Embed Size (px)
Citation preview

Chemical Physics 99 (1985) 73-85 73 North-Holland, Amsterdam
D I A T O M I C S - I N - M O L E C U L E S P O T E N T I A L ENERGY SURFACES FOR T H E C O L L I S I O N - I N D U C E D D I S S O C I A T I O N OF EXCITED L i 2 ( t I I . ) BY H E L I U M A T O M S
Christian C. C H A N G and Philip J. K U N T Z
Hahn - Meitner- Institut fur Kernforschung Berlin, Glienicker Strasse 100, D - 1000 Berlin 39, FRG
Received 25 March 1985
The 1 ~A", 21A", 31A', 41A', and 51A' potential energy surfaces (PES) relevant to the collision-induced dissociation (CID) process Li 2 ( t 17I. ) + He( 1Sg ) ~ Li( 2 S~ ) + Li( 2 Pu ) + He( 1Sg ) are approximated by three dia tomics-in-molecules models differing from each other in the,source of diatomic input. The semi-empirical model, the most realistic of the three, shows the CID to depend critically on the position of avoided crossings on the PES and on the magnitude of the non-adiabatic coupling, which is estimated numerically. The 1A" calculations support a qualitative two-step mechanism recently proposed by Poppe and Whitton. The IA' surfaces exhibit a degree of complexity which precludes a simple picture of the CID process. The results suggest that only a quan tum scattering treatment or perhaps a very detailed surface-hopping trajectory calculation will be adequate for the interpretation of the experimental observations on this reaction, and the estimation of the cross sections.
1. Introduction
Numerous studies on the transfer reaction A + BC ~ AB + C over the last two decades have re- sulted in a sound framework for the interpretation of experimental observations in terms of the fea- tures of potential energy surfaces (PES) [1-4]. This is particularly true for reactions governed by a single PES. Difficulties arise when several surfaces are energetically accessible. The possibility of non-adiabatic transitions [5,6] between surfaces complicates the trajectories of A, B, and C enor- mously and interpretation of experimental ob- servations in terms of a single-surface theory then leads to considerable confusion.
Recognizing just when non-adiabatic effects may be important is not always easy. They nearly always need to be taken into account if the re- actants are clearly in a different electronic state than the products, as in Penning ionization, He* + H 2 ~ He + H~- + e , or in electronic to vibra- tional energy conversion processes, Na (2po)+ H2(o ) ~ Na(2Sg)+ H2(v ' ) [7,8]. Even if the elec- tronic state of reactants and products is the same, a dramatic change in the character of the wave- function, as for example in reactions producing
ionic products from covalent reactants (e.g. K + Br 2 ~ K + B r - + Br) [9], may signal the presence of non-adiabaticity.
Somewhat more difficult to assess are ion molecule reactions, such as Ne + H e ~ - - , N e H e + + H e , where the behaviour of the elec- tronic wavefunction in going from reactants to products is not obvious [10-13]. Finally, most confusing of all are processes which at first sight seem to proceed on a single PES because of the clear correlation of the reactants with the products [14,15]: e.g.
Li2 ( ' I I ~ ) + He('Sg )
Li(2Sg) + Li(2pu) + He(1Sg). (1)
Non-adiabatic effects can sometimes be identi- fied by the presence of a real curve crossing in the BC coordinate in the asymptotic region A + BC. As atom A approaches BC, the diatomic symmetry is broken and the crossing becomes an avoided crossing. In ion-molecule reactions, the crossing arises from the near degeneracy of charge-transfer states such as A + BC and A + + BC . The posi- tion of the crossing for a given BC molecule
0301-0104/85/$03.30 © Elsevier Science Publishers B.V. (North-Holland Physics Publishing Division)
74 C.C. Chang, P.J. Kun tz / C1D of excited Li, ( IfI , ,) by He atoms
3.0
2.5
2.0
"-' 1.5
1.0
0.5
Liz VB
- - - - - OK
/ /
V / /
/
i i i
5 10 15 R [au]
20
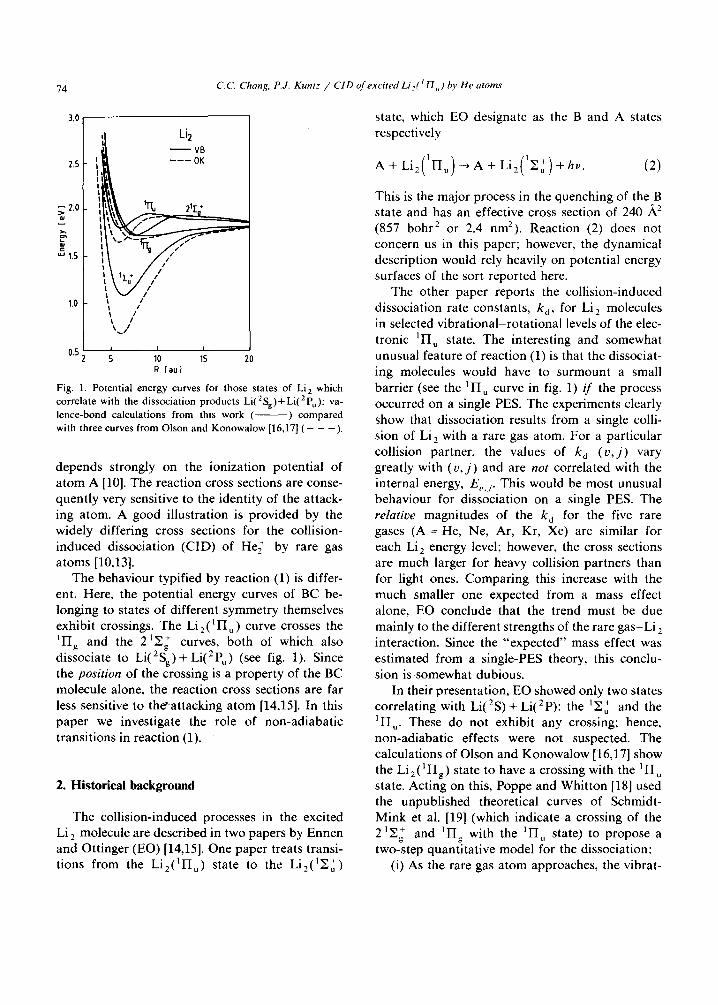
Fig. 1. Po ten t ia l e n e r g y curves for those s ta tes of Li 2 wh ich
co r re l a t e wi th the d i s soc ia t ion p r o d u c t s Li( 2 Sg) + Li( 2 Pu ): va-
l e n c e - b o n d ca l cu l a t i ons f rom this w o r k ( ) c o m p a r e d
wi th three curves f r o m O l s o n a n d K o n o w a l o w [16,17] ( - - - ) .
depends strongly on the ionization potential of a tom A [10]. The reaction cross sections are conse- quently very sensitive to the identity of the attack- ing atom. A good illustration is provided by the widely differing cross sections for the collision- induced dissociation (CID) of He~ by rare gas atoms [10,13].
The behaviour typified by reaction (1) is differ- ent. Here, the potential energy curves of BC be- longing to states of different symmetry themselves exhibit crossings. The Li2( l I Iu) curve crosses the l IIg and the 21Y + 2 g curves, both of which also dissociate to Li( Sg)+ Li(2P,) (see fig. 1). Since the position of the crossing is a property of the BC molecule alone, the reaction cross sections are far less sensitive to the'attacking atom [14,15]. In this paper we investigate the role of non-adiabatic transitions in reaction (1).
2. Historical background
The collision-induced processes in the excited Li 2 molecule are described in two papers by Ennen and Ottinger (EO) [14,15]. One paper treats transi- tions from the L iz (1H. ) state to the Li2(~Y,,)+
state, which EO designate as the B and A states respectively
1 +
This is the major process in the quenching of the B state and has an effective cross section of 240 ~2 (857 bohr 2 or 2.4 nm2). Reaction (2) does not concern us in this paper; however, the dynamical description would rely heavily on potential energy surfaces of the sort reported here.
