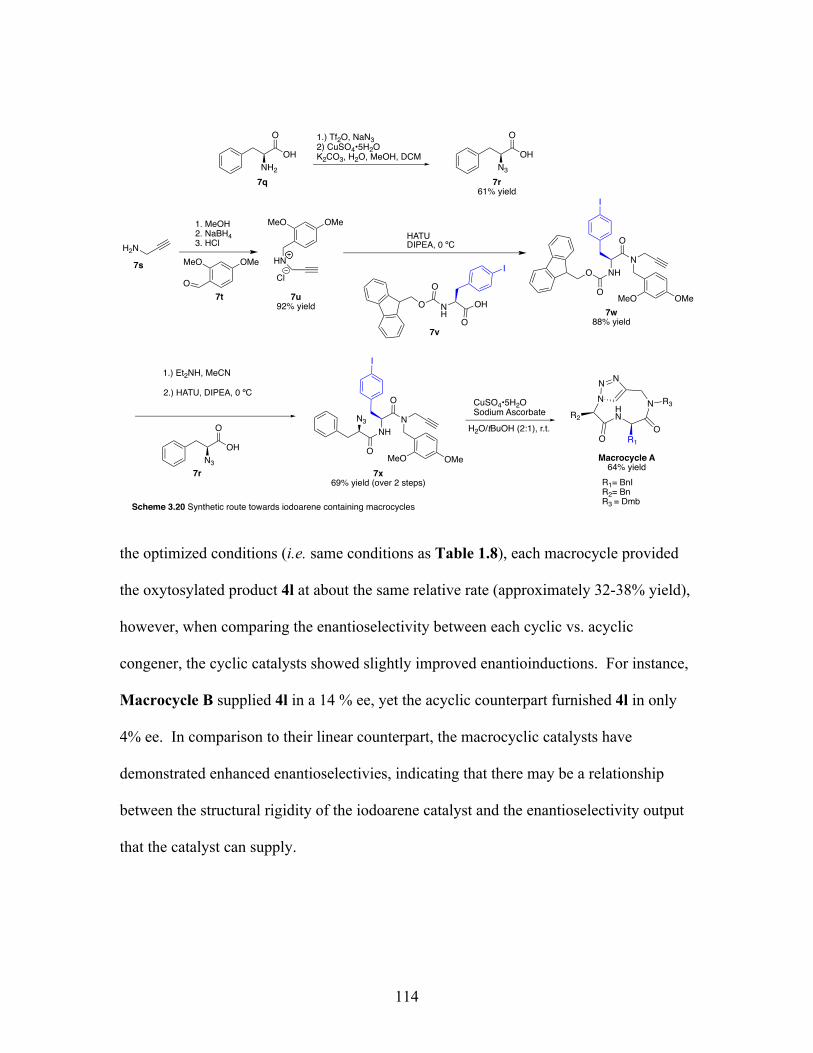
114
the optimized conditions (i.e. same conditions as Table 1.8), each macrocycle provided
the oxytosylated product 4l at about the same relative rate (approximately 32-38% yield),
however, when comparing the enantioselectivity between each cyclic vs. acyclic
congener, the cyclic catalysts showed slightly improved enantioinductions. For instance,
Macrocycle B supplied 4l in a 14 % ee, yet the acyclic counterpart furnished 4l in only
4% ee. In comparison to their linear counterpart, the macrocyclic catalysts have
demonstrated enhanced enantioselectivies, indicating that there may be a relationship
between the structural rigidity of the iodoarene catalyst and the enantioselectivity output
that the catalyst can supply.
N
O
R3
R1
R2
NNN
O
HN
OMeMeO
HNH2N
1. MeOH2. NaBH43. HCl
ONH
I
O
O OH
N
O
NH
I
O
O
HATUDIPEA, 0 ºC
N
O
NH
I
O
N3
7w88% yield
2.) HATU, DIPEA, 0 ºC
OH
O
N3
R1= BnIR2= BnR3 = Dmb
7x69% yield (over 2 steps)
OMeMeO
OMeO OMe
MeO OMe
Cl
H2O/tBuOH (2:1), r.t.
CuSO4•5H2OSodium Ascorbate
Macrocycle A64% yield
7s
7t 7u92% yield
7v
7r
1.) Et2NH, MeCN
Scheme 3.20 Synthetic route towards iodoarene containing macrocycles
OH
O
NH2
1.) Tf2O, NaN32) CuSO4•5H2OK2CO3, H2O, MeOH, DCM OH
O
N3
7r61% yield
7q

115
Current efforts are geared toward synthesizing macrocyclic iodoarene-containing
derivatives that can further improve the steroinduction process, and corroborate the
structure-stereoselectivity relationship. Figure 3.17 exemplifies some of the derivatives
that could be constructed in order to better understand the role that iodoarene cyclic
catalysts play in the ⍺-oxytosylation of propiophenone. One cogent modification
involves the use of starting materials with the opposite chiral configuration in order to
furnish enantiomeric (Figure 3.17, 8a) or diastereomeric (8b and 8c) versions of the
parent macrocycle (Macrocycle A, Figure3.16). Another idea would be to vary the
electronics and sterics around the ring that bears the iodine (R group highlighted in pink)
and/or the chiral substituent that is appended at the gamma position (R group highlighted
in green) in relation to the active site (Figure 3.17, 8d). To probe the effect of the
dimethoxy benzene substituent, one could deprotect the macrocyclic backbone with a
known protocol, such as with a solution of trifluoroacetic acid, trimethylsilyltriflate and
anisole (8e converted to 8f).329 A known concept in click-chemistry is that the clickable
units can favor dimerization, oligomerization, and/or polymerization, thus if the reaction
conditions are altered so that there is a higher concentration of the azide alkyne (i.e. 7x,
Figure 3.20) in the reaction mixture, one could form a dimer/oligomer/polymeric species
(8g).306, 311, 327, 334-336 Another added element of diversity would be to attach the aryl iodo
active site alpha, rather than delta, to the triazole (8j); this can be envisioned by applying
a copper(II)-catalyzed diazo transfer to a substrate that already harbors an iodoarene,
such as the commercially available 4-iodo-L-phenylalanine (Figure 3.17, 8h).
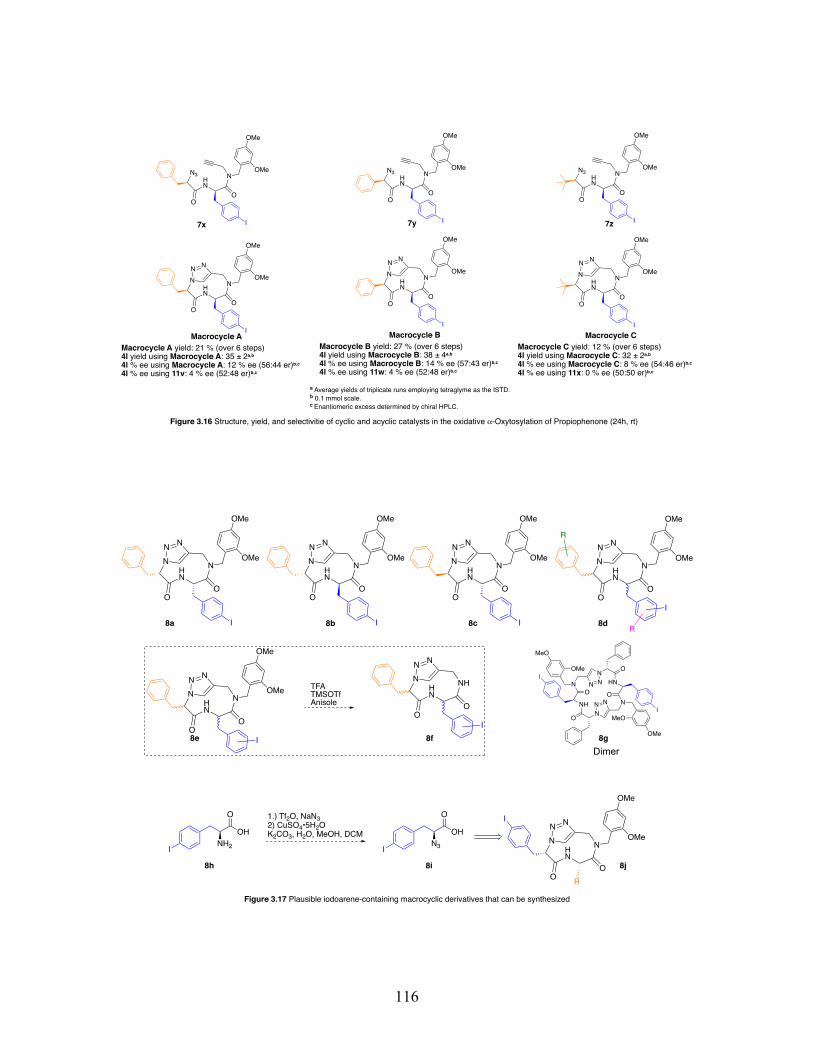
116
I
OH
O
NH2
1.) Tf2O, NaN32) CuSO4•5H2OK2CO3, H2O, MeOH, DCM
I
OH
O
N3
8i
N
O
NNN
O
HN
OMe
OMe
I
N
O
NNN
O
HN
OMe
OMe
I
N
O
NNN
O
HN
OMe
OMe
I
N
O
NNN
O
HN
R
OMe
OMe
I
NH
O
NNN
O
HN
I
TFATMSOTfAnisoleN
O
NNN
O
HN
OMe
OMe
I
NNN
O
OMe
MeO
I
NH
ONN N
O
HN
IN
O
MeO
OMe
N
Dimer
Figure 3.17 Plausible iodoarene-containing macrocyclic derivatives that can be synthesized
N
O
NNN
O
HN
OMe
OMe
I
R
R
8a 8b 8c 8d
8e 8f 8g
8h 8j
N
O
NNN
O
HN
OMe
OMe
I
N
O
NNN
O
HN
OMe
OMe
I
Macrocycle C yield: 12 % (over 6 steps)4l yield using Macrocycle C: 32 ± 2a,b
4l % ee using Macrocycle C: 8 % ee (54:46 er)b,c
4l % ee using 11x: 0 % ee (50:50 er)b,c
Macrocycle B yield: 27 % (over 6 steps)4l yield using Macrocycle B: 38 ± 4a,b
4l % ee using Macrocycle B: 14 % ee (57:43 er)b,c
4l % ee using 11w: 4 % ee (52:48 er)b,c
a Average yields of triplicate runs employing tetraglyme as the ISTD.b 0.1 mmol scale.c Enantiomeric excess determined by chiral HPLC.
Macrocycle CMacrocycle B
N
O
NNN
O
HN
OMe
OMe
I
Macrocycle A yield: 21 % (over 6 steps)4l yield using Macrocycle A: 35 ± 2a,b
4l % ee using Macrocycle A: 12 % ee (56:44 er)b,c
4l % ee using 11v: 4 % ee (52:48 er)b,c
Macrocycle A
Figure 3.16 Structure, yield, and selectivitie of cyclic and acyclic catalysts in the oxidative α-Oxytosylation of Propiophenone (24h, rt)
N
O
N3
O
HN
OMe
OMe
I 7z7y7x
N
O
N3
O
HN
OMe
OMe
I
N
O
N3
O
HN
OMe
OMe
I

117
3.7 CONCLUSIONS
The more eco-friendly nature of hypervalent iodine compounds has attracted attention
from the scientific community as they can be utilized as sustainable alternatives to more
toxic, heavy metal and halogen-based reagents. Recent advancements in the field of
hypervalent iodine have demonstrated that polyvalent iodine reagents are essential tools
in synthetic organic chemistry. Nevertheless, this field of chemistry is still in its infancy
and has proven to be a demanding discipline. The presented work has assisted in trying to
improve upon the difficulties associated with hypervalent iodine chemistry. A
particularly efficient catalyst framework has been disclosed in which the aryl iodo
catalytic site sustains an electron rich nature by orienting the iodine meta to the amide,
and situating an electron donating group para to the iodine. We can attach this highly
proficient iodoarene active unit anywhere along a peptide scaffold through a
straightforward amide linkage. By incorporating iodoarene-containing pre-catalysts into
small peptide sequences, we have revealed a unique strategy that can potentially render
hypervalent iodine mediated reactions highly reactive and enantioselective. Furthermore,
this approach demonstrates synthetic value due to the modular aspect of the designed
catalyst scaffolds. Iodoarene-containing macrocycles have also emerged as a new
paradigm toward hypervalent-iodine-mediated asymmetric transformations. The
macrocycles provide a rigid framework that can potentially induce enantioselectivity, and
have shown to outperform their acyclic analogs. Chemists can apply these versatile
chiral iodine(III) reagents in various synthetic transformations, and due to their easily
iterative nature, they can be tuned and optimized in regard to the reaction at hand. This

118
research has assisted in progressing the hypervalent iodine field by designing new and
efficient methodologies that be used with broad applicability.

119
3.8 EXPERIMENTAL
General Experimental Information
All reagents were purchased from commercial suppliers and used without further
purification. Purification via flash column chromatography was done using silica gel SDS
60 C.C. 40−63 μm. All known compounds and starting materials had 1H NMR and 13C
NMR spectra consistent with previous literature reports. 1H NMR and 13C NMR spectra
were collected at ambient temperature on a 500 or 300 MHz NMR spectrometer.
Chemical shifts are reported in parts per million (ppm) and referenced to residual solvent
peaks (i.e., δ 7.28 ppm for 1H NMR, 77 ppm for 13C NMR) in CDCl3, MeOD (i.e., δ 3.31
ppm for 1H NMR, 49 ppm for 13C NMR), or (CDC3)2SO (i.e., δ 2.50 ppm for 1H NMR,
39.5 ppm for 13C NMR). Data are presented as follows: chemical shift, integration,
multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, br = broad, m = multiplet),
and coupling constants (J, in hertz). Infrared (IR) spectra, reported in cm−1, were
collected using a Fourier transform spectrophotometer. Bands are characterized as strong
(s), medium (m), weak (w), and broad (br). Gas chromatograph (GC) analyses were
conducted using an autosampler and a flame ionization detector (FID). The GC was
equipped with a 30 m × 0.32 mm × 0.25 μm GC column with an HP-5 stationary phase.
GC analyses were carried out within the following parameters: inlet temperature: 250.0
˚C; split injection (4 µL) at 280 mL min-1; column flow: 2.8 mL min-1, constant pressure;
carrier gas: helium; FID temperature: 300 ˚C; temperature program: 100 ˚C for 3 min.;
ramp to 320 ˚C at 20 ˚C min-1, hold for 3 min. Enantiomeric excess (% ee) of selective
products were determined using chiral high performance liquid chromatography (HPLC).

120
Chiral samples were run on an Agilent 1260 Infinity system equipped with a diode array
detector (G4212-60008), and an Agilent 1260 Infinity auto sampler. The chiral stationary
column used was a Chiralpak-AD (250mm X 4.6 mm). The mobile phase consisted of
iso-propyl alcohol/hexanes mixture (50:50). The flow rate was set at 1 mL/min and the
elution of analytes was monitored at 254 nm.
General Procedure for Preparation of the N-Butyl-iodobenzamides (Figure 3.5,
Catalysts 1-12 and refer to Scheme 3.13 as the general synthesis is illustrated for
Catalyst 3). N-Butyl-iodobenzamide Catalysts 1-12 were synthesized from their
corresponding carboxylic acids via a conventional amidation reaction by the formation of
the acid chloride and subsequent displacement by n-butyl amine.
A representative procedure for the synthesis of n-butyl-4-iodobenzamide
(Catalyst 3) is as follows337: The substrate 4-iodobenzoic acid (0.2 g, 0.66 mmol, 1.0
equiv.) and triethylamine (0.18 mL, 1.32 mmol, 2.0 equiv.) were dissolved in anhydrous
DCM (2 mL) and stirred for 5 min at room temperature under nitrogen in a flame-dried
round-bottom flask. The solution was cooled to 0 °C. Thionyl chloride (0.053 mL, 0.726
mmol, 1.1 equiv.) was slowly added (dropwise over 20 min), and the resulting solution
was allowed to stir at 0 °C for 1 hour. The resulting mixture was brought to room
temperature and stirred for 30 min. This solution was concentrated under vacuum and
then redissolved in anhydrous DCM (2 mL). To this solution was added (dropwise over
10 min) 1-butylamine (0.072 mL, 0.726 mmol, 1.1 equiv.), anhydrous DCM (2 mL), and
triethylamine (0.18 mL, 1.32 mmol, 2.0 equiv.). The resulting solution was stirred

121
overnight at room temperature under nitrogen. The crude mixture was concentrated by
rotary evaporation, diluted in DCM, and washed with 1 M HCl, and the aqueous layers
were extracted with DCM (×3). The organic layers were washed with H2O (×2) and
saturated brine (×1) and dried over sodium sulfate. The resulting product was filtered and
concentrated by rotary evaporation. The crude product was purified by column
chromatography on silica gel (20:80 EtOAc:hexanes) to give Catalyst 3 as an orange
solid (0.2819 g, 93%), mp 121−122 °C. IR (neat): 3312 (br), 3075 (w), 2959 (w), 2929
(w), 2862 (w), 1632 (s), 1587 (w), 1543, 1468 (w), 1008 (w) cm−1. 1H NMR (500 MHz,
CDCl3): δ 0.97−0.99 (3H, t, J = 7.5 Hz), 1.39−1.47 (2H, m), 1.59−1.65 (2H, m), 3.44−
3.48 (2H, m), 6.14 (1H, s, br), 7.50, 7.51 (2H, d, J = 8.5 Hz), 7.79, 7.81 (2H, d, J = 8.0
Hz) ppm. 13C NMR (500 MHz, CDCl3): δ 13.7, 20.1, 31.7, 39.9, 98.1, 128.5, 134.3,
137.8, 166.7 ppm. HRMS (ESI): m/z found 304.0200, calcd for C11H15INO (M + H)+
304.0198.
General Procedure for α-Oxytosylation of Propiophenone313, 338 (Scheme 3.11, eq. 1
and Tables 1.1-1.5, Tables 1.7-11, 4l). The relative rates of the prepared Catalysts 1-12,
Catalysts 1a-3d, and Catalysts 4a-4h were evaluated in the α-oxytosylation of
propiophenone using the following representative procedure with Catalyst 1.
Propiophenone 4k (13.3 μL, 0.1 mmol, 1.0 equiv), p-toluenesulfonic acid monohydrate
(57.1 mg, 0.3 mmol, 3 equiv), m-chloroperoxybenzoic acid (51.8 mg, 0.3 mmol, 3 equiv),
and N-butyliodobenzamide Catalyst 1 (3.0 mg, 0.01 mmol, 0.1 equiv) were dissolved in
acetonitrile (1 mL) and stirred for the appropriate time (48, 24, 4, or 1 h) and temperature

122
(room temperature or 50 °C). After the prescribed time, the reaction was quenched upon
addition of dimethyl sulfide (3 equiv). This solution was concentrated by rotary
evaporation, diluted in DCM, and washed with aqueous saturated sodium bicarbonate
(×3). The organic layers were dried over sodium sulfate, filtered, and concentrated by
rotary evaporation. The resulting product and a 0.05 mmol aliquot of tetraglyme [as
internal standard (ISTD)] were added to 1 mL volumetric flasks, and the final volume
was taken to 1 mL with DCM. All trials were run in triplicate and injected into the GC in
order to evaluate percent yields and their standard deviations (see procedure details
below).
General Procedure for Preparation of GC Standard Curves. Stock solutions (1 M) of
the desired tosylated propiophenone 4l and ISTD (tetraglyme) were prepared in DCM.
Varying amounts of desired product (0.005, 0.01, 0.02, 0.03, 0.04, 0.05, 0.06, 0.07, 0.08,
0.09, and 0.100 mmol) and a constant 0.05 mmol aliquot of ISTD were added to 1 mL
volumetric flasks. The final volume was taken to 1 mL with DCM. These samples were
injected into the GC, and the peak area ratio [(product area)/(ISTD area)] was calculated.
This ratio was plotted versus the concentration of the desired product to yield a linear
relationship (r2 values greater than 0.997), thus correlating GC chromatogram peak areas
with product concentration. Reported yields and standard deviations were collected from
averaged triplicate runs with tetraglyme (0.05 M) employed as the ISTD.

123
General Procedure for Percent Yield Determination via GC Analysis. GC analyses
were carried out within the following parameters: inlet temperature, 250 °C; 20:1 split at
130 mL/min; column flow, 6.5 mL/min; constant pressure; carrier gas, helium; FID
temperature, 300 °C; temperature program, 100 °C for 3 min, 20 °C/ min ramp to 320 °C.
Reactions were quenched with dimethyl sulfide (3 equiv) at prescribed times followed by
GC analysis. Yields were calculated from a standard curve of the product in
concentrations ranging from 0.005 to 0.1 M. Reported yields and standard deviations
were collected from averaged triplicate runs with tetraglyme (0.05 M) employed as the
ISTD.
General Procedure for Scaleup and Isolated Yield Determination (i.e., Data for
Table 1.7). Utilizing our best catalyst, Catalyst 11, scaled-up syntheses (1 mmol scales)
of α-oxtosylated propiophenone 4l and a brief substrate scope were conducted to give
isolated yields for 4l as well as compounds 18-23.
Representative procedure for the synthesis of 1-oxo-1-p-tolylpropan-2-yl 4-
methylbenzenesulfonate 18 (Table 1.7)277: 4′-methylpropiophenone (149 μL, 1.0 mmol,
1.0 equiv), p-toluenesulfonic acid monohydrate (571 mg, 3 mmol, 3 equiv), m-
chloroperoxybenzoic acid (518 mg, 3 mmol, 3 equiv.), and iodobenzamide Catalyst 11
(33 mg, 0.1 mmol, 0.1 equiv.) were dissolved in acetonitrile (10 mL) and stirred for 36 h
at 50 °C. The reaction was immediately quenched with dimethyl sulfide (3 equiv.). This
solution was concentrated by rotary evaporation, diluted in DCM, and washed with
aqueous saturated sodium bicarbonate (×3). The organic layers were dried over sodium

124
sulfate, filtered, and concentrated by rotary evaporation. The crude product was purified
by column chromatography on silica gel (20:80 EtOAc:hexanes) to give 18 as a white
solid (0.2783 g, 87%). Rf = 0.29. 1H NMR (300 MHz, CDCl3): δ 1.59, 1.61 (3H, d, J =
6.9 Hz), 2.44 (6H, s, br), 5.79 (1H, q, J = 7.2 Hz), 7.30−7.27 (4H, m), 7.82−7.63 (4H, m)
ppm. 13C NMR (90 MHz, CDCl3): δ 18.8, 21.6, 21.7, 76.6, 128.0, 128.9, 129.5, 129.8,
131.1, 133.5, 144.9, 145.0, 194.3 ppm.
General Procedure for NMR Kinetic Profiles (i.e., Figure 3.6). The activity of
Catalysts 11, 8, and 4 was evaluated by monitoring oxidation rates of the catalysts from
their iodine(I) state to their corresponding iodine(III) state. The kinetic profiles were
generated by dissolving m-chloroperoxybenzoic acid (51.8 mg, 0.3 mmol, 3 equiv) and
the respective iodobenzamide catalyst (0.01 mmol, 0.1 equiv) in CD3CN (1 mL). Due to
rapid oxidation of Catalyst 11 and 8, 1H NMR spectra were recorded every 5 min for 1 h
and then were recorded every hour after the first hour (recorded spectra up to 12 h).
Catalyst 4 had a slower oxidation rate; thus,1H NMR spectra were recorded every hour
starting at 0 h (recorded spectra up to 12 h). The percent conversion of the I(I) catalyst to
its corresponding I(III) was determined via changes in well-resolved NMR signal
integration values as a function of time.
Characterization Data for All Reported Compounds.
N-Butyl-2-iodobenzamide, Catalyst 1339 Brown solid (0.2839 g, 94%), mp 90−91 °C. IR
(neat): 3287 (br), 3055 (w), 2953 (s), 2865 (m),1649 (s), 1585 (m), 1539 (s), 1463 (m),

125
1015 (w) cm−1 . 1H NMR (500 MHz, CDCl3): δ 0.97−1.00 (3H, t, J = 7.0 Hz), 1.40−1.47
(2H, m), 1.59− 1.65 (2H, m), 3.45−3.49 (2H, m), 6.11 (1H, s, br), 7.18−7.21 (1H, t, J =
8.0 Hz), 7.73−7.74 (1H, d, J = 8.0 Hz), 7.84 (1H, q, J = 8.0 Hz), 8.10−8.11 (1H, t, J = 1.5
Hz) ppm. 13C NMR (125 MHz, CDCl3): δ 13.8, 20.2, 31.5, 39.9, 92.4, 128.2, 128.3,
131.0, 139.8, 142.5, 169.4 ppm. HRMS (ESI): m/z found 304.0204, calcd for C11H15INO
(M + H)+ 304.0198.
N-Butyl-3-iodobenzamide, Catalyst 2340 White solid (0.2776 g, 92%), mp 70−71 °C. IR
(neat): 3280 (br), 3063 (m), 2958 (s), 2930 (s), 2870 (m), 1634 (s), 1592 (m), 1555 (s),
1467 (s), 1062 (w) cm−1; 1H NMR (500 MHz, CDCl3): δ 0.97−1.00 (3H, t, J = 7.5 Hz),
1.40−1.47 (2H, m), 1.59−1.65 (2H, m), 3.45−3.49 (2H, m), 6.13 (1H, s, br), 7.17−7.20
(1H, t, J = 7.5 Hz), 7.72, 7.74 (1H, d, J = 10.5 Hz), 7.83, 7.85 (1H, d, J = 8.0 Hz), 8.11
(1H, s) ppm. 13C NMR (125 MHz, CDCl3): δ 13.8, 20.2, 31.7, 40.0, 94.3, 126.1, 130.2,
135.9, 137.0, 140.2, 166.0 ppm. HRMS (ESI): m/z found 304.0201, calcd for C11H15INO
(M + H)+ 304.0198.
N-Butyl-4-iodobenzamide, Catalyst 3 as an orange solid (0.2819 g, 93%), mp 121−122
°C. IR (neat): 3312 (br), 3075 (w), 2959 (w), 2929 (w), 2862 (w), 1632 (s), 1587 (w),
1543, 1468 (w), 1008 (w) cm−1. 1H NMR (500 MHz, CDCl3): δ 0.97−0.99 (3H, t, J = 7.5
Hz), 1.39−1.47 (2H, m), 1.59−1.65 (2H, m), 3.44− 3.48 (2H, m), 6.14 (1H, s, br), 7.50,
7.51 (2H, d, J = 8.5 Hz), 7.79, 7.81 (2H, d, J = 8.0 Hz) ppm. 13C NMR (500 MHz,

126
CDCl3): δ 13.7, 20.1, 31.7, 39.9, 98.1, 128.5, 134.3, 137.8, 166.7 ppm. HRMS (ESI): m/z
found 304.0200, calcd for C11H15INO (M + H)+ 304.0198.
N-Butyl-2-iodo-4,5-dimethoxybenzamide, Catalyst 4.341 Brown solid (0.5207 g, 88%),
mp 104−106 °C; IR (neat): 3264 (br), 2957 (m), 2930 (m), 2869 (w), 1638 (s), 1593 (m),
1543 (m), 1501 (m), 1025 (w) cm−1; 1H NMR (500 MHz, CDCl3) δ 0.97−1.00 (3H, t, J =
7.0 Hz), 1.43−1.51 (2H, m), 1.62−1.67 (2H, m), 3.45−3.49 (2H, m), 3.90 (6H, s, br), 5.87
(1H, s, br), 7.03 (1H, s), 7.22 (1H, s) ppm. 13C NMR (125 MHz, CDCl3) δ 13.8, 20.3,
31.4, 40.0, 56.1, 56.3, 80.8, 112.0, 122.0, 134.6, 149.2, 150.4, 168.9 ppm. HRMS (ESI):
m/z found 364.0411, calcd for C13H19INO3 (M + H)+ 364.0410.
N-Butyl-2-iodo-5-methoxybenzamide, Catalyst 5. Brown solid (0.2699 g, 90%), mp
80−81 °C. IR (neat): 3287 (br), 3069 (w), 2957 (m), 2932 (m), 2862 (w) 1642 (s), 1584
(m), 1542 (m), 1467 (m), 1009 (w) cm−1. Rf = 0.12 (20:80 EtOAc:hexanes). 1H NMR
(500 MHz, CDCl3): δ 0.97−1.00 (3H, t, J = 7.0 Hz), 1.43−1.50 (2H, m), 1.62− 1.68 (2 H,
m), 3.45−3.49 (2H, m), 3.81 (3H, s), 5.82 (1H, s, br), 6.69−6.71 (1H, m), 6.98, 6.99 (1H,
d, J = 3.0 Hz), 7.70−7.71 (1H, d, J = 9.0 Hz) ppm. 13C NMR (125 MHz, CDCl3): δ 13.7,
20.2, 31.4, 39.9, 55.6, 80.6, 114.2, 117.6, 140.4, 143.2, 159.7, 169.1 ppm. HRMS (ESI):
m/z found 334.0310, calcd for C12H17INO2 (M + H)+ 334.0304.
N-Butyl-2-iodo-5-methylbenzamide, Catalyst 6. White solid (0.2250 g, 93%), mp
106−107 °C. IR (neat): 3292 (br), 2957 (w), 2925 (w), 2864 (w) 1643 (s), 1541 (m), 1458

127
(w), 1012 (w) cm−1. Rf = 0.44 (20:80 EtOAc:hexanes). 1H NMR (500 MHz, CDCl3): δ
0.98−1.00 (3H, t, J = 7.5 Hz), 1.43−1.51 (2H, m), 1.62−1.68 (2H, m), 2.33 (3H, s),
3.45−3.49 (2H, m), 5.76 (1H, s, br), 6.92−6.94 (1H, m), 7.23− 7.24 (1H, d, J = 2.0 Hz),
7.72−7.73 (1H, d, J = 8.0 Hz) ppm. 13C NMR (125 MHz, CDCl3): δ 13.8, 20.2, 20.9,
31.5, 39.8, 88.2, 129.2, 132.0, 138.4, 139.6, 142.3, 169.5 ppm. HRMS (ESI): m/z found
318.0374, calcd for C12H17INO (M + H)+ 318.0355.
N-Butyl-2-iodo-3-methylbenzamide, Catalyst 7. Yellow solid (0.2153 g, 89%), mp
68−69 °C. IR (neat): 3283 (br), 2957 (m), 2686 (w) 1640 (s), 1546 (m), 1476 (m), 1036
(w) cm−1. Rf = 0.19 (20:80 EtOAc:hexanes). 1H NMR (500 MHz, CDCl3): δ 0.97−1.00
(3H, t, J = 3.5 Hz), 1.42−1.49 (2H, m), 1.61−1.67 (2H, m), 2.50 (3H, s), 3.45−3.49 (2H,
m), 5.75 (1H, br), 7.10−7.13 (1H, m), 7.26−7.27 (2H, t, J = 2.0 Hz) ppm. 13C NMR (125
MHz, CDCl3): δ 13.8, 20.2, 29.2, 31.4, 39.8, 99.3, 125.0, 128.1, 130.3, 142.8, 144.3,
170.4 ppm. HRMS (ESI): m/z found 318.0377, calcd for C12H17INO (M + H)+ 318.0355.
N-Butyl-3-iodo-2-methylbenzamide, Catalyst 8. Light brown solid (0.2178 g, 90%),
mp 98−99 °C. IR (neat): 3286 (br), 2955 (w), 2928 (w), 2862 (w), 1634 (s), 1581 (w),
1536 (m), 1433 (w), 999 (w) cm−1. Rf = 0.28 (20:80 EtOAc:hexanes). 1H NMR (500
MHz, CDCl3): δ 0.97−1.00 (3H, t, J = 7.5 Hz), 1.40−1.47 (2H, m), 1.58− 1.64 (2H, m),
2.52 (3H, s), 3.43−3.48 (2H, m), 5.75 (1H, s, br), 6.90−6.93 (1H, t, J = 7.5 Hz), 7.30 (1H,
d, J = 0.50 Hz), 7.88−7.90 (1H, m) ppm. 13C NMR (125 MHz, CDCl3) δ 13.8, 20.1, 25.6,

128
31.6, 39.7, 103.4, 126.5, 127.3, 138.2, 138.4, 140.5, 169.6 ppm. HRMS (ESI): m/z found
318.0377, calcd for C12H17INO (M + H)+ 318.0355.
N-Butyl-3-iodo-4-methylbenzamide, Catalyst 9. Brown oil (0.0871 g, 72%). IR (neat):
3300 (br), 2957 (m), 2929 (m), 2870 (w), 1636 (s), 1600 (w), 1547 (s), 1481 (m), 1032
(w) cm−1. Rf = 0.33 (20:80 EtOAc:hexanes). 1H NMR (500 MHz, CDCl3): δ 0.97−1.00
(3H, t, J = 7.5 Hz), 1.40−1.47 (2H, m), 1.59−1.65 (2H, m), 2.48 (3H, s), 3.44−3.48 (2H,
m), 6.10 (1H, s, br), 7.30 (1H, s), 7.65−7.66 (1H, m), 8.20 (1H, d, J = 2.0 Hz) ppm. 13C
NMR (125 MHz, CDCl3): δ 13.8, 20.2, 28.1, 31.7, 39.9, 100.9, 126.7, 129.6, 134.0,
137.4, 145.0, 165.8 ppm. HRMS (ESI): m/z found 318.0377, calcd for C12H17INO (M +
H)+ 318.0355.
N-Butyl-3-iodo-4-methoxybenzamide, Catalyst 10. Brown solid (0.4105 g, 69%), mp
71−72 °C. IR (neat): 3315 (br), 2958 (m), 2932 (m), 2870 (w), 1631 (s), 1595 (s), 1548
(s), 1487 (s), 1016 (w) cm−1. Rf = 0.32 (20:80 EtOAc:hexanes). 1H NMR (500 MHz,
CDCl3): δ 0.95−0.99 (3H, t, J = 7.5 Hz), 1.38−1.45 (2H, m), 1.58−1.64 (2H, m), 3.42−
3.46 (2H, m), 3.93 (3H, s), 6.20 (1H, s, br), 6.82−6.84 (1H, d, J = 0.50 Hz), 7.78−7.80
(1H, m), 8.17−8.18 (1H, d, J = 2.5 Hz) ppm. 13C NMR (125 MHz, CDCl3): δ 13.8, 20.2,
31.8, 39.9, 56.6, 85.6, 110.2, 128.9, 129.0, 138.1, 160.4, 165.7 ppm. HRMS (ESI): m/z
found 334.0333, calcd for C12H17INO2 (M + H)+ 334.0304.

129
N-Butyl-5-iodo-2-methoxybenzamide, Catalyst 11. White solid (0.2240 g, 75%), mp
41−42 °C. IR (neat): 3405 (br), 2957 (m), 2930 (m), 2870 (w), 1643 (s), 1586 (m), 1534
(s), 1477 (s), 1019 (m) cm−1. Rf = 0.31 (20:80 EtOAc:hexanes). 1H NMR (500 MHz,
CDCl3): δ 0.94−0.97 (3H, t, J = 7.0 Hz), 1.37−1.44 (2H, m), 1.56−1.62 (2H, m), 3.42−
3.46 (2H, m), 3.94 (3H, s), 6.72,6.74 (1H, d, J = 9.0 Hz), 7.66−7.69 (1H, m), 7.75 (1H, s,
br), 8.45−8.46 (1H, d, J = 2.0 Hz) ppm. 13C NMR (125 MHz, CDCl3): δ 13.8, 17.6, 20.2,
31.6, 39.6, 56.2, 83.7, 113.7, 123.9, 140.7, 157.2, 163.7 ppm. HRMS (ESI): m/z found
334.0333, calcd for C12H17INO2 (M + H)+ 334.0304.
N-Butyl-5-iodo-2-methoxy-4-methylbenzamide, Catalyst 12. Brown solid (0.1164 g,
98%), mp 117−118 °C. IR (neat): 3401 (br), 2955 (m), 2935 (m), 2859 (w), 1646 (s),
1593 (m), 1530 (s), 1460 (m), 1033 (w) cm−1. Rf = 0.22 (20:80 EtOAc:hexanes). 1H
NMR (500 MHz, CDCl3): δ 0.97−0.99 (3H, t, J = 7.0 Hz), 1.39−1.47 (2H, m), 1.58− 1.64
(2H, m), 2.47 (3H, s), 3.45−3.49 (2H, m), 3.96 (3H, s), 6.86 (1H, s), 7.76 (1H, s, br), 8.58
(1H, s) ppm. 13C NMR (125 MHz, CDCl3): δ 13.8, 20.3, 28.4, 31.6, 39.5, 56.1, 90.6,
113.0, 121.1, 142.0, 146.1, 157.3, 163.7 ppm. HRMS (ESI): m/z found 348.0491, calcd
for C13H19INO2 (M + H)+ 348.0460.
1-oxo-1-p-tolylpropan-2-yl 4-methylbenzenesulfonate,18 (Table 1.7).
White solid (0.2783 g, 87%). Rf = 0.29 (20:80 EtOAc:hexanes). 1H NMR (300 MHz,
CDCl3): δ 1.59, 1.61 (3H, d, J = 6.9 Hz), 2.44 (6H, s, br), 5.79 (1H, q, J = 7.2 Hz),

130
7.30−7.27 (4H, m), 7.82−7.63 (4H, m) ppm. 13C NMR (90 MHz, CDCl3): δ 18.8, 21.6,
21.7, 76.6, 128.0, 128.9, 129.5, 129.8, 131.1, 133.5, 144.9, 145.0, 194.3 ppm.
1-(4-Methoxyphenyl)-1-oxopropan-2-yl 4-Methylbenzenesulfonate, 19 (Table 1.7).313
White solid (0.2408 g, 72%). Rf = 0.183 (20:80 EtOAc:hexanes). 1H NMR (300 MHz,
CDCl3): δ 1.59, 1.61 (3H, d, J = 6.9 Hz) 2.43 (3H, s), 3.89 (3H, s), 5.76 (1H, q, J = 6.9),
7.27−7.30 (2H, m), 7.76, 7.79 (2H, d, J = 8.4), 7.88−7.93 (2H, m) 7.89−7.91 ppm (2H,
m) ppm. 13C NMR (90 MHz, CDCl3): δ 18.9, 21.7, 55.6, 76.8, 114.0, 126.5, 128.0, 129.7,
131.2, 133.5, 145.0, 164.1, 193.1 ppm.
1-(4-Fluorophenyl)-1-oxopropan-2-yl 4-Methylbenzenesulfonate, 20 (Table 1.7).277
White solid (0.3030 g, 94%). Rf = 0.33 (20:80 EtOAc:hexanes). 1H NMR (500 MHz,
CDCl3): δ 1.60, 1.61 (3H, d, J = 7.0 Hz), 2.44 (3H, s), 5.72 (1H, q, J = 7.0 Hz), 7.14−7.17
(2H, t, J = 8.0 Hz), 7.29− 7.31 (2H, m), 7.76−7.78 (2H, m), 7.95−7.98 (2H, m) ppm. 13C
NMR (125 MHz, CDCl3): δ 18.6, 21.7, 76.8, 115.9, 116.1, 128.0, 129.8, 131.6, 131.7,
133.4, 145.2, 165.1, 167.1, 193.4 ppm.
1-(4-Chlorophenyl)-1-oxopropan-2-yl 4-Methylbenzenesulfonate, 21(Table 1.7).277
White solid (0.2464 g, 75%). Rf = 0.32 (20:80 EtOAc:hexanes). 1H NMR (500 MHz,
CDCl3): δ 1.60, 1.61 (3H, d, J = 7.0 Hz), 2.45 (3H, s), 5.71 (1H, q, J = 7.0 Hz), 7.30,
7.31(2H, d, J = 8.0 Hz), 7.44, 7.46 (2H, d, J = 8.5 Hz), 7.75, 7.77 (2H, d, J = 8.5 Hz),

131
7.85, 7.87 (2H, d, J = 9.0 Hz) ppm. 13C NMR (125 MHz, CDCl3): δ 18.6. 21.7, 76.8,
128.0, 129.1, 129.8, 130.3, 132.0, 133.3, 140.4, 145.2, 194.0 ppm.
1-(4-Bromophenyl)-1-oxopropan-2-yl 4-Methylbenzenesulfonate, 22 (Table 1.7).338
White solid (0.3266 g, 85%). Rf = 0.37 (20:80 EtOAc:hexanes). 1H NMR (300 MHz,
CDCl3): δ 1.59, 1.61 (3H, d, J = 6.9 Hz), 2.44 (3H, s), 5.69 (1H, q, J = 6.9 Hz), 7.31 (2H,
s), 7.59− 7.63 (2H, m), 7.74−7.80 (4H, m) ppm. 13C NMR (90 MHz, CDCl3): δ 18.6,
21.7, 76.8, 127.9, 129.2, 129.9, 130.3, 132.1, 132.4, 133.3, 145.2, 194.1 ppm.
α-Tosyloxyacetophenone, 23 (Table 1.7).338 White solid (0.2729 g, 94%). Rf = 0.24
(20:80 EtOAc:hexanes). 1H NMR (500 MHz, CDCl3): δ 2.47 (3H, s), 5.29 (2H, s), 7.36,
7.38 (2H, d, J = 8.0 Hz), 7.48−7.51 (2H, m), 7.61−7.64 (1H, m), 7.85−7.88 (4H, m) ppm.
13C NMR (125 MHz, CDCl3): δ 21.7, 70.0, 128.0, 128.2, 128.9, 129.9, 132.7, 133.8,
134.2, 145.3, 190.3 ppm.
General Procedure for Preparation of the Iodoarene Amino Methyl Esters
(Catalysts 1a-3d, Figure 3.8 and Scheme 3.15).
Iodoarene Nα-Z-amino methyl ester Catalysts 1a-3d were prepared from their
corresponding Nα-protected Z-diamino acids and iodobenzoic acids. The Z-diamino
acids, L-diaminopropanoic acid (Dap), L-diaminobutanoic acid (Dab), L-ornithine (Orn)
and L-lysine (Lys), were first converted to their corresponding methyl esters and without

132
further purification the free amine side chain was coupled to appropriate iodoarene active
site (cf. Scheme 3.15).
A representative two step procedure for the synthesis of Catalyst 1a is as follows:
Step 1: To a cooled (0 °C) suspension of Nα -Cbz-L-2,3-Diaminopropionic acid (Z-L-
DAP-OH) (5 g, 21 mmol, 1 equiv.) in 100 mL of MeOH was added thionyl chloride (23
mmol, 1.1 equiv.) over 20 min (dropwise). The resulting solution was allowed to stir at
room temperature overnight. After approximately 18 h, the solvent was removed in
vacuo, and the residual solid was stirred with 150 mL Et2O for 0.5 h. The resulting crude
compound was obtained following rotary evaporation to give Nα-Z-diaminopropanoic
acid as a white solid, (5.29 g, quant. yield).
Step 2: A solution of 3-iodobenzoic acid (4.6 g, 18.5 mmol, 1.0 equiv) and
trimethylamine (3.78 g, 37.1 mmol, 2 equiv) in anhydrous dichloromethane (150 mL)
was allowed to stir at room temperature for 10 min while under nitrogen atmosphere.
The mixture was cooled to 0°C, and after 30 min, thionyl chloride (20.4 mmol, 1.1 equiv)
was added dropwise and allowed to stir for 1.5 hours. The reaction mixture was brought
to room temperature, with continued stirring for 30 minutes followed by rotary
evaporation to dryness. The reaction mixture was then redissolved in anhydrous DCM
(150 mL). A separate solution of the Z-diamino methyl ester (5.15 g, 20.4 mmol, 1.1
equiv) was prepared by dissolving this substrate in anhydrous DCM (100 mL) followed
by dropwise addition of triethylamine (3.78 g, 37.1 mmol, 2 equiv). This freshly
prepared solution was then slowly added to the flask containing the 3-iodobenzoic acid
and allowed to stir for 18 h at room temperature under nitrogen. The resulting crude

133
mixture was concentrated by rotary evaporation, diluted in DCM and washed with 1M
HCl. The aqueous layers were extracted with DCM (3X). Organic layers were washed
with H2O (2X), saturated brine (1X), and dried over sodium sulfate. Following filtration
and rotary evaporation, the resulting crude product was purified by column
chromatography on silica gel (gradient to 1:9 MeOH:DCM) to produce the dark brown
oil, Catalyst 1a (7.5 g, 84%); Rf = 0.79 (1:9 MeOH:DCM) ; IR (neat): 3337 (br), 3068
(m), 3031 (m), 2953 (m), 2930 (m), 1759 (s), 1748 (s), 1643 (m), 1551 (m), 1543 (m),
1438 (s), 1270 (w), 1066 (w) cm-1; 1H NMR (500 MHz, CDCl3): δ 3.75 (3 H, s), 3.81-
3.84 (2 H, t), 4.51-4.58 (1 H, q), 5.09 (2H, s), 5.30 (Residual DCM solvent peak), 6.19-
6.21 (1 H, d), 7.07-7.13 (1H, t), 7.27-7.39 (5H, m), 7.67-7.70 (1H, d), 7.78-7.81 (1H, d),
8.11 (1H, s) ppm; 13C NMR (500 MHz, CDCl3) δ 42.3, 53.0, 54.3, 67.3, 94.3, 126.3,
128.1, 128.3, 128.5, 130.2, 135.8, 135.9, 136.2, 140.6, 156.6, 166.8, 170.8 ppm.
3-Iodoarene-Z-L-Dab-Amino Methyl Ester, Catalyst 1b: dark brown oil, (0.148 g, 74%).
Rf = 0.75 ; IR (neat): 3385 (br), 3066 (m), 3040 (m), 2953 (m), 2922 (w), 1760 (s), 1745
(s), 1644 (s), 1556 (m), 1540 (m), 1454 (w), 1448 (w), 1267 (w), 1062 (w) cm-1; 1H NMR
(500 MHz, CDCl3) : 1.82-1.83 (1 H, m), 2.20-2.23 (1 H, m), 3.18-3.21 (1 H, m), 3.69
(3H, s), 3.86-3.88 (1 H, m), 4.46-4.49 (1H, m), 5.14 (2H, s), 5.97 (1H, s, br), 7.13-7.16
(1H, m), 7.32-7.35 (5H, m), 7.52 (1H, s, br), 7.79-7.81 (2H, m), 8.22 (1H, s) ppm; 13C
NMR (500 MHz, CDCl3) δ 32.8, 36.1, 51.6, 52.8, 67.4, 94.4, 126.2, 128.1, 128.3, 128.6,
130.2, 136.0, 136.1, 136.3, 140.4, 156.8, 166.0, 172.6 ppm.

134
3-Iodoarene-Z-L-Lys-Amino Methyl Ester, Catalyst 1c: dark brown oil, (0.167 g, 81%).
Rf =0.74 (1:9 MeOH:DCM); IR (neat): 3377 (br), 3070 (m), 3063 (m), 2939 (m), 2926
(m), 2854 (m), 1758 (s), 1714 (s), 1643 (s), 1566 (m), 1538 (m), 1454 (m), 1380 (w),
1266 (m), 1063 (w) cm-1; 1H NMR (500 MHz, CDCl3) : 1.68-1.72 (2 H, m), 1.78-1.95 (2
H, m), 3.41-3.56 (2 H, m), 3.75 (3H, s), 4.39-4.42 (1 H, m), 5.13 (2H, s), 5.58-5.61 (1H,
d), 6.73 (1H, br), 7.13-7.18 (1H, t), 7.35 (5H, m), 7.74-7.77 (1H, d), 7.80-7.83 (1H, d),
8.14 (1H, s) ppm; 13C NMR (500 MHz, CDCl3) δ 25.3, 30.4, 39.4, 52.6, 53.3, 67.2, 94.2,
126.3, 128.1, 128.3, 128.6, 130.2, 136.0, 136.1, 136.5, 140.3, 156.1, 166.1, 172.6 ppm.
3-Iodoaryl-Z-L-Orn-Amino Methyl Ester, Catalyst 1d: dark brown oil, (0.182 g, 86%). Rf
= 0.78 (1:9 MeOH:DCM); IR (neat): 3366 (br), 3063 (m), 3042 (m) 2940 (m), 2933 (m),
2861 (m), 1751 (s), 1720 (s), 1692 (s), 1644 (s), 1556 (m), 1538 (m), 1463 (m), 1455 (m),
1267 (m), 1193 (w), 1052 (w) cm-1; 1H NMR (500 MHz, CDCl3) : 1.45-1.67 (2 H, m),
1.71-1.87 (2 H, m), 2.05-2.09 (2 H, m), 3.37-3.44 (2 H, m), 3.75 (3H, s), 4.40 (1H, br),
5.04-5.12 (2H, m), 5.53-5.54 (1H, d), 6.51 (1H, br), 7.11-7.14 (1H, t), 7.34-7.36 (5H, m),
7.74-7.76 (1H, d), 7.79-7.81 (1H, d), 8.13 (1 h, br) ppm; 13C NMR (500 MHz, CDCl3) δ
22.5, 28.7, 32.3, 39.7, 52.5, 53.5, 67.1, 94.2, 126.3, 128.1, 128.2, 128.6, 130.2, 136.0,
136.2, 136.7, 140.2, 156.2, 166.2, 172.9 ppm.
5-Iodo-2-Methoxy-arene-Z-L-Dap-Amino Methyl Ester, Catalyst 2a: dark brown oil,
(0.175 g, 95%). Rf = 0.76 (1:9 MeOH:DCM); IR (neat): 3337 (br), 3068 (m), 3031 (m),
2953 (m), 2930 (m), 1759 (s), 1748 (s), 1643 (), 1551 (), 1543 (m), 1438 (s), 1270 (w),

135
1066 (w) cm-1; 1H NMR (500 MHz, CDCl3): δ 3.40-3.41 (2 H, m), 3.70 (3 H, s), 3.88 (3
H, s), 4.33-4.34 (1 H, br), 5.08 (2H, s), 5.59-5.62 (1 H, d), 6.67-6.70 (1H, d), 7.26-7.42
(5H, m), 7.64-7.67 (1H, d), 7.76 (1H, d), 8.43 (1H, s) ppm; 13C NMR (500 MHz, CDCl3)
δ 29.0, 39.3, 52.4, 56.2, 66.9, 83.7, 113.7, 123.6, 128.0, 128.1, 128.5, 136.3, 140.6, 141.1,
156.1, 157.2, 163.9, 172.9 ppm.
5-Iodo-2-methoxy-arene-Z-L-Dab-Amino Methyl Ester, Catalyst 2b: dark brown oil,
(0.129 g, 68%). Rf = 0.75 (1:9 MeOH:DCM); IR (neat): 3385 (br), 3066 (m), 3040 (m),
2953 (m), 2922 (w), 1760 (s), 1745 (s), 1644 (s), 1556 (m), 1540 (m), 1454 (w), 1448
(w), 1267 (w), 1062 (w) cm-1; 1H NMR (500 MHz, CDCl3) : 1.82-1.87 (1 H, m), 2.18-
2.22 (1 H, m), 3.21-3.23 (1 H, m), 3.68 (3H, s), 3.83-3.87 (1 H, m), 3.95 (3H, s), 4.47-
4.50 (1H, m), 5.13 (2H, s), 5.82-5.84 (1H, d), 6.72-6.74 (1H, d), 7.33-7.36 (5H, m), 7.68-
7.70 (1H, d), 8.34 (1H, s, br), 8.46 (1H, s) ppm; 13C NMR (500 MHz, CDCl3) δ 32.9,
35.7, 51.7, 52.6, 56.1, 67.1, 83.4, 113.7, 123.4, 128.0, 128.2, 128.6, 136.2, 140.6, 141.3,
156.5, 157.5, 164.1, 172.8 ppm.
5-Iodo-2-methoxy-Iodoarene-Z-L-Lys-Amino Methyl Ester, Catalyst 2c: dark brown oil,
(0.140 g, 72%). Rf = 0.73 (1:9 MeOH:DCM); IR (neat): 3398 (br), 3074 (m), 3052 (m),
2942 (m), 2932 (m), 2882 (m), 1772 (s), 1728 (s), 1640 (s), 1560 (m), 1544 (m), 1426
(m), 1378 (w), 1263 (m), 1060 (w) cm-1; 1H NMR (500 MHz, CDCl3) : 1.67-1.78 (2 H,
m), 1.94-1.97 (2 H, m), 3.48-3.49 (2 H, m), 3.77 (3H, s), 3.95 (3 H, s), 4.43-4.46 (1H,
m), 5.13 (2H, s), 5.46-5.47 (1H, d), 6.75-6.77 (1H, d), 7.34-7.38 (5H, m), 7.73-7.74 (1H,

136
d), 7.80 (1H, br), 8.48 (1H, s) ppm; 13C NMR (500 MHz, CDCl3) δ 25.6, 30.1, 39.2, 52.6,
53.7, 56.2, 67.1, 83.8, 113.7, 123.5, 128.1, 128.2, 128.6, 136.2, 140.8, 141.2, 155.9,
157.2, 163.9, 172.7 ppm.
5-Iodo-2-methoxy-Iodoarene-Z-L-Orn-Amino Methyl Ester, Catalyst 2d: dark brown oil,
(0.140 g, 70%). Rf = 0.76 (1:9 MeOH:DCM); IR (neat): 3378 (br), 3058 (m), 3048 (m)
2936 (m), 2940 (m), 2858 (m), 1760 (s), 1728 (s), 1700 (s), 1640 (s), 1562 (m), 1530 (m),
1458 (m), 1458 (m), 1270 (m), 1190 (w), 1048 (w) cm-1; 1H NMR (500 MHz, CDCl3) :
1.41-1.48 (2H, m), 1.61-1.67 (2H, m), 1.72-1.79 (1H, m), 1.89-1.93 (1H, m), 3.42-3.50
(2H, m), 3.75 (3H, s), 3.94 (3H, s), 4.36-4.41 (1H, m), 5.12 (2H, s), 5.47-5.48 (1H, d),
6.73-6.75 (1H, d), 7.36 (5H, m), 7.70-7.72 (1H, d), 7.77 (1H, s, br), 8.49 (1H, s) ppm; 13C
NMR (500 MHz, CDCl3) δ 22.6, 29.1, 32.1, 39.3, 52.4, 53.8, 56.2, 66.9, 83.8, 113.7,
123.6, 128.1, 128.2, 128.5, 136.3, 140.8, 141.2, 156.0, 157.2, 163.9, 172.9 ppm.
5-Iodo-2-methoxy-4-mehtyl-Z-L-Dap-Amino Methyl Ester, Catalyst 3a: dark brown oil,
(0.149 g, 83%). %). Rf = 0.76 (1:9 MeOH:DCM); IR (neat): 3348 (br), 3072 (m), 3026
(m), 2950 (m), 2936 (m), 1760 (s), 1748 (s), 1640 (s), 1551 (s), 1538 (m), 1440 (s), 1262
(w), 1060 (w) cm-1; 1H NMR (500 MHz, CDCl3): 2.45 (3 H, s), 3.79 (3 H, s), 3.80-3.88
(2H, m), 3.89 (3H, s), 4.54-4.55 (1H, d), 5.09-5.16 (2H, m), 6.01-6.03 (1H, d), 6.84 (1H,
s), 7.31-7.35 (5H, m), 8.07 (1H, s, br), 8.52 (1H, s) ppm; 13C NMR (500 MHz, CDCl3) δ
17.6, 28.5, 41.5, 54.8 (Residual DCM solvent peak), 54.8, 56.1, 67.0, 90.6, 113.0, 120.2,
128.0, 128.2, 128.5, 136.2, 142.1, 146.9, 156.1, 157.5, 164.9, 171.1 ppm.

137
5-Iodo-2-methoxy-4-mehtyl -Z-L-Dab-Amino Methyl Ester, Catalyst 3b: dark brown oil,
(0.152 g, 82%). %). Rf = 0.74 (1:9 MeOH:DCM); IR (neat): 3390 (br), 3062 (m), 3040
(m), 2948 (m), 2926 (w), 1760 (s), 1752 (s), 1642 (s), 1560 (m), 1548 (m), 1468 (w),
1442 (w), 12664 (w), 1058 (w) cm-1; 1H NMR (500 MHz, CDCl3) : 1.84-1.89 (1H, m),
2.16-2.25 (1H, m), 2.45 (3H, s), 3.19-3.24 (1H, m), 3.68 (3H, s), 3.83-3.87 (1H, m), 3.96
(3H, s), 4.48-4.49 (1H, d), 5.14 (2H, s,), 5.73-5.74 (1H, d), 6.85 (1H, s), 7.32-7.39 (5H,
m), 8.30 (1H, s, br), 8.56 (1H, s) ppm; 13C NMR (500 MHz, CDCl3) δ 17.7, 28.5, 33.0,
35.6, 51.7, 55.9, 67.1, 90.4, 112.9, 120.7, 128.0, 128.2, 128.6, 136.2, 141.9, 146.4, 156.4,
157.6, 164.1, 172.8 ppm.
5-Iodo-2-methoxy-4-mehtyl -Z-L-Lys-Amino Methyl Ester, Catalyst 3c: dark brown oil,
(0.165 g, 87%). Rf = 0.76 (1:9 MeOH:DCM); IR (neat): 3390 (br), 3076 (m), 3060 (m),
2944 (m), 2924 (m), 2849 (m), 1760 (s), 1712 (s), 1648 (s), 1572 (m), 1544 (m), 1456
(m), 1388 (w), 1264 (m), 1066 (w) cm-1; 1H NMR (500 MHz, CDCl3) : 1.60-1.68 (2H,
m), 1.72-1.95 (2H, m,), 2.46 (3H, s), 3.46-3.47 (2H, d), 3.76 (3H, s), 3.94 (3 H, s), 4.42-
4.43 (1H, d), 5.12 (2H, s), 5.48-5.49 (1H, br), 6.86 (1H, s), 7.37 (5H, m), 7.79 (1H, s, br),
8.56 (1H, s) ppm; 13C NMR (500 MHz, CDCl3) δ 25.7, 28.5, 30.1, 39.1, 52.5, 53.7, 56.1,
67.0, 90.7, 112.9, 120.8, 128.1, 128.2, 128.6, 136.2, 142.1, 146.4, 155.9, 157.4, 163.9,
172.7 ppm.

138
5-Iodo-2-methoxy-4-mehtyl -Z-L-Orn-Amino Methyl Ester, Catalyst 3d: dark brown oil,
(0.187 g, 96%). Rf = 0.77 (1:9 MeOH:DCM); IR (neat): 3398 (br), 3078 (m), 3052 (m)
2956 (m), 2940 (m), 2858 (m), 1750 (s), 1728 (s), 1690 (s), 1648 (s), 1554 (m), 1532 (m),
1464 (m), 1442 (m), 1266 (m), 1190 (w), 1040 (w) cm-1; 1H NMR (500 MHz, CDCl3) :
1.19-1.25 (2H, m), 1.65-1.77 (2H, m), 1.91 (2H, m), 2.42 (3H, s). 3.42-3.44 (2H, m,) 3.72
(3H, s), 3.90 (3H, s), 4.38-4.40 (1H, d), 5.09 (2H, s), 5.64-5.67 (1H, d), 6.82 (1H, s),
7.24-7.38 (5H, m), 7.78 (1H, s, br), 8.50 (1H, s) ppm; 13C NMR (500 MHz, CDCl3) δ
25.7, 28.5, 29.5, 29.9, 39.1, 52.5, 53.7, 56.1, 66.9, 90.6, 112.9, 120.8, 128.0, 128.2, 128.5,
136.2, 141.9, 146.3, 156.0, 157.3, 163.9, 172.7 ppm.
Synthesis of 3-iodoarene-Nα-Fmoc-L-diaminopropionic acid, 6l, occurred over 3 steps
including:
Hydrolysis of Iodoarene Amino Methyl Ester Catalyst 1a. The iodoarene amino methyl
ester Catalyst 1a (10 g, 20.7 mmol) was dissolved in dioxane (50-100 mL) and cooled to
0 °C. Sodium hydroxide (2N) (10 equiv.) was slowly added and the resulting mixture was
allowed to stir for 18 hours at room temperature. The organic solvent was evaporated off,
and then H2O (mL) was added. The solution was acidified to pH ≈ 6 with 3M HCl. This
solution was extracted with EtOAc (x4) and the organic layers were washed with brine
(x1) and dried over Na2SO4. The crude product was obtained following rotary
evaporation, and carried on to next step without purification.

139
Cbz deprotection of Catalyst 1a303 While under at argon atmosphere, the hydrolyzed
product (2.5 g, 5.3 mmol) was dissolved in anhydrous DCM (60 mL). The solution was
cooled to 0 °C, and trimethylsilane (11.7 mmol, 2.2 equiv.) was slowly added. This
mixture was stirred at 0 °C for 3 h, and then 12 h at room temperature. The mixture was
concentrated to dryness and the residue was taken up in water. The pH was adjusted to ≈
6 with 0.2N NaOH, and the pale-yellow precipitate was collected, dried via high pump
vacuum, and carried on to the next step without purification.
N-Fmoc Protection (6l)304 A round-bottomed flask was charged with the amino acid
(1.34 g, 4 mmol), H2O (100 mL), and diisopropylethylamine (DIPEA) (0.78 g, 6.02
mmol, 1.5 equiv). The reaction was cooled in an ice bath, followed by the addition of
Fmoc-OSu (1.47 g, 4.37 mmol, 1.1 equiv) in MeCN (100 mL) and was stirred for 30
minutes. The reaction was stirred at room temperature overnight. The solvent was
rotovapped off, and the resulting mixture was dissolved in water (100 mL) and then
acidified with 2N HCl to pH ~ 3. The aqueous layer was extracted with ethyl acetate
(x3), and dried over Na2SO4. The crude product was purified via column
chromatography (gradient from 100% DCM to 1:9 MeOH/DCM) to give 6l: off white
solid, (1.8 g, 81% over 3 steps); Rf = 0.14 (Gradient 1:9 MeOH:DCM); IR (neat): 3480
(br), 3180 (w), 2571 (br), 1697 (br), 1635 (m), 1601 (br), 1558 (m), 1447 (m), 1423 (m),
1404 (m), 1354 (m), 1288 (w), 1249 (w), 736 (s), (m), 1426 (m), 1378 (w), 1263 (m),
1060 (w) cm-1; 1H NMR (500 MHz, (MeOD) : 3.39-3.76 (1H, m), 3.82-3.85 (1H, dd),
4.18-4.20 (1H, m), 4.23-4.27 (1H, m), 4.37-4.40 (2H, m), 7.18-7.20 (1H, d), 7.21-7.27

140
(2H, m), 7.28 (1H, br), 7.35-7.39 (2H, m ), 7.63-7.64 (2H, m), 7.77-7.86 (4H, m), 8.17
(1H, s) ppm; 13C NMR (500 MHz, MeOD) δ 42.1, 42.3, 54.4, 66.7, 93.3, 119.5, 124.9,
126.3, 126.8, 127.3, 129.9, 136.1, 136.3, 140.2, 141.1, 143.9, 157.1, 167.5, 174.4 ppm.
Crystal Structure of Catalyst 2a (C20H21IN2O6)
Crystal Structure Report for D8_2036_MS163_EtOH
A specimen of C20H21IN2O6, approximate dimensions 0.027 mm x 0.029 mm x 0.388
mm, was used for the X-ray crystallographic analysis. The X-ray intensity data were
measured.
The integration of the data using an orthorhombic unit cell yielded a total of 16298
reflections to a maximum θ angle of 25.50° (0.83 Å resolution), of which 3958 were
independent (average redundancy 4.118, completeness = 99.5%, Rint = 3.30%, Rsig =
3.06%) and 3792 (95.81%) were greater than 2σ(F2). The final cell constants of a =
4.8220(2) Å, b = 13.9582(6) Å, c = 31.7890(14) Å, volume = 2139.60(16) Å3, are based
upon the refinement of the XYZ-centroids of reflections above 20 σ(I). The calculated
minimum and maximum transmission coefficients (based on crystal size) are 0.8771 and
1.0000.
The structure was solved and refined using the Bruker SHELXTL Software Package,
using the space group P 21 21 21, with Z = 4 for the formula unit, C20H21IN2O6. The final
anisotropic full-matrix least-squares refinement on F2 with 268 variables converged at R1
141
= 2.31%, for the observed data and wR2 = 5.49% for all data. The goodness-of-fit was
1.123. The largest peak in the final difference electron density synthesis was 0.380 e-/Å3
and the largest hole was -0.994 e-/Å3 with an RMS deviation of 0.087 e-/Å3. On the basis
of the final model, the calculated density was 1.590 g/cm3 and F(000), 1024 e-.
142

143
Table 1. Sample and crystal data for D8_2036_MS163_EtOH. Identification code D8_2036_MS163_EtOH Chemical formula C20H21IN2O6 Formula weight 512.29 g/mol Temperature 140(2) K Wavelength 0.71073 Å Crystal size 0.027 x 0.029 x 0.388 mm Crystal system orthorhombic Space group P 21 21 21 Unit cell dimensions a = 4.8220(2) Å α = 90° b = 13.9582(6) Å β = 90° c = 31.7890(14) Å γ = 90° Volume 2139.60(16) Å3 Z 4 Density (calculated) 1.590 g/cm3 Absorption coefficient 1.534 mm-1 F(000) 1024 Table 2. Data collection and structure refinement for D8_2036_MS163_EtOH. Theta range for data collection 2.41 to 25.50°
Index ranges -5<=h<=5, -16<=k<=16, -38<=l<=38 Reflections collected 16298 Independent reflections 3958 [R(int) = 0.0330]
Max. and min. transmission 1.0000 and 0.8771
Structure solution technique direct methods
Structure solution program SHELXT-2014 (Sheldrick 2014)
Refinement method Full-matrix least-squares on F2 Refinement program SHELXL-2014 (Sheldrick 2014) Function minimized Σ w(Fo2 - Fc2)2 Data / restraints / parameters 3958 / 2 / 268
Goodness-of-fit on F2 1.123 Δ/σmax 0.002

144
Final R indices 3792 data; I>2σ(I) R1 = 0.0231, wR2 = 0.0542
all data R1 = 0.0249, wR2 = 0.0549
Weighting scheme w=1/[σ2(Fo2)+(0.0280P)2+0.1408P] where P=(Fo2+2Fc2)/3
Absolute structure parameter -0.0(0)
Largest diff. peak and hole 0.380 and -0.994 eÅ-3
R.M.S. deviation from mean 0.087 eÅ-3
Table 3. Atomic coordinates and equivalent isotropic atomic displacement parameters (Å2) for D8_2036_MS163_EtOH. U(eq) is defined as one third of the trace of the orthogonalized Uij tensor. x/a y/b z/c U(eq) I1 0.79969(5) 0.51368(2) 0.84588(2) 0.03603(10) O1 0.2988(6) 0.36512(15) 0.70696(8) 0.0313(6) O2 0.0091(5) 0.66481(18) 0.58967(9) 0.0304(6) O3 0.3672(5) 0.5705(2) 0.61011(9) 0.0329(7) O4 0.7479(11) 0.3631(2) 0.54951(12) 0.0906(16) O5 0.0397(9) 0.2617(2) 0.58074(10) 0.0547(9) O6 0.9626(6) 0.63738(18) 0.70624(8) 0.0356(7) N1 0.9333(7) 0.4510(2) 0.68548(10) 0.0284(7) N2 0.9346(6) 0.5114(2) 0.59928(9) 0.0244(6) C1 0.0008(8) 0.4135(2) 0.61069(12) 0.0254(8) C2 0.8597(7) 0.3844(2) 0.65225(12) 0.0279(8) C3 0.1616(8) 0.4393(2) 0.70849(11) 0.0232(8) C4 0.2450(7) 0.5185(2) 0.73803(10) 0.0224(7) C5 0.1532(8) 0.6143(2) 0.73659(11) 0.0259(8) C6 0.2565(9) 0.6807(2) 0.76521(12) 0.0327(10) C7 0.4453(8) 0.6535(3) 0.79560(12) 0.0312(9) C8 0.5329(8) 0.5594(3) 0.79791(11) 0.0264(8) C9 0.4360(7) 0.4932(2) 0.76886(10) 0.0244(7) C10 0.1249(8) 0.5807(3) 0.60082(12) 0.0241(8) C11 0.1987(9) 0.7463(2) 0.58822(13) 0.0337(9)
145
x/a y/b z/c U(eq) C12 0.0578(8) 0.8230(2) 0.56285(12) 0.0271(8) C13 0.8394(8) 0.8757(2) 0.57918(12) 0.0297(9) C14 0.7114(10) 0.9464(3) 0.55561(13) 0.0373(9) C15 0.8026(11) 0.9651(3) 0.51527(13) 0.0427(10) C16 0.0182(10) 0.9139(3) 0.49849(13) 0.0430(12) C17 0.1467(9) 0.8422(3) 0.52222(12) 0.0353(10) C18 0.9123(10) 0.3451(3) 0.57604(13) 0.0369(10) C19 0.9662(16) 0.1877(3) 0.55087(17) 0.079(2) C20 0.8745(11) 0.7350(3) 0.70233(14) 0.0444(12) Table 4. Bond lengths (Å) for D8_2036_MS163_EtOH. I1-C8 2.094(4) O1-C3 1.230(4) O2-C10 1.347(4) O2-C11 1.460(5) O3-C10 1.213(5) O4-C18 1.184(5) O5-C18 1.324(5) O5-C19 1.447(6) O6-C5 1.371(5) O6-C20 1.433(4) N1-C3 1.331(5) N1-C2 1.451(5) N1-H1 0.83(2) N2-C10 1.335(5) N2-C1 1.449(5) N2-H2 0.84(2) C1-C18 1.518(5) C1-C2 1.540(5) C1-H1A 1.0 C2-H2A 0.99 C2-H2B 0.99 C3-C4 1.505(5) C4-C9 1.390(5) C4-C5 1.409(5) C5-C6 1.391(5) C6-C7 1.380(6) C6-H6 0.95 C7-C8 1.383(5) C7-H7 0.95 C8-C9 1.387(5) C9-H9 0.95 C11-C12 1.503(5) C11-H11A 0.99 C11-H11B 0.99 C12-C13 1.385(6) C12-C17 1.387(5) C13-C14 1.385(5) C13-H13 0.95 C14-C15 1.381(6) C14-H14 0.95 C15-C16 1.370(6) C15-H15 0.95 C16-C17 1.398(6) C16-H16 0.95 C17-H17 0.95 C19-H19A 0.98 C19-H19B 0.98 C19-H19C 0.98 C20-H20A 0.98 C20-H20B 0.98 C20-H20C 0.98

146
Table 5. Bond angles (°) for D8_2036_MS163_EtOH. C10-O2-C11 115.3(3) C18-O5-C19 116.1(4) C5-O6-C20 118.9(3) C3-N1-C2 121.6(3) C3-N1-H1 117.(3) C2-N1-H1 120.(3) C10-N2-C1 121.6(3) C10-N2-H2 118.(3) C1-N2-H2 119.(3) N2-C1-C18 110.4(3) N2-C1-C2 111.4(3) C18-C1-C2 109.4(3) N2-C1-H1A 108.5 C18-C1-H1A 108.5 C2-C1-H1A 108.5 N1-C2-C1 110.4(3) N1-C2-H2A 109.6 C1-C2-H2A 109.6 N1-C2-H2B 109.6 C1-C2-H2B 109.6 H2A-C2-H2B 108.1 O1-C3-N1 121.7(3) O1-C3-C4 120.0(3) N1-C3-C4 118.3(3) C9-C4-C5 118.1(3) C9-C4-C3 115.5(3) C5-C4-C3 126.3(3) O6-C5-C6 122.9(3) O6-C5-C4 117.2(3) C6-C5-C4 119.9(3) C7-C6-C5 120.7(3) C7-C6-H6 119.6 C5-C6-H6 119.6 C6-C7-C8 120.0(3) C6-C7-H7 120.0 C8-C7-H7 120.0 C7-C8-C9 119.6(3) C7-C8-I1 121.1(3) C9-C8-I1 119.3(3) C8-C9-C4 121.6(3) C8-C9-H9 119.2 C4-C9-H9 119.2 O3-C10-N2 125.8(4) O3-C10-O2 124.4(4) N2-C10-O2 109.7(3) O2-C11-C12 106.8(3) O2-C11-H11A 110.4 C12-C11-H11A 110.4 O2-C11-H11B 110.4 C12-C11-H11B 110.4 H11A-C11-H11B 108.6 C13-C12-C17 118.8(4) C13-C12-C11 121.4(3) C17-C12-C11 119.8(4) C14-C13-C12 121.0(4) C14-C13-H13 119.5 C12-C13-H13 119.5 C15-C14-C13 119.7(4) C15-C14-H14 120.2 C13-C14-H14 120.2 C16-C15-C14 120.3(4) C16-C15-H15 119.8 C14-C15-H15 119.8 C15-C16-C17 120.0(4) C15-C16-H16 120.0 C17-C16-H16 120.0 C12-C17-C16 120.3(4) C12-C17-H17 119.9 C16-C17-H17 119.9 O4-C18-O5 125.2(4) O4-C18-C1 124.9(4) O5-C18-C1 109.9(4) O5-C19-H19A 109.5 O5-C19-H19B 109.5

147
H19A-C19-H19B 109.5 O5-C19-H19C 109.5 H19A-C19-H19C 109.5 H19B-C19-H19C 109.5 O6-C20-H20A 109.5 O6-C20-H20B 109.5 H20A-C20-H20B 109.5 O6-C20-H20C 109.5 H20A-C20-H20C 109.5 H20B-C20-H20C 109.5 Table 6. Anisotropic atomic displacement parameters (Å2) for D8_2036_MS163_EtOH. The anisotropic atomic displacement factor exponent takes the form: -2π2[ h2 a*2 U11 + ... + 2 h k a* b* U12 ] U11 U22 U33 U23 U13 U12
I1 0.03463(14) 0.04674(15) 0.02670(13) -0.00231(11) -0.00304(10)
-0.00369(11)
O1 0.0403(15) 0.0195(12) 0.0343(14) -0.0048(10) -0.0065(13) 0.0047(13) O2 0.0202(14) 0.0244(13) 0.0465(18) 0.0080(12) -0.0037(12) -0.0041(11) O3 0.0165(14) 0.0351(15) 0.0470(18) 0.0040(13) -0.0014(12) -0.0002(11) O4 0.161(5) 0.0463(18) 0.065(2) -0.0160(17) -0.073(3) 0.018(3) O5 0.090(3) 0.0275(15) 0.047(2) -0.0080(14) -0.007(2) 0.0066(17) O6 0.0535(19) 0.0239(14) 0.0293(15) 0.0009(11) -0.0023(14) 0.0127(13) N1 0.0320(18) 0.0263(16) 0.0268(17) -0.0040(14) -0.0028(15) 0.0036(14) N2 0.0180(14) 0.0236(16) 0.0315(16) 0.0035(14) -0.0048(13) -0.0004(14) C1 0.027(2) 0.0226(18) 0.027(2) -0.0001(16) -0.0052(17) 0.0016(16) C2 0.031(2) 0.0291(17) 0.0236(18) -0.0010(16) -0.0021(17) -0.0055(14) C3 0.028(2) 0.0228(17) 0.0189(18) 0.0012(14) 0.0092(16) -0.0007(17) C4 0.0250(19) 0.0213(15) 0.0208(16) 0.0008(13) 0.0066(13) -0.0009(15) C5 0.032(2) 0.0225(17) 0.0228(19) 0.0028(14) 0.0046(16) 0.0023(16) C6 0.050(3) 0.0167(15) 0.031(2) -0.0005(14) 0.0083(19) 0.0020(17) C7 0.044(2) 0.0234(19) 0.026(2) -0.0076(16) 0.0058(18) -0.0100(17) C8 0.027(2) 0.0300(19) 0.0222(19) 0.0010(15) 0.0035(15) -0.0067(16) C9 0.0280(17) 0.0204(17) 0.0248(18) -0.0018(15) 0.0058(14) -0.0007(15) C10 0.026(2) 0.027(2) 0.0194(18) 0.0030(15) 0.0016(15) 0.0008(16) C11 0.0254(19) 0.0292(18) 0.047(2) 0.0026(16) -0.003(2) -0.0099(18) C12 0.029(2) 0.0209(18) 0.031(2) 0.0001(15) -0.0038(17) -0.0116(16) C13 0.034(2) 0.0301(18) 0.025(2) -0.0001(15) 0.0012(17) -0.0107(18) C14 0.041(2) 0.0285(17) 0.042(2) -0.0021(17) 0.000(2) -0.002(2) C15 0.058(3) 0.029(2) 0.041(2) 0.0072(17) -0.014(2) -0.007(2) C16 0.062(3) 0.044(3) 0.024(2) 0.0060(19) 0.003(2) -0.018(2) C17 0.040(3) 0.033(2) 0.033(2) -0.0041(17) 0.0063(18) -0.0077(18) C18 0.057(3) 0.029(2) 0.026(2) 0.0027(17) -0.005(2) -0.0030(19) C19 0.154(6) 0.032(2) 0.051(3) -0.017(2) -0.004(4) 0.000(3)
148
U11 U22 U33 U23 U13 U12 C20 0.064(3) 0.030(2) 0.039(3) 0.0063(18) 0.005(2) 0.018(2) Table 7. Hydrogen atomic coordinates and isotropic atomic displacement parameters (Å2) for D8_2036_MS163_EtOH. x/a y/b z/c U(eq)
H1 -0.141(7) 0.5044(19) 0.6864(13) 0.03
H2 -0.232(5) 0.527(3) 0.5959(12) 0.03
H1A 0.2062 0.4082 0.6144 0.031 H2A -0.3441 0.3841 0.6484 0.033 H2B -0.0815 0.3189 0.6602 0.033 H6 0.1965 0.7455 0.7638 0.039 H7 0.5150 0.6996 0.8149 0.037 H9 0.5018 0.4291 0.7700 0.029 H11A 0.3759 0.7276 0.5748 0.04 H11B 0.2381 0.7697 0.6170 0.04 H13 -0.2234 0.8631 0.6070 0.036 H14 -0.4385 0.9820 0.5672 0.045 H15 -0.2846 1.0137 0.4991 0.051 H16 0.0804 0.9270 0.4707 0.052 H17 0.2954 0.8065 0.5105 0.042 H19A 0.0715 0.1294 0.5571 0.119 H19B -0.2328 0.1742 0.5529 0.119 H19C 0.0101 0.2095 0.5223 0.119 H20A -0.2609 0.7403 0.6795 0.067 H20B -0.2110 0.7559 0.7288 0.067 H20C 0.0351 0.7757 0.6961 0.067 Table 8. Hydrogen bond distances (Å) and angles (°) for D8_2036_MS163_EtOH. Donor-
H Acceptor-H
Donor-Acceptor Angle
N1-H1...O6 0.83(2) 2.02(3) 2.688(4) 137.(4) N2-H2...O3 0.84(2) 2.08(3) 2.879(4) 160.(4) C2-H2A...O1 0.99 2.55 3.227(5) 125.6

149
Donor-H
Acceptor-H
Donor-Acceptor Angle
C7-H7...O1 0.95 2.58 3.202(4) 123.7 C20-H20B...O1 0.98 2.58 3.508(5) 157.2
Synthesis of Ac-Ile-3-I-Dap -Pro-D-Ala-Ala-Ala Peptide, Figure 3.9:
Compound 6l was installed into a peptide as described: Following standard Fmoc-SPPS
protocols, peptide precatalyst 6l, with the sequence, Ac-Ile-3-I-Dap -Pro-D-Ala-Ala-Ala,
was prepared on solid support using the commercially available Rink Amide MBHA
resin (200 mg, 0.3–0.8 meq/g, 200-400 mesh). Rink Amide MBHA resin comes pre-
loaded with an Fmoc-protected amine for the preparation of C-terminal amide peptides.
Therefore, amino acid residues were built on the solid support beginning at the C-
terminus, more specifically Ala-Ala-D-Ala-Pro-3-I-Dap-Ile. Double couplings were
achieved using a 5 equiv. excess of the appropriate Fmoc-amino acid, HCTU as the
coupling agent, and DIPEA in DMF as the solvent. Fmoc-deprotections were completed
upon triple treatments with a basic solution of 20% 4-methylpiperidine in DMF. A step-
wise procedure for the Fmoc-solid-phase synthesis of the peptide is given below.
Resin Swelling: The Rink Amide MBHA resin (200 mg) was added to the barrel of a 10
mL fritted syringe and swelled by a 4 mL DCM wash, followed by soaking for 5 min. in
an additional 5 mL of DCM. The residual DCM was purged from the syringe and the
resin was washed with DMF (3 x 4 mL).

