Embed Size (px)
Citation preview

INTERNATIONAL JOURNAL OF ONCOLOGY 40: 711-720, 2012
Abstract. Histone deacetylase inhibitors (HDACi) are prom-ising epigenetic cancer chemotherapeutics rapidly approaching clinical use. In this study, we tested using in vitro and in vivo models the differential biological effects of a novel HDAC inhibitor [belinostat (PXD101)], in a wide panel of androgen-sensitive and androgen-independent tumor cells. Belinostat significantly increased acetylation of histones H3 and H4. Belinostat potently inhibited the growth of prostate cancer cell lines (IC50 range from 0.5 to 2.5 µM) with cytotoxic activity preferentially against tumor cells. This agent induced G2/M arrest and increased significantly the percentage of apoptosis mainly in androgen-sensitive tumor cells confirming its growth-inhibitory effects. The cell death mechanisms were studied in three different prostate cancer cell lines with different androgen dependence and expression of androgen receptor; LAPC-4 and 22rv1 (androgen-dependent and expressing androgen receptor) and PC3 (androgen-independent not expressing androgen receptor). Belinostat induced the expression of p21 and p27, acetylation of p53 and G2/M arrest associated with Bcl2 and Bcl-Xl downmodulation and significant reduction of survivin, IAPs and Akt/pAkt and increased caspase-8 and -9 expres-sion/activity. Belinostat effectiveness was dependent on the androgen receptor (AR), since the stable transfection of AR greatly increased the efficacy of this HDAC inhibitor. These observations were correlated using in vivo models. We demon-strated that belinostat preferentially resulted in antitumor effect in androgen-dependent tumor cells expressing AR. Our findings provide evidence that belinostat may be a promising anticancer
drug for prostate cancer expressing AR, supporting its clinical role in prostate cancer.
Introduction
Prostate cancer is a major health problem and is the second highest cause of deaths from cancer each year in males in the European countries. The hormone therapy for prostate cancer targets the ligand-binding domain of the androgen receptor using drugs that reduce the serum level of testosterone. This is often administered in combination with competitive androgen receptor (AR) antagonists. The outcome of such a treatment follows a predictable course comprised of an initial response, a period of quiescence during which the tumor does not prolif-erate, followed by progression of the disease. This indicates that ligand directed anti-androgen therapy alone is not sufficient for achieving long-term remission, thus identification of other targets that can be used in conjunction with the current hormonal treatment is warranted. Histone deacetylases (HDACs) are a group of co-repressors of transcriptional activators, including AR (1). These set of proteins regulate gene expression by altering nucleosome conformation at the chromatin level and the stability of several large complexes of transcription factors. Prostate cancer begins as a hormone-sensitive tumor and its growth is stimulated by the activation of AR in conjunction with multiple nuclear co-regulatory proteins such as histone deacetylases. The histone deacetylases modulate transcrip-tional activity of hormonal receptors including AR by altering the stability of the transcriptional pre-initiation complex and/or modifying the chromatin structure. HDACs associate with other co-repressors and form large protein complexes including Sin3 (Sin3A/Sin3B/HDAC1/HDAC2), HDAC3/GPS/TBL/NCoR/SMRT, and the HDAC4/HDAC5/SMRT/NCoR complex (2). Wang and co-workers (3) demonstrated that HDAC1 is overexpressed in 35% of prostate cancer, in metastatic tumors and in androgen-independent cell lines. These findings were consistent with the previously reported increase in HDAC1 activity, mRNA expression, and protein expression in prostate
Differential effects of PXD101 (belinostat) on androgen-dependent and androgen-independent prostate cancer modelsGIOVANNI LUCA GRAVINA1,2, FRANCESCO MARAMPON1,2, ILARIA GIUSTI3, ELEONORA CAROSA4,
STEFANIA DI SANTE4, ENRICO RICEVUTO5, VINCENZA DOLO3, VINCENZO TOMBOLINI6, EMMANUELE A JANNINI4 and CLAUDIO FESTUCCIA2
1Department of Experimental Medicine, Division of Radiotherapy, 2Department of Experimental Medicine, Laboratory of Radiobiology, Departments of 3Health Sciences and 4Experimental Medicine, Course of Endocrinology and
Medical Sexology and 5Experimental Medicine, Division of Clinical Oncology, University of L'Aquila, I-67100 L'Aquila; 6Department of Radiological Sciences, University of Rome
‘La Sapienza’ Spencer-Lorillard Foundation, I-00100 Rome, Italy
Received July 7, 2011; Accepted August 29, 2011
DOI: 10.3892/ijo.2011.1270
Correspondence to: Dr Claudio Festuccia, Department of Experi-mental Medicine, Via Vetoio, Coppito-2, I-67100 L'Aquila, ItalyE-mail: [email protected]
Key words: prostate cancer, histone deacetylation, epigenetic, belinostat

GRAVINA et al: BELINOSTAT AND PROSTATE CANCER712
cancer cell lines (1) and cancers that are refractory to hormone treatment (5). HDAC2 had been found to be increased in human colon cancer and overexpressed in about 30% of patients with prostate cancer (3). HDAC4 was also overexpressed in 23% of prostate cancer specimens (1,3) whereas an altered intracellular localization of HDAC4 in prostate cancers was described in prostate cancer that had become refractory to hormone treat-ment (6), suggesting that HDAC4 may be involved in the late events of prostate cancer progression. HDAC5 expression was increased in 57% of prostate cancer specimens (3). In addition, it has been demonstrated that HDAC5 is able to associate with known AR co-repressors such as SMRT and N-CoR (7), it may also play a role in tumorigenesis of prostate cancer.