The other paper reports the collision-induced dissociation rate constants, k d, for Li 2 molecules in selected vibrational-rotat ional levels of the elec- tronic 1H u state. The interesting and somewhat unusual feature of reaction (1) is that the dissociat- ing molecules would have to surmount a small barrier (see the l IIu curve in fig. 1) if the process occurred on a single PES. The experiments clearly show that dissociation results from a single colli- sion of Li 2 with a rare gas atom. For a particular collision partner, the values of k~ (u , j ) vary greatly with ( o , j ) and are not correlated with the internal energy, E,,, I. This would be most unusual behaviour for dissociation on a single PES. The relatioe magnitudes of the k~ for the five rare gases (A = He, Ne, Ar, Kr, Xe) are similar for each Li 2 energy level; however, the cross sections are much larger for heavy collision partners than for light ones. Comparing this increase with the much smaller one expected from a mass effect alone, EO conclude that the trend must be due mainly to the different strengths of the rare gas-Li 2 interaction. Since the "expected" mass effect was estimated from a single-PES theory, this conclu- sion is somewhat dubious.
In their presentation, EO showed only two states 1 + correlating with Li (2S)+Li(zP) : the Y,o and the
11-Io. These do not exhibit any crossing; hence, non-adiabatic effects were not suspected. The calculations of Olson and Konowalow [16,17] show the Li2(~Hg) state to have a crossing with the ~H u state. Acting on this, Poppe and Whitton [18] used the unpublished theoretical curves of Schmidt- Mink et al. [19] (which indicate a crossing of the
1 + 2 Y~g and ]I-[g with the ~Hu state) to propose a two-step quantitative model for the dissociation:
(i) As the rare gas atom approaches, the vibrat-

C C. Chang, P.J. Kuntz / CID of excited Li z(I H,) by He atoms 75
ing Li2 molecule, instead of following a diabat ic potent ial corresponding to ~FI, symmetry , remains on the adiabatic surface which correlates with
1 + either the 2 Y.g or ~ Hg state of Li 2. (ii) The Li 2 molecule is then able to stretch to a
much larger turning point (see fig. 1) present ing a bigger target to the collision partner. Prestretching of the L i -L i bond facilitates dissociation in the ensuing direct collision, especially since the I/-Ig curve has no barr ier to dissociation. It is clear that the first step in this mechanism depends in a compl ica ted way on the internal energy of the Li 2 molecule, E , , r precluding a correlat ion of k d with E,w. The model accounts nicely for the unex- pected behaviour of the experimental ly determined rate constants.
3. T h e D I M m o d e l
3.1. Overview
Motiva ted by Poppe and Whi t ton ' s arguments , we a t t empt here to provide a more quant i ta t ive picture of the C I D process by est imating the PES and associated n o n - a d i a b a t i c coupling by the me thod of diatomics- in-molecules ( D I M ) [20-22]. React ion (1) is rather demanding for D I M since it requires the es t imat ion of weakly bound excited states, which often means a large D I M basis. It is indeed fortunate that a basis derived solely f rom He(1Sg), Li( 2 Sg) and Li(2 p~ ) a tomic states appears to be adequate, judged by a project ion analysis [23] of the relevant Li 2 states, and that the neces- sary dia tomic f ragment matr ices could be obta ined f rom valence-bond (VB) calculations. Our results demons t ra te the utility of Poppe and Whi t ton ' s suggestions and form the basis for a future trajec- tory surface-hopping est imate of the rate con- stants.
The D I M tr iatomic basis set is obta ined f rom a direct product of the a tomic eigenfunctions for the states He(ISg), Li(2Sg) and Li(2pu). Four state groups [24] determine the structure of the model:
(1) LiLiHe
(2) Li*LiHe
(3) LiLi*He
(4) Li*Li*He,
where Li stands for Li(2Sg), Li* for L i (2p , ) and He for He(1Sg). These give rise to ten basis func- tions for the 1A' and six basis functions for the 1A" states of Li2He. Each function can also be ex- pressed as a product of the He(1Sg) a tomic ei- genfunct ion multipl ied by a Li 2 basis funct ion having ~Z, ~FI or ~A symmetry. The Li 2 functions are listed in table 1.
The D I M model is precisely defined only after the numerical values of the elements of the a tomic and dia tomic f ragment matrices implied by the model structure have been specified. Since there are many ways of doing this, a single model struc- ture may spawn a variety of numerical representa- tions. In this paper, three models are considered: (a) The dia tomic f ragment matrices are computed in the VB approximat ion using a basis large enough to ensure a g o o d value for the Li(2Sg ~ 2Pu) exci- tation. (b) The matr ices f rom model (a) are mod- ified by replacing the eigenvalues for the Li 2 (X1 + 1 + 1 t Z g , Y.~, H~ and l-[g) states by those of Olson and Konowalow (OK) [17]. In addition, the
1 + VB 2 Eg curve is shifted to mainta in the same relative posit ion with respect to the ~U curves as
Table 1 DIM basis functions for the diatomic fragments ~)
Fragment N Atomic states Basis functions 1 + Li 2 YZg 16 (1) Li(ES) Li(2S) ~oIsg- ssl
(2) Li(2S) Li(2P) ¢oZlsE-gz-zX+Ysl (3) Li(2P) Li(Ep) ~o21y~-y,y+xX-~x[ (4) Li( 2 p) Li(2 p) ~01 z,~ - ~.z I
ly+ 10 (1) Li(2S)Li(2p) ~o2ls~-~z+zg-~s[ IFI u 20 (1)Li(ZS)Li(2P) ~o2[sX-sx+xg-Xs]
(2) Li( 2 p) Li( 2 p) 21 x~. - ~z - zY + ,~x I 1Fig 20 (1) Li(ZS)Li(2P) ~o2lsX-gx-xg+Xsl
(2) Li(2P) Li(2P) (o2[x~-2z+z.~-~x[ lAg 6 (1) Li(2p) Li(2P) (oZ[~y -~y -xX+~x l tZ~ 6 (1)Li(2P)Li(zP) ~o21xP-Xy-yX+)x l
Lille 2y+ 36 (1) Li(2S) He(tS) sHe (2) Li(2P) He(IS) zHe
2H 18 (1)Li(ZP)He(IS) xHe
~) N is number of basis functions in the VB calculations, s is Li(2S) atomic eigenfunction with Ms=l /2 . x, y, z are Li(2p) atomic eigenfunctions with M s = 1/2. w is 2 1/z.

76 C.C. Chang, P.J. Kuntz / C1D of excited Li e ( I H , ) by He atoms
1 + in model (a). (c) The 2 Eg curve of model (b) is shifted downwards to a position similar to that shown in fig. 1 of Poppe and Whitton [18]. We regard model (c) as the most realistic.
3.2. The valence-bond calculations
The program MULTIBOND A of Balint-Kurti and Yardley [25] was used for all of our VB calculations. For the helium atom, the 10s gaus- sian orbital contractions of Huzinaga [26] were employed. These yielded -2.8616692 hartree for the energy of He(1Sg) (cf. -2.903724375 hartree of Pekeris [27]). For the ls and 2s orbitals of Li we used the 10s contractions of Huzinaga [26] and for the 3s, 2p and 3p orbitals the contractions in table 2. The resulting energies were -7.43250455 for Li(2S~), - 7 . 3 6 4 8 4 5 6 2 for L i (2pu) and -7.23624047 hartree for Li + (lSg). This corre- sponds to an ionization potential for Li of 5.34 eV (cf. 5.39 eV from experiment [28]) and an excita- tion energy Li(2S--, 2p) of 1.84 eV (cf. 1.85 eV from experiment [28]). From these atomic energies the (diagonal) atomic fragment matrices could be constructed. An energy of zero was assigned to Li(2Sg) and ne(1Sg).