150
Fmoc-Deprotection: The initial Fmoc-deprotection of the Rink Amide resin was
performed by three sequential one-min treatments with 2 mL of 20% 4-methylpiperidine
in DMF solution. The deprotected resin was then washed three times with 5 mL of DMF,
resulting in a free amine ready for the first amino acid coupling.
Amino acid couplings: The peptide was assembled on the solid-support beginning at the
C-terminus. Therefore, 5 equiv. (relative to resin loading) of Fmoc-L-alanine, HCTU, and
DIPEA were added to a 3 ml screw-top vial. The solids were then dissolved in 1 mL of
DMF. This prepared mixture was then taken up into the fritted syringe, containing the
resin, and agitated for 2 min. This first coupling mixture was then ejected. The resin was
subsequently treated with a second aliquot of the Fmoc-L-alanine amino acid mixture for
2 min. Following this double amino acid coupling procedure, the resin was once again
washed with DMF (3 x 5 mL). The Nα-Fmoc group was then removed following the
above described Fmoc-deprotection protocol in order to achieve the next amino acid
residue coupling of another Fmoc-L-alanine residue. This deprotection-coupling pattern
was repeated until the desired amino acid residues were assembled as follows: Resin-Ala-
Ala-D-Ala-Pro-3-I-Dap-Ile. Where the 3-I-Dap amino acid residue represents the
incorporation of Catalyst 1a as its Fmoc-SPPS ready derivative 3-iodoarene-Nα-Fmoc-L-
diaminopropionic acid, 6l.
N-terminal Acetate Capping: The fully assembled peptide, sequence Ile-3-I-Dap -Pro-D-
Ala-Ala-Ala, was N-caped with an acetate group following double, 5 min, treatments

151
with a 4 mL aliquot of a 5% acetic anhydride in DMF solution. After the allotted time for
the double treatment, the solution was ejected from the syringe into a waste container and
the resin was washed with DMF (3 x 5 mL).
Peptide Cleavage and Isolation: Following N-terminal acetate capping of the resin-bound
peptide, the resin was washed with DCM (3 x 5 mL), and methanol (3 x 5 mL). The resin
was dried in a vacuum oven at room temperature for 24 h. The resin-bound peptide was
cleaved from and R-side chain deprotected via a 2 h treatment with an acidic cleavage
cocktail of trifluoroacetic acid: H2O: triisopropylsilane (95:4.5:0.5). Following,
evaporation of the cleavage cocktail and precipitation with ice cold ether, the peptide was
isolated as a white powdery solid and characterized by MALDI-TOF mass spectrometry.
Ac-Ile-3-I-Dap -Pro-D-Ala-Ala-Ala peptide precatalyst as shown in Figure 3.9: white
solid, (0.140 g, 72%)
A representative procedure for the n-butyl amidation of Catalysts 4a-4h is as follows,
using the starting materials for Catalyst 4a as an example337:
To a flame dried round bottom flask was added 3-amino-2-iodobenzoic acid 4aa
(1.1 g, 4.18 mmol, 1.0 equiv) and triethylamine (1.17 mL, 8.4 mmol, 2.0 equiv) in
anhydrous DCM (40 mL) while under nitrogen. This homogeneous solution was stirred
I
OH
OH2N
I
NH
OH2N
4ab74%
1.) NEt3, Dry DCMSOCl2 (0ºC)
2.) NEt3, Dry DCM
NH2
I
NH
OHN
OCatalyst 4a
94%
Acetic anhydridepyridine, r.t.
4aa

152
for 5 min at room temperature under nitrogen. The solution was cooled to 0 °C. Thionyl
chloride (0.34 mL, 4.6 mmol, 1.1 equiv) was slowly added (dropwise over 20 min), and
the resulting solution was allowed to stir at 0 °C for 1 h. The resulting mixture was
brought to room temperature and stirred for 30 min. This solution was concentrated under
vacuum and then redissolved in anhydrous DCM (40 mL). To this solution was added
(dropwise over 10 min) 1-butylamine (0.45 mL, 4.6 mmol, 1.1 equiv), anhydrous DCM
(20 mL), and triethylamine (1.17 mL, 8.4 mmol, 2.0 equiv). The resulting solution was
stirred overnight at room temperature under nitrogen. The crude mixture was
concentrated by rotary evaporation, diluted in DCM, and washed with 1 M HCl, and the
aqueous layers were extracted with DCM (×3). The organic layers were washed with H2O
(×2) and saturated brine (×1) and dried over sodium sulfate. The resulting product was
filtered and concentrated by rotary evaporation. The crude product was purified by
column chromatography on silica gel (Gradient to 95:5 DCM:MeOH + 2% NEt3) to give
3-amino-N-butyl-2-iodobenzamide (4ab) as a yellow solid (0.98 g, 74%). 1H NMR (500
MHz, MeOD): δ 0.98−1.01 (3H, t), 1.46−1.52 (2H, m), 1.60−1.66 (2H, m), 3.26−3.30
(2H, q), 6.58-6.60 (1H, dd), 6.82-6.84 (1H, d), 7.12-7.15 (1H, d) ppm. 13C NMR (500
MHz, CDCl3): δ 13.8, 20.1, 31.3, 39.7, 46.3 (residual DCM), 82.3, 115.3, 116.9, 129.0,
143.8, 147.8, 170.4 ppm.
A representative procedure for the acetylation of Catalysts 4a-4h is as follows,
using Catalyst 4a as an example337:

153
3-amino-N-butyl-2-iodobenzamide 4ab (0.50 g, 1.06 mmol) in tetrahydrofuran
(THF) (10 mL) was mixed with a mixture of acetic anhydride (1 mL, 10.6 mmol) and
pyridine (86 μL, 1.06 mmol), and the mixture was stirred overnight at RT. The reaction
mixture was then acidified with 1% HCl and the final product was extracted into ethyl
acetate (3 X 20 mL). The organic phase was washed with brine, dried over sodium
sulfate and concentrated in vacuo to give the crude product. The crude product was
purified by column chromatography on silica gel (Gradient to 50:50 EtOAc:Hex) to give
Catalyst 4a as a yellow solid (0.53 g, 94%). IR (neat): 3264 (br), 2920 (s), 2851 (m),
1643 (s), 1543 (m), 1454 (m), 1257 (m), 1092 (w), 1018 (s), 799 (s) cm-1. 1H NMR (500
MHz, CDCl3): δ 0.95−0.98 (3H, t), 1.41−1.50 (2H, m), 1.60−1.63 (2H, m), 2.33 (3H, s),
3.41-3.43 (2H, t), 6.28 (1H, s, br), 6.98-6.99 (1H, d), 7.20-7.29 (1H, m), 7.87-7.94 (1H,
d) ppm. 13C NMR (500 MHz, CDCl3): δ 13.8, 20.2, 24.6, 31.4, 39.8, 90.5, 123.6, 128.9,
129.4, 139.0, 144.0, 168.8, 169.7 ppm.
General Synthesis of Catalyst 4b (Org. Lett. 2018, 20, 2345-348):
A representative procedure for diazotization/iodination is described using 4ba as
the substrate to produce 4bb:
I
OH
O
4bc90%
O2N
I
OH
O
H2N
I
OH
O
4bd90%
1.) NEt3, Dry DCMSOCl2 (0ºC)
2.) NEt3, Dry DCM
NH2
I
NH
O
NH
O
NH Catalyst 4b
79%
NH2
OH
O
O2N
NaNO2 (0ºC)KI (90ºC)
HCl/H2O4ba 4bb
72%
Fe powder, NH4ClH2O/EtOH (60ºC)
Acetic anhydridepyridine, r.t. O

154
2-amino-4-nitro-carboxylic acid 4ba (1.82 g, 10 mmol) was added to a 100 mL
round bottom flask. To this flask was added conc. HCl (14 mL) and water (14 mL). The
suspension was cooled in an ice bath to 0 ℃. When cooled, sodium nitrite (0.83 g, 12
mmol, 1.2 equiv.) in 5 mL of water was slowly added. The reaction was stirred for 30
minutes at 0 ℃. Subsequently, KI (3.32 g, 20 mmol, 2 equiv.) in in 5 mL of water, was
slowly (over 10 minutes) added to the solution. The reaction was heated to 90 ℃ for 90
minutes. The reaction was cooled and quenched with a saturated solution of sodium
sulfite, and then transferred to a 500 mL separatory funnel with ethyl acetate. This was
acidified with conc. HCl (pH~2). The product was extracted with EtOAc (3x) then dried
over magnesium sulfate and filtered. The filtrate was concentrated and used without
further purification to give 2-iodo-4-nitrobenzoic acid (4bb) as a yellow solid (0.98 g,
72%). 1H NMR (500 MHz, MeOD): δ 5.04 (1H, br), 7.91-7.93 (1H, d), 8.28-8.29 (1H, d),
8.75 (1H, s) ppm. 13C NMR (500 MHz, MeOD): δ 92.3, 122.5, 130.4, 135.1, 142.6,
148.6, 167.4 ppm.
A representative procedure for iron assisted nitro reduction is described using 4bb
as the substrate (Org. Lett. 2011, 13, 24, 6488-6491):
4-amino-2-iodobenzoic acid, 4bc:
To a two-neck round bottom flask was added a solution NH4Cl (0.547 g, 10.2
mmol, 3 equiv.) in H2O (10 mL) and EtOH (10 mL). To this was added iron powder
(1.14 g, 20.5 mmol, 6 equiv.), and the suspension was warmed to 60 ℃ for 30 minutes.
2-iodo-4-nitrobenzoic acid (4bb) (1g, 3.41 mmol, 1 equiv.) was then added in small

155
portions, and the reaction temperature was elevated to 80 ℃. The reaction was
monitored via TLC, and when the reaction was complete, the mixture was cooled in an
ice bath (0 ℃) and basified with NaOH (5N, pH>7) and filtered to remove the excess
iron powder. The filtrate was evaporated via rotary evaporation, and the residue was
acidified by 6N HCl (pH~3). The mixture was then filtered again and washed with
MeOH/DCM (v/v = 1/1). The filtrate was concentrated to give 4-amino-2-iodobenzoic
acid, 4bc (0.81 g, 90%). 1H NMR (500 MHz, MeOD): δ 6.63-6.65 (1H, d), 7.32 (1H, s),
7.74-7.75 (1H, d) ppm. 13C NMR (500 MHz, MeOD): δ 95.8, 112.5, 120.5, 126.2, 132.7,
152.7, 167.9 ppm.
4-acetamido-2-iodobenzoic acid, 4bd:
4bd was synthesized using the representative procedure for the acetylation as
stated above. By using 4-amino-2-iodobenzoic acid (4bc) as the starting material, 4-
acetamido-2-iodobenzoic acid (4bd) was synthesized in a 90% yield (0.85 g). 1H NMR
(500 MHz, MeOD): δ 2.15 (3H, s), 7.65-7.66 (1H, d), 7.84-7.86 (1H, d), 8.35 (1H, s)
ppm. 13C NMR (500 MHz, MeOD): δ 22.6, 93.7, 118.0, 129.8, 131.3, 131.4, 142.1,
167.7, 170.54 ppm.
4-acetamido-N-butyl-2-iodobenzoic acid, Catalyst 4b:

156
Catalyst 4b was synthesized using the representative procedure for the n-butyl
amidation as stated above by using 4-acetamido-2-iodobenzoic acid (4bd) as the starting
material. Catalyst 4b as a yellow solid (0.53 g, 79%). IR (neat): 3290 (br), 3263 (br),
2928 (s), 2859 (w), 1636 (s), 1589 (m), 1519 (m), 1369 (m), 1300 (w), 1258 (m), 833 (s)
cm-1. 1H NMR (500 MHz, CDCl3): δ 0.93−0.96 (3H, t), 1.40−1.44 (2H, m), 1.60−1.63
(2H, m), 2.07 (3H, s), 3.36-3.40 (2H, t), 6.86 (1H, s, br), 7.00-7.02 (1H, d), 7.29-7.30
(1H, d), 7.75 (1H, s), 9.40 (1H, br) ppm. 13C NMR (500 MHz, CDCl3): δ 13.8, 20.2, 24.4,
31.4, 39.9, 92.2, 119.7, 127.9, 130.7, 137.3, 140.1, 169.7, 169.9 ppm.
General Synthesis of Catalyst 4c:
5-amino-2-iodobenzoic acid, 4cc:
By using 4ca as the substrate, and applying the representative procedure for
diazotization/iodination, followed by the iron assisted nitro reduction, as described above,
gave the product (without purification) 5-amino-2-iodobenzoic acid (4cc) in a 65% yield
(1.87 g) over the two steps. 1H NMR (500 MHz, MeOD): δ 8.01-8.05 (1H, d), 8.29-8.32
(1H, d), 8.57 (1H, s) ppm. 13C NMR (500 MHz, MeOD): δ 101.6, 124.4, 125.7, 137.6,
142.7, 147.8, 166.5 ppm.
5-amino-N-butyl-2-iodobenzamide, 4cd:
NH2
OH
O
NO2
I
OH
O
NO2
4cb
I
OH
O
NH2
Fe powder, NH4ClH2O/EtOH (60ºC)
4cc65% (Over 2 steps)
I
NH
O
NH2
4cd76%
1.) NEt3, Dry DCMSOCl2 (0ºC)
2.) NEt3, Dry DCM
NH2
NaNO2 (0ºC)KI (90ºC)
HCl/H2O
I
NH
O
HN
OCatalyst 4c
84%
Acetic anhydridepyridine, r.t.
4ca

157
By using 4cc as the substrate and applying the representative procedure for the n-
butyl amidation as described above, 5-amino-N-butyl-2-iodobenzamide (4cd) was
synthesized (76%, 0.92 g). 1H NMR (500 MHz, MeOD): δ 0.99-1.05 (3H, m), 1.38-1.47
(2H, m), 1.52-1.67 (2H, m), 3.31-3.51 (2H, m), 7.25 (1H, br), 7.82-7.89 (2H, m), 8.22
(1H, s) ppm. 13C NMR (500 MHz, MeOD): δ 12.8, 19.8, 31.2, 39.5, 93.3, 126.1, 129.9,
135.9, 136.5, 140.1, 166.9 ppm.
5-acetamido-N-butyl-2-iodobenzoic acid, Catalyst 4c:
By using 4cd as the substrate and applying the representative procedure for the
acetylation as described above, Catalyst 4c was synthesized (84%, 0.92 g). IR (neat):
3746 (w), 2955 (w), 2924 (w), 2866 (w), 1670 (s), 1589 (s), 1466 (m), 1385 (w), 829 (m)
cm-1. 1H NMR (500 MHz, CDCl3): δ 0.98−1.00 (3H, t), 1.44−1.50 (2H, m), 1.64−1.68
(2H, m), 2.18 (3H, s), 3.44-3.48 (2H, t), 6.09 (1H, s, br), 7.18-7.20 (1H, d), 7.42-7.43
(1H, d), 7.86 (1H, s), 8.38 (1H, br) ppm. 13C NMR (500 MHz, CDCl3): δ 13.8, 20.2, 24.5,
31.4, 39.9, 92.3, 119.8, 128.2, 130.8, 137.5, 139.9, 169.2, 169.6 ppm.
General Synthesis of Catalyst 4d:
O
O
4db99%
I
H2N
CH3 OH
OI
H2N
NH
O
4dc68%
I
H2N
NH2
NH
OI
NH
O
Catalyst 4d82%
Acetic anhydridepyridine, r.t.1.) NEt3, Dry DCM
SOCl2 (0ºC)
2.) NEt3, Dry DCM
KOH, MeOH/H2O75 ºC
4da

158
A representative procedure for the hydrolysis of methyl esters is illustrated with -
4-amino-3-iodobenzoic acid (4db) (J. Comb. Chem. 2004, 6, 3, 407-413.):
To a 150 mL round bottom flask was added KOH (6.9 mmol, 0.39 g, 1 equiv.)
in 50 mL of MeOH/H2O (v/v = 1/1). To this solution was added 4da (6.9 mmol, 1.9 g,
1 equiv.). The resulting mixture was heated at 75 °C for 20 h. The reaction mixture was
concentrated in vacuo to one-half of its original volume. The residue was acidified
(pH~4) with 1 N HCl. The white precipitate was collected by filtration and then dried
under vacuum to give 4db (99%, 1.78 g) as an off-white powder. 1H NMR (500 MHz,
MeOD): δ 6.77-6.78 (1H, d), 7.76-7.77 (1H, d), 8.27 (1H, s). 13C NMR (150 MHz,
MeOD) δ 80.2, 112.5, 119.8, 130.9, 141.1, 152.6, 167.6 ppm.
4-amino-N-butyl-3-iodobenzamide, 4dc:
By using 4db as the substrate and applying the representative procedure for the n-
butyl amidation as described above, 5-amino-N-butyl-2-iodobenzamide (4dc) was
synthesized (84%, 1.02 g). 1H NMR (500 MHz, CDCl3): δ 0.86-0.87 (3H, m), 1.31 (2H,
m), 1.49-1.50 (2H, m), 3.32 (2H, m), 4.51 (2H, s), 6.47 (1H, br), 6.62-6.64 (1H, d), 7.50-
7.52 (1H, d), 8.04 (1H, s) ppm. 13C NMR (500 MHz, CDCl3): δ 13.8, 20.2, 31.8, 39.8,
82.8, 113.4, 125.6, 128.4, 138.2, 149.7, 166.1 ppm.
4-acetamido-N-butyl-3-iodobenzamide, Catalyst 4d:
By using 4db as the substrate and applying the representative procedure for the
acetylation as described above, Catalyst 4d was synthesized (84%, 1.02 g). IR (neat):

159
3259 (br), 2955 (w), 2924 (w), 1663 (s), 1632 (s), 1516 (s), 1373 (m), 1296 (m), 1038
(m), 883 (w) cm-1. 1H NMR (500 MHz, CDCl3): δ 0.96-0.98 (3H, m), 1.38-1.46 (2H, m),
1.58-1.64 (2H, m), 2.28 (3H, s), 3.43-3.47 (2H, m), 6.33 (1H, br), 7.63 (1H, s), 7.66-7.68
(1H, d), 8.26-8.29 (1H, d) ppm. 13C NMR (500 MHz, CDCl3): δ 13.8, 20.2, 25.0, 31.7,
39.6, 89.3, 120.6, 127.4, 131.8, 138.1, 140.7, 165.4, 168.5 ppm.
General Synthesis of Catalyst 4e (Chem. Eur. J. 2009, 15, 9424 – 9433):
3-amino-5-nitribenzoic Acid, 6p:
In a 100 ml round bottom flask was added 3,5- dinitrobenzoic acid 6o (1 g, 4.7
mmol) and 20 ml glacial acetic acid. This heterogeneous mixture was slowly heated up to
120 °C, and turned to homogeneous as the temperature increased. After the temperature
of solution increased to 120 °C, Fe powder (0.77 g, 13.8 mmol) was added with stirring
(this was added over ten portions within 30 minutes). The reaction mixture was kept at
120 °C for 15 min, and then poured over ice and water, followed by extracted with ethyl
acetate (x3). The organic phase was washed saturated aqueous Na2CO3 (x3) and brine
Scheme 3.17 Synthetic operations to access the aniline amide Catalyst 4e
6o
COOH
NO2O2N
COOH
NH2O2N
Fe PowderGlacial acetic acid (120 ºC)
COOH
IO2N
conc. HClNaNO2, H2O, 0 ºCKI (90 ºC)
6p45%
6q59%
conc. NH4OHMohr’s salt, H2O
COOH
IH2N INH
O NH2
6r68%
pyridine, r.t. INH
OHN
O
6s73%
Catalyst 4e64%
O
O O
O
1.) NEt3, Dry DCM SOCl2, 0 ºC
2.) NEt3, Dry DCM
NH2

160
(x1), dried over sodium sulfate and concentrated in vacuo to give 6p as a yellow powder
(0.39 g, 45%). 1H NMR (500 MHz, MeOD): δ 8.65 (1H, s), 8.72 (1H, s), 8.74 (1H, s)
ppm. 13C NMR (500 MHz, CDCl3): δ 92.9, 123.2, 133.8, 135.5, 143.7, 148.5, 164.5 ppm.
3-iodo-5-nitrobenzoic acid, 6q:
By using 6p as the substrate, and applying the representative procedure for
diazotization/iodination, as described above, the product 6q was synthesized in a 59%
yield (2.6 g). 1H NMR (500 MHz, DMSO): δ 7.49 (1H, s), 8.59-8.61 (1H, s), 8.70 (1H, s)
ppm. 13C NMR (500 MHz, DMSO): δ 93.5, 123.8, 134.4, 135.5, 144.2, 148.4, 164.8
ppm.
3-amino-5-iodobenzoic acid, 6r:
Compound 6q (1.3 g, 4.4 mmol) was dissolved in 20 ml conc. ammonia. To this
solution, (NH4)2Fe(SO4)2 (10.5 g, 26.7 mmol) in 20 ml H2O was added. After refluxing
for 10 min, the mixture was filtered through celite and the solution was cooled down to
ambient temperature. The pH of the solution was adjusted to ~4 with conc. HCl and then
extracted with ethyl acetate (x3). The combined organic layers were washed with brine
(x1), dried over sodium sulfate and evaporated in vacuo to give 6r as a yellow solid (1.2
g, 68%). 1H NMR (500 MHz, MeOD): δ 7.24 (1H, s), 7.29 (1H, s), 7.57 (1H, s) ppm. 13C
NMR (500 MHz, MeOD): δ 61.5, 94.8, 116.2, 127.9, 128.2, 134.2, 151.0 ppm.

161
3-acetamido-5-iodobenzoic acid, 6s:
6s was synthesized using the representative procedure for the acetylation as stated
above. By using 6q as the starting material, 6s was synthesized in a 73% yield (0.85 g).
1H NMR (500 MHz, MeOD): δ 2.01 (3H, s), 8.03 (1H, s), 8.13 (1H, s), 8.30 (1H, s) ppm.
13C NMR (500 MHz, MeOD): δ 23.9, 94.1, 121.1, 133.4, 134.3, 134.7, 141.4, 167.8,
171.9 ppm.
3-acetamido-N-butyl-5-iodobenzamide, Catalyst 4e:
Catalyst 4e was synthesized using the representative procedure for the n-butyl
amidation as stated above by using 6s as the starting material. Catalyst 4e (0.53 g, 64%).
IR (neat): 3279 (br), 2955 (w), 2928 (w), 2866 (w), 1635 (m), 1589 (m), 1535 (s), 1312
(m), 1280 (m), 1258 (m), 867 (m) cm-1. 1H NMR (500 MHz, CDCl3): δ 0.88−0.91 (3H,
t), 1.32−1.37 (2H, m), 1.53−1.57 (2H, m), 2.13 (3H, s), 3.34-3.36 (2H, m), 7.13 (1H, s,
br), 7.68 (1H, s), 7.87 (1H, s), 8.14 (1H, s), 9.50 (1H, br) ppm. 13C NMR (500 MHz,
CDCl3): δ 0.88−0.91 (3H, t), 1.32−1.37 (2H, m), 1.53−1.57 (2H, m), 2.13 (3H, s), 3.34-
3.36 (2H, m), 7.13 (1H, s, br), 7.68 (1H, s), 7.87 (1H, s), 8.14 (1H, s), 9.50 (1H, br) ppm
General Synthesis of Catalyst 4f:
OH
O
NH2
O
4fb87% Catalyst 4f
84%
Acetic anhydridepyridine, r.t. II
NHNH2
NH
OI
NH
O4fa O
1.) NEt3, Dry DCMSOCl2 (0ºC)
2.) NEt3, Dry DCM
NH2

162
2-amino-N-butyl-5-iodobenzamide, 4fb: 4fb was synthesized using the representative procedure for the acetylation as
stated above. By using 4fa as the starting material, 4fb was synthesized in 87% yield
(0.53 g). 1H NMR (500 MHz, MeOD): δ 2.21 (3H, s), 7.85-7.87 (1H, d), 8.36-8.39 (2H,
m) ppm. 13C NMR (500 MHz, MeOD): δ 25.1, 85.9, 119.4, 123.3, 140.9, 142.1, 143.8,
169.9, 171.4 ppm.
2-acetamido-N-butyl-5-iodobenzamide, Catalyst 4f:
Catalyst 4f was synthesized using the representative procedure for the n-butyl
amidation as stated above by using 4fb as the starting material. Catalyst 4f (0.53 g,
64%). IR (neat): 2955 (w), 2932 (w), 2870 (w), 1670 (s), 1589 (s), 1466 (m), 1385 (w),
1339 (w), 1278 (w), 829 (m) cm-1. 1H NMR (500 MHz, CDCl3): δ 0.98−1.01 (3H, t),
1.42−1.50 (2H, m), 1.68−1.74 (2H, m), 2.64 (3H, s), 4.06-4.09 (2H, m), 7.32-7.34 (1H,
d), 7.95-7.97 (1H, d), 8.57 (1H, s) ppm. 13C NMR (500 MHz, CDCl3): δ 13.9, 20.2, 24.4,
31.5, 40.2, 94.1, 118.0, 130.9, 131.2, 136.9, 139.9, 166.6, 169.8 ppm.
General Synthesis of Catalyst 4g:
1.) NEt3, Dry DCMSOCl2 (0ºC)
2.) NEt3, Dry DCMO
O
4gb99%
H2N
I
CH3 OH
OH2N
I
NH
O
4gc74%
H2N
I
KOH, MeOH/H2O75 ºC
NH2
NH
OHN
Catalyst 4g86%
Acetic anhydridepyridine, r.t.
IO
4ga

163
3-amino-4-iodobenzoic acid, 4gb:
Using the representative procedure for the hydrolysis of methyl esters, 4gb was
synthesized (99%, 1.75 g) as an off yellow powder. 1H NMR (500 MHz, MeOD): δ
7.03-7.04 (1H, d), 7.44 (1H, s), 7.71-7.72 (1H, d). 13C NMR (150 MHz, MeOD) δ 88.3,
114.9, 119.2, 131.6, 138.8, 148.3, 168.4 ppm.
3-amino-N-butyl-4-iodobenzamide, 4gc:
By using 4gb as the substrate and applying the representative procedure for the n-
butyl amidation as described above, 5gc was synthesized (74%, 0.98 g). 1H NMR (500
MHz, CDCl3): δ 0.92-0.97 (3H, m), 1.34-1.39 (2H, m), 1.41-1.59 (2H, m), 3.37-3.44 (2H,
m), 4.31 (2H, s), 6.34 (1H, br), 6.74-6.78 (1H, d), 7.21 (1H, s), 7.63-7.66 (1H, d) ppm.
13C NMR (500 MHz, CDCl3): δ 13.8, 20.2, 31.7, 39.8, 87.3, 113.2, 117.2, 136.2, 139.0,
147.2, 167.1 ppm.
3-acetamido-N-butyl-4-iodobenzamide, Catalyst 4g:
By using 4gb as the substrate and applying the representative procedure for the
acetylation as described above, Catalyst 4g was synthesized (86%, 0.82 g). IR (neat):
2955 (w), 2924 (w), 2867 (w), 2465 (br), 2403 (br), 1628 (s), 1558 (m), 1447 (s), 1396
(w), 1280 (m), 1014 (s), 752 (s) cm-1. 1H NMR (500 MHz, MeOD): δ 0.96-0.99 (3H, m),
1.39-1.44 (2H, m), 1.59-1.62 (2H, m), 2.21 (3H, s), 3.35-3.81 (2H, m), 7.39-7.42 (1H, d),
7.88 (1H, s), 7.97-7.99 (1H, d) ppm. 13C NMR (500 MHz, MeOD): δ 12.8, 19.8, 21.9,
31.2, 39.5, 98.9, 125.6, 126.0, 135.5, 139.3, 139.5, 167.4, 170.8 ppm.

164
General Synthesis of Catalyst 4h:
2-amino-N-butyl-4-iodobenzamide, 4hb:
By using 4ha as the substrate and applying the representative procedure for the n-
butyl amidation as described above, 4hb was synthesized (76%, 0.67 g). 1H NMR (500
MHz, CDCl3): δ 0.98-1.01 (3H, m), 1.42-1.47 (2H, m), 1.60-1.65 (2H, m), 3.44-3.48 (2H,
m), 6.24 (1H, br), 7.10-7.11(1H, d), 7.17-7.18 (1H, d), 7.36 (1H, s), 744.-7.46 (1H, m)
ppm. 13C NMR (500 MHz, CDCl3): δ 13.8, 20.2, 31.4, 39.9, 99.3, 114.5, 119.9, 130.3,
131.7, 150.6, 167.5 ppm.
2-acetamido-N-butyl-4-iodobenzamide, Catalyst 4h:
By using 4hb as the substrate and applying the representative procedure for the
acetylation as described above, Catalyst 4h was synthesized (90%, 0.82 g). IR (neat):
3340 (br), 2958 (m), 2870 (w), 1682 (s), 1543 (m), 1501 (s), 1396 (s), 1273 (m), 1153
(w), 775 (w) cm-1. 1H NMR (500 MHz, CDCl3): δ 0.98-1.01 (3H, m), 1.42-1.48 (2H, m),
1.62-1.67 (2H, m), 2.20 (3H, s), 3.44-3.47 (2H, m), 6.39 (1H, br), 7.12-7.14 (1H, d), 7.39
(1H, s), 7.41-7.42 (1H, d), 8.99 (1H, br) ppm. 13C NMR (500 MHz, CDCl3): δ 13.8, 20.2,
25.3, 31.4, 39.9, 99.2, 119.6, 127.4, 130.0, 131.7, 140.1, 168.5, 169.1 ppm.
4ha
NH2
OH
O
I
NH2
NH
O
4hb76%
1.) NEt3, Dry DCMSOCl2 (0ºC)
2.) NEt3, Dry DCM
NH2
NH
NH
O
IICatalyst 4h
90%
Acetic anhydridepyridine, r.t.
O

165
General procedure for azido acid synthesis by diazo transfer330, 342:
Triflyl azide preparation using 7r as an example:
To a round bottom flask was added sodium azide (3.94 g, 6.05 mmol) was added
distilled H2O (15 mL) and DCM (30 mL). This solution was cooled to 0 ℃, and stirred
for 20 minutes. Triflyl anhydride (2.04 mL, 12.1 mmol) was added slowly over 10 min
with stirring continued for 2 h. The mixture was placed in a separatory funnel and the
DCM phase removed. The aqueous portion was extracted with DCM, and the organic
fractions, containing the triflyl azide, were combined and washed with saturated Na2CO3
(x1) and used without further purification.
In a separate round-bottomed flask was added the L-phenylalanine 7q (1.0 g, 6.05
mmol), K2CO3 (1.67 g, 12.1 mmol), Cu(II)SO4 pentahydrate (9.66 mg, 60.5 μmol),
distilled H2O (9 mL) and MeOH (18 mL). The triflyl azide from the previous step was
dissolved in DCM (15 mL) and was added to this separate flask. The mixture was stirred
at ambient temperature and pressure overnight. Subsequently, the organic solvents were
removed under reduced pressure and the aqueous slurry was diluted with H2O (50 mL).
This was acidified to pH 6 with conc. HCl and diluted with 0.25 M, pH 6.2 phosphate
OH
O
NH2
1.) Tf2O, NaN32) CuSO4•5H2OK2CO3, H2O, MeOH, DCM OH
O
N3
7r61% yield
7q

166
buffer (50 mL) and extracted with EtOAc to remove sulfonamide by-product. The
aqueous phase was then acidified to pH 2 with conc. HCl. The product was obtained from
another round of EtOAc extractions. The EtOAc extracts were combined, dried (Na2SO4)
and evaporated to dryness giving 0.71 g of the pale oil (7r) in 61% yield with no need for
further purification.
1H NMR (300 MHz, CDCl3) δ 11.4 (1H, br), 7.31-7.40 (m, 5H), 3.25-3.24 (dd, J = 5.0,
8.8 Hz, 1H), 3.05-3.05 (dd, J = 5.0, 13.9 Hz, 1H) ppm.
13C NMR (100 MHz, CDCl3) δ 174.3, 136.0, 129.3, 128.7, 127.3, 63.1, 37.4 ppm.
The procedure above was used to generate L-phenylglycine (8k) and L-tert-Leucine (8l)
azide derivatives.329
L-phenylglycine azide (8k),Yellow oil, in 75%: 1H NMR (300 MHz, CDCl3) δ
11.9 (1H, br), 7.50-7.46 (m, 5H), 5.13 (s, 1H) ppm.13C NMR (100 MHz, CDCl3) δ 174.9,
133.3, 129.7, 129.3, 127.8, 65.2 ppm.
OH
O
NH2
1.) Tf2O, NaN32) CuSO4•5H2OK2CO3, H2O, MeOH, DCM OH
O
N3
8k75% yield

167
L-tert-Leucine azide (8l), clear oil in 73%: 1H NMR (300 MHz, CDCl3) δ 11.66
(1H, br), 3.75 (s, 1H), 1.08 (s, 9H); 13C NMR (100 MHz, CDCl3) δ 175.6, 71.7, 35.7,
26.5.
N-(2,4-dimethoxybenzyl)propargylamine hydrochloride (7u)329: Propargylamine (2.0 g,
36.2 mmol) and 2,4-dimethoxybenzaldehyde (5.0 g, 30.2 mmol) were dissolved in
anhydrous MeOH (100 mL) and stirred for 2 h. To the resulting clear solution was added
sodium borohydride (2.0 g, 54.0 mmol) in several portions. The suspension was stirred
for 2 h. After dilution with ether (200 mL), 2 M NaOH (40 mL) was added to quench the
reaction. The organic layer was washed with water (x2), brine, and dried over Na2SO4.
After filtration the volatiles were evaporated in vacuo and the sticky residue was
dissolved in methanol (50 mL). To this 12 M HCl (4 mL) was added and the volatiles
were removed under reduced pressure. The residue was dissolved in MeOH/EtOAc (1:4,
80 mL) followed by addition of hexanes (ether can be added to induce crystallization).
The resulting crystals were collected by filtration and dried to give 7u (6.7 g, 92%) as its
HCl salt. 1H NMR (500 MHz, MeOD) δ 7.31-7.30 (1H, d, J = 8 Hz), 6.67 (1H, s), 6.61-
6.60 (1H, d, J = 8 Hz), 4.25 (2H, s), 3.93 (3H, s), 3.90 (d, J = 3 Hz, 2H), 3.85 (3H, s),
OH
O
NH2
1.) Tf2O, NaN32) CuSO4•5H2OK2CO3, H2O, MeOH, DCM OH
O
N3
8l73% yield

168
3.29-3.28 (t, J = 3 Hz, 1H); 13C NMR (100 MHz, MeOD) δ 161.1, 157.8, 132.2, 108.6,
103.9, 97.5, 78.7, 71.9, 57.2, 55.8, 44.7, 35.7.
General procedure for amino acid coupling toward macrocycles329: In a round bottom
flask was added the carboxylic acid component (1.1 eq) and DCM/DMF (4:1) at a
concentration ~0.4 M. To this solution was added DIPEA (2.2 eq). The solution was
cooled to 0ºC and HATU (1 eq) was added. This solution was stirred at 0 ºC for 10
minutes at, and then a solution of the amine component (1 eq) in DMF at a concentration
of ~0.5 M was added. The reactions were reacted at 0 ºC and followed by TLC until
complete. Work-up was accomplished by diluting the reaction mixture with EtOAc
followed by successive washing with 0.5 M KHSO4 (3×) and brine. After drying with
Na2SO4 and filtration, the solvents were removed via rotary evaporation and the residue
was purified by flash chromatography (gradient up to 50:50 EtOAc/Hexanes) or moved
on to the next step without purification.
General procedure for the Fmoc deprotection: In a round bottom flask was added the
Fmoc-protected peptide. This was dissolved in acetonitrile / diethylamine (2:1)
(concentration ~0.1 M) and then stirred for 1 hr, and concentrated under vacuum. The
resulting residue was exchanged with acetonitrile (x3) to remove excess diethylamine and
used directly in the next coupling step.
General procedure for Copper(II) catalyzed macrocyclization via Click reaction331-333:

169
Azido-alkyne 7x (1 equiv) was dissolved in H2O and tert-butanol (2:1) at a concentration
of 1 mM. An aqueous solution of CuSO4∙5H2O (1M, 20 mL) and sodium ascorbate (1 M,
100 mL) were added. After stirring at room temperature for 12 h, water (50 mL) was
added and the mixture was extracted with ethyl acetate (3 x 20 mL). The combined
organic layers were washed with brine (20 mL), dried with Na2SO4, concentrated, and the
residue was purified by flash column chromatography (gradient up to 1:9 MeOH/DCM)
to produce Macrocycle A.
170
Oxytosylated propiophenone, 4l
O
OTs
171
N-butyl-2-Iodobenzamide, Catalyst 1
O
NH
I
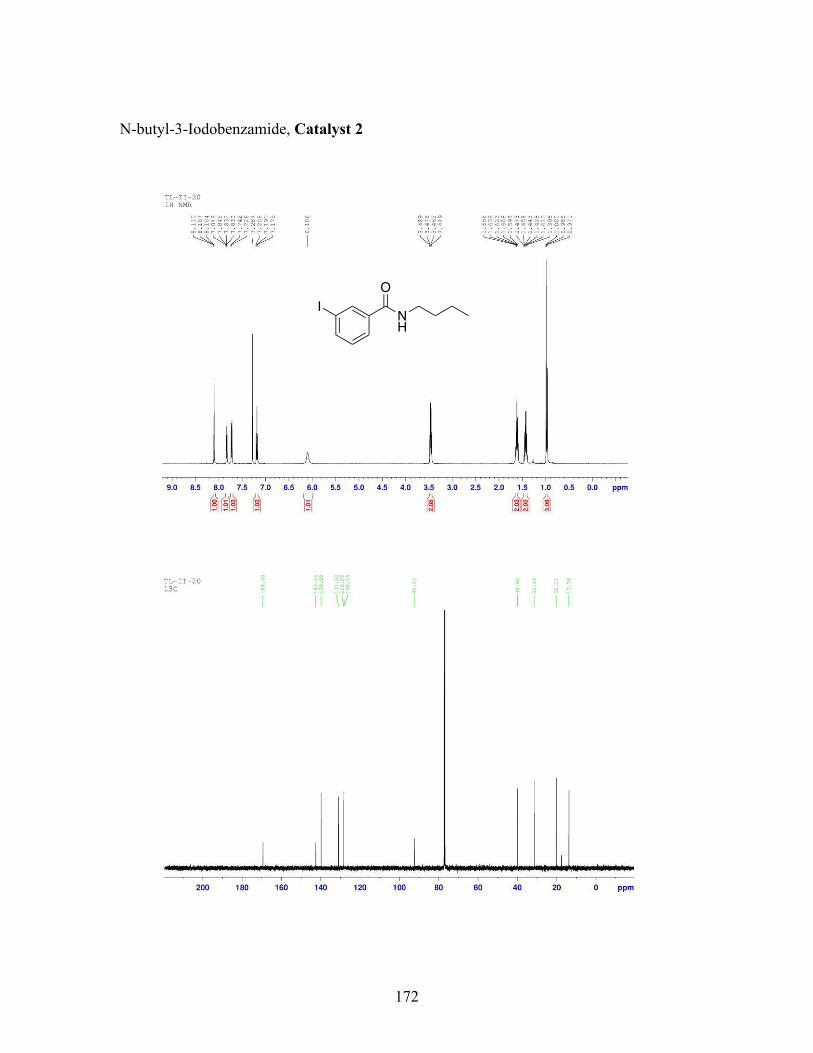
172
N-butyl-3-Iodobenzamide, Catalyst 2
O
NH
I
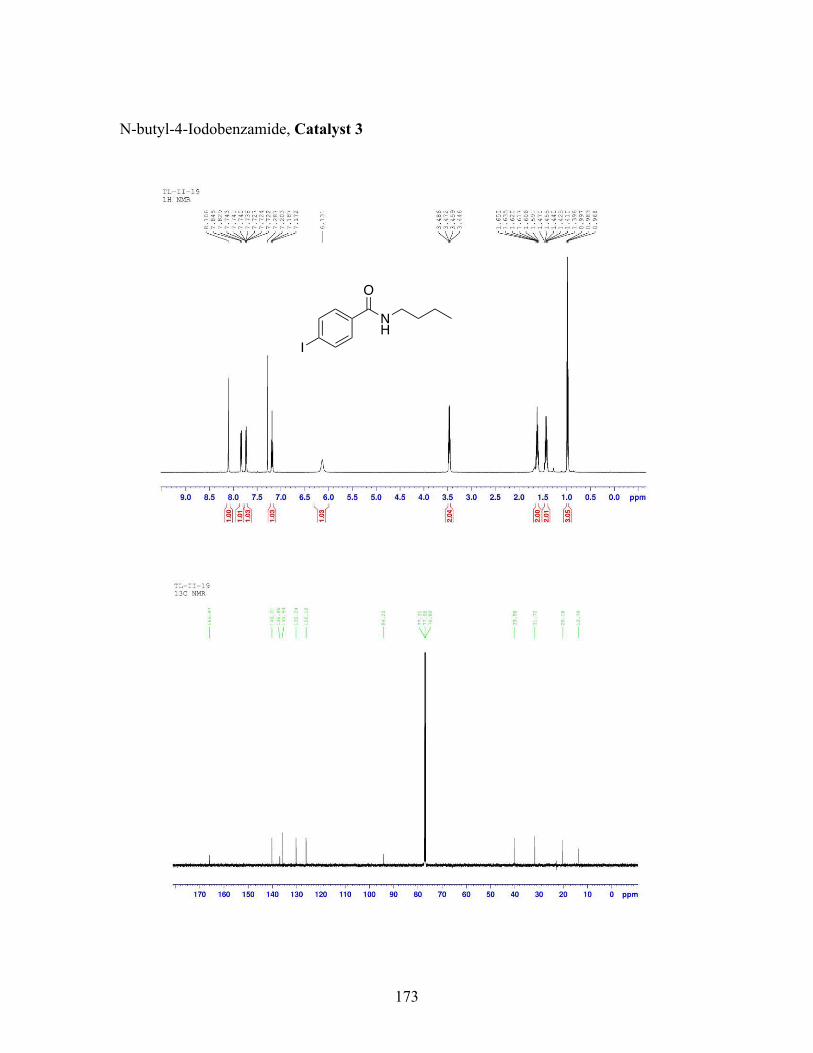
173
N-butyl-4-Iodobenzamide, Catalyst 3
O
NH
I
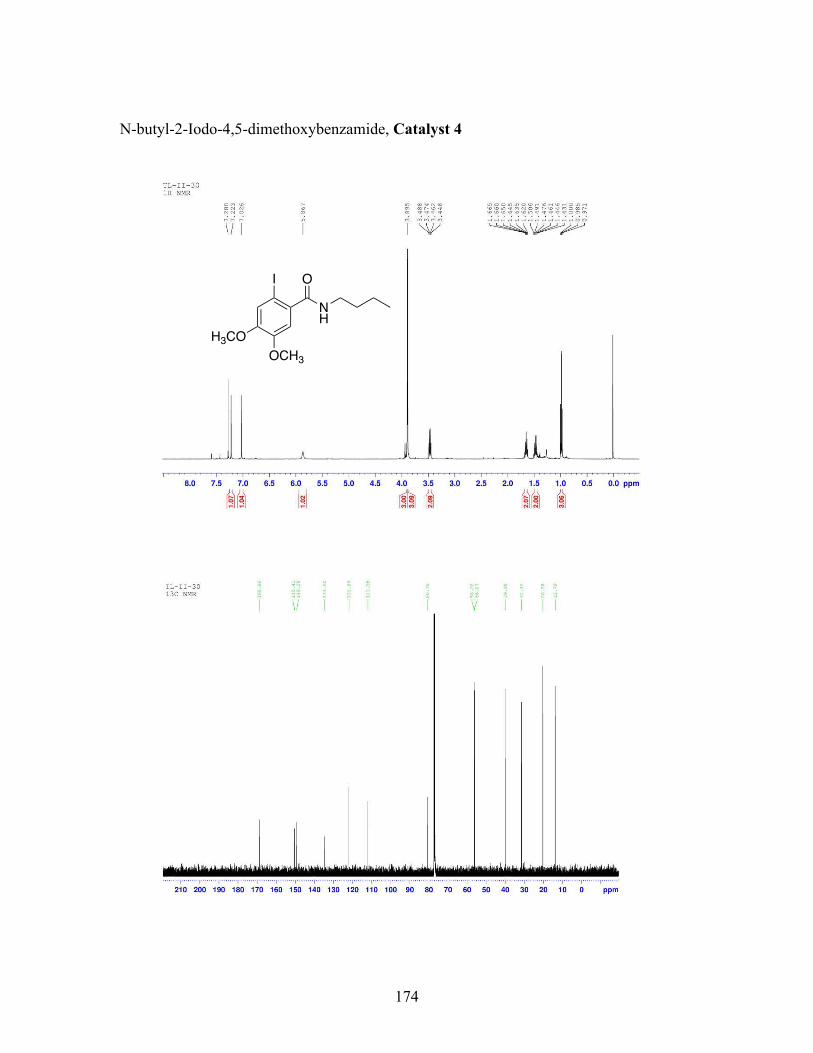
174
N-butyl-2-Iodo-4,5-dimethoxybenzamide, Catalyst 4
O
NH
H3CO
I
OCH3
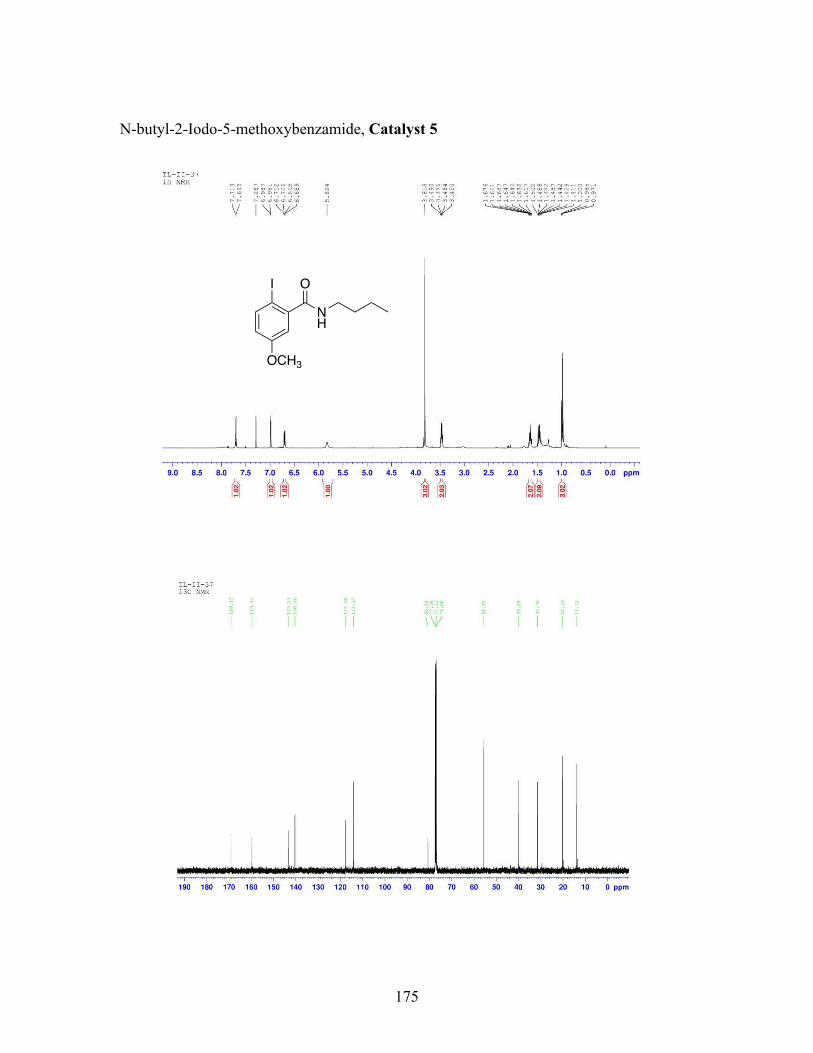
175
N-butyl-2-Iodo-5-methoxybenzamide, Catalyst 5
O
NH
I
OCH3
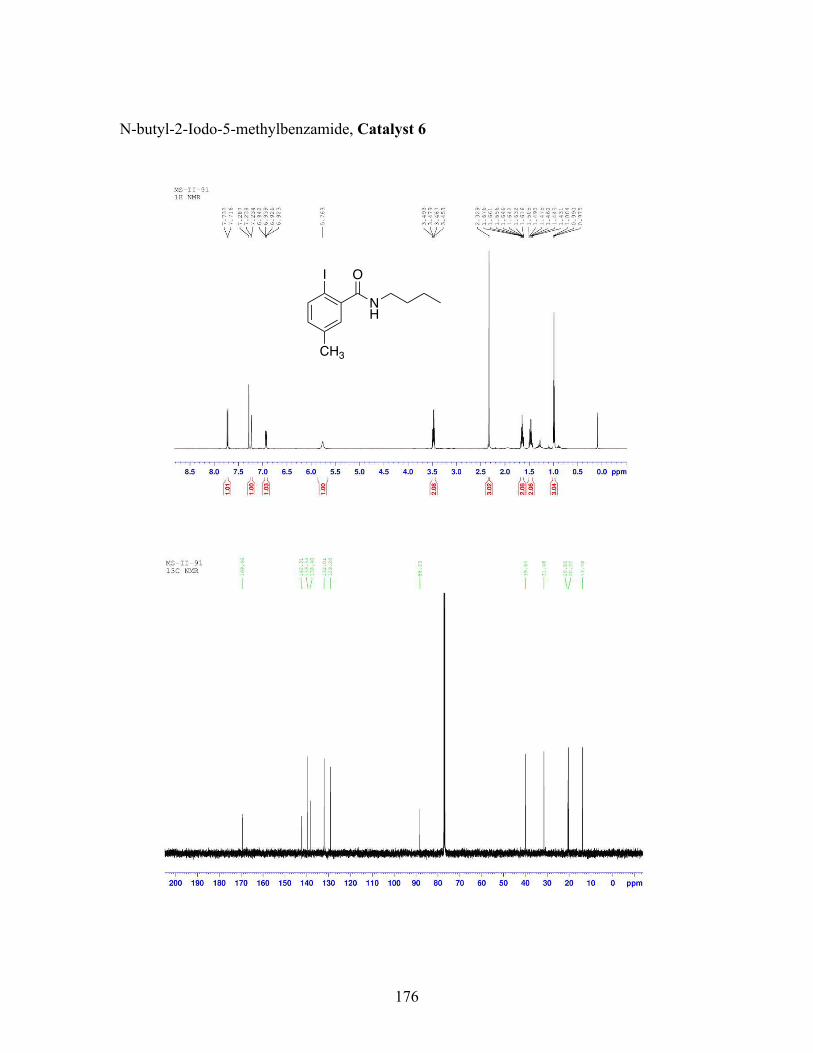
176
N-butyl-2-Iodo-5-methylbenzamide, Catalyst 6
O
NH
I
CH3
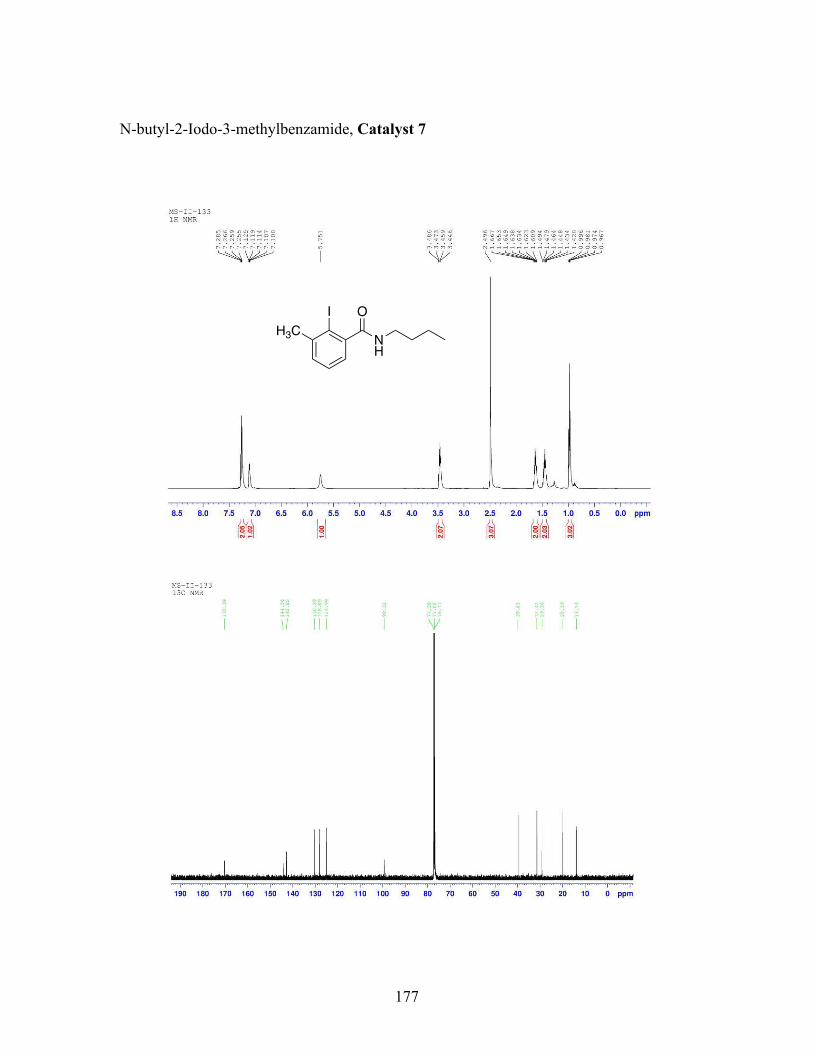
177
N-butyl-2-Iodo-3-methylbenzamide, Catalyst 7
O
NH
IH3C
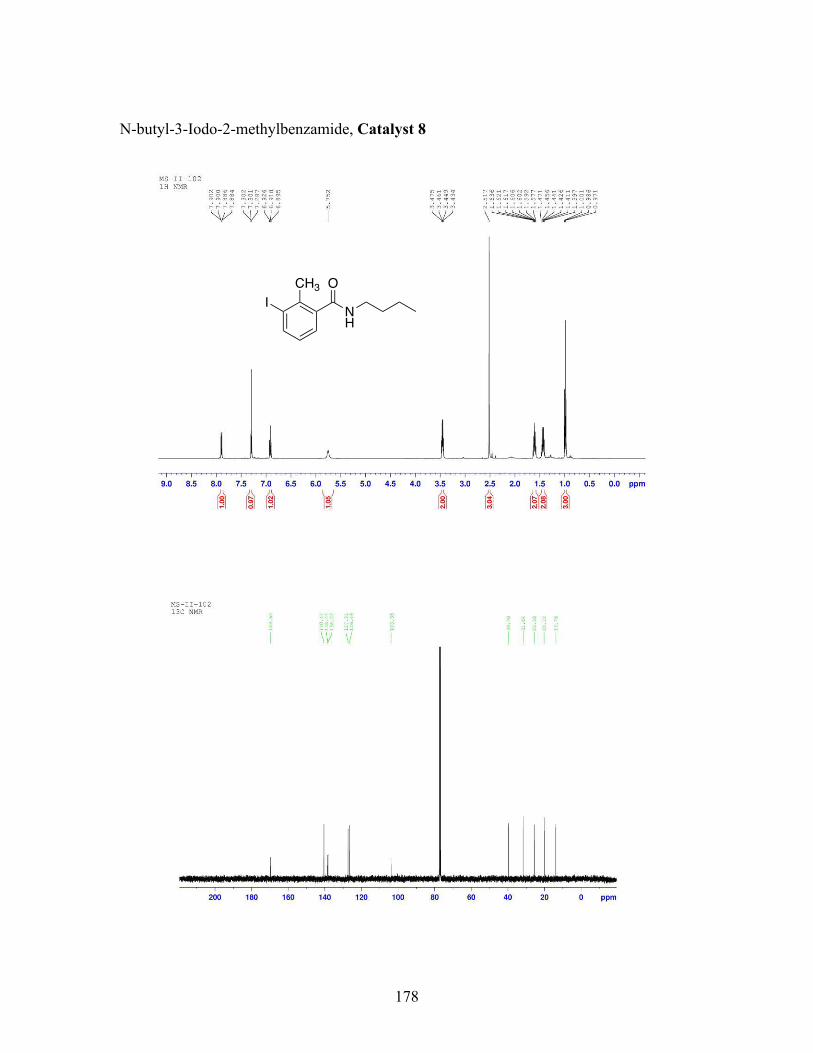
178
N-butyl-3-Iodo-2-methylbenzamide, Catalyst 8
O
NH
ICH3
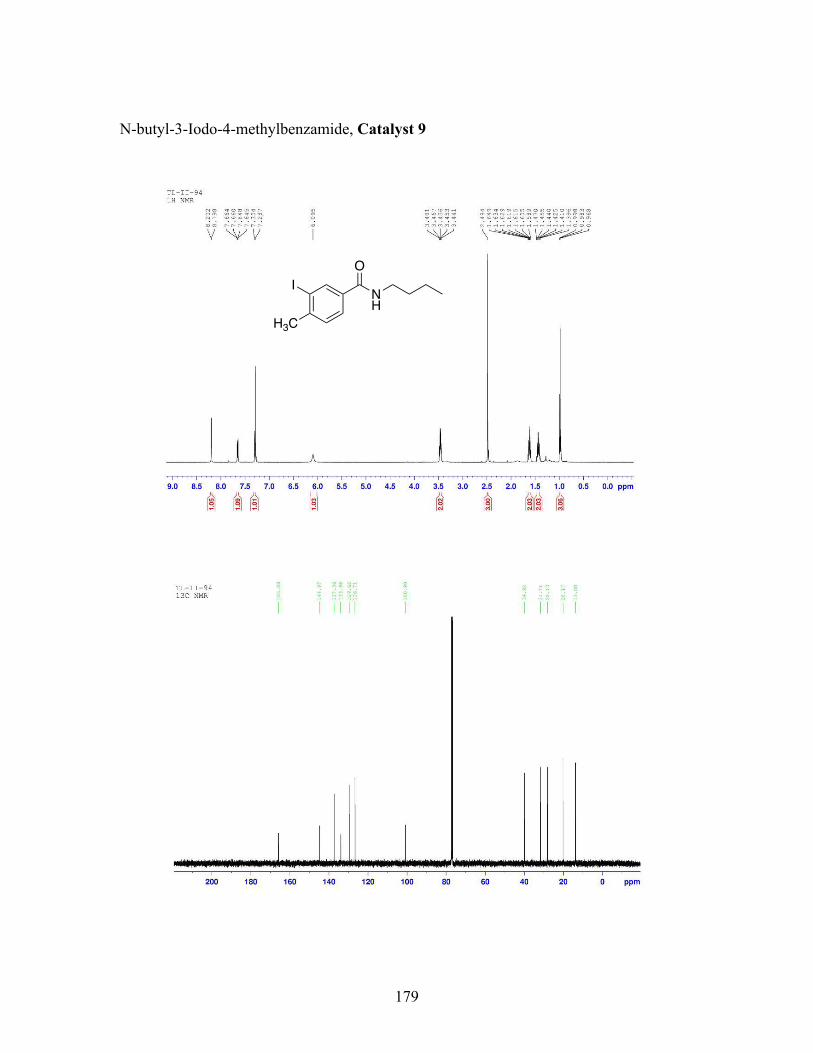
179
N-butyl-3-Iodo-4-methylbenzamide, Catalyst 9
O
NH
I
H3C
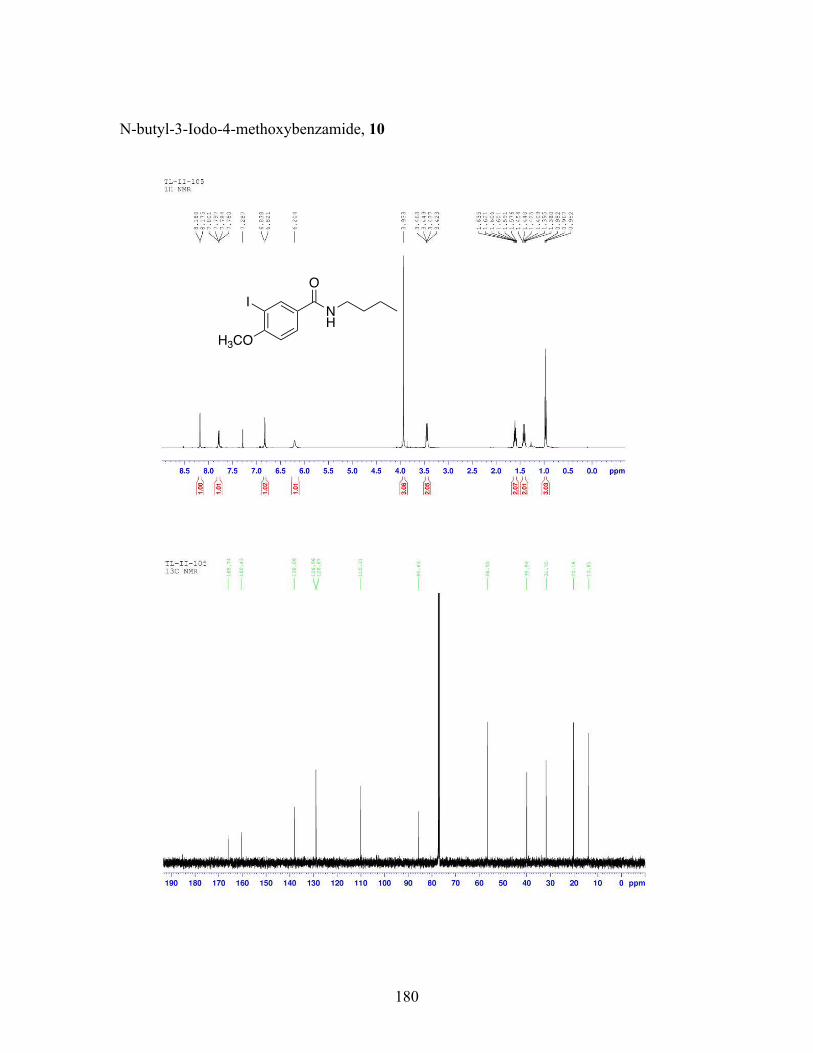
180
N-butyl-3-Iodo-4-methoxybenzamide, 10
O
NH
H3CO
I
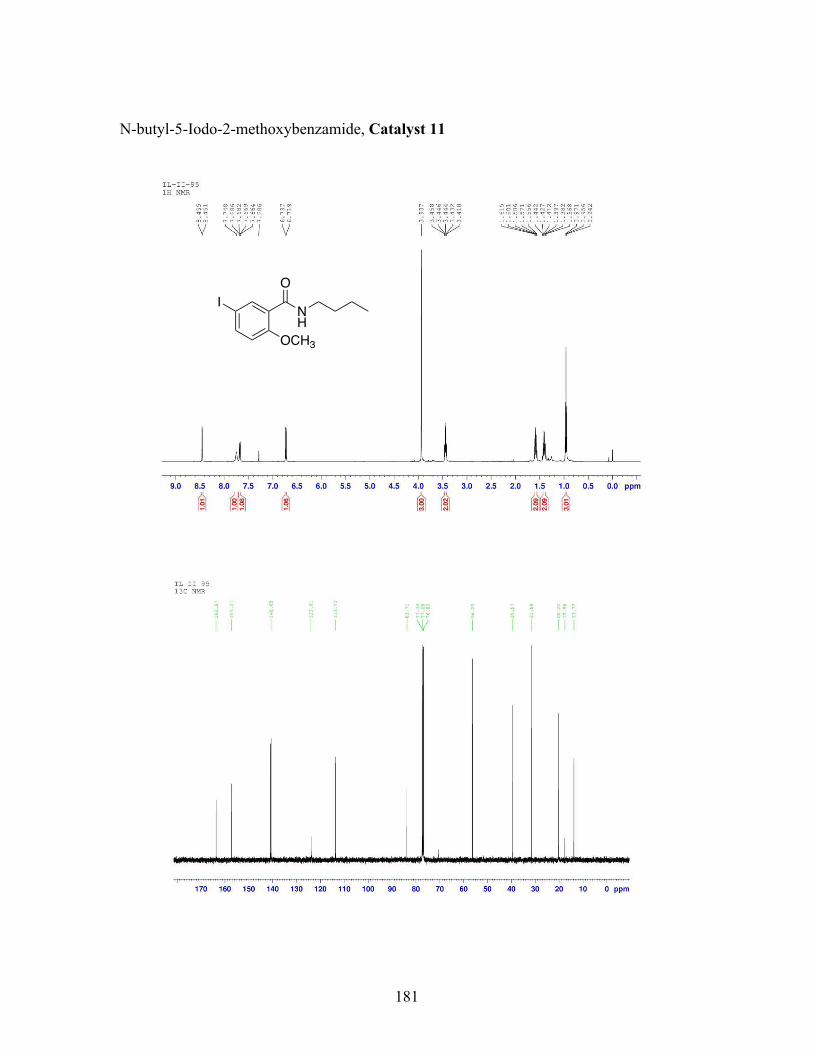
181
N-butyl-5-Iodo-2-methoxybenzamide, Catalyst 11
O
NH
I
OCH3
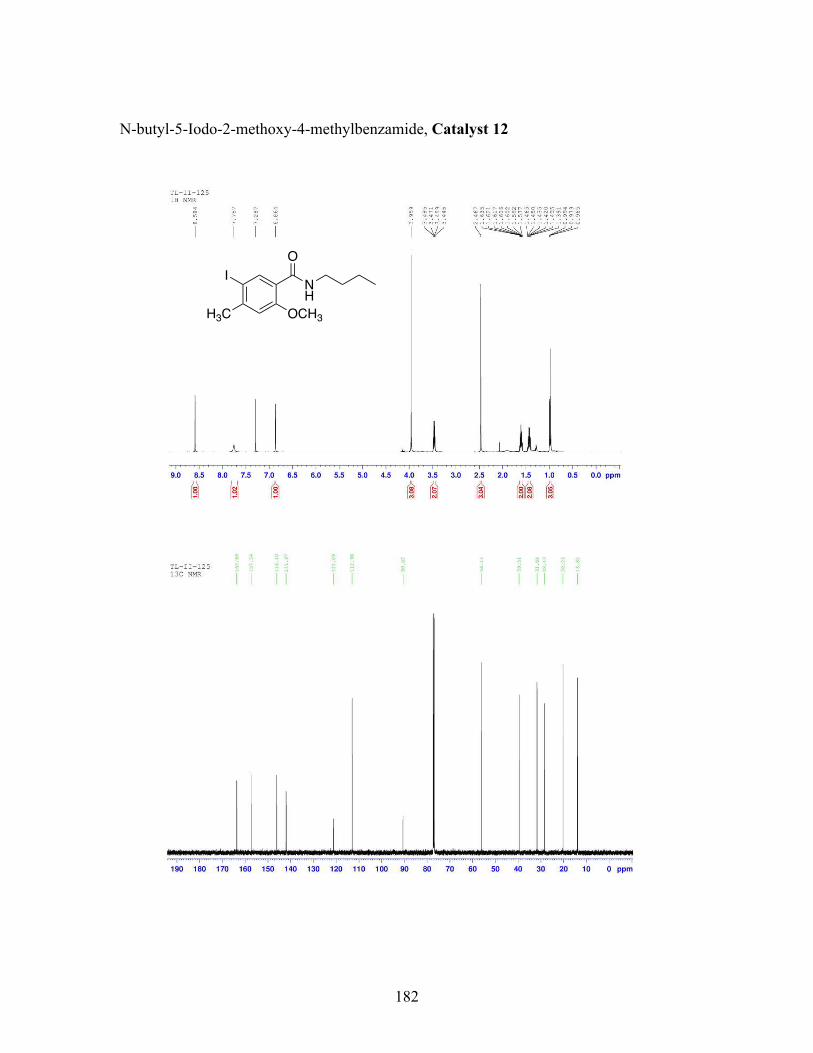
182
N-butyl-5-Iodo-2-methoxy-4-methylbenzamide, Catalyst 12
O
NH
I
OCH3
O
NH
H3C
I
OCH3
183
1-oxo-1-p-tolylpropan-2-yl 4-methylbenzenesulfonate, Product 18
O
OTsH3C
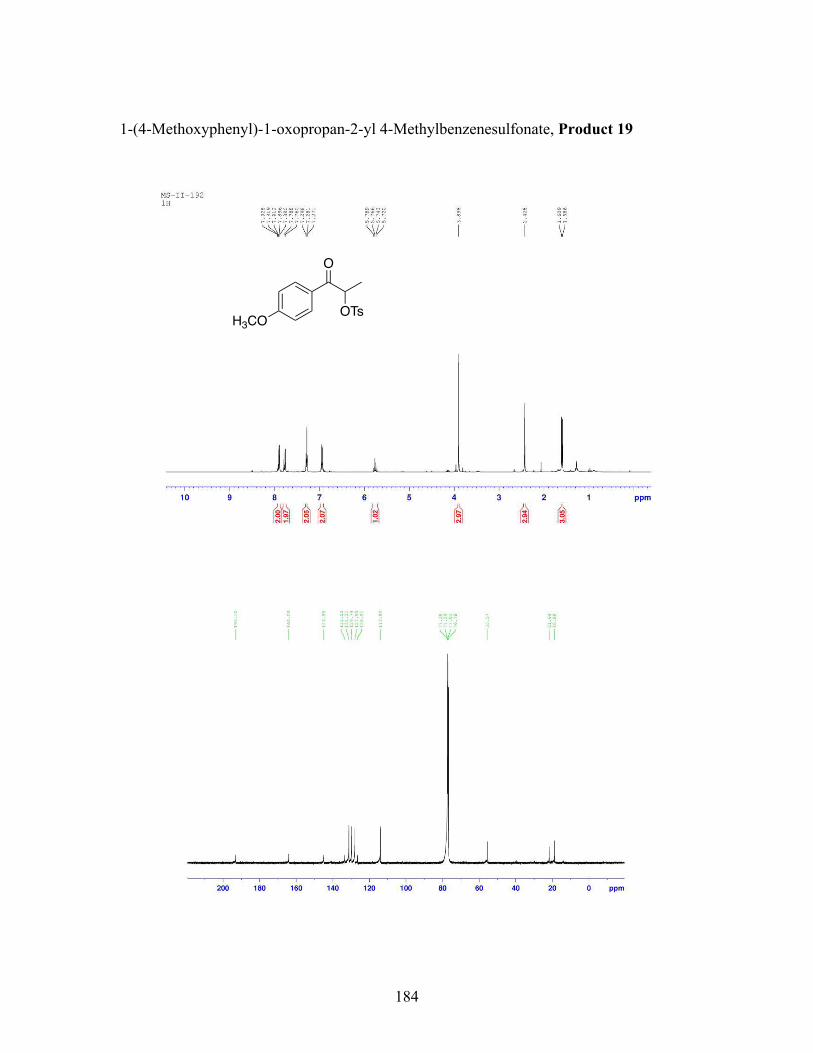
184
1-(4-Methoxyphenyl)-1-oxopropan-2-yl 4-Methylbenzenesulfonate, Product 19
O
OTsH3CO
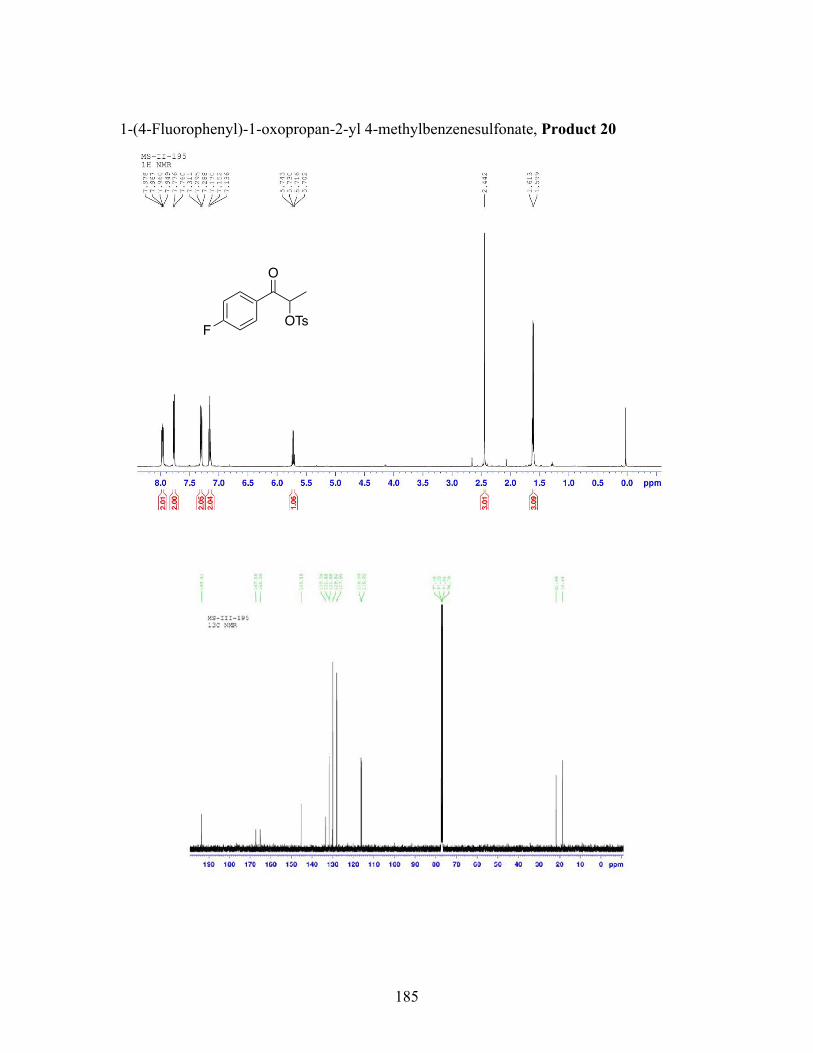
185
1-(4-Fluorophenyl)-1-oxopropan-2-yl 4-methylbenzenesulfonate, Product 20
O
OTsF
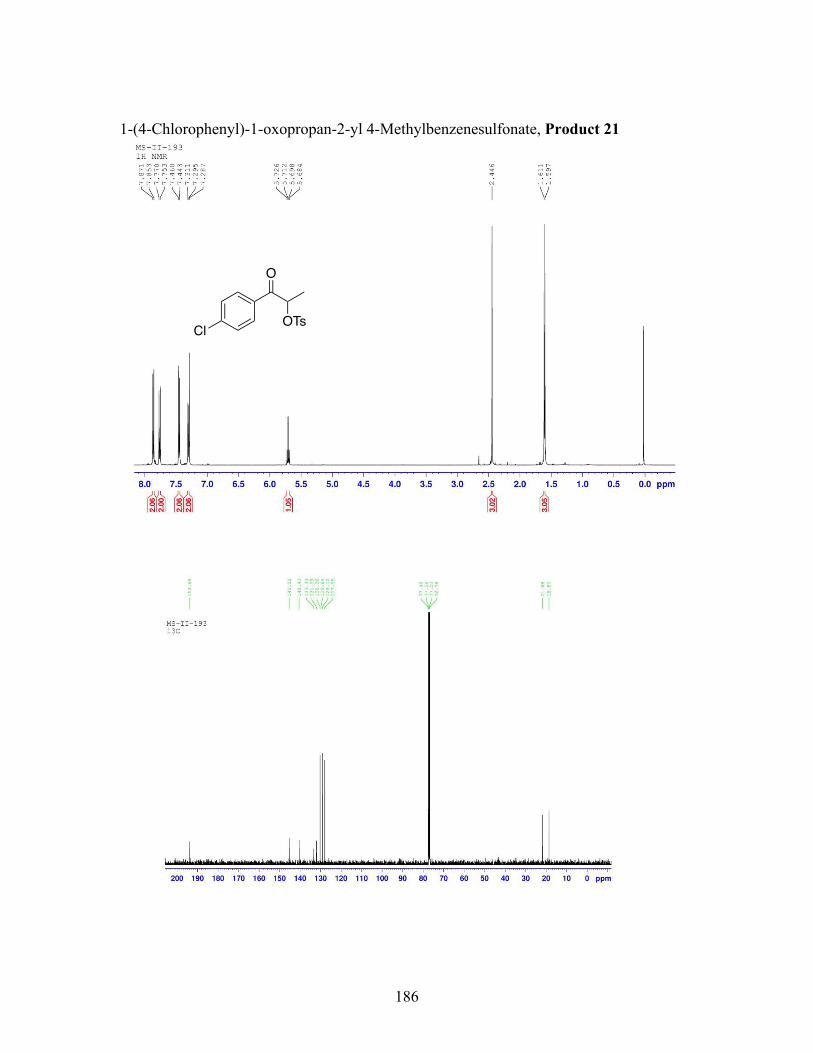
186
1-(4-Chlorophenyl)-1-oxopropan-2-yl 4-Methylbenzenesulfonate, Product 21
O
OTsCl
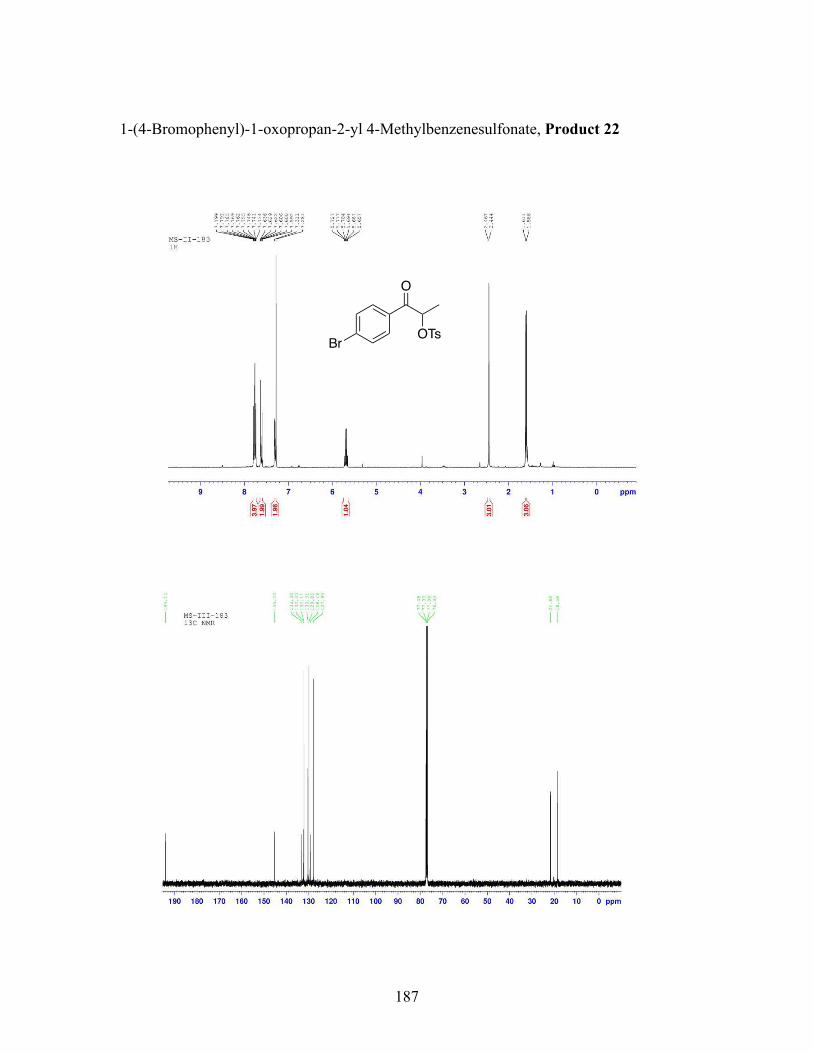
187
1-(4-Bromophenyl)-1-oxopropan-2-yl 4-Methylbenzenesulfonate, Product 22
O
OTsBr
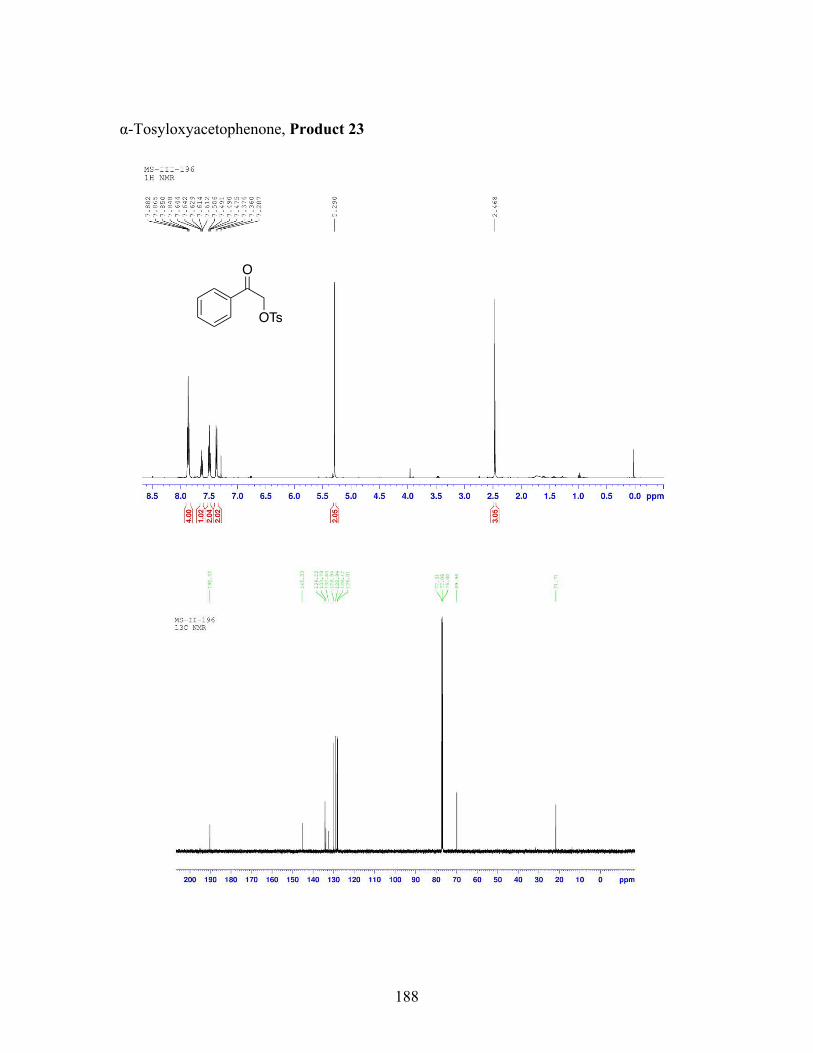
188
α-Tosyloxyacetophenone, Product 23
O
OTs
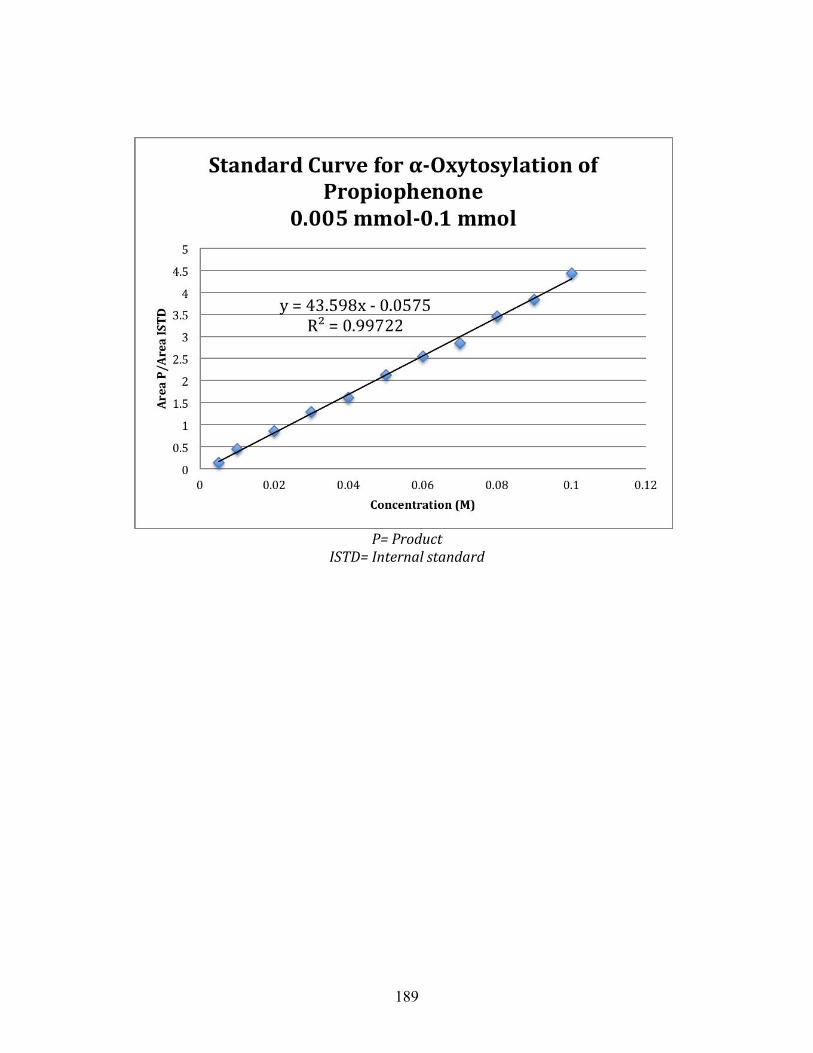
189
P=ProductISTD=Internalstandard
190
3-Iodoarene-arene-Z-L-Dap-Amino Methyl Ester, Catalyst 1a
HN
O
ONH
O
O
O
I