Several HDAC inhibitors have been developed for treatment of ematological and solid tumors. These agents represent a rela-tively new and interesting class of anticancer agents. Histones play an important role in maintaining chromatin structure, and posttranslational modifications to these molecules serve to regulate chromatin density. As a result of HDAC-inhibition, histones become hyper-acetylated and DNA is maintained in a relatively open conformation that is conducive to interaction with transcription factors. Increased levels of histone acetylation lead to relaxation of the chromatin structure, allowing access of transcription factors and increased transcription, while decreased levels of acetylation are associated with repressed transcription. Consistent with this scenario, HDAC inhibitors have been shown to alter the transcription of a number of genes. In addition, these compounds have been shown to mediate tumor cell differentia-tion, growth-inhibition and death, and a number of such HDAC inhibitors are in clinical trials. In contrast to the relatively detailed understanding that has been attained regarding the role that histones and their modifications play in maintaining chromatin structure, the mechanism(s) by which HDAC inhibitors mediate anticancer effects are poorly understood.
While histones clearly represent a predominant class of proteins which exhibit HDAC inhibitors-mediated hyper-acetylation, a number of non-histone proteins have also been shown to be hyper-acetylated and functionally influenced as a consequence of HDAC inhibitors. Thus, HDAC inhibitors mediate a variety of transcriptional and non-transcriptional alterations in cancer cells. The ability of HDAC inhibitors to affect the activity of a variety of disparate proteins and path-ways in tumor cells makes them attractive anticancer agents and also makes the understanding of their mechanism of action a daunting task.
We have been studying a novel hydroxamate, belinostat (previously called PXD101), which inhibits HDAC activity in cancer cell lysates with nM IC50 potency and induces the hyper-acetylation of proteins such as histones and tubulin in tumor cells (8). This compound, which is considered a pan-HDAC inhibitor on account of its inhibitory activity on a number of purified recombinant HDACs, was previously shown to inhibit tumor cell growth in vitro and in animal models both as mono-therapy (9,10) as well as in combination with other anticancer agents and (11,12) it is currently being evaluated as an anticancer agent in clinical trials (reviewed in ref. 13). To support the clinical development of belinostat in prostate cancer, we tested, in vitro and in vivo models, the differential biological effects of this agent in a wide panel of androgen-sensitive and androgen-independent tumor cells.
Materials and methods
Reagents. All the materials for tissue culture were purchased from Hyclone (Cramlington, NE, USA). Plastic-ware was obtained from Nunc (Roskilde, Denmark). Antibodies were purchased from Santa Cruz (Santa Cruz, CA, USA) unless otherwise indicated. PXD101 (Belinostat) was kindly provided from TopoTarget A/S (Copenhagen, Denmark).
Cell lines. One non-tumor prostate epithelial cell line (BPH-1), 14 PCa cell lines [CWR22 (14), CWR22R-2152 (14), CWR22R-2272 (14), CWR22R-2274 (14), CWR22R-2524 (14), 22rv1 (14), LAPC-4 (15), LnCaP, LnCaP-C81 (16), C4-2B (17), PC3, PTEN-transfected PC3 (18), AR-transfected PC3 (19), DU145, AR-transfected DU145, named DU-AR, (20), VCaP (21) and DuCaP (21) cells were used (a complete characterization of the above mentioned cells is reviewed in Pienta et al (22). CWR22, LAPC-4, LnCaP, VCaP and DuCaP cell lines are androgen-dependent whereas CWR22R derivatives, 22rv1, LnCaP-C-81, C4-2B, PC3 and DU145 cell lines are androgen-independent (22).