The diatomic fragment matrices were con- structed by projection [29] from an extended-basis VB calculation. Such a basis was necessary in order to test the adequacy of the DIM model (see section 3.3) and was built by augmenting the basis He(ls), Li(ls, 2s, 2p) with the 3s and 3p contrac- tions in table 2 and with the single GTOs in table 3. The exponents a of the single GTOs were optimized a t RLiLi = 6 bohr. A comparison of our
Table 2 Li 3s, 2p and 3p orbital contractions (atomic units)
Li(3s) orbital Li(2p, 3p)"
exponent C(3s) exponent C(2p) C(3p)
0.22271 -0.00571 0.057156 0.31796 0.0086456 - 1.16887 0.0047430 -0 .10520
3.79963112 -0 .0069 -0 .00413 0.817154884 -0 .03415 - 0.02425 0.237145007 -0 .12362 -0 .06809 0.0887045264 -0 .32298 -0 .31862 0.0377507210 -0 .46454 -0 .04424 0.0176130086 -0 .19507 -1.26411 0.012 -0 .0335 2.09610
Table 3 Additional GTOs
Molecule State ~4s(Li) Ot4p(Li ) a2s(He ) Ot2p(He ) I + Li 2 Zg 0.032 l y + 0.03 lAg 0.025
1Z~ 0.015 1 H ° 0.028 0.02 1Fl 0.031
Lil le 2 Z +~ 0.035 0.035 0.03 1.2 z H 0.035 0.035 0.03 1.0
VB energies with ab initio results [16,17,30] is shown in figs. 1-3.
3.3. Adequacy of the D I M model
The extended-basis VB calculations reproduce the qualitative behaviour of the first five states of the Li 2 molecule and can therefore serve as a standard against which the DIM basis can be tested. The projection, p, of each Li 2 eigenfunc- tion onto the space spanned by the DIM diatomic basis is a satisfactory measure of adequacy: a DIM basis for which p > 0.85 is deemed accept- able [23].
To calculate the projection p, for a particular VB eigenvector I E,) onto the space spanned by the set of DIM basis functions [q,), it is necessary to know the overlaps u = (q,I E,) and $ = (q'l@).
1.0 i 1 - - VB il OK
~\ LilZs) ÷ Li(2S)
i\ .. ' , \ / , " ~\ / / XIZ *
-1.0 x J ' g I I I , I I
2 ~ 6 8 10 12 R[au]
Fig. 2. Potential energy curves for the Li2(X ly.~ ) ground state as calculated from the valence-bond program ( ) and from Olson and Konowalow [16,17] ( - - - ) .
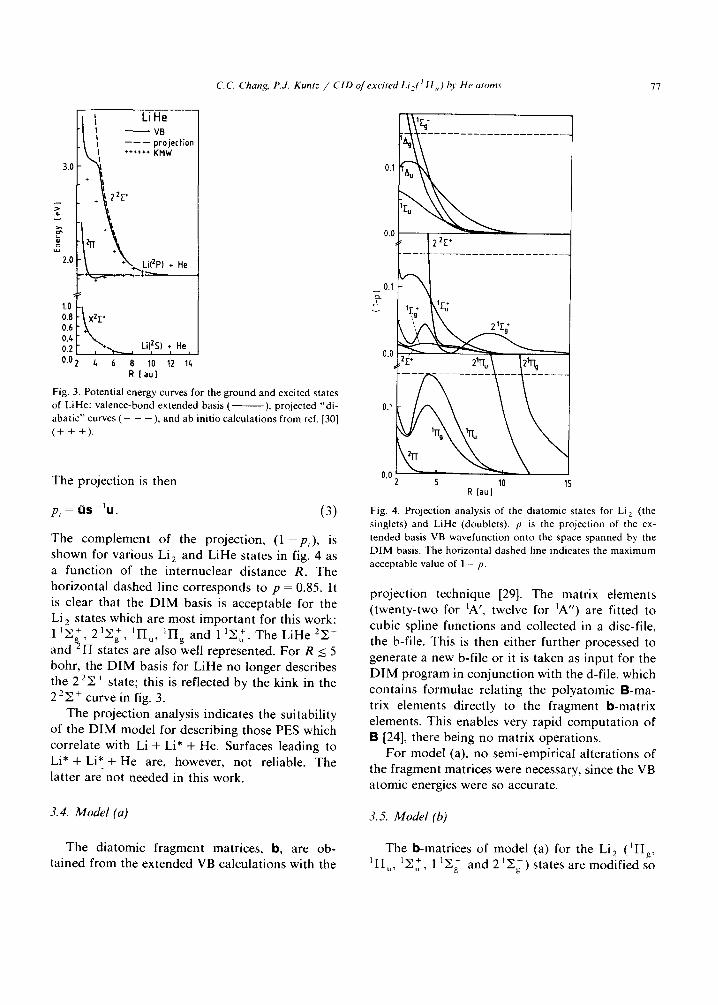
C, C. Chang, P.J. Kuntz / CID of excited Li f t I [I , ) hy He atoms 77
Lille | VB It ------ projection 1 ****++ KHW
3.0 ~ ~ ,
,-°t\ 0.8 XZE •
" 0.4 Li(2S) He 0.2[ , *--,.__~ ~ , 0.02 ~ 6 8 10 12 I~
R [ a u ]
Fig. 3. Potential energy curves for the ground and excited states of Lille: valence-bond extended basis ( ), projected "di- abatic'" curves ( - - - ), and ab initio calculations from ref. [30] (+ + +).
The projection is then
p, = o s - ' u . (3)
The complement of the projection, ( 1 - P i ) , is shown for various Li 2 and Li l le states in fig. 4 as a function of the internuclear distance R. The horizontal dashed line corresponds to p = 0.85. It is clear that the D I M basis is acceptable for the Li 2 states which are most important for this work: 11~+ 91y+ IYI u, 1Fig a n d 1 ]E~. The Li l le 2E+ and ~ FI states are also well represented. For R ~< 5 bohr, the D I M basis for Li l le no longer describes t h e 22y . + state; this is reflected by the kink in the 2 2 ~ + c u r v e in fig. 3.
The projection analysis indicates the suitability of the D I M model for describing those PES which correlate with Li + Li* + He. Surfaces leading to L i * + Li* + He are, however, not reliable. The latter are not needed in this work.
01 ~~ '~- . . . . . . . . . . . . . . . . . . . . .
0.0 : 2_2_E L . . . . . . . . . . . . . . . . . .
0.1 ~ i
O.C 5 10 15 R [au ]
Fig. 4. Projection analysis of the diatomic states for Li2 (the singlets) and Lille (doublets). p is the projection of the ex- tended basis VB wavefunction onto the space spanned by the DIM basis. The horizontal dashed line indicates the maximum acceptable value of 1 - p.
projection technique [29]. The matrix elements ( twenty-two for ]A', twelve for ]A") are fitted to cubic spline functions and collected in a disc-file, the b-file. This is then either further processed to generate a new b-file or it is taken as input for the D I M program in conjunct ion with the d-file, which contains formulae relating the polyatomic B-ma- trix elements directly to the fragment b-matrix elements. This enables very rapid computa t ion of B [24], there being no matrix operations.
For model (a), no semi-empirical alterations of the fragment matrices were necessary, since the VB atomic energies were so accurate.
