Embed Size (px)
Citation preview

A
w(a(twps©
K
1
lnifapssc
R2f
0d
Vaccine 25 (2007) 2497–2506
Development of lactococcal GEM-based pneumococcal vaccines
Sandrine A.L. Audouy a,1, Saskia van Selm b,1, Maarten L. van Roosmalen a, Eduard Post a,Rolf Kanninga a, Jolanda Neef a, Silvia Estevao c, Edward E.S. Nieuwenhuis c,
Peter V. Adrian c, Kees Leenhouts a, Peter W.M. Hermans b,∗a BiOMaDe Technology Foundation, Groningen, The Netherlands
b Laboratory of Pediatric Infectious Diseases, Radboud University Nijmegen Medical Centre, Nijmegen, The Netherlandsc Laboratory of Pediatrics, Erasmus Medical Center, Rotterdam, The Netherlands
Available online 18 September 2006
bstract
We report the development of a novel protein-based nasal vaccine against Streptococcus pneumoniae, in which three pneumococcal proteinsere displayed on the surface of a non-recombinant, killed Lactococcus lactis-derived delivery system, called Gram-positive Enhancer Matrix
GEM). The GEM particles induced the production of the proinflammatory cytokine tumour necrosis factor-alpha (TNF-�) by macrophagess well as the maturation of dendritic cells. The pneumococcal proteins IgA1 protease (IgA1p), putative proteinase maturation protein APpmA) and streptococcal lipoprotein A (SlrA) were anchored in trans to the surface of the GEM particles after recombinant production ofhe antigens in L. lactis as hybrids with a lactococcal cell wall binding domain, named Protein Anchor domain (PA). Intranasal immunisation
ith the SlrA-IgA1p or trivalent vaccine combinations without additional adjuvants showed significant protection against fatal pneumococcalneumonia in mice. The GEM-based trivalent vaccine is a potential pneumococcal vaccine candidate that is expected to be easy to administer,afe and affordable to produce.2006 Elsevier Ltd. All rights reserved.
ncer Ma
ppdpgSe
cv
eywords: Nasal vaccine; Streptococcus pneumoniae; Gram-positive Enha
. Introduction
Streptococcus pneumoniae (the pneumococcus) is theeading etiological agent of severe infections such as pneumo-ia, meningitis and septicaemia. Young children, elderly andmmunocompromised individuals are particularly vulnerableor pneumococcal diseases, which results in high morbiditynd mortality [1]. The currently available vaccines againstneumococcal infections are based on serotype-specific cap-ular polysaccharides. These include a vaccine containing
olely polysaccharides of 23 serotypes and a conjugate vac-ine consisting of polysaccharides of the 7 most prevalent∗ Corresponding author at: Laboratory of Pediatric Infectious Diseases,adboud University Nijmegen Medical Centre, P.O. Box 9101 (Internal Post24), 6500 HB Nijmegen, The Netherlands. Tel.: +31 24 3666406;ax: +31 24 3666352.
E-mail address: [email protected] (P.W.M. Hermans).1 Both authors contributed equally to this paper.
aaoceyoda
264-410X/$ – see front matter © 2006 Elsevier Ltd. All rights reserved.oi:10.1016/j.vaccine.2006.09.026
trix (GEM)
ediatric serotypes conjugated to an immunogenic carrierrotein. The latter vaccine was introduced for the use in chil-ren under the age of 5, since their immune response to pureolysaccharides is inadequate. The introduction of this conju-ate vaccine in the national vaccination program in the Unitedtates has had a major effect on invasive pneumococcal dis-ase incidence [2].
Since at least 90 different polysaccharide structures areurrently known within the species, polysaccharide-basedaccines only protect against a limited number of serotypes,nd hence, replacement by non-vaccine serotypes remains
threat for vaccine efficacy [3]. High production costsf the conjugate vaccines make their use in developingountries less feasible. The development of an affordableffective vaccine against invasive pneumococcal disease in
oung children and elderly will have major benefits in termsf reducing disease burden and health care costs in botheveloped and developing countries. Immunogenic surfacentigens of pneumococcal origin that are conserved amongst
2498 S.A.L. Audouy et al. / Vaccine 25 (2007) 2497–2506
Table 1Primers used for the cloning of the pneumococcal genes
Primer sequence Gene
CGGTCTCACATGTCGAAAGGGTCAGAAGGTGCAGACC PpmACGGTCTCGAATTGCTTCGTTTGATGTACTACTGCTTGAG PpmACCGTCTCCCATGGTCCAACGCAGTCTGCGT SlrACCGTCTCGAATTCCAGATTTAAAATCGTAGTCTTTCACCA SlrACGTCTCCCATGGGACAAACAGAACCAGAGCCATCAAACGG IgA1pC
R ences h
npsstpd[pppitm
eiacfeppgptttisiathne
bla[aGvp
prvacGTp
2
2
dCi(suuC
2
tTicpsaliPmPhP
CGTCTCGAATTCTAGCCAGTCCAGCATC
estriction enzyme recognition sites are depicted in italics or bold, and sequ
umerous serotypes would be desirable for conferringrotection against infections caused by a broad range oferotypes. Much research effort is currently invested inearch for pneumococcal proteins with protective potentialo be included in future vaccines. Several pneumococcalroteins have been investigated as potential vaccine candi-ates, such as the toxoid derivative of pneumolysin (PdB)4–6], pneumococcal surface protein A (PspA) [4,5,7–9],neumococcal surface adhesin A (PsaA) [7], choline bindingrotein A (CbpA) [6], BVH-3 [10], PiuA and PiaA [11] or theneumococcal protective protein A (PppA) [12]. Preliminarymmunisation studies with these proteins in mice have shownhat they have the potential to protect against infection with
ultiple serotypes and/or prevent nasopharyngeal carriage.Mucosal immunisation offers several advantages over par-
nteral immunisation, such as ease of administration andnduction of immunity via the same routes pathogens initi-te infection. Commensal and non-pathogenic bacteria areurrently being explored for the use as delivery vehiclesor mucosal vaccines, and data on the use of geneticallyngineered bacteria producing heterologous antigens areromising [13–16]. Lactococcus lactis is a food grade non-athogenic, non-colonising Gram-positive bacterium that isenerally recognised as safe (GRAS). Many heterologousroteins have been expressed in L. lactis and immunisa-ion with these strains elicited immune responses specifico the heterologous antigens [17–23]. Mucosal immunisa-ion with L. lactis expressing tetanus toxin fragment Cnduced a TH1/TH2 response characterised by mixed IgGubclasses and T-helper responses, while a TH2 biasedmmune response was obtained when the lactococci weredministered parentally [16]. Although based on a safe bac-erium, this approach still suffers from the disadvantage ofaving recombinant DNA in the final vaccine formulation,amely the risk of dissemination of recombinant DNA in thenvironment.
In this study, we used a non-recombinant lactococcal-ased vaccine displaying pneumococcal antigens. Theactococcal-derived bacterial shaped particles are non-livingnd are designated Gram-positive Enhancer Matrix (GEM)24]. These GEM particles are deprived of surface proteins
nd the intracellular content is largely degraded [25]. TheEM particles were used as an anchoring and deliveryehicle for three pneumococcal proteins, namely putativeroteinase maturation protein A (PpmA) [26,27], IgA1Eco(
IgA1p
omologous with the target are underlined.
rotease (IgA1p) [26,28] and the streptococcal lipoproteinotamase A (SlrA) [26]. Of these proteins, PpmA was pre-iously shown to elicit species-specific opsonophagocyticntibodies that were cross-reactive with various pneumo-occal strains [27]. In the current study, we show that theEM particles display immune stimulatory properties.he vaccine potential of the GEM particles exposing theneumococcal proteins was investigated.