191
3-Iodoarene-Z-L-Dab-Amino Methyl Ester, Catalyst 1b
8.0 7.5 7.0 6.5 6.0 5.5 5.0 4.5 4.0 3.5 3.0 2.5 2.0 1.5 1.0 0.5 0.0 ppm
1.823
1.832
2.199
2.208
2.216
2.225
2.234
3.184
3.192
3.201
3.210
3.687
3.856
3.869
3.881
4.455
4.463
4.473
4.479
4.489
5.142
5.974
7.132
7.147
7.162
7.317
7.326
7.333
7.350
7.517
7.786
7.800
7.814
8.219
0.98
1.05
1.05
3.03
1.05
1.01
2.00
1.01
1.09
5.05
0.92
2.01
1.02
1H, CDCl3TL−−VI−3b�
DCM H−grease
NH
OI
HN
O
O
O
O
180 170 160 150 140 130 120 110 100 90 80 70 60 50 40 30 20 10 0 ppm
32.75
36.07
51.62
52.75
67.39
94.35
126.17
128.14
128.34
128.59
130.22
135.99
136.12
136.34
140.36
156.84
166.01
172.64
13C, CDCl3�TL−VI−3b�
192
3-Iodoarene-Z-L-Lys-Amino Methyl Ester, Catalyst 1c
9.0 8.5 8.0 7.5 7.0 6.5 6.0 5.5 5.0 4.5 4.0 3.5 3.0 2.5 2.0 1.5 1.0 0.5 0.0 ppm
1.678
1.698
1.718
1.776
1.800
1.868
1.909
1.933
1.952
3.409
3.431
3.453
3.474
3.497
3.518
3.539
3.562
3.751
4.399
4.420
5.127
5.584
5.609
6.731
7.129
7.155
7.181
7.352
7.744
7.769
7.803
7.830
8.142
2.22
2.04
2.00
3.08
0.99
2.06
0.95
0.97
1.06
5.05
1.05
1.08
1.00
1H, CDCl3�TL−VI−3C�
H−grease
HN
O
ONH
O
O
O
I
190 180 170 160 150 140 130 120 110 100 90 80 70 60 50 40 30 20 10 0 ppm
25.26
30.40
39.42
52.62
53.34
67.18
94.21
126.29
128.13
128.27
128.56
130.22
136.00
136.09
136.45
140.29
156.10
166.08
172.61
13C, CDCl3TL−VI−3C�
H−grease
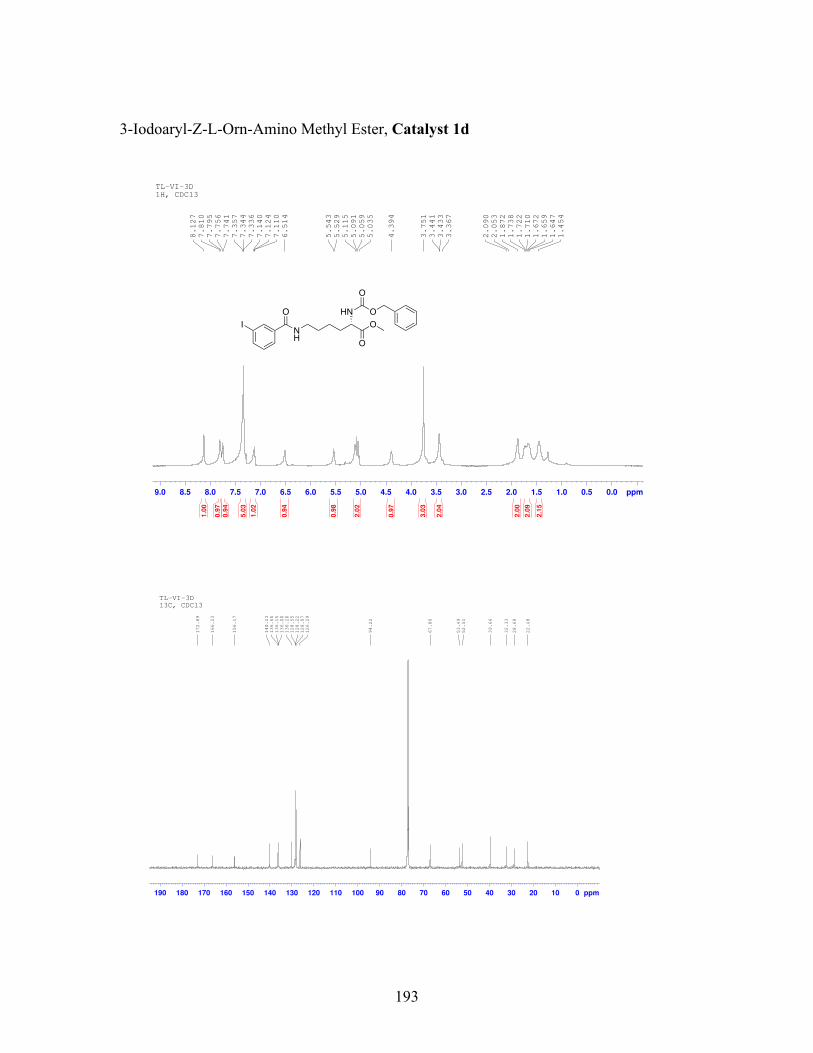
193
3-Iodoaryl-Z-L-Orn-Amino Methyl Ester, Catalyst 1d
190 180 170 160 150 140 130 120 110 100 90 80 70 60 50 40 30 20 10 0 ppm
22.48
28.68
32.33
39.66
52.51
53.49
67.06
94.22
126.29
128.07
128.22
128.55
130.20
136.00
136.15
136.65
140.23
156.17
166.23
172.89
13C, CDCl3�TL−VI−3D�
9.0 8.5 8.0 7.5 7.0 6.5 6.0 5.5 5.0 4.5 4.0 3.5 3.0 2.5 2.0 1.5 1.0 0.5 0.0 ppm
1.454
1.647
1.659
1.672
1.710
1.722
1.738
1.872
2.053
2.090
3.367
3.433
3.441
3.751
4.394
5.035
5.059
5.091
5.115
5.529
5.543
6.514
7.110
7.124
7.140
7.336
7.344
7.357
7.741
7.756
7.795
7.810
8.127
2.15
2.09
2.00
2.04
3.03
0.97
2.02
0.98
0.94
1.02
5.03
0.94
0.97
1.00
1H, CDCl3TL−VI−3D�
NH
OI
HN
O
O
O
O
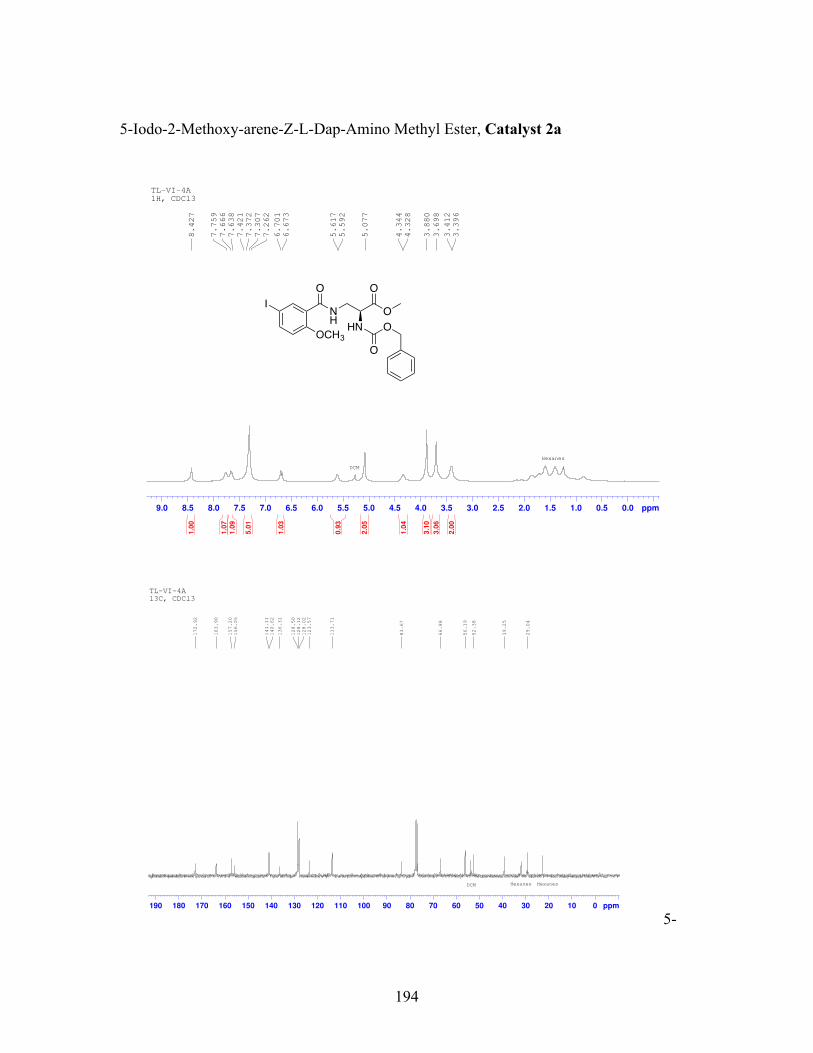
194
5-Iodo-2-Methoxy-arene-Z-L-Dap-Amino Methyl Ester, Catalyst 2a
5-
9.0 8.5 8.0 7.5 7.0 6.5 6.0 5.5 5.0 4.5 4.0 3.5 3.0 2.5 2.0 1.5 1.0 0.5 0.0 ppm
3.396
3.412
3.698
3.880
4.328
4.344
5.077
5.592
5.617
6.673
6.701
7.262
7.307
7.372
7.421
7.638
7.666
7.759
8.427
2.00
3.06
3.10
1.04
2.05
0.93
1.03
5.01
1.09
1.07
1.00
1H, CDCl3TL−VI−4A�
Hexanes
DCM
HN
O
ONH
O
O
O
I
OCH3
190 180 170 160 150 140 130 120 110 100 90 80 70 60 50 40 30 20 10 0 ppm
29.04
39.25
52.38
56.19
66.88
83.67
113.71
123.57
128.02
128.12
128.50
136.31
140.62
141.11
156.05
157.20
163.90
172.92
13C, CDCl3�TL−VI−4A�
DCM Hexanes Hexanes
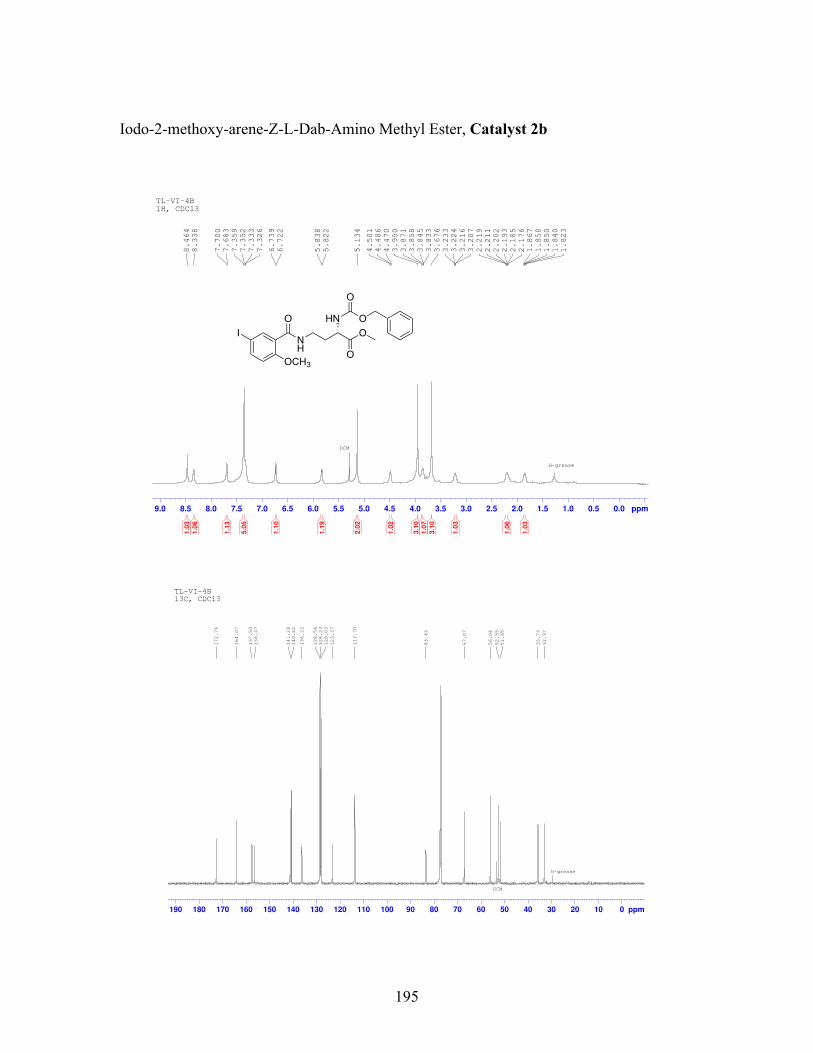
195
Iodo-2-methoxy-arene-Z-L-Dab-Amino Methyl Ester, Catalyst 2b
9.0 8.5 8.0 7.5 7.0 6.5 6.0 5.5 5.0 4.5 4.0 3.5 3.0 2.5 2.0 1.5 1.0 0.5 0.0 ppm
1.823
1.840
1.850
1.858
1.867
2.176
2.185
2.193
2.202
2.211
2.219
3.207
3.216
3.224
3.233
3.676
3.833
3.845
3.858
3.871
3.950
4.470
4.486
4.501
5.134
5.822
5.838
6.722
6.739
7.326
7.333
7.352
7.359
7.683
7.700
8.338
8.464
1.03
1.06
1.03
3.10
1.07
3.10
1.02
2.02
1.19
1.10
5.05
1.13
1.06
1.03
1H, CDCl3TL−VI−4B�
DCM
H−grease
190 180 170 160 150 140 130 120 110 100 90 80 70 60 50 40 30 20 10 0 ppm
32.97
35.74
51.65
52.55
56.06
67.07
83.44
113.70
123.37
128.03
128.23
128.56
136.23
140.62
141.26
156.47
157.50
164.07
172.76
13C, CDCl3�TL−VI−4B�
H−grease
DCM
NH
OI
HN
O
O
O
O
OCH3
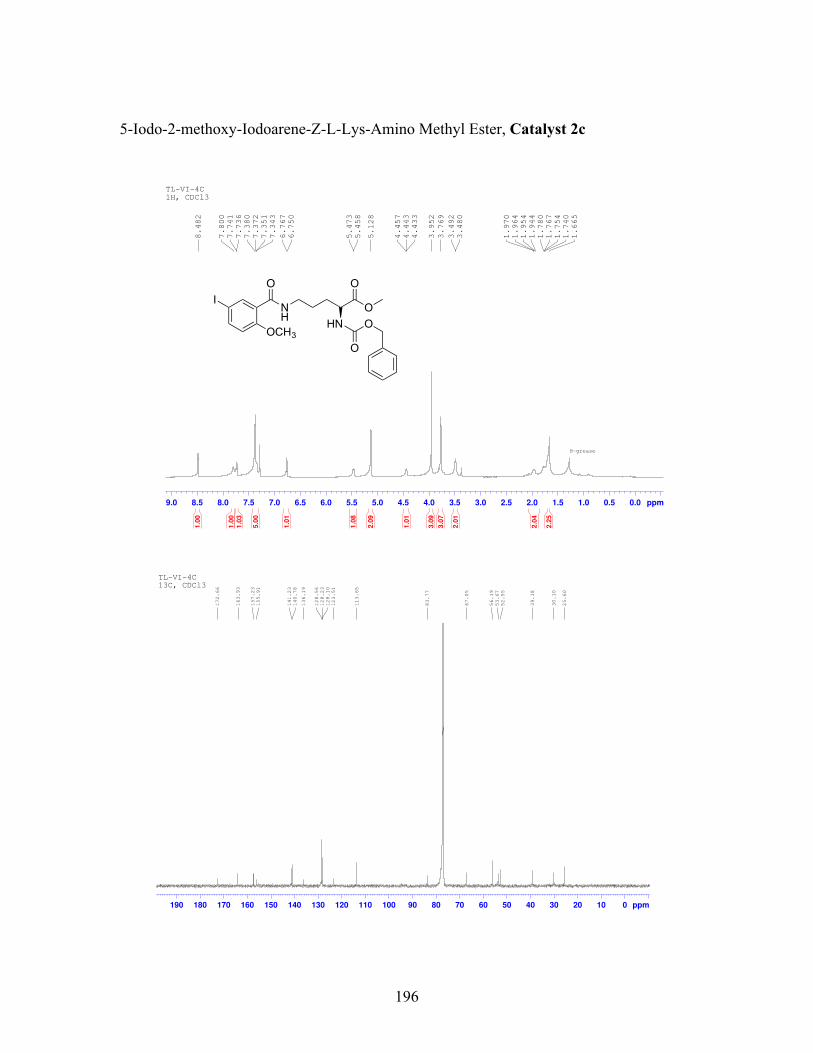
196
5-Iodo-2-methoxy-Iodoarene-Z-L-Lys-Amino Methyl Ester, Catalyst 2c
9.0 8.5 8.0 7.5 7.0 6.5 6.0 5.5 5.0 4.5 4.0 3.5 3.0 2.5 2.0 1.5 1.0 0.5 0.0 ppm
1.665
1.740
1.754
1.767
1.780
1.944
1.954
1.964
1.970
3.480
3.492
3.769
3.952
4.433
4.443
4.457
5.128
5.458
5.473
6.750
6.767
7.343
7.351
7.372
7.380
7.736
7.741
7.800
8.482
2.25
2.04
2.01
3.07
3.09
1.01
2.09
1.08
1.01
5.00
1.03
1.00
1.00
1H, CDCl3TL−VI−4C�
H−grease
NH
OI
OCH3HN
O
O
O
O
190 180 170 160 150 140 130 120 110 100 90 80 70 60 50 40 30 20 10 0 ppm
25.60
30.10
39.18
52.55
53.67
56.19
67.05
83.77
113.65
123.51
128.10
128.23
128.56
136.19
140.78
141.23
155.91
157.23
163.93
172.6613C, CDCl3�
TL−VI−4C�
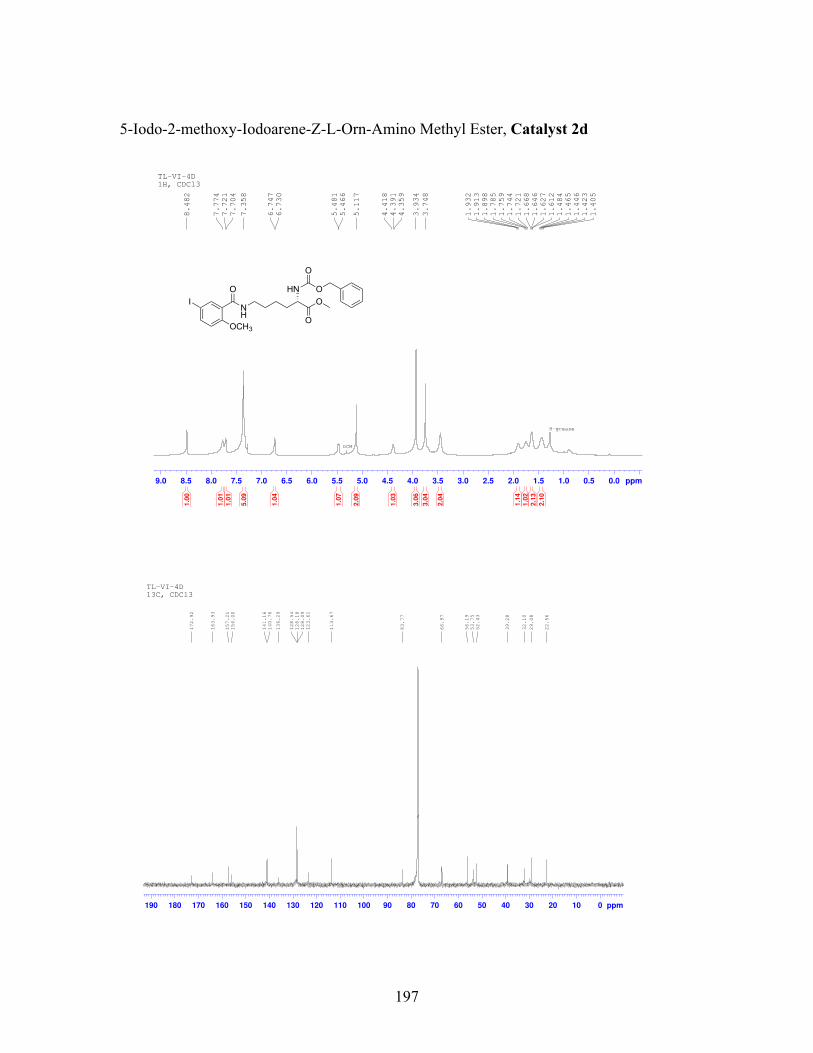
197
5-Iodo-2-methoxy-Iodoarene-Z-L-Orn-Amino Methyl Ester, Catalyst 2d
9.0 8.5 8.0 7.5 7.0 6.5 6.0 5.5 5.0 4.5 4.0 3.5 3.0 2.5 2.0 1.5 1.0 0.5 0.0 ppm
1.405
1.423
1.446
1.465
1.484
1.612
1.627
1.646
1.668
1.721
1.744
1.759
1.785
1.898
1.913
1.932
3.748
3.934
4.359
4.391
4.418
5.117
5.466
5.481
6.730
6.747
7.358
7.704
7.721
7.774
8.482
2.10
2.13
1.02
1.14
2.04
3.04
3.06
1.03
2.09
1.07
1.04
5.09
1.01
1.01
1.00
1H, CDCl3TL−VI−4D�
DCM
H−grease
NH
OI
HN
O
O
O
O
OCH3
190 180 170 160 150 140 130 120 110 100 90 80 70 60 50 40 30 20 10 0 ppm
22.56
29.08
32.10
39.28
52.43
53.75
56.19
66.97
83.77
113.67
123.61
128.09
128.18
128.54
136.29
140.76
141.16
156.00
157.21
163.93
172.92
13C, CDCl3�TL−VI−4D�
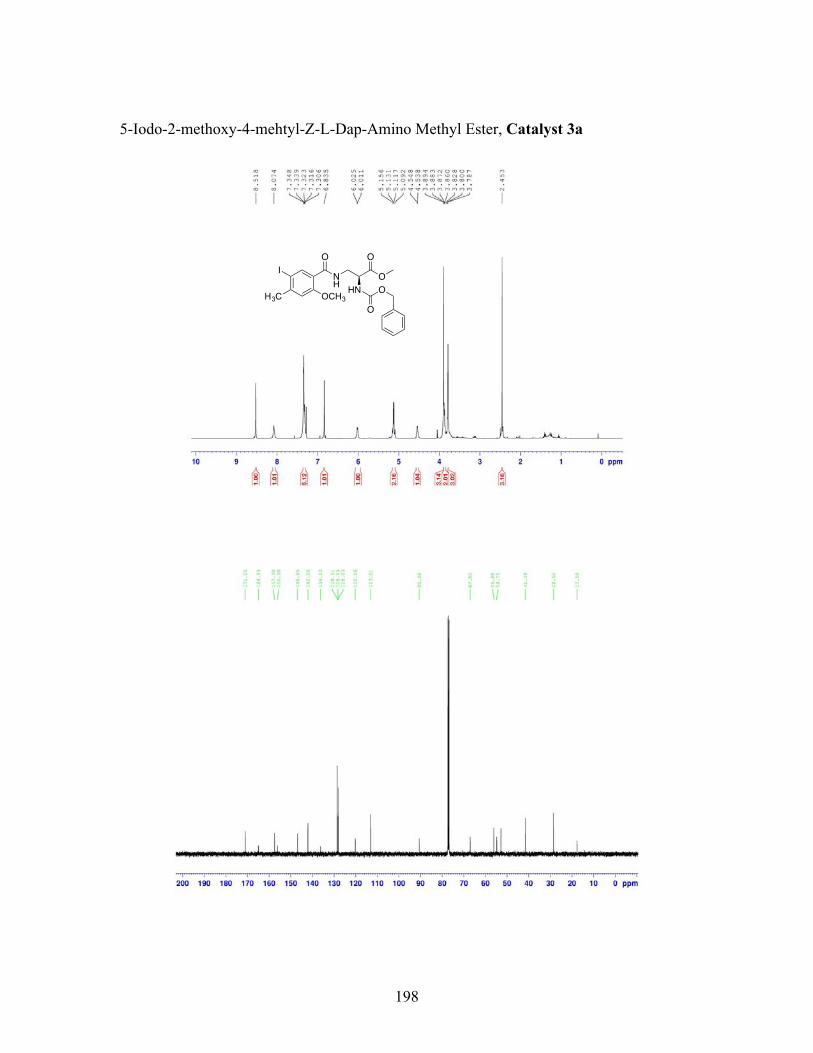
198
5-Iodo-2-methoxy-4-mehtyl-Z-L-Dap-Amino Methyl Ester, Catalyst 3a
HN
O
ONH
O
O
O
I
OCH3H3C
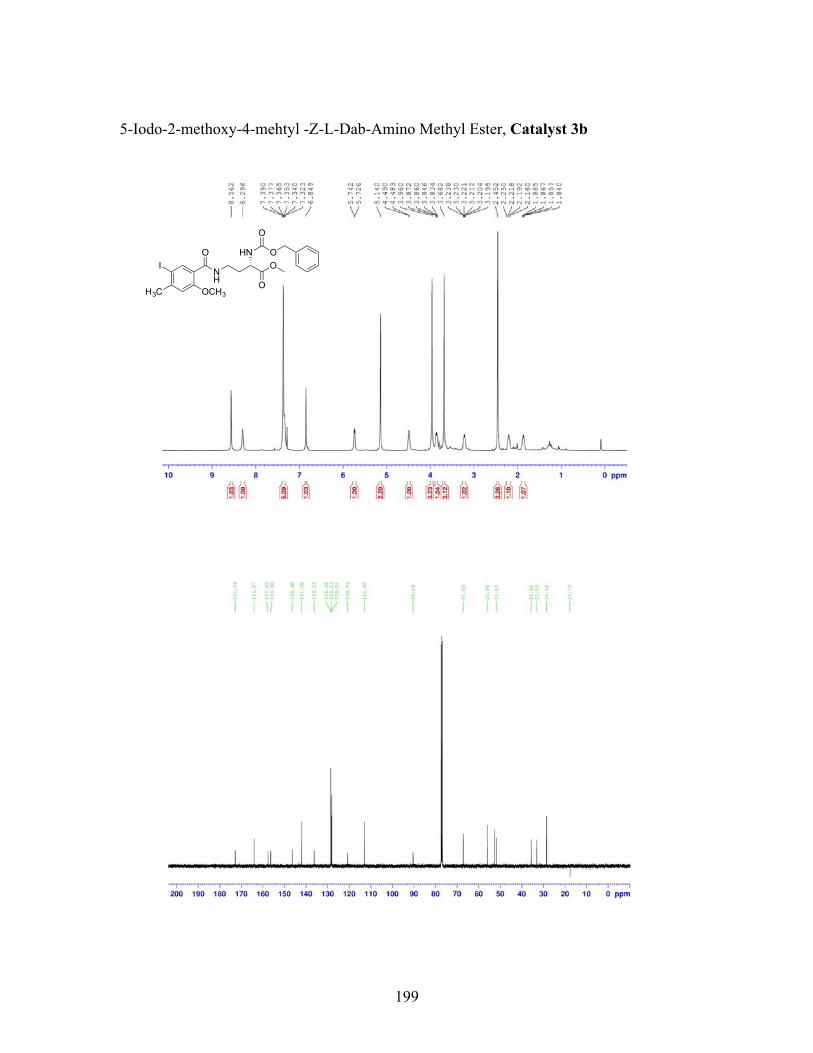
199
5-Iodo-2-methoxy-4-mehtyl -Z-L-Dab-Amino Methyl Ester, Catalyst 3b
NH
OI
HN
O
O
O
O
OCH3H3C
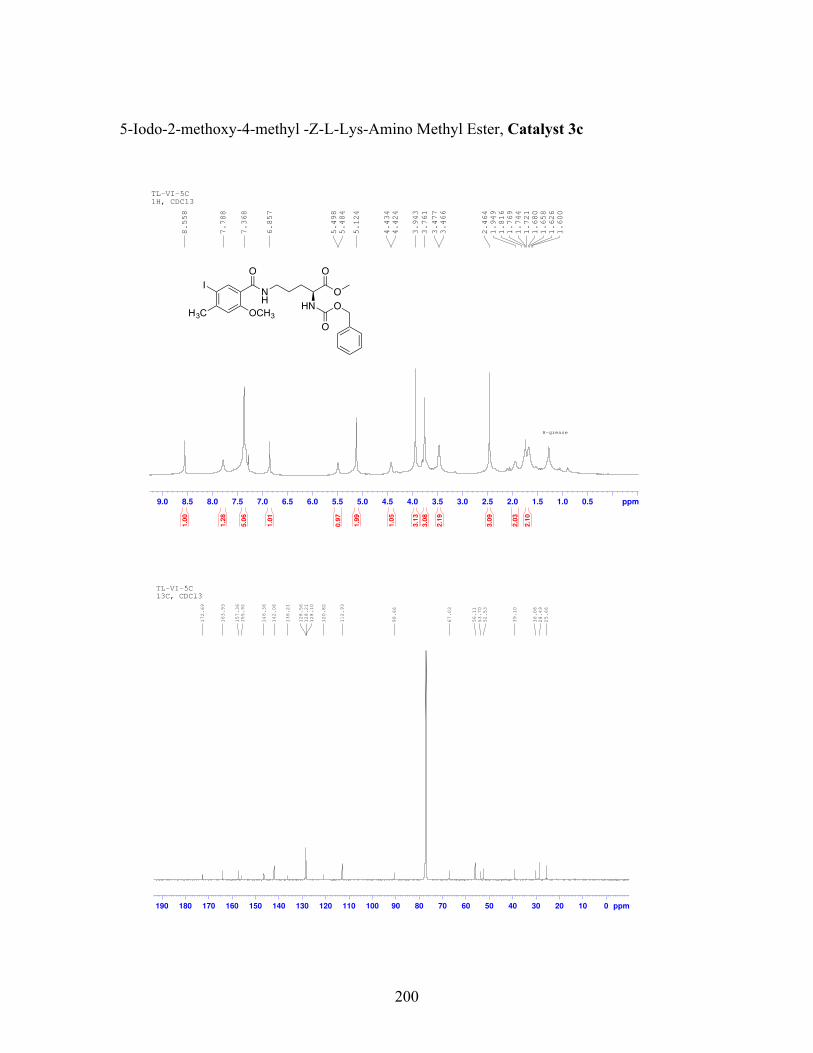
200
5-Iodo-2-methoxy-4-methyl -Z-L-Lys-Amino Methyl Ester, Catalyst 3c
9.0 8.5 8.0 7.5 7.0 6.5 6.0 5.5 5.0 4.5 4.0 3.5 3.0 2.5 2.0 1.5 1.0 0.5 ppm
1.600
1.626
1.658
1.680
1.721
1.744
1.769
1.816
1.949
2.464
3.466
3.477
3.761
3.943
4.424
4.434
5.124
5.484
5.498
6.857
7.368
7.788
8.558
2.10
2.03
3.09
2.19
3.08
3.13
1.05
1.99
0.97
1.01
5.06
1.28
1.00
1H, CDCl3TL−VI−5C�
H−grease
190 180 170 160 150 140 130 120 110 100 90 80 70 60 50 40 30 20 10 0 ppm
25.66
28.49
30.08
39.10
52.53
53.70
56.11
67.03
90.66
112.93
120.82
128.10
128.21
128.56
136.21
142.06
146.36
155.92
157.36
163.93
172.69
13C, CDCl3�TL−VI−5C�
HN
O
ONH
O
O
O
I
H3C OCH3
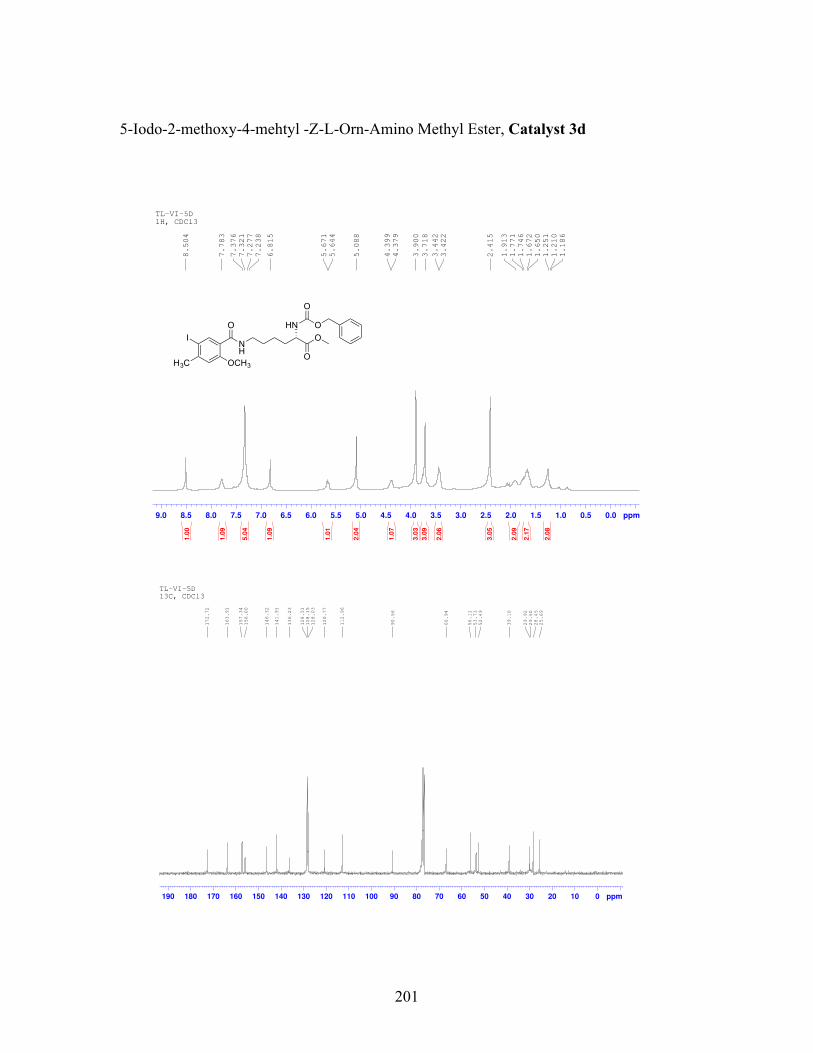
201
5-Iodo-2-methoxy-4-mehtyl -Z-L-Orn-Amino Methyl Ester, Catalyst 3d
9.0 8.5 8.0 7.5 7.0 6.5 6.0 5.5 5.0 4.5 4.0 3.5 3.0 2.5 2.0 1.5 1.0 0.5 0.0 ppm
1.186
1.210
1.251
1.650
1.672
1.746
1.771
1.913
2.415
3.422
3.442
3.718
3.900
4.379
4.399
5.088
5.644
5.671
6.815
7.238
7.277
7.321
7.376
7.783
8.504
2.08
2.17
2.09
3.05
2.06
3.09
3.03
1.07
2.04
1.01
1.09
5.04
1.09
1.00
1H, CDCl3�TL−VI−5D�
190 180 170 160 150 140 130 120 110 100 90 80 70 60 50 40 30 20 10 0 ppm
25.69
28.45
29.50
29.92
39.10
52.49
53.73
56.11
66.94
90.56
112.96
120.77
128.03
128.15
128.51
136.23
141.91
146.32
156.00
157.34
163.91
172.72
13C, CDCl3�TL−VI−5D�
NH
OI
HN
O
O
O
O
OCH3H3C
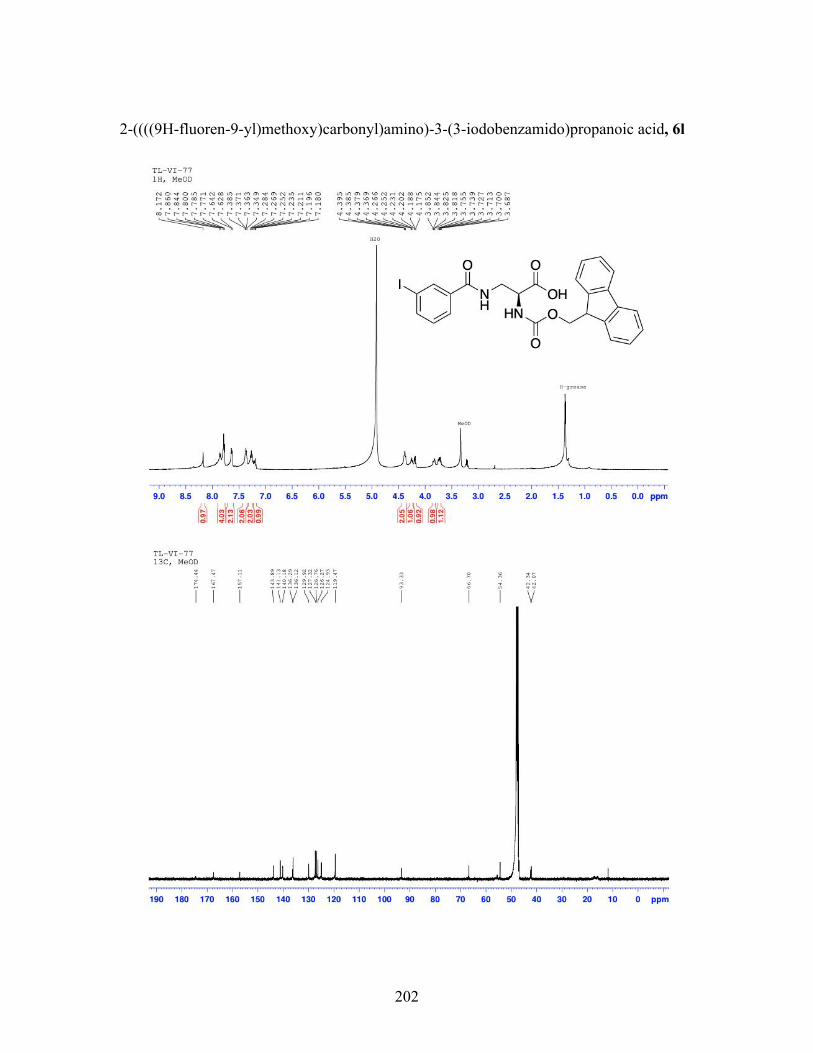
202
2-((((9H-fluoren-9-yl)methoxy)carbonyl)amino)-3-(3-iodobenzamido)propanoic acid, 6l
203
3-amino-N-butyl-2-iodobenzamide, 4ab
I
NH
OH2N
204
3-acetamido-N-butyl-2-iodobenzamide, Catalyst 4a
I
NH
OHN
O
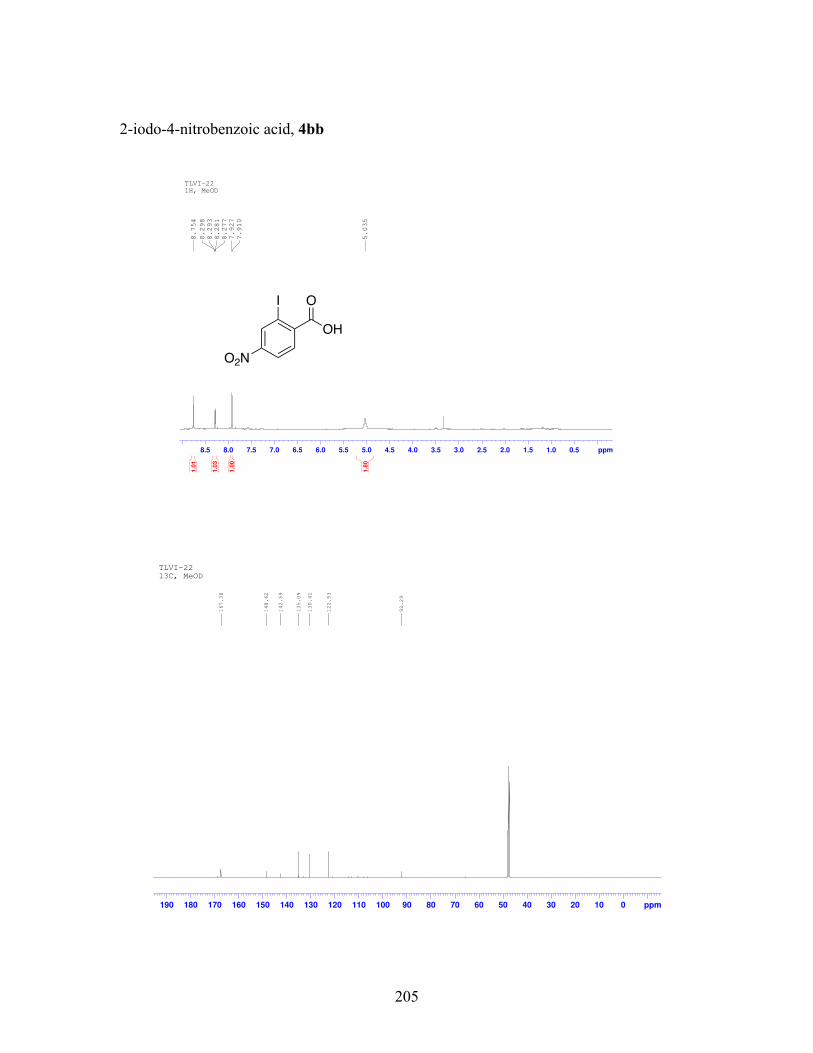
205
2-iodo-4-nitrobenzoic acid, 4bb
190 180 170 160 150 140 130 120 110 100 90 80 70 60 50 40 30 20 10 0 ppm
92.29
122.53
130.41
135.09
142.59
148.62
167.38
13C, MeOD�TLVI−22�
8.5 8.0 7.5 7.0 6.5 6.0 5.5 5.0 4.5 4.0 3.5 3.0 2.5 2.0 1.5 1.0 0.5 ppm
5.035
7.910
7.927
8.277
8.281
8.293
8.298
8.754
1.80
1.00
1.03
1.01
1H, MeODTLVI−22�
I
OH
O
O2N
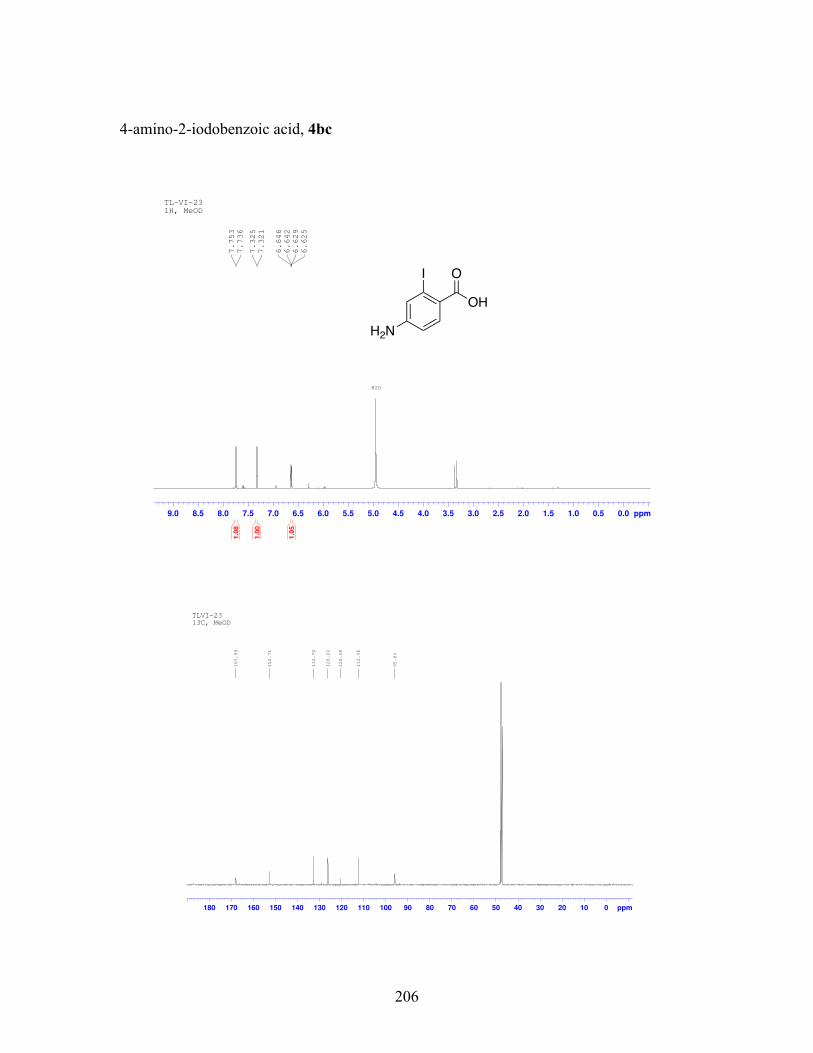
206
4-amino-2-iodobenzoic acid, 4bc
9.0 8.5 8.0 7.5 7.0 6.5 6.0 5.5 5.0 4.5 4.0 3.5 3.0 2.5 2.0 1.5 1.0 0.5 0.0 ppm
6.625
6.629
6.642
6.646
7.321
7.325
7.736
7.753
1.05
1.00
1.08
1H, MeODTL−VI−23�
H2O
180 170 160 150 140 130 120 110 100 90 80 70 60 50 40 30 20 10 0 ppm
95.84
112.46
120.48
126.22
132.70
152.71
167.99
13C, MeOD�TLVI−23�
I
OH
O
H2N
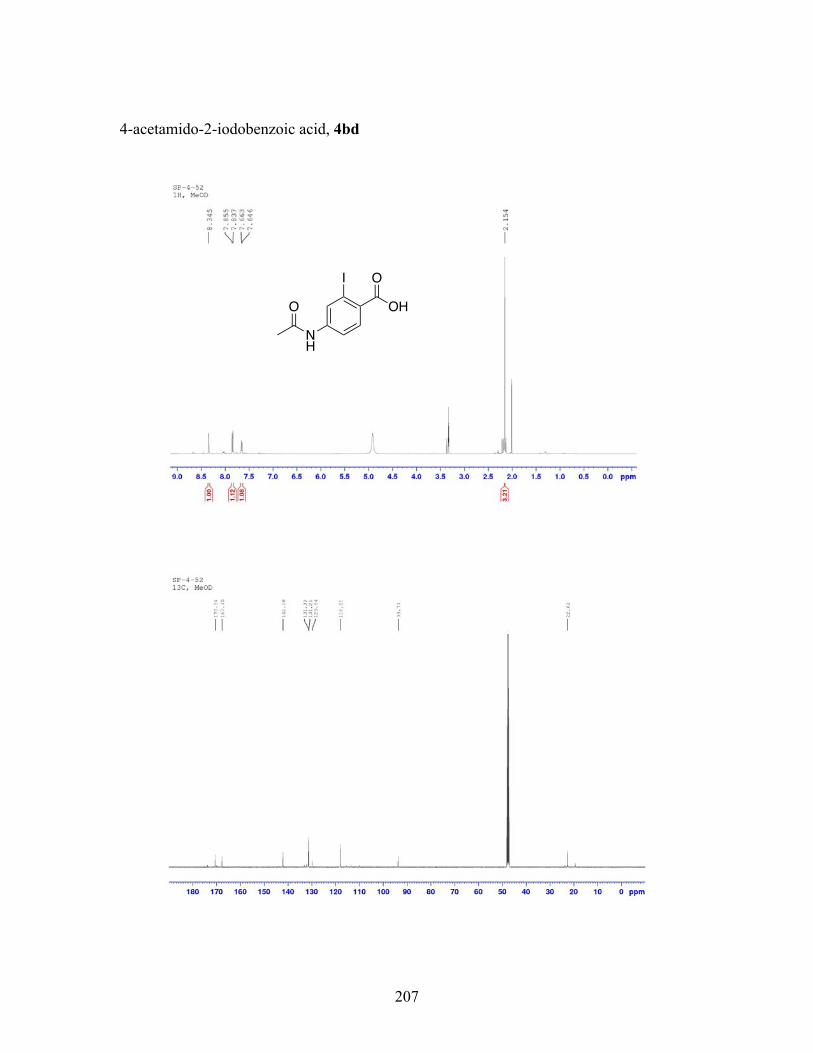
207
4-acetamido-2-iodobenzoic acid, 4bd
I
OH
O
NH
O
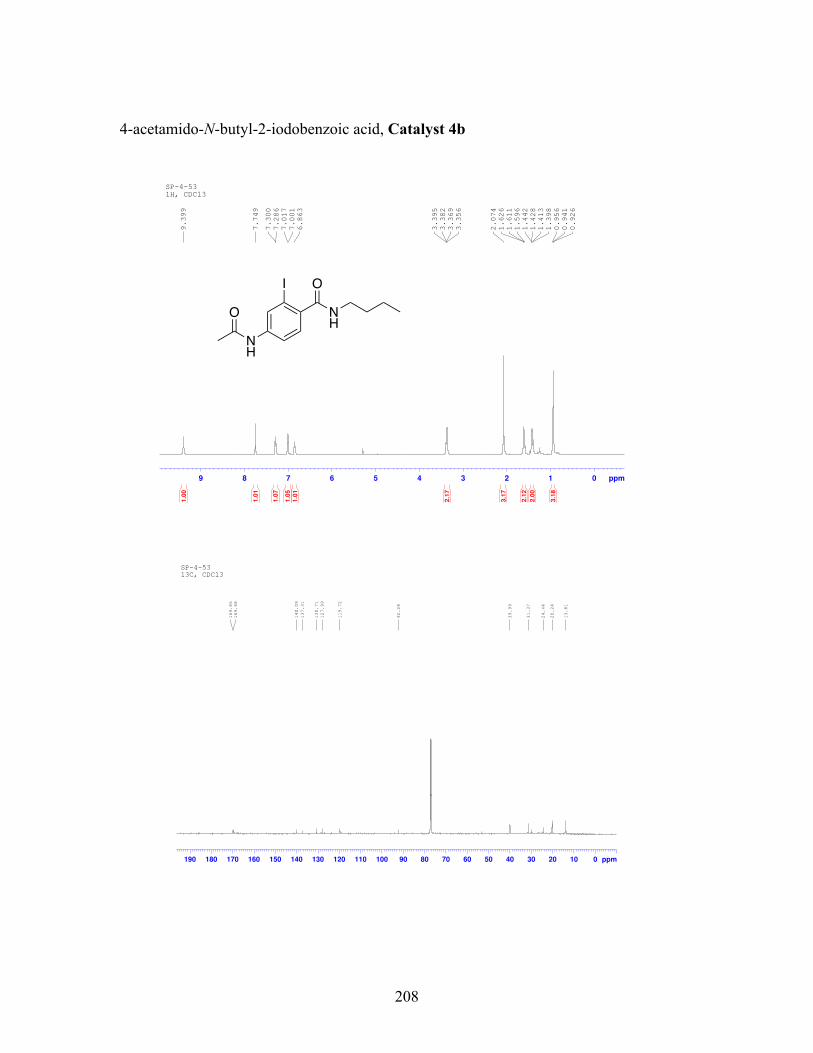
208
4-acetamido-N-butyl-2-iodobenzoic acid, Catalyst 4b
9 8 7 6 5 4 3 2 1 0 ppm
0.926
0.941
0.956
1.398
1.413
1.428
1.442
1.596
1.611
1.626
2.074
3.356
3.369
3.382
3.395
6.863
7.001
7.017
7.286
7.300
7.749
9.399
3.18
2.00
2.12
3.17
2.17
1.01
1.05
1.07
1.01
1.00
1H, CDCl3SP−4−53�
190 180 170 160 150 140 130 120 110 100 90 80 70 60 50 40 30 20 10 0 ppm
13.81
20.24
24.44
31.37
39.99
92.24
119.72
127.99
130.71
137.31
140.09
169.68
169.85
13C, CDCl3�SP−4−53�
I
NH
O
NH
O