3.4. Model (a) 3.5. Model (b)
The diatomic fragment matrices, b, are ob- tained from the extended VB calculations with the
The b-matrices of model (a) for the Li 2 ( ] H g , l H u ' 1 + 1 + E u , 1 Eg and 2 l e g ) states are modified so
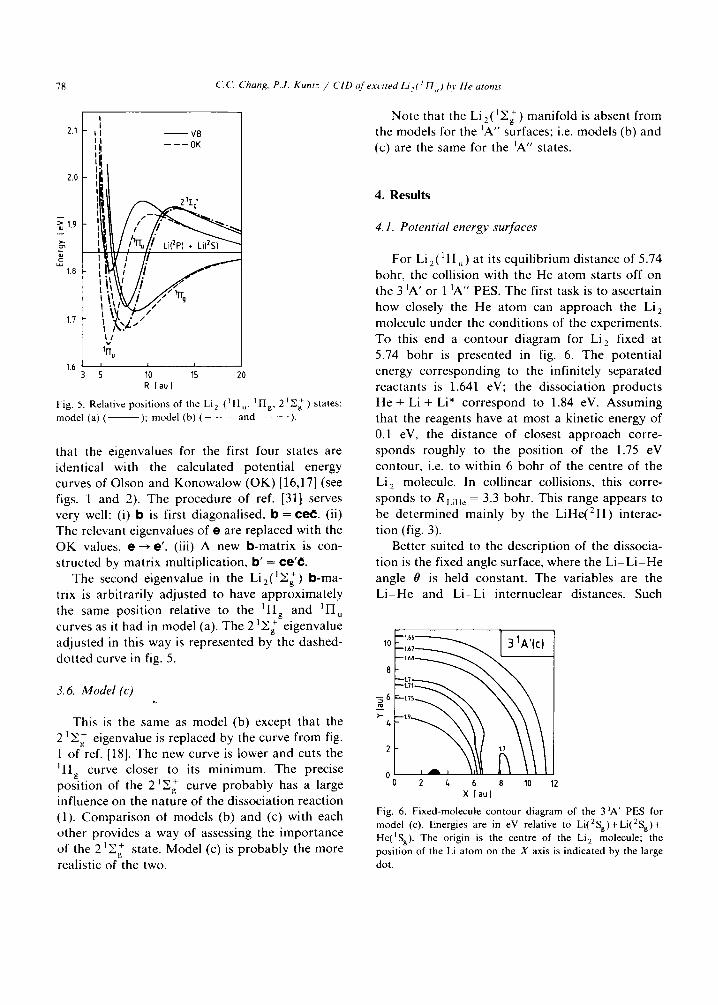
78 C.C. Chang, P.J. Kuntz / CID of excited Li, ( /II, ,) l~v He atoms
2.1
2.0
~1.9
taa 1.8
1.7
I il
ii, /I
i)d\' l# , t ',.,XL,.'
v
- - V B
OK
1 .6 3 5 10 15 20
R laul
Fig. 5. Relative posi t ions of the Li 2 ( I H , , l]qg, 2 1 Z ~ ) states: model (a) ( ); model (b) ( - - - and . . . . ).
that the eigenvalues for the first four states are identical with the calculated potent ial energy curves of Olson and Konowalow (OK) [16,17] (see figs. 1 and 2). The procedure of ref. [31] serves very well: (i) b is first diagonalised, b = e e l . (ii) The relevant eigenvalues of e are replaced with the O K values, e--+ e ' . (iii) A new b-mat r ix is con- structed by matr ix multiplication, b ' = ¢e'¢;.
The second eigenvalue in the Li2( ~ + Eg ) b -ma- trix is arbitrari ly adjusted to have approx imate ly the same posit ion relative to the l I Ig and ~II u curves as it had in model (a). The 1 + 2 Y.g eigenvalue adjusted in this way is represented by the dashed- dot ted curve in fig. 5.
3.6. Model (c)
This is the same as model (b) except that the I + 2 Eg eigenvalue is replaced by the curve f rom fig.
1 of ref. [18]. The new curve is lower and cuts the ~Hg curve closer to its min imum. The precise
I + posit ion of the 2 Zg curve p robab ly has a large influence on the nature of the dissociation reaction (1). Compar i son ol models (b) and (c) with each other provides a way of assessing the impor tance of the 1 + 2 £g state. Model (c) is p robab ly the more realistic of the two.
+ Note that t h e L i 2 ( i Y ~ g ) manifold is absent f rom
the models for the 1A" surfaces; i.e. models (b) and (c) are the same for the 1A" states.
4. Results
4.1. Potential energy surfaces
For Li2(1Hu) at its equil ibrium distance of 5.74 bohr, the collision with the He a tom starts off on the 3 1A' or 1 1A" PES. The first task is to ascertain how closely the He a tom can approach the Li 2 molecule under the condit ions of the experiments . To this end a contour d iagram for Li 2 fixed at 5.74 bohr is presented in fig. 6. The potent ial energy corresponding to the infinitely separated reactants is 1.641 eV; the dissociation products He + Li + Li* cor respond to 1.84 eV. Assuming that the reagents have at most a kinetic energy of 0.1 eV, the dis tance of closest approach corre- sponds roughly to the position of the 1.75 eV contour, i.e. to within 6 bohr of the centre of the Li 2 molecule. In collinear collisions, this corre- sponds to RL~H~ = 3.3 bohr. This range appears to be determined mainly by the L i H e ( 2 H ) interac- tion (fig. 3).
Better suited to the description of the dissocia- tion is the fixed angle surface, where the L i - L i - H e angle 0 is held constant . The variables are the L i - H e and L i -L i internuclear distances. Such
10 ii 3Ac
0 0 2 l, 6 8 10 12
x [aul
Fig. 6. Fixed-molecule con tour d iagram of the 31A' PES for model (c). Energies are in eV relative to L i (2Sg)+Li (2Sg)+ He(ISg). The origin is the centre of the Li 2 molecule; the posi t ion of the Li a t om on the X axis is indicated by the large dot.

C.C. Chang. P.J. Kuntz / CID of excited Lie( i I I . ) t~v He atoms 79
20 [ ,.9 ,., [ 3 1A'(c)
15
1.75
10 1.7Z
1.7
1.65 L62
,
5 10 15 20 RkiHe [ a u ]
Fig. 7. Contour diagram of the 31A' PES for model (c). The Li-Li-He angle is 135 °. Units as in fig. 6.
20
15
m
lO
2.1 195 5 1A'(c)
1.89 19 1.91 192
1.92
t ~ / tss , , 5
10 15 20
•LiHe [ au ]
Fig. 9. Contour diagram of the 5 tA' PES for model (c). • LiLiHe = 135 °. Units as in fig. 6.
surfaces are shown in figs. 7 -9 for the third, fourth and fifth %' states (model c) and in figs. 10 and 11 for the first and second %" states (model b) at 0 = 135 ° . The IA" and 3 %' surfaces show similar behaviour: There are two minima in the entrance channel (i.e. in the Li-Li coordinate) which are separated by a ridge. The upper mini- mum on the ~A" PES corresponds to Li2(~H~) but on the ~A' PES it occurs at the minimum of the Li2( 1 + Y.g ) curve. A t L i l l e d i s t a n c e s sma l l er than
5 bohr, the ridge disappears. Surfaces 2 ~A" and
20
10
, I
L~ IA'(c)
1,89
1.9
1.9
1.85
1.8 1.75 1.7
i
5 10 15 20 RLiHe [ aU ]
Fig. 8. Contour diagram of the 41A ' PES for model (c). • LiLiHe =135 ° Units as in fig. 6.
5 1A' are also similar in that they possess a broad ridge at REin ~ ----- 10-13 bohr, the barrier to dissoci- ation. Surface 4 %' has two ridges and shares some of the properties of both the 3 %' and 5 %' PES.
A trajectory leading from the reactants to the dissociation products would enter from the bottom right of figs. 7-11, tracing out a zig-zag path because of the Li-Li vibrational motion. At large Lille distances, the trajectory would "hop" from 3 A ' to 4 A ' to 5 A ' (or f rom 1 A " to 2 A " ) w i t h
every outward swing of the Li~ vibration; i.e. a diabatic p a t h w o u l d be f o l l o w e d . T h i s is m o r e easily seen from vertical cuts through the fixed-
20
15
g J
1 1A"(b)
1.75
10 15 Rl_iHe [ at/]
Fig. 10. Contour diagram of the 1 IA" PES for models (b) and (c). A LiLiHe =135 °. Uni ts as in fig. 6.