. Materials and methods
.1. Bacterial strains and growth conditions
L. lactis NZ9000 and L. lactis PA1001 were grown asescribed elsewhere [24]. S. pneumoniae D39 (NTTC 7466;entral Public Health Laboratory, London, UK) was grown
n Todd–Hewitt broth supplemented with 0.5% yeast extractTHY) at 37 ◦C or on Colombia agar supplemented with 5%heep blood (BD medical systems, Erembodegem, Belgium)nder oxygen-poor conditions. S. pneumoniae R6 [29], anncapsulated variant of D39 was a kind gift from Jean-Pierrelaverys. It was grown as S. pneumoniae D39.
.2. Plasmid constructs
S. pneumoniae D39 was used as template for subcloninghe genes encoding PpmA and SlrA, and S. pneumoniaeIGR4 was used as template for subcloning the gene encod-
ng IgA1p. PCR fragments of the antigen genes were sub-loned in pPA3 [25] and the obtained plasmids, namedPA32, pPA162 and pPA152, were used to express andecrete the recombinant fusion proteins PpmA-PA, SlrA-PAnd IgA1p-PA, respectively. The primers used for PCR areisted in Table 1. Plasmid pPA32 was constructed by ligat-ng a NcoI- and EcoRI-cleaved PCR-amplified fragment ofpmA into the NcoI and EcoRI sites of plasmid pPA3. Plas-id pPA162 was constructed by ligating an Esp3I-cleavedCR-amplified fragment of SlrA into the corresponding over-anging ends of the NcoI and EcoRI sites of plasmid pPA3.lasmid pPA152 was constructed by ligating a NcoI- and
coRI-cleaved PCR-amplified fragment of IgA1p into theorresponding sites of plasmid pPA3. Electrotransformationf L. lactis PA1001 was performed by using a gene pulserBio-Rad, The Netherlands) as described before [30].
accine
2
L((iH
2
iotwdudoCt1iwtabdw
2m
bZNwRsw3s
2m
ccac(pm
3Tks
2
[fai2rlTd20wCi(o0wflp
2
HfNiioncatcicgo1vam
S.A.L. Audouy et al. / V
.3. Production and purification of antigens
Purified Protein Anchor (PA) was produced by using strain. lactis PA1001 [pPA3] as described before [31]. SlrA-HispET11-SlrA), PpmA-His (pET11-PpmA) and IgA1p-HispET11-IgA1p) with a C-terminal his-tag [26] were producedn E. coli BL21 (de3) using IPTG induction and purified byis-tag isolation for ELISA.
.4. Vaccine preparation
Preparation of GEM particles and vaccines is describedn details elsewhere [24]. Briefly, GEM particles werebtained by boiling freshly grown L. lactis NZ9000 in 0.6 Mrichloroacetic acid (pH 1) for 30 min, followed by extensiveashing with phosphate buffered saline (PBS). The proce-ure results in non-living particles, i.e. zero colony-formingnits in a vaccine dose of 2.5 × 109 GEM particles. Pro-uction of the antigen-PA fusions was induced by additionf nisin to cultures of the suitable L. lactis PA1001 strains.ulture supernatants containing the fusion recombinant pro-
eins were concentrated with a VivaFlow (VivaFlow200,0,000 Da cut-off, Vivascience, Hannover, Germany). Bind-ng of antigens was achieved by mixing the concentratesith GEM particles under gentle agitation for 30 min at room
emperature, followed by extensive washing with PBS. Themount of bound antigen-PA was estimated using Coomassierilliant blue stained gels and compared to BSA protein stan-ards. Immunisation with ethanol-inactivated R6 bacteriaas used as a positive control for protection [32].
.5. Incubation of fluorescent GEM particles withacrophages
For fluorescent labelling, GEM particles were incu-ated with fluorescein isothiocyanate, FITC (Sigma–Aldrich,wijndrecht, The Netherlands), in reaction buffer (0.1 MaHCO3, pH 9) for 2 h at room temperature, followed byashing with water. The murine macrophage cell line ATCCAW 264.7 (TIB-71) was cultured in RPMI 1640 medium
upplemented with l-glutamine and 10% FCS. RAW cellsere incubated with fluorescent GEM particles for 2 h at7 ◦C and were examined using an epifluorescence micro-cope (Nikon Eclipse E800).
.6. Isolation and stimulation of murine peritonealacrophages (PMs)
Resident macrophages were isolated from the peritonealavity of CD-1 mice as described before [33]. Cells wereollected by intraperitoneal injection of RPMI 1640 mediumnd plated in 12-well flat-bottomed plates to 106 viable
ells/well. After overnight culture at 37 ◦C, live L. lactis cells2.5 × 107 CFU/well), GEM particles (2.5 × 107 well−1) orurified PA protein (0.05 �g/well) were added to theacrophages, followed by another overnight incubation at2
P
25 (2007) 2497–2506 2499
7 ◦C. Supernatants were collected for determination ofNF-� concentrations using a sandwich ELISA detectionit according to the manufacturer’s instructions (BD Bio-ciences, Erembodegem, Belgium).
.7. DC maturation assay
Dendritic cells (DCs) were prepared as described before34]. Briefly, bone marrow was flushed with RPMI 1640rom femurs and tibiae of BALB/c mice. Cells were washednd plated in bacteriological 100-mm diameter Petri dishes,n RPMI 1640 supplemented with gentamycin (60 �g/ml),-mercaptoethanol (50 �M; Sigma–Aldrich), 5% FCS andecombinant mouse granulocyte macrophage colony stimu-ating factor (GM-CSF) (200 IU/ml; kindly provided by K.hielemans, University of Brussels, Brussels, Belgium). Atay 9 of the culture, DCs were pulsed overnight with either.5 × 107 GEM particles/ml, 2.5 × 107 L. lactis CFU/ml or.05 �g/ml PA. Cells were harvested and the maturation stateas determined by staining for 30 min with monoclonal antiD-11c-allophycocyanin, anti-MHC-II-FITC (M5/114.5.2),
n combination with anti-CD40-PE (3/23), anti-CD80-PE16-10A1), and anti-CD86-PE (GL-1) antibodies (allbtained from BD Biosciences) diluted in PBS containing.5% BSA and 0.01% sodium azide. Maturation of DCsas determined on CD11c-allophycocyanin-gated cells byow cytometry. Dead cells and debris were excluded usingropidium iodide.