209
5-amino-2-iodobenzoic acid, 4cc
9.0 8.5 8.0 7.5 7.0 6.5 6.0 5.5 5.0 4.5 4.0 3.5 3.0 2.5 2.0 1.5 1.0 0.5 0.0 ppm
8.009
8.018
8.037
8.047
8.289
8.318
8.568
8.577
1.02
1.02
1.00
1H, MeODTL−VI−10�
H2O
190 180 170 160 150 140 130 120 110 100 90 80 70 60 50 40 30 20 10 0 ppm
101.63
124.44
125.71
137.59
142.72
147.76
166.45
13C, MeOD�TL−VI−10�
I
OH
O
NH2
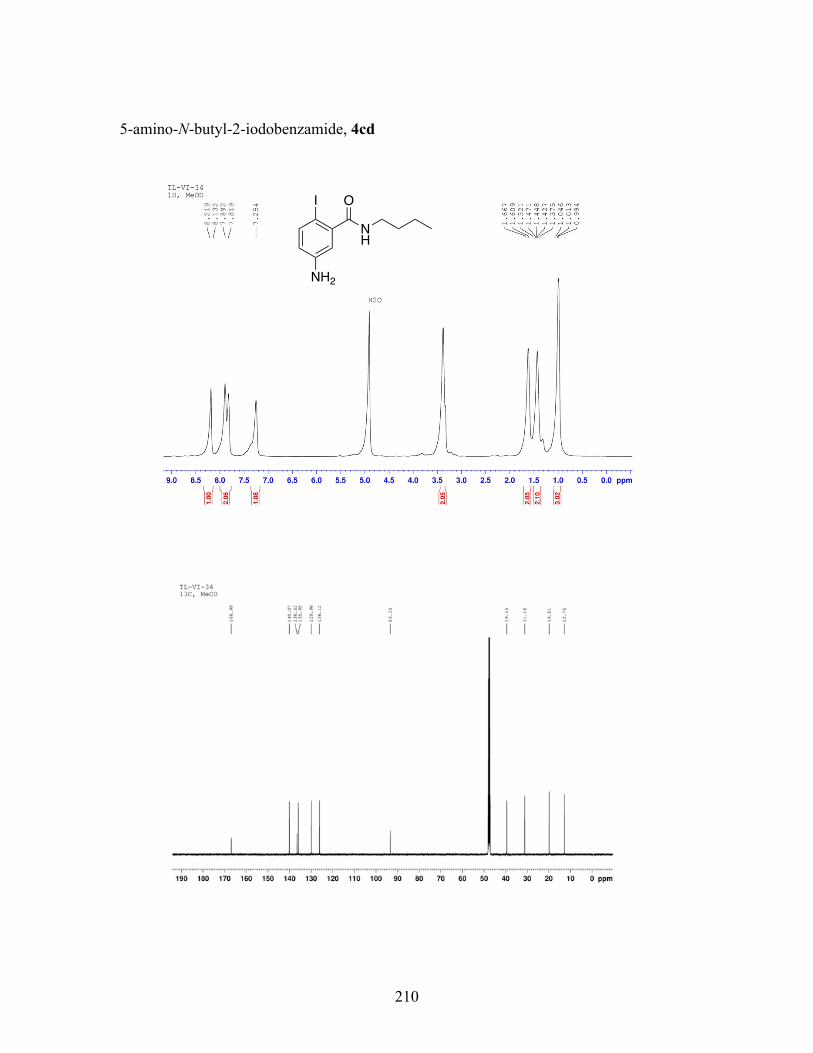
210
5-amino-N-butyl-2-iodobenzamide, 4cd
I
NH
O
NH2
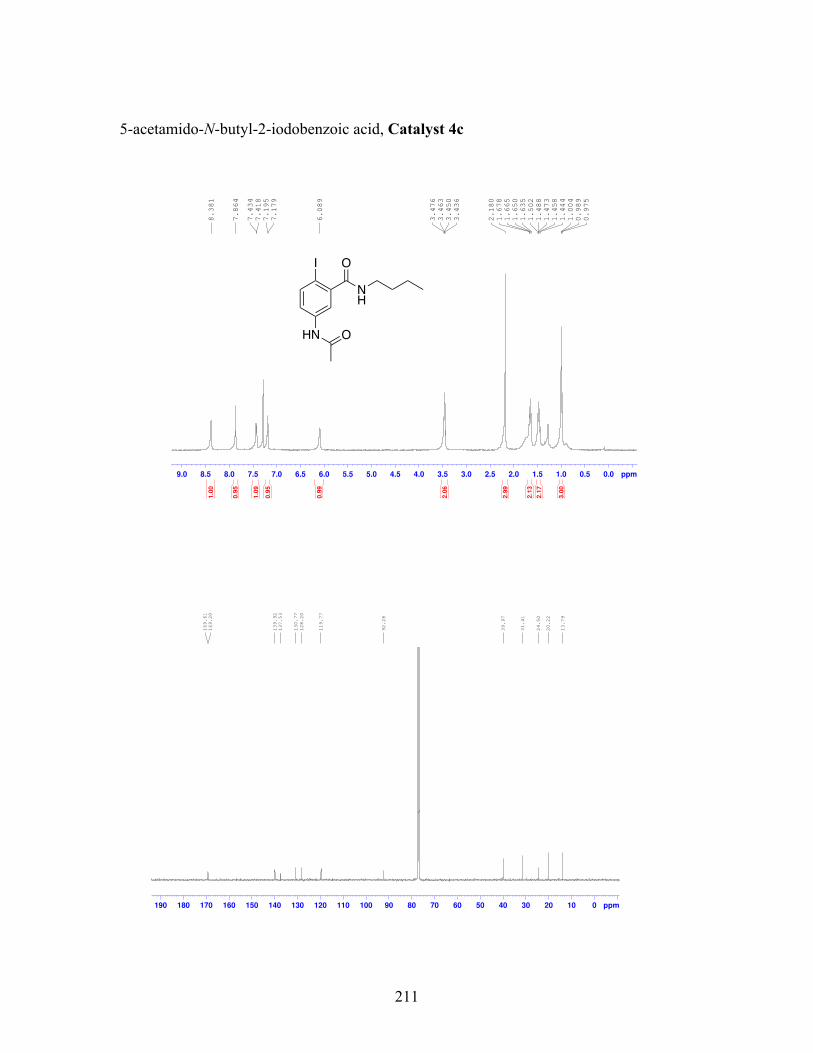
211
5-acetamido-N-butyl-2-iodobenzoic acid, Catalyst 4c
9.0 8.5 8.0 7.5 7.0 6.5 6.0 5.5 5.0 4.5 4.0 3.5 3.0 2.5 2.0 1.5 1.0 0.5 0.0 ppm
0.975
0.989
1.004
1.444
1.458
1.473
1.488
1.502
1.635
1.650
1.665
1.678
2.180
3.436
3.450
3.463
3.476
6.089
7.179
7.195
7.418
7.434
7.864
8.381
3.00
2.17
2.13
2.99
2.06
0.99
0.95
1.09
0.95
1.00
1H, CDCl3TL−VI−45�
I
NH
O
HN O
190 180 170 160 150 140 130 120 110 100 90 80 70 60 50 40 30 20 10 0 ppm
13.79
20.22
24.50
31.41
39.97
92.28
119.77
128.20
130.77
137.53
139.92
169.20
169.61
13C, CDCl3�TL−VI−45�
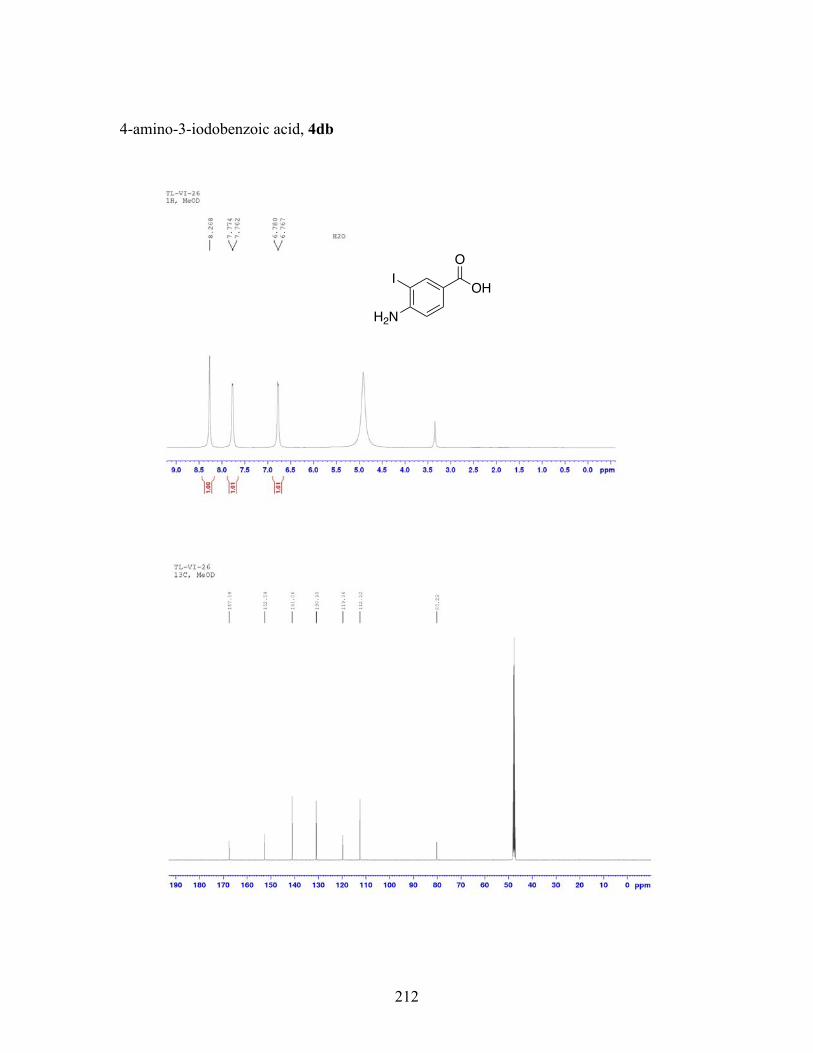
212
4-amino-3-iodobenzoic acid, 4db
OH
OI
H2N
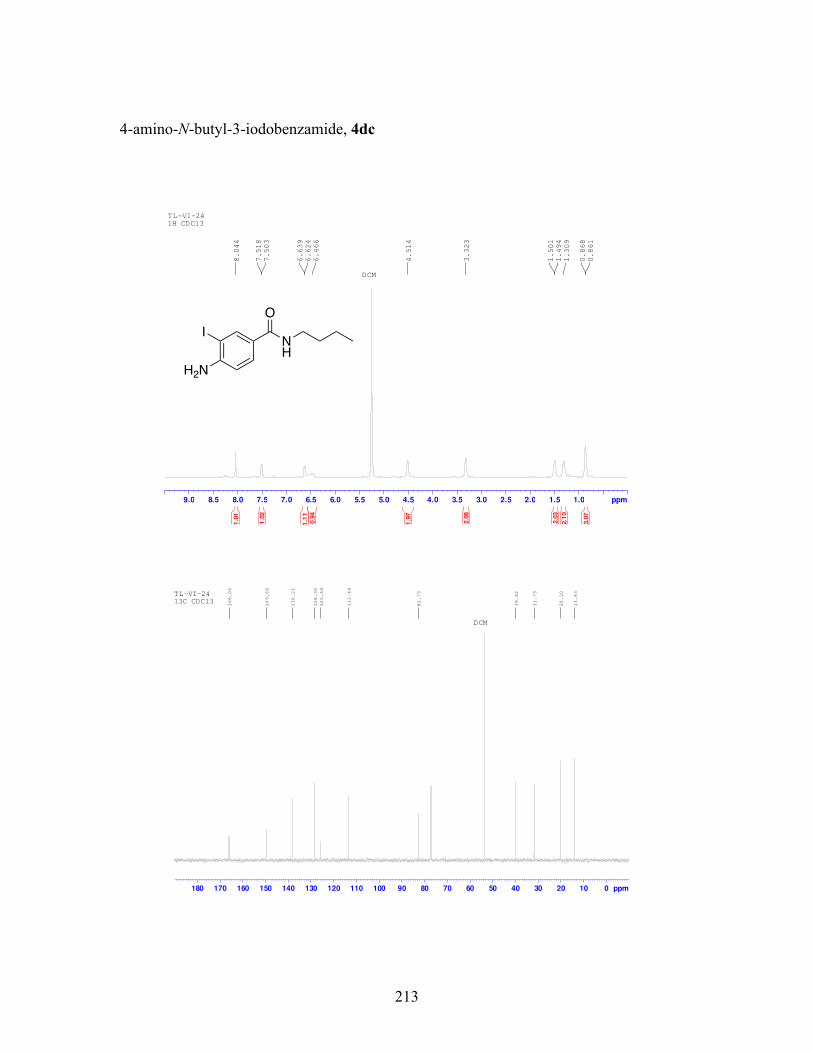
213
4-amino-N-butyl-3-iodobenzamide, 4dc
NH
OI
H2N
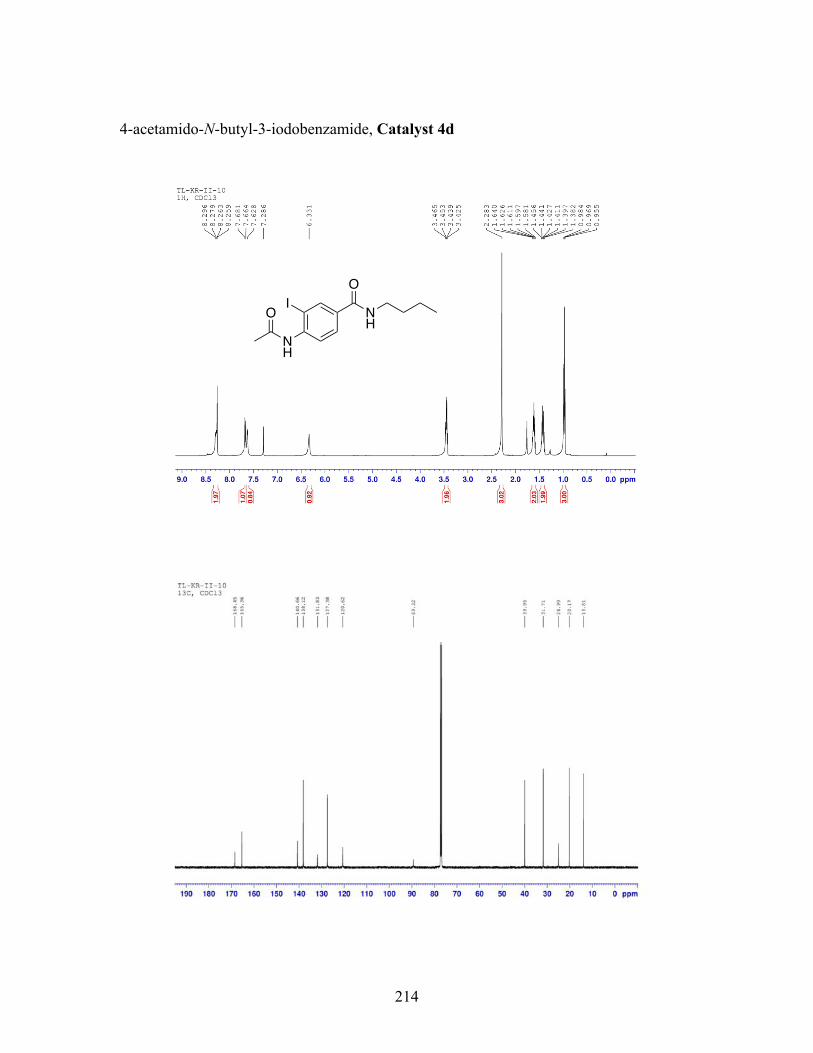
214
4-acetamido-N-butyl-3-iodobenzamide, Catalyst 4d
NH
OI
NH
O
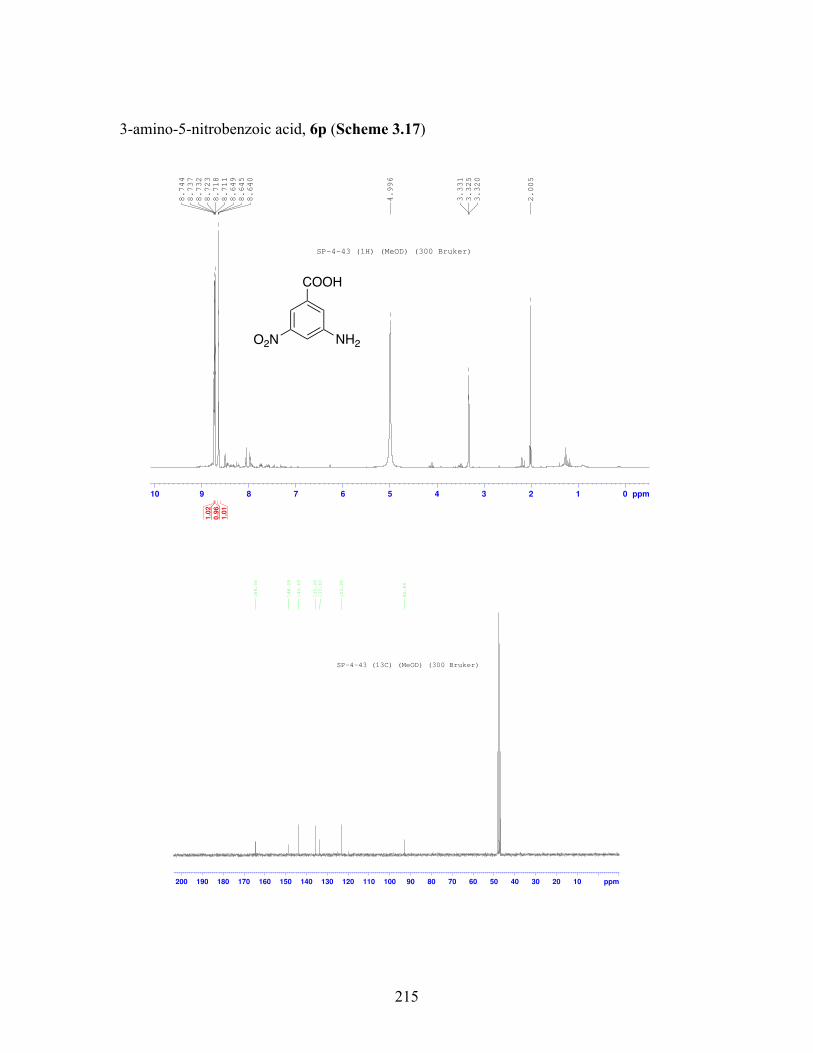
215
3-amino-5-nitrobenzoic acid, 6p (Scheme 3.17)
10 9 8 7 6 5 4 3 2 1 0 ppm
2.005
3.320
3.325
3.331
4.996
8.640
8.645
8.649
8.711
8.718
8.723
8.732
8.737
8.744
1.01
0.96
1.02
SP−4−43 (1H) (MeOD) (300 Bruker)
200 190 180 170 160 150 140 130 120 110 100 90 80 70 60 50 40 30 20 10 ppm
92.89
123.20
133.83
135.49
143.69
148.49
164.45
SP−4−43 (13C) (MeOD) (300 Bruker)�
COOH
NH2O2N
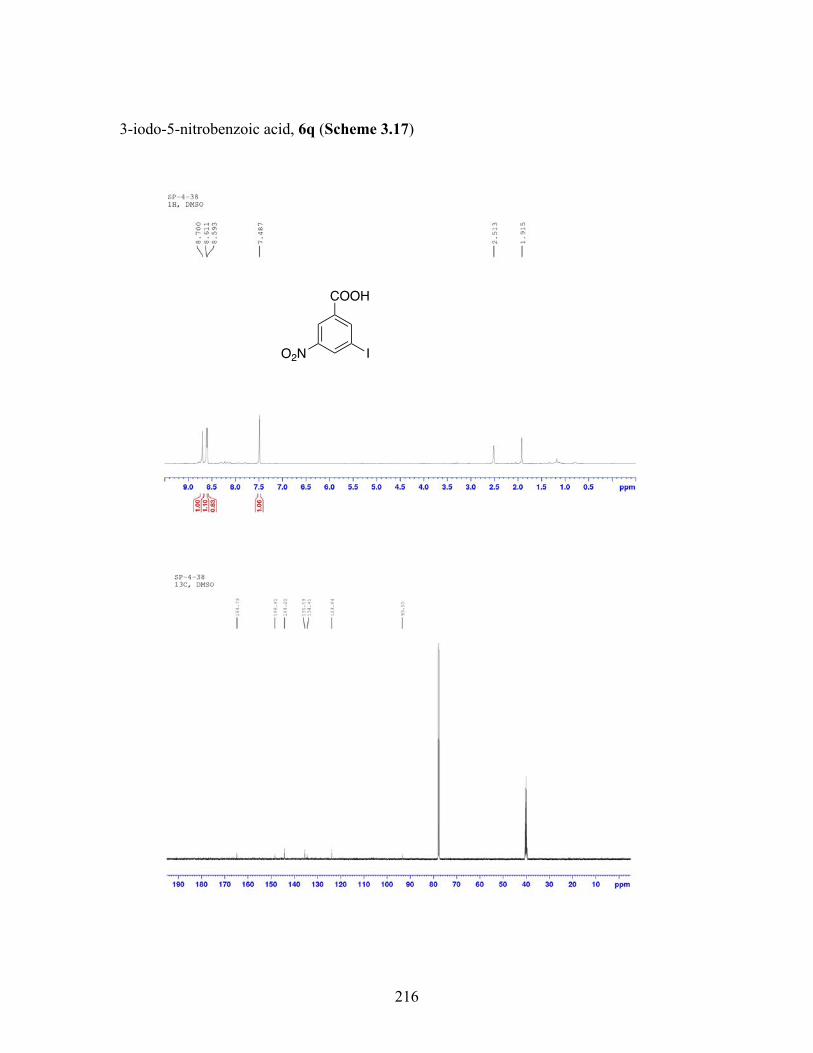
216
3-iodo-5-nitrobenzoic acid, 6q (Scheme 3.17)
COOH
IO2N
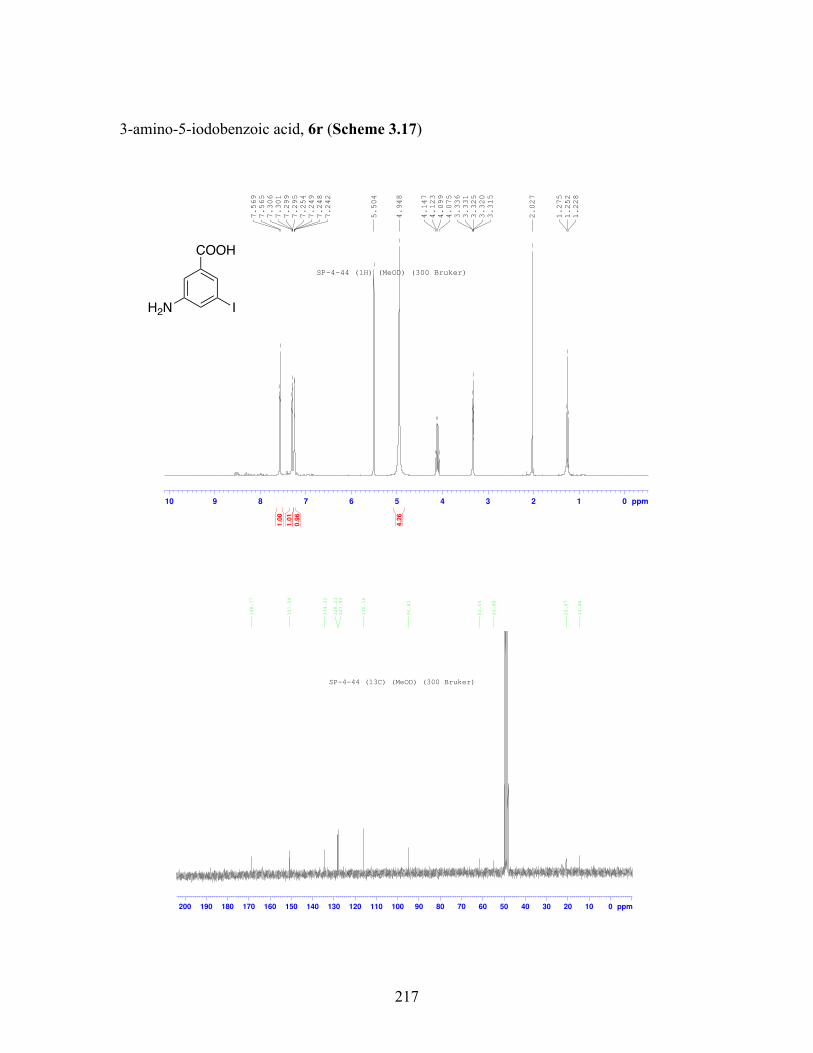
217
COOH
IH2N
3-amino-5-iodobenzoic acid, 6r (Scheme 3.17)
10 9 8 7 6 5 4 3 2 1 0 ppm
1.228
1.252
1.275
2.027
3.315
3.320
3.325
3.331
3.336
4.075
4.099
4.123
4.147
4.948
5.504
7.242
7.248
7.249
7.254
7.295
7.299
7.301
7.306
7.565
7.569
4.26
0.96
1.01
1.00
SP−4−44 (1H) (MeOD) (300 Bruker)�
200 190 180 170 160 150 140 130 120 110 100 90 80 70 60 50 40 30 20 10 0 ppm
14.46
20.87
54.80
61.54
94.82
116.16
127.93
128.23
134.22
151.04
168.77
SP−4−44 (13C) (MeOD) (300 Bruker)�
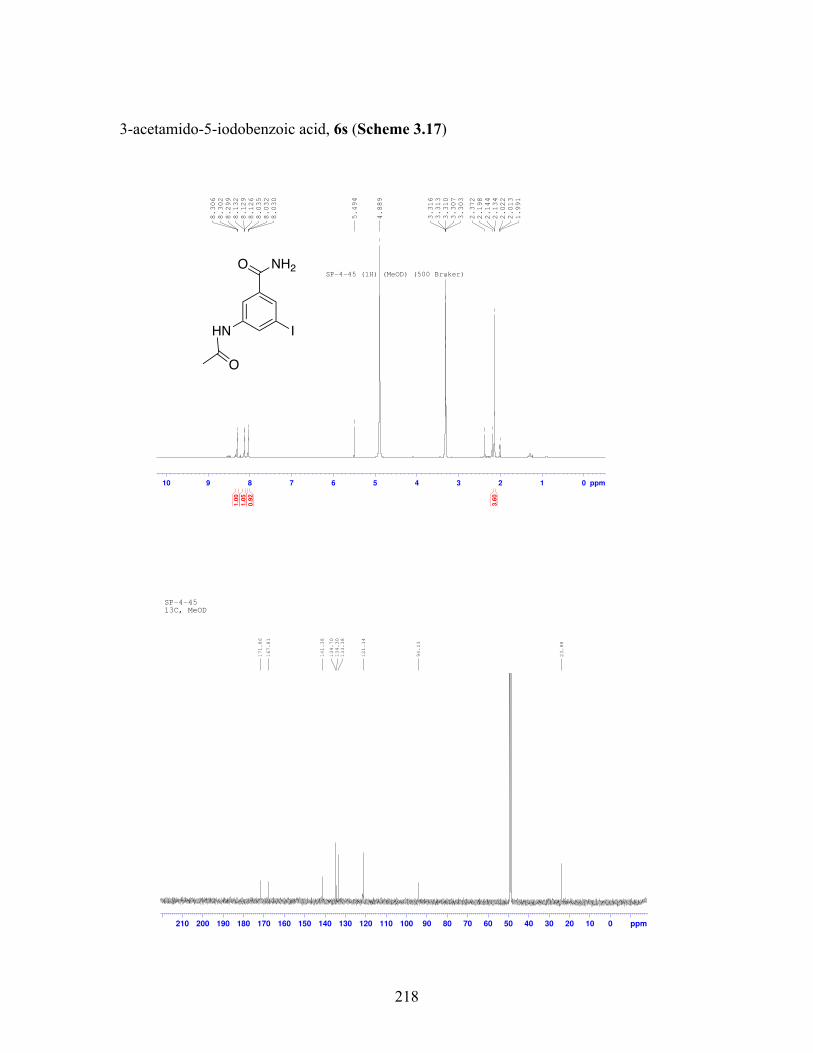
218
3-acetamido-5-iodobenzoic acid, 6s (Scheme 3.17)
10 9 8 7 6 5 4 3 2 1 0 ppm
1.991
2.013
2.022
2.134
2.144
2.198
2.372
3.303
3.307
3.310
3.313
3.316
4.889
5.494
8.030
8.032
8.035
8.126
8.129
8.132
8.299
8.302
8.306
3.60
0.92
1.05
1.00
SP−4−45 (1H) (MeOD) (500 Bruker)
210 200 190 180 170 160 150 140 130 120 110 100 90 80 70 60 50 40 30 20 10 0 ppm
23.88
94.13
121.14
133.38
134.30
134.70
141.38
167.81
171.86
13C, MeOD�SP−4−45�
IHN
O NH2
O
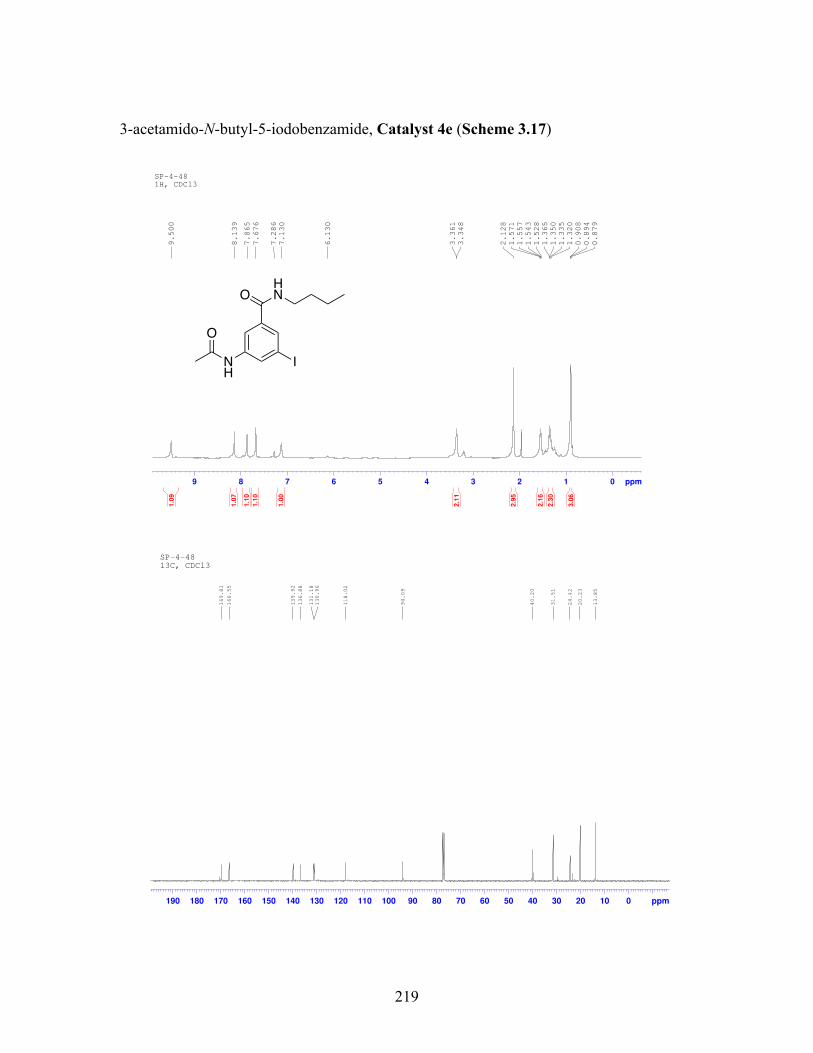
219
3-acetamido-N-butyl-5-iodobenzamide, Catalyst 4e (Scheme 3.17)
9 8 7 6 5 4 3 2 1 0 ppm
0.879
0.894
0.908
1.320
1.335
1.350
1.365
1.528
1.543
1.557
1.571
2.128
3.348
3.361
6.130
7.130
7.286
7.676
7.865
8.139
9.500
3.06
2.30
2.16
2.95
2.11
1.00
1.10
1.10
1.07
1.09
1H, CDCl3SP−4−48�
190 180 170 160 150 140 130 120 110 100 90 80 70 60 50 40 30 20 10 0 ppm
13.85
20.23
24.42
31.51
40.20
94.09
118.02
130.96
131.18
136.88
139.92
166.55
169.81
13C, CDCl3�SP−4−48�
INH
OHN
O
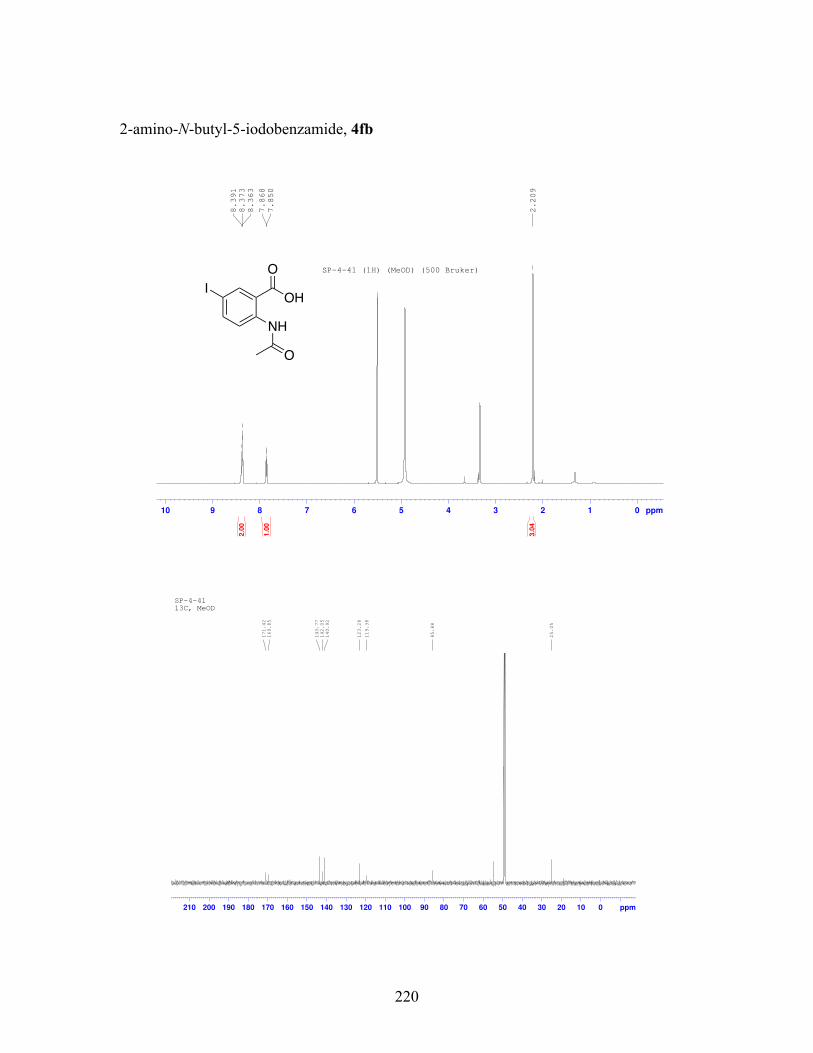
220
2-amino-N-butyl-5-iodobenzamide, 4fb
10 9 8 7 6 5 4 3 2 1 0 ppm
2.209
7.850
7.868
8.363
8.373
8.391
3.04
1.00
2.00
SP−4−41 (1H) (MeOD) (500 Bruker)
210 200 190 180 170 160 150 140 130 120 110 100 90 80 70 60 50 40 30 20 10 0 ppm
25.05
85.88
119.38
123.28
140.92
142.05
143.77
169.85
171.42
13C, MeOD�SP−4−41�
OH
OI
NH
O
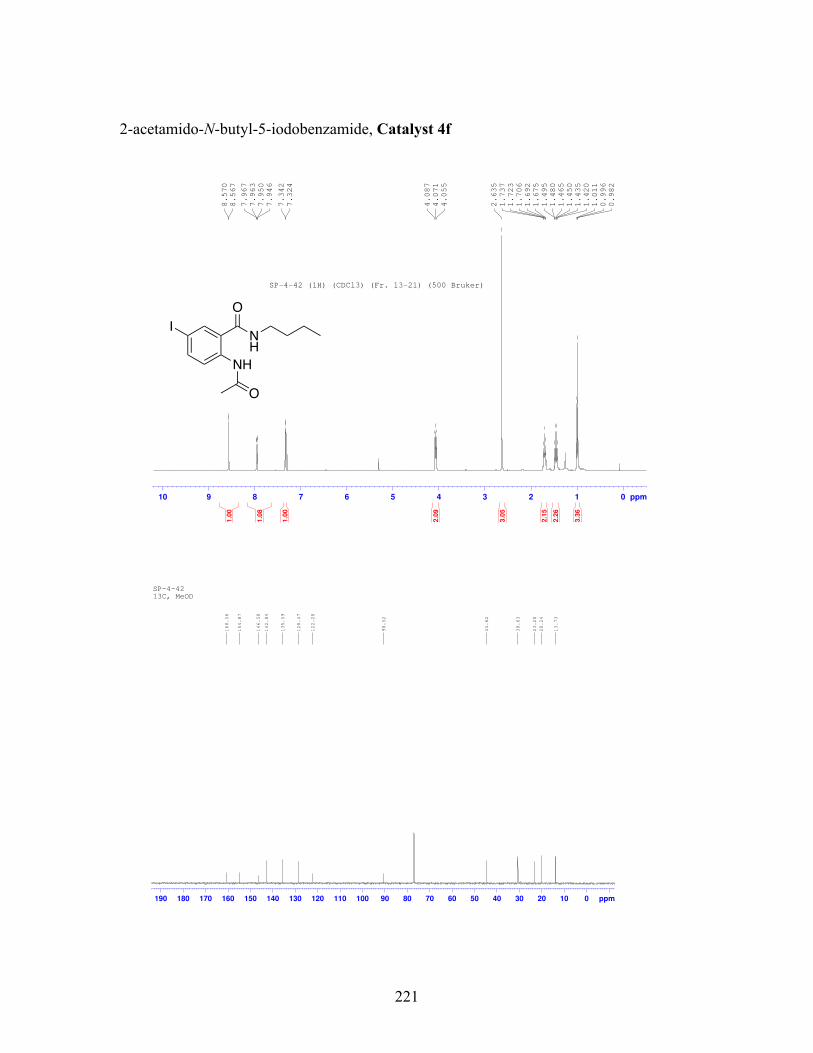
221
2-acetamido-N-butyl-5-iodobenzamide, Catalyst 4f
10 9 8 7 6 5 4 3 2 1 0 ppm
0.982
0.996
1.011
1.420
1.435
1.450
1.465
1.480
1.495
1.675
1.692
1.706
1.723
1.737
2.635
4.055
4.071
4.087
7.324
7.342
7.946
7.950
7.963
7.967
8.567
8.570
3.36
2.26
2.15
3.05
2.09
1.00
1.08
1.00
SP−4−42 (1H) (CDCl3) (Fr. 13−21) (500 Bruker)
190 180 170 160 150 140 130 120 110 100 90 80 70 60 50 40 30 20 10 0 ppm
13.73
20.24
23.20
30.63
44.62
90.52
122.20
128.47
135.59
142.84
146.50
154.87
160.56
13C, MeOD�SP−4−42�
NH
OI
NH
O
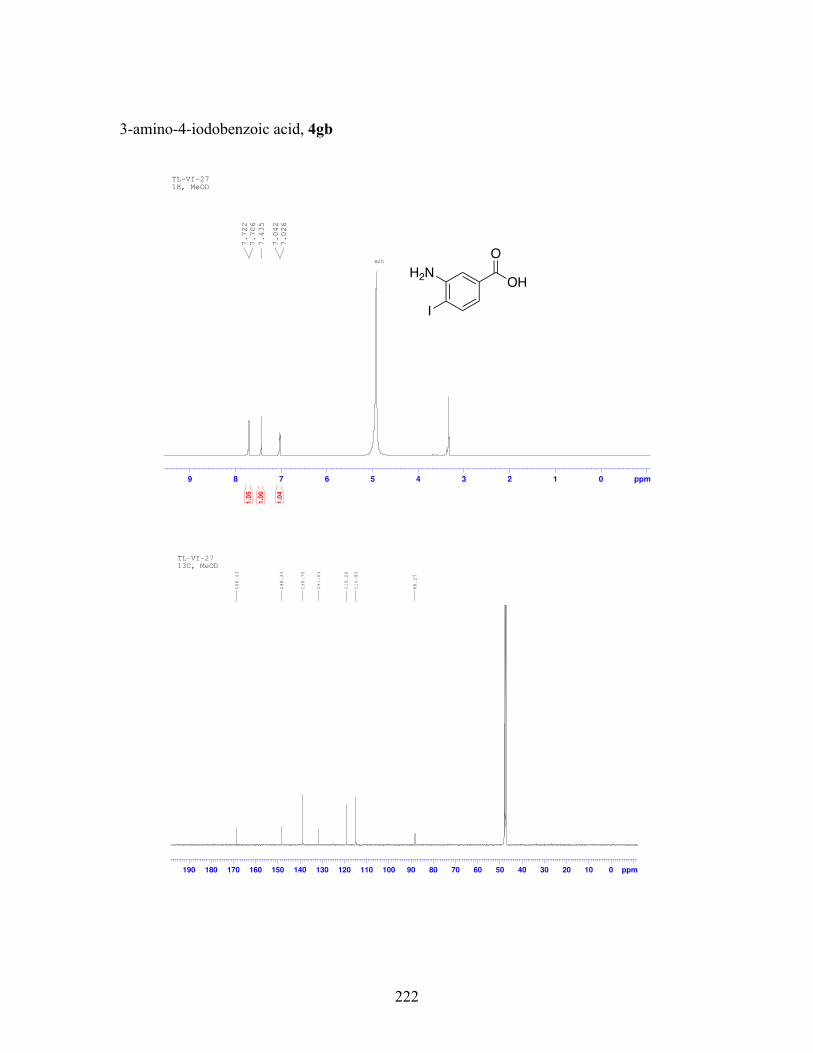
222
3-amino-4-iodobenzoic acid, 4gb
9 8 7 6 5 4 3 2 1 0 ppm
7.026
7.042
7.435
7.706
7.722
1.04
1.00
1.05
1H, MeODTL−VI−27�
H2O
190 180 170 160 150 140 130 120 110 100 90 80 70 60 50 40 30 20 10 0 ppm
88.27
114.85
119.20
131.63
138.76
148.33
168.43
13C, MeOD�TL−VI−27�
OH
OH2N
I
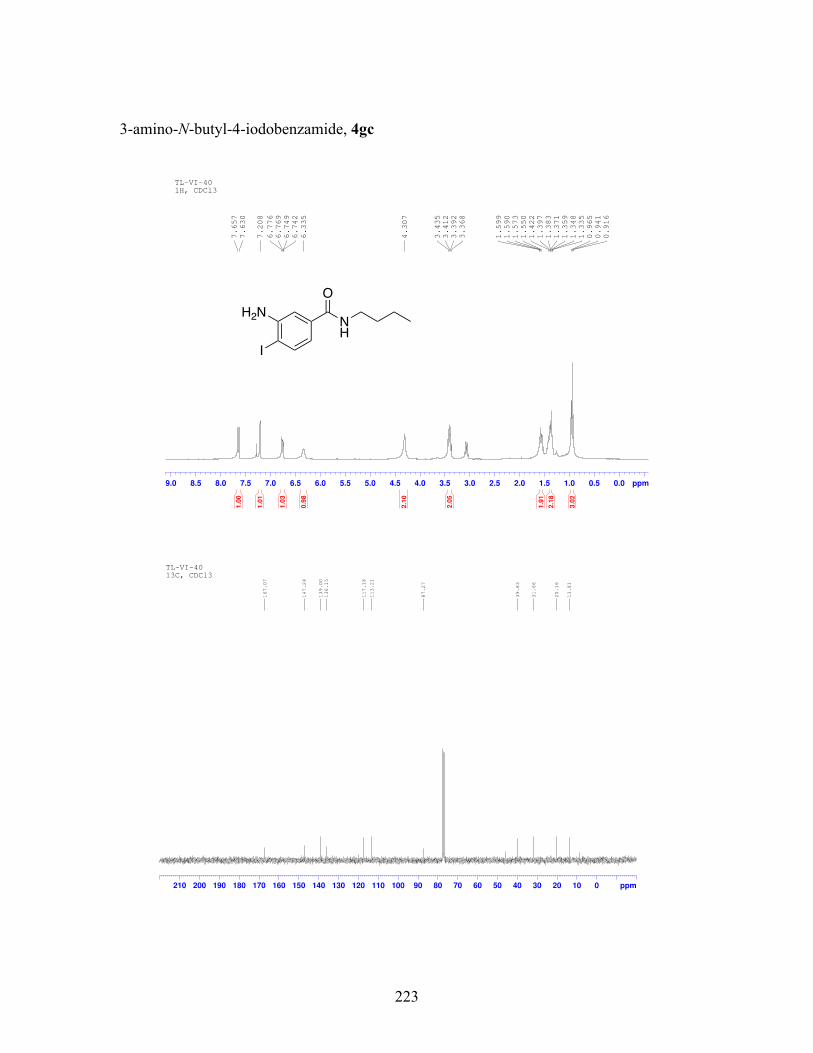
223
3-amino-N-butyl-4-iodobenzamide, 4gc
9.0 8.5 8.0 7.5 7.0 6.5 6.0 5.5 5.0 4.5 4.0 3.5 3.0 2.5 2.0 1.5 1.0 0.5 0.0 ppm
0.916
0.941
0.965
1.335
1.348
1.359
1.371
1.383
1.397
1.422
1.550
1.573
1.590
1.599
3.368
3.392
3.412
3.435
4.307
6.335
6.742
6.749
6.769
6.776
7.208
7.630
7.657
3.02
2.18
1.91
2.05
2.10
0.98
1.03
1.01
1.00
1H, CDCl3�TL−VI−40�
210 200 190 180 170 160 150 140 130 120 110 100 90 80 70 60 50 40 30 20 10 0 ppm
13.81
20.16
31.66
39.83
87.27
113.21
117.18
136.15
139.00
147.24
167.07
13C, CDCl3�TL−VI−40�
NH
OH2N
I
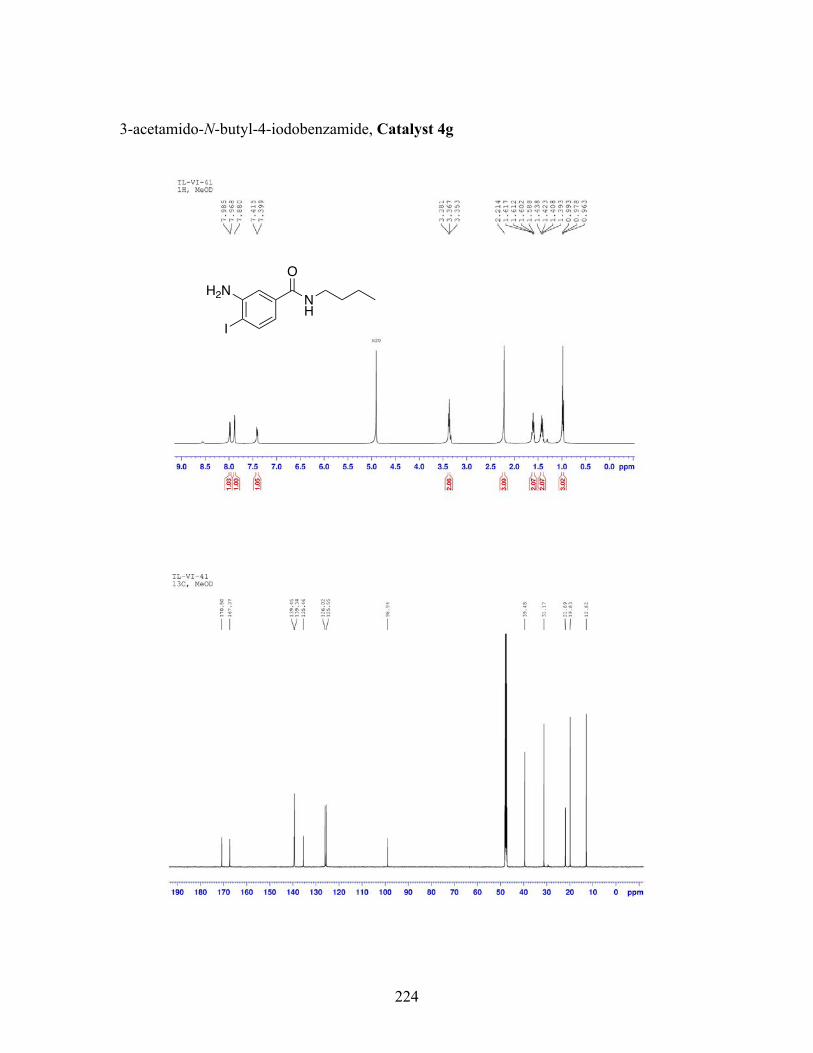
224
3-acetamido-N-butyl-4-iodobenzamide, Catalyst 4g
NH
OH2N
I
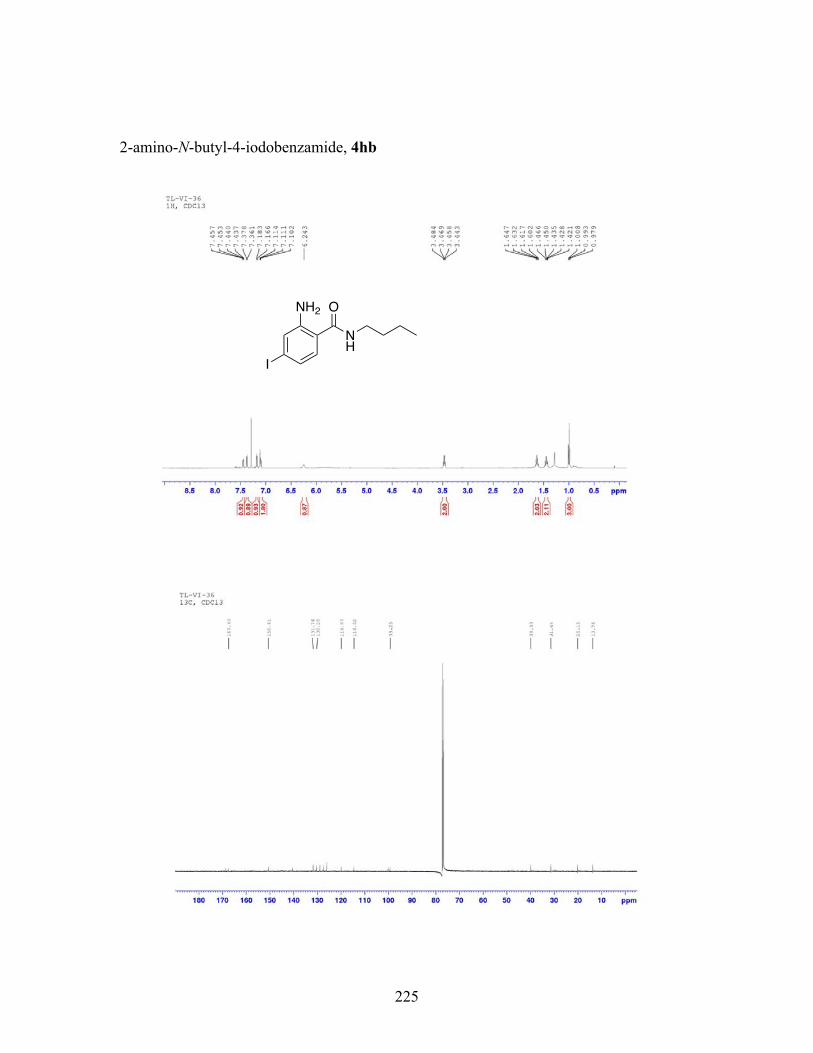
225
2-amino-N-butyl-4-iodobenzamide, 4hb
NH2
NH
O
I
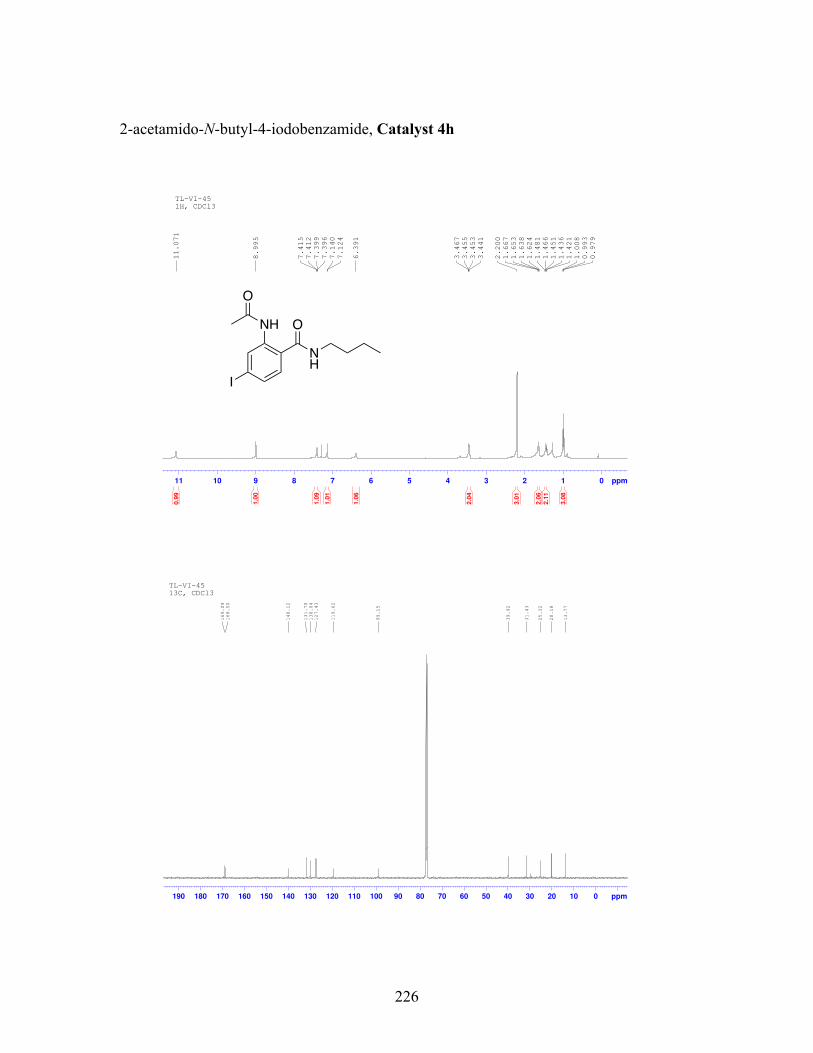
226
2-acetamido-N-butyl-4-iodobenzamide, Catalyst 4h
11 10 9 8 7 6 5 4 3 2 1 0 ppm
0.979
0.993
1.008
1.421
1.436
1.451
1.466
1.481
1.624
1.638
1.653
1.667
2.200
3.441
3.453
3.455
3.467
6.391
7.124
7.140
7.396
7.399
7.412
7.415
8.995
11.071
3.08
2.11
2.06
3.01
2.04
1.06
1.01
1.09
1.00
0.99
1H, CDCl3TL−VI−45�
NH
NH
O
I
O
190 180 170 160 150 140 130 120 110 100 90 80 70 60 50 40 30 20 10 0 ppm
13.77
20.18
25.32
31.43
39.92
99.15
119.62
127.43
130.04
131.70
140.12
168.50
169.09
13C, CDCl3�TL−VI−45�
227
7r
190 180 170 160 150 140 130 120 110 100 90 80 70 60 50 40 30 20 10 0 ppm
37.44
63.05
127.31
128.71
129.29
136.02
174.33
13C, CDCl3�TL−VI−49�
EtOAc
EtOAc
EtOAc
EtOAc
12 11 10 9 8 7 6 5 4 3 2 1 0 ppm
3.053
3.064
3.242
3.253
7.306
7.346
7.359
11.411
0.87
0.86
4.11
1.00
1H, CDCl3TL−VI−49�
EtOAc
EtOAc
EtOAc
OH
O
N3
228
7u
8.5 8.0 7.5 7.0 6.5 6.0 5.5 5.0 4.5 4.0 3.5 3.0 2.5 2.0 1.5 1.0 0.5 0.0 ppm
3.285
3.289
3.295
3.848
3.909
3.914
3.926
4.246
6.592
6.596
6.608
6.613
6.662
6.667
7.297
7.314
1.09
3.00
2.06
3.03
2.04
1.09
1.15
1.16
1H, MeODTL−IV−13�
H2O
190 180 170 160 150 140 130 120 110 100 90 80 70 60 50 40 30 20 10 0 ppm
35.731
44.669
55.819
57.201
71.851
78.710
97.526
103.933
108.615
132.191
157.832
161.141
13C, CDCl3/MeODTL−VI−13�
CDCl3
MeOD
OMeMeO
HN
Cl
229
8k
OH
O
N3
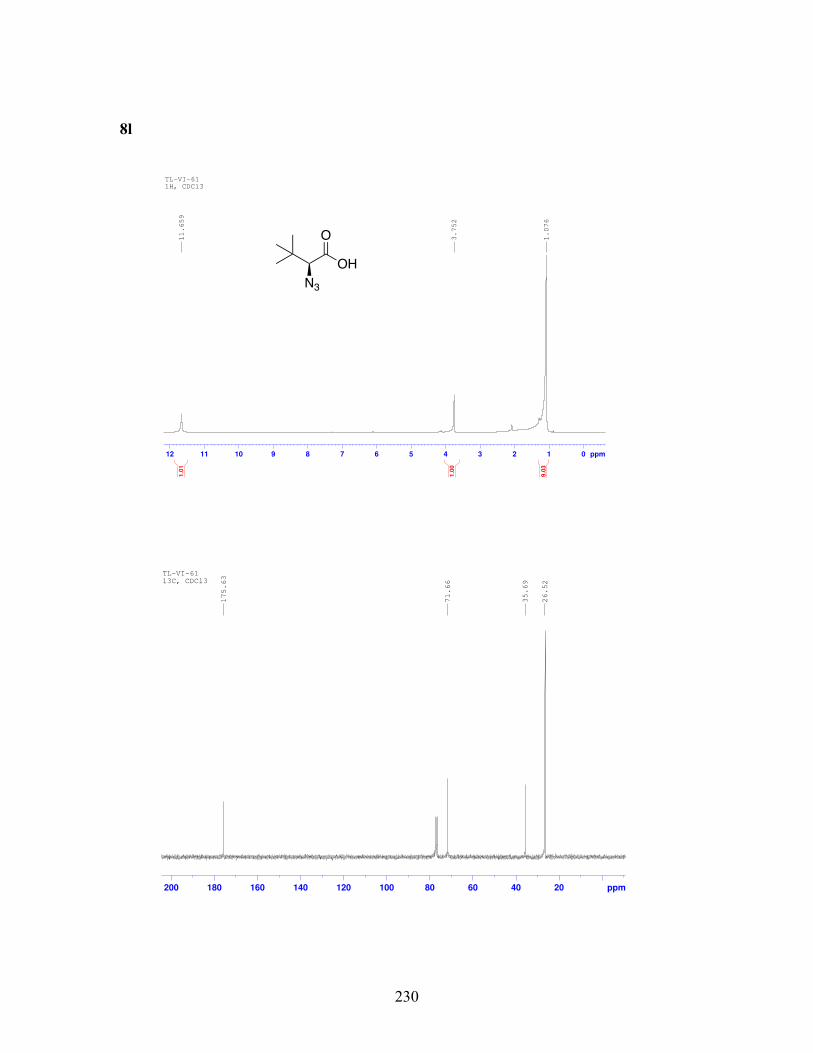
230
8l
12 11 10 9 8 7 6 5 4 3 2 1 0 ppm
1.076
3.752
11.659
9.03
1.00
1.01
1H, CDCl3�TL−VI−61�
200 180 160 140 120 100 80 60 40 20 ppm
26.52
35.69
71.66
175.6313C, CDCl3�
TL−VI−61�
OH
O
N3

231
3.9 REFERENCES
217. Ghosh, S.; Pradhan, S.; Chatterjee, I., A survey of chiral hypervalent iodine reagents in asymmetric synthesis. Beilstein J. Org. Chem. 2018, 14, 1244-1262. 218. Berthiol, F., Reagent and Catalyst Design for Asymmetric Hypervalent Iodine Oxidations. Synthesis-Stuttgart 2015, 47, 587-603. 219. Parra, A.; Reboredo, S., Chiral Hypervalent Iodine Reagents: Synthesis and Reactivity. Chem.: Eur. J 2013, 19, 17244-17260. 220. Claraz, A.; Masson, G., Asymmetric iodine catalysis-mediated enantioselective oxidative transformations. Org. Bio. Chem. 2018, 16, 5386-5402. 221. Hirt, U. H.; Spingler, B.; Wirth, T., New chiral hypervalent iodine compounds in asymmetric synthesis. J. Org. Chem. 1998, 63, 7674-7679. 222. Gonzalez, D. F.; Benfatti, F.; Waser, J., Asymmetric Organocatalysis Meets Hypervalent Iodine Chemistry for the alpha-Functionalization of Carbonyl Compounds. Chemcatchem 2012, 4, 955-958. 223. Flores, A.; Cots, E.; Berges, J.; Muniz, K., Enantioselective Iodine(I/III) Catalysis in Organic Synthesis. Adv. Synth. Catal 2019, 361, 2-25. 224. Merritt, E. A.; Olofsson, B., alpha-Functionalization of Carbonyl Compounds Using Hypervalent Iodine Reagents. Synthesis-Stuttgart 2011, 517-538. 225. Yoshimura, A.; Zhdankin, V. V., Advances in Synthetic Applications of Hypervalent Iodine Compounds. Chem. Rev. 2016, 116, 3328-3435. 226. Dong, D. Q.; Hao, S. H.; Wang, Z. L.; Chen, C., Hypervalent iodine: a powerful electrophile for asymmetric alpha-functionalization of carbonyl compounds. Org. Bio. Chem. 2014, 12, 4278-4289. 227. Wirth, T., IBX - New reactions with an old reagent. Angew. Chem. Int. Ed. 2001, 40, 2812-2814. 228. Zhdankin, V. V., Hypervalent iodine(III) reagents in organic synthesis. Arkivoc 2009, 1-62. 229. Brown, M.; Kumar, R.; Rehbein, J.; Wirth, T., Enantioselective Oxidative Rearrangements with Chiral Hypervalent Iodine Reagents. Chem.: Eur. J 2016, 22, 4030-4035. 230. Farid, U.; Malmedy, F.; Claveau, R.; Albers, L.; Wirth, T., Stereoselective Rearrangements with Chiral Hypervalent Iodine Reagents. Angew. Chem. Int. Ed. 2013, 52, 7018-7022. 231. Li, X.; Chen, P. H.; Liu, G. S., Recent advances in hypervalent iodine(III)-catalyzed functionalization of alkenes. Beilstein J. Org. Chem. 2018, 14, 1813-1825. 232. Fujita, M.; Ookubo, Y.; Sugimura, T., Asymmetric cycloetherification based on a chiral auxiliary for 4-acyloxy-1-butene substrates during oxidation with iodosylbenzene via a 1,3-dioxan-2-yl cation. Tetrahedron Lett. 2009, 50, 1298-1300. 233. Fujita, M., Mechanistic aspects of alkene oxidation using chiral hypervalent iodine reagents. Tetrahedron Lett. 2017, 58, 4409-4419.

232
234. Mu, R. H.; Liu, Z. Q.; Yang, Z. J.; Liu, Z. G.; Wu, L. M.; Liu, Z. L., An efficient catalytic aerobic oxidation of alcohols in water using hypervalent iodine(V). Adv. Synth. Catal. 2005, 347, 1333-1336. 235. Herrerias, C. I.; Zhang, T. Y.; Li, C. J., Catalytic oxidations of alcohols to carbonyl compounds by oxygen under solvent-free and transition-metal-free conditions. Tetrahedron Lett. 2006, 47, 13-17. 236. Richardson, R. D.; Wirth, T., Hypervalent iodine goes catalytic. Angew. Chem. Int. Ed. 2006, 45, 4402-4404. 237. Thayer, A. M., Catalyst Suppliers Face Changing Industry. Chem. Eng. News 1992, 70, 27-49. 238. Han, J. L.; Soloshonok, V. A.; Klika, K. D.; Drabowicz, J.; Wzorek, A., Chiral sulfoxides: advances in asymmetric synthesis and problems with the accurate determination of the stereochemical outcome. Chem. Soc. Rev. 2018, 47, 1307-1350. 239. Mellah, M.; Voituriez, A.; Schulz, E., Chiral sulfur ligands for asymmetric catalysis. Chem. Rev. 2007, 107, 5133-5209. 240. Bentley, R., Role of sulfur chirality in the chemical processes of biology. Chem. Soc. Rev. 2005, 34, 609-624. 241. Pribram, R., Justus Liebigs Ann. Chem. 1907, 351, 481-485. 242. Amey, R. L. M., J. C., Synthesis and reaction of substituted arylalkoxyiodinanes: formation of stable bromoarylalkoxy and aryldialkoxy heterocyclic derivatives of tricoordinate organoiodine(III). J. Org. Chem. 1979, 44, 1779-1784. 243. Imamoto, T.; Koto, H., Asymmetric oxidation of sulfides to sulfoxides with trivalent iodine reagents. Chem. Lett. 1986, 967-968. 244. Ray, D. G.; Koser, G. F., Iodinanes with chiral ligands. Synthesis and structure of iodine(III) dibenzoyl tartrates. J. Org. Chem. 1992, 57, 1607-1610. 245. Hatzigrigoriou, E.; Varvoglis, A.; Bakola-Christianopoulou, M., Preparation of [Hydroxy(((+)-10-camphorsulfonyl)oxy)iodo]benzene and Its Reactivity toward Carbonyl Compounds. J. Org. Chem. 1990, 55, 315-318. 246. Min, X.; Chen, Z. C., Hypervalent iodine in synthesis .24. A facile method for the preparation of arylsulfinic esters: Oxidation of disulfides or thiophenols by phenyliodine(III) bis(trifluoroacetate) in the presence of alcohols. Synth. Commun. 1997, 27, 1321-1326. 247. Ray, D. G.; Koser, G. F., Iodinanes with iodine(III)-bound homochiral alkoxy ligands: preparation and utility for the synthesis of alkoxysulfonium salts and chiral sulfoxides. J. Am. Chem. Soc. 1990, 112, 5672-5673. 248. Roche, S. P.; Porco, J. A., Dearomatization Strategies in the Synthesis of Complex Natural Products. Angew. Chem. Int. Ed. 2011, 50, 4068-4093. 249. Pape, A. R.; Kaliappan, K. P.; Kundig, E. P., Transition-metal-mediated dearomatization reactions. Chem. Rev. 2000, 100, 2917-2940. 250. Ortiz, F. L.; Iglesias, M. J.; Fernandez, I.; Sanchez, C. M. A.; Gomez, G. R., Nucleophilic dearomatizing (DNAr) reactions of aromatic C,H-Systems. A mature paradigm in organic synthesis. Chem. Rev. 2007, 107, 1580-1691. 251. Zhuo, C. X.; Zhang, W.; You, S. L., Catalytic Asymmetric Dearomatization Reactions. Angew. Chem. Int. Ed. 2012, 51, 12662-12686.

233
252. Siegel, A.; Antony, F., Monatsh. Chem. 1955, 86, 292-300. 253. Dohi, T.; Maruyama, A.; Takenaga, N.; Senami, K.; Minamitsuji, Y.; Fujioka, H.; Caemmerer, S. B.; Kita, Y., A chiral hypervalent iodine(III) reagent for enantioselective dearomatization of phenols. Angew Chem. Int. Ed. 2008, 47, 3787-3790. 254. Swenton, J. S.; Callinan, A.; Chen, Y.; Rohde, J. J.; Kerns, M. L.; Morrow, G. W., Intramolecular anodic carbon-carbon bond formation from oxidized phenol intermediates. Effect of oxygenated substituents on the yields of spiro dienones in electrochemical and iodobenzene diacetate oxidations. J.Org. Chem. 1996, 61, 1267-1274. 255. Kita, Y.; Arisawa, M.; Gyoten, M.; Nakajima, M.; Hamada, R.; Tohma, H.; Takada, T., Oxidative intramolecular phenolic coupling reaction induced by a hypervalent iodine(III) reagent: Leading to galanthamine-type amaryllidaceae alkaloids. J. Org. Chem. 1998, 63, 6625-6633. 256. Pelter, A.; Hussain, A.; Smith, G.; Ward, R. S., The synthesis of 8a-methoxy-2H,6H-chromen-6-ones and corresponding 2H-chromenes by a unique process utilising phenolic oxidation. Tetrahedron 1997, 53, 3879-3916. 257. Dohi, T.; Ito, M.; Yamaoka, N.; Morimoto, K.; Fujioka, H.; Kita, Y., Hypervalent iodine(III): selective and efficient single-electron-transfer (SET) oxidizing agent. Tetrahedron 2009, 65, 10797-10815. 258. Tohma, H.; Morioka, H.; Takizawa, S.; Arisawa, M.; Kita, Y., Efficient oxidative biaryl coupling reaction of phenol ether derivatives using hypervalent iodine(III) reagents. Tetrahedron 2001, 57, 345-352. 259. Kita, Y.; Tohma, H.; Hatanaka, K.; Takada, T.; Fujita, S.; Mitoh, S.; Sakurai, H.; Oka, S., Hypervalent Iodine-Induced Nucleophilic Substitution of para-Substituted Phenol Ethers. Generation of Cation Radicals as Reactive Intermediates. J. Am. Chem. Soc. 1994, 116, 3684-3691. 260. Harned, A. M., Asymmetric oxidative dearomatizations promoted by hypervalent iodine(III) reagents: an opportunity for rational catalyst design? Tetrahedron Lett. 2014, 55, 4681-4689. 261. Dohi, T.; Kita, Y.; Maruyama, A. 2009. 262. Dohi, T.; Maruyama, A.; Yoshimura, M.; Morimoto, K.; Tohma, H.; Kita, Y., Versatile hypervalent-iodine(III)-catalyzed oxidations with m-chloroperbenzoic acid as a cooxidant. Angew. Chem. Int. Ed. 2005, 44, 6193-6196. 263. Dohi, T.; Takenaga, N.; Nakae, T.; Toyoda, Y.; Yamasaki, M.; Shiro, M.; Fujioka, H.; Maruyama, A.; Kita, Y., Asymmetric Dearomatizing Spirolactonization of Naphthols Catalyzed by Spirobiindane-Based Chiral Hypervalent Iodine Species. J.Am. Chem.Soc. 2013, 135, 4558-4566. 264. Uyanik, M.; Yasui, T.; Ishihara, K., Chiral hypervalent iodine-catalyzed enantioselective oxidative Kita spirolactonization of 1-naphthol derivatives and one-pot diastereo-selective oxidation to epoxyspirolactones. Tetrahedron 2010, 66, 5841-5851. 265. Uyanik, M.; Yasui, T.; Ishihara, K., Enantioselective Kita Oxidative Spirolactonization Catalyzed by In Situ Generated Chiral Hypervalent Iodine(III) Species. Angew. Chem. Int. Ed. 2010, 49, 2175-2177.

234
266. Quideau, S.; Lyvinec, G.; Marguerit, M.; Bathany, K.; Ozanne-Beaudenon, A.; Buffeteau, T.; Cavagnat, D.; Chenede, A., Asymmetric Hydroxylative Phenol Dearomatization through In Situ Generation of Iodanes from Chiral Iodoarenes and m-CPBA. Angew. Chem. Int. Ed. 2009, 48, 4605-4609. 267. Volp, K. A.; Harned, A. M., Chiral aryl iodide catalysts for the enantioselective synthesis of para-quinols. ChemComm. 2013, 49, 3001-3003. 268. Mizar, P.; Wirth, T., Flexible Stereoselective Functionalizations of Ketones through Umpolung with Hypervalent Iodine Reagents. Angew. Chem. Int. Ed. 2014, 53, 5993-5997. 269. Miyata, O.; Miyoshi, T.; Ueda, M., Umpolung reactions at the alpha-carbon position of carbonyl compounds. Arkivoc 2013, 60-81. 270. Ochiai, M.; Kitagawa, Y.; Takayama, N.; Takaoka, Y.; Shiro, M., Synthesis of chiral diaryliodonium salts, 1,1 '-binaphthyl-2-yl(phenyl)iodonium tetrafluoroborates: Asymmetric alpha-phenylation of beta-keto ester enolates. J. Am. Chem. Soc. 1999, 121, 9233-9234. 271. Jalalian, N.; Olofsson, B., Design and asymmetric synthesis of chiral diaryliodonium salts. Tetrahedron 2010, 66, 5793-5800. 272. Brown, M.; Delorme, M.; Malmedy, F.; Malmgren, J.; Olofsson, B.; Wirth, T., Synthesis of New Chiral Diaryliodonium Salts. Synlett 2015, 26, 1573-1577. 273. Uyanik, M.; Okamoto, H.; Yasui, T.; Ishihara, K., Quaternary Ammonium (Hypo)iodite Catalysis for Enantioselective Oxidative Cycloetherification. Science 2010, 328, 1376-1379. 274. Ochiai, M.; Takeuchi, Y.; Katayama, T.; Sueda, T.; Miyamoto, K., Iodobenzene-catalyzed alpha-acetoxylation of ketones. In situ generation of hypervalent (diacyloxyiodo)benzenes using m-chloroperbenzoic acid. J. Am. Chem. Soc. 2005, 127, 12244-12245. 275. Dohi, T.; Kita, Y., Kagaku 2006, 61, 68 – 69. 276. Yamamoto, Y.; Togo, H., PhI-catalyzed alpha-tosyloxylation of ketones with m-chloroperbenzoic acid and p-toluenesulfonic acid. Synlett 2006, 798-800. 277. Guilbault, A. A.; Basdevant, B.; Wanie, V.; Legault, C. Y., Catalytic Enantioselective alpha-Tosyloxylation of Ketones Using Iodoaryloxazoline Catalysts: Insights on the Stereoinduction Process. J. Org. Chem. 2012, 77, 11283-11295. 278. Guilbault, A. A.; Legault, C. Y., Drastic Enhancement of Activity in Iodane-Based alpha-Tosyloxylation of Ketones: Iodine(III) Does the Hypervalent Twist. ACS Catalysis 2012, 2, 219-222. 279. Nieland, O.; Karele, B., J. Org. Chem. USSR (Engl. Transl.) 1970, 6, 889. 280. Uyanik, M.; Yasui, T.; Ishihara, K., Hypervalent iodine-catalyzed oxylactonization of ketocarboxylic acids to ketolactones. Bioorg. Med. Chem. Lett. 2009, 19, 3848-3851. 281. Mizukami, F.; Ando, M.; Tanaka, T.; Imamura, J., Bull. Chem. Soc. Jap. 1978, 51, 335-336. 282. Beaulieu, S.; Legault, C. Y., Mechanistic Insights on the Iodine(III)-Mediated alpha-Oxidation of Ketones. Chem.: Eur. J. 2015, 21, 11206-11211.

235
283. Moriarty, R. M.; Vaid, R. K., Carbon-Carbon Bond Formation Via Hypervalent Iodine Oxidations. Synthesis-Stuttgart 1990, 431-447. 284. Brown, M.; Farid, U.; Wirth, T., Hypervalent Iodine Reagents as Powerful Electrophiles. Synlett 2013, 24, 424-431. 285. Hirt, U. H.; Schuster, M. F. H.; French, A. N.; Wiest, O. G.; Wirth, T., Chiral hypervalent organo-iodine(III) compounds. European Journal of Organic Chemistry 2001, 1569-1579. 286. Richardson, R. D.; Page, T. K.; Altermann, S.; Paradine, S. M.; French, A. N.; Wirth, T., Enantioselective alpha-oxytosylation of ketones catalysed by iodoarenes. Synlett 2007, 538-542. 287. Altermann, S. M.; Richardson, R. D.; Page, T. K.; Schmidt, R. K.; Holland, E.; Mohammed, U.; Paradine, S. M.; French, A. N.; Richter, C.; Bahar, A. M.; Witulski, B.; Wirth, T., Catalytic Enantioselective alpha-Oxysulfonylation of Ketones Mediated by Iodoarenes. Eur. J. Org. Chem. 2008, 5315-5328. 288. Farooq, U.; Schafer, S.; Shah, A. A.; Freudendahl, D. M.; Wirth, T., Synthesis of New Enantiomerically Pure Organoiodine Catalysts and Their Application in the alpha-Functionalization of Ketones. Synthesis-Stuttgart 2010, 1023-1029. 289. Rodriguez, A.; Moran, W. J., Chiral Aryl Iodide-Catalyzed Enantioselective alpha-Oxidation of Ketones. Synthesis-Stuttgart 2012, 44, 1178-1182. 290. Yu, J.; Cui, J.; Hou, X. S.; Liu, S. S.; Gao, W. C.; Jiang, S.; Tian, J.; Zhang, C., Enantioselective alpha-tosyloxylation of ketones catalyzed by spirobiindane scaffold-based chiral iodoarenes. Tetrahedron-Asymmetry 2011, 22, 2039-2055. 291. Brenet, S.; Berthiol, F.; Einhorn, J., 3,3 '-Diiodo-BINOL-Fused Maleimides as Chiral Hypervalent Iodine(III) Organocatalysts. J. Eur. J. Org. Chem. 2013, 2013, 8094-8096. 292. Basdevant, B.; Legault, C. Y., Enantioselective Iodine(III)-Mediated Synthesis of alpha-Tosyloxy Ketones: Breaking the Selectivity Barrier. Org. Lett. 2015, 17, 4918-4921. 293. Fields, G. B.; Noble, R. L., Solid phase peptide synthesis utilizing 9‐fluorenylmethoxycarbonyl amino acids. Int. J. Pept. Protein Res. 1990, 35, 161-214. 294. Imperiali, B.; Kapoor, T. M., The Reverse Turn as a Template for the Metal Coordination. Tetrahedron 1993, 49, 3501-3510. 295. Aubry, A.; Cung, M. T.; Marraud, M., .beta.I and .beta.II-turn conformations in model dipeptides with the Pro-Xaa sequences. J. Am. Chem. Soc. 1985, 107, 7640-7647. 296. Lex, T. R.; Swasy, M. I.; Whitehead, D. C., Relative Rate Profiles of Functionalized Iodoarene Catalysts for Iodine(III) Oxidations. J. Org. Chem. Soc. 2015, 80, 12234-12243. 297. Su, J. T.; Goddard, W. A., Enhancing 2-iodoxybenzoic acid reactivity by exploiting a hypervalent twist. J. Am. Chem. Soc. 2005, 127, 14146-14147. 298. Rao, Y. S., Recent advances in the chemistry of unsaturated lactones. Chem. Rev. 1976, 76, 625-694. 299. Shiina, I., Total synthesis of natural 8-and 9-membered lactones: Recent advancements in medium-sized ring formation. Chem. Rev. 2007, 107, 239-273.