1,66
t
2O
1.72
1.69
1.69
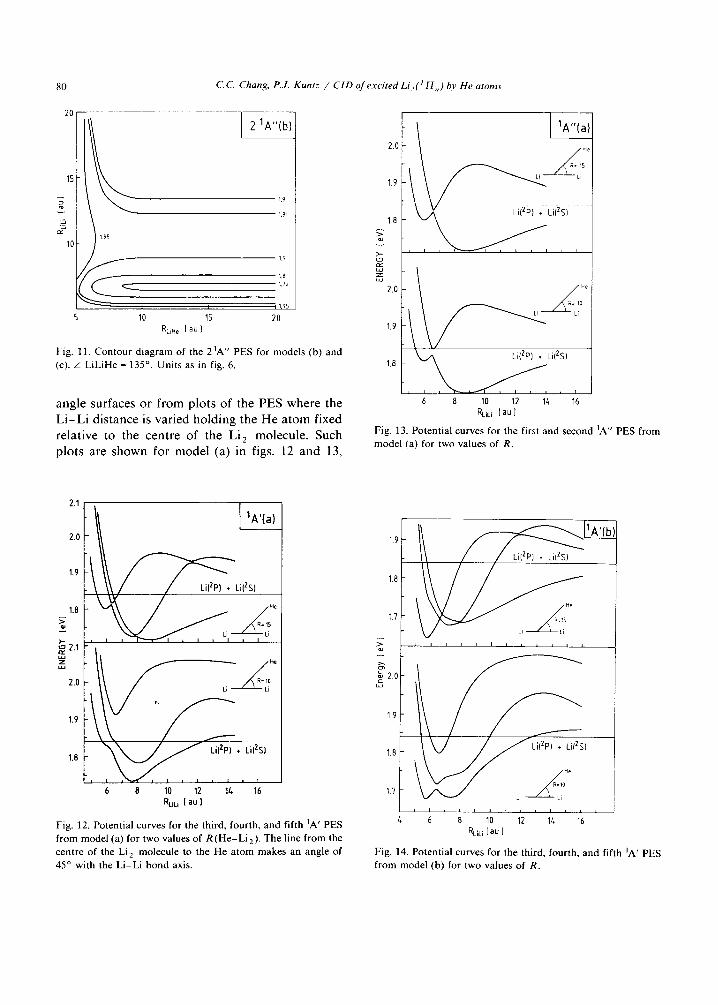
80 C.C. Chang, P.J. Kuntz / CID of excited Li , ( / H,, ) by He atoms
t 9
L8 1,72
5 10 15 20 RLiHe [ au ]
Fig. 11. Contour diagram of the 21A" PES for models (b) and (c). • LiLiHe = 135 °. Units as in fig. 6.
angle surfaces or from plots of the PES where the Li -Li distance is varied holding the He atom fixed relative to the centre of the Li 2 molecule. Such plots are shown for model (a) in figs. 12 and 13,
1A"(a ) 2.0
1.9
_~ 1.8 ~ ~ Li(zs)
L I ~ " ' - ' r " " ~ I I [ I I i I
C l !
2.0 He
6 8 10 12 14 16 RLiLi [ au ]
Fig. 13. Potential curves for the first and second 1A" PES from model (a) for two values of R.
2.1 Ill 1A'(a)
1.9
Li(2S)
~ ,
w
2.0 ki
1.9
I ~ , ' r i I i I I I i t
8 10 12 14 16 R L i L i [ a u ]
Fig. 12. Potential curves for the third, fourth, and fifth 1A' PES from model (a) for two values of R(He-Li2). The line from the centre of the Li 2 molecule to the He atom makes an angle of 45 ° with the Li-Li bond axis.
19
1.8
H e
1.7 ~=~s
I i I I ] J i L I i I oJ
~ 2.0
1.9 ~
F 1.8 Li(ZP) + Li(ZS)
I i i I , i i l i I i
/* 6 8 10 12 14 16 RLILI [ au ]
Fig. 14. Potential curves for the third, fourth, and fifth 1A' PES from model (b) for two values of R.
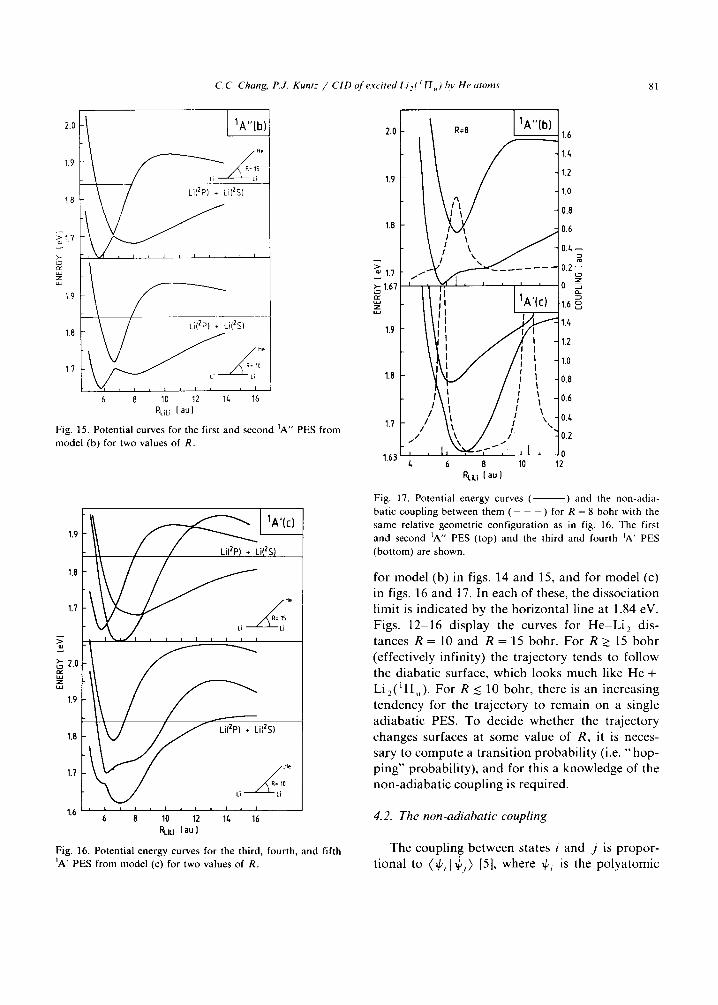
C.C. Chang, P.J. Kuntz / CID of excited Li e(Y H,) I~v He atoms 81
2.0
1.9
1.8
~1.7
u~
1A, , (b )
I i I
1.8 Lil2P) *L i (2 S)
1.7 \ / / - ~ _ ~ L [~ v l , I i I i I , I i I 6 8 10 12 14 16
RLiLi [ au]
Fig. 15. Potential curves for the first and second 1A" PES from model (b) for two values of R.
1.9
He
1.7
Li ~ Li
~ L0
I.u
1.9
1.8 Li(Tp) * LilZS)
1.7 0
Li
1.6 ~ 6 8 10 12 14 16
RLiki [ au ]
Fig. 16. Potential energy curves for the third, fourth, and fifth 1A' PES from model (c) for two values of R.
2.0
1.9
1.8
I . I
= 1.7 >- 1.67 t . u Z t ~
1.9
1.8
1.7
1.63 t, 6 8 RL~ i [ au ]
Fig. 17. Potential energy curves (
1.6
1.4
1.2
1.0
0.8
0.6
0. t , - -
0 .2 - -
° 2 1.6 S
1.4
1.2
1.0
0.8
0.6
0.4
0.2
0 10 12
) and the non-adia- batic coupling between them ( - - - ) for R = 8 bohr with the same relative geometric configuration as in fig. 16. The first and second 1A" PES (top) and the third and fourth 1A' PES (bottom) are shown.
for m o d e l (b) in figs. 14 and 15, and for m o d e l (c)
in figs. 16 and 17. In each of these, the d i s soc ia t ion
l imi t is ind ica ted by the ho r i zon ta l l ine at 1.84 eV.