.8. Immunisations
Female CD-1 mice (6 weeks old) were purchased fromarlan (Gannat, France). Animal experiments were per-
ormed with approval of the University of Groningen (Theetherlands) Committee for Animal Ethics. For intranasal
mmunisation, CD-1 mice were lightly anaesthetised withsoflurane (2.5%) over oxygen and nitrous oxyde, heldn their back and 20 �l of vaccine or control (PBS as aegative control and 108 ethanol-inactivated R6 as a positiveontrol) was administered into the nostrils. No additionaldjuvant was included in the tested formulations, includinghe positive control. Prevnar was included as a negativeontrol for vaccination and administered by subcutaneousnjection of 50 �l in the neck. Complete immunisationonsisted of three doses given at 10-day intervals. Eachroup consisted of 10 mice. Monovalent vaccines consistedf 2.5 × 109 GEM particles and contained 6 �g IgA1p-PA,0 �g PpmA-PA or 50 �g SlrA-PA, per dose. The divalentaccines and the trivalent vaccine contained, respectively,half or a third of the amount of antigen present in theonovalent vaccines for each antigen.
.9. Detection of antigen specific IgG by ELISA
To determine the concentrations of antibodies againstpmA, SlrA and IgA1p, an ELISA procedure was used.

2500 S.A.L. Audouy et al. / Vaccine 25 (2007) 2497–2506
F d with flw open. (N
HdH0wiaaaI1n0rcw
2
ipWht4cS5hltc(lbao
ima(Iuc
3
3
ipcTm
sfamcPtpoaG(ot
ig. 1. Uptake of GEM particles by macrophages. RAW cells were incubateith channel for fluorescence source and conventional light simultaneouslyindicates a cell nucleus.
igh-binding capacity microtitreplates (Greiner, Alphen aane Rijn, The Netherlands) were coated with purified SlrA-is, PpmA-His (0.2 �g/well) or IgA1p-His (1 �g/well) in.05 M carbonate buffer (pH 9.6) overnight at 4 ◦C. Platesere washed with PBS (pH 7.4) with 0.02% Tween 20, then
ncubated 1 h with 5% BSA in PBS/Tween. Sera were dilutedppropriately and added to the plates in three-fold dilutionsnd incubated for 2 h at room temperature. After washing, thelkaline phosphatase secondary antibody directed to mousegG-Fc (Sigma–Aldrich) was incubated for 1.5 h at a dilution:5000. Colorimetric reaction was obtained by addition of p-itrophenyl phosphate substrate (Sigma–Aldrich) diluted in.05 M carbonate buffer (pH 9.6) + 1 �M MgCl2. Enzymaticeaction was stopped with NaOH and read at 405 nm. Con-entrations were calculated from a calibration curve madeith purified mouse IgG (Sigma–Aldrich).
.10. Animal model of pneumococcal pneumonia
Prior to use in mice, D39 was passaged by intraperitonealnjection in CD-1 mice to maintain virulence as describedreviously [35]. Aliquots of bacteria were stored at −80 ◦C.hen required, aliquots of bacteria were rapidly thawed,
arvested by centrifugation and resuspended in sterile PBSo 106 CFU/50 �l. Pneumococcal challenge was performed
weeks after immunisation was completed. Challenge wasarried out under inhalation anaesthesia as described underection 2.8 and the bacteria were administered by pipetting0 �l of inoculum onto the nostrils of the mice, which wereeld in upright position to administer the inoculum in theungs. Body weight was determined before challenge and athe time of termination. Animals were terminated by cervi-al dislocation 40 h post-challenge after scoring health statushealthy means that none of the symptoms were observed,
ightly ill: light piloerection, ill: strong piloerection, hunchedack, very ill: strong piloerection, hunched back, and reducedctivity). This was done by two persons, without knowledgef the immunisation treatment. The number of viable bacteriasapT
uorescently labelled GEM particles for 2 h at 37 ◦C. (A) Microscopic viewB) Microscopic view of the same field with fluorescent light source. Each
n nasal lavages, blood and homogenised lungs was deter-ined by culturing serial dilutions on agar plates overnight
t 37 ◦C. Data were expressed as log 10 colony-forming unitsCFU). Statistical analyses were performed using GraphPadnStat3. Differences between groups were assessed with annpaired one-tail Mann–Whitney test. P-values < 0.05 wereonsidered to be statistically significant.
. Results
.1. GEM particles activate macrophages
Interaction of GEM particles with macrophages was stud-ed using fluorescence microscopy with FITC labelled GEMarticles. Association of GEM particles by RAW cells waslearly observed after 2 h of incubation at 37 ◦C (Fig. 1).he pictures suggest that GEM particles are internalised byacrophages.Proinflammatory properties of the GEM particles were
tudied by measuring the production of tumour necrosisactor-� (TNF-�), an early phase cytokine and a marker forctivation of the innate immune response. Murine peritonealacrophages were incubated overnight with either lacto-
occal GEM particles, intact live L. lactis cells, or purifiedrotein Anchor (PA) and TNF-� release was determined in
he culture supernatant. Macrophages stimulated with GEMarticles and live L. lactis cells produced high concentrationsf TNF-�, 977 and 3560 pg/ml, respectively (Fig. 2). Themount of TNF-� produced was dependent on the dose ofEM particles or live L. lactis cells added to the macrophages
data not shown). Purified PA alone did not induce activationf macrophages as the TNF-� production was equivalent tohat of non-stimulated macrophages (60 and 54 pg/ml mea-
ured, respectively). These data indicate that live lactococcind lactococcal GEM particles induce the production of theroinflammatory cytokine and innate immune system markerNF-�.
S.A.L. Audouy et al. / Vaccine
Fig. 2. TNF-� response to GEM particles or L. lactis in peritonealm(A
3m
DirgmtlowlbcnuDDc
3a
vpaowicwnlr(PtnwPai
rsdoppO3twaa
FCl
acrophages. TFN-� was determined in supernatants of unstimulated PMscontrol) or cells incubated with L. lactis, GEM particles (GEM) or Proteinnchor (PA). Bars represent average of triplicate measurements and S.D.
.2. GEM particles and live L. lactis cells induceaturation of dendritic cells (DCs)
Maturation of specialised antigen presenting cells likeCs is an important step toward the development of a specific
mmune response as antigen presentation to lymphocytesequires mature DCs. The GEM particles were further investi-ated by testing their capacity to induce dendritic cells (DCs)aturation in vitro. To this aim, bone marrow derived imma-
ure DCs were pulsed overnight with either GEM particles,ive L. lactis cells or purified PA. The induced expressionf the maturation specific markers CD40, CD80, and CD86as monitored using flow cytometry. Both GEM particles and
ive L. lactis cells induced maturation of DCs as determinedy elevated expression levels of CD40, CD80, and CD86 asompared to non-stimulated DCs (Fig. 3). Purified PA didot induce maturation of DCs since the expression of mat-
ration specific markers was comparable to non-stimulatedCs. Our data demonstrate that live L. lactis cells induceC maturation in vitro and that GEM particles retained thatapacity.
wopd
ig. 3. Maturation of bone marrow-derived DCs induced by GEM particles or L. lacD80, and CD86 on CD11c-gated cells measured by flow cytometry. Immature DC
ive L. lactis (light grey area) or GEM particles (dark grey area).