Growth αssays. Briefly, PCa cells (3x104 cells/well) were seeded in 24-well tissue culture plates (Costar, Corning, NY) and left to attach and grow for 24 h. Cells were incubated with belinostat at 0.05-5 µM for 2-72 h dependent on the read out. After this time, cells were maintained in the appropriate culture condi-tions. Morphological controls were performed every day with an inverted phase-contrast photomicroscope (Nikon Diaphot, Tokyo, Japan), before cell trypsinization and counting. Cells trypsinised and resuspended in 1.0 ml of saline were counted using the NucleoCounter™ NC-100 (automated cell counter systems, Chemotec, Cydevang, Denmark). All experiments were conducted in triplicate. NucleoCounter NC-100 allows to determine also the number of dead cells present in a cell sample. Therefore we considered viable and dead cells as separated enti-ties. IC50 values were calculated by the GraFit method (Erithacus Software Limited, Staines, UK) considering the slopes of inhibi-tion curves obtained for each group of tests. The effect on cell proliferation was measured by taking the mean cell number respect to controls in the time for the different treatment groups.
Apoptosis and cell cycle analysis. Cells (1x106) were fixed for 30 min in 70% ethanol, pelleted by centrifugation (720 g; 5 min). After removal of ethanol by centrifugation, cells were incu-bated and resuspended in 1 ml of DNA staining solution (PBS containing 200 mg/ml RNase A, 20 mg/ml propidium iodide plus 0.1% Triton X-100) and left at room temperature for 60 min. All cells were then measured on a FACScan flow cytometer (Becton-Dickinson, UK) and analysed using Cell Quest software (Becton-Dickinson). Resulting DNA distributions were analyzed for the proportion of cells in G1, S and G2-M phases of the cell cycle. Apoptotic cells were detected by both the quantifiable peak in sub-G1 phase, corresponding to the red fluorescence light emitted by sub-diploid nuclei of cells. For statistical comparisons apoptosis was quantified by using a quantitative ELISA tests for DNA fragmentation (Trevigen, Gaithersburg, MD).
Preparation of cell lysates and Western blot analysis. Following treatments, cells grown in 90-mm diameter Petri dishes were

INTERNATIONAL JOURNAL OF ONCOLOGY 40: 711-720, 2012 713
washed with cold PBS and immediately lysed with 1 ml lysis buffer containing a proteinase and phosphatase inhibitor cocktail. Lysates were electrophoresed in 7% SDS-PAGE, and separated proteins transferred to nitrocellulose and probed with the appropriate antibodies using the conditions recommended by the suppliers.
In vivo experiments. Male CD1 nude mice (Charles River, Milan, Italy) were kept in line with University guidelines (University of L'Aquila, Medical School and Science and Technology School Board Regulations, in compliance with Italian government regulation no. 116 of January 27, 1992 on the use of laboratory animals). Before any invasive manipulation, mice were anesthe-tized with a ketamine (25 mg/ml)/xylazine (5 mg/ml) mixture. All mice received s.c. flank injections of 1x106 in 250 µl of 12 mg/ml Matrigel (Becton-Dickinson, Franklin Lakes, NJ, USA) of 22Rv1 and PC3 cells. Treatments were started when a mean tumor volume of 100 mm3 was reached. Before tumor inoculation mice were randomly assigned to 4 experimental groups of 12 mice each. For each cell line one control group received an intraperi-toneal (i.p.) injection of 100 µl PBS for 18 consecutive days and one group received 20 and 40 mg/kg/dose bid for 18 consecutive days. The effects on tumor growth of different treatments were evaluated by: i) tumor volume measured during and at the end of experiment by biweekly measurement of tumor diameters with a Vernier caliper (length x width). Tumor volume was calculated according to the formula: TV (mm3) = π x d2 x D/2, where d and D are the shortest and longest diameters, respectively (23). ii) Tumor weight, measured at the end of experiment. Experiments were stopped 30 sec/day after the first day of treatment, mice were sacrificed by carbon dioxide inhalation and tumors were removed surgically. Tumor growth delay (TGD) was determined as % TGD = ((T_C)/C)_100, where T and C are the mean times in days required to reach the same fixed tumor volume in the treated group and control group, respectively (24,25). In vivo drug combination studies were evaluated by CalcuSyn [Biosoft, Cambridge, UK). For the calculation of CI, the values of cell kill for a fixed tumor volume were considered (determined by the log cell kill (LCK)]. Log cell kill was determined as LCK = (T-C)/(3.3-Td), where Td represents the mean control group doubling time required to reach a fixed tumor volume, expressed in days, whereas T and C are the same values as described above (24,25).