Figs. 1 2 - 1 6 d i sp lay the curves for H e - L i 2 dis-
t ances R = 10 and R = 15 bohr . F o r R > 15 b o h r
(e f fec t ive ly inf ini ty) the t r a j ec to ry tends to fo l low
the d iaba t i c surface, wh ich looks m u c h like H e +
L i2 (~Hu) . F o r R < 10 bohr , there is an inc reas ing
t e n d e n c y for the t r a j ec to ry to r e m a i n on a s ingle
ad i aba t i c PES. To dec ide w h e t h e r the t r a j ec to ry
changes surfaces at s o m e va lue of R, it is neces-
sary to c o m p u t e a t r ans i t i on p r o b a b i l i t y (i.e. " h o p -
p i n g " p robab i l i ty ) , a n d for this a k n o w l e d g e of the
n o n - a d i a b a t i c c o u p l i n g is requi red .
4.2. The non-adiabatic coupling
T h e coup l ing b e t w e e n states i and j is p r o p o r -
t iona l to {~b ]~)/) [5], where +, is the p o l y a t o m i c
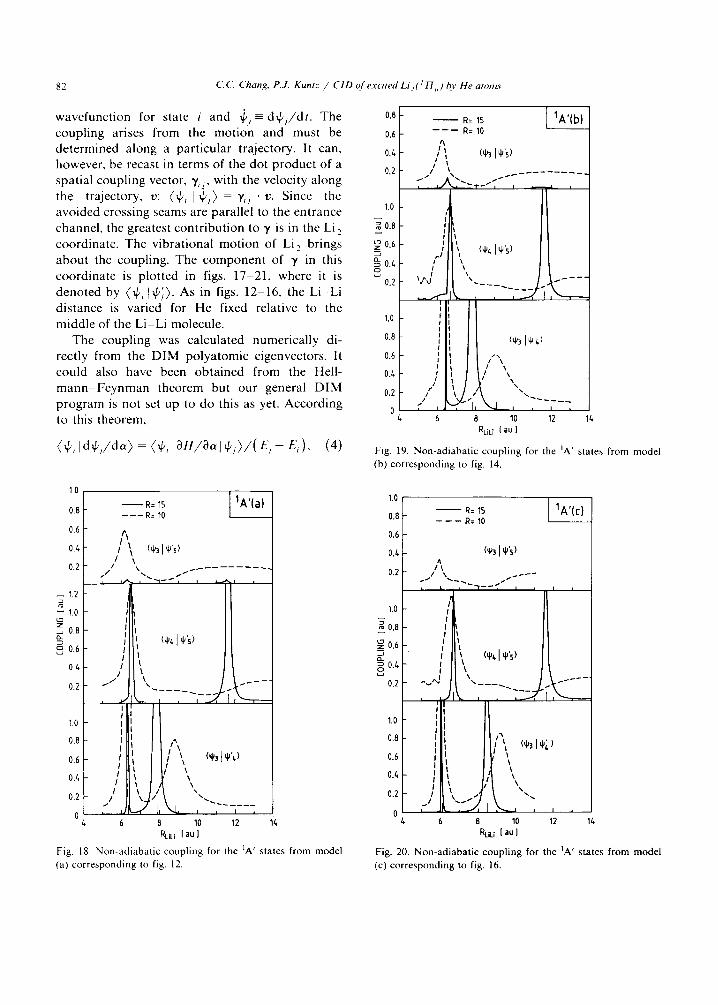
82 C. C. Chang. P.J. Kuntz / CID of excited Li_,( ~ H ,) t~v He atoms
wavefunction for state i and (pj=-d+l/dt. The coupling arises from the motion and must be determined along a particular trajectory. It can, however, be recast in terms of the dot product of a spatial coupling vector, y,z, with the velocity along the t r a j e c t o r y , v : ( ' L 14' , ) = 7 , , • v. S i n c e the avoided crossing seams are parallel to the entrance channel, the greatest contribution to 7 is in the Li 2 coordinate. The vibrational motion of Li 2 brings about the coupling. The component of 7 in this coordinate is plotted in figs. 17-21, where it is denoted by (~b , [~) . As in figs. 12-16, the Li-Li distance is varied for He fixed relative to the middle of the Li-Li molecule.
The coupling was calculated numerically di- rectly from the DIM polyatomic eigenvectors. It could also have been obtained from the Hell- m a n n - F e y n m a n theorem but our general DIM program is not set up to do this as yet. According to this theorem,
(~, I d ~ J d a ) = (~, I OH/3a ] ~ , ) / ( E, - E, ), (4)
o.8 R: is 1A'(b) 0.6 R= 10 ' [
rt
o.~ / ', ~*~ 1.'5) ' '
0.2 / ~ . . . . . . . . .
,0 - 1 ~ o . ~ ]
~o.6 I ', 1.'5> ~ o.z, F ' \
02
l 1.0 - I
0.8 - I (q,3 I*'~> /1%\
I OJ,-
r I i \ 0.2 " ~ / "h . . . . .
0 , I , , i t. 6 8 10 12 1&
RLiLT [ au ]
Fig. 19. Non-adiabatic coupling for the IA' states from model (b) corresponding to fig. 14.
0.2 , / I ~\\ ,( ,31,S~1 ~ . . . . . . . . . 1
/11\ / /
1.0
0.8 / ~
I / ~ (% I * ' b 0.6 I I .I \~\
0"t' / i / ~ xx 0.2 . , / . , ~ . .
6 10 12 14 RLILi [ aU ]
Fig. 18. Non-adiabatic coupling for the 1A' states from model (a) corresponding to fig. 12.
1.0 R= 15 [ 1A ' ( c )
0.8 - - - - R= 10
0.6
0./.,
0.2
1.0
~= o.e z__ 0.6
O- N 0.4
0.2
1.0
0.8
0.6
0.4
0.2
0
('.~ [ ¢5)
I
f
I
!
, /
I !
/
6
k \ ~*,1"5) t
\ ~__~l ~.1~-- ".----
/ ~\ (*3 I*',.)
/ \ , \ I " ,
B 10 12 1
RL~ i ( au )
Fig. 20. Non-adiabatic coupling for the 1A' states from model (c) corresponding to fig. 16.
C.C. Chang, P.J. Kuntz / CID of excited Li_,(1II,,) 12V He atoms 83
1.2! 1.0
0.8
0.6
"~ 03,
.~0.2 o-
~_ 1.0
~ 0 . 8
~ 0.6
/
- - R = 15 [ 1A"(b) - - - - - R = 10
I I l I
~lA"la )
0.4 I I
I , 0.2 /
0 .~ i ~" -i.- ,1- t 6 B 10 12 lt~
RLiLi [ au ]
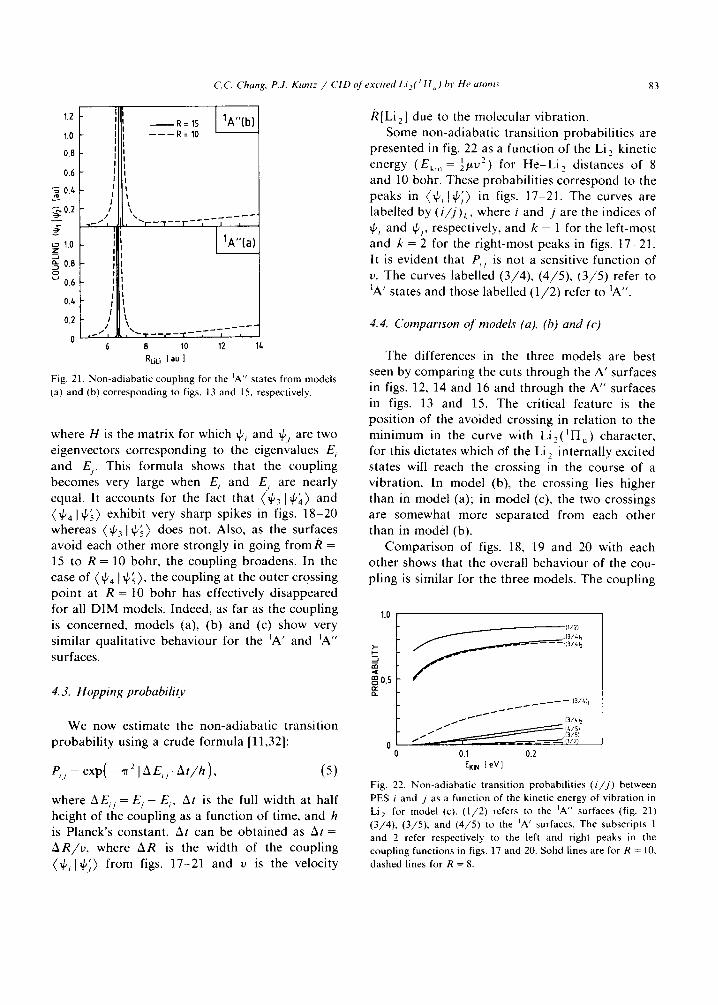
Fig. 21. Non-ad iaba t i c coupl ing for the ]A" states from models
(a) and (b) co r respond ing to figs. 13 and 15, respectively.