25 (2007) 2497–2506 2501
.3. Pneumococcal GEM-based vaccines protect micegainst pneumococcal disease
The protective potential of the pneumococcal GEM-basedaccines was tested in an animal model of pneumococcalneumonia. In a first experiment, the three pneumococcalntigens or the Protein Anchor were displayed individuallyn the GEM particles and the obtained monovalent vaccinesere administered intranasally. The unencapsulated, ethanol
nactivated, S. pneumoniae strain R6 was used as a positiveontrol, PBS as a negative control. Although specific IgGsere induced against the antigen present in the vaccine (dataot shown), none of the monovalent GEM-based vaccinesed to a significant reduction of pneumococcal disease aseflected by the bacterial counts in the lungs, blood and noseTable 2). A trivalent vaccine was then prepared, containingpmA, SlrA and IgA1p bound to the GEM particles and the
hree divalent vaccines were made, containing all two combi-ations of these antigens. Mice were immunised intranasallyith the trivalent or divalent vaccines, with inactivated R6 andBS as controls. Prevnar was given subcutaneously as a neg-tive control since serotype 2 polysaccharide is not includedn its formulation.
Prior to challenge with S. pneumoniae, the antibodyesponse was evaluated by measuring specific IgG levels inerum. Results are shown in Table 3. No specific IgGs wereetected in pre-immune serum and immune serum containednly IgGs against the vaccine antigens. All three antigens,resent in different amounts in the vaccines, induced theroduction of systemic IgGs following nasal immunisation.ne dose of the trivalent vaccine contained 2 �g of IgA1p,.5 �g of PpmA and 17 �g of SlrA. After immunisation withhis vaccine, the anti-IgA1p IgG concentration was 12 �g/ml,hile the anti-PpmA and anti-SlrA IgG levels were 343
nd 238 �g/ml, respectively. Interestingly, levels of IgGntibodies induced against a specific antigen were similar
hen this antigen was formulated either a divalent vacciner in a trivalent, in which 1.5 time less of this antigen isresent. In other terms, the way the antigens were combinedid not affect their intrinsic immunogenicity, indicatingtis. Histograms show the expression of maturation-specific markers CD40,s were either not stimulated (solid line) or stimulated with PA (dotted line),

2502 S.A.L. Audouy et al. / Vaccine 25 (2007) 2497–2506
Table 2Pneumococcal load in lungs, blood and nose 40 h after intranasal pneumococcal challenge
Vaccine treatment Lungs Blood Nose
PBS 7.55 (0.47) 5.81 (1.13) 5.20 (0.10)GEM-PA 6.55 (0.80), P = 0.35 5.96 (1.32), P = 0.24 4.56 (0.31), P = 0.09GEM-SlrA 7.01 (0.58), P = 0.39 3.20 (1.47), P = 0.36 4.53 (0.39), P = 0.10GEM-PpmA 6.36 (0.67), P = 0.50 3.40 (1.41), P = 0.40 4.93 (0.23), P = 0.14GEM-IgA1p 7.89 (0.54), P = 0.17R6 3.91 (0.58), P = 0.003
Median values of CFU/ml (S.E.M.) and the P-values of the comparison with the PB
Table 3Post-vaccination concentrations (�g/ml) of specific IgG antibodies againstPpmA, SlrA and IgA1p
Vaccine Antigens used in ELISA
PpmA SlrA IgA1p
GEM-PpmA-SlrA 222.8 (66.4) 302.6 (51.6) N.D.GEM-PpmA-IgA1p 321.5 (97) N.D. 8.2 (5.2)GEM-SlrA-IgA1p N.D. 462.5 (173) 8.1 (3)GEM-PpmA-SlrA-IgA1p 342.6 (72) 237.7 (52.3) 12.1 (1.2)
Average of 10 mice per group and (S.E.M.) are shown. No specific IgGw(O
ta
tatiSar
Fwptwwoyiw*
bsAgPrcibdPtavsrotrivalent vaccine showed only minor weight loss (0.9% ofinitial weight on average), comparable to that seen in the mice
ere detected in pre-immune serum pools (not shown). N.D.: not detectablesamples are considered negative for the presence of specific antibodies whenD405 ≤ 0.1).
hat there was no antagonistic effect between the threentigens.
The health status, weight of the mice, together withhe bacterial counts in the lungs, blood and nose, 40 hfter challenge with serotype 2 D39 S. pneumoniae, werehe parameters for evaluation of vaccine protection. Mice
mmunised with the trivalent GEM vaccine or the combinedlrA-IgA1p GEM-vaccine showed the best health conditiont the time of termination. In these groups, 7 out of 10 miceemained healthy meaning that no symptoms of illness couldig. 4. Health status 40 h after intranasal pneumococcal challenge. Miceere immunised intranasally with the indicated antigens bound to GEMarticles (trivalent vaccine: all three antigens bound to GEM particles), inac-ivated R6 S. pneumoniae or PBS as control. The negative control Prevnaras given subcutaneously. Four weeks after the third immunisation, the miceere challenged with 106 D39 S. pneumoniae bacteria. The health conditionf the mice was scored as described in the Section 2. For statistical anal-sis a number was assigned to the health score (healthy = 1, lightly ill = 2,ll = 3, very ill = 4, dead = 5) and comparison between PBS and treated groupsas performed with an unpaired one-tail Mann–Whitney test. **P < 0.01,
**P < 0.001.
im
Fmlfgtvt
7.95 (1.11), P = 0.02 5.08 (0.30), P = 0.420.22 (0.99), P = 0.07 4.39 (0.14), P = 0.0007
S group, obtained with an unpaired Mann–Whitney test, are indicated.
e detected (Fig. 4). This improvement was statisticallyignificant when comparing with the PBS treated group.s expected, almost all mice in the PBS (9 out of 10)roups showed signs of disease. In the PpmA-IgA1p andpmA-SlrA divalent vaccine groups, four and five mice,espectively, remained healthy, suggesting an improvementompared to the PBS group, although not statistically signif-cant. In each of these two groups, two mice died of diseaseefore the termination time. Immunisation with Prevnarid not improve the health status of the mice compared toBS, with 8 mice out of 10 presenting signs of disease at= 40 h. Although Prevnar elicited significant levels of serumntibody responses against the serotypes represented in thisaccine (data not shown), the lack of protection was expectedince serotype 2, the serotype of the challenge strain, is notepresented in Prevnar. Loss of weight is a reliable indicationf disease or discomfort in mice. Mice immunised with the
mmunised with inactivated R6 S. pneumoniae (1.3%), whileice in the PBS control and the Prevnar groups had lost 6.3
ig. 5. Weight loss 40 h after intranasal pneumococcal challenge. Treat-ent groups are the same as in Fig. 4. %Weight loss = weight prior chal-
enge × 100/(weight prior challenge − weight at termination). Weight lossor individual mice are shown (diamond symbols) as well as the median perroup (light grey bars) (n = 10). The health status of the mice is indicated byhe color code used in Fig. 4. Dead mice were arbitrary attributed the highestalue measured in the PBS treatment group (n = 10). **P < 0.01, comparedo PBS treatment with an unpaired one-tail Mann–Whitney test.