236
300. Devon, T. K.; Scott, A. I. Handbook of Naturally Occuring Compounds. Academic Press New York, 1975; Vol. 1. 301. Mayr, H.; Ofial, A. R., The reactivity-selectivity principle: an imperishable myth in organic chemistry. Angew. Chem. Int. Ed. 2006, 45, 1844-1854. 302. Lex, T. R.; Swasy, M. I.; Giambalvo, L. N.; Gross, K. G.; McMillen, C. D.; \Whitehead, D. C., Design and Synthesis of Fmoc-SPPS-ready Iodoarene Amino Acids and their Reactivity in the Catalytic α-Oxytosylation of Ketones. (In review) 2019. 303. Urwyler, S.; Floersheim, P.; Roy, B. L.; Koller, M., Drug Design, in Vitro Pharmacology, and Structure-Activity Relationships of 3-Acylamino-2-aminopropionic Acid Derivatives, a Novel Class of Partial Agonists at the Glycine Site on the N-Methyl-D-aspartate (NMDA) Receptor Complex. J. Med. Chem. 2009, 52, 5093-5107. 304. Kretsinger, J. K.; Schneider, J. P., Design and application of basic amino acids displaying enhanced hydrophobicity. J. Am. Chem. Soc. 2003, 125, 7907-7913. 305. Wang, Y.; Bluhm, L. H.; Li, T. Y., Identification of chiral selectors from a 200-member parallel combinatorial library. Anal. Chem. 2000, 72, 5459-5465. 306. Wang, Y.; Li, F.; Han, Y. M.; Wang, F. Y.; Jiang, H., Folding and Aggregation of Cationic Oligo(aryl-triazole)s in Aqueous Solution. Chem.: Eur. J. 2009, 15, 9424-9433. 307. O'Brien, N. J.; Brzozowski, M.; Wilson, D. J. D.; Deady, L. W.; Abbott, B. M., Synthesis and biological evaluation of substituted 2-anilino-7H-pyrrolopyrimidines as PDK1 inhibitors. Tetrahedron 2014, 70, 4947-4956. 308. Cui, L. D.; Dong, Z. L.; Liu, K.; Zhang, C., Design, Synthesis, Structure, and Dehydrogenation Reactivity of a Water-Soluble o-lodoxybenzoic Acid Derivative Bearing a Trimethylammonium Group. Org. Lett. 2011, 13, 6488-6491. 309. Vaidyanathan, G.; Shankar, S.; Zalutsky, M. R., Synthesis of ring- and side-chain-substituted m-iodobenzylguanidine analogues. Bioconjugate Chem. 2001, 12, 786-797. 310. Uyanik, M.; Akakura, M.; Ishihara, K., 2-Iodoxybenzenesulfonic Acid as an Extremely Active Catalyst for the Selective Oxidation of Alcohols to Aldehydes, Ketones, Carboxylic Acids, and Enones with Oxone. J. Am. Chem. Soc. 2009, 131, 251-262. 311. Debets, M. F.; Prins, J. S.; Merkx, D.; van Berkel, S. S.; van Delft, F. L.; van Hest, J. C. M.; Rutjes, F. P. J. T., Synthesis of DIBAC analogues with excellent SPAAC rate constants. Org. Bio. Chem. 2014, 12, 5031-5037. 312. Whyte, A.; Olson, M. E.; Lautens, M., Palladium-Catalyzed, Norbornene-Mediated, ortho-Amination ipso-Amidation: Sequential C-N Bond Formation. Organic Lett. 2018, 20, 345-348. 313. Basdevant, B.; Legault, C. Y., Study of the Reactivity of Hydroxy(tosyloxy)iodo benzene Toward Enol Esters to Access alpha-Tosyloxy Ketones. Journal of Organic Chemistry 2015, 80, 6897-6902. 314. Moriarty, R. M.; Penmasta, R.; Awasthi, A. K.; Epa, W. R.; Prakash, I., Reaction of [hydroxy(tosyloxy)iodo]benzene and [hydroxy(mesyloxy)iodo]benzene with trimethylsilyl enol ethers. A new general method for .alpha.-sulfonyloxylation of carbonyl compounds. J. Org. Chem. 1989, 54, 1101-1104.

237
315. Evans, D. A.; Nelson, J. V.; Vogel, E.; Taber, T. R., Stereoselective Aldol Condensations via Boron Enolates. J. Am. Chem. Soc 1981, 103, 3099-3111. 316. Brown, H. C.; Dhar, R. K.; Bakshi, R. K.; Pandiarajan, P. K.; Singaram, B., Major effect of the leaving group in dialkylboron chlorides and triflates in controlling the stereospecific conversion of ketones into either [E]- or [Z]-enol borinates. J. Am. Chem. Soc. 1989, 111, 3441-3442. 317. Weber, E.; Vögtle, F.; Burrell, A. K., Macrocycles. Springer-Verlag: Berlin ; London, 1992; p 278 p. 318. Mallinson, J.; Collins, I., Macrocycles in new drug discovery. Future Med. Chem 2012, 4, 1409-1438. 319. Zhang, X. X.; Bradshaw, J. S.; Izatt, R. M., Enantiomeric recognition of amine compounds by chiral macrocyclic receptors. Chem. Rev. 1997, 97, 3313-3361. 320. Marsault, E.; Peterson, M. L., Macrocycles Are Great Cycles: Applications, Opportunities, and Challenges of Synthetic Macrocycles in Drug Discovery. J. Med. Chem 2011, 54, 1961-2004. 321. Tejeda, A.; Oliva, A. I.; Simon, L.; Grande, M.; Caballero, M. C.; Moran, J. R., A macrocyclic receptor for the chiral recognition of hydroxycarboxylates. Tetrahedron Lett. 2000, 41, 4563-4566. 322. Godoy-Alcantar, C.; Nelen, M. I.; Eliseev, A. V.; Yatsimirsky, A. K., Molecular recognition by natural macrocycles. Part II. Esterolytic activity and chiral discrimination of amino acid derivatives by the zwitterionic form of (+)-tubocurarine. J. Chem. Soc., Perkin Trans. 2 1999, 353-361. 323. Gross, Z.; Ini, S., Asymmetric catalysis by a chiral ruthenium porphyrin: Epoxidation, hydroxylation, and partial kinetic resolution of hydrocarbons. Org. Lett. 1999, 1, 2077-2080. 324. Collman, J. P.; Wang, Z.; Straumanis, A.; Quelquejeu, M.; Rose, E., An efficient catalyst for asymmetric epoxidation of terminal olefins. J. Am. Chem. Soc, 1999, 121, 460-461. 325. De Rosa, M.; La Manna, P.; Talotta, C.; Soriente, A.; Gaeta, C.; Neri, P., Supramolecular Organocatalysis in Water Mediated by Macrocyclic Compounds. Front. Chem . 2018, 6. 326. Ema, T.; Yokoyama, M.; Watanabe, S.; Sasaki, S.; Ota, H.; Takaishi, K., Chiral Macrocyclic Organocatalysts for Kinetic Resolution of Disubstituted Epoxides with Carbon Dioxide. Org. Lett. 2017, 19, 4070-4073. 327. Angell, Y.; Burgess, K., Ring closure to beta-turn mimics via copper-catalyzed azide/alkyne cycloadditions. J. Org. Chem. 2005, 70, 9595-9598. 328. Still, W. C.; Galynker, I., Chemical Consequences of Conformation in Macrocyclic Compounds. Tetrahedron 1981, 37, 3981-3996. 329. van Maarseveen, J. H.; Horne, W. S.; Ghadiri, M. R., Efficient route to C-2 symmetric heterocyclic backbone modified cyclic peptides. Org. Lett. 2005, 7, 4503-4506. 330. Lundquist, J. T.; Pelletier, J. C., Improved solid-phase peptide synthesis method utilizing alpha-azide-protected amino acids. Org. Lett. 2001, 3, 781-783.

238
331. Paul, A.; Bittermann, H.; Gmeiner, P., Triazolopeptides: chirospecific synthesis and cis/trans prolyl ratios of structural isomers. Tetrahedron 2006, 62, 8919-8927. 332. Pasini, D., The Click Reaction as an Efficient Tool for the Construction of Macrocyclic Structures. Molecules 2013, 18, 9512-9530. 333. Barlow, T. M. A.; Tourwe, D.; Ballet, S., Cyclisation To Form Small, Medium and Large Rings by Use of Catalysed and Uncatalysed Azide-Alkyne Cycloadditions (AACs). Eur. J. Org. Chem. 2017, 4678-4694. 334. Haldon, E.; Nicasio, M. C.; Perez, P. J., Copper-catalysed azide-alkyne cycloadditions (CuAAC): an update. Org. Bio. Chem. 2015, 13, 9528-9550. 335. Chandrasekaran, S., Click reactions in organic synthesis. Wiley-VCH,: Weinheim, 2016; p 1 online resource. 336. Kolb, H. C.; Finn, M. G.; Sharpless, K. B., Click chemistry: Diverse chemical function from a few good reactions. Angew. Chem. Int. Ed. 2001, 40, 2004-+. 337. Wang, T.; Yuan, L.; Zhao, Z. G.; Shao, A. L.; Gao, M.; Huang, Y. F.; Xiong, F.; Zhang, H. L.; Zhao, J. F., Direct oxidative amidation between methylarenes and amines in water. Green Chemistry 2015, 17, 2741-2744. 338. John, O. R. S.; Killeen, N. M.; Knowles, D. A.; Yau, S. C.; Bagley, M. C.; Tomkinson, N. C. O., Direct alpha-oxytosylation of carbonyl compounds: One-pot synthesis of heterocycles. Org. Lett. 2007, 9, 4009-4012. 339. Schroder, N.; Wencel-Delord, J.; Glorius, F., High-Yielding, Versatile, and Practical [Rh(III)Cp*]-Catalyzed Ortho Bromination and Iodination of Arenes. J. Am. Chem. Soc. 2012, 134, 8298-8301. 340. Wang, L.; Zhong, C.; Xue, P.; Fu, E. Q., Fluorescent beta-Cyclodextrins Modified by Isomeric Aminobenzamides: Synthesis, Conformational Analysis, and Fluorescent Behaviors. J. Org. Chem. 2011, 76, 4874-4883. 341. Madich, Y.; Denis, J. G.; Ortega, A.; Martinez, C.; Matrane, A.; Belachemi, L.; de Lera, A. R.; Alvarez, R.; Aurrecoechea, J. M., A Practical Protocol for Three-Component, One-Pot, Stepwise Sonogashira-Heterocyclization-Heck Couplings. Synthesis 2013, 45, 2009-2017. 342. Alper, P. B.; Hung, S. C.; Wong, C. H., Metal catalyzed diazo transfer for the synthesis of azides from amines. Tetrahedron Lett. 1996, 37, 6029-6032.

239
CHAPTER FOUR
IODOARENE-CONTAINING BIODEGRADABLE X-RAY MATERIALS
4.1 INTRODUCTION
Synthetic materials exhibiting contrast imaging properties have become vital to
the field of biomedical imaging. However, polymeric biomaterials typically lack imaging
contrast properties for deep tissue imaging. This chapter details the synthesis and
characterization of a suite of aryl-iodo monomers, which were used to prepare iodinated
polyesters using a pre-functionalization approach. Commercially available 4-iodo-
phenylalanine or 4-iodobenzyl bromide served as the starting materials for the synthesis
of three iodinated monomeric moieties (a modified lactide, morpholine-2,5-dione, and
caprolactone), which under a tin-mediated ring-opening polymerization (ROP), generated
their respective polyesters (PE) or poly(ester amides) (PEA). An increase in X-ray
intensity of all synthesized iodine-containing polymers, in comparison to noniodinated
poly(lactic acid) (PLA), validated their functionality as radio-opaque materials. The
iodinated-poly(lactic acid) (iPLA) material was visualized through varying thicknesses of
chicken tissue, thus demonstrating its potential as a radio-opaque biomaterial (Figure
4.1).
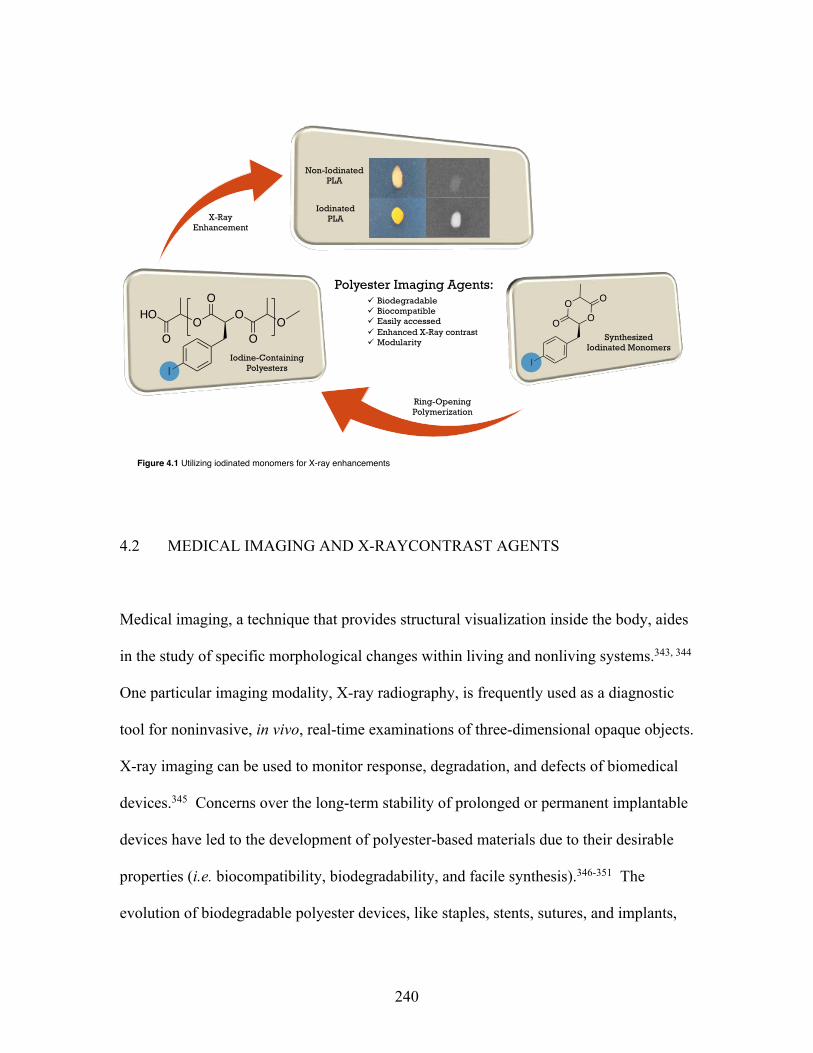
240
4.2 MEDICAL IMAGING AND X-RAYCONTRAST AGENTS
Medical imaging, a technique that provides structural visualization inside the body, aides
in the study of specific morphological changes within living and nonliving systems.343, 344
One particular imaging modality, X-ray radiography, is frequently used as a diagnostic
tool for noninvasive, in vivo, real-time examinations of three-dimensional opaque objects.
X-ray imaging can be used to monitor response, degradation, and defects of biomedical
devices.345 Concerns over the long-term stability of prolonged or permanent implantable
devices have led to the development of polyester-based materials due to their desirable
properties (i.e. biocompatibility, biodegradability, and facile synthesis).346-351 The
evolution of biodegradable polyester devices, like staples, stents, sutures, and implants,
HO O O O
O
O
I
O
PLA
iP
CL6
1
2 2
4PL
A
iPCL
6
12
24
Iodinated PLA
Non-Iodinated PLA
Ring-Opening Polymerization
X-Ray Enhancement
Synthesized Iodinated Monomers
Iodine-Containing Polyesters
ü Biodegradableü Biocompatibleü Easily accessedü Enhanced X-Ray contrastü Modularity
Polyester Imaging Agents:O
O
O
O
I
Figure 4.1 Utilizing iodinated monomers for X-ray enhancements

241
have had a significant impact on the biomedical field.346, 352, 353 Perhaps the most
noteworthy advantage is their ability to be degraded and excreted from the body,
obviating the need for their removal or surgical revision. This can be vital in major
surgical procedures such as fracture fixation, spinal fixation, and abdominal wall
repair.346, 353 While commercially available polyester devices have seen a considerable
amount of use in the biomedical field, their in vivo performance can be difficult to predict
and evaluate due to the complex biological environment associated with tissues.353, 354
Therefore, the real-time monitoring of these devices is critical in order to understand their
performance and fate in the body. The major drawback of polyester-based devices is that
they lack inherent contrast imaging properties, making it difficult to visualize the area of
interest. Imaging techniques are useful only when the intensity of a signal is sufficient
enough to distinguish the target from surrounding tissues or materials. This issue
becomes even more evident when imaging materials through deep tissue or when
monitoring minor defects in biomaterials.355 Recent advancements have addressed such
problems through the improvement of contrast-enhancing agents, which can enrich the
quality of images.346 Iodine-containing small organic compounds remain at the forefront
as contrast media due to the ability of heavy iodine atoms to highly absorb X-rays. With
this overall strategy in mind, Van Horn, Whitehead, and Alexis have generated
degradable iodine-bearing polyesters by utilizing conjugation strategies356 and oxime
“Click” ligation reactions.357 More recently, this group has conducted a post-
polymerization modification reaction between poly(e-caprolactones) and iodinated
hydroxylamines.358 The modified poly(e-caprolactones) displayed an increase in X-ray

242
contrast when compared to previously reported monoiodinated materials. Our strategy
builds upon these reports, in which we explored the ring-opening polymerization of
mono-iodinated monomers, omitting the need for post-polymerization modification.
Further, in contrast to the previous studies, the strategy employed herein hinges on the
irreversible incorporation of the iodine-containing motif through the formation of key
carbon-carbon bonds. This report details the design, synthesis, and applicability of
iodine-containing monomers for the synthesis of biocompatible polyesters as X-ray
contrast imaging agents.
4.3 DESIGNING BIODEGRADABLE X-RAY CONTRAST AGENTS
We designed four target monomers each bearing a 4-iodobenzyl moiety to impart X-ray
opacity of the resulting polymer materials. Specifically, we designed lactide 4c,
morpholinediones 4e and 4f, and caprolactone 4j (Scheme 4.1). These monomers were
designed with the twin goals of maintaining high reactivity in the tin-catalyzed
polymerization reaction, but also incorporated the aryl-iodo motif by the formation of a
stable, non-reversible carbon-carbon bond. The syntheses of lactide 4c and
morpholinediones 4e and 4f all commenced with commercially available 4-
iodophenylalanine (4a). Lactide 4c was synthesized in two steps.
Diazotization/hydrolysis of 4a to provide the a-hydroxy carboxylic acid 4b was followed
by acylation/substitution with 2-bromopropionyl chloride, thus resulting in 4-iodo-benzyl
lactide 4c (34–43% over two steps). The methyl substituted morpholinedione 4e (42–48%
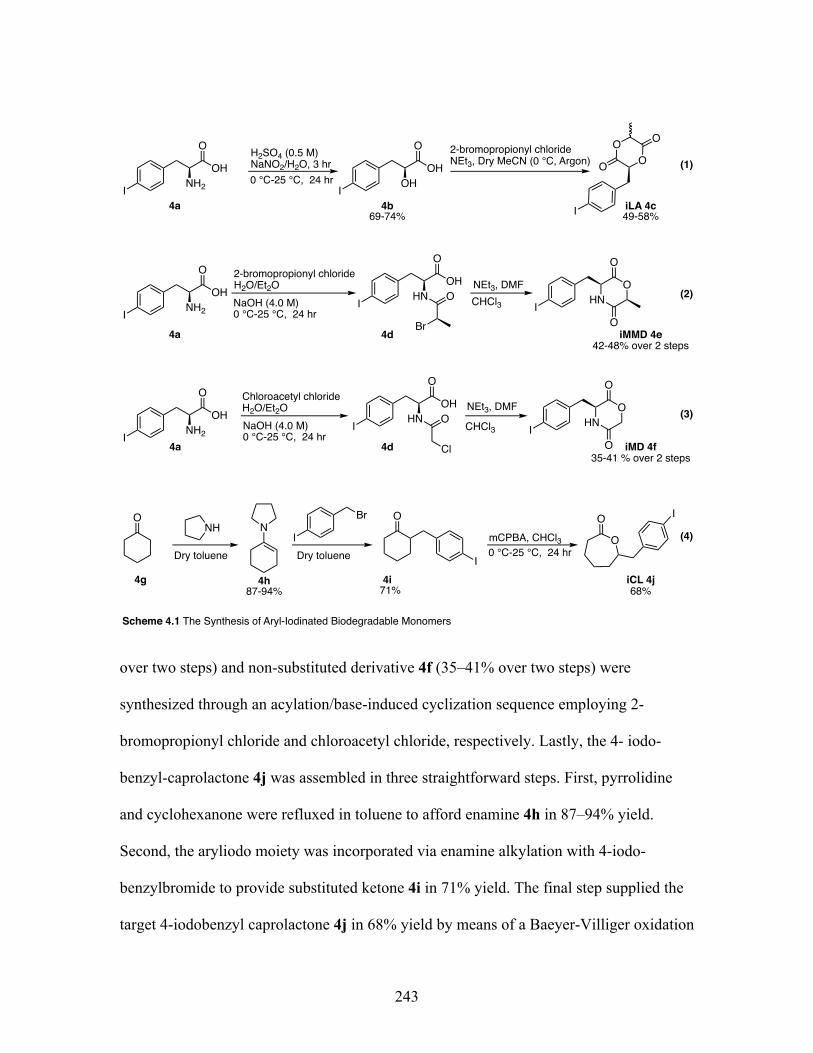
243
over two steps) and non-substituted derivative 4f (35–41% over two steps) were
synthesized through an acylation/base-induced cyclization sequence employing 2-
bromopropionyl chloride and chloroacetyl chloride, respectively. Lastly, the 4- iodo-
benzyl-caprolactone 4j was assembled in three straightforward steps. First, pyrrolidine
and cyclohexanone were refluxed in toluene to afford enamine 4h in 87–94% yield.
Second, the aryliodo moiety was incorporated via enamine alkylation with 4-iodo-
benzylbromide to provide substituted ketone 4i in 71% yield. The final step supplied the
target 4-iodobenzyl caprolactone 4j in 68% yield by means of a Baeyer-Villiger oxidation
I
O
OHNH2 I
O
OHOH
H2SO4 (0.5 M)NaNO2/H2O, 3 hr
2-bromopropionyl chlorideNEt3, Dry MeCN (0 °C, Argon)
OO
O
O
I4b69-74%
iLA 4c49-58%
I
O
OHNH2
I
O
OHHN
2-bromopropionyl chlorideH2O/Et2O NEt3, DMF
4d iMMD 4e42-48% over 2 steps
O
Br
0 °C-25 °C, 24 hr
NaOH (4.0 M)0 °C-25 °C, 24 hr I HN
O
O
O
I
O
OHNH2
I
O
OHHN
Chloroacetyl chlorideH2O/Et2O NEt3, DMF
4d iMD 4f35-41 % over 2 steps
ONaOH (4.0 M)0 °C-25 °C, 24 hr I HN
O
O
OCl
CHCl3
CHCl3
O
Dry toluene
NNH
Dry tolueneI
Br
I
O
mCPBA, CHCl30 °C-25 °C, 24 hr
O
O I
4h87-94%
4i 71%
iCL 4j68%
Scheme 4.1 The Synthesis of Aryl-Iodinated Biodegradable Monomers
(1)
(2)
(3)
(4)
4a
4a
4a
4g

244
with m-chloroperbenzoic acid. The corresponding polyesters were synthesized using
standard ring-opening polymerization techniques (Scheme 4.2).
To target a predictable ratio of copolymers, a conventional thermal ring-opening
polymerization with tin(II) 2-ethylhexanoate was exploited.346, 359 Isolation of the final
products was carried out by means of precipitation in cold methanol followed by
centrifugation at –9 ˚C for 5 min followed by freeze-drying for 3 days. The polymers
were then stored –20 ˚C before use. The resulting iodinated polyesters were characterized
by 1H NMR and FTIR spectroscopy. An illustrative example of the 1H NMR and FTIR
spectra comparing the iodinated and non-iodinated poly(lactic) acid, along with its
monomeric precursor 4c, can be seen in Figure 4.2. The presence of the 4-iodobenzyl
moiety in monomeric aryl-iodo lactide 4c is readily apparent based on the downfield
resonances at 7.2 and 7.7 ppm, which are still present in the polymerized (iPLA) 1H
OO
O
O
I
Sn(II), Lactic Acid
110 °CHO O O O
O
O
I
O
Iodinated Lactide (iLA 4c) Iodinated Polylactide (iPLA)
Scheme 4.2 Tin-mediated ring opening polymerization
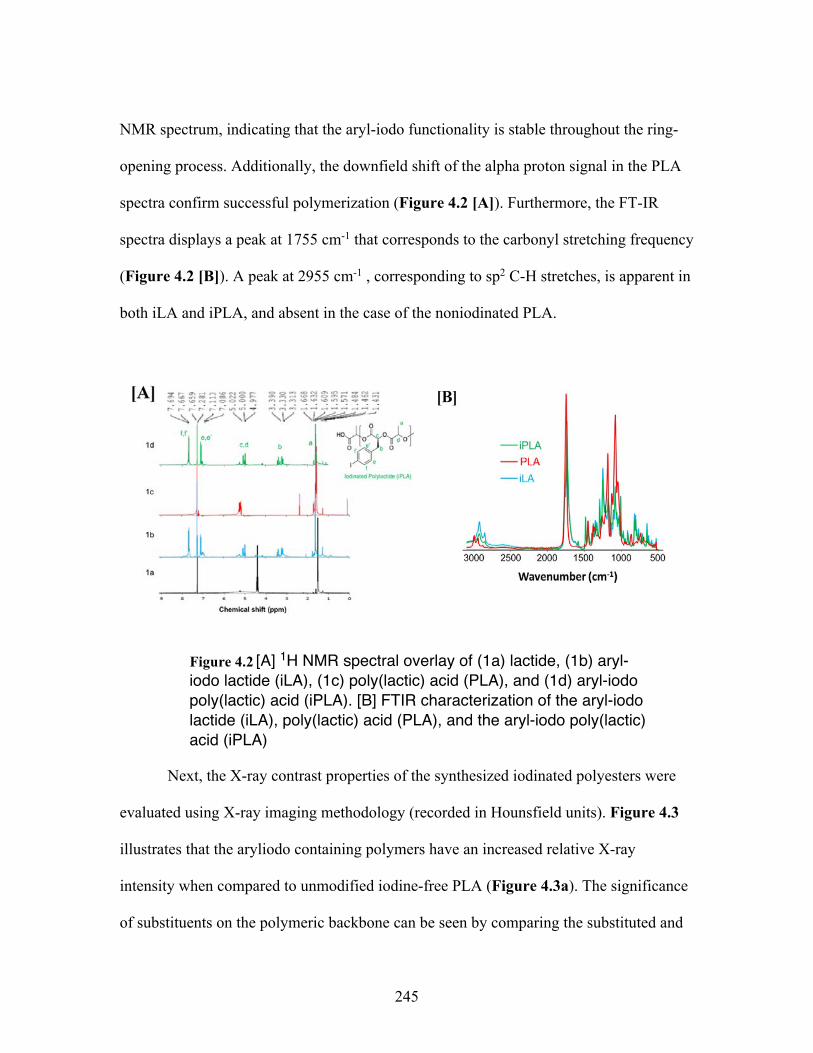
245
NMR spectrum, indicating that the aryl-iodo functionality is stable throughout the ring-
opening process. Additionally, the downfield shift of the alpha proton signal in the PLA
spectra confirm successful polymerization (Figure 4.2 [A]). Furthermore, the FT-IR
spectra displays a peak at 1755 cm-1 that corresponds to the carbonyl stretching frequency
(Figure 4.2 [B]). A peak at 2955 cm-1 , corresponding to sp2 C-H stretches, is apparent in
both iLA and iPLA, and absent in the case of the noniodinated PLA.
Next, the X-ray contrast properties of the synthesized iodinated polyesters were
evaluated using X-ray imaging methodology (recorded in Hounsfield units). Figure 4.3
illustrates that the aryliodo containing polymers have an increased relative X-ray
intensity when compared to unmodified iodine-free PLA (Figure 4.3a). The significance
of substituents on the polymeric backbone can be seen by comparing the substituted and
Figure 1 [A] 1H NMR spectral overlay of (1a) lactide, (1b) aryl-iodo lactide (iLA), (1c) poly(lactic) acid (PLA), and (1d) aryl-iodo poly(lactic) acid (iPLA). [B] FTIR characterization of the aryl-iodo lactide (iLA), poly(lactic) acid (PLA), and the aryl-iodo poly(lactic) acid (iPLA)
Figure 4.2

246
non-substituted morpholinedione polyesters. The polymer of the iodine-containing
modified morpholinedione (iPMMD), which bears an additional methyl-substituent,
contains a lower iodine-weight concentration than the non-substituted iodinated polymer
of morpholinedione (iPMD), and in theory should have a lower X-ray intensity.
However, to our surprise, iPMMD is able to absorb X-rays more efficiently than the non-
methylated analog (iPMD). The iodinated lactide and substituted morpholinedione
polyesters exhibited comparable X-ray intensities, which can be attributed to their similar
chemical structures and molecular weights. The 4-iodobenzylcaprolactone (iCL)
monomer, provided an iodine-containing poly(-e-caprolactone) (iPCL). Unlike
commonly used PLA/PLG polymers, PCL polymers slowly degrade into less acidic, low
molecular weight by-products, preventing the generation of a toxic environment.360
Although the relative X-ray intensity of iPCL was lower than the iPLA and iPMMD
materials, the access to a class of bioresorbable X-ray enhancing polycaprolactones may
be useful for some medical applications.346
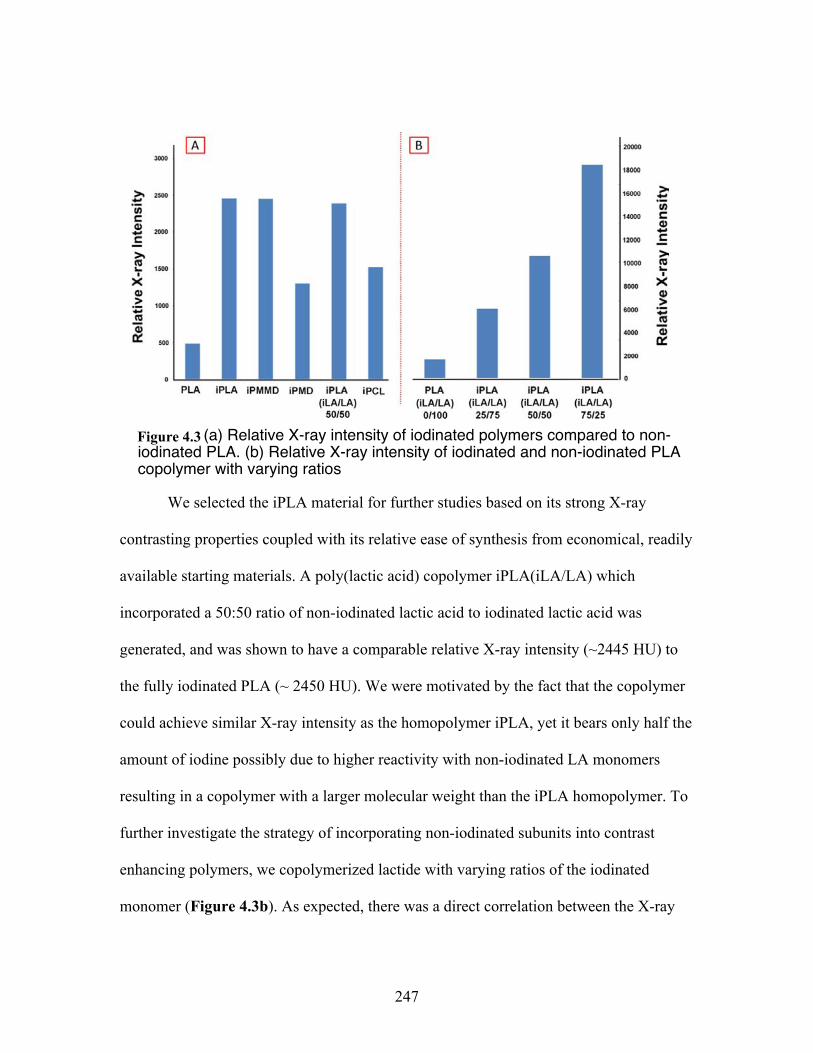
247
We selected the iPLA material for further studies based on its strong X-ray
contrasting properties coupled with its relative ease of synthesis from economical, readily
available starting materials. A poly(lactic acid) copolymer iPLA(iLA/LA) which
incorporated a 50:50 ratio of non-iodinated lactic acid to iodinated lactic acid was
generated, and was shown to have a comparable relative X-ray intensity (~2445 HU) to
the fully iodinated PLA (~ 2450 HU). We were motivated by the fact that the copolymer
could achieve similar X-ray intensity as the homopolymer iPLA, yet it bears only half the
amount of iodine possibly due to higher reactivity with non-iodinated LA monomers
resulting in a copolymer with a larger molecular weight than the iPLA homopolymer. To
further investigate the strategy of incorporating non-iodinated subunits into contrast
enhancing polymers, we copolymerized lactide with varying ratios of the iodinated
monomer (Figure 4.3b). As expected, there was a direct correlation between the X-ray
Figure 2 (a) Relative X-ray intensity of iodinated polymers compared to non-iodinated PLA. (b) Relative X-ray intensity of iodinated and non-iodinated PLA copolymer with varying ratios
Figure 4.3

248
intensity and the amount of iodinated monomer used. The resulting intensity values in
Figure 4.3b are consistent with a gradual increase of iLA monomer in the
copolymerization process of poly(lactic) acid. PLA (100% LA), PLA (iLA: LA; 25:75),
PLA (iLA: LA; 50:50), PLA (iLA: LA; 75:25) showed ~ 1800 HU, ~ 6000 HU, ~ 10700,
~ 18550 HU respectively. To validate the successful incorporation of iLA into the
copolymer, we probed the relative X-ray intensity of the copolymerization at 6, 12, and
24-hour time points (Figure 4.4a).
The increase in X-ray intensity over time confirms the effective incorporation of
iLA into the resulting copolymer. Figure 4.4b depicts the generated polymeric pellets
used in the copolymerization time study, taking the noniodinated PLA with low contrast
as a control showing the gradual increase in the intensity over reaction time. persistent
obstacle frequently encountered in medical imaging is the visualization of biomedical
devices through deep tissue. In order to explore the potential of the synthesized polymeric
materials to help overcome this obstacle, in vitro imaging of iPLA powder was performed
through varying thicknesses (i.e. 0, 2, and 5 cm) of chicken tissue. As seen in Figure
4.4c, effective visualization of the iPLA material was observed through 5 cm-thick
chicken tissue, confirming the potential of the polymeric material for deep tissue imaging
applications.
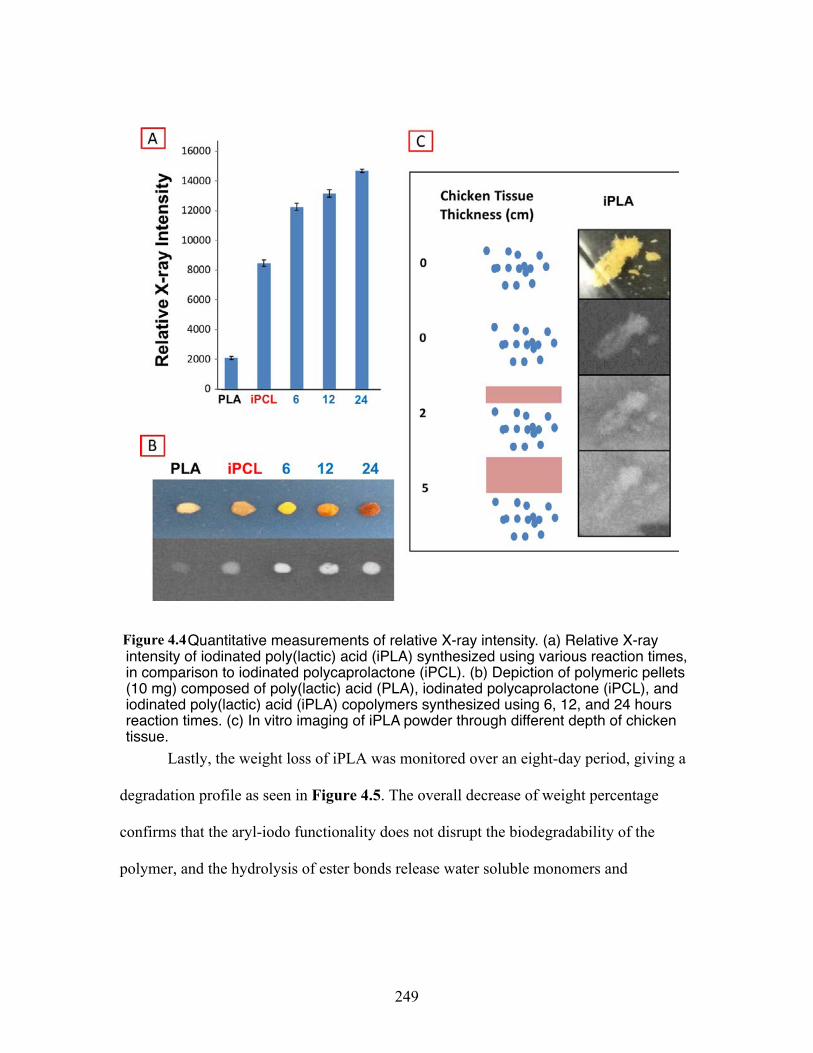
249
Lastly, the weight loss of iPLA was monitored over an eight-day period, giving a
degradation profile as seen in Figure 4.5. The overall decrease of weight percentage
confirms that the aryl-iodo functionality does not disrupt the biodegradability of the
polymer, and the hydrolysis of ester bonds release water soluble monomers and
Figure 3 Quantitative measurements of relative X-ray intensity. (a) Relative X-ray intensity of iodinated poly(lactic) acid (iPLA) synthesized using various reaction times, in comparison to iodinated polycaprolactone (iPCL). (b) Depiction of polymeric pellets (10 mg) composed of poly(lactic) acid (PLA), iodinated polycaprolactone (iPCL), and iodinated poly(lactic) acid (iPLA) copolymers synthesized using 6, 12, and 24 hours reaction times. (c) In vitro imaging of iPLA powder through different depth of chicken tissue.
Figure 4.4
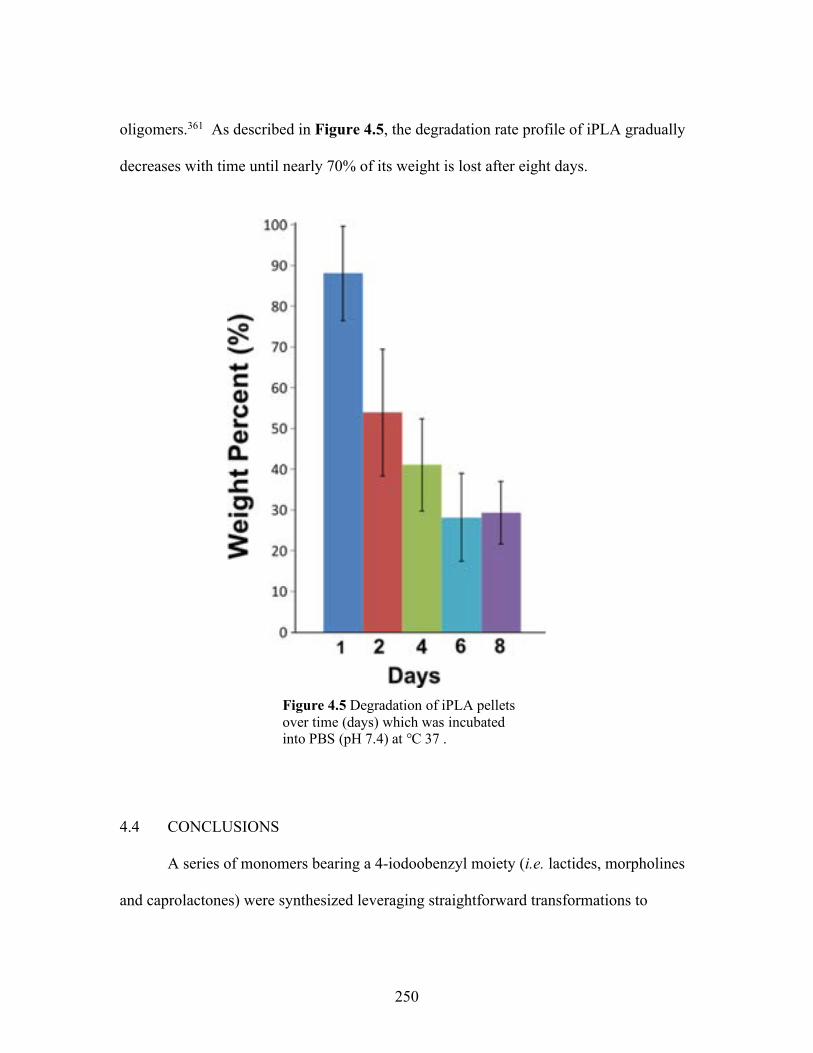
250
oligomers.361 As described in Figure 4.5, the degradation rate profile of iPLA gradually
decreases with time until nearly 70% of its weight is lost after eight days.
4.4 CONCLUSIONS
A series of monomers bearing a 4-iodoobenzyl moiety (i.e. lactides, morpholines
and caprolactones) were synthesized leveraging straightforward transformations to
Figure 4 Degradation of iPLA pellets over time (days) which was incubated into PBS (pH 7.4) at 37 oC
Figure 4.5 Degradation of iPLA pellets over time (days) which was incubated into PBS (pH 7.4) at ℃ 37 .

251
prepare biomaterials with X-ray contrast properties. Unlike post-polymerization
functionalization methodology of biomaterials such as polyesters, it incorporates the
critical iodine atom into each monomeric unit, therefore maximizing iodine content
within the resulting polymer. The modified polyesters were evaluated as viable contrast
X-ray imaging agents, with emphasis on their potential use in deep tissue imaging. The
iodine-containing polyesters that exhibited the highest
X-ray intensities were the modified iodo-morpholinedione polymer (iPMMD) and the
iodopoly(lactic) acid (iPLA). Lastly, we probed the degradation profile of iPLA, and
confirmed that the covalently bound iodine does not perturb the biodegradability of the
polyester. Iodine containing X-ray contrast agents have demonstrated clinical relevance
within the field of medical imaging. Iodinated polymeric contrast-enhancing materials,
being biodegradable, and therefore less toxic, have the potential to further improve
medical imaging capabilities. This work demonstrates a highly concise and easily
modified strategy to synthesize biodegradable and biocompatible X-ray visible materials.
4.5 FUTURE WORK
Another potential application is to use polymers prepared from related functional
monomers as nanoparticle drug carriers.362-365 Polyester-based nanoparticles can serve as
a vehicle for drug delivery as they can supply therapeutic agents in a controlled manner,
and biodegrade from the body after the drug is released.365, 366 Furthermore, polyesters
are tunable scaffolds, in which many properties (i.e. thermal transitions, mechanical
strength, degradability, crystallinity) can be tailored for various applications.366
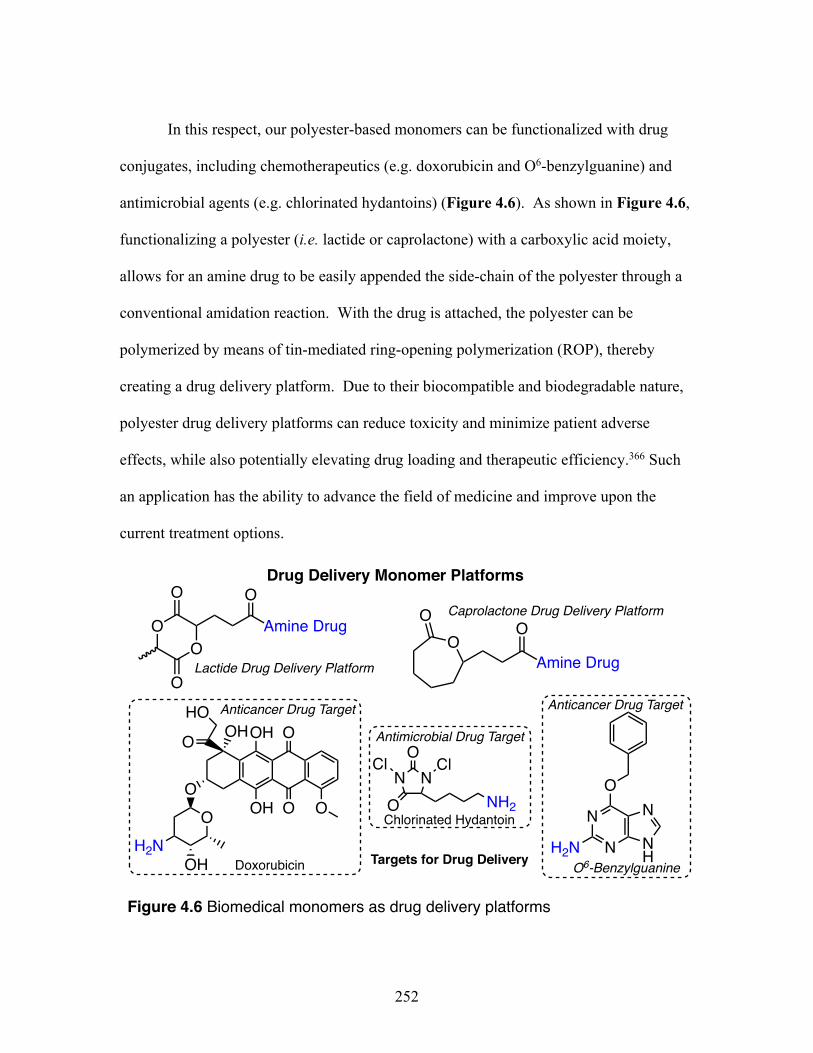
252
In this respect, our polyester-based monomers can be functionalized with drug
conjugates, including chemotherapeutics (e.g. doxorubicin and O6-benzylguanine) and
antimicrobial agents (e.g. chlorinated hydantoins) (Figure 4.6). As shown in Figure 4.6,
functionalizing a polyester (i.e. lactide or caprolactone) with a carboxylic acid moiety,
allows for an amine drug to be easily appended the side-chain of the polyester through a
conventional amidation reaction. With the drug is attached, the polyester can be
polymerized by means of tin-mediated ring-opening polymerization (ROP), thereby
creating a drug delivery platform. Due to their biocompatible and biodegradable nature,
polyester drug delivery platforms can reduce toxicity and minimize patient adverse
effects, while also potentially elevating drug loading and therapeutic efficiency.366 Such
an application has the ability to advance the field of medicine and improve upon the
current treatment options.
OO
Amine Drug
OAmine Drug
O
OO
O
O
NNO
O NH2
Cl
Drug Delivery Monomer Platforms
Figure 4.6 Biomedical monomers as drug delivery platforms
Chlorinated Hydantoin
H2N N
NNH
NO
H2NO
OH
O
O
O
OH
OH
OHO
HO
O
O6-BenzylguanineDoxorubicin
Cl
Anticancer Drug Target Anticancer Drug Target
Antimicrobial Drug Target
Targets for Drug Delivery
Caprolactone Drug Delivery Platform
Lactide Drug Delivery Platform

253
4.6 Experimental Section
Materials: All reagents and chemicals were obtained from commercial sources and used
without further purification unless stated otherwise. Water was purified on a Millipore
Direct-Q S Water Purification System. Acetonitrile was dried by refluxing over
phosphorous pentoxide (P2O5) and distilling under nitrogen prior to use. Lactic acid,
synthesized monomers, and Na2SO4 were vacuum-dried overnight in the reaction vessel
before use. All isolated products were purified by flash column chromatography using
silica gel SDS 60 C.C. 40−63 μm. All known compounds and starting materials had 1H
NMR and 13C NMR spectra consistent with previous literature reports. 1H and 13C NMR
spectra were recorded at ambient temperature on a 500 MHz NMR spectrometer
(Bruker). Proton and carbon chemical shifts were reported in parts per million (ppm)
downfield from tetramethylsilane (TMS) with reference to the deuterated solvent as the
internal standard (i.e., δ 7.26 ppm for 1H NMR, 77 ppm for 13C NMR in CDCl3). Data are
presented as follows: chemical shift, integration, multiplicity (s = singlet, d = doublet, t =
triplet, q = quartet, br = broad, m = multiplet), and coupling constants (J, in Hertz).
Infrared (IR) spectra, reported in cm−1, were collected using a Shimadzu IRAffinity-1S
Fourier transform spectrophotometer. Bands are characterized as strong (s), medium (m),
weak (w), and broad (br). Melting points were recorded on a DigiMelt MPA160
apparatus.
A Tingle 325MVET X-ray machine was used to perform the X-ray imaging (51 kVp,
300 mA, and 5 millisecond exposure time). Due to its lack of radio-opaque properties and
frequent use in biomedical applications, PLA was chosen as the control. All X-ray

254
images were processed, and image intensities quantified using ImageJ (NIH) normalized
to unmodified polymer.
Synthesis:
α-hydroxy-4-iodo-benzenepropionic acid (2); Typical Procedure[19]
To a flame dried round bottom flask equipped with a stir bar was added 4-Iodo-L-
phenylalanine (17.2 mmol, 5.0 g, 1.0 equiv) and 0.5 M H2SO4 (34.4 mmol, 2.0 equiv).
The reaction was stirred for 10 minutes or until the solution was homogeneous. The
solution was cooled to 0 ºC and a solution of sodium nitrite (7.1 g, 103 mmol, 6.0 equiv)
in H2O (50 mL) was added dropwise. The reaction was stirred at 0 ºC for an additional 4
h, and then allowed to warm to room temperature. The resulting solution was stirred at
room temperature for 24 hours (monitored completion by TLC). The reaction mixture
was extracted with diethyl ether (3 x 50 mL), and the organic phases were combined and
washed with brine (50 mL) and dried over anhydrous sodium sulfate. The drying agent
was filtered off, and the filtrate was concentrated in vacuo. The crude product was
recrystallized from hexane-diethyl ether to give α-hydroxy-4-iodo-benzenepropionic acid
as a yellowish white solid (74%, 3.7 g).
1H NMR (500 MHz, DMSO-d6): δ 2.75 (dd, 1H, 1J = 13.92 , 2J = 4.32 Hz), 2.93 (dd, 1H,
1J = 13.7 , 2J = 8.3 Hz), 4.16 (dd, 1H, 1J = 7.9 , 2J = 4.4 Hz),7.06 (d, 2H, 1J = 7.8 Hz),
7.62 (d, 2H, 1J = 7.8 Hz) ppm.
13C NMR (125 MHz, DMSO-d6): δ 64.9, 70.6, 91.9, 131.9, 136.7, 137.9, 174.9 ppm.

255
(3S)-3-(4-iodobenzyl)-6-methyl-1,4-dioxane-2,5-dione (3); Typical Procedure[20]
To a flame dried round bottom flask equipped with a stir bar and under argon was added
α-hydroxy-4-iodo-benzenepropionic acid (5.1 mmol, 1.5 g, 1.0 equiv), triethylamine
(0.79 mL, 5.6 mmol, 1.1 equiv), and dry MeCN (25 mL) while under argon. This
solution was cooled to 0 ºC, followed by the dropwise addition of 2-bromopropionyl
chloride (0.57 mL, 5.6 mmol, 1.1 equiv). The mixture was stirred for 30 min.
Triethylamine (0.79 mL, 5.6 mmol, 1.1 equiv) was added and the reaction was stirred at
70 ºC for 3-5 h. The reaction was cooled to room temperature, quenched with 1M HCl
(25 mL) and extracted with EtOAc (3 x 25 mL). The combined organic phases were
washed with H2O (25 mL), brine (25 mL), and dried over anhydrous sodium sulfate. The
drying agent was filtered off, and the filtrate was concentrated in vacuo. The crude
product was purified by column chromatography (gradient 70:30 hexanes:EtOAc) as a
yellow oil, which solidified upon storage in the freezer (M.p. 121-122 ºC, 58 %, 1.03 g).
Rf = 0.26 (70:30 hexanes:EtOAc)
1H NMR (500 MHz, CDCl3): δ 1.60 (d, 3H, 1J = 6.7 Hz), 3.19 (dd, 1H, 1J = 14.8 , 2J =
7.5 Hz ), 3.40 (dd, 1H, 1J = 14.9, 2J = 4.0 Hz ) 4.96 (q, 1H 1J = 6.7 Hz), 5.06 (dd, 1H, 1J
= 3.9 , 2J = 7.7 Hz), 7.08 (d, 1J = 8.3 Hz), 7.66 (d, 1J = 8.3 Hz) ppm.
13C NMR (125 MHz, CDCl3): δ 15.9, 35.7, 72.5, 76.1, 93.1, 131.9, 134.2, 137.8, 166.0,
166.7 ppm.
IR (neat): 2995 (w), 2932 (w), 1769 (s), 1738 (s), 1485 (w), 1250 (s) cm−1.

256
(S)-2-((R)-2-bromopropanamido)-3-(4-iodophenyl)propionic acid (4); Typical
Procedure[21-23]
To a flame dried round bottom flask, equipped with a stir bar was added 4-Iodo-
L-phenylalanine (6.9 mmol, 2.0 g, 1.0 equiv), and 20 mL H2O:Et2O (1:1 (V/V)). To this
solution was added 4 M NaOH (8 mL) and stirred until all solids dissolved; this solution
was then cooled to 0 ºC. In a separate flame dried vial was added 2-bromopropionyl
chloride (0.76 mL, 7.6 mmol, 1.1 equiv) and 4 M NaOH (8 mL). The vial solution was
added (dropwise) to the round bottom flask while maintaining a temperature of 0 ºC and a
pH of 11 throughout the addition (4 M NaOH added to maintain pH via dropping funnel)
. After completion of the reaction (monitored by TLC), the reaction was warmed to room
temperature and the ether layer was separated in a separatory funnel. The aqueous layer
was acidified with concentrated HCl to pH of 1, and extracted with ethyl acetate (4 x 25
mL). The combined organic phases were washed with brine and dried over anhydrous
sodium sulfate. The drying agent was filtered off, and the filtrate was concentrated in
vacuo. The white, solid crude product was placed under high vacuum and used in next
step without further purification.
(3S,6S)-3-(4-iodobenzyl)-6-methylmorpholine-2,5-dione (5); Typical Procedure[21-24]
To a flame dried round bottom flask equipped with a stir bar was added crude (S)-2-((R)-
2-bromopropanamido)-3-(4-iodophenyl)propionic acid (6.8 mmol, 2.9 g, 1 equiv) and
DMF (25-30 mL) followed by triethylamine (1.04 mL, 7.5 mmol, 1.1 equiv). The
mixture was heated to 90 ºC for 12 h under N2. When reaction was completed

257
(monitored by TLC), the mixture was allowed to stand overnight in the freezer. The
crystallized salt and DMF/TEA were filtered, and the filtrate was concentrated in vacuo.
Residual DMF was removed by adding toluene to the sample, followed by rotary
evaporation (repeated 4 times). Further purification was performed by recrystallizing in
chloroform and cold Et2O to furnish a white solid (M.p. 147-148 ºC, 1.12 g, 48 % over 2
steps).
1H NMR (500 MHz, DMSO-d6): δ 1.13 (d, 3H, 1J = 6.9 Hz), 3.02 (d, 2H, 1J = 5.1 Hz),
4.67 (t, 1H, 1J = 5.0 Hz ), 4.99 (q, 1H, 1J = 6.8 Hz), 7.09 (d, 2H, 1J = 8.1 Hz), 7.68 (d,
2H, 1J = 8.2 Hz) ppm.
13C NMR (125 MHz, CDCl3): δ 16.8, 36.1, 54.2, 74.6, 93.3, 132.7, 136.5, 137.4, 168.5,
168.6 ppm.
IR (neat): 3202 (br), 1747 (s), 1694 (s), 1483 (w), 1375 (m), 1315 (m) cm−1.
(S)-2-(2-chloroacetamido)-3-(4-iodophenyl)propionic acid (6)[21-24]
Following the typical procedure of 4 was obtained by using chloroacetyl chloride in place
of 2-bromopropionyl chloride. The yellow colored crude product was used in next step
without further purification.
(S)-3-(4-iodobenzyl)morpholine-2,5-dione (7)[21-24]
Following the typical procedure of 5, but using 6 as the starting material, the product was
obtained as an off-white solid. (M.p. - ºC, 1.12 g, 41 % over 2 steps).

258
1H NMR (500 MHz, DMSO-d6): δ 1.13 (d, 3H, 1J = 6.90 Hz), 3.02 (d, 2H, 1J = 5.10 Hz),
4.67 (t, 1H, 1J = 4.98 Hz ), 4.99 (q, 1H, 1J = 6.84 Hz), 7.09 (d, 2H, 1J = 8.10 Hz), 7.68
(d, 2H, 1J = 8.15 Hz) ppm.
13C NMR (125 MHz, CDCl3): δ 16.8, 36.1, 54.2, 74.6, 93.3, 132.7, 136.5, 137.4, 168.5,
168.6 ppm.
1-Cyclohexenylpyrrolidine (9); Typical Procedure[25]
While under N2, a flame dried round bottom flask equipped with a stir bar and dean stark
trap (with activated 4 Å molecular sieves) was charged with cyclohexanone (4.22 mL,
40.8 mmol, 1 equiv), dry toluene (20 mL), and pyrrolidine (6.02 mL, 73.4 mmol, 1.8
equiv). The mixture was refluxed under N2 for 5-7 h. The solvent and excess pyrrolidine
were removed in vacuo. The crude product was used without further purification to afford
the colorless product. (5.8 g, 94%).
1H NMR (500 MHz, CDCl3): δ 1.38-1.43 (m, 2H), 1.52-1.55 (m, 2H), 1.66-1.68 (m, 4H),
1.95-1.96 (m, 2H), 2.02-2.03 (m, 2H), 2.84-2.85 (m, 4H), 4.10-4.16 (m, 1H) ppm.
13C NMR (125 MHz, CDCl3, with traces of toluene present): δ 22.9, 23.3, 24.5, 26.9,
27.4, 47.2, 93.3, 142.9 ppm.
2-(4-iodobenzyl)cyclohexan-1-one (10); Typical Procedure[26]
While under N2, a flame dried round bottom flask equipped with a stir bar was charged
with 4-iodobenzyl bromide (4.9 g , 16.5 mmol, 1 equiv) and dry toluene (30 mL). When
homogeneous solution formed, the flask was charged with 1-cyclohexenylpyrrolidine

259
(2.5 g, 16.5 mmol , 1 equiv). The mixture was refluxed under N2 for 18 h. H2O (30 mL)
was added and the solution was heated for an additional hour. The solvent was
evaporated under vacuum, and the residue was extracted with diethyl ether. The ether
phase was washed consecutively with 5% HCl, 5% NaHCO3 solution, and water, then
dried, and evaporated. The residue was purified via silica gel chromatography eluted with
hexanes/ethyl acetate (9:1) to provide a white solid (3.7 g, 71 %).
1H NMR (500 MHz, CDCl3): δ 1.15-3.02 (m, 11H), 6.79 (d, 2H, 1J = 8.3 Hz), 7.42 (d,
2H, 1J = 8.3 Hz) ppm.
13C NMR (125 MHz, CDCl3): δ 25.1, 27.9, 33.5, 35.1, 42.1, 52.0, 91.2, 131.4, 137.2,
140.1, 211.4 ppm.
7-(4-iodobenzyl)oxepan-2-one (11); Typical Procedure[27. 28]
While under N2, a flame dried round bottom flask equipped with a stir bar was charged
with 2-(4-iodobenzyl)cyclohexan-1-one (1.5 g, 4.8 mmol, 1 equiv) and CHCl3 (50 mL).
The solution was cooled to 0 ºC, and to this solution was added mCPBA (77% reagent)
(14.3 mmol, 3 equiv). The mixture was stirred at room temperature for 2-3 d (monitored
by TLC), and the reaction mixture was quenched with an sat. Na2S2O3 aqueous solution.
The aqueous layer was extracted with CHCl3 (x3). The combined organic layer was
washed with sat. NaHCO3 aqueous solution and brine. The organic layers were dried
over Na2SO4, filtered, and concentrated in vacuo. The residue was purified via silica gel
chromatography eluted with a gradient of hexanes:ethyl acetate (2:8) to provide a
colorless oil (1.07 g, 68%). Rf = 0.69 (1:9 hexanes:EtOAc)

260
1H NMR (500 MHz, CDCl3): δ 1.33-2.78 (m, 10H), 4.28-4.3 (m, 1H), 6.85 (d, 2H, 1J =
8.1 Hz), 7.40 (d, 2H, 1J = 8.2 Hz) ppm.
13C NMR (125 MHz, CDCl3): δ 22.9, 28.3, 33.8, 34.9, 42.1, 80.7, 92.2, 131.7, 136.9,
137.6, 175.2 ppm.
IR (neat): 2924 (w), 2857 (w), 1726 (s), 1483 (w), 1173 (m), 1007 (w) cm−1.
Example Procedure for Conventional Ring-Opening Polymerization: Polymers were synthesized using ring opening polymerization with lactic acid as the
initiator and tin (II) 2-ethylhexanoate as the catalyst. Prior to use, the monomer, lactic
acid, sodium sulfate, and stir bar were vacuum-dried overnight in the reaction vessel. The
reaction vessel was equipped with a reflux condenser, and the reagents were dissolved in
anhydrous toluene while under a nitrogen atmosphere. When the reaction reached 120 ºC,
tin(II) 2-ethylhexanoate was added, and the reaction was allowed to stir at this
temperature for 24 h. The resulting polymer was partitioned between chloroform and
water, and the chloroform phase was collected (3x). The chloroform phases were
combined and dried over MgSO4. The filtrate was collected and the desired polymer
precipitated out by using cold methanol.
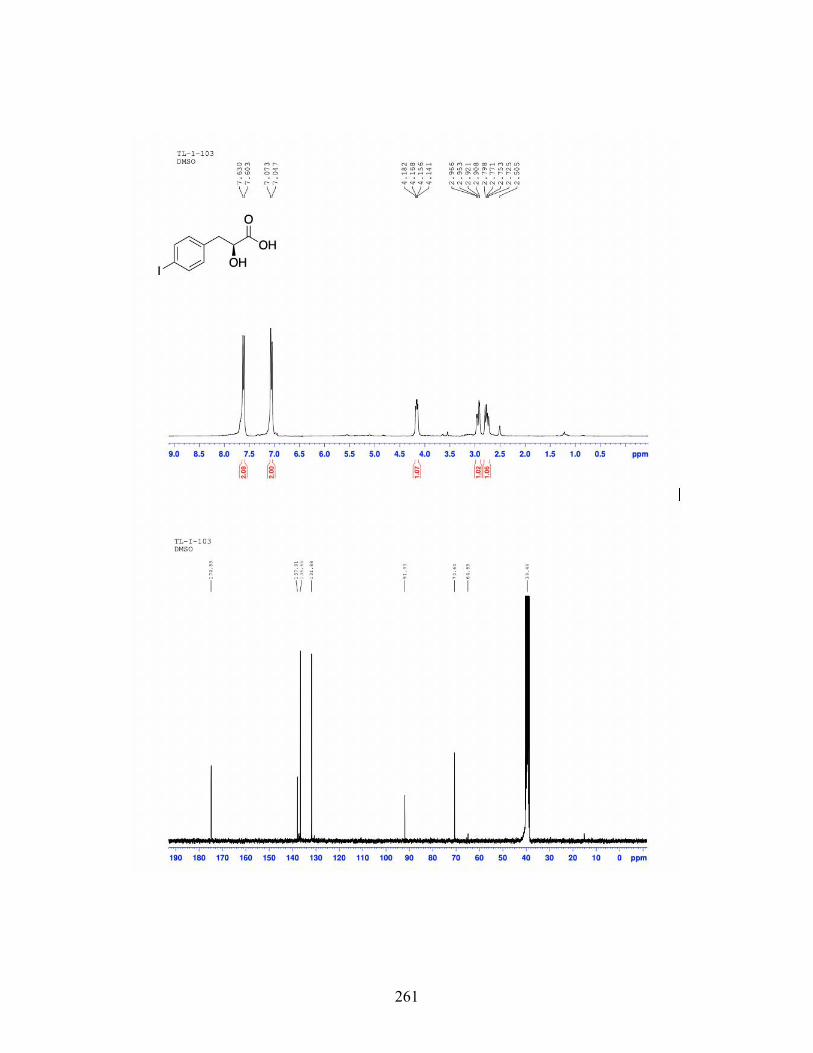
261
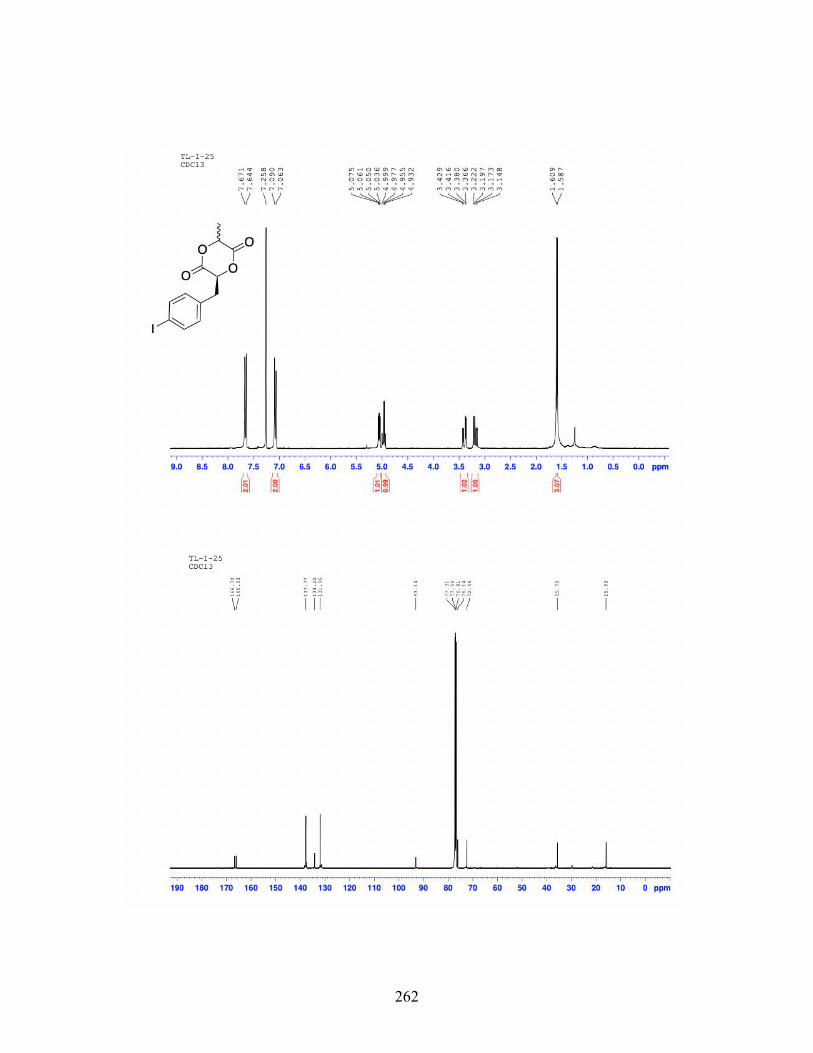
262
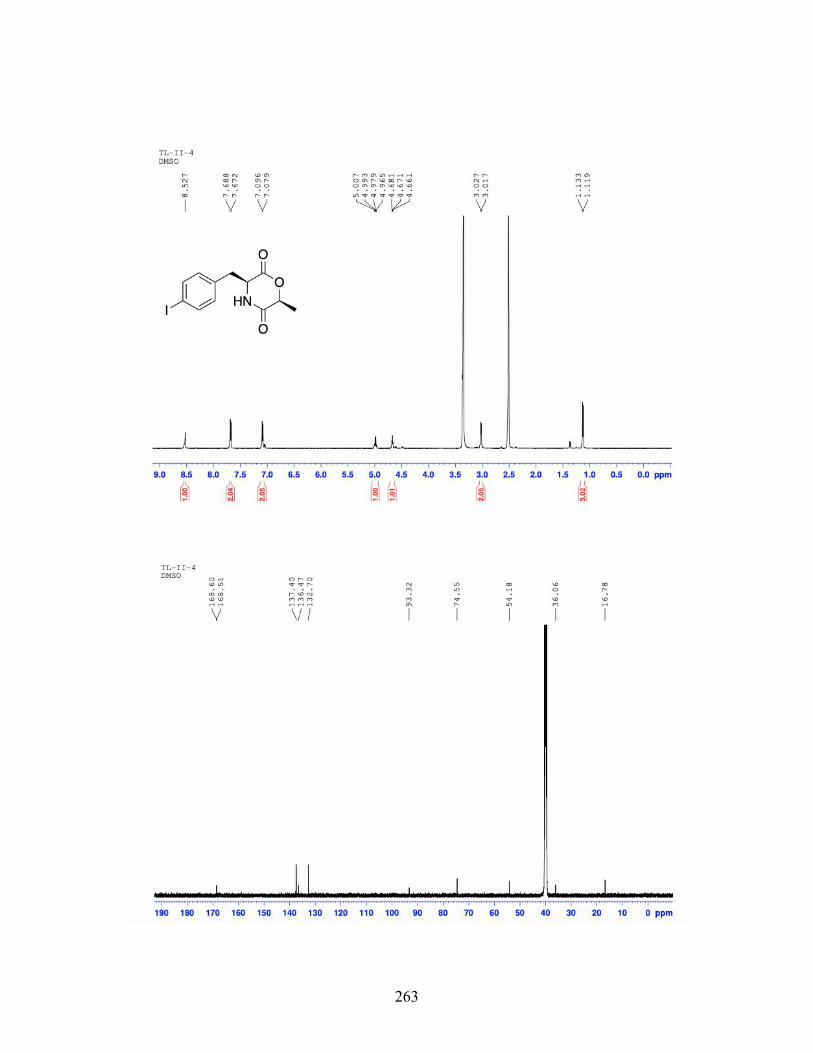
263
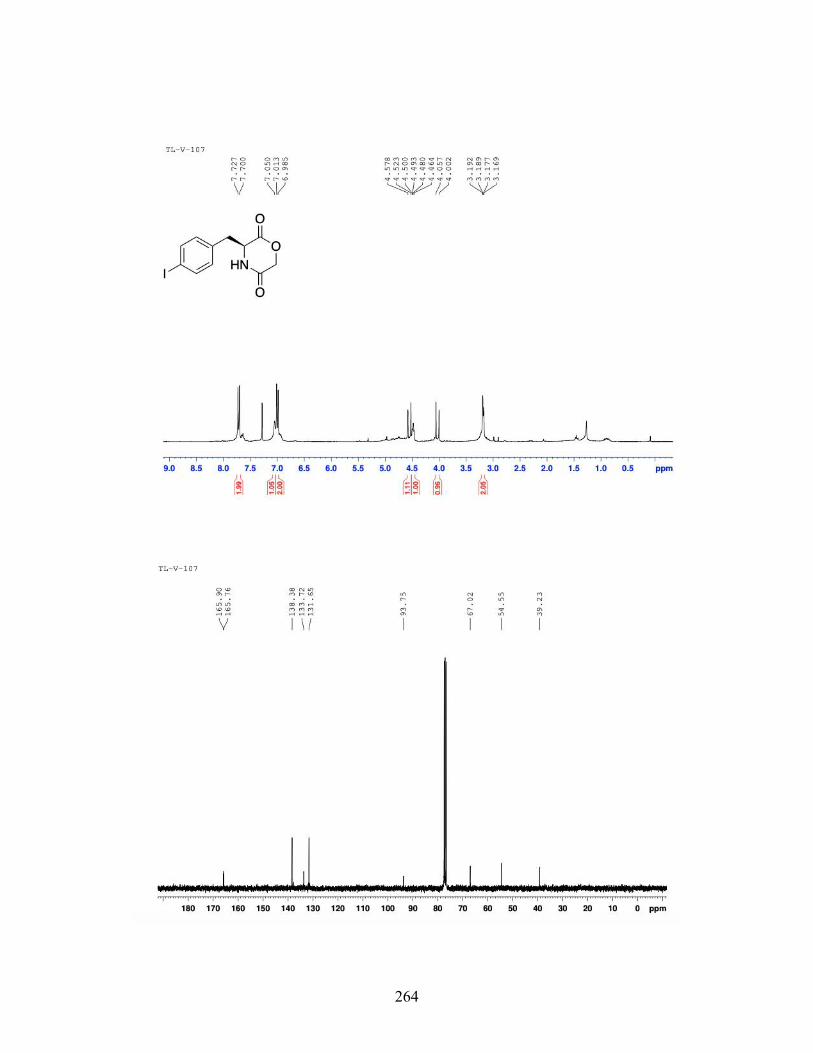
264
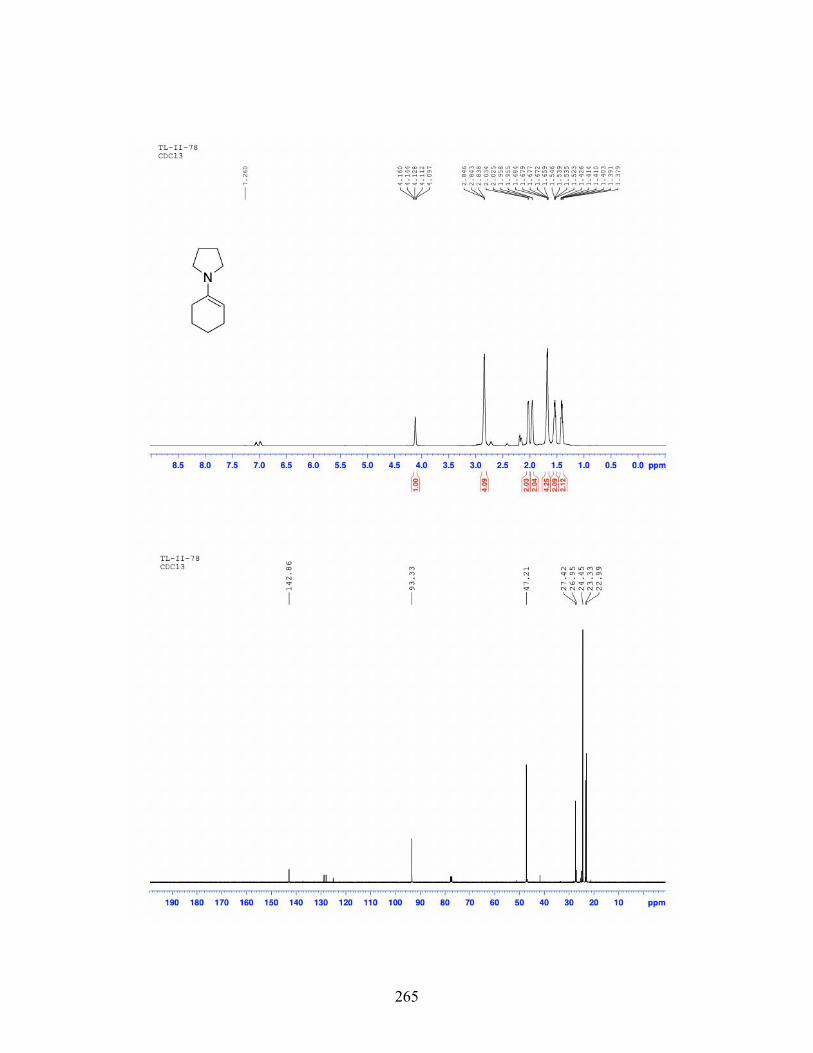
265
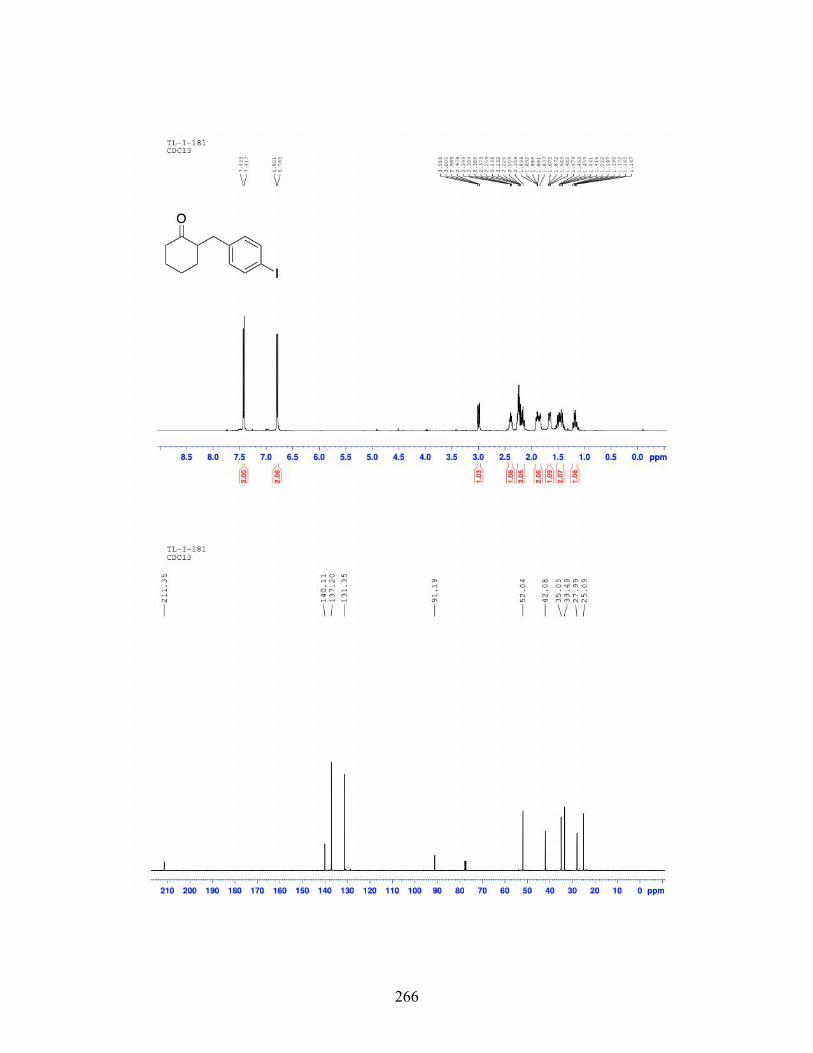
266
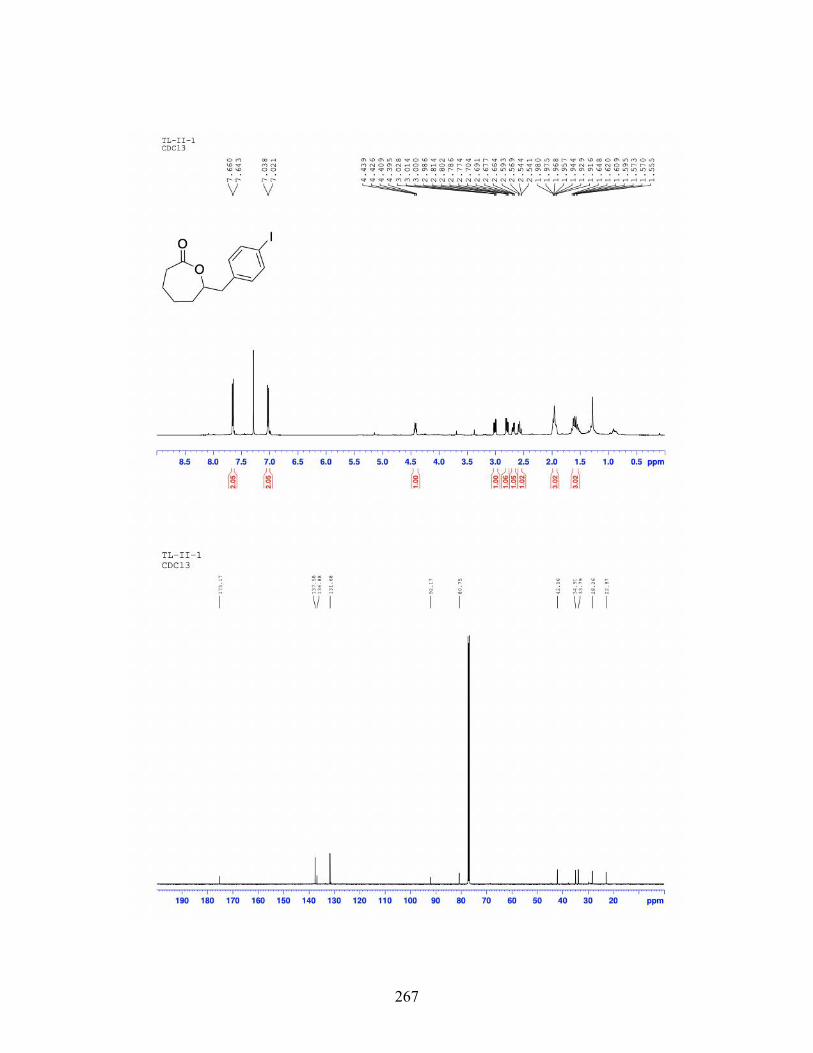
267

268
4.7 REFERENCES
343. Alikacem, N.; Stroman, P. W.; Marois, Y.; Jakubiec, B.; Roy, R.; Guidoin, R., Noninvasive follow-up of tissue encapsulation of foreign materials - Are magnetic resonance imaging and spectroscopy breakthroughs? Asaio Journal 1995, 41, M617-M624. 344. Ehlerding, E. B.; Grodzinski, P.; Cai, W. B.; Liu, C. H., Big Potential from Small Agents: Nanoparticles for Imaging-Based Companion Diagnostics. ACS Nano 2018, 12, 2106-2121. 345. Hemonnot, C. Y. J.; Koster, S., Imaging of Biological Materials and Cells by X-ray Scattering and Diffraction. ACS Nano 2017, 11, 8542-8559. 346. Attia, M. F.; Brummel, B. R.; Lex, T. R.; Van Horn, B. A.; Whitehead, D. C.; Alexis, F., Recent Advances in Polyesters for Biomedical Imaging. Adv. Healthc. Mater. 2018, 7. 347. Nottelet, B.; Darcos, V.; Coudane, J., Aliphatic polyesters for medical imaging and theranostic applications. Eur. J. Pharm. Biopharm. 2015, 97, 350-370. 348. El Habnouni, S.; Darcos, V.; Coudane, J., Synthesis and Ring Opening Polymerization of a New Functional Lactone, alpha-Iodo-epsilon-caprolactone: A Novel Route to Functionalized Aliphatic Polyesters. Macromol. Rapid Commun. 2009, 30, 165-169. 349. Benabdillah, K. M.; Coudane, J.; Boustta, M.; Engel, R.; Vert, M., Synthesis and characterization of novel degradable polyesters derived from D-gluconic and glycolic acids. Macromolecules 1999, 32, 8774-8780. 350. Samuel, R.; Girard, E.; Chagnon, G.; Dejean, S.; Favier, D.; Coudane, J.; Nottelet, B., Radiopaque poly(epsilon-caprolactone) as additive for X-ray imaging of temporary implantable medical devices. RSC Adv. 2015, 5, 84125-84133. 351. Attia, M. F.; Brummel, B. R.; Lex, T. R.; Van Horn, B. A.; Whitehead, D. C.; Alexis, F., Recent Advances in Polyesters for Biomedical Imaging. Adv. Healthcare Mater. 2018, 7. 352. Boase, N. R. B.; Blakey, I.; Thurecht, K. J., Molecular imaging with polymers. Polym. Chem. 2012, 3, 1384-1389. 353. He, W.; Feng, Y.; Ma, Z. W.; Ramakrishna, S.; Mahapatro, A.; Kulshrestha, A. S., Polymers for Tissue Engineering. Polymers For Biomedical Applications 2008, 977, 310-335. 354. Francis, R.; Kumar, D. S., Biomedical Applications of Polymeric Materials and Composites. 1st ed. Wiley-VCH,2016; p 1 online resource (400 pages). 355. Helmchen, F.; Denk, W., Deep tissue two-photon microscopy. Nature Methods 2005, 2, 932-940. 356. Olsen, T. R.; Davis, L. L.; Nicolau, S. E.; Duncan, C. C.; Whitehead, D. C.; Horn, B. A.; Alexis, F., Non-invasive deep tissue imaging of iodine modified poly(caprolactone-co-1-4-oxepan-1,5-dione) using X-ray. Acta Biomaterialia 2015, 20, 94-103.