where H is the matrix for which ~Pi and 4'j are two eigenvectors corresponding to the eigenvalues E, and E~. This formula shows that the coupling becomes very large when Ei and E/ are nearly equal. It accounts for the fact that (~p3 I~p~) and ~l.p4 I~p.~) exhibit very sharp spikes in figs. 18-20 whereas (~p31tp~) does not. Also, as the surfaces avoid each other more strongly in going f r o m R = 15 to R = 10 bohr, the coupling broadens. In the case of ~P4 I IPS~ ' the coupling at the outer crossing point at R = 10 bohr has effectively d isappeared for all D I M models. Indeed, as far as the coupling is concerned, models (a), (b) and (c) show very similar qualitative behaviour for the 1A' and IA" surfaces.
4.3. Hopping probability
We now est imate the non-adiabat ic transit ion probabi l i ty using a crude formula [11,32]:
P, , = ex p( - I A E , , I At/h), (5)
where AE,~ = E / - E,, At is the full width at half height of the coupling as a function of time, and h is Planck 's constant. At can be obta ined as At = AR/v , where AR is the width of the coupling (~P, lq'~) f rom figs. 17-21 and v is the velocity
/~[Li2] due to the molecular vibration. Some non-adiabat ic transit ion probabil i t ies are
presented in fig. 22 as a function of the Li 2 kinetic energy (Ek i n = ½/.tU 2) for H e - L i 2 distances of 8 and 10 bohr. These probabil i t ies correspond to the peaks in (~p, I~p~) in figs. 17-21. The curves are labelled by (i/j)~, where i and j are the indices of ~p, and ~p~, respectively, and k = 1 for the left-most and k = 2 for the r ight-most peaks in figs. 17-21. It is evident that P,/ is not a sensitive function of v. The curves labelled (3 /4) , (4 /5) , (3 /5 ) refer to 1A' states and those labelled (1 /2 ) refer to 1A".
4.4. Comparison of models (a), (b) and (c)
The differences in the three models are best seen by compar ing the cuts through the A' surfaces in figs. 12, 14 and 16 and through the A" surfaces in figs. 13 and 15. The critical feature is the posit ion of the avoided crossing in relation to the min imum in the curve with Li2(~Hu) character, for this dictates which of the Li 2 internally excited states will reach the crossing in the course of a vibration. In model (b), the crossing lies higher than in model (a); in model (c), the two crossings are somewhat more separated from each other than in model (b).
Compar i son of figs. 18, 19 and 20 with each other shows that the overall behaviour of the cou- pling is similar for the three models. The coupling
1.0
>-
~ 0.5
- - 1 1 / 2 1
~ _ - ~ 13/t,,q
~ 13/4Y z
/ / [3/51 0 I (I/2)
0.1 0.2 EKI N [ eV ]
F ig. 22. N o n - a d i a b a t i c t r ans i t i on p robab i l i t i e s ( i / j ) be tween
PES i and j as a funct ion of the kinet ic energy of v ibra t ion in
Li 2 for model (c). ( 1 / 2 ) refers to the IA" surfaces (fig. 21) (3 /4 ) , (3 /5 ) , and ( 4 / 5 ) to the 1A' surfaces. The subscr ipts 1 and 2 refer respect ively to the left and right peaks in the coup l ing funct ions in figs. 17 and 20. Solid lines are for R = 10,
dashed lines for R = 8.
84 C.C. Chang, P.J. Kuntz / CID of excited Li2(l II,,) by He atoms
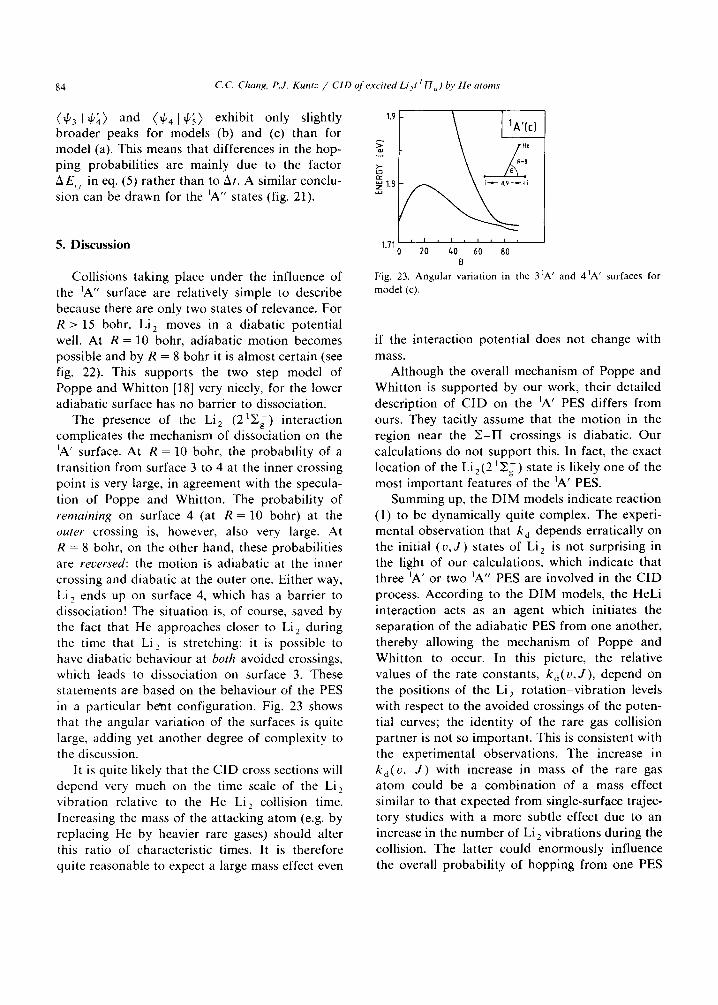
(4'3 14,~) and (4,414,.~) exhibit only slightly broader peaks for models (b) and (c) than for model (a). This means that differences in the hop- ping probabilities are mainly due to the factor AE, i in eq. (5) rather than to At. A similar conclu- sion can be drawn for the ~A" states (fig. 21).
5. Discussion
Collisions taking place under the influence of the ~A" surface are relatively simple to describe because there are only two states of relevance. For R > 15 bohr, Li 2 moves in ad iaba t i c potential well. At R = 10 bohr, adiabatic motion becomes possible and by R = 8 bohr it is almost certain (see fig. 22). This supports the two step model of Poppe and Whitton [18] very nicely, for the lower adiabatic surface has no barrier to dissociation.