accine
a(Sctlw
FiFwtPiabt
lctT
S.A.L. Audouy et al. / V
nd 5.3% of body weight, respectively, 40 h post-challengeFig. 5). The loss of weight (2.5%) in the group receiving thelrA-IgA1p divalent GEM vaccine was significantly reducedompared to the PBS group (P < 0.005). Taken together,hese results indicate that the trivalent and SlrA-IgA1p diva-
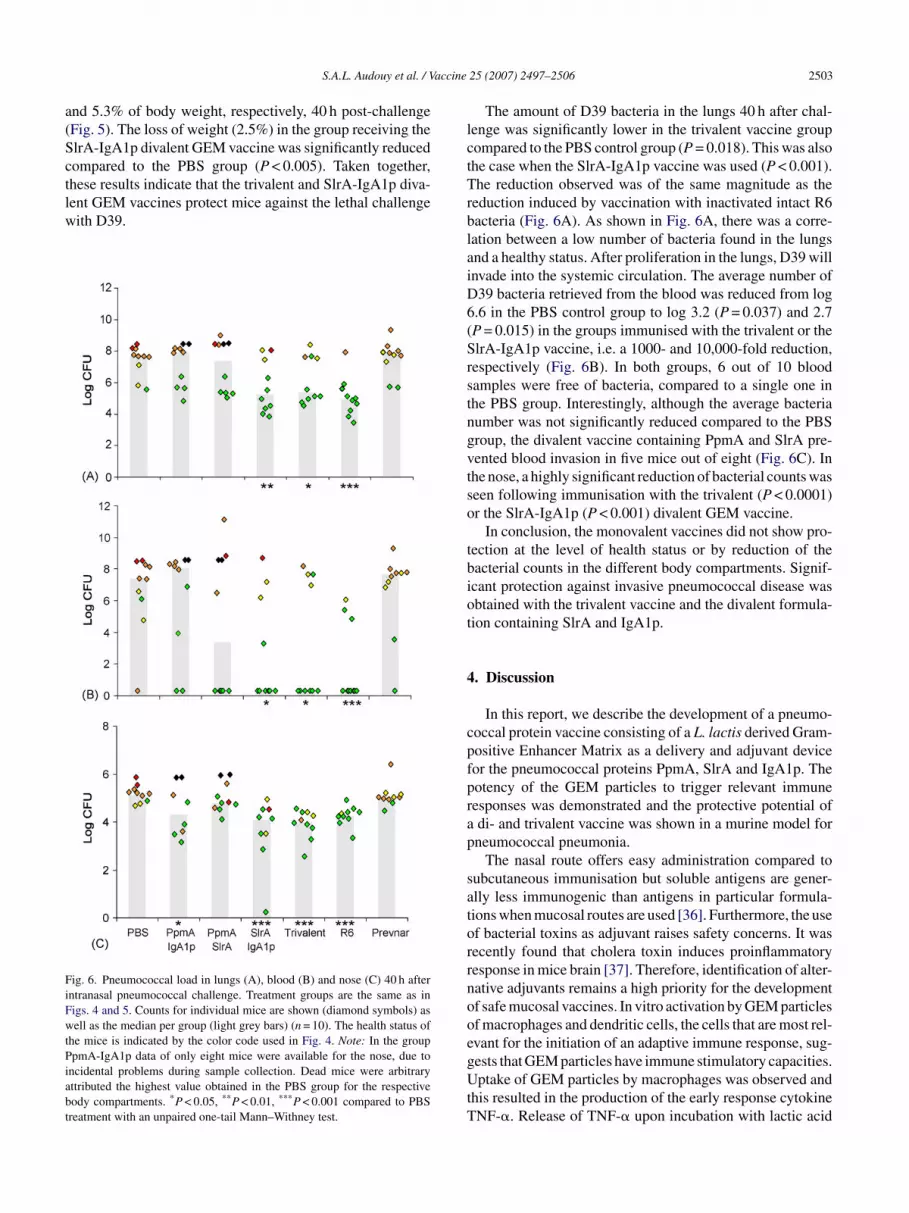
ent GEM vaccines protect mice against the lethal challengeith D39.ig. 6. Pneumococcal load in lungs (A), blood (B) and nose (C) 40 h afterntranasal pneumococcal challenge. Treatment groups are the same as inigs. 4 and 5. Counts for individual mice are shown (diamond symbols) asell as the median per group (light grey bars) (n = 10). The health status of
he mice is indicated by the color code used in Fig. 4. Note: In the grouppmA-IgA1p data of only eight mice were available for the nose, due to
ncidental problems during sample collection. Dead mice were arbitraryttributed the highest value obtained in the PBS group for the respectiveody compartments. *P < 0.05, **P < 0.01, ***P < 0.001 compared to PBSreatment with an unpaired one-tail Mann–Withney test.
rblaiD6(Srstngvtso
tbiot
4
cpfprap
satorrnooegUtT
25 (2007) 2497–2506 2503
The amount of D39 bacteria in the lungs 40 h after chal-enge was significantly lower in the trivalent vaccine groupompared to the PBS control group (P = 0.018). This was alsohe case when the SlrA-IgA1p vaccine was used (P < 0.001).he reduction observed was of the same magnitude as the
eduction induced by vaccination with inactivated intact R6acteria (Fig. 6A). As shown in Fig. 6A, there was a corre-ation between a low number of bacteria found in the lungsnd a healthy status. After proliferation in the lungs, D39 willnvade into the systemic circulation. The average number of39 bacteria retrieved from the blood was reduced from log.6 in the PBS control group to log 3.2 (P = 0.037) and 2.7P = 0.015) in the groups immunised with the trivalent or thelrA-IgA1p vaccine, i.e. a 1000- and 10,000-fold reduction,espectively (Fig. 6B). In both groups, 6 out of 10 bloodamples were free of bacteria, compared to a single one inhe PBS group. Interestingly, although the average bacteriaumber was not significantly reduced compared to the PBSroup, the divalent vaccine containing PpmA and SlrA pre-ented blood invasion in five mice out of eight (Fig. 6C). Inhe nose, a highly significant reduction of bacterial counts waseen following immunisation with the trivalent (P < 0.0001)r the SlrA-IgA1p (P < 0.001) divalent GEM vaccine.
In conclusion, the monovalent vaccines did not show pro-ection at the level of health status or by reduction of theacterial counts in the different body compartments. Signif-cant protection against invasive pneumococcal disease wasbtained with the trivalent vaccine and the divalent formula-ion containing SlrA and IgA1p.
. Discussion
In this report, we describe the development of a pneumo-occal protein vaccine consisting of a L. lactis derived Gram-ositive Enhancer Matrix as a delivery and adjuvant deviceor the pneumococcal proteins PpmA, SlrA and IgA1p. Theotency of the GEM particles to trigger relevant immuneesponses was demonstrated and the protective potential ofdi- and trivalent vaccine was shown in a murine model forneumococcal pneumonia.