269
357. Nicolau, S. E.; Davis, L. L.; Duncan, C. C.; Olsen, T. R.; Alexis, F.; Whitehead,D. C.; Van Horn, B. A., Oxime functionalization strategy for iodinated poly(epsilon-caprolactone) X-ray opaque materials. J. Polym. Sci. A 2015, 53, 2421-2430.358. Van Horn, B. A.; Davis, L. L.; Nicolau, S. E.; Burry, E. E.; Bailey, V. O.; Guerra,F. D.; Alexis, F.; Whitehead, D. C., Synthesis and conjugation of a triiodohydroxylaminefor the preparation of highly X-ray opaque poly(epsilon-caprolactone) materials. J.Polym. Sci. A 2017, 55, 787-793.359. Dechy-Cabaret, O.; Martin-Vaca, B.; Bourissou, D., Controlled ring-openingpolymerization of lactide and glycolide. Chem. Rev. 2004, 104, 6147-6176.360. Benoit, M. A.; Baras, B.; Gillard, J., Preparation and characterization of protein-loaded poly(epsilon-caprolactone) microparticles for oral vaccine delivery. Int. J. Pharm.1999, 184, 73-84.361. Wilbur, D. S. Iodinated borane cage molecules as X-ray contrast media. 1996.362. Andronescu, E.; Grumezescu, A. M., Nanostructures for oral medicine. In Microand nano technologies series Elsevier,: Amsterdam, 2017; p 1 online resource.363. Janib, S. M.; Moses, A. S.; MacKay, J. A., Imaging and drug delivery usingtheranostic nanoparticles. Adv. Drug Delivery Rev. 2010, 62, 1052-1063.364. Bikiaris, D.; Karavelidis, V.; Karavas, E., Novel Biodegradable Polyesters.Synthesis and Application as Drug Carriers for the Preparation of Raloxifene HCl LoadedNanoparticles. Molecules 2009, 14, 2410-2430.365.Shalgunov, V.; Zaytseva-Zotova, D.; Zintchenko, A.; Levada, T.; Shilov, Y.;Andreyev, D.; Dzhumashev, D.; Metelkin, E.; Urusova, A.; Demin, O.; McDonnell, K.;Troiano, G.; Zale, S.; Safarova, E., Comprehensive study of the drug delivery propertiesof poly(L-lactide)-poly (ethylene glycol) nanoparticles in rats and tumor-bearing mice. JControl Release. 2017, 261, 31-42.366. Washington, K. E.; Kularatne, R. N.; Karmegam, V.; Biewer, M. C.; Stefan, M.C., Recent advances in aliphatic polyesters for drug delivery applications. WileyInterdiscip. Rev. Nanomed. Nanobiotechnol. 2017, 9.

270
CHAPTER FIVE
SYNTHESIS OF DIAZACYCLOBUTENES VIA A FORMAL [2 + 2] CYCLOADDITION
5.1 PREFACE
This abridged chapter describes a formal [2 + 2] cycloaddition between electron-rich
alkynes and triazolinediones to generate rather unexplored chemical moieties known as
diazacyclobutenes. The research described in this chapter was conducted in collaboration
with a fellow colleague, Chandima J. Narangoda (Clemson University, advisor: Dr.
Daniel C. Whitehead) and will therefore only be described briefly. For a more detailed
account, the reader is encouraged to read the current and future published peer reviewed
articles that the Whitehead group publishes.367, 368
5.2 INTRODUCTION
The field of organic chemistry perpetually challenges and motivates scientists to design,
develop and implement new synthetic methodologies in order to construct target
compounds. One common chemical moiety that chemists continually aspire to create are
cyclic frameworks. Cyclic compounds, which contain at least one connected series of
atoms that form a ring, have proven to be important in all aspects of life. In particular,
cyclic species have found significance as biologically and pharmaceutically relevant
compounds (i.e. agrochemicals, medicines, dyes, optical materials etc.).369 One
important synthetic strategy to assemble cyclized products has been the use of

271
cycloaddition reactions.369, 370 Cycloadditions allow for two or more new bond
formations to manifest from a single manipulation. Cycloadditions have been utilized by
chemists to create various cyclic moieties, including nitrogen containing heterocyclic
compounds.
Diazacyclobutenes are a unique class of nitrogen-containing heterocycles
containing two nitrogen atoms and a carbon-carbon double bond constrained within a
four-membered ring (Scheme 5.1, 5d).
This scaffold has gained some attention in the scientific community due to their apparent
non-aromatic character, even though they formally obey Hückel’s (4n+2) rule of
aromaticity.371-381 Nevertheless, the diazacyclobutene motif has been relatively
inaccessible due to the limited number of synthetic methodologies reported in the
literature. In fact, the known synthetic routes to generate diazacyclobutenes have been
R1 YR2R1
1.) nBuLi, THF, -78 ℃2.) S8, -78 ℃, 1 h3.) R2X, THF, 0 ℃, 4 h
Condition A
Y = S or Se1.) nBuLi, THF, -78 ℃3.) R2-Y-Y-R2, THF, -78 ℃ to rt, 1 h
Condition Bor
N N
N OO
MeCN, reflux, 24 h5a 5b
5c
Scheme 5.1 General synthetic route toward diazacyclobutenes
N N
N OO
5dYR1 R2

272
rather ineffective, and consequently, less than 10 total examples of this molecular
scaffold have been disclosed (Figure 5.2).373, 378-381
Our group has recently designed a convenient [2 + 2] cycloaddition between
electron-rich alkynes (5a) and a triazolinedione derivative (5c, 4-phenyl-1,2,4-
triazoline-3,5-dione or PTAD) to construct diazacyclobutene derivatives (5d, Scheme
5.1).367 It should be noted that sulfide and selenide alkynes were employed due to their
electron donating ability, thereby creating a more electron rich alkyne.
The production of the diazacyclobutenes began with the synthesis of sulfur-
and/or selenium-containing alkynes (5b). These alkynes were synthesized by means of an
n-butyl lithium (nBuLi)-assisted deprotonation of a terminal alkyne (5a), producing a
lithium acetylide, which can supply the targeted alkyne (5b) with use of elemental sulfur
(Condition A) or an appropriate dialkyl sulfide or selenide (Condition B).367, 382, 383
With optimized reaction conditions, the generated alkynyl sulfides or selenides (5b)
underwent the desired cycloaddition upon refluxing in the presence of acetonitrile
(MeCN) and PTAD (5c), thus furnishing the diazacyclobutene adduct (5d).367
N NArO2S
NRR
5e51-77% (1 step)
7 examples(Effenberger, 1966)
N N
5f5% (3 steps)
t1/2 (20 ℃) = 6.9 h(Warrener, 1972)
OOO
OCH3H3C
N N
N OO
R
5g21-26% (1 step)
2 examples(Greene, 1984)
N N
N OO
CH3
5h11% (2 steps)(Breton, 2001)
Figure 5.1 Historical examples of diazacyclobutene motifs
Ar Ar

273
To investigate this methodology, an assortment of alkynyl sulfides and selenides
were synthesized and treated with PTAD to create a small library of diazacyclobutene
derivatives (Figure 5.2). By using sulfur phenylacetylene derivatives, we first varied the
substituent located on the sulfur/selenium atom (R2). The carbon chain length was
altered, and shorter, as well as longer alkyl chains, supplied the corresponding product in
moderate to good yields (Figure 5.2, 5i-5n, 7789% yields). A benzyl (5o) and aryl (5p)
functional group residing on the sulfur atom provided diazacyclobutene derivatives in 62
and 85% yields, respectively. Compound 5p was also produced in an 81% yield when
carried out at a 6 mmol scale. Next, we modified the R1 substituent with an aryl moiety.
Arenes that contain either a para-positioned electron donating (5q–5s) or electron
withdrawing (5t and 5u) group yielded products with good efficiency (74–92%). When
the R1 and R2 substituents were both alkyl groups (5v–5x), the transformation was also
successful, ranging from moderate to good yields (56–94%). Finally, selenide alkynes
were used in the [2 + 2] cycloaddition, and proceeded to give good diazacyclobutene
yields (5y and 5z, 80 and 93%).
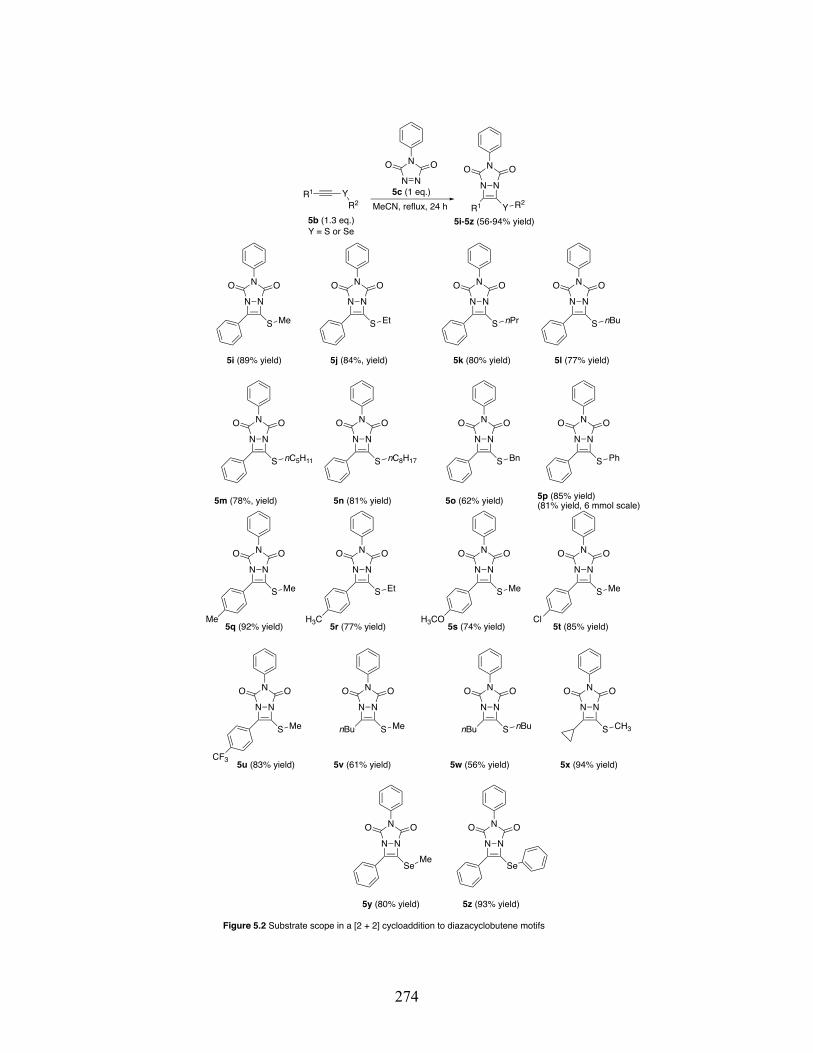
274
N N
N OO
5i (89% yield) 5j (84%, yield) 5k (80% yield) 5l (77% yield)
5m (78%, yield) 5n (81% yield) 5o (62% yield) 5p (85% yield)(81% yield, 6 mmol scale)
5q (92% yield) 5r (77% yield) 5s (74% yield) 5t (85% yield)
5u (83% yield) 5v (61% yield) 5w (56% yield) 5x (94% yield)
5y (80% yield) 5z (93% yield)
Figure 5.2 Substrate scope in a [2 + 2] cycloaddition to diazacyclobutene motifs
R1 YR2
Y = S or Se
N N
N OO
MeCN, reflux, 24 h5b (1.3 eq.)
5c (1 eq.)
5i-5z (56-94% yield)
S Me
N N
N OO
S Et
N N
N OO
S nPr
N N
N OO
S nBu
N N
N OO
S nC5H11
N N
N OO
S nC8H17
N N
N OO
S Bn
N N
N OO
S Ph
N N
N OO
S Me
N N
N OO
S Et
N N
N OO
S Me
N N
N OO
S Me
N N
N OO
S Me
N N
N OO
nBu S Me
N N
N OO
S nBu
N N
N OO
S CH3
N N
N OO
Se
N N
N OO
Se
Me H3C H3CO
CF3
nBu
Cl
N N
N OO
YR1 R2
Me

275
The previous work demonstrated a simple, yet efficient protocol that involves a formal [2
+ 2] cycloaddition between electron rich alkynes and PTAD to generate
diazacyclobutenes. This chemistry has been extended to synthesize another rarely
accessible molecular scaffold, 2-imidothioimidates (Scheme 5.2, 5ac).368
A reaction between sulfide or selenide alkynes (5b) with open-chain
azodicarboxylates (5aa) form a [2 + 2] cycloadduct (5ab), which quickly experiences a
4p conrotatory cycloreversion to provide N,N-dicarbamoyl 2-iminothioimidates (Scheme
5.2, 5ac). The substrate scope involving the use of diethyl azodicarboxylate (DEAD,
5ad) and sulfide/selenide alkynes (5b) is shown in Figure 5.3; it should be noted that
although it is not described in this text, we have been successful with employing other
azodicarboxylates using this strategy, and the reader is directed to our publication for
further details.368 Next, the alkyl chain (R2) situated on the sulfur/selenium atom was
explored. Shorter (5ae-5ah) and longer (5ai-5aj) alkyl chains supplied the desired 2-
imidothioimidates in good to great yields (73–99%). By using aryl and benzyl moieties
at the R2 position, phenyl (5ak) and benzyl (5al) substituted N,N-dicarbamoyl 2-
iminothioimidates could be produced in good yields (77 and 83%, respectively). Further
N N CO2R3R3O2C
MeCN (reflux)24 h
R1 YR2
N
N
O
O R3
R1Y R2N N
R1 Y R2
OO
OR3R3O 4π conrotatory
cycloreversion
40 - 99% yield
5bY = S, Se
O
OR3
Scheme 5.2 General synthetic route toward N,N-dicarbamoyl 2-iminothioimidates
5ab5ac
5aa

276
studies involved substituting arenes at the R1 position of the alkyne, while maintaining a
methyl sulfide motif (5am-5aq). Electron donating groups in the para-position of the
arene were highly effective using these reaction conditions (5am and 5an), however
withdrawing donating groups in the aryl para-position gave mediocre yields (5ao and
5ap). Modifying R1 with alkyl substituents such as n-butyl gave lower yields (40% for
5aq and 45% for 5ar), while a cyclopropyl substituent gave a moderate yield of 77%
(5as). Finally, selenide 2-imidothioimidate derivatives could be produced using this
protocol, and supplied products in good yields (76% for 5at and 73% for 5au).
The work discussed herein demonstrates the high synthetic potential for sulfur
and selenium-containing alkynes to undergo a [2 + 2] cycloaddition with both cyclic (i.e.
PTAD) or acyclic (i.e. DEAD) azodicarboxylates to create novel chemical moieties. The
rarely accessible diazacyclobutene and 2-imidimidates motifs were synthesized in a
simple, facile manner with moderate to good yields. These protocols have the ability to
enrich the synthetic applications of cycloadditions and provide new strategies for
obtaining unique molecular scaffolds.
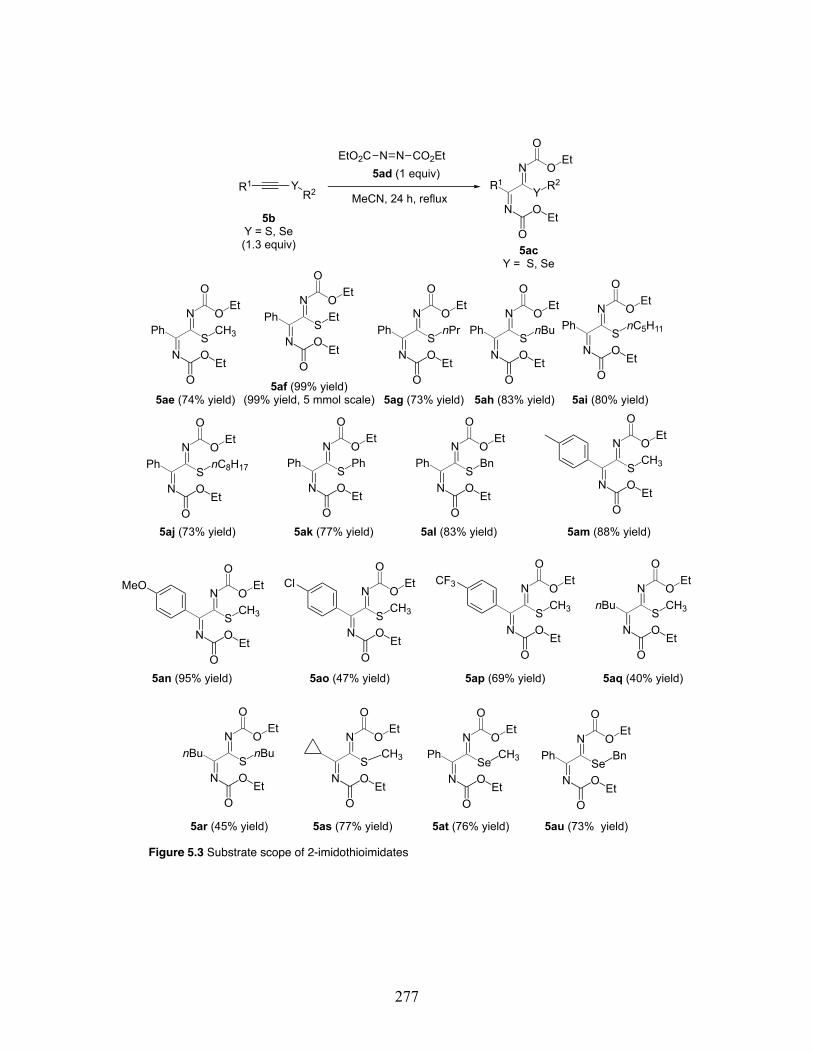
277
N
N
O
OEt
PhS
CH3
5ae (74% yield)
N
N
O
OEt
PhS
Et
5af (99% yield)(99% yield, 5 mmol scale)
N
N
O
OEt
PhSnPr
5ag (73% yield)
N
N
O
OEt
PhSnBu
5ah (83% yield)
N
N
O
OEt
PhSnC5H11
5ai (80% yield)
N
N
O
OEt
PhSnC8H17
5aj (73% yield)
N
N
O
OEt
PhS
Ph
5ak (77% yield)
N
N
O
OEt
SCH3
5am (88% yield)
N
N
O
OEt
SCH3
5an (95% yield)
MeO
N
N
O
OEt
SCH3
5ao (47% yield)
Cl
N
N
O
OEt
SCH3
5ap (69% yield)
CF3
N
N
O
OEt
nBuS
CH3
5aq (40% yield)
N
N
O
OEt
nBuSnBu
5ar (45% yield)
N
N
O
OEt
S
5as (77% yield)
N
N
O
OEt
PhSe
5at (76% yield)
N
N
O
OEt
PhS
Bn
5al (83% yield)
N N CO2EtEtO2C
MeCN, 24 h, refluxR1 Y
R2
N
N
O
OEt
R1Y R2
CH3CH3
5bY = S, Se(1.3 equiv)
5ad (1 equiv)
5acY = S, Se
O
OEt
O
OEt O
OEt
O
OEt
O O
O
O
OOEt Et
EtO
OEt
O
OEt
O
OEt
O
OEt
O
OEt
O
OEt
O
OEt
O
OEt
O
OEt
O
OEt N
N
O
OEt
PhSe
5au (73% yield)
Bn
O
OEt
Figure 5.3 Substrate scope of 2-imidothioimidates
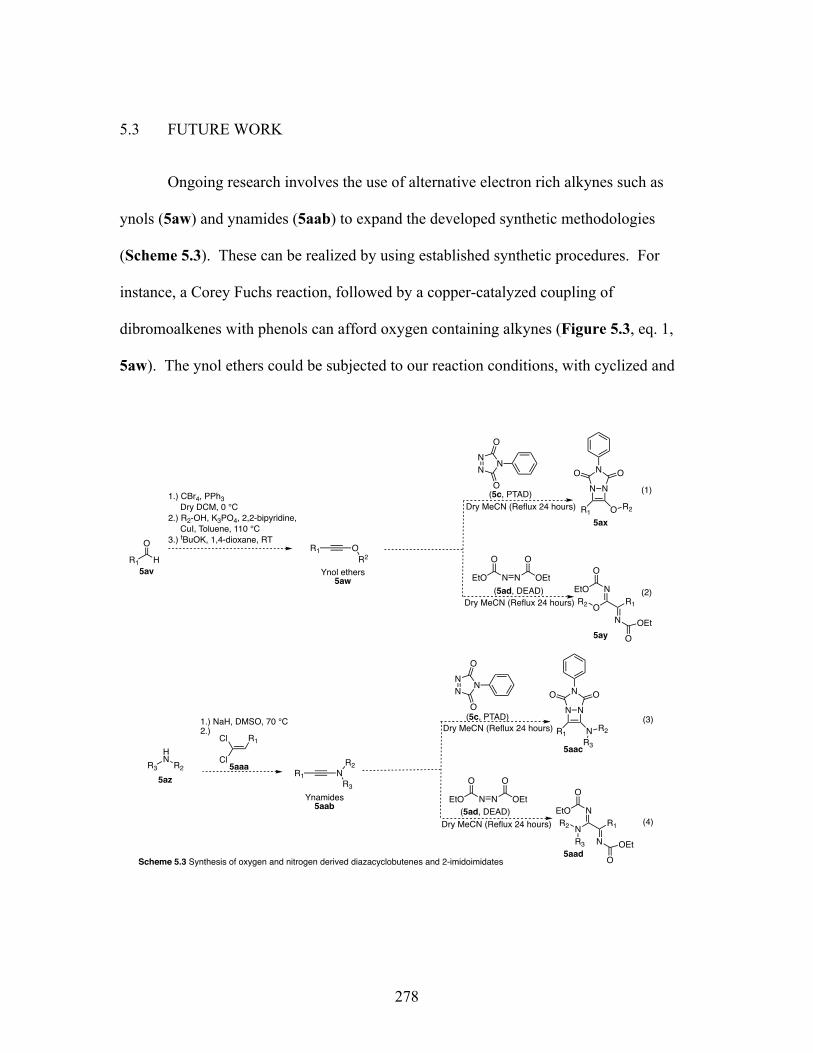
278
5.3 FUTURE WORK
Ongoing research involves the use of alternative electron rich alkynes such as
ynols (5aw) and ynamides (5aab) to expand the developed synthetic methodologies
(Scheme 5.3). These can be realized by using established synthetic procedures. For
instance, a Corey Fuchs reaction, followed by a copper-catalyzed coupling of
dibromoalkenes with phenols can afford oxygen containing alkynes (Figure 5.3, eq. 1,
5aw). The ynol ethers could be subjected to our reaction conditions, with cyclized and
1.) NaH, DMSO, 70 °C2.)
R3
HN R2
Cl
Cl R1
1.) CBr4, PPh3 Dry DCM, 0 °C2.) R2-OH, K3PO4, 2,2-bipyridine, CuI, Toluene, 110 °C3.) tBuOK, 1,4-dioxane, RT
Ynol ethers5aw
R1 H
O R1 O
Ynamides5aab
R1 NR3
R2
N N
R1 O
N OO
R2
NN N
O
O
EtO
O
N
ON
R1
OOEt
Dry MeCN (Reflux 24 hours)
Dry MeCN (Reflux 24 hours)
EtO
O
N N
O
OEt
(5c, PTAD)
(5ad, DEAD)R2
N N
R1 N
N OO
R2
NN N
O
O
EtO
O
N
NN
R1
OOEt
Dry MeCN (Reflux 24 hours)
Dry MeCN (Reflux 24 hours)
EtO
O
N N
O
OEt
(5c, PTAD)
R2
R3
R3
R2
(1)
(2)
(3)
(4)
Scheme 5.3 Synthesis of oxygen and nitrogen derived diazacyclobutenes and 2-imidoimidates
5av
5ax
5ay
5az
5aac
5aad
(5ad, DEAD)
5aaa

279
open-chain azodicarboxylates, to afford oxygen-containing diazacyclobutenes and 2-
imidimidates (Figure 5.3, eq. 1, 5ax, eq. 2, 5ay).384-387 Similarly, nitrogen-containing
alkynes, called ynamides (5aab), can be synthesized from commercially available, cheap,
vinyl dichlorides (5aaa).388, 389 These are relatively unexplored chemical scaffolds, and
thus have the potential to be useful in many areas of science, including medicinal and
synthetic chemistry.

280
5.4 REFERENCES
367. Narangoda, C. J.; Lex, T. R.; Moore, M. A.; McMillen, C. D.; Kitaygorodskiy,A.; Jackson, J. E.; Whitehead, D. C., Accessing the Rare Diazacyclobutene Motif. Org.Lett. 2018, 20, 8009-8013.368. Narangoda, C. J.; Kakeshpour, T.; Lex, T. R.; Redden, B. K.; Moore, M. A.;Frank, E. M.; McMillen, C. D.; Wiskur, S. L.; Kitaygorodskiy, A.; Jackson, J. E.;Whitehead, D. C., A cycloaddition/electrocyclic ring opening sequence between alkynylsulfides and azodicarboxylates to provide N,N-dicarbamoyl 2-iminothioimidates. (Justaccepted DOI:10.1021/acs.joc.9b01515) 2019.369. Nishiwaki, N., Methods and applications of cycloaddition reactions in organicsyntheses. John Wiley & Sons: Hoboken, New Jersey, 2014; p xii, 659 pages.370. Quadrelli, P., Modern applications of cycloaddition chemistry. Elsevier,:Amsterdam, 2019; p 1 online resource.371. Hassner, A., Small ring heterocycles. Wiley: New York ; Chichester, 1983.372. Vol'pin, M. E., Non-benzenoid aromatic compounds and the concept ofaromaticity. Russ. Chem. Rev. 1960, 29, 129-160.373. Warrener, R. N.; Nunn, E. E.; Paddon-Row, M. N., Aust. J. Chem. 1979, 32,2659-2674.374. Budzelaar, P. H. M.; Cremer, D.; Wallasch, M.; Würthwein, E.-U.; Schleyer, P.;R., v., DIOXETENES AND DIAZETINES - NONAROMATIC 6-PI-SYSTEMS IN 4-MEMBERED RINGS. J.Am. Chem. Soc. 1987, 109, 6290-6299.375. Mo, O.; Ya ́ ń ̃ez, M.; Elguero, J., J. Mol. Struct.: THEOCHEM 1989, 201, 17-37.376. Bachrach, S. M.; Liu, M., J. Org. Chem. 1992, 57, 2040-2047.377. Breton, G. W.; Martin, K. L., Are 1,2-dihydrodiazetes aromatic? An experimentaland computational investigation. J. Org. Chem. 2002, 67, 6699-6704.378. Effenberger, F.; Maier, R., CYCLOADDITION OF AZOSULFONES ANDSULFONYLIMINES. Angew. Chem., Int. Ed. Engl. 1966, 5, 416-417.379. Nunn, E. E.; Warrener, R. N., J. Chem. Soc., Chem. Commun. 1972, 818-819.380. Cheng, C.-C.; Greene, F. D.; Blount, J. F., REACTION OFTRIAZOLINEDIONES WITH ACETYLENES - ELECTROPHILIC ADDITION. J.Org. Chem. 1984, 49, 2917-2922.381. Breton, G. W.; Shugart, J. H.; Hughey, C. A.; Perala, S. M.; Hicks, A. D.,Synthesis of Delta(1)-1,2-diazetines via a Diels-Alder cycloaddition approach. Org. Lett.2001, 3, 3185-3187.382. Alcaide, B.; Almendros, P.; Aparicio, B.; Lazaro-Milla, C.; Luna, A.; Faza, O. N.,Gold-Photoredox-Cocatalyzed Tandem Oxycyclization/Coupling Sequence of Allenolsand Diazonium Salts with Visible Light Mediation. Adv. Synth. Catal. 2017, 359, 2789-2800.383. Lang, H.; Keller, H.; Imhof, W.; Martin, S., SYNTHESIS ANDCOORDINATION ABILITY OF SELENOALKYNES R-SE-CC-R'. Chem. Ber. 1990,123, 417-422.

281
384. Jouvin, K.; Bayle, A.; Legrand, F.; Evano, G., Copper-Catalyzed Coupling of 1,1-Dibromo-1-alkenes with Phenols: A General, Modular, and Efficient Synthesis of YnolEthers, Bromo Enol Ethers, and Ketene Acetals. Org. Lett. 2012, 14, 1652-1655.385. Rao, M. L. N.; Dasgupta, P., A concise route to functionalized benzofuransdirectly from gem-dibromoalkenes and phenols. RSC Adv. 2015, 5, 65462-65470.386. Rao, M. L. N.; Jadhav, D. N.; Dasgupta, P., Pd-Catalyzed Domino Synthesis ofInternal Alkynes Using Triarylbismuths as Multicoupling Organometallic Nucleophiles.Org. Lett. 2010, 12, 2048-2051.387. Cao, W.; Chen, P.; Wang, L.; Wen, H.; Liu, Y.; Wang, W. S.; Tang, Y., A HighlyRegio- and Stereoselective Syntheses of alpha-Halo Enamides, Vinyl Thioethers, andVinyl Ethers with Aqueous Hydrogen Halide in Two-Phase Systems. Org. Lett. 2018, 20,4507-4511.388. Alcaide, B.; Almendros, P.; Lazaro-Milla, C., Regioselective Synthesis ofHeteroatom-Functionalized Cyclobutene-triflones and Cyclobutenones. Adv. Synth.Catal. 2017, 359, 2630-2639.389. Tu, Y. L.; Zeng, X. Z.; Wang, H.; Zhao, J. F., A Robust One-Step Approach toYnamides. Org. Lett. 2018, 20, 280-283.

282
5.5 EXPERIMENTAL
General Information:
All reagents were purchased from commercial sources and used without
purification. THF and acetonitrile were dried prior to use over sodium/benzophenone and
phosphorous pentoxide, respectively. 1H and 13C NMR spectra were collected on Bruker
Avance 300 MHz, 500 MHz and JOEL Eclipse 500 MHz NMR spectrometers using
CDCl3. Chemical shifts are reported in parts per million (ppm). Spectra are referenced to
residual solvent peaks. Infrared spectroscopy data were collected using a Shimadzu
IRAffinity-1S instrument (with MIRacle 10 single reflection ATR accessory) operating
over the range of 400 to 4000 cm-1 . Flash silica gel (40-63 µm) was used for column
chromatography. All known compounds were characterized by 1H and 13C NMR and are
in complete agreement with samples reported elsewhere. All new compounds were
characterized by 1H and 13C NMR, ATR-FTIR, HRMS, XRD, and melting point (where
appropriate). TGA analysis was performed using TGA instrument SDT Q600 V20.9
Build 20 over the range of 200 – 800 ˚C, with a ramp rate of 20 ˚C/min under a flow of
nitrogen (100 mL/min).
5b

283
Condition A:
In a flame dried 2-neck round bottom flask was added a stir bar and a solution of
the terminal alkyne (1 equiv) in THF (5 mL for 1 mmol of alkyne) under argon
atmosphere. A septum was placed over the round bottom inlets and the solution was
cooled to -78 ˚C. A solution of n-BuLi (1.1 equiv., 1.6 M in hexane) was added. The
reaction solution was stirred for 30 min, and 1 equiv. of finely ground sulfur powder was
added by briefly removing one of the septa and then replacing it. The resulting mixture
was stirred for 1 hour at -78 ˚C. The resulting mixture was allowed to gradually warm to
0 ˚C until the sulfur was completely consumed, thus producing the lithium alkynyl
thiolate (red color). The corresponding alkyl halide (5.0 mmol, 1 equiv.) was then added
in a dropwise fashion. After 4 h the reaction mixture was quenched with saturated
aqueous NH4Cl (5 mL for 1 mmol of alkyne). The reaction mixture was then poured into
a separatory funnel, and the aqueous layer was extracted with diethyl ether (3 x 5 mL for
1 mmol of alkyne). The combined ether extracts were washed with saturated aqueous
brine (5 mL for 1 mmol of alkyne). The organic layer was then dried over Na2SO4,
decanted, and the solvent was evaporated by rotary evaporation. The crude residue was
then purified by flash chromatography (silica gel, hexane) to afford the desired alkynyl
sulfide.
Condition B:
To a flame dried round bottom flask, equipped with a magnetic stir bar and a
septum, was added a solution of the terminal alkyne (1 equiv.) dissolved in THF (1 mL

284
for 1 mmol of alkyne). The solution was then cooled to -78 ˚C and n-BuLi (1.1 equiv.,
1.6 M in hexane) was added dropwise. This solution was stirred for 10 min after which
dimethyl disulfide (1.2 equiv.) was added at -78 ˚C. The solution was then allowed to
warm to room temperature and stirred for 1 h. The reaction mixture was quenched with
saturated aqueous NH4Cl (5 mL for 1 mmol of alkyne) and extracted with ethyl acetate (3
x 5 mL for 1 mmol of alkyne). The combined organic layers were dried over Na2SO4,
decanted, and concentrated by rotary evaporation. The crude mixture was purified by
flash chromatography (silica gel, hexanes) to yield the desired alkynyl sulfide.
General procedure for the synthesis of diazacyclobutenes:
To a flame dried round bottom flask equipped with a stir bar was added a solution
of 4-phenyl1,2,4-triazoline-3,5-dione (PTAD) (1 equiv.) in dry acetonitrile (5 mL for 1
mmol of PTAD). To this stirring solution was added dropwise a solution of alkynyl
sulfide or alkynyl selenide (1.3 equiv.) in dry acetonitrile (5 mL for 1.3 mmol of alkynyl
substrate). Then the round bottom flask was attached to a water-cooled condenser and the
mixture was refluxed for 24 hours. The resultant mixture was concentrated under reduced
pressure and purified via flash chromatography with hexane and ethyl acetate (gradient
from 100% hexane to 80:20 hexane/ethyl acetate) to afford the corresponding
diazacyclobutene.