The presence of the Li 2 (21Y.g) interaction complicates the mechanism of dissociation on the ~A' surface. At R = 10 bohr, the probability of a transition from surface 3 to 4 at the inner crossing point is very large, in agreement with the specula- tion of Poppe and Whitton. The probability of remaining on surface 4 (at R = 10 bohr) at the outer crossing is, however, also very large. At R = 8 bohr, on the other hand, these probabilities are reversed: the motion is adiabatic at the inner crossing and diabatic at the outer one. Either way, Li 2 ends up on surface 4, which has a barrier to dissociation! The situation is, of course, saved by the fact that He approaches closer to Li~_ during the time that Li 2 is stretching: it is possible to have diabatic behaviour at both avoided crossings, which leads to dissociation on surface 3. These statements are based on the behaviour of the PES in a particular bent configuration. Fig. 23 shows that the angular variation of the surfaces is quite large, adding yet another degree of complexity to the discussion.
It is quite likely that the CID cross sections will depend very much on the time scale of the Li 2 vibration relative to the He-Li 2 collision time. Increasing the mass of the attacking atom (e.g. by replacing He by heavier rare gases) should alter this ratio of characteristic times. It is therefore quite reasonable to expect a large mass effect even
_ 1.9 \ iA,(c)
~1.8 ' ~ - - ~ " laJ
i i i i 1.71
20 ~.0 60 80 0
Fig. 23. Angu la r var ia t ion in the 31A ' and 41A ' surfaces for
model (c).
if the interaction potential does not change with mass.
Although the overall mechanism of Poppe and Whitton is supported by our work, their detailed description of CID on the 1A' PES differs from ours. They tacitly assume that the motion in the region near the E - H crossings is diabatic. Our calculations do not support this. In fact, the exact location of the Li2(2 1 + Eg ) state is likely one of the most important features of the ~A' PES.
Summing up, the DIM models indicate reaction (1) to be dynamically quite complex. The experi- mental observation that k a depends erratically on the initial ( v , J ) states of Li 2 is not surprising in the light of our calculations, which indicate that three ~A' or two ~A" PES are involved in the CID process. According to the DIM models, the HeLi interaction acts as an agent which initiates the separation of the adiabatic PES from one another, thereby allowing the mechanism of Poppe and Whitton to occur. In this picture, the relative values of the rate constants, k j ( v , J ) , depend on the positions of the Li 2 rotation-vibration levels with respect to the avoided crossings of the poten- tial curves; the identity of the rare gas collision partner is not so important. This is consistent with the experimental observations. The increase in ka(v, J ) with increase in mass of the rare gas atom could be a combination of a mass effect similar to that expected from single-surface trajec- tory studies with a more subtle effect due to an increase in the number of Li 2 vibrations during the collision. The latter could enormously influence the overall probability of hopping from one PES

c.c. Chang, P.J. Kuntz / CID of excited Li:( I H,) by He atoms 85
to ano the r , i.e. a l ong co l l i s ion t ime a l lows the
v i b r a t i n g mo lecu l e several chances to f ind a
f a v o u r a b t e pa th to d i ssoc ia t ion . Of course , in the
e x p e r i m e n t s [14,15], it is i m p o s s i b l e to d i s t ingu ish
the mass ef fec t f r o m changes in the i n t e r a c t i o n
po t en t i a l ; n o t h i n g shor t of an i so tope e x p e r i m e n t
or a b e a m e x p e r i m e n t cou ld test a p u r e mass effect .
T h e ma jo r conc lu s ion f r o m this w o r k is that the
C I D process is far too c o m p l e x to a l low specu la -
t ion abou t the de t a i l ed m e c h a n i s m . A d y n a m i c a l
ca l cu l a t i on t ak ing a c c o u n t o f the va r ious p o t e n t i a l
su r faces and thei r i n t e r ac t i ons is ab so lu t e ly essen- tial.
Acknowledgement
W e are g ra te fu l to Dr. W . N . W h i t t o n and Dr.
D. P o p p e for he lp fu l d i scuss ions a n d to Mrs. G.
Snoei , Mrs. H. G a d e w o l t z , Mrs . K. G f r 0 r e r and
Miss K. Char i s ius for thei r he lp in p r e p a r i n g the m a n u s c r i p t .
References
[1] M. Baer, ed., The theory of chemical reaction dynamics (CRC Press, Ceveland), to be published.
[2] R.B. Bernstein, Chemical dynamics via molecular beam and laser techniques (Clarendon Press, Oxford, 1982).
[3] W.H. Miller, ed., Dynamics of molecular collisions (Plenum Press, New York, 1976).
[4[ R.B. Bernstein, ed., Atom-molecule collision theory (Plenum Press, New York, 1979).
[5] J.C. Tully, in: Dynamics of molecular collisions, ed. W.H. Miller (Plenum Press, New York, 1976) ch. 5.
[6] M.S. Child, in: Atom-molecular collision theory, ed. R.B. Bernstein (Plenum Press, New York, 1979) ch. 13.
[7] I.V. Hertel, Advan. Chem. Phys. 45 (1981) 341. [8] D.G. Truhlar, J.W. Duff, N.C. Blais, J.C. Tully and B.C.
Garrett, J. Chem. Phys. 77 (1982) 764. [9] K. Lacmann, Advan. Chem. Phys. 42 (1980) 513.
[10] P.J. Kuntz and W.N. Whitton, Chem. Phys. 16 (1976) 301. [11] P.J. Kuntz, J. Kendrick and W,N. Whitton, Chem. Phys.
38 (1979) 147. [12] J. Kendrick and P.J. Kuntz, J. Chem. Phys. 70 (1979) 736. [13] W.B. Maier II, J. Chem. Phys. 62 (1975) 4615. [14] G. Ennen and Ch. Ottinger, Chem. Phys. 40 (1979) 127. [15] G. Ennen and Ch. Ottinger, J. Chem. Phys. 76 (1982)
5812. [16] D.D. Konowalow and M.L. Olson, J. Chem. Phys. 71
(1979) 450. [17] M.L.Olson and D.D. Konowalow, Chem. Phys. 21 (1977)
393; 22 (1977) 29; Chem. Phys. Letters 39 (1976) 281. [18] D. Poppe and W.N. Whitton, Chem. Phys. Letters 107
(1984) 304. [19] I. Schmidt-Mink, W. Mi~ller and W. Meyer, Chem. Phys.
92 (1985) 263. [20] F.O. Ellison, J.Am. Chem. Soc. 85 (1963) 3540. [21] J.C. Tully, in: Semi-empirical methods of electronic struc-
ture calculations, ed. G.A. Segal (Plenum Press, New York, 1977).
[22] P.J. Kuntz, in: Atom-molecule collision theory, ed. R.B. Berustein (Plenum Press, New York, 1979) ch. 2.
[23] J.L. Schreiber and P.J. Kuntz, J. Chem. Phys. 76 (1982) 1872.
[24] R. Polfik, I. Paidarovfi and P.J. Kuntz, J. Chem. Phys. 82 (1985) 2352.
[25] G.G. Balint-Kurti and R.N. Yardly, QCPE, Program 335 (MULT1BOND A) (1974).
[26] S. Huzinaga, J. Chem. Phys. 42 (1965) 1293. [27] C.L. Pekeris, Phys. Rev. 115 (1959) 1216. [28] C.E. Moore, Atomic energy levels, NBS Circular No. 467
(Natl. Bur. Std., Washington, 1949). [29] P.J. Kuntz and J.L. Schreiber, J. Chem. Phys. 76 (1982)
4120. [30] M. Krauss, P. Maldonado and A.C. Wahl, J. Chem. Phys.
54 (1971) 4944. [31] P.J. Kuntz and C.C. Chang, Chem. Phys. 75 (1983) 79. [32] W.H. Miller, private communication.





