The nasal route offers easy administration compared toubcutaneous immunisation but soluble antigens are gener-lly less immunogenic than antigens in particular formula-ions when mucosal routes are used [36]. Furthermore, the usef bacterial toxins as adjuvant raises safety concerns. It wasecently found that cholera toxin induces proinflammatoryesponse in mice brain [37]. Therefore, identification of alter-ative adjuvants remains a high priority for the developmentf safe mucosal vaccines. In vitro activation by GEM particlesf macrophages and dendritic cells, the cells that are most rel-vant for the initiation of an adaptive immune response, sug-
ests that GEM particles have immune stimulatory capacities.ptake of GEM particles by macrophages was observed andhis resulted in the production of the early response cytokineNF-�. Release of TNF-� upon incubation with lactic acid

2 accine
biwttatcohoGtihacpra
cabaGAsrtts
nntfaciiPskipupIiMwptbs
afmecGptvptvagwtgotGopaifFcog
osAoivtattnoicaaahscce
504 S.A.L. Audouy et al. / V
acteria and Gram-positive bacteria was observed by othersn vitro with human monocytes and DCs [38–40]. The resultse obtained with intact L. lactis fits within these observa-
ions. Interestingly, we show that L. lactis GEM particleshat are deprived almost completely of DNA, intact proteinsnd cell wall components like lipoteichoic acids [25] retainedhe ability to induce TNF-� release. A possible explanationould be that the activity is induced by peptidoglycan presentn the surface of the GEM particles. Alternatively, we canypothesise that this activity relates to the particular naturef the bacteria [40]. Maturation of DCs by incubation withEM particles was also observed. The upregulation of cos-
imulatory molecules, crucial for the induction of an adaptivemmune response, suggests that L. lactis GEM particles couldelp the development of a specific immune response whendministered together with an antigen. This assumption wasorroborated by in vivo results showing that mixing GEMarticles with an antigen significantly enhance the humoralesponse against this antigen compared to the antigen givenlone, following nasal immunisation [41].
An important advantage of the pneumococcal subunit vac-ines used here is the combination of presenting the antigenss a bacterial delivery particle without the presence of recom-inant DNA. Because of the particular nature of the vaccinend its dimensions comparable to that of the pathogen, theEM-based vaccines are likely to be efficiently taken up byPCs in the NALT [42,43], and may activate the immune
ystem in a similar manner as intact S. pneumoniae bacte-ia do. Finally, when pneumococcal antigens were bound tohe GEM particles and delivered intranasally, high concentra-ions of circulating specific IgG were detected, showing thatuch a formulation is suitable for mucosal immunisation.
The classical approach for vaccination against S. pneumo-iae is based on the use of capsular polysaccharides. Immu-isation against capsular polysaccharides can provide protec-ion by parenteral [44] or nasal [45,46] administration. A dif-erent approach represents the use of pneumococcal proteinss antigens. Several studies report protection in infection andarriage models following subcutaneous or intraperitonealmmunisation with a protein-based vaccine [4,5,10,11]. Nasalmmunisation could also reduce carriage when PsaA andspA were formulated with cholera-toxin B subunit [7]. Theame finding was made using PppA as antigen [12]. To ournowledge, protection against a lethal infection by nasalmmunisation with a protein vaccine was reported only oncereviously [9]. BALB/c mice were immunised with PspA,sing cholera toxin fragment B as an adjuvant. Protection wasrovided against intravenous, i.p. and intratracheal challenge.n our challenge model, the bacteria were delivered directlynto the lungs, as confirmed using a dye (data not shown).
ice were sacrified 40 h post-challenge and the health status,eight and pneumococcal load were determined. Based on
revious experiments, the 40 h time point was set to includehe development of pneumococcal disease, but was also setefore the non-vaccinated mice die or reach the moribundtate (data not shown). Mice showing signs of severe diseasettWt
25 (2007) 2497–2506
nd pneumococcal load at this time point will not recoverrom pneumococcal disease, while low numbers of pneu-ococci in the lungs and blood reflect protection and an
xpected prolonged survival time. In the present study, theombination of three pneumococcal proteins formulated in aEM-based vaccine showed the ability to protect mice fromneumococcal disease, as revealed by mice welfare and bac-erial growth in different body compartments. Of the divalentaccines included in this study, the SlrA-IgA1p vaccine alsorovided significant protection. Monovalent formulations ofhese three antigens with the GEM particles did not pro-ide protection in this model indicating that combination ofntigens present a great potential for vaccination above sin-le antigen vaccines. These observations are in agreementith the work of Ogunniyi et al. [5]. These authors similarly
ested combinations of three different pneumococcal anti-ens and found protection only with a trivalent vaccine andne of the divalent vaccine combinations, in a i.p. immunisa-ion/i.p. challenge model. The trivalent pneumococcal proteinEM-based vaccine approach showed that the combinationf a limited number of surface proteins is able to providerotection to a similar extent as whole inactivated bacteriagainst pneumococcal disease. This finding is particularlynteresting regarding safety issues and considering the needor well-defined vaccines to replace inactivated pathogens.urthermore, the use of protein antigens instead of polysac-harides is expected to confer protection against a broad rangef serotypes, which remains to be investigated for the anti-ens used in the present study.
Reduction of the bacterial numbers in the lungs is readilybtained with parenteral immunisation [4,10] implying thatystemic antibodies are sufficient to clear infection at this site.lthough we cannot exclude a role for the mucosal response,ur observations suggest that the reduction of bacterial countsn the lungs obtained after immunisation with GEM-basedaccines is mediated by the systemic response. Also, bac-eremia could be reduced in our infection model. Serum IgGntibodies are likely to play a role in the observed preven-ion of invasive disease. Serum IgG antibodies are thoughto be the first mediator of protection against S. pneumo-iae. However, we found no correlation between the levelf protection and the serum concentration of IgGs for thendividual mice. Measurement of opsonophacytic IgG titersould provide valuable information as opsonophagocytic IgGntibodies are relevant for bacterial neutralisation and clear-nce. The lipoprotein PpmA was already shown to inducentibodies with opsonophagocytic activity [27]. On the otherand, Gor et al. did not find binding of anti-PpmA serum to 11erotypes of S. pneumoniae, using flow cytometry [47]. Thisould indicate that other mechanisms are involved in bacterialearance when a protein vaccine is used, as suggested by oth-rs [47,48]. In the nasal cavity, the site of pathogen challenge,
he number of bacteria was also reduced after immunisa-ion with the SlrA-IgA1p divalent or the trivalent vaccine.e anticipate that the IgA subclass of antibody mediatedhis reduction. Indeed, Sun et al. demonstrated that secretory

accine
Iscniv
oscpotmmmba
t[gtnIteip
mvae
A
sasGbe
R
[
[
[
[
[
[
[
[
[
S.A.L. Audouy et al. / V
gAs do not play a crucial role for protection against inva-ive disease, but that they are essential for protection againstarriage [49]. Furthermore, production of specific IgA in theose cavity was demonstrated when GEM vaccines are givenntranasally [41]. This could be an indication that the GEMaccines provide protection against carriage.
Although good protection and bacterial reduction werebtained when PpmA was not included in the vaccine, thepecific role of each antigen in protection remains to be elu-idated. SlrA is thought to be located on the surface of S.neumoniae as a lipoprotein [26], and may similarly inducepsonophagocytic antibodies. IgA1p can circumvent the pro-ective effects of the IgA1-mediated host mucosal defense
echanism by proteolytic cleavage of IgA1 [28]. However,ouse IgAs are not susceptible to cleavage by IgA1p [50],eaning that the response against IgA1p provides protection
y other means than just simple inactivation of the enzymaticctivity by binding.
PpmA genes of various serotypes show hardly any varia-ion as determined by sequencing and comparative genomics51]. Comparative genomics between currently sequencedenomes for the genes encoding SlrA and IgA1p revealedhat SlrA is a conserved protein [26] while IgA1p shows sig-ificant variation [52]. However, the most variable parts ofgA1p are located in a parallel repeat region N-terminally ofhe enzymatic domain. In our study, the strongly conservednzymatic domain is used in the GEM-based vaccine. Hence,t is conceivable that IgA1p, PpmA and SlrA confer cross-rotective immunity.