285
Characterization of alkynyl sulfides and selenides:

286

287

288

289
Characterization of diazacyclobutenes:
5i
5j
5k

290
5l
5m
5n

291
5l
5m
5n
5o
5p
5q
292
5r
5s
293
5t
5u

294
5v
5w
5x

295
5y
5z
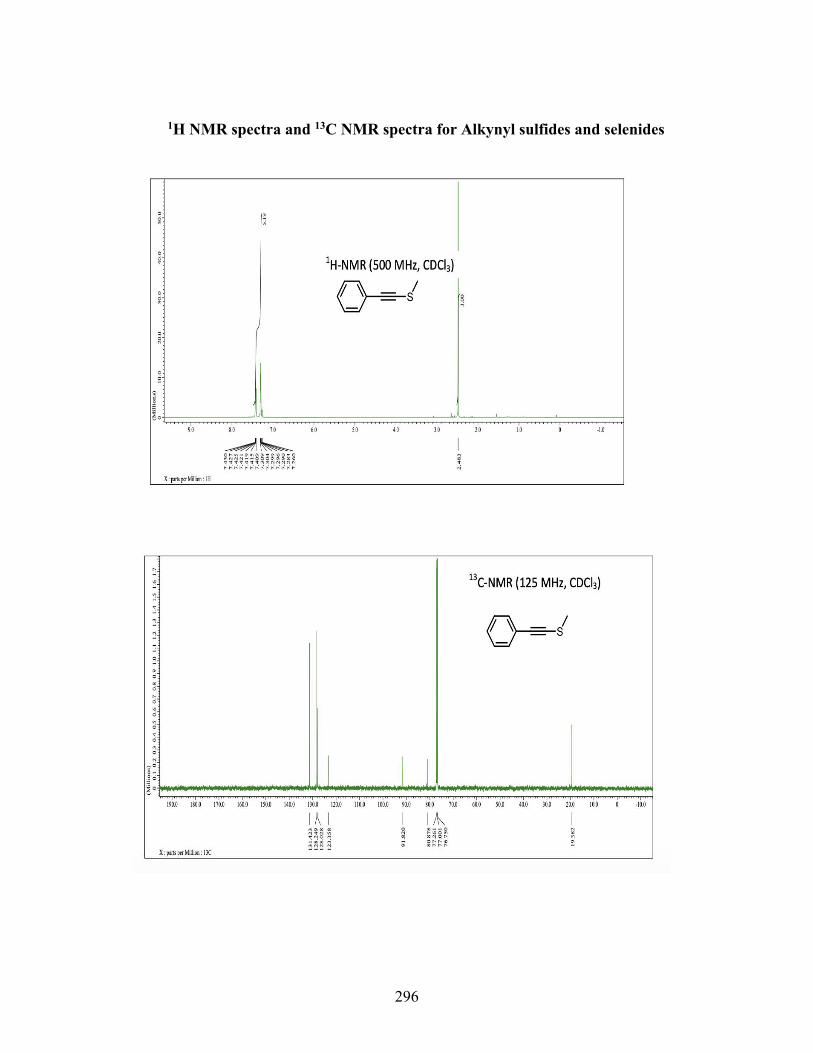
296
1H NMR spectra and 13C NMR spectra for Alkynyl sulfides and selenides
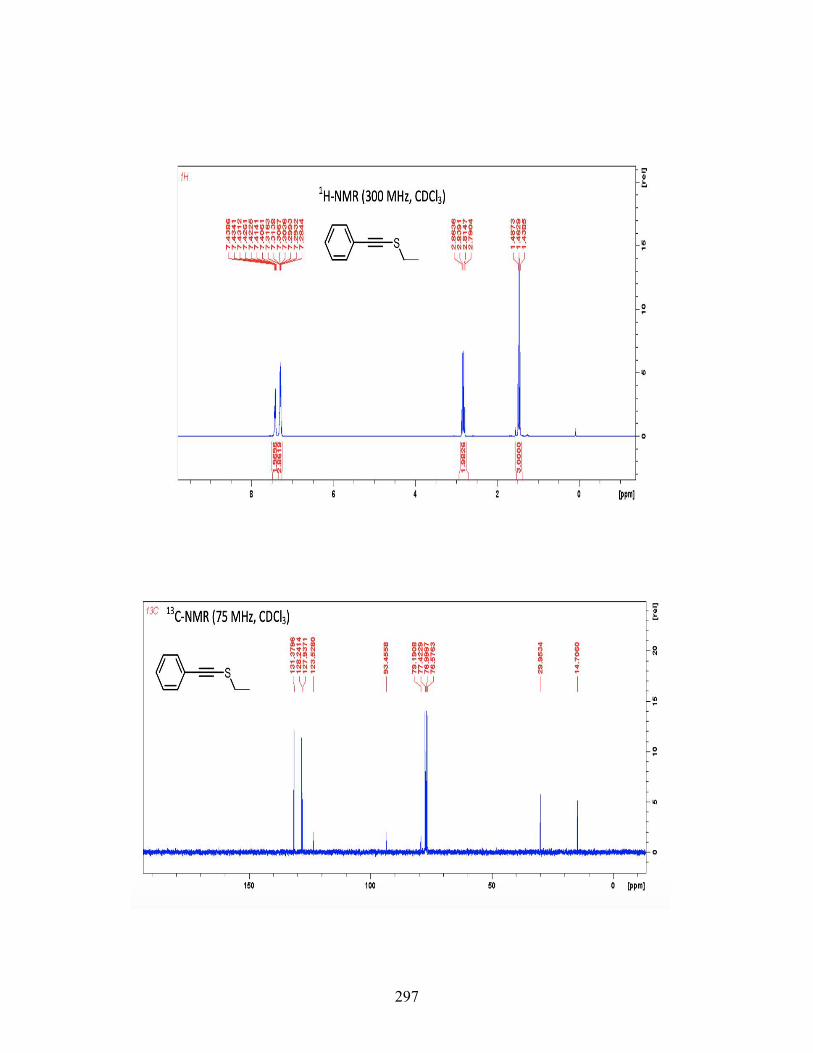
297
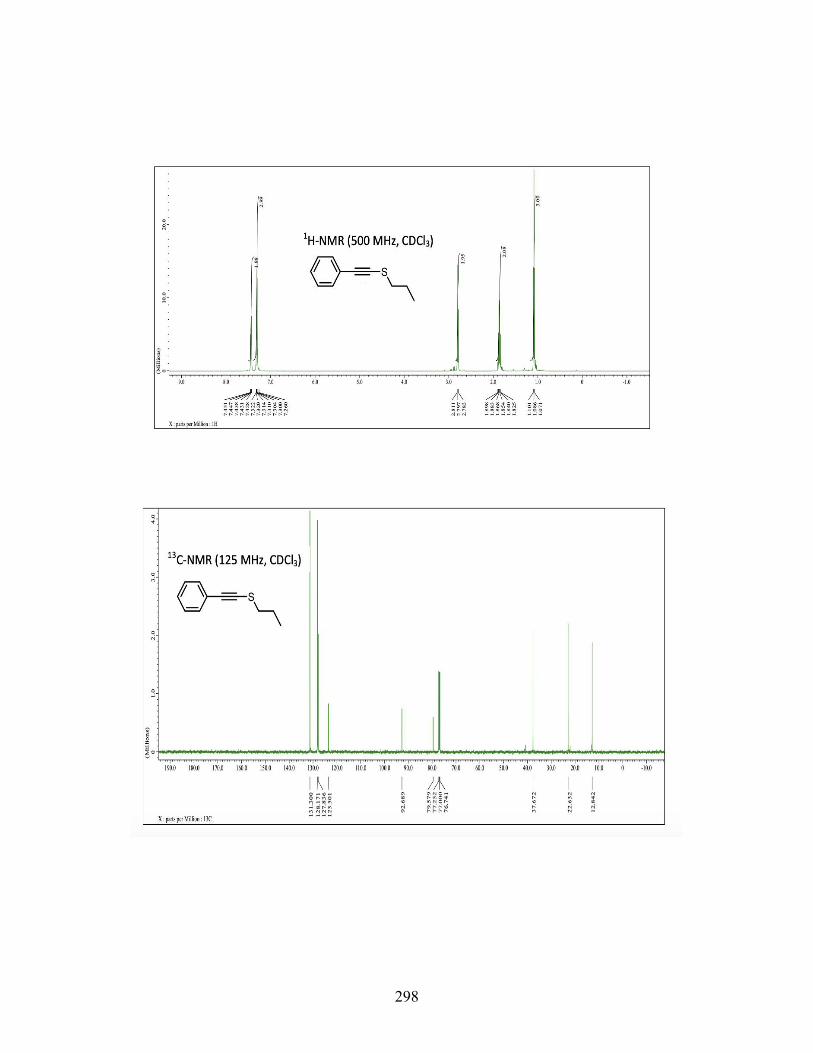
298
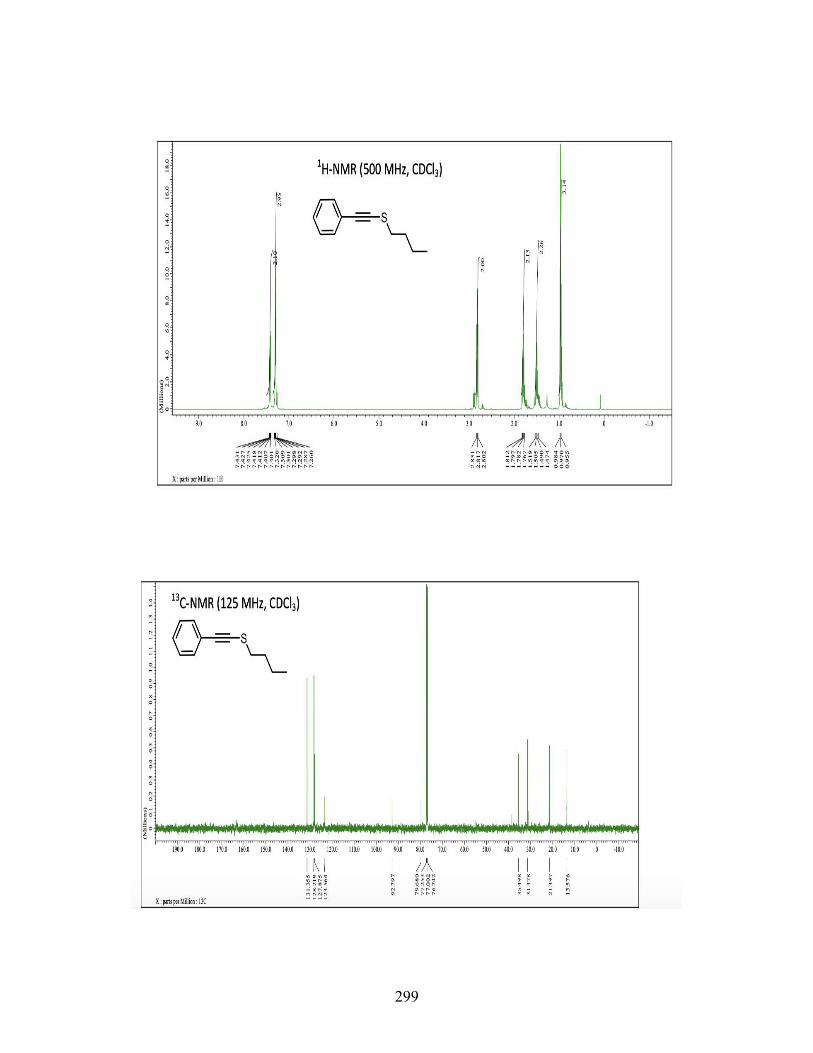
299
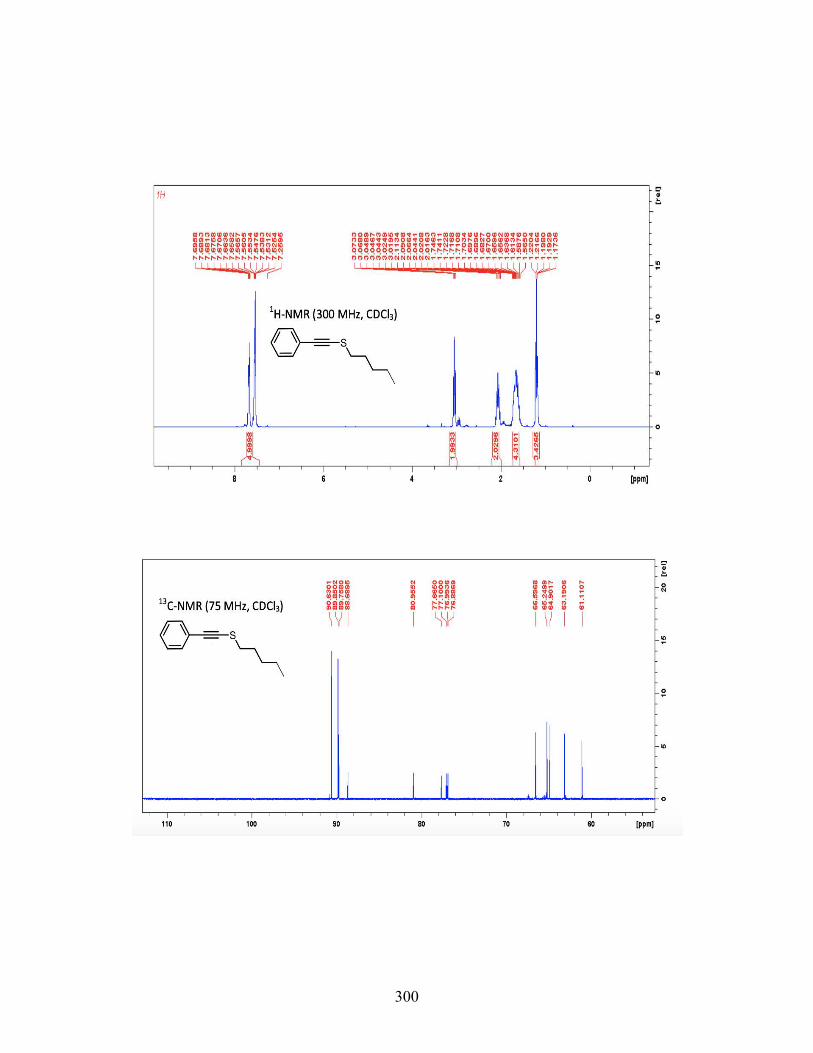
300
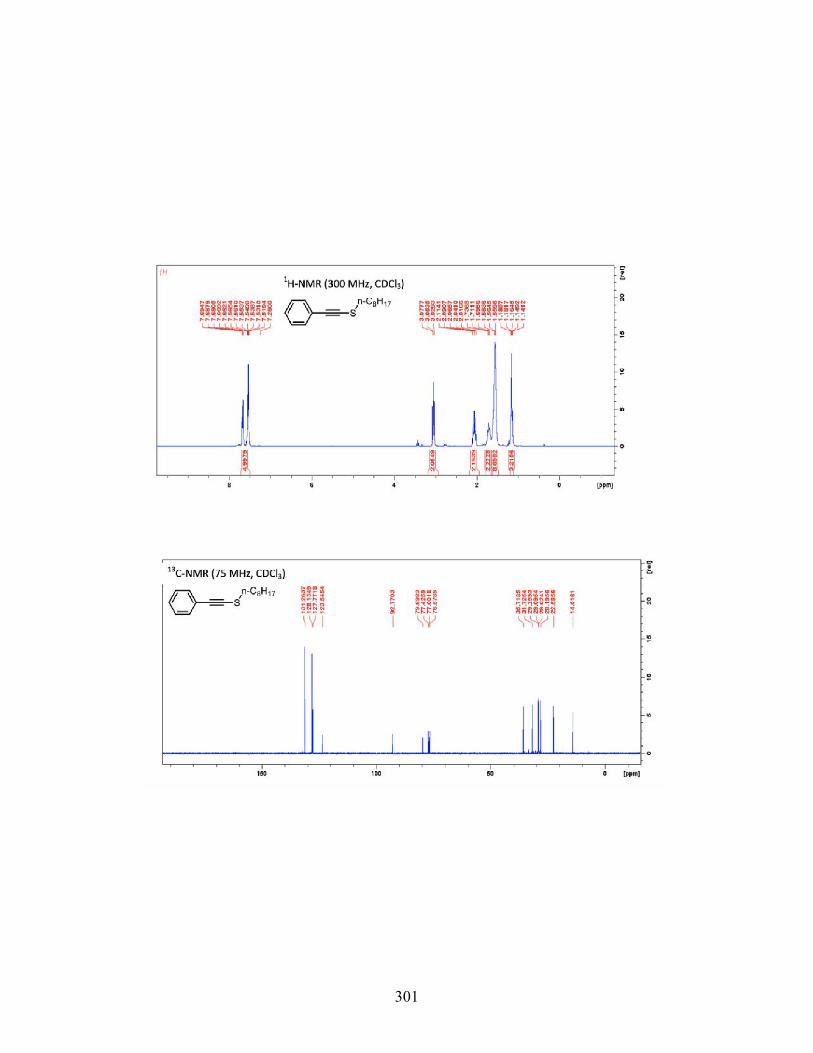
301
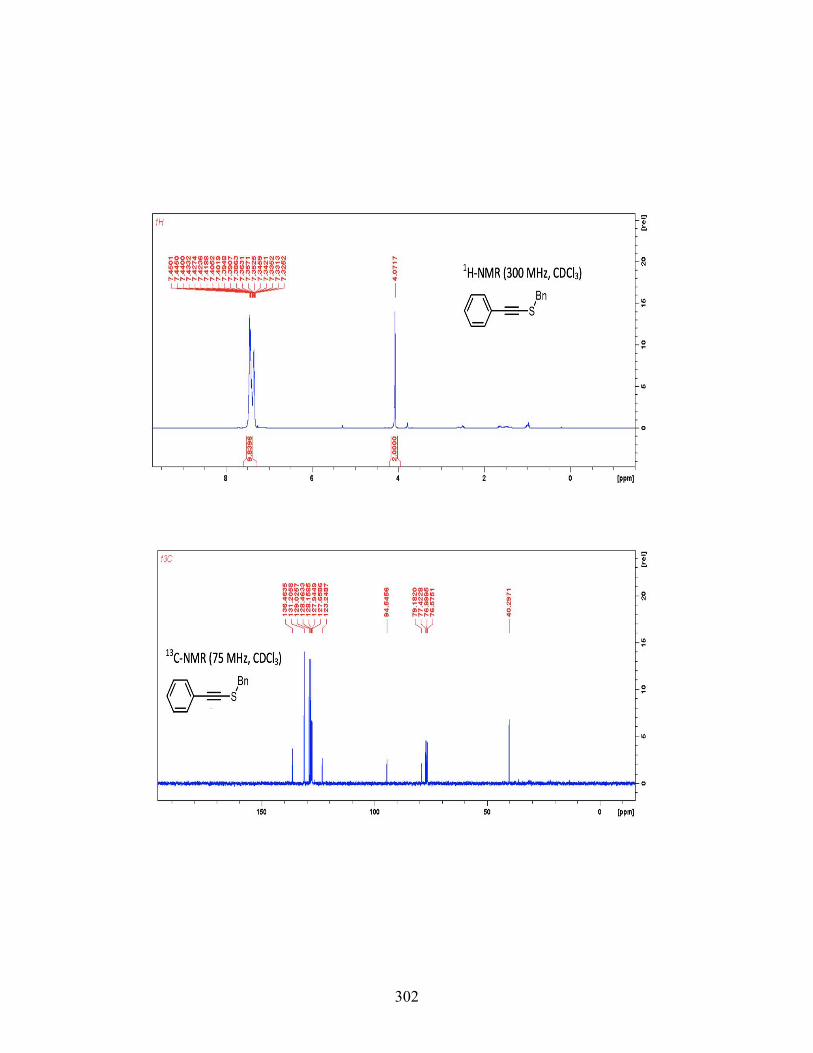
302
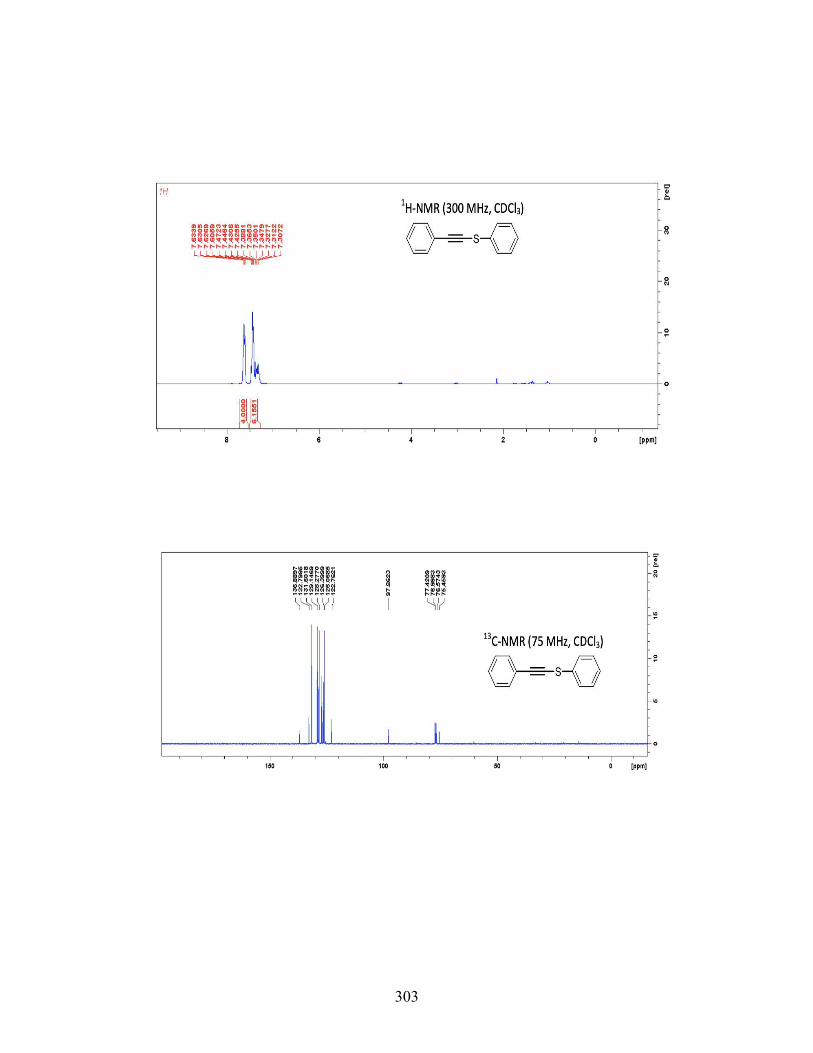
303
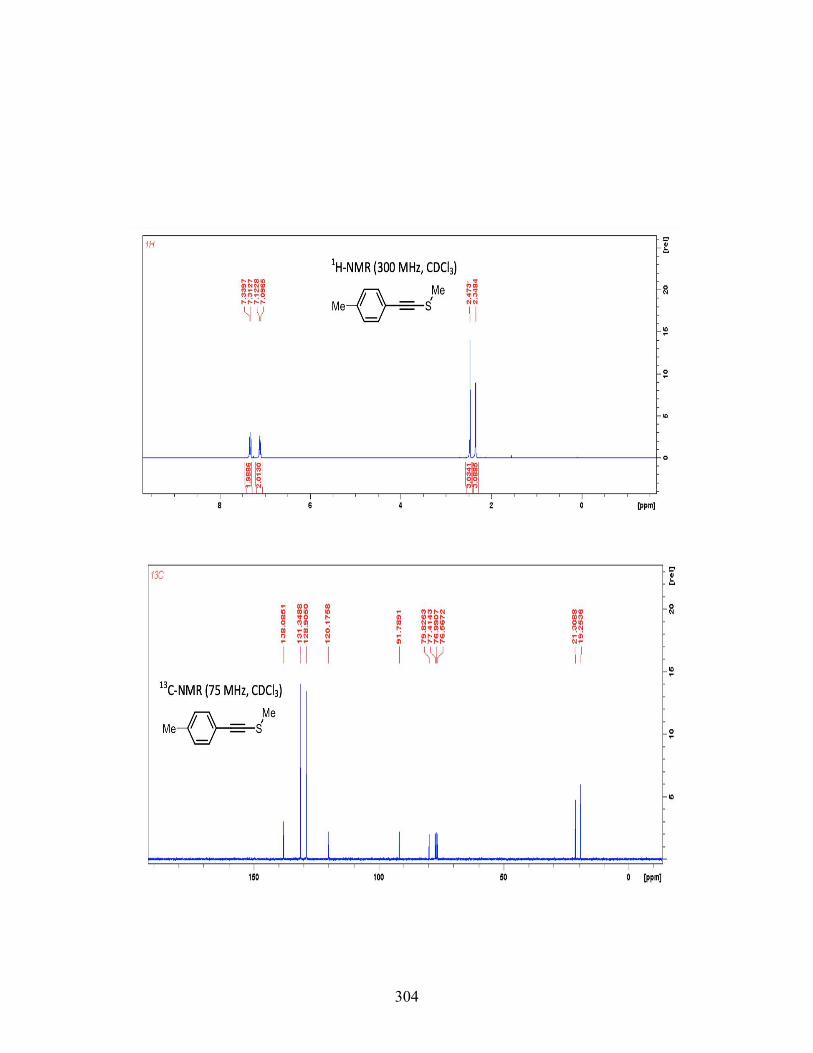
304
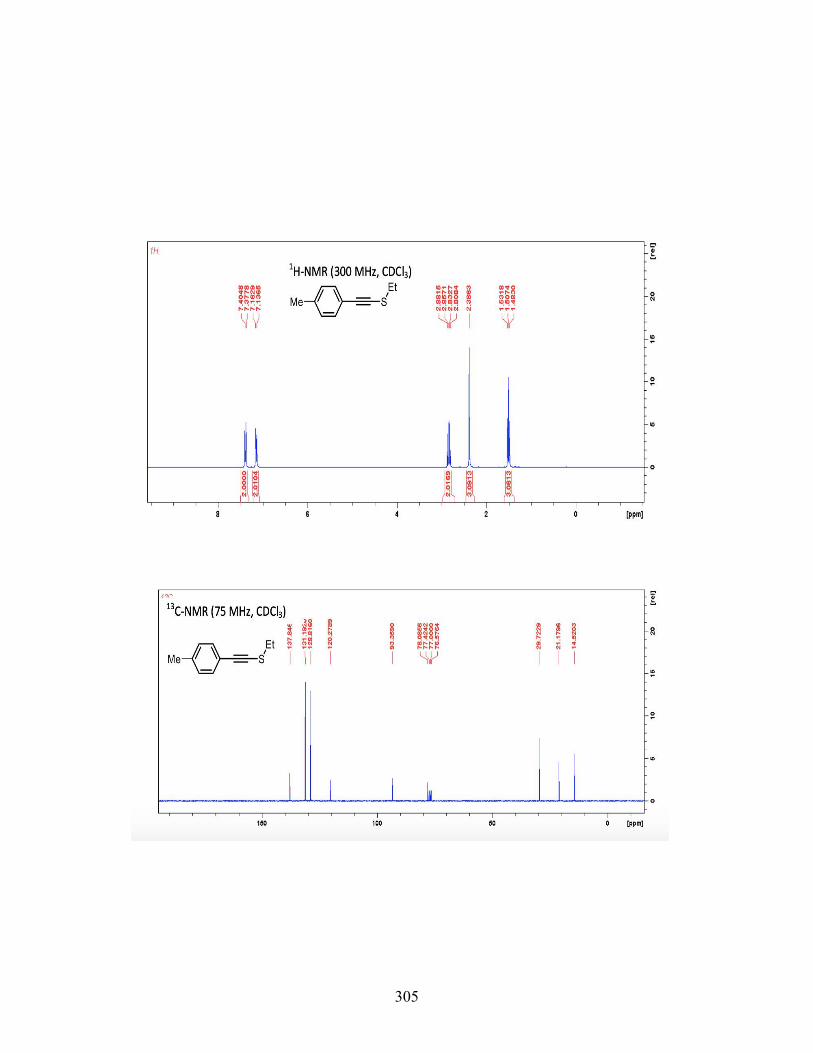
305
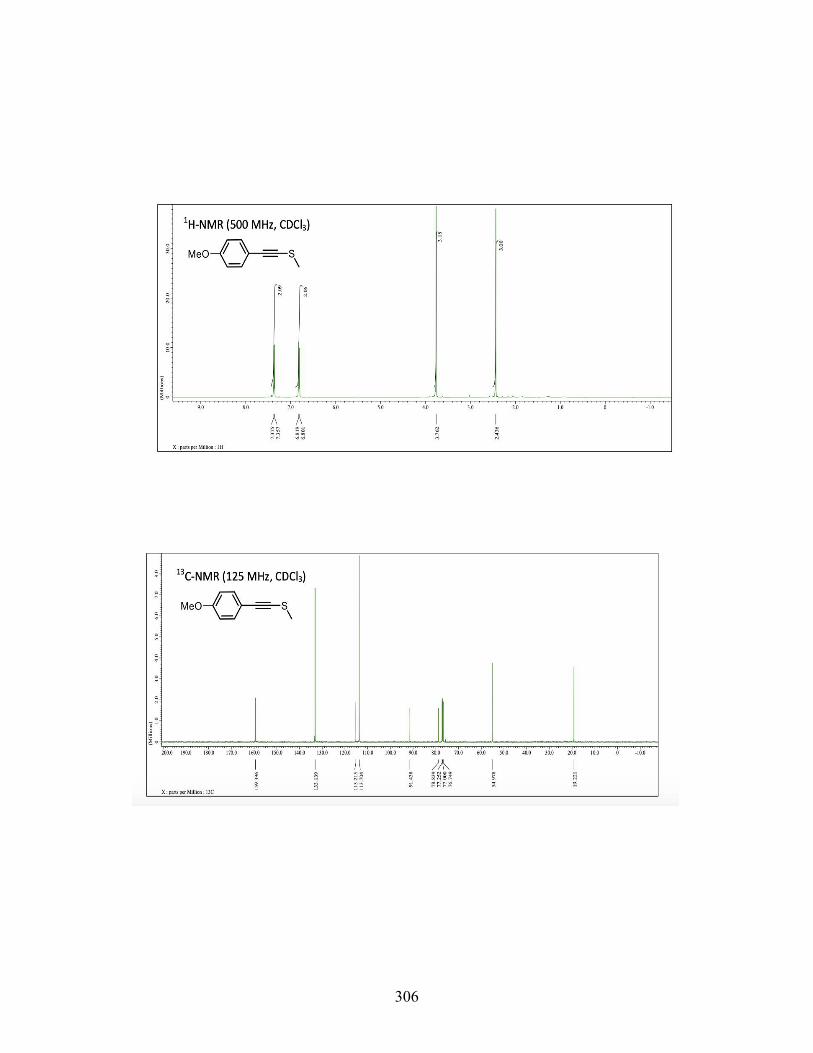
306
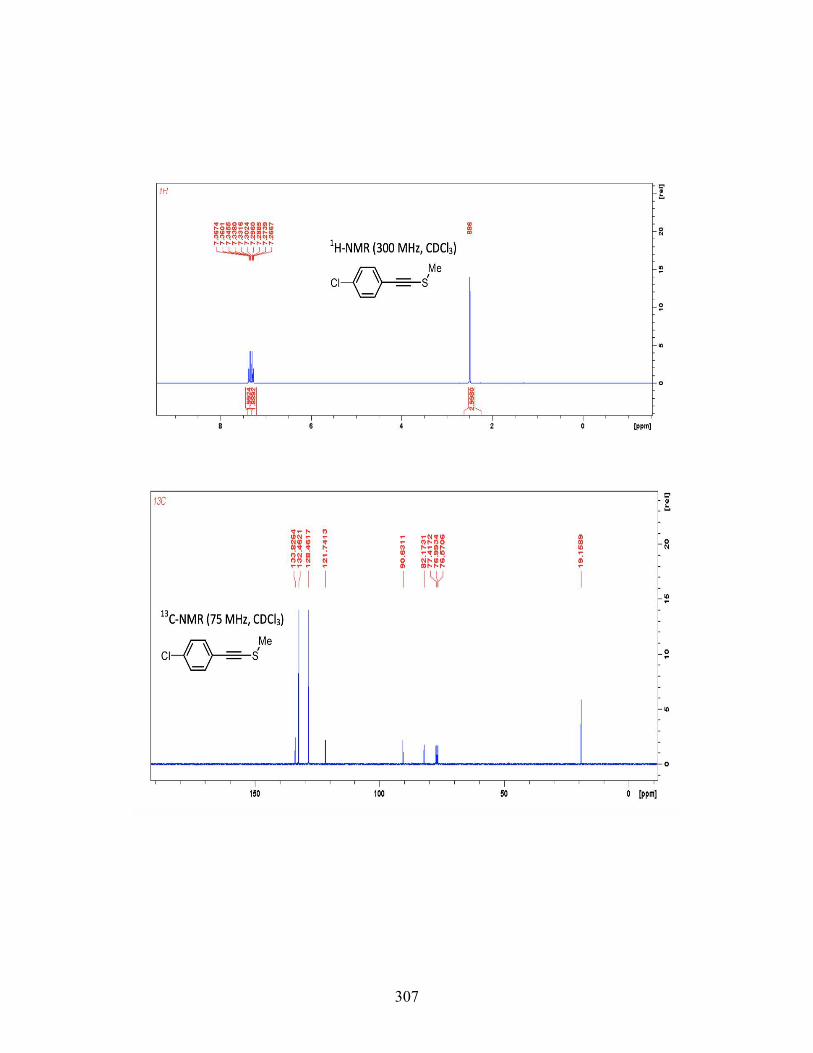
307
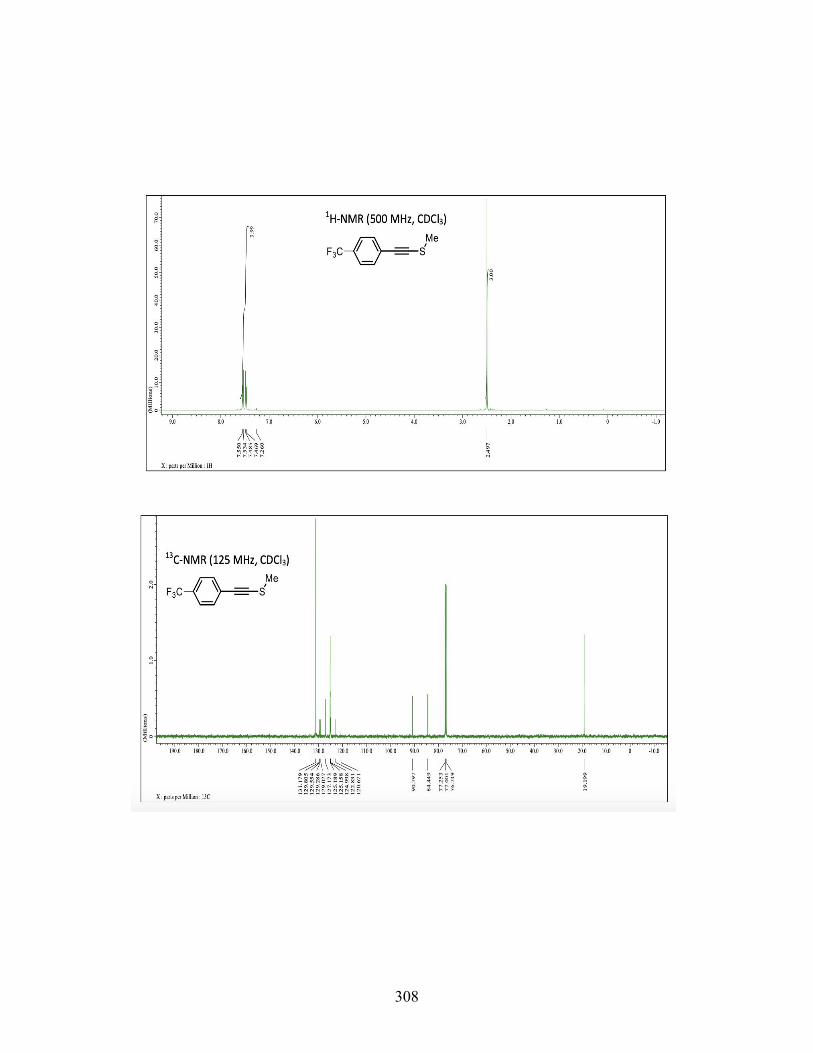
308
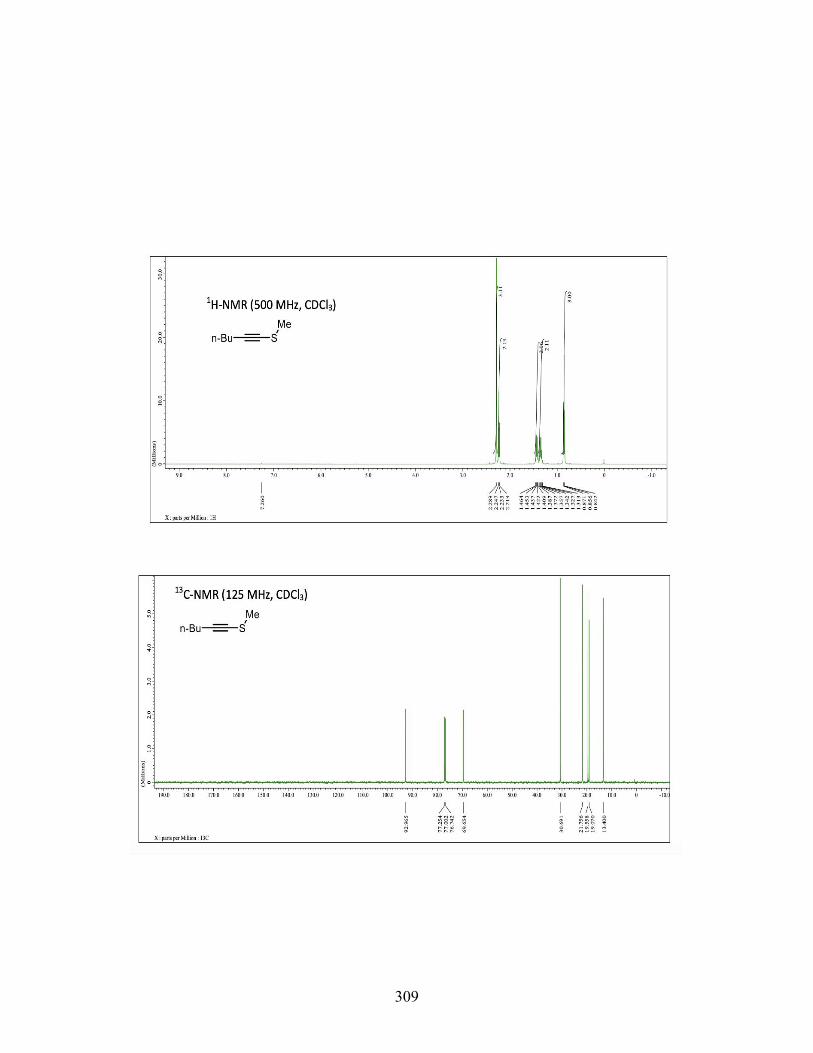
309
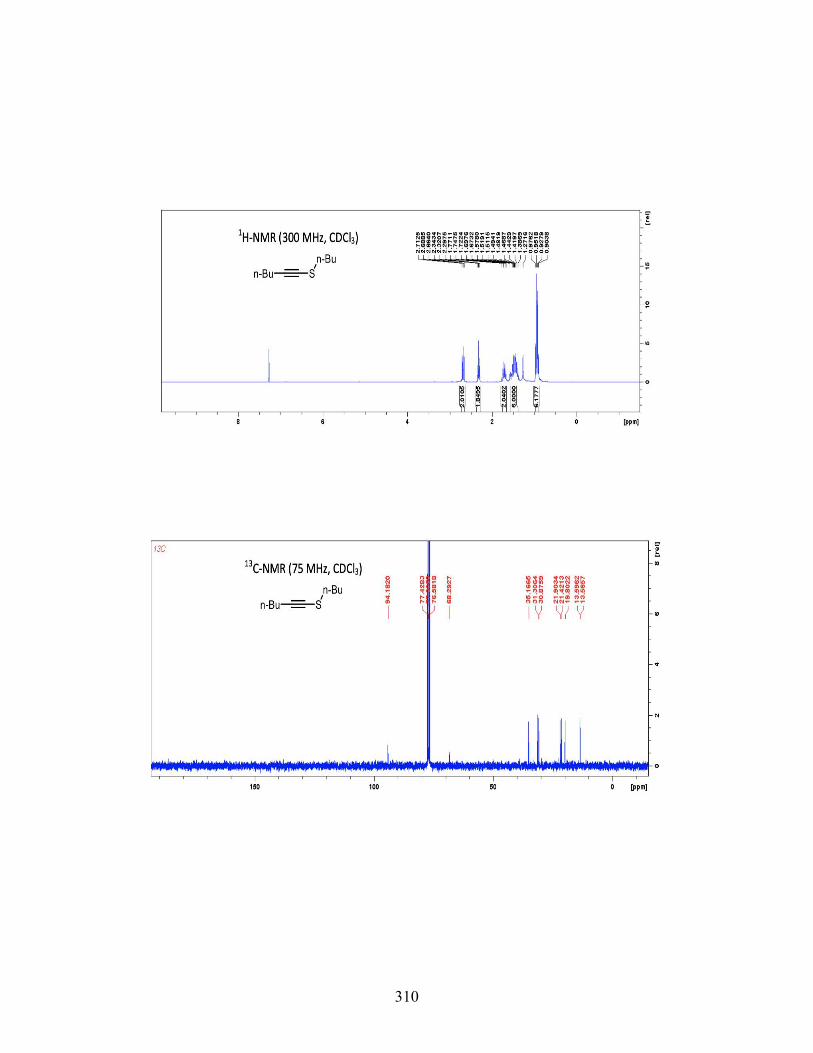
310
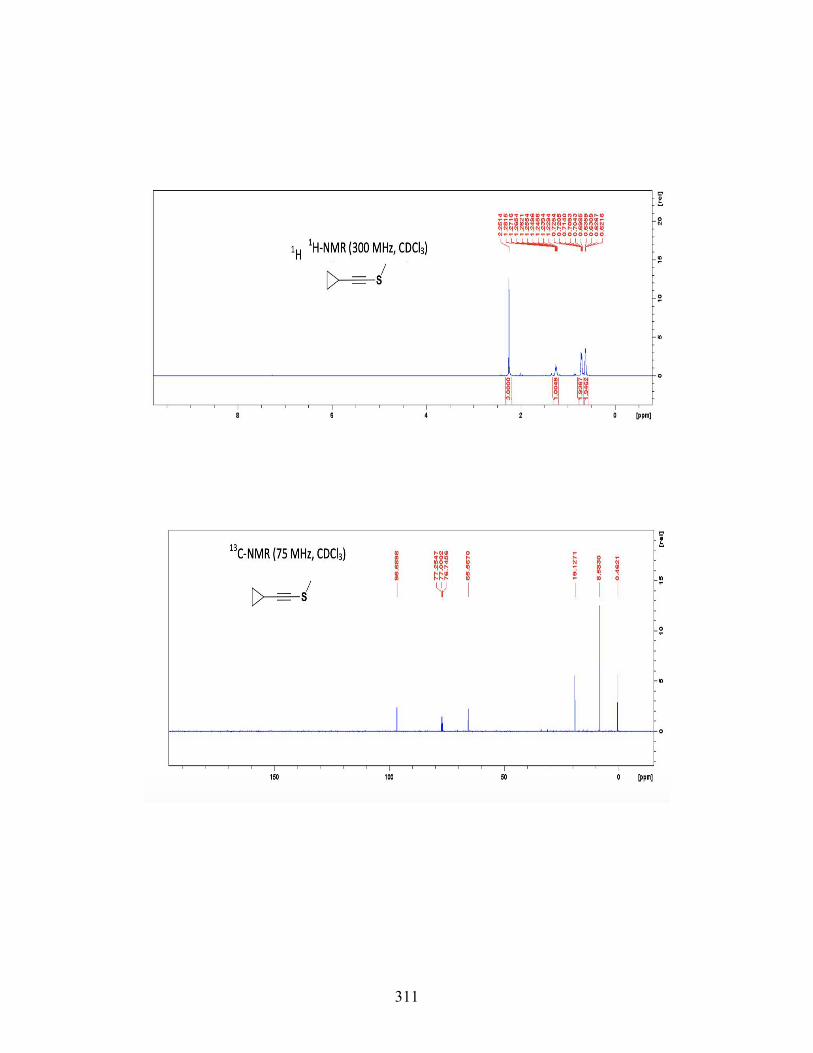
311
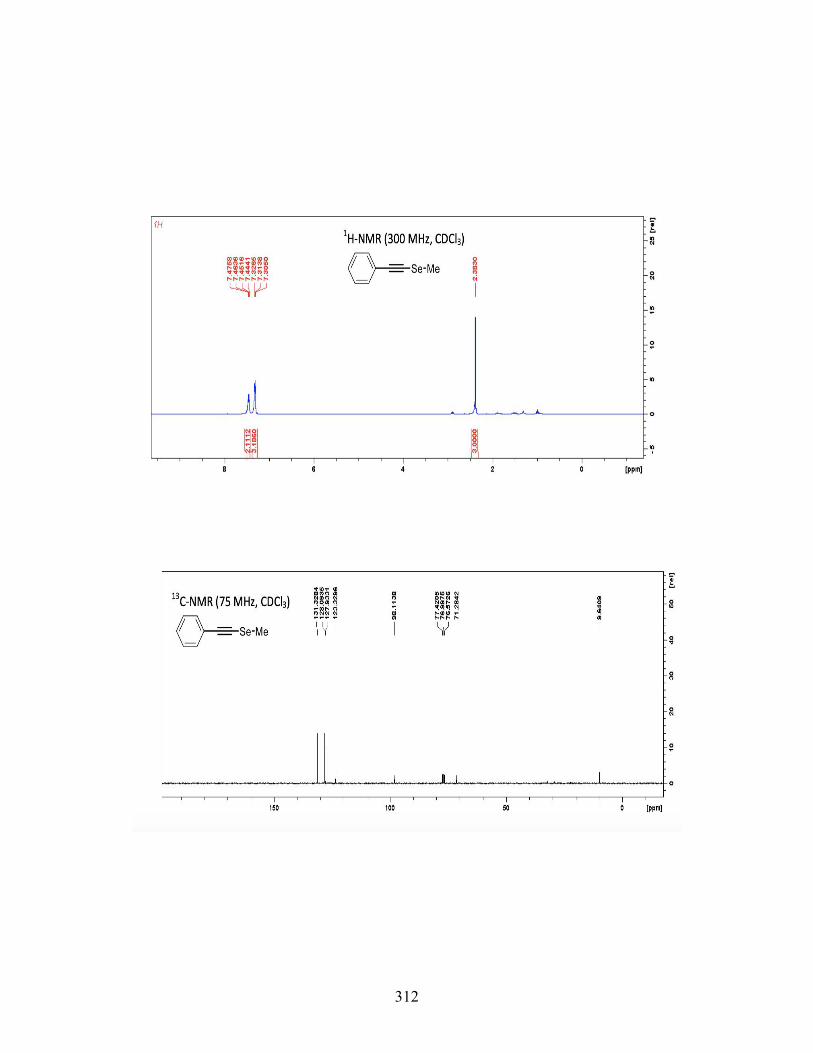
312
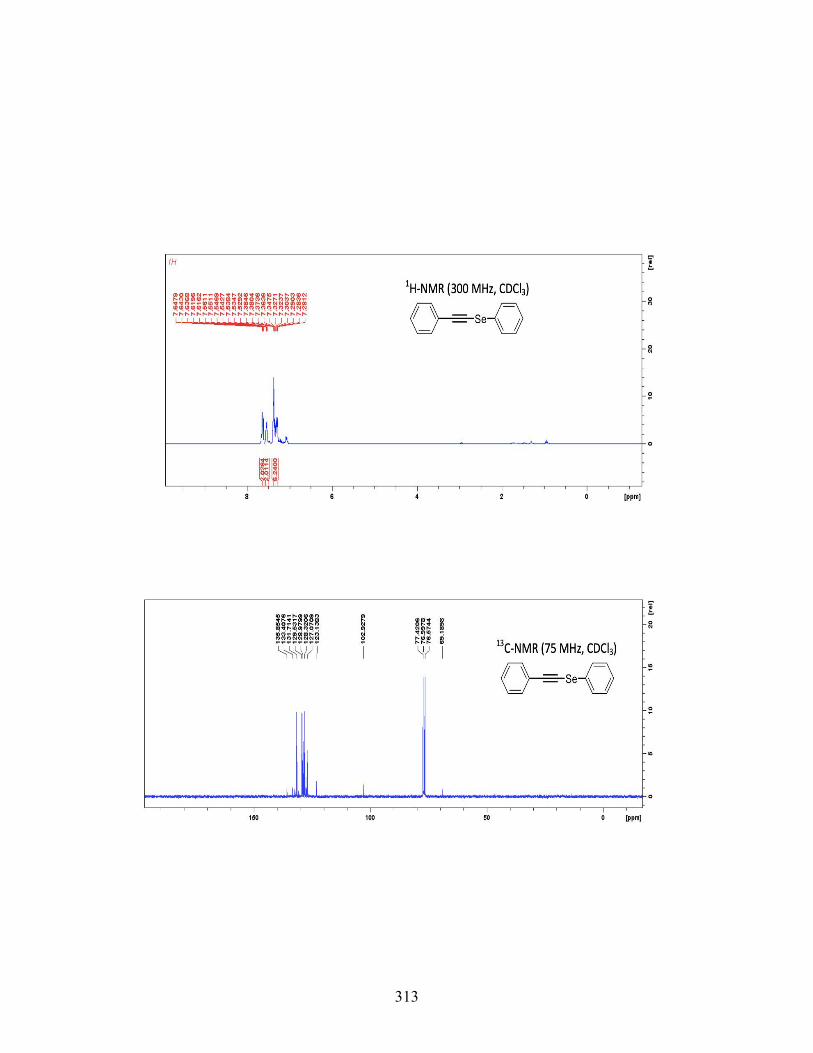
313
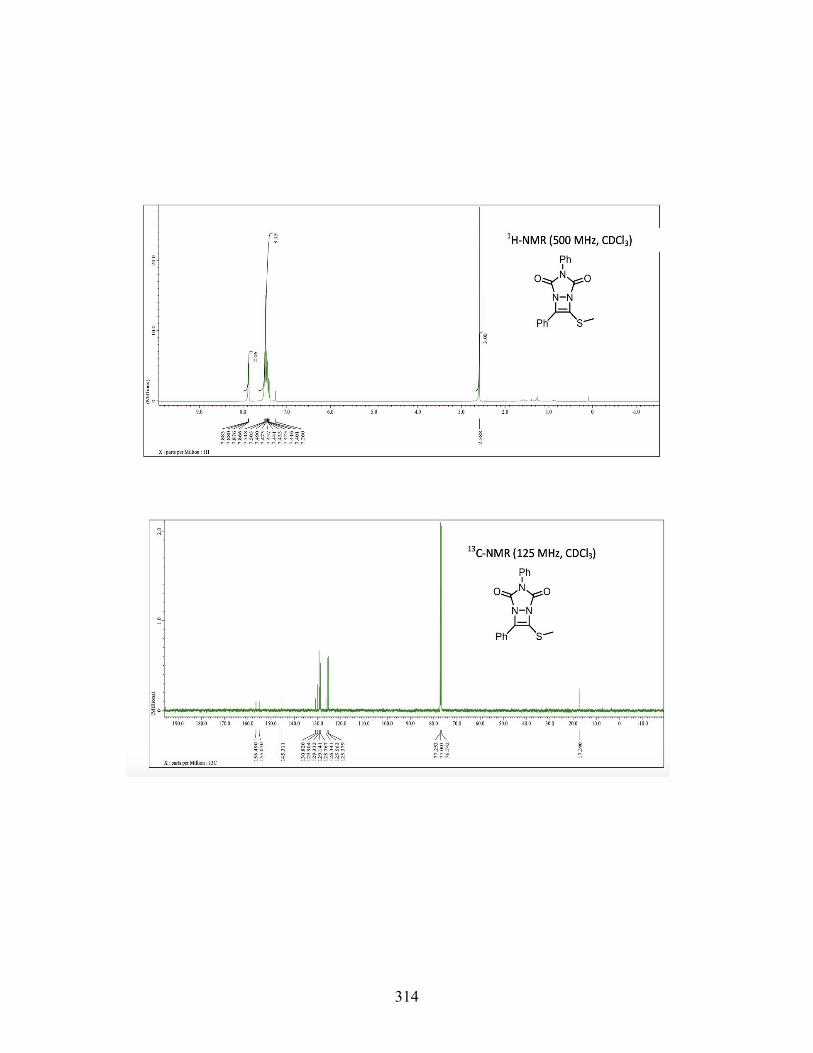
314
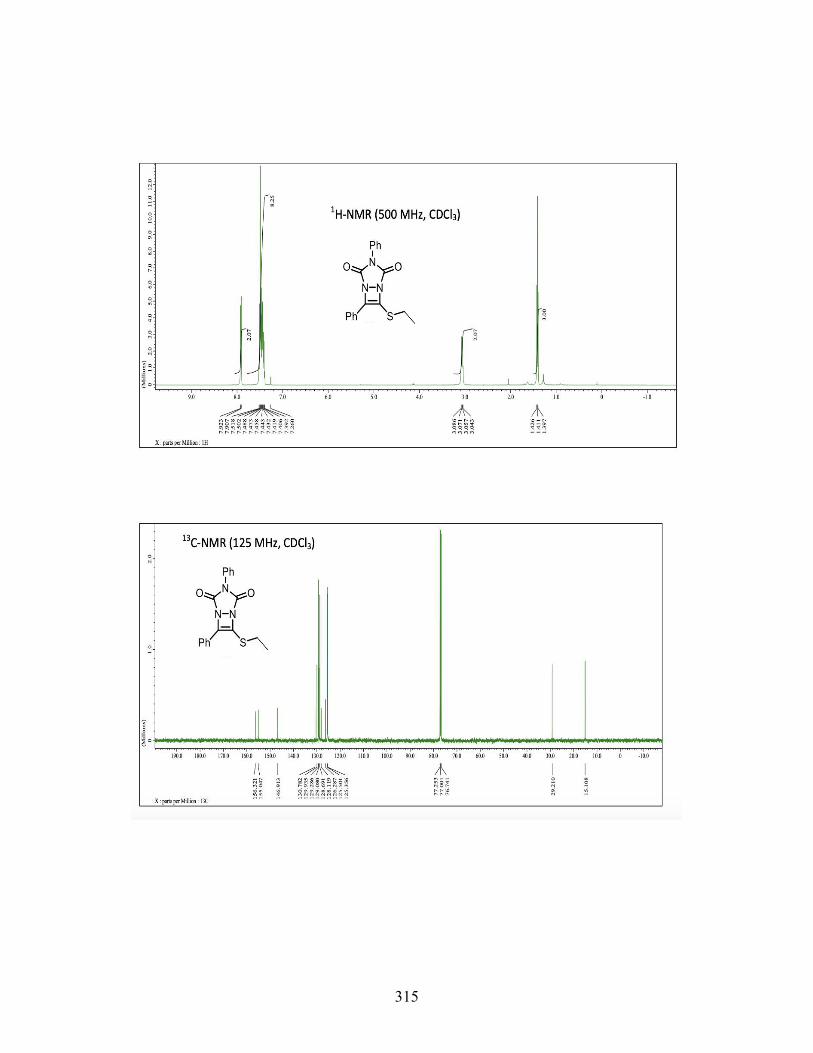
315
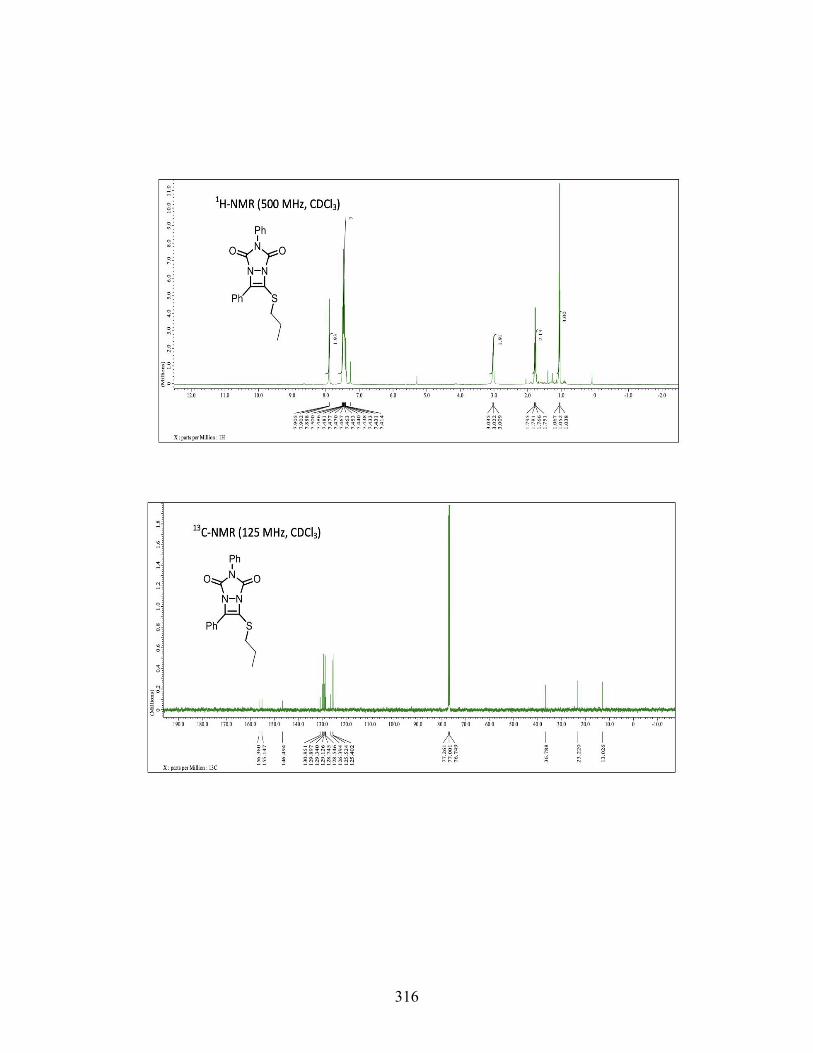
316
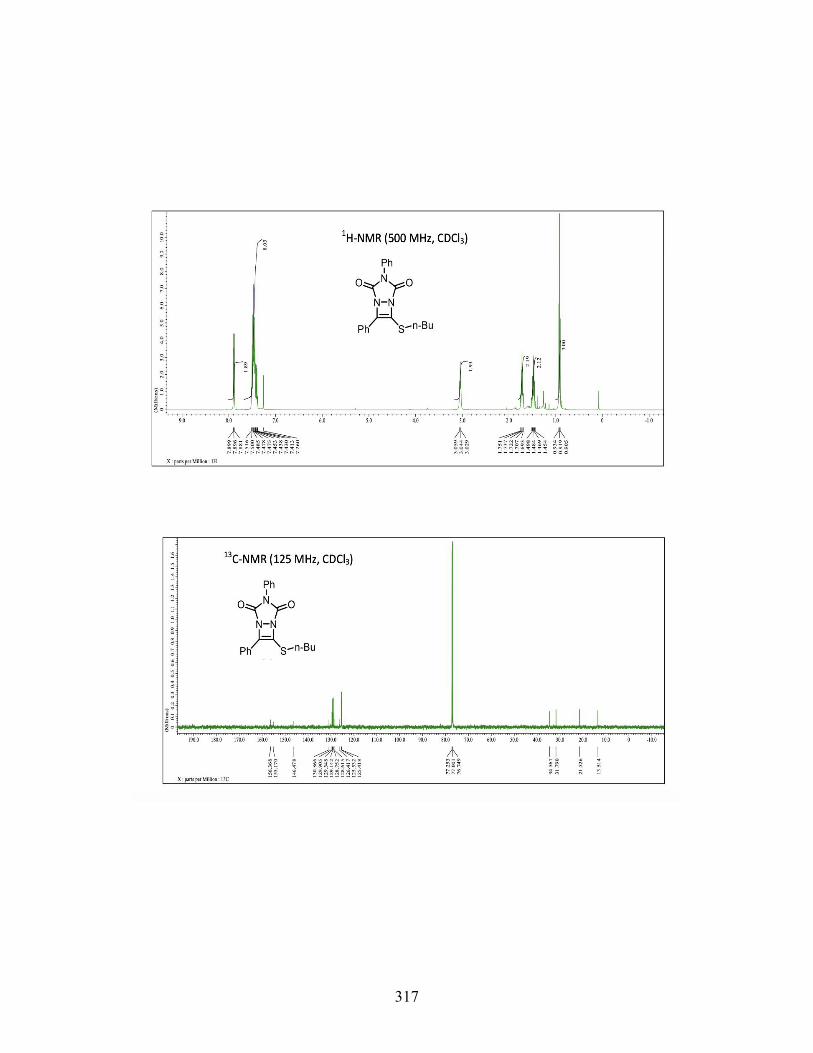
317
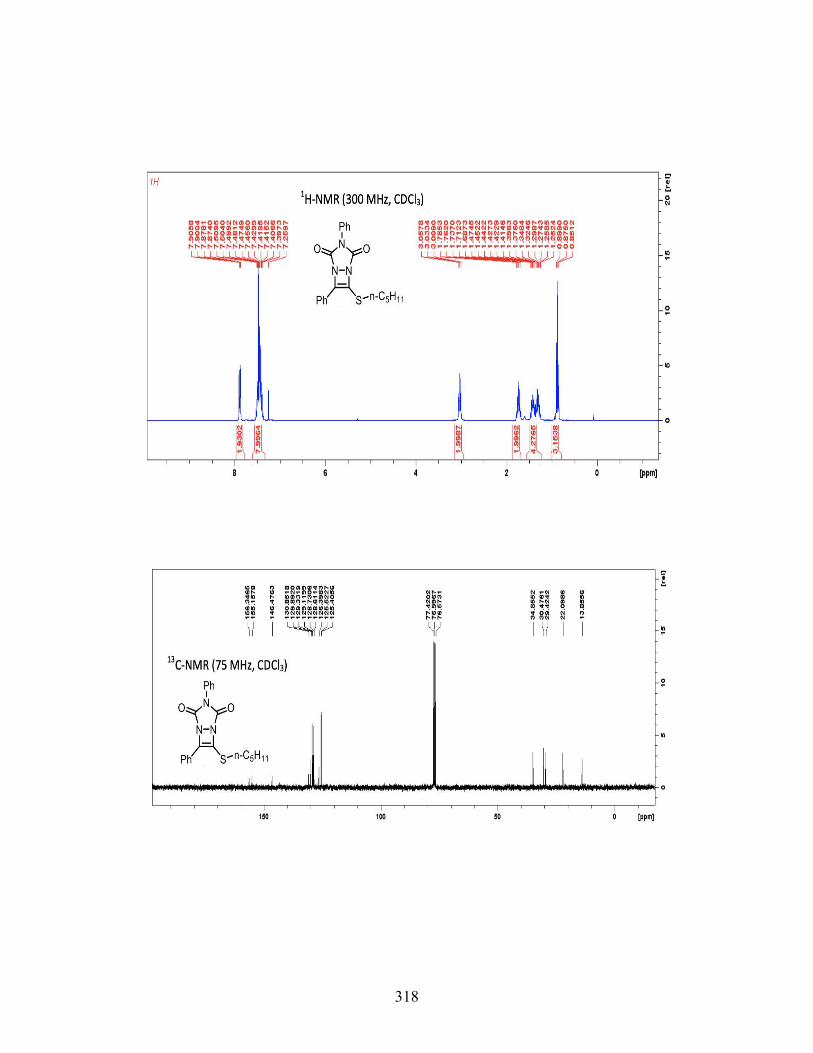
318
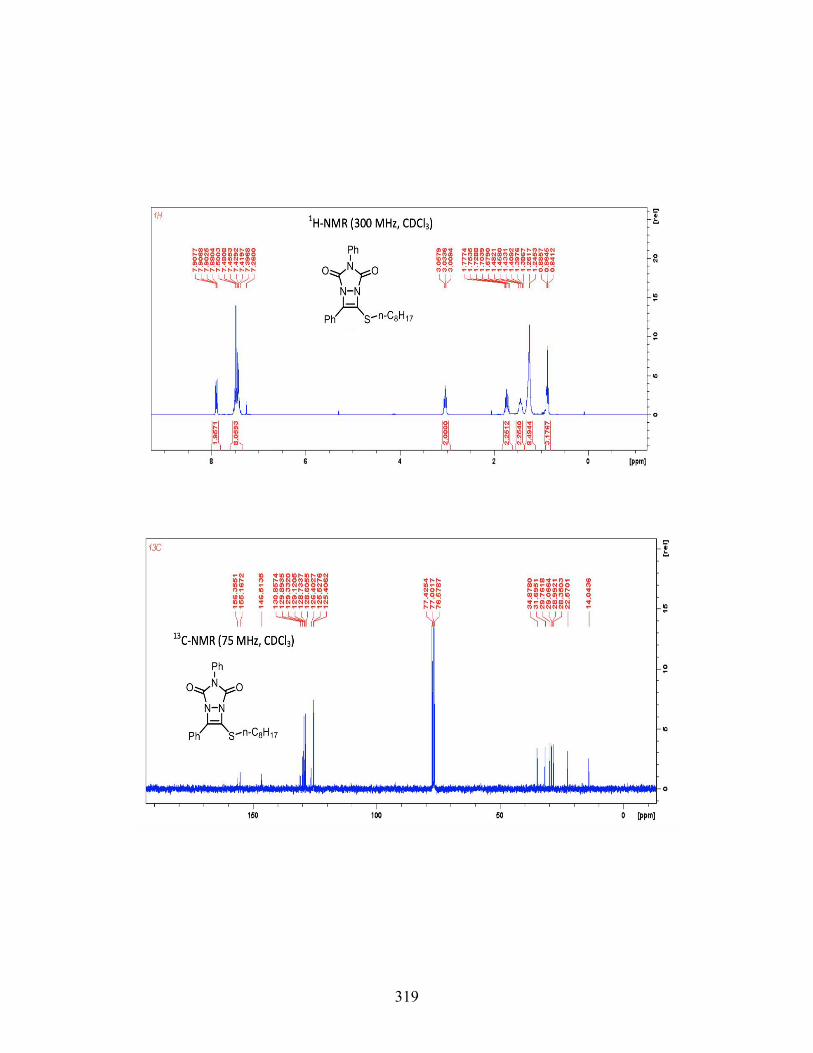
319
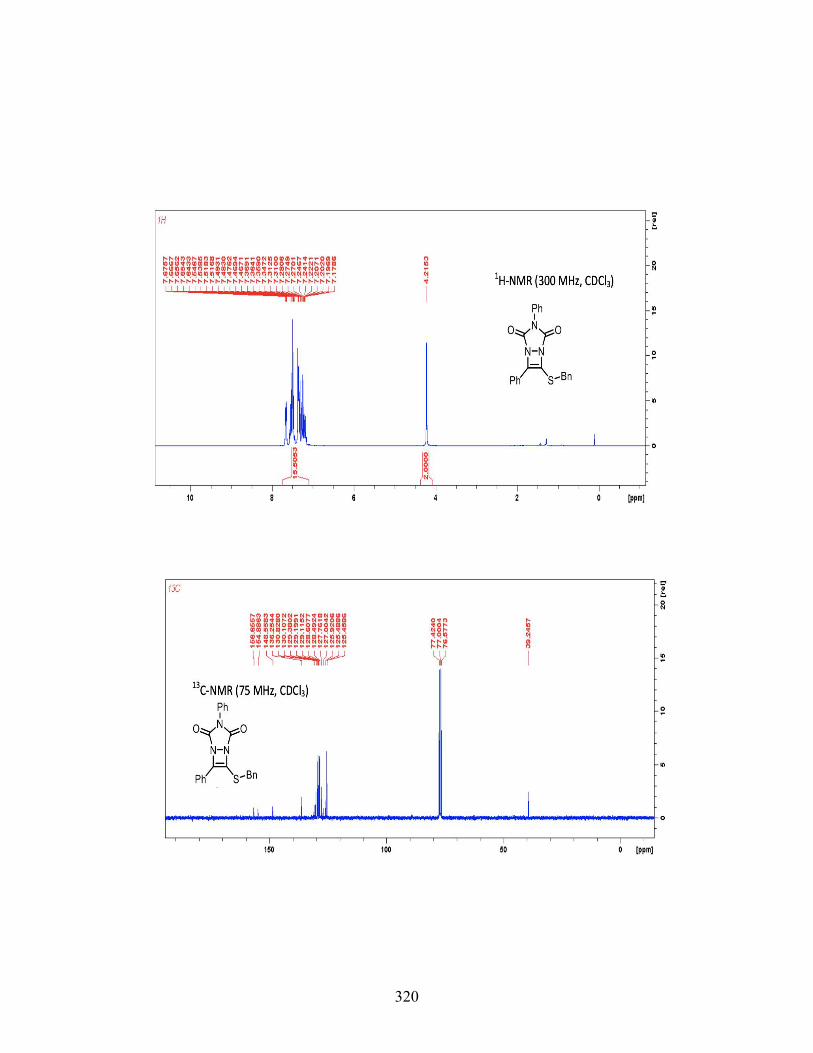
320
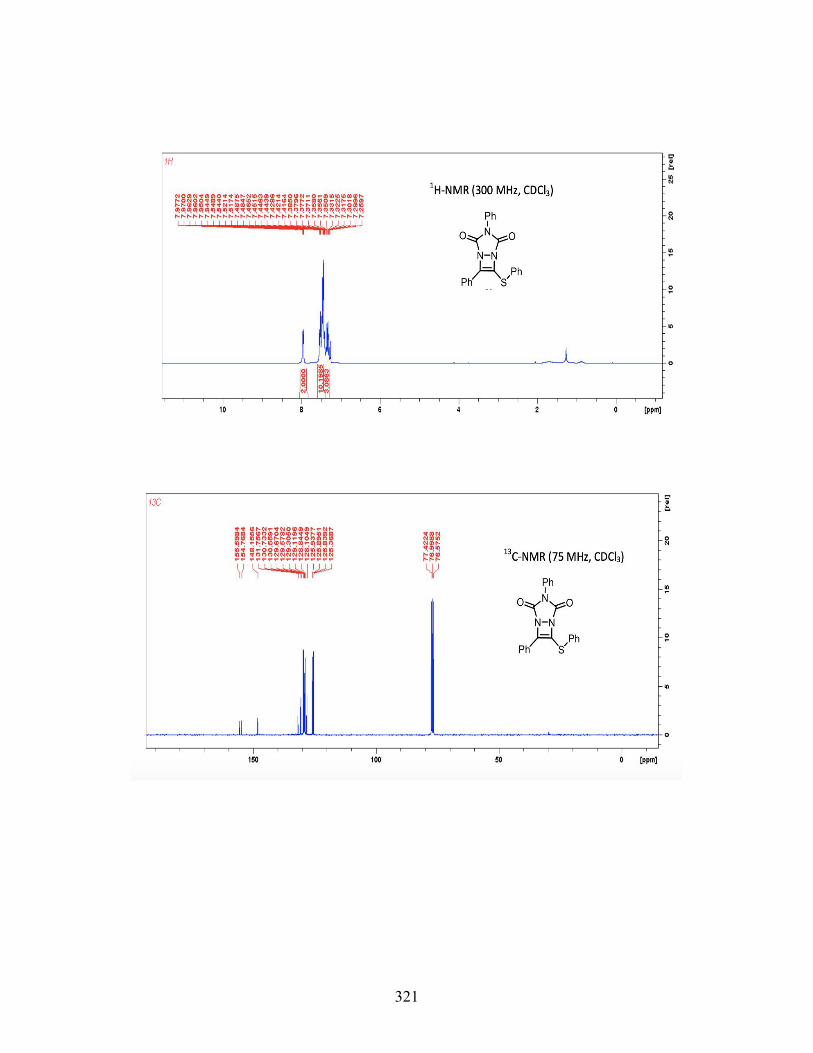
321
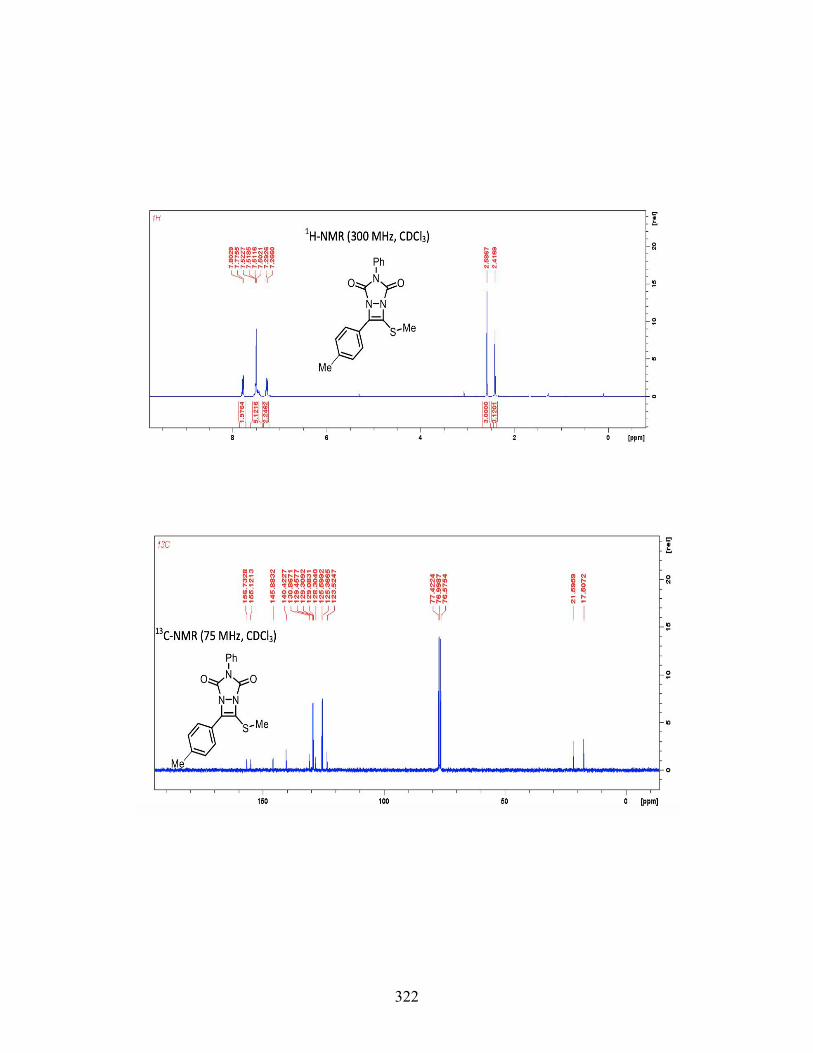
322
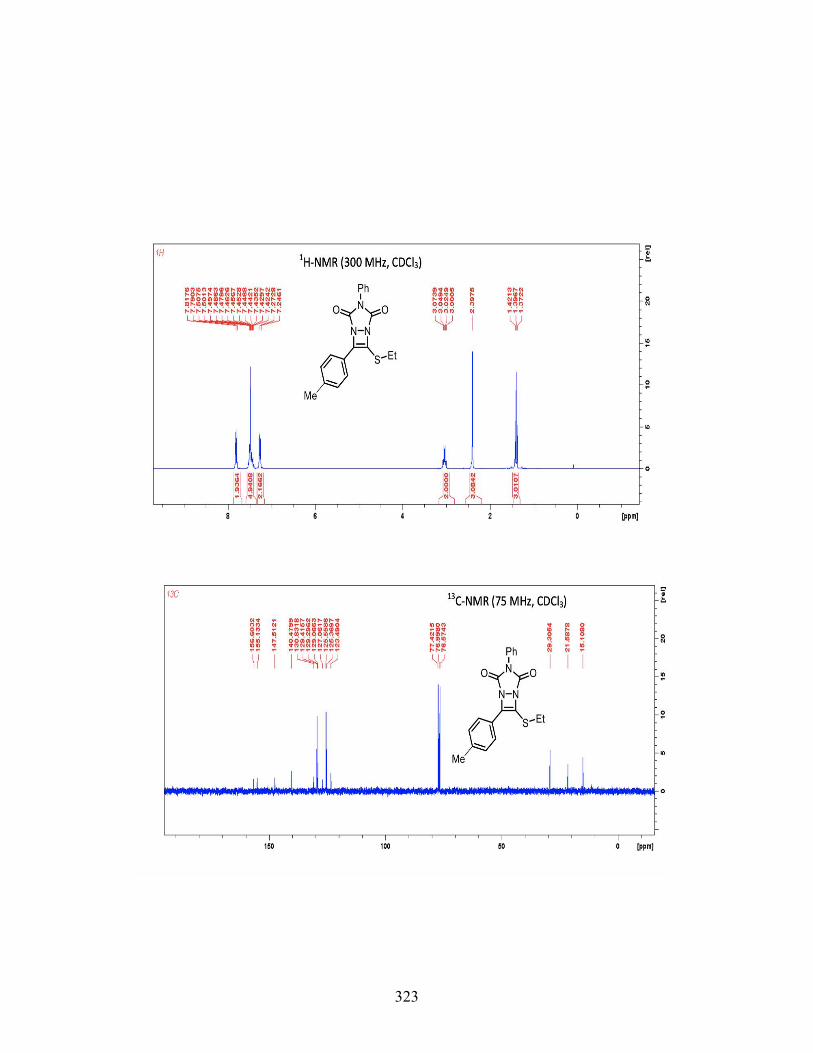
323
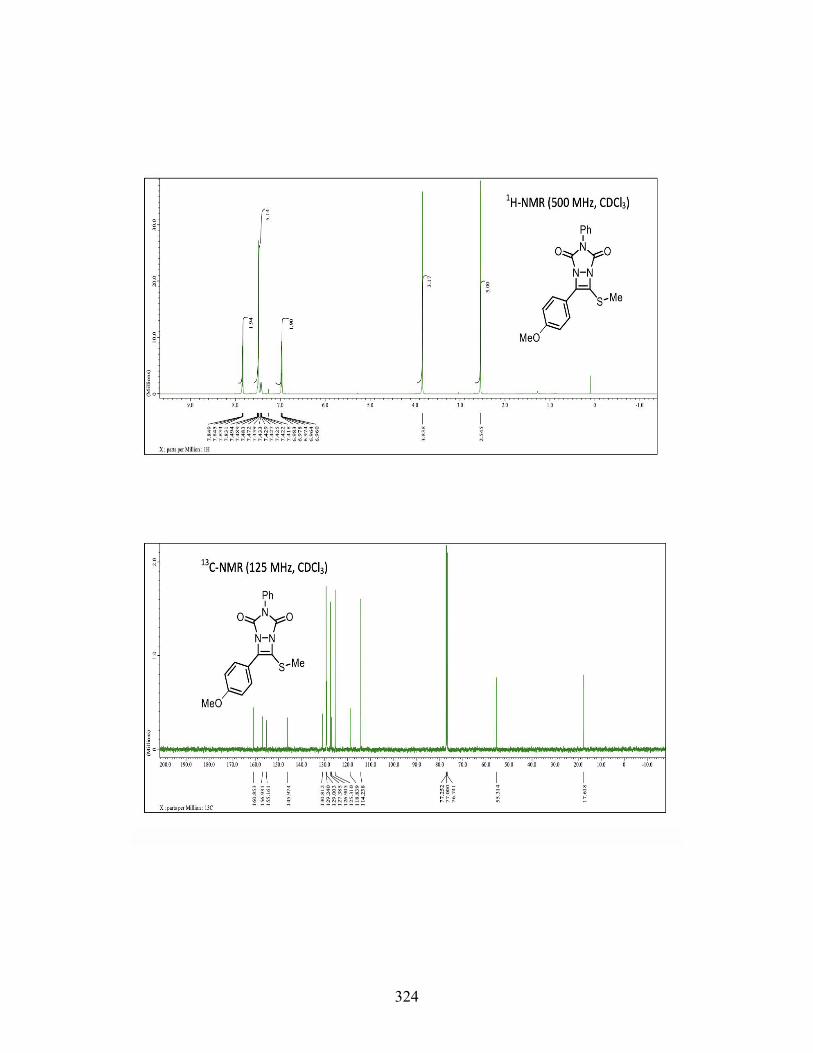
324
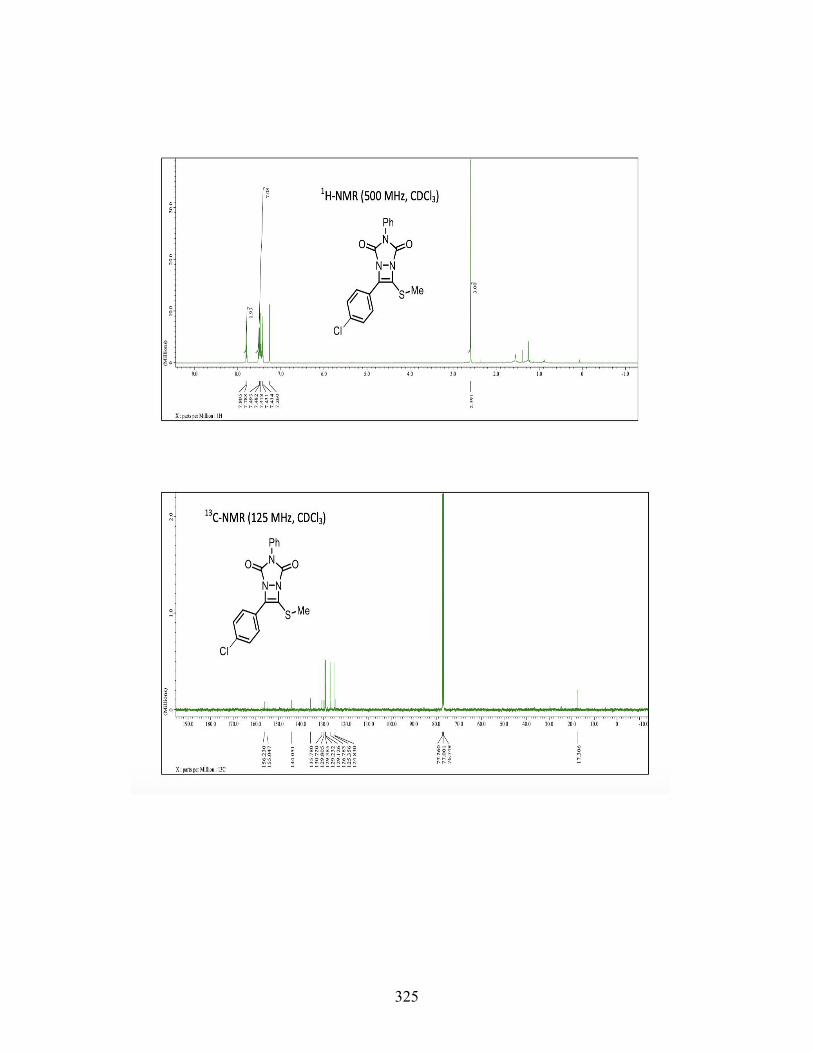
325
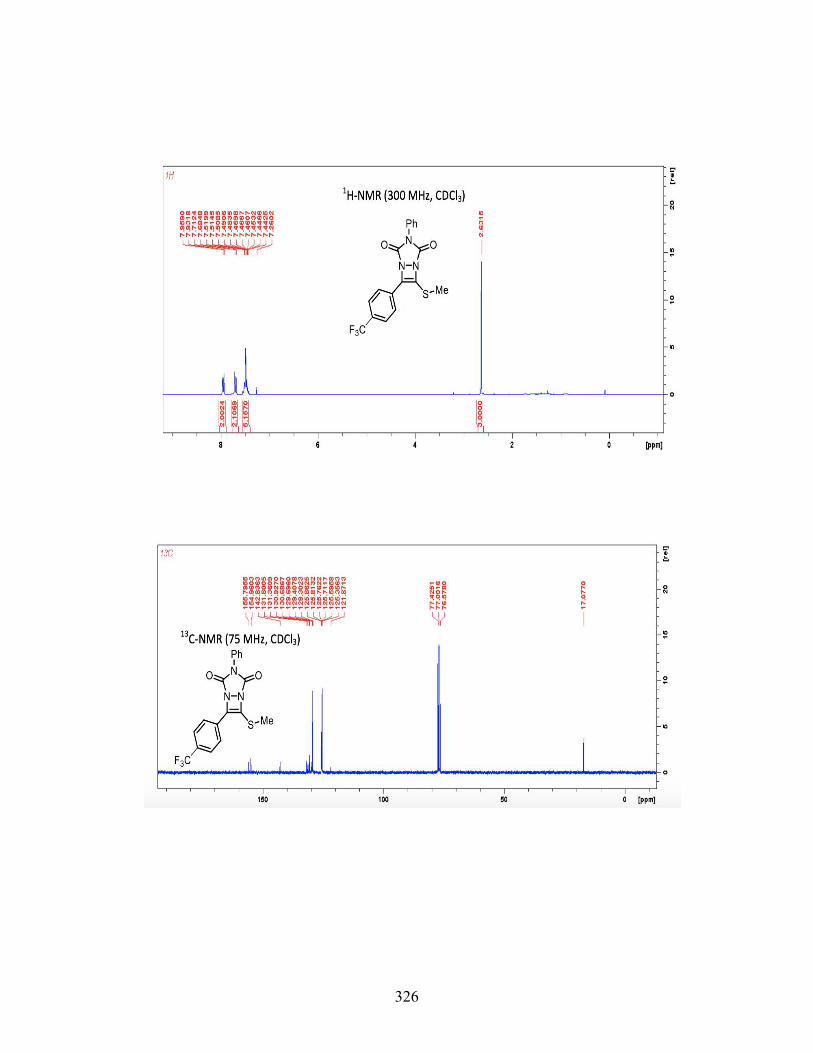
326
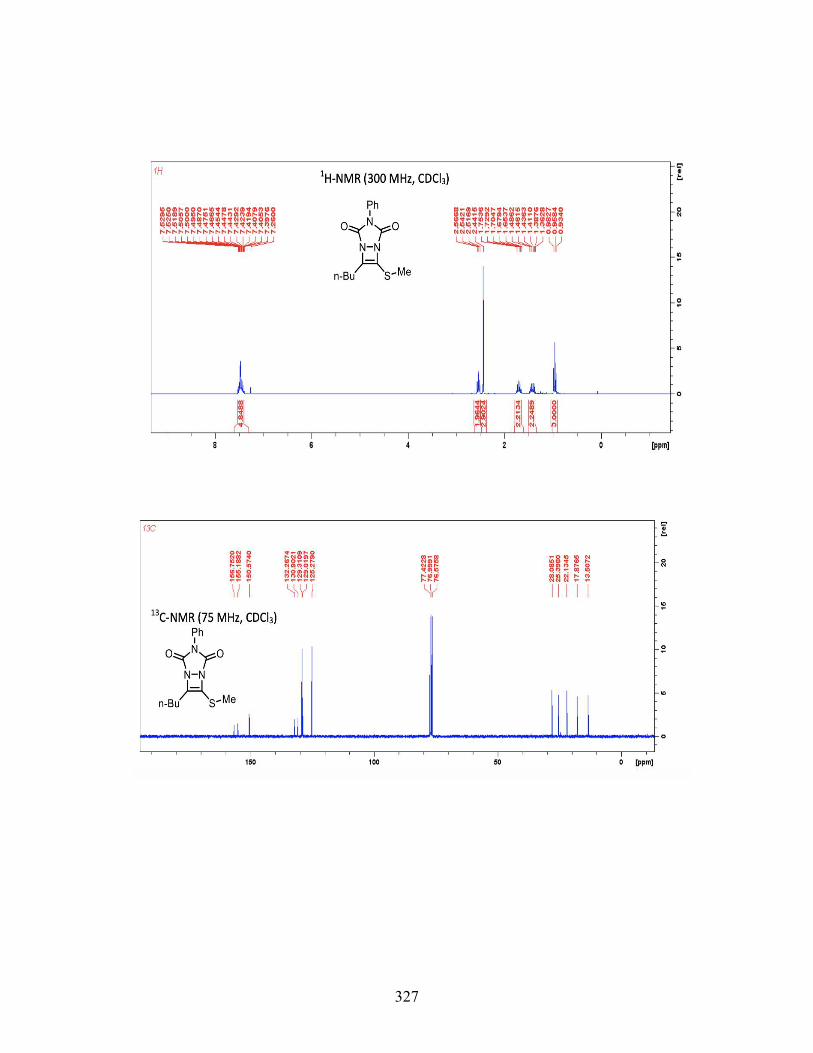
327
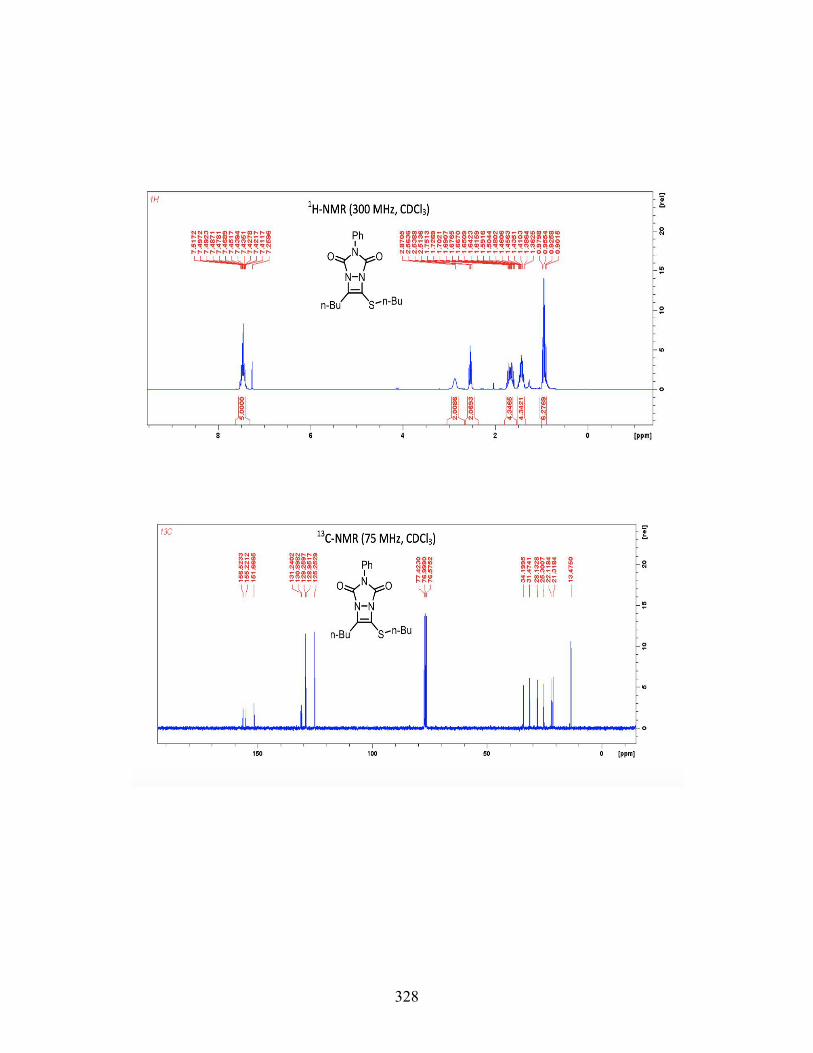
328
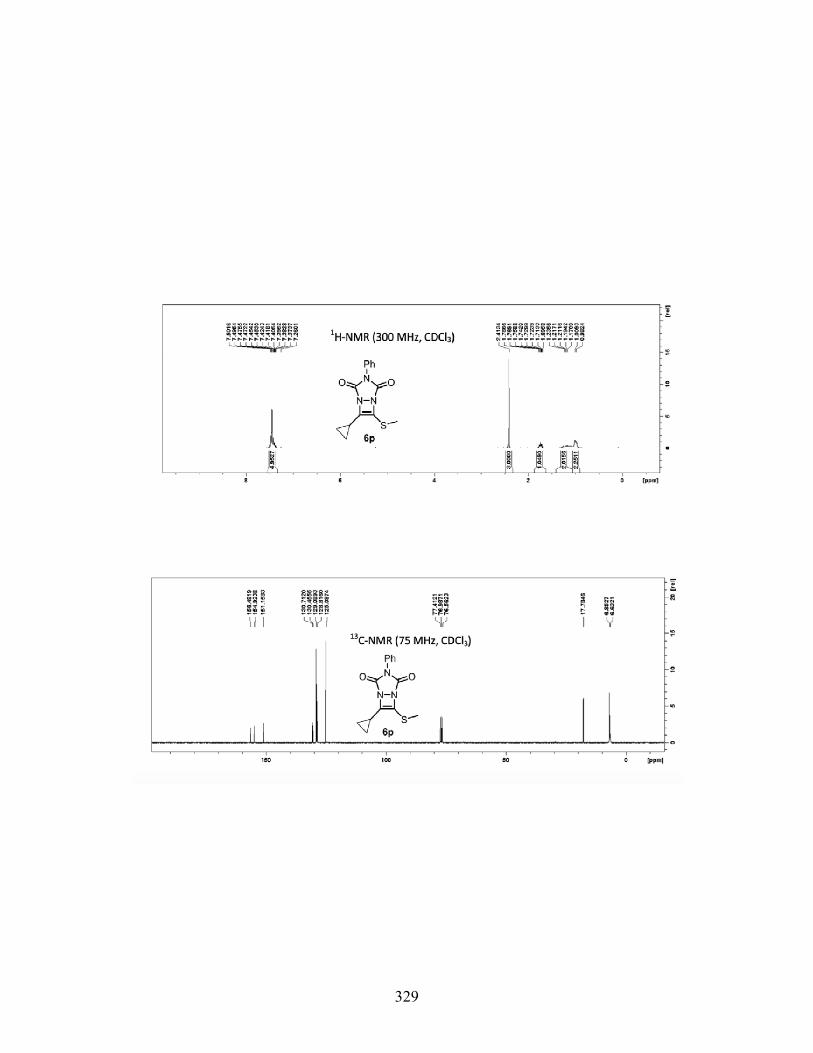
329
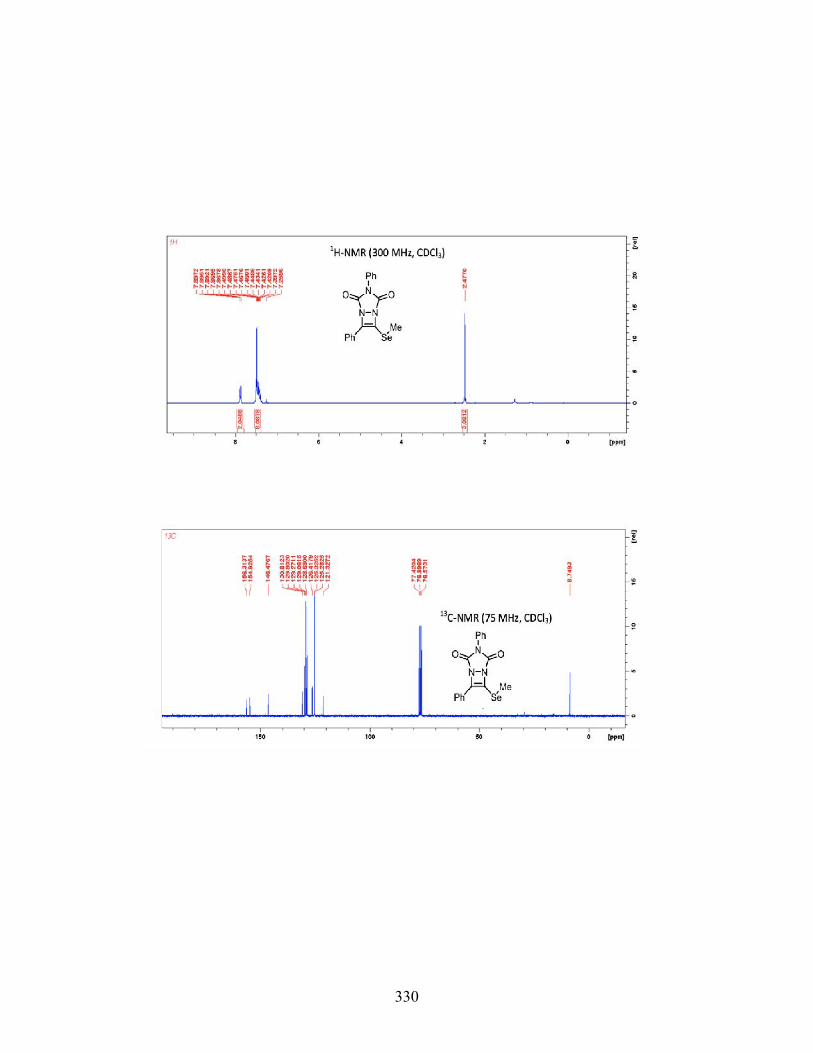
330
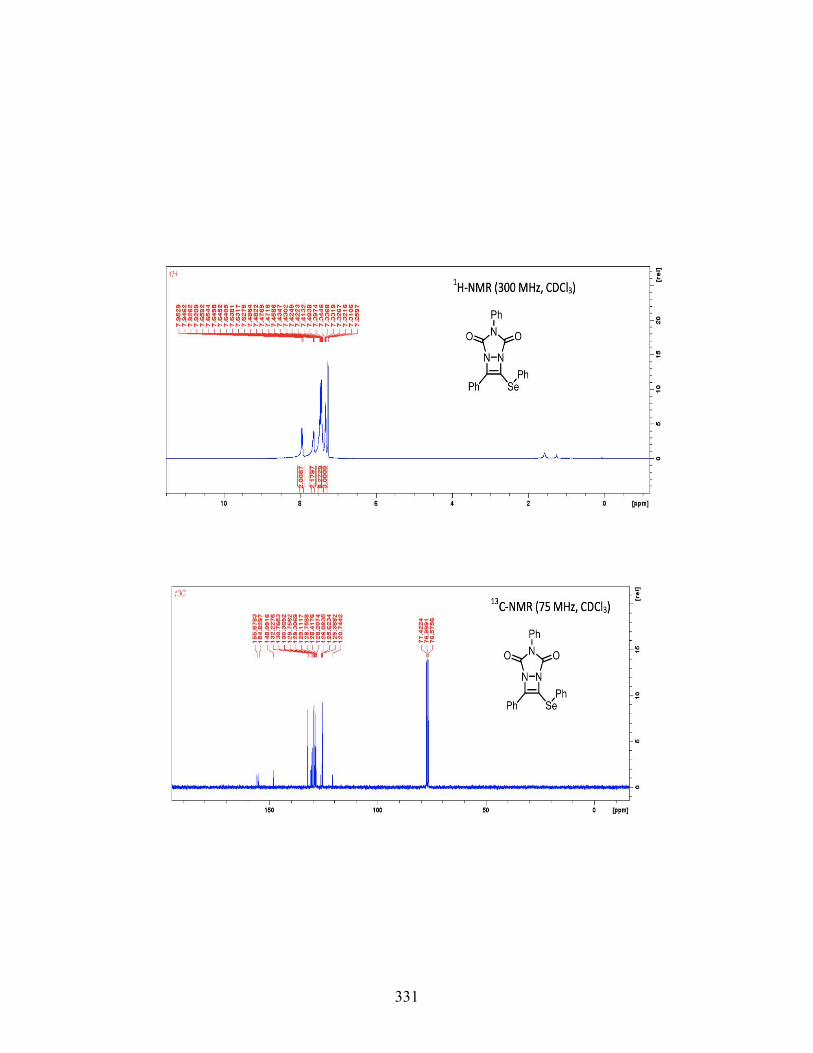
331

332
General Procedure for the synthesis of N,N-dicarbamoyl 2-iminothioimidates
To a flame-dried round bottom flask equipped with a water-cooled condenser and a
stir bar was added a solution of azodicarboxylate (1 mmol,1 equiv.) and 5 mL of dry
acetonitrile under nitrogen. To this stirring solution was added dropwise a solution of
alkynyl sulfide (1.3mmol, 1.3 equiv.) in 5 mL of dry acetonitrile. The mixture was then
refluxed for 24 hours. The resultant mixture was concentrated under reduced pressure
and purified via flash chromatography with hexanes and ethyl acetate (100% hexanes to
80:20 hexanes/ethyl acetate) to afford the corresponding α-imidothioimidate product.
Characterization data for N,N-dicarbamoyl 2-iminothioimidates
5ae Colorless liquid; Yield: 74% (239 mg); IR (neat): 2981 (w), 2931 (w), 1716 (s), 1627 (m), 1577 (m), 1446 (m), 1365 (m), 1207 (s), 1091 (m), 1033 (m), 864 (w), 690 (m) cm-
1;1H-NMR (300 MHz, CDCl3) δ = 7.83 (d, J = 6.6 Hz, 2H), 7.52 (t, J = 7.4 Hz, 1H), 7.42 (t, J = 7.3 Hz, 2H), 4.27 (q, J = 7.1 Hz, 2H), 4.08 (q, J = 6.8 Hz, 2H), 2.47 (s, 3H), 1.30 (t, J = 7.1 Hz, 3H), 1.15 (t, J = 7.1 Hz, 3H); 13C NMR (75 MHz, CDCl3) δ = 175.6, 166.2, 160.8, 159.2, 133.0, 132.4, 128.7, 128.7, 63.0 (2C), 14.3, 14.1, 14.0; HRMS (ESI+): Calcd for C15H19N2O4S, [M+H]+323.1066 Found m/z 323.1087.
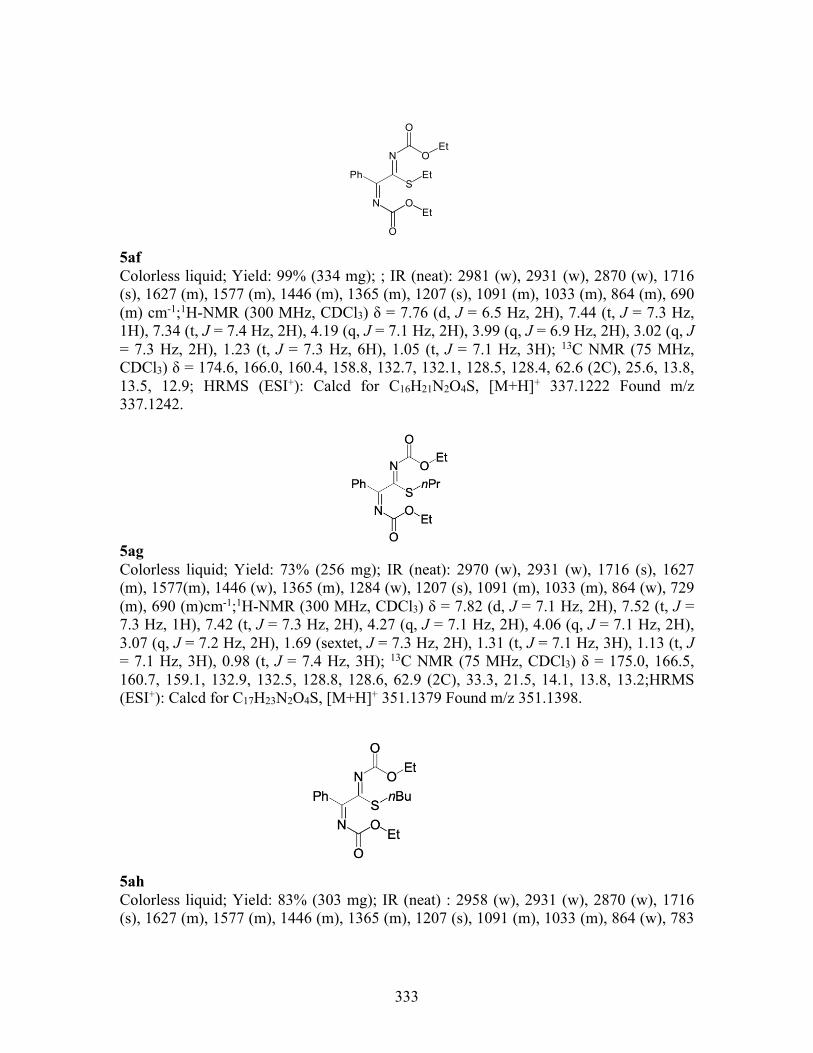
333
5af Colorless liquid; Yield: 99% (334 mg); ; IR (neat): 2981 (w), 2931 (w), 2870 (w), 1716 (s), 1627 (m), 1577 (m), 1446 (m), 1365 (m), 1207 (s), 1091 (m), 1033 (m), 864 (m), 690 (m) cm-1;1H-NMR (300 MHz, CDCl3) δ = 7.76 (d, J = 6.5 Hz, 2H), 7.44 (t, J = 7.3 Hz, 1H), 7.34 (t, J = 7.4 Hz, 2H), 4.19 (q, J = 7.1 Hz, 2H), 3.99 (q, J = 6.9 Hz, 2H), 3.02 (q, J = 7.3 Hz, 2H), 1.23 (t, J = 7.3 Hz, 6H), 1.05 (t, J = 7.1 Hz, 3H); 13C NMR (75 MHz, CDCl3) δ = 174.6, 166.0, 160.4, 158.8, 132.7, 132.1, 128.5, 128.4, 62.6 (2C), 25.6, 13.8, 13.5, 12.9; HRMS (ESI+): Calcd for C16H21N2O4S, [M+H]+ 337.1222 Found m/z 337.1242. 5ag Colorless liquid; Yield: 73% (256 mg); IR (neat): 2970 (w), 2931 (w), 1716 (s), 1627 (m), 1577(m), 1446 (w), 1365 (m), 1284 (w), 1207 (s), 1091 (m), 1033 (m), 864 (w), 729 (m), 690 (m)cm-1;1H-NMR (300 MHz, CDCl3) δ = 7.82 (d, J = 7.1 Hz, 2H), 7.52 (t, J = 7.3 Hz, 1H), 7.42 (t, J = 7.3 Hz, 2H), 4.27 (q, J = 7.1 Hz, 2H), 4.06 (q, J = 7.1 Hz, 2H), 3.07 (q, J = 7.2 Hz, 2H), 1.69 (sextet, J = 7.3 Hz, 2H), 1.31 (t, J = 7.1 Hz, 3H), 1.13 (t, J = 7.1 Hz, 3H), 0.98 (t, J = 7.4 Hz, 3H); 13C NMR (75 MHz, CDCl3) δ = 175.0, 166.5, 160.7, 159.1, 132.9, 132.5, 128.8, 128.6, 62.9 (2C), 33.3, 21.5, 14.1, 13.8, 13.2;HRMS (ESI+): Calcd for C17H23N2O4S, [M+H]+ 351.1379 Found m/z 351.1398. 5ah Colorless liquid; Yield: 83% (303 mg); IR (neat) : 2958 (w), 2931 (w), 2870 (w), 1716 (s), 1627 (m), 1577 (m), 1446 (m), 1365 (m), 1207 (s), 1091 (m), 1033 (m), 864 (w), 783
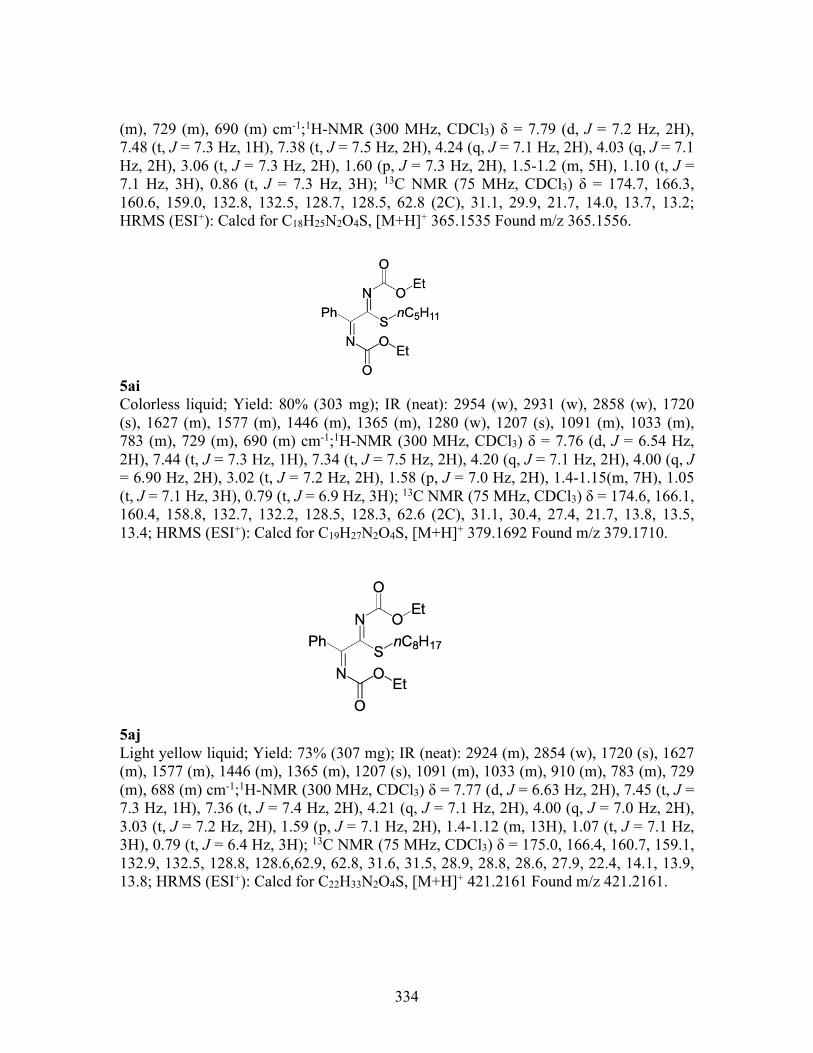
334
(m), 729 (m), 690 (m) cm-1;1H-NMR (300 MHz, CDCl3) δ = 7.79 (d, J = 7.2 Hz, 2H), 7.48 (t, J = 7.3 Hz, 1H), 7.38 (t, J = 7.5 Hz, 2H), 4.24 (q, J = 7.1 Hz, 2H), 4.03 (q, J = 7.1 Hz, 2H), 3.06 (t, J = 7.3 Hz, 2H), 1.60 (p, J = 7.3 Hz, 2H), 1.5-1.2 (m, 5H), 1.10 (t, J = 7.1 Hz, 3H), 0.86 (t, J = 7.3 Hz, 3H); 13C NMR (75 MHz, CDCl3) δ = 174.7, 166.3, 160.6, 159.0, 132.8, 132.5, 128.7, 128.5, 62.8 (2C), 31.1, 29.9, 21.7, 14.0, 13.7, 13.2; HRMS (ESI+): Calcd for C18H25N2O4S, [M+H]+ 365.1535 Found m/z 365.1556. 5ai Colorless liquid; Yield: 80% (303 mg); IR (neat): 2954 (w), 2931 (w), 2858 (w), 1720 (s), 1627 (m), 1577 (m), 1446 (m), 1365 (m), 1280 (w), 1207 (s), 1091 (m), 1033 (m), 783 (m), 729 (m), 690 (m) cm-1;1H-NMR (300 MHz, CDCl3) δ = 7.76 (d, J = 6.54 Hz, 2H), 7.44 (t, J = 7.3 Hz, 1H), 7.34 (t, J = 7.5 Hz, 2H), 4.20 (q, J = 7.1 Hz, 2H), 4.00 (q, J = 6.90 Hz, 2H), 3.02 (t, J = 7.2 Hz, 2H), 1.58 (p, J = 7.0 Hz, 2H), 1.4-1.15(m, 7H), 1.05 (t, J = 7.1 Hz, 3H), 0.79 (t, J = 6.9 Hz, 3H); 13C NMR (75 MHz, CDCl3) δ = 174.6, 166.1, 160.4, 158.8, 132.7, 132.2, 128.5, 128.3, 62.6 (2C), 31.1, 30.4, 27.4, 21.7, 13.8, 13.5, 13.4; HRMS (ESI+): Calcd for C19H27N2O4S, [M+H]+ 379.1692 Found m/z 379.1710. 5aj Light yellow liquid; Yield: 73% (307 mg); IR (neat): 2924 (m), 2854 (w), 1720 (s), 1627 (m), 1577 (m), 1446 (m), 1365 (m), 1207 (s), 1091 (m), 1033 (m), 910 (m), 783 (m), 729 (m), 688 (m) cm-1;1H-NMR (300 MHz, CDCl3) δ = 7.77 (d, J = 6.63 Hz, 2H), 7.45 (t, J = 7.3 Hz, 1H), 7.36 (t, J = 7.4 Hz, 2H), 4.21 (q, J = 7.1 Hz, 2H), 4.00 (q, J = 7.0 Hz, 2H), 3.03 (t, J = 7.2 Hz, 2H), 1.59 (p, J = 7.1 Hz, 2H), 1.4-1.12 (m, 13H), 1.07 (t, J = 7.1 Hz, 3H), 0.79 (t, J = 6.4 Hz, 3H); 13C NMR (75 MHz, CDCl3) δ = 175.0, 166.4, 160.7, 159.1, 132.9, 132.5, 128.8, 128.6,62.9, 62.8, 31.6, 31.5, 28.9, 28.8, 28.6, 27.9, 22.4, 14.1, 13.9, 13.8; HRMS (ESI+): Calcd for C22H33N2O4S, [M+H]+ 421.2161 Found m/z 421.2161.