Half of the tumor was directly frozen in liquid nitrogen for protein analysis and the other half fixed in paraformalde-hyde overnight for immunohistochemical analyses. Masson's trichrome staining, CD31, Ki67, K18, VEGF, pAkt (Ser473) and Her2 expression was evaluated in 4 µm tissue sections cut from blocks selected for the presence of representative tumor tissue. Negative controls were incubated only with universal negative control antibodies under identical conditions, processed, and mounted. Expression of K18, VEGF, pAkt (Ser473) and Her2 was subjectively graded and quantified as 0, 1, 2, 3, 4, and 5 representing not detectable, weak, moderate, strong, and very strong staining, respectively. Tissue disorganization and vessel distribution were also analyzed using a variation of Masson trichrome staining (MBS-HS443, HD supplies, Aylesbury, UK).
Statistical analysis. Continuous variables were summarized as mean and standard deviation (SD) and were compared by the
unpaired Student's t-test. Dichotomous variables were summa-rized by absolute and/or relative frequencies and were compared by the exact Fisher's test or χ2 test when appropriate. For multiple comparisons the level of significance was corrected by multi-plying the P-value by the number of comparisons performed (n) according to Bonferroni correction. All tests were two-sided and were determined by Monte Carlo significance. P-values <0.05 were considered statistically significant. All statistical analyses were performed using the SPSS® statistical analysis software package, version 10.0.
Results
Belinostat results in acetylation of histonic and non-histonic proteins in vitro. It has been demonstrated that HDAC inhibi-tors increase histone H3 and H4 acetylation. In Fig. 1A we demonstrate that belinostat resulted in acetylation of H3 and H4 histones in LnCaP cell line in a dose-dependent manner. At the same time, we also demonstrate that belinostat was able to induce acetylation of the non-histonic protein p53 involved in cell proliferation and apoptosis (Fig. 1). As shown in Fig. 1B acetylation of histones occurred in other cell models (PC3, 22rv1, LAPC-4 and LnCaP cell lines) with kinetics and EC20 and EC50 (concentration of belinostat able to result in acetyla-tion of 20 and 50% of H3 and H4 histones) with characteristics typical of the different cellular models. Fig. 1C demonstrates that 0.1 µM Belinostat, responsible for a sub-total acetylation of histones, induced acetylation of H3 and H4 histones as well as of p53 within 8 h of treatment, raising maximal values after 16-24 h of culture. Histone acetylation was maintained at high levels until at least 72 h.
Belinostat results in differential antiproliferative and cytotoxic effects in androgen-sensitive and androgen-independent tumor cells in vitro. Dose-dependent experiments with belinostat ranging from 0.05 to 5.00 μM were used to calculate the IC20 and IC50 values for proliferation, viability and apoptosis in the different prostate cancer cell models. In Fig. 2A we show the growth inhibition measured after 24-, 48- and 72-h incubation at 0.05-1 µM belinostat in a panel of cancer cell lines with high hormone sensitivity (LAPC-4 and LnCaP) and low hormone sensitivity (22rv1 and PC3) and in a non-tumor cell line (BPH-1). In these cellular models, belinostat inhibited cell proliferation in a dose-dependent manner in cancer cells. Overall the antiprolif-erative affects of belinostat was greater in androgen-dependent than androgen-independent tumor cells with IC20 and IC50 values significantly lower for androgen-dependent models. In non-tumor cell line BPH-1 belinostat resulted in antiproliferative effect only at the higher drug concentrations (Fig. 2A). Using the NucleoCounter NC-100 we determined also the number of dead cells as marker of cytotoxic effect of Belinostat. This drug induced cell toxicity in a time-dependent manner (Fig. 2B). The increment of mortality was strongly dose-dependent and clearly documentable in cancer cells but not in BPH-1 cells. As in the case of the antiproliferative effects, belinostat were significantly more effective in AD than AI cell lines with EC20 and EC50 values significantly lower for androgen-dependent models (Fig. 2B).
The transfection of androgen receptor in androgen-independent tumor cells enhances the sensitivity to belinostat. To determine

GRAVINA et al: BELINOSTAT AND PROSTATE CANCER714
Figure 1. (A) Dose-dependent effects of belinostat measured after 24 h of treatment in LnCaP cell cultures. (B) Summarized effects of histone H3 and H4 acetylation on PC3, 22rv1, LnCaP, DU145, C4-2B and LAPC-4 cell lines. (C) Time-dependent effects of 0.1 µM belinostat administered to LnCaP cell culture. Each data are representative of three individual experiments. Each Western blot lane was loaded with 40 µg of proteins and normalized vs actin.