In summary, protection against pneumococcal disease inice makes a multivalent pneumococcal protein GEM-based
accine a promising candidate for the development of a safend affordable mucosal vaccine against pneumococcal dis-ases.
cknowledgments
The authors thank Hendrik Jan de Heer, Heidi Met-elaar and Louwe de Vries for technical assistance. Theuthors also thank Marijke Haas and Bart Jan Kroesen fortimulating discussions, George Robillard and Ronald deroot for supporting this work. This work was supportedy NWO-STIGON (STIchting Geneesmiddelenonderzoekn ONdernemerschap).
eferences
[1] Hausdorff WP, Feikin DR, Klugman KP. Epidemiological differencesamong pneumococcal serotypes. Lancet Infect Dis 2005;5:83–93.
[2] Withney CG, Farley MM, Hadler J, Harrison LH, Bennett NM, LynfieldR, et al. Decline in invasive pneumococcal disease after the intro-
duction of protein–polysaccharide conjugate vaccine. N Engl J Med2003;348:1737–46.[3] Bogaert D, Veenhoven RH, Sluijter M, Wannet WJ, Rijkers GT,Mitchell TJ, et al. Molecular epidemiology of pneumococcal colo-nization in response to pneumococcal conjugate vaccination in chil-
[
25 (2007) 2497–2506 2505
dren with recurrent acute otitis media. J Clin Microbiol 2005;43:74–83.
[4] Briles DE, Hollingshead SK, Paton JC, Ades EW, Novak L, van GinkelFW, et al. Immunizations with pneumococcal surface protein A andpneumolysin are protective against pneumonia in a murine modelof pulmonary infection with Streptococcus pneumoniae. J Infect Dis2003;188:339–48.
[5] Ogunniyi AD, Folland RL, Briles DE, Hollingshead SK, Paton JC.Immunization of mice with combinations of pneumococcal virulenceproteins elicits enhanced protection against challenge with Streptococ-cus pneumoniae. Infect Immun 2000;68:3028–33.
[6] Ogunniyi AD, Woodrow MC, Poolman JT, Paton JC. Protection againstStreptococcus pneumoniae elicited by immunization with pneumolysinand CbpA. Infect Immun 2001;69:5997–6003.
[7] Briles DE, Ades E, Paton JC, Sampson JS, Carlone GM, HuebnerRC, et al. Intranasal immunization of mice with a mixture of thepneumococcal proteins PsaA and PspA is highly protective againstnasopharyngeal carriage of Streptococcus pneumoniae. Infect Immun2000;68:796–800.
[8] Swiatlo E, King J, Nabors GS, Mathews B, Briles DE. Pneumococcalsurface protein A is expressed in vivo, and antibodies to PspA areeffective for therapy in a murine model of pneumococcal sepsis. InfectImmun 2003;71:7149–53.
[9] Wu HY, Nahm MH, Guo Y, Russell MW, Briles DE. Intranasal immu-nization of mice with PspA (Pneumococcal surface protein A) canprevent intranasal carriage, pulmonary infection, and sepsis with Strep-tococcus pneumoniae. J Infect Dis 1997;175:839–46.
10] Hamel J, Charland N, Pineau I, Ouellet C, Rioux S, Martin D, et al.Prevention of pneumococcal disease in mice immunized with conservedsurface-accessible proteins. Infect Immun 2004;72:2659–70.
11] Brown JS, Ogunniyi AD, Woodrow MC, Holden DW, Paton JC. Immu-nization with components of two iron uptake ABC transporters pro-tects mice against systemic Streptococcus pneumoniae infection. InfectImmun 2001;69:6702–6.
12] Green BA, Zhang Y, Masi AW, Barniak V, Wetherell M, Smith RP,et al. PppA, a surface-exposed protein of Streptococcus pneumoniae,elicits cross-reactive antibodies that reduce colonization in a murineintranasal immunization and challenge model. Infect Immun 2005;73:981–9.
13] Grangette C, Muller-Alouf H, Goudercourt D, Geoffroy M-C, TurneerM, Mercenier A. Mucosal immune responses and protection againsttetanus toxin after intranasal immunization with recombinant Lacto-bacillus plantarum. Infect Immun 2001;69:1547–53.
14] Hanniffy S, Wiedermann U, Repa A, Mercenier A, Daniel C, Fio-ramonti J, et al. Potential and opportunities for use of recombinantlactic acid bacteria in human health. Adv Appl Microbiol 2004;56:1–64.
15] Norton PM, Brown HWG, Wells JM, Macpherson AM, Wilson PW,Le Page RWF. Factors affecting the immunogenicity of tetanus toxinfragment C expressed in Lactococcus lactis. FEMS Immunol MedMicrobiol 1996;14:167–77.
16] Robinson K, Chamberlain LM, Lopez MC, Rush CM, Marcotte H, LePage RWF, et al. Mucosal and cellular immune responses elicited byrecombinant Lactococcus lactis strains expressing tetanus toxin frag-ment C. Infect Immun 2004;72:2753–61.
17] Bermudez-Humaran LG, Cortes-Perez NG, Le Loir Y, Alcocer-Gonzalez JM, Tamez-Gurra RS, Montes de Oca-Luna R, et al. Aninducible surface presentation system improves cellular immunityagainst human papillomavirus type 16 E7 antigen in mice afternasal administration with recombinant lactococci. J Med Microbiol2004;53:427–33.
18] Dieye Y, Hoekman AJ, Clier F, Juillard V, Boot HJ, Piard JC. Abil-
ity of Lactococcus lactis to export viral capsid antigens: a cru-cial step for development of live vaccines. Appl Environ Microbiol2003;69:7281–8.19] Lee MH, Roussel Y, Wilks M, Tabaqchali S. Expression of Helicobacterpylori urease subunit B gene in Lactococcus lactis MG1363 and its use

2 accine
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
506 S.A.L. Audouy et al. / V
as a vaccine delivery system against H. pylori infection in mice. Vaccine2001;19:3927–35.
20] Norton PM, Wells JM, Brown HWG, Macpherson AM, Le Page RWF.Protection against tetanus toxin in mice nasally immunized with recom-binant Lactococcus lactis expressing tetanus toxin fragment C. Vaccine1997;15:616–9.
21] Pei H, Liu J, Cheng Y, Sun C, Wang C, Lu Y, et al. Expression of SARS-coronavirus nucleocapsid in Escherichia coli and Lactococcus lactisfor serodiagnosis and mucosal vaccination. Appl Microbiol Biotechnol2005;68:220–7.
22] Pontes DS, Dorella FA, Ribeiro LA, Miyosi A, Le Noir Y, Gruss A, etal. Induction of partial protection in mice after oral administration ofLactococcus lactis producing Brucella abortus L7/L12 antigen. J DrugTarget 2003;11:489–93.
23] Xin K-Q, Hoshino Y, Toda Y, Igimi S, Kojima Y, Jounai N, et al.Immunogenicity and protective efficacy of orally administered recom-binant Lactococcus lactis expressing surface-bound HIV env. Blood2003;102:223–8.
24] van Roosmalen ML, Kanninga R, El Khatabbi M, Neef J, AudouyS, Bosma T, et al. Mucosal vaccine delivery of antigens tightlybound to an adjuvant particle made from food-grade bacteria. Meth-ods 2006;38:144–9.