335
5ak Light yellow solid; Yield: 77% (296 mg); Mp: 70-71˚C;IR (neat) : 2981 (w), 1724 (s), 1712 (s), 1631 (m), 1612 (m), 1577 (w), 1477 (m), 1442 (m), 1207 (s), 1095 (w), 1022 (m), 968 (m), 860 (w), 752 (s), 686 (s) cm-1; 1H-NMR (300 MHz, CDCl3) δ = 7.54 (d, J = 7.4 Hz, 2H), 7.50-7.20 (m, 6H), 7.15 (t, J = 7.5 Hz, 2H), 4.38 (q, J = 7.1 Hz, 2H), 4.28 (broad q, J = 7.1 Hz, 2H), 1.41 (t, J = 7.1 Hz, 3H), 1.35 (t, J = 7.1 Hz, 3H); 13C NMR (125 MHz, CDCl3) δ = 173.9, 165.5, 161.3, 159.6, 135.8, 133.2, 132.5, 130.3, 129.0, 128.4, 128.2, 126.1, 63.23, 63.16, 14.2, 14.1; HRMS (ESI+): Calcd for C20H21N2O4S, [M+H]+ 385.1222 Found m/z 385.1240. 5al Colorless liquid; Yield: 83% (330 mg); IR (neat):2978 (w), 1716 (s), 1627 (w), 1577 (m), 1492 (w), 1450 (w), 1207 (s), 1087 (m), 1026 (m), 910 (w), 729 (w), 690 (w) cm-1; 1H-NMR (300 MHz, CDCl3) δ = 7.81 (d, J = 6.2 Hz, 2H), 7.51 (t, J = 7.0 Hz, 1H), 7.40 (t, J = 7.7 Hz, 2H), 7.35-7.16 (m, 5H), 4.34 (s, 2H), 4.23 (q, J = 7.1 Hz, 2H), 4.10 (q, J = 7.1 Hz, 2H), 1.27 (t, J = 7.1 Hz, 3H), 1.15 (t, J = 7.1 Hz, 3H); 13C NMR (75 MHz, CDCl3) δ = 174.2, 165.9, 160.6, 158.9, 134.6, 132.9, 132.3, 130.4, 129.1, 128.7, 128.5, 127.7, 62.88, 62.86,35.8, 14.0, 13.7; HRMS (ESI+): Calcd for C21H23N2O4S, [M+H]+ 399.1379 Found m/z 399.1391.
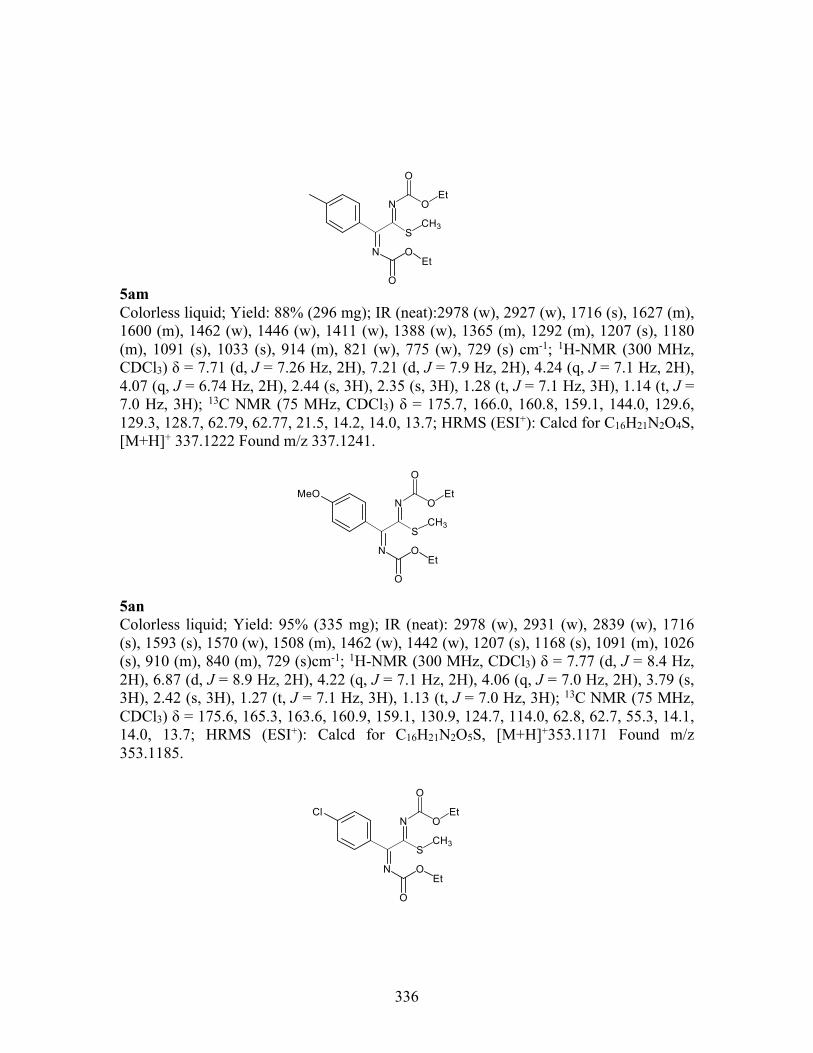
336
5am Colorless liquid; Yield: 88% (296 mg); IR (neat):2978 (w), 2927 (w), 1716 (s), 1627 (m), 1600 (m), 1462 (w), 1446 (w), 1411 (w), 1388 (w), 1365 (m), 1292 (m), 1207 (s), 1180 (m), 1091 (s), 1033 (s), 914 (m), 821 (w), 775 (w), 729 (s) cm-1; 1H-NMR (300 MHz, CDCl3) δ = 7.71 (d, J = 7.26 Hz, 2H), 7.21 (d, J = 7.9 Hz, 2H), 4.24 (q, J = 7.1 Hz, 2H), 4.07 (q, J = 6.74 Hz, 2H), 2.44 (s, 3H), 2.35 (s, 3H), 1.28 (t, J = 7.1 Hz, 3H), 1.14 (t, J = 7.0 Hz, 3H); 13C NMR (75 MHz, CDCl3) δ = 175.7, 166.0, 160.8, 159.1, 144.0, 129.6, 129.3, 128.7, 62.79, 62.77, 21.5, 14.2, 14.0, 13.7; HRMS (ESI+): Calcd for C16H21N2O4S, [M+H]+ 337.1222 Found m/z 337.1241.
5an Colorless liquid; Yield: 95% (335 mg); IR (neat): 2978 (w), 2931 (w), 2839 (w), 1716 (s), 1593 (s), 1570 (w), 1508 (m), 1462 (w), 1442 (w), 1207 (s), 1168 (s), 1091 (m), 1026 (s), 910 (m), 840 (m), 729 (s)cm-1; 1H-NMR (300 MHz, CDCl3) δ = 7.77 (d, J = 8.4 Hz, 2H), 6.87 (d, J = 8.9 Hz, 2H), 4.22 (q, J = 7.1 Hz, 2H), 4.06 (q, J = 7.0 Hz, 2H), 3.79 (s, 3H), 2.42 (s, 3H), 1.27 (t, J = 7.1 Hz, 3H), 1.13 (t, J = 7.0 Hz, 3H); 13C NMR (75 MHz, CDCl3) δ = 175.6, 165.3, 163.6, 160.9, 159.1, 130.9, 124.7, 114.0, 62.8, 62.7, 55.3, 14.1, 14.0, 13.7; HRMS (ESI+): Calcd for C16H21N2O5S, [M+H]+353.1171 Found m/z 353.1185.

337
5ao Colorless liquid; Yield: 47% (168 mg); IR (neat): 2978 (w), 2927 (w), 1716 (s), 1627 (m), 1589 (m), 1400 (w), 1365 (w), 1207 (s), 1087 (s), 1029 (s), 864 (w), 783 (w), 725 (m) cm-1;1H-NMR (300 MHz, CDCl3) δ = 7.77 (d, J = 8.0 Hz, 2H), 7.40 (d, J = 8.6 Hz, 2H), 4.26 (q, J = 7.1 Hz, 2H), 4.10 (q, J = 7.1 Hz, 2H), 2.47 (s, 3H), 1.30 (t, J = 7.1 Hz, 3H), 1.17 (t, J = 7.1 Hz, 3H); 13C NMR (75 MHz, CDCl3) δ = 175.4, 165.4, 160.6, 159.0, 139.5, 130.9, 130.0, 129.0, 63.1(2C), 14.3, 14.1, 13.8; HRMS (ESI+): Calcd for C15H18ClN2O4S, [M+H]+ 357.0676 Found m/z 357.0686.
5ap Colorless liquid; Yield: 69% (270 mg); IR (neat): 2981 (w), 1724 (s), 1631 (w), 1577 (w), 1411 (w), 1323 (s), 1211 (s), 1168 (m), 1126 (m), 1064 (s), 1033 (w), 1014 (w), 848 (m), 771 (w), 748 (w)cm-1; 1H-NMR (300 MHz, CDCl3) δ = 7.95 (d, J = 7.4 Hz, 2H), 7.68 (d, J = 8.4 Hz, 2H), 4.27 (q, J = 7.1 Hz, 2H), 4.09 (q, J = 6.9 Hz, 2H), 2.49 (s, 3H), 1.30 (t, J = 7.1 Hz, 3H), 1.16 (t, J = 7.1 Hz, 3H); 13C NMR (75 MHz, CDCl3) δ = 175.5, 165.5, 160.4, 159.1, 135.8, 134.3 (q, 2JCF = 32.8 Hz), 129.1, 125.7 (q, 3JCF = 3.7 Hz), 123.5 (q, 1JCF = 272.7 Hz), 63.33, 63.25, 14.3, 14.1, 13.9; HRMS (ESI+): Calcd for C16H18F3N2O4S, [M+H]+ 391.0939 Found m/z 391.0963. 5aq Light yellow liquid; Yield: 40% (121 mg) ; IR (neat): 2958 (w), 2931 (w), 1716 (s), 1662 (w), 1608 (w), 1500 (w), 1465 (w), 1365 (w), 1215 (s), 1095 (w), 1033 (m), 948 (w), 914 (w), 732 (m) cm-1;1H-NMR (300 MHz, CDCl3) δ = 4.4-4.1 (m, 4H), 2.48 (t, J = 7.8 Hz, 2H), 2.41 (s, 3H), 1.56 (p, J = 7.6 Hz, 2H), 1.40-1.23 (m, 8H), 0.86 (t, J = 7.3 Hz, 3H); 13C NMR (75 MHz, CDCl3) δ = 171.2, 161.7, 160.6, 159.7, 62.9, 62.8, 27.8, 23.3, 15.1,
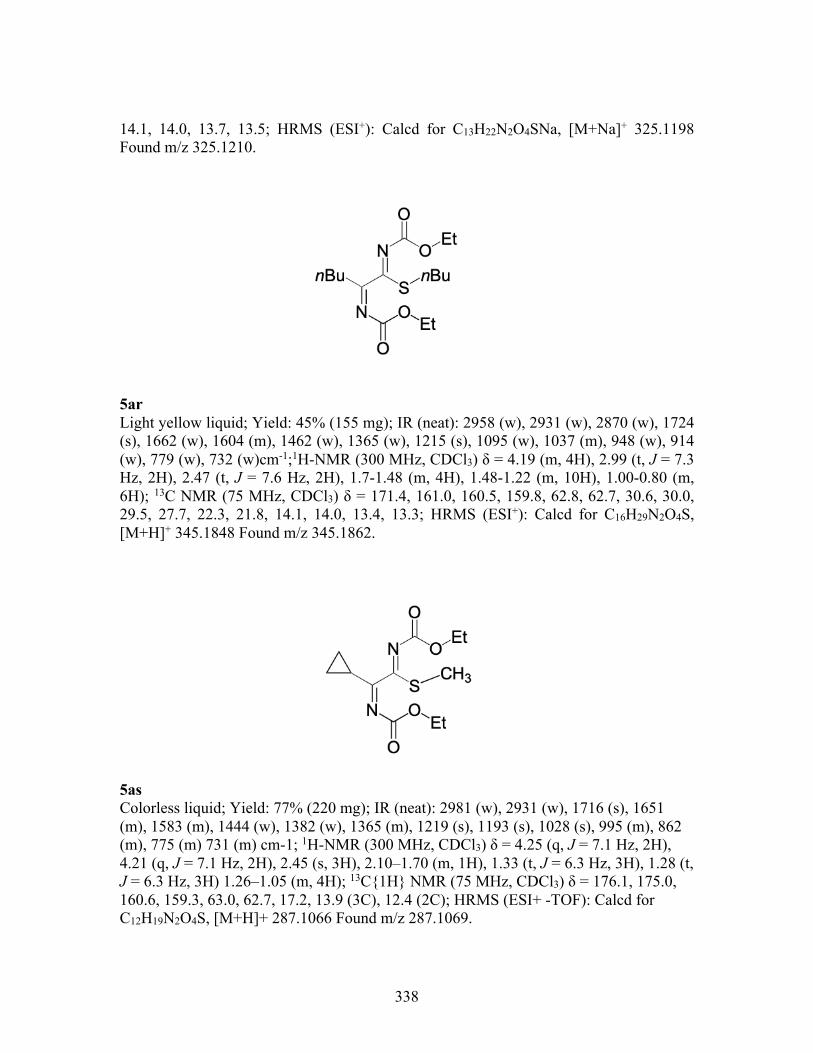
338
14.1, 14.0, 13.7, 13.5; HRMS (ESI+): Calcd for C13H22N2O4SNa, [M+Na]+ 325.1198 Found m/z 325.1210.
5ar Light yellow liquid; Yield: 45% (155 mg); IR (neat): 2958 (w), 2931 (w), 2870 (w), 1724 (s), 1662 (w), 1604 (m), 1462 (w), 1365 (w), 1215 (s), 1095 (w), 1037 (m), 948 (w), 914 (w), 779 (w), 732 (w)cm-1;1H-NMR (300 MHz, CDCl3) δ = 4.19 (m, 4H), 2.99 (t, J = 7.3 Hz, 2H), 2.47 (t, J = 7.6 Hz, 2H), 1.7-1.48 (m, 4H), 1.48-1.22 (m, 10H), 1.00-0.80 (m, 6H); 13C NMR (75 MHz, CDCl3) δ = 171.4, 161.0, 160.5, 159.8, 62.8, 62.7, 30.6, 30.0, 29.5, 27.7, 22.3, 21.8, 14.1, 14.0, 13.4, 13.3; HRMS (ESI+): Calcd for C16H29N2O4S, [M+H]+ 345.1848 Found m/z 345.1862.
5as Colorless liquid; Yield: 77% (220 mg); IR (neat): 2981 (w), 2931 (w), 1716 (s), 1651 (m), 1583 (m), 1444 (w), 1382 (w), 1365 (m), 1219 (s), 1193 (s), 1028 (s), 995 (m), 862 (m), 775 (m) 731 (m) cm-1; 1H-NMR (300 MHz, CDCl3) δ = 4.25 (q, J = 7.1 Hz, 2H), 4.21 (q, J = 7.1 Hz, 2H), 2.45 (s, 3H), 2.10–1.70 (m, 1H), 1.33 (t, J = 6.3 Hz, 3H), 1.28 (t, J = 6.3 Hz, 3H) 1.26–1.05 (m, 4H); 13C{1H} NMR (75 MHz, CDCl3) δ = 176.1, 175.0, 160.6, 159.3, 63.0, 62.7, 17.2, 13.9 (3C), 12.4 (2C); HRMS (ESI+ -TOF): Calcd for C12H19N2O4S, [M+H]+ 287.1066 Found m/z 287.1069.
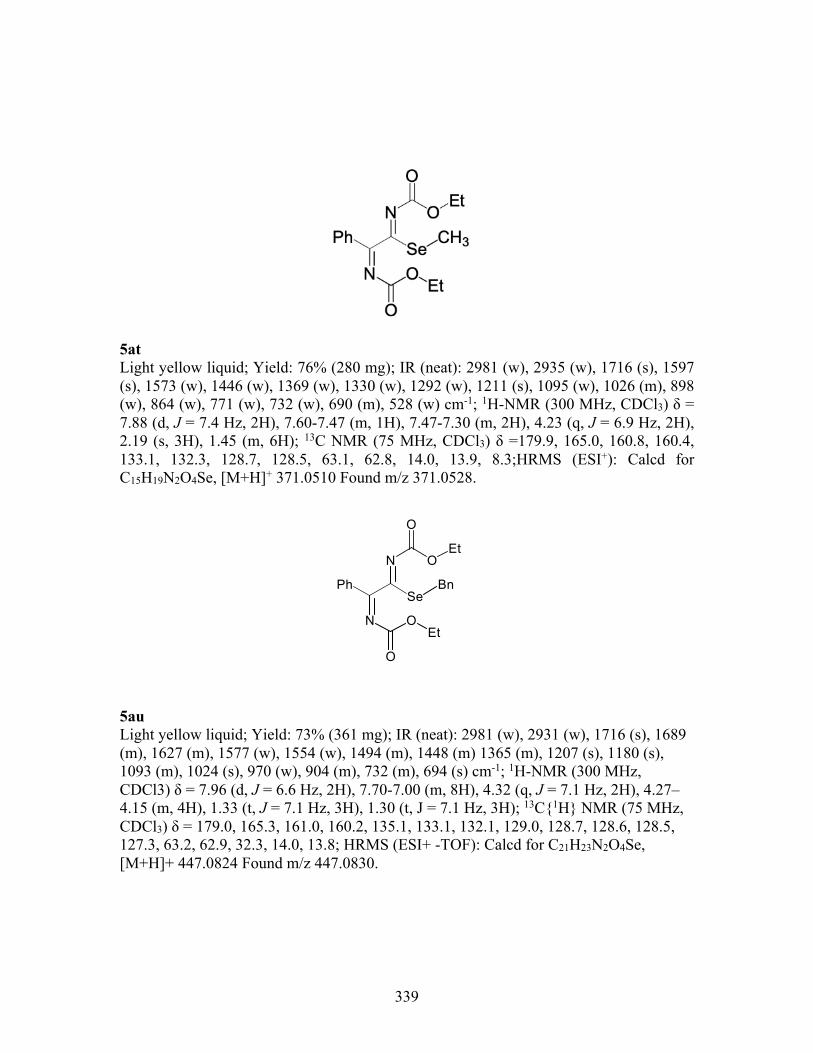
339
5at Light yellow liquid; Yield: 76% (280 mg); IR (neat): 2981 (w), 2935 (w), 1716 (s), 1597 (s), 1573 (w), 1446 (w), 1369 (w), 1330 (w), 1292 (w), 1211 (s), 1095 (w), 1026 (m), 898 (w), 864 (w), 771 (w), 732 (w), 690 (m), 528 (w) cm-1; 1H-NMR (300 MHz, CDCl3) δ = 7.88 (d, J = 7.4 Hz, 2H), 7.60-7.47 (m, 1H), 7.47-7.30 (m, 2H), 4.23 (q, J = 6.9 Hz, 2H), 2.19 (s, 3H), 1.45 (m, 6H); 13C NMR (75 MHz, CDCl3) δ =179.9, 165.0, 160.8, 160.4, 133.1, 132.3, 128.7, 128.5, 63.1, 62.8, 14.0, 13.9, 8.3;HRMS (ESI+): Calcd for C15H19N2O4Se, [M+H]+ 371.0510 Found m/z 371.0528.
5au Light yellow liquid; Yield: 73% (361 mg); IR (neat): 2981 (w), 2931 (w), 1716 (s), 1689 (m), 1627 (m), 1577 (w), 1554 (w), 1494 (m), 1448 (m) 1365 (m), 1207 (s), 1180 (s), 1093 (m), 1024 (s), 970 (w), 904 (m), 732 (m), 694 (s) cm-1; 1H-NMR (300 MHz, CDCl3) δ = 7.96 (d, J = 6.6 Hz, 2H), 7.70-7.00 (m, 8H), 4.32 (q, J = 7.1 Hz, 2H), 4.27– 4.15 (m, 4H), 1.33 (t, J = 7.1 Hz, 3H), 1.30 (t, J = 7.1 Hz, 3H); 13C{1H} NMR (75 MHz, CDCl3) δ = 179.0, 165.3, 161.0, 160.2, 135.1, 133.1, 132.1, 129.0, 128.7, 128.6, 128.5, 127.3, 63.2, 62.9, 32.3, 14.0, 13.8; HRMS (ESI+ -TOF): Calcd for C21H23N2O4Se, [M+H]+ 447.0824 Found m/z 447.0830.
340
1H NMR spectra and 13C NMR spectra for Alkynyl sulfides and selenides
341
1H NMR and 13C NMR spectra for N,N-dicarbamoyl 2-iminothioimidates
1H-NMR (300 MHz, CDCl3)
13C-NMR (75 MHz, CDCl3)
N
N
O
OEt
PhSCH3
5aeO
OEt
342
1H-NMR (300 MHz, CDCl3)
13C-NMR (75 MHz, CDCl3)
N
N
O
OEt
PhSEt
5afO
OEt
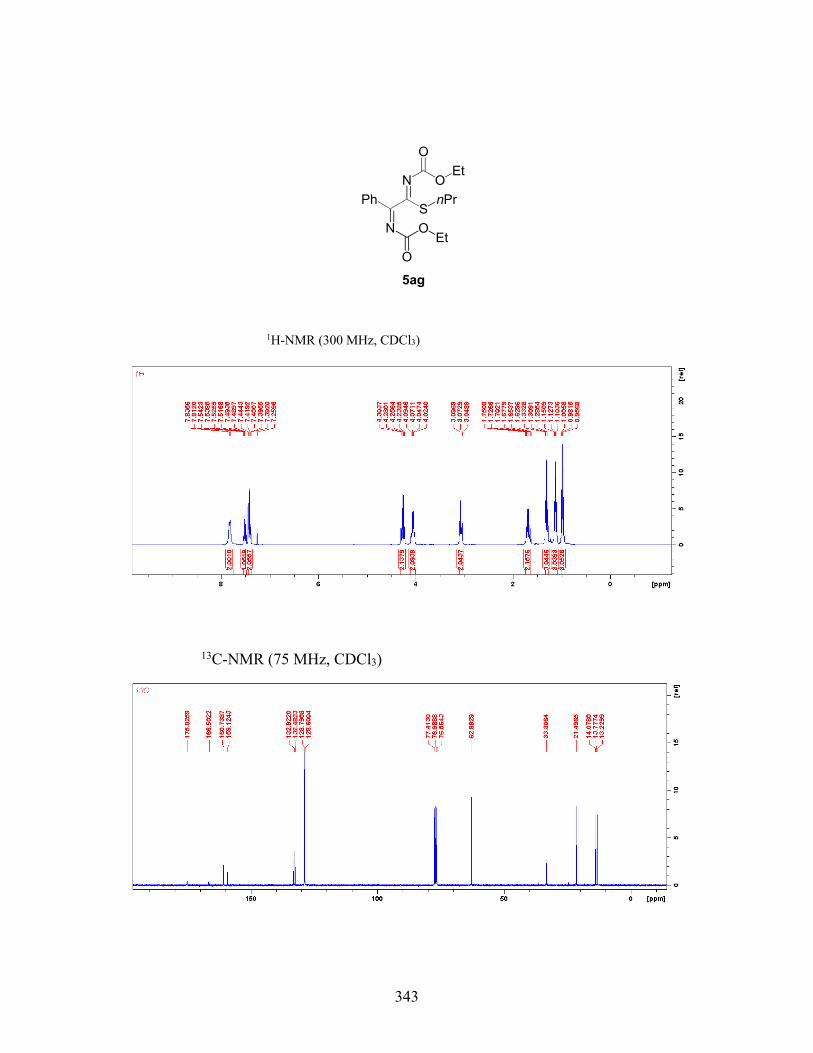
343
1H-NMR (300 MHz, CDCl3)
13C-NMR (75 MHz, CDCl3)
N
N
O
OEt
PhSnPr
5agO
OEt
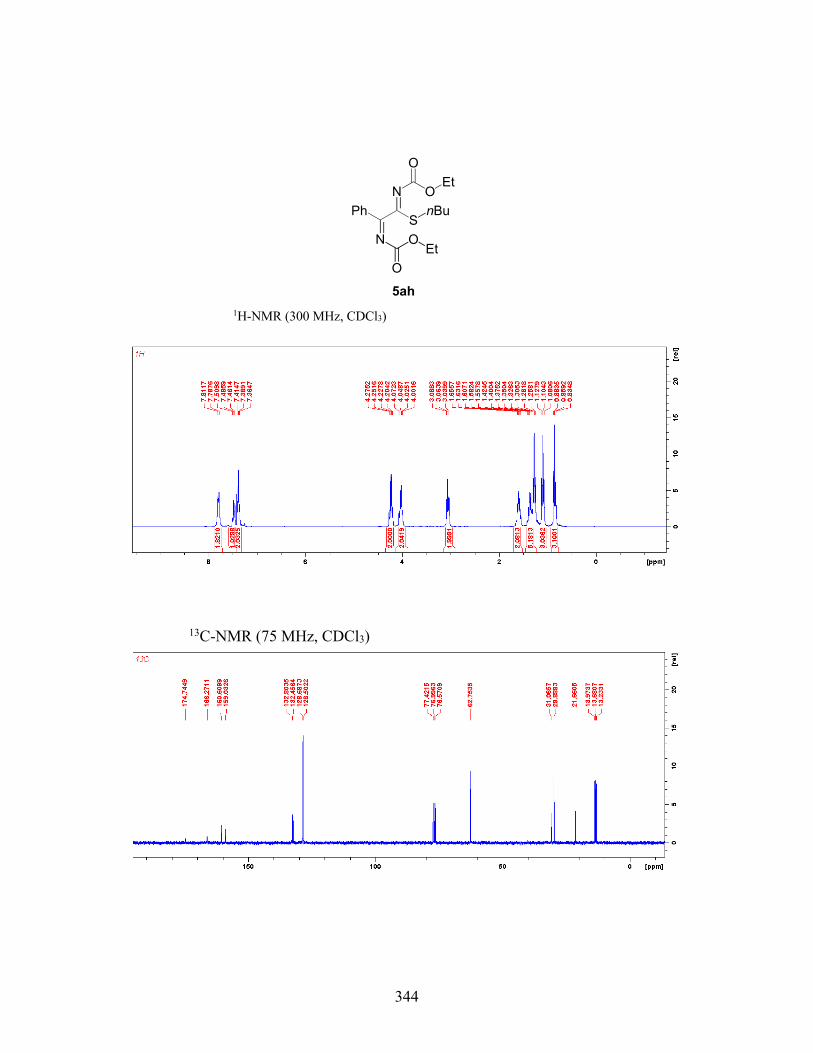
344
1H-NMR (300 MHz, CDCl3)
13C-NMR (75 MHz, CDCl3)
N
N
O
OEt
PhSnBu
5ah
O
OEt
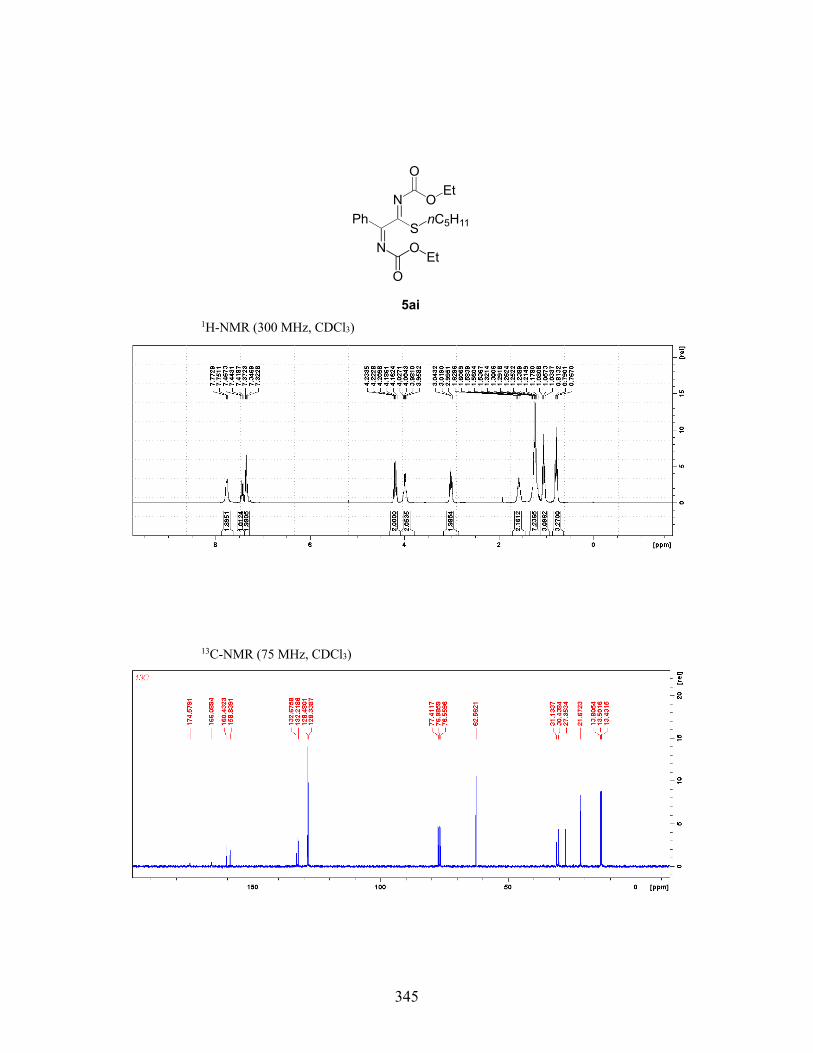
345
N
N
O
OEt
PhSnC5H11
5ai
O
OEt
1H-NMR (300 MHz, CDCl3)
13C-NMR (75 MHz, CDCl3)
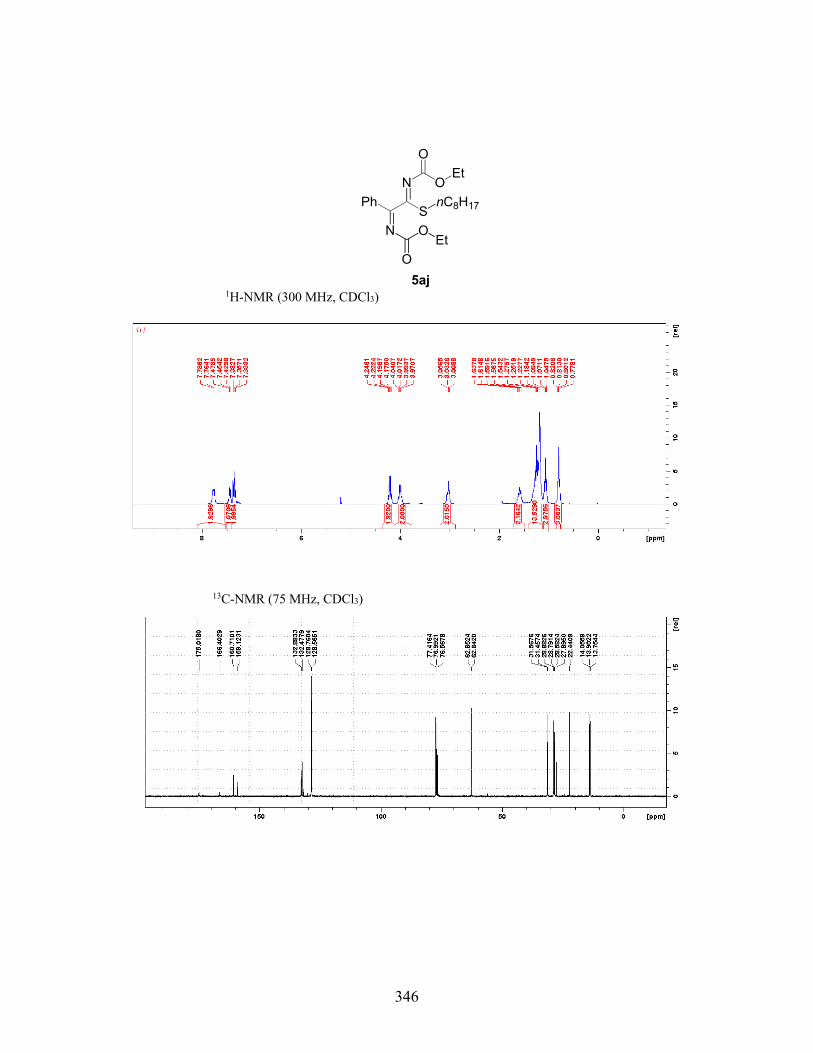
346
1H-NMR (300 MHz, CDCl3)
13C-NMR (75 MHz, CDCl3)
N
N
O
OEt
PhSnC8H17
5ajO
OEt
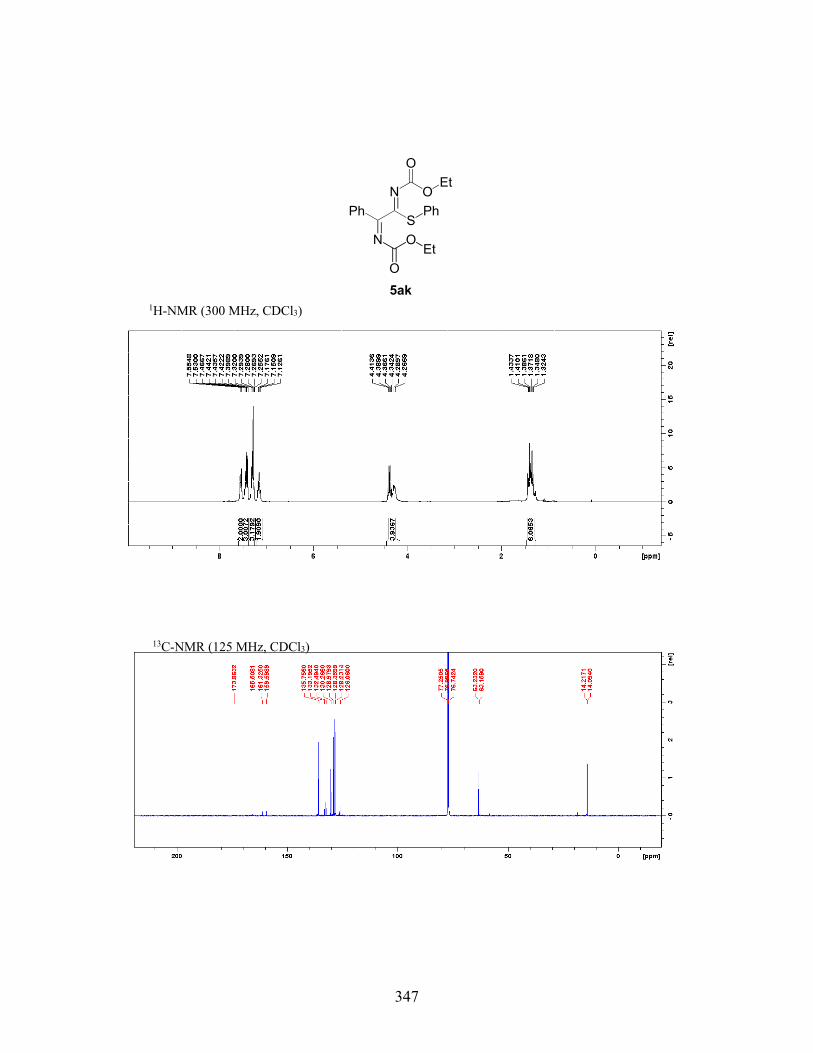
347
1H-NMR (300 MHz, CDCl3)
13C-NMR (125 MHz, CDCl3)
N
N
O
OEt
PhSPh
5akO
OEt
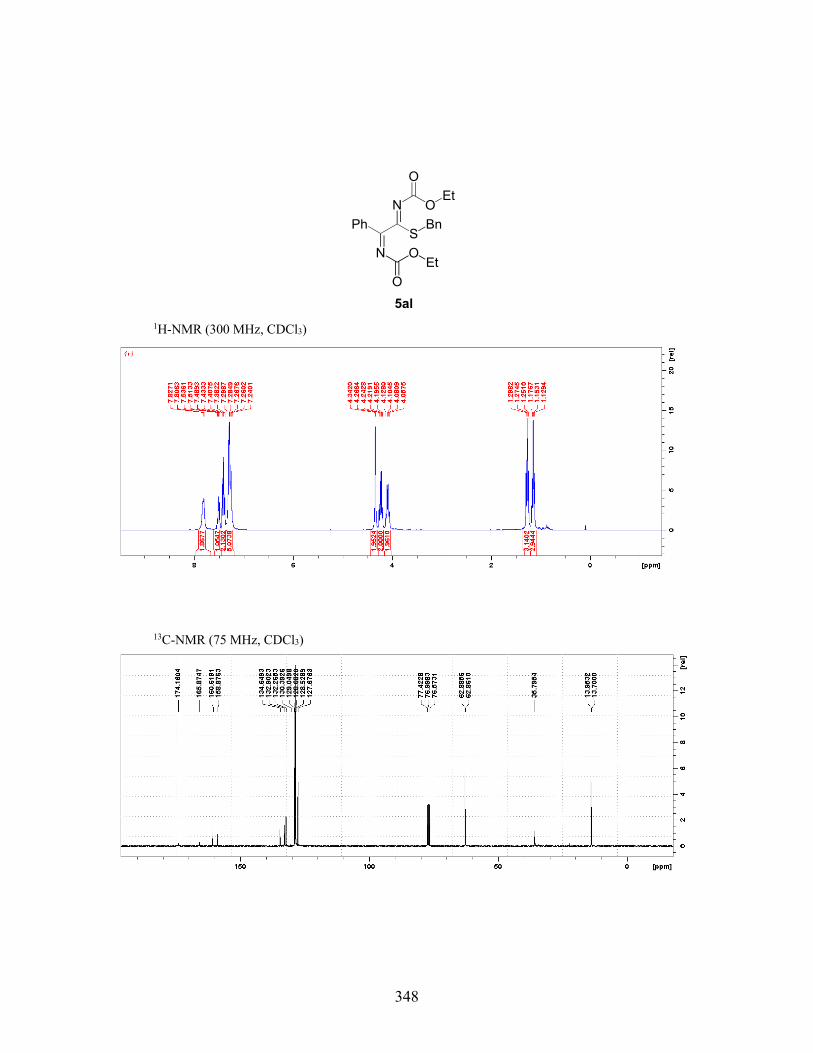
348
13C-NMR (75 MHz, CDCl3)
1H-NMR (300 MHz, CDCl3)
N
N
O
OEt
PhSBn
5alO
OEt
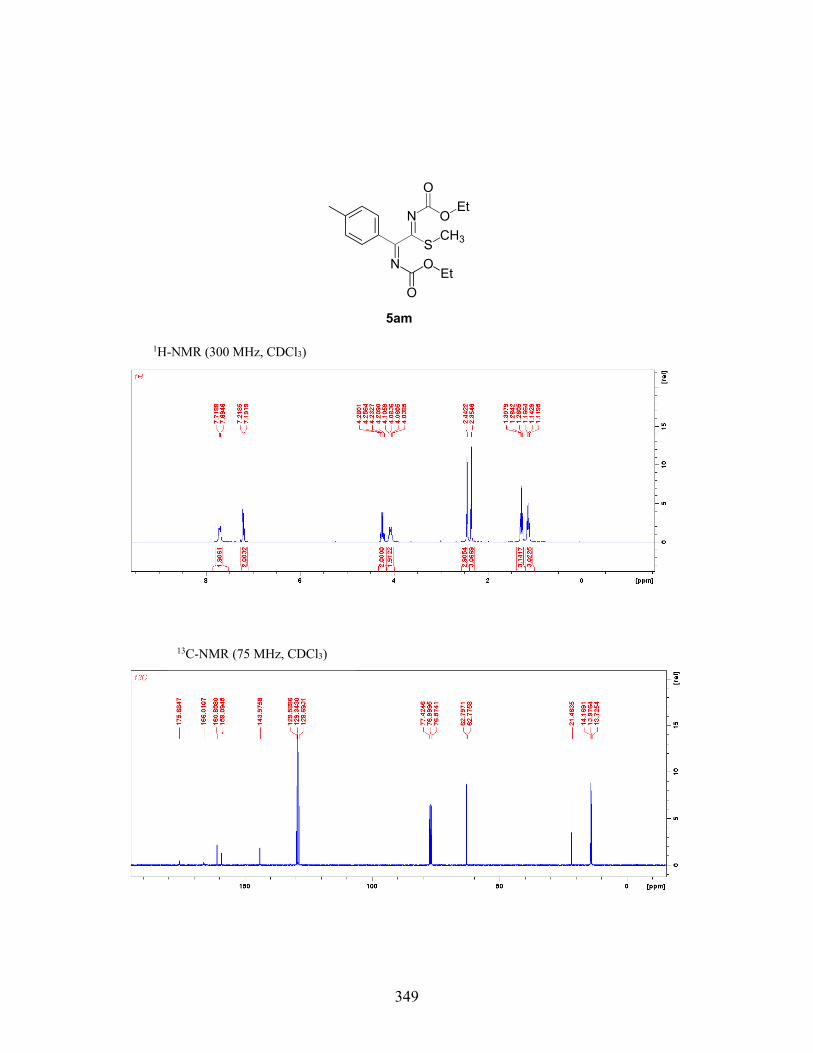
349
N
N
O
OEt
SCH3
5am
O
OEt
1H-NMR (300 MHz, CDCl3)
13C-NMR (75 MHz, CDCl3)
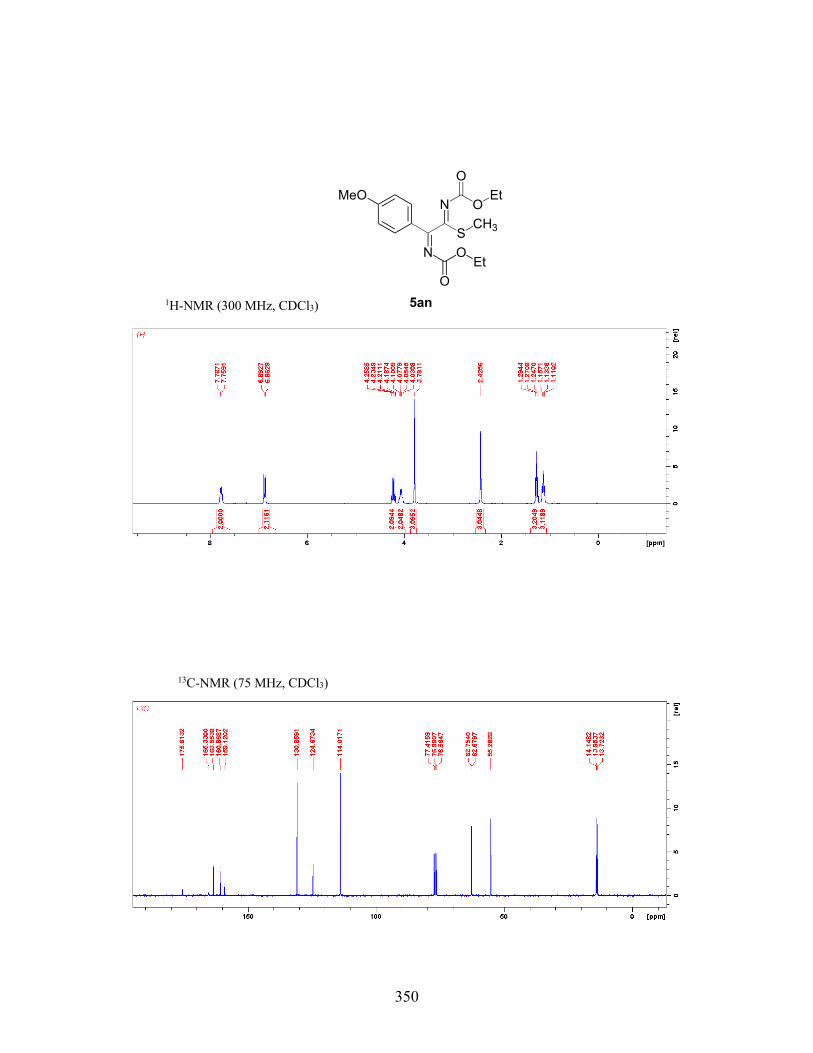
350
N
N
O
OEt
SCH3
5an
MeO
O
OEt
1H-NMR (300 MHz, CDCl3)
13C-NMR (75 MHz, CDCl3)
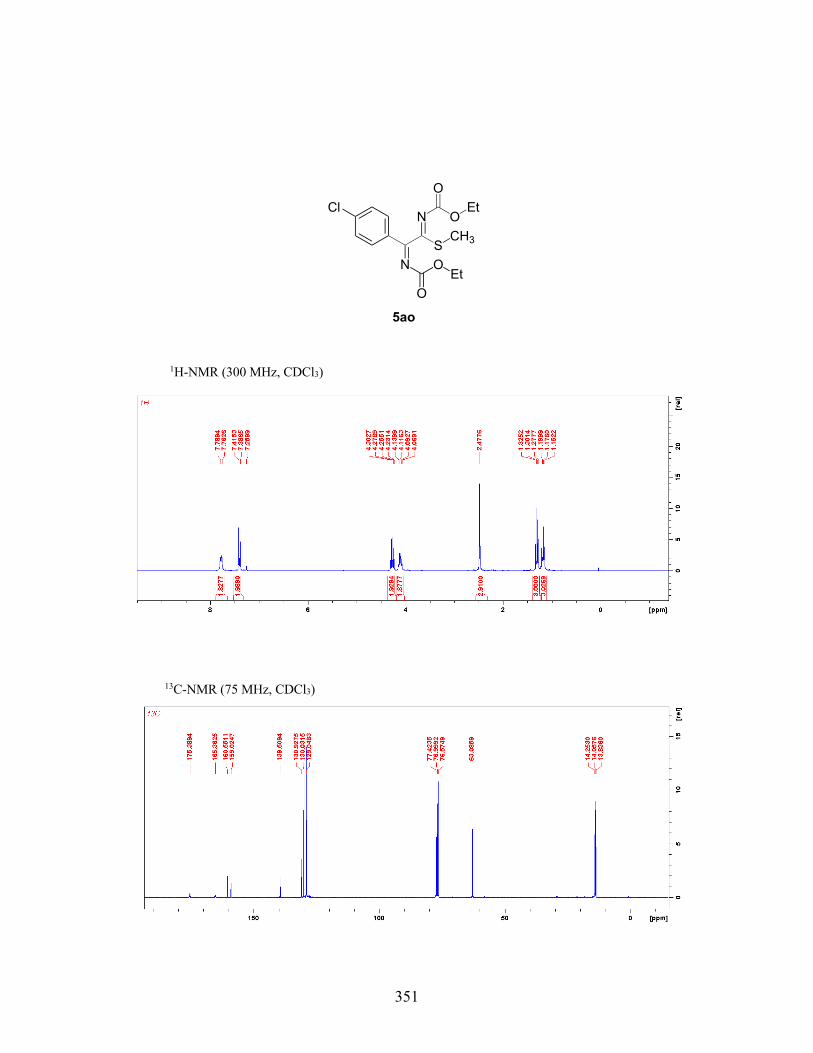
351
N
N
O
OEt
SCH3
5ao
Cl
O
OEt
1H-NMR (300 MHz, CDCl3)
13C-NMR (75 MHz, CDCl3)
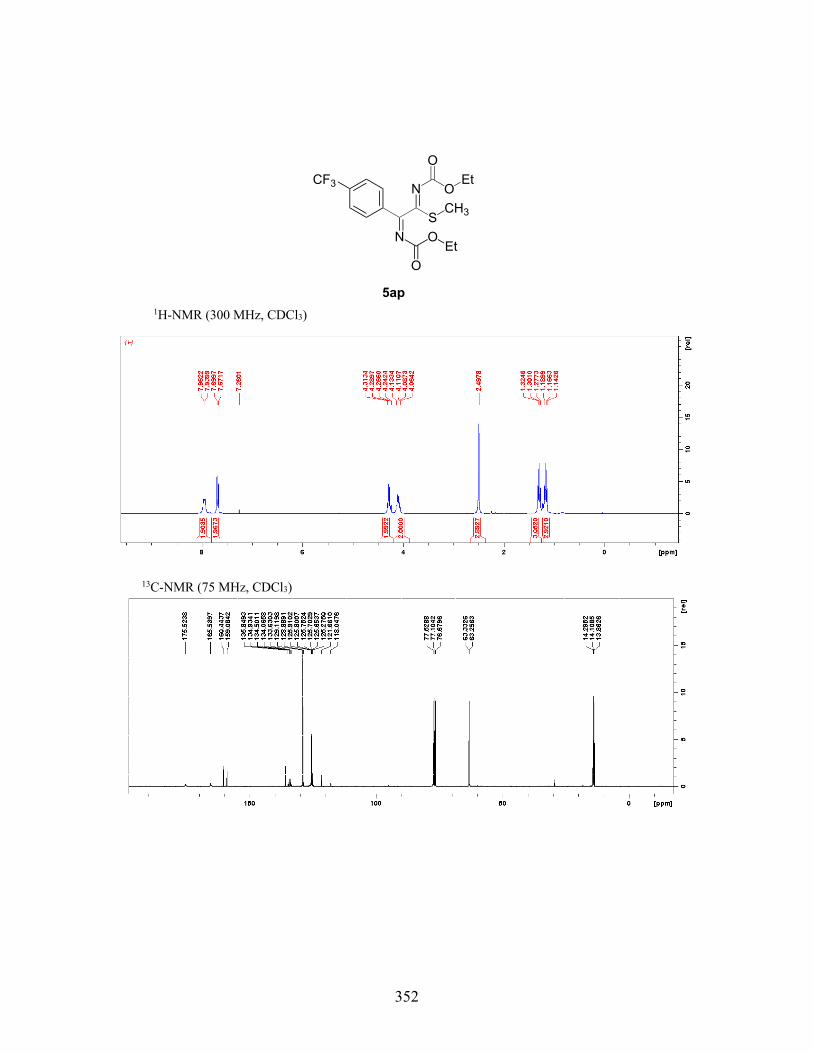
352
N
N
O
OEt
SCH3
5ap
CF3
O
OEt
1H-NMR (300 MHz, CDCl3)
13C-NMR (75 MHz, CDCl3)
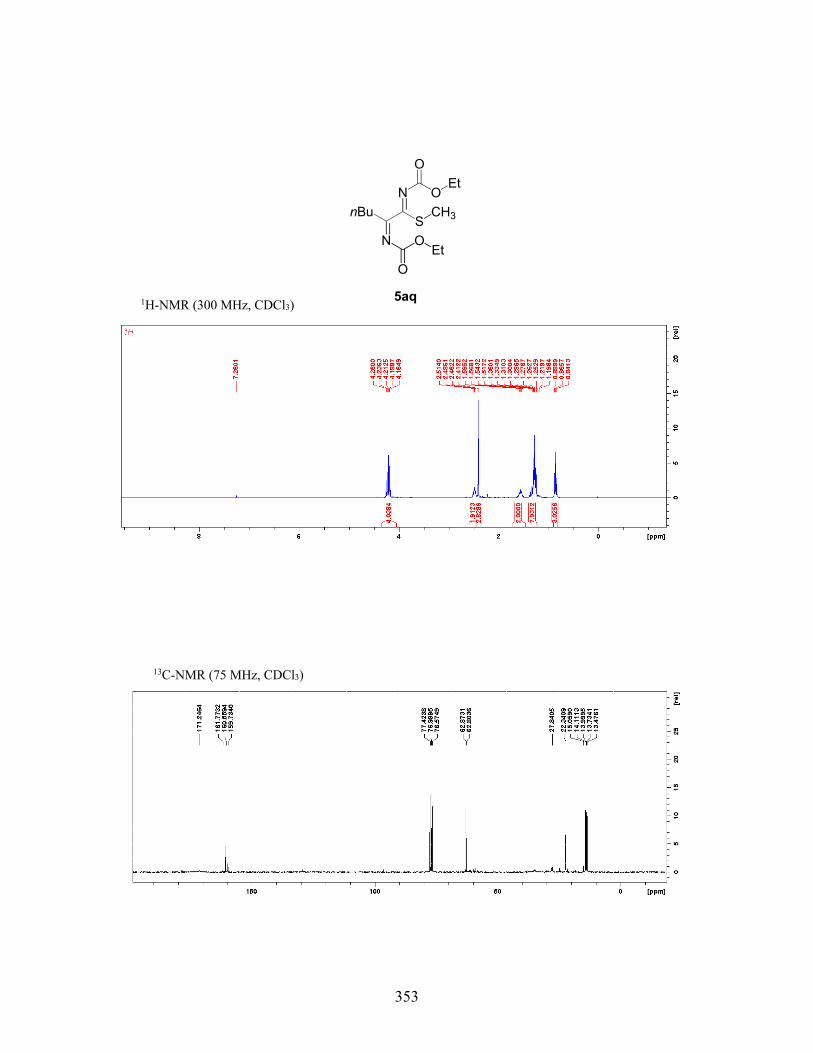
353
N
N
O
OEt
nBuSCH3
5aq
O
OEt
1H-NMR (300 MHz, CDCl3)
13C-NMR (75 MHz, CDCl3)
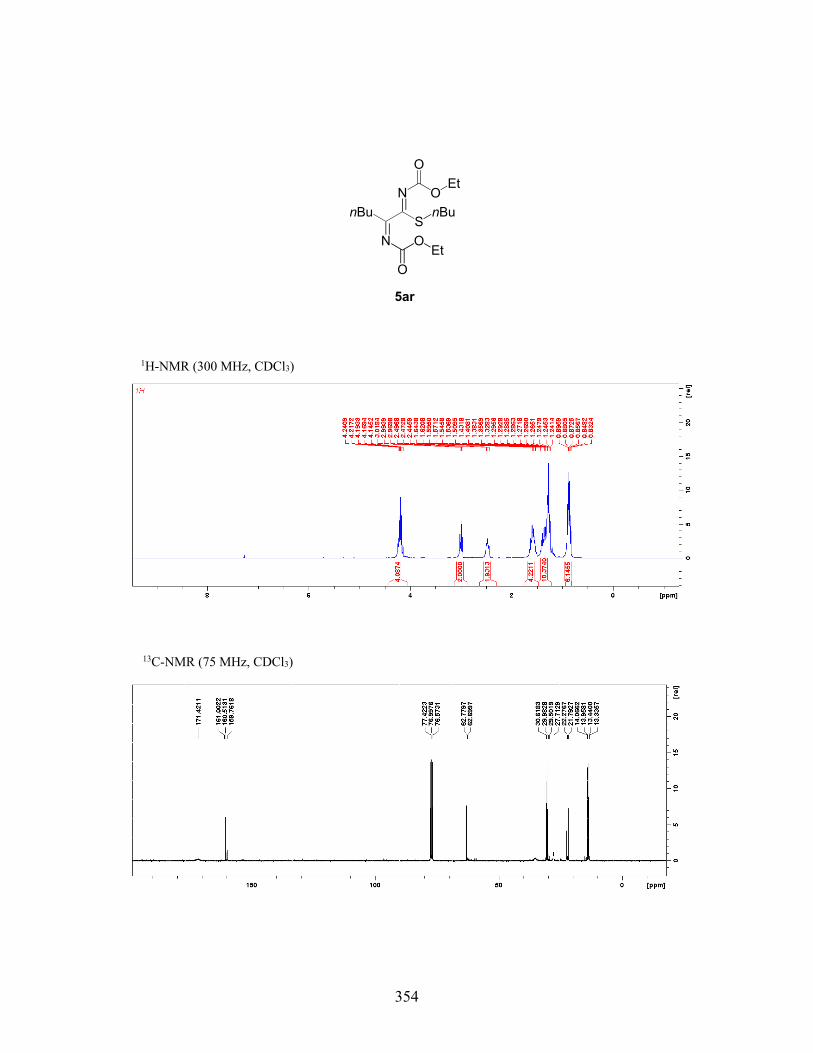
354
N
N
O
OEt
nBuSnBu
5ar
O
OEt
1H-NMR (300 MHz, CDCl3)
13C-NMR (75 MHz, CDCl3)
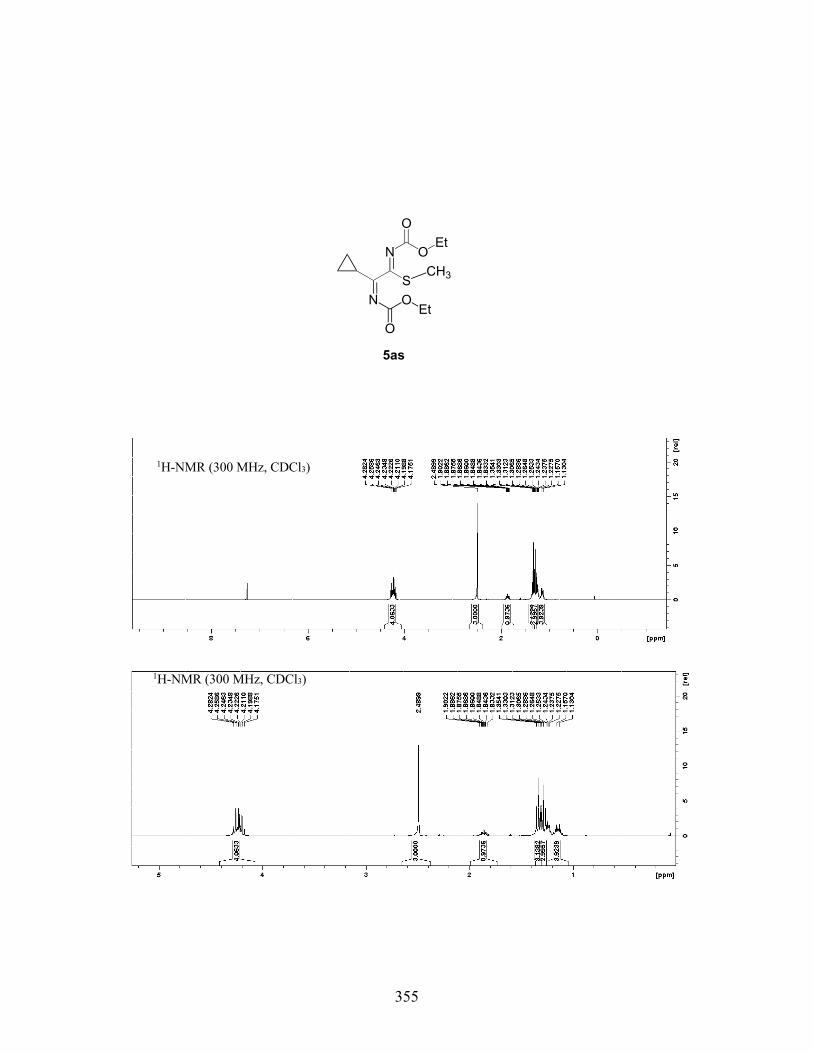
355
N
N
O
OEt
S
5as
CH3
O
OEt
1H-NMR (300 MHz, CDCl3)
1H-NMR (300 MHz, CDCl3)
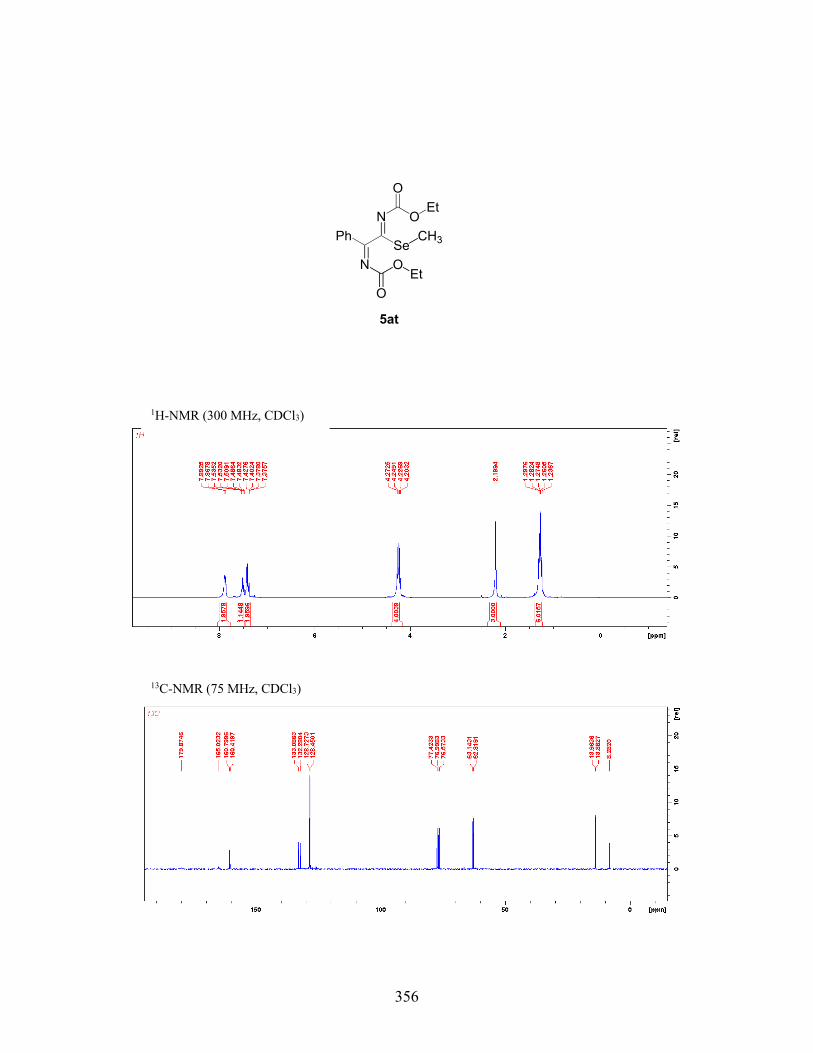
356
N
N
O
OEt
PhSe
5at
CH3
O
OEt
1H-NMR (300 MHz, CDCl3)
13C-NMR (75 MHz, CDCl3)
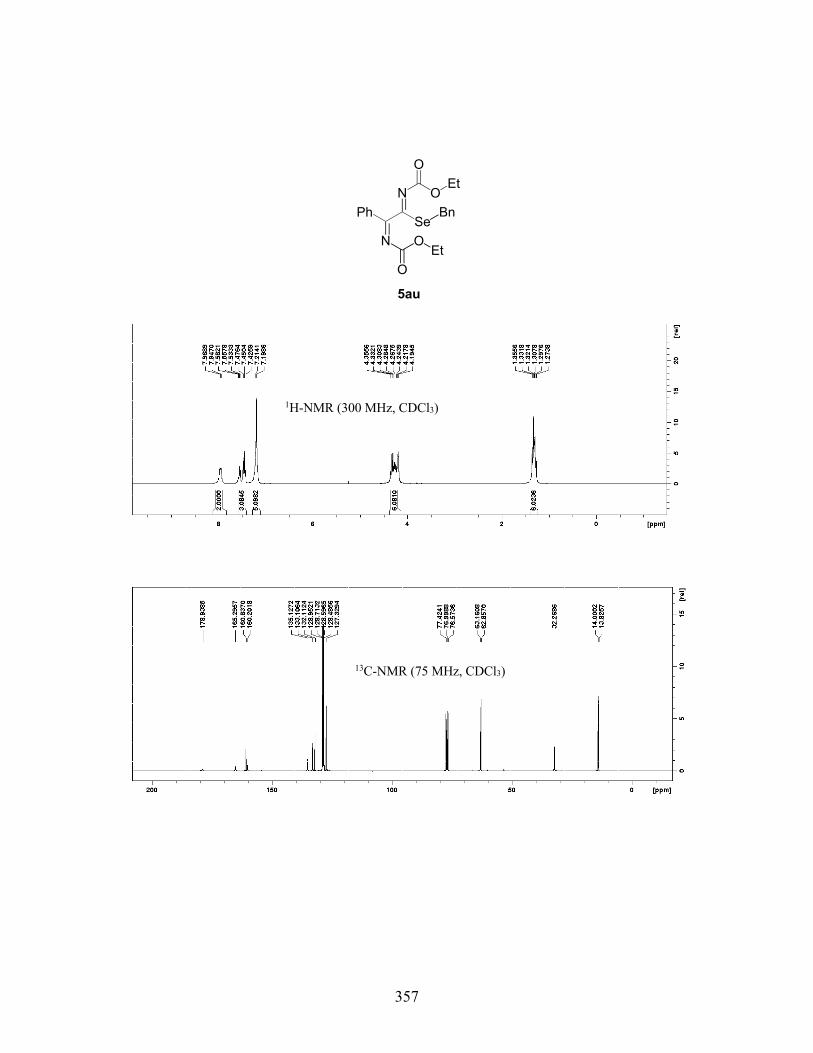
357
N
N
O
OEt
PhSe
5au
Bn
O
OEt
1H-NMR (300 MHz, CDCl3)
13C-NMR (75 MHz, CDCl3)





