25] Bosma T, Kanninga R, Neef J, Audouy SAL, van Roosmalen ML,Steen A, et al. Novel surface display system for proteins on non-genetically modified Gram-positive bacteria. Appl Environ Microbiol2006;72:880–9.
26] Adrian PV, Bogaert D, Oprins M, Rapola S, Lahdenkari M, KilpiT, et al. Development of antibodies against pneumococcal proteinsalpha-enolase, immunoglobulin A1 protease, streptococcal lipoproteinrotamase A, and putative proteinase maturation protein A in rela-tion to pneumococcal carriage and otitis media. Vaccine 2004;22:2737–42.
27] Overweg K, Kerr A, Sluijter M, Jackson MH, Mitchell TJ, de JongAP, et al. The putative proteinase maturation protein A of Streptococ-cus pneumoniae is a conserved surface protein with potential to elicitprotective immune responses. Infect Immun 2000;68:4180–8.
28] Weiser JN, Bae D, Fasching C, Scamurra RW, Ratner AJ, Janoff EN.Antibody-enhanced pneumococcal adherence requires IgA1 protease.Proc Natl Acad Sci USA 2003;100:4215–20.
29] Hoskins J, Alborn Jr WE, Arnold J, Blaszczak LC, Burgett S, DeHoffBS, et al. Genome of the bacterium Streptococcus pneumoniae strainR6. J Bacteriol 2001;183:5709–17.
30] Holo H, Nes IF. Transformation of Lactococcus by electroporation.Methods Mol Biol 1995;47:195–9.
31] Steen A, Buist G, Leenhouts KJ, El Khatabbi M, Grijpstra F, ZomerAL, et al. Cell wall attachment of a widely distributed peptidoglycanbinding domain is hindered by cell wall constituents. J Biol Chem2003;278:23874–81.
32] Malley R, Lipsitch M, Stack A, Saladino R, Fleisher G, Pelton S, et al.Intranasal immunization with killed unencapsulated whole cells pre-vents colonization and invasive disease by capsulated pneumococci.Infect Immun 2001;69:4870–3.
33] Speert DP, Gordon S. Phagocytosis of unopsonized Pseudomonasaeruginosa by murine macrophages is a two-step process requiringglucose. J Clin Invest 1992;90:1085–92.
34] Visser L, Jan de Heer H, Boven LA, van Riel D, van Meurs M, MeliefMJ, et al. Proinflammatory bacterial peptidoglycan as a cofactor for thedevelopment of central nervous system autoimmune disease. J Immunol
2005;174:808–16.35] Alexander JE, Lock RA, Peeters CC, Poolman JT, Andrew PW, MitchellTJ, et al. Immunization of mice with pneumolysin toxoid confers asignificant degree of protection against at least nine serotypes of Strep-tococcus pneumoniae. Infect Immun 1994;62:5683–8.
[
25 (2007) 2497–2506
36] Grangette C, Muller-Alouf H, Hols P, Goudercourt D, Delcour J,Turneer M, et al. Enhanced mucosal delivery of antigens withcell wall mutants of lactic acid bacteria. Infect Immun 2004;72:2731–7.
37] Armstrong ME, Lavelle EC, Loscher CE, Lynch MA, Mills KH. Proin-flammatory responses in the murine brain after intranasal delivery ofcholera toxin: implications for the use of AB toxins as adjuvants inintranasal vaccines. J Infect Dis 2005;192:1628–33.
38] Zeuthen LJ, Christensen HR, Frøkiær H. Lactic acid bacteria induc-ing a weak Interleukin-12 and tumor necrosis factor alpha response inhuman dendritic cells inhibit strongly stimulating lactic acid bacteriabut act synergically with Gran-negative bacteria. Clin Vaccine Immunol2006;13:365–75.
39] Miettinen M, Vuopio-Varkila J, Varkila K. Production of human tumornecrosis factor alpha, Interleukin-6 and Interleukin-10 is induced bylactic acid bacteria. Infect Immun 1999;64:5403–5.
40] Hessle CC, Andersson B, Wold AE. Gram-positive and Gram-negativebacteria elicit different patterns of pro-inflammatory cytokines inhuman monocytes. Cytokine 2005;30:311–8.
41] Audouy SA, van Roosmalen ML, Neef J, Kanninga R, Post E,van Deemter M, et al. Lactococcus lactis GEM particles dis-playing pneumococcal antigens induce local and systemic immuneresponses following intranasal immunization. Vaccine 2006;24:5424–41.
42] Bienenstock J, McDermott MR. Bronchus- and nasal-associated lym-phoid tissues. Immunol Rev 2005;206:22–31.
43] Kiyono H, Fukuyama S. NALT- versus Peyer’s-patch-mediatedmucosal immunity. Nat Rev Immunol 2004;4:699–710.
44] Libon C, Haeuw JF, Crouzet F, Mugnier C, Bonnefoy JY, CorvaiaN. Streptococcus pneumoniae polysaccharides conjugated to the outermembrane protein A from Klebsiella pneumoniae elicit protective anti-bodies. Vaccine 2002;20:2174–80.
45] Jakobsen H, Saeland E, Gizurarson S, Schuldz D, Jonsdottir I. Intranasalimmunization with pneumococcal polysaccharide conjugate vaccinesprotects mice against invasive pneumococcal infections. Infect Immun1999;67:4128–33.
46] Jakobsen H, Schuldz D, Pizza M, Rappuoli R, Jonsdottir I. Intranasalimmunization with pneumococcal polysaccharide conjugate vaccineswith nontoxic mutants of Escherichia coli heat-labile enterotoxinsas adjuvants protects mice against invasive pneumococcal infections.Infect Immun 1999;67:5892–7.
47] Gor DO, Ding X, Briles DE, Jacobs MR, Greenspan NS. Relation-ship between surface accessibility for PpmA, PsaA, and PspA andantibody-mediated immunity to systemic infection by Streptococcuspneumoniae. Infect Immun 2005;73:1304–12.
48] Miyaji EN, Dias WO, Tanizaki MM, Leite LC. Protective efficacy ofPspA (pneumococcal surface protein A)-based DNA vaccines: contri-bution of both humoral and cellular responses. FEMS Immunol MedMicrobiol 2003;37:53–7.
49] Sun K, Johansen F-E, Eckmann L, Metzger D. An important rolefor polymeric Ig receptor-mediated transport of IgA in protectionagainst Streptococcus pneumoniae nasopharyngeal carriage. J Immunol2004;173:4576–81.
50] Qiu J, Brackee GP, Plaut AG. Analysis of the specificity of bacterialimmunoglobin A (IgA) protease by a comparative study of ape serumIgAs as substrates. Infect Immun 1996;64:933–7.
51] Overweg K, Pericone CD, Verhoef GG, Weiser JN, Meiring HD, deJong AP, et al. Differential protein expression in phenotypic variants of
Streptococcus pneumoniae. Infect Immun 2000;68:4604–10.52] Poulsen K, Reinholdt J, Jespersgaard C, Boye K, Brown TA, Hauge M,et al. A comprehensive genetic study of streptococcal immunoglobulinA1 proteases: evidence for recombination within and between species.Infect Immun 1998;66:181–90.





