Figure 2. (A) Variation in cell viable number in the time and at different drug concentration in the PC3, 22rv1, LnCaP and LAPC-4 cell lines: the number of cells are normalized as percent of inhibition vs control to calculate IC20 and IC50 values, representation of growth inhibition curves in non-tumor BPH-1 (IC50 > 5 µM) cell line and neoplastic PC3 (IC50 = 1.35±0.32 µM), 22rv1 (IC50 = 1.23±0.45 µM), LnCaP (IC50 = 0.84±0.16) and LAPC-4 (IC50 = 0.68±0.15 µM) cell line. (B) Variation in cell death number in the time and at different drug concentration in PC3, 22rv1, LnCaP and LAPC-4 cell lines: the number of cells/hour are normalized as percent vs control to calculate efficacy concentration (EC20 and IC50) values, comparison of the effects in prostate cancer cell lines vs non-tumor/BPH-1 cell line.
INTERNATIONAL JOURNAL OF ONCOLOGY 40: 711-720, 2012 715
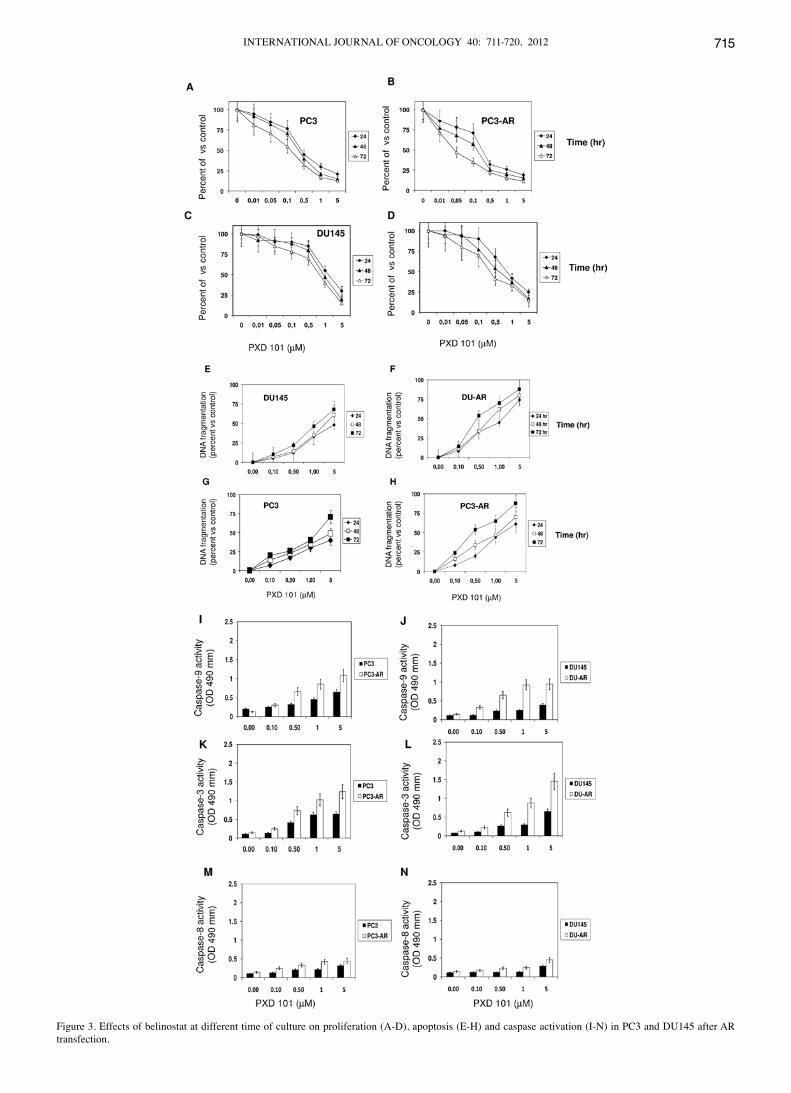
Figure 3. Effects of belinostat at different time of culture on proliferation (A-D), apoptosis (E-H) and caspase activation (I-N) in PC3 and DU145 after AR transfection.
GRAVINA et al: BELINOSTAT AND PROSTATE CANCER716
if AR modified belinostat sensitivity, two androgen-indepen-dent and AR-negative tumor cells were stably transfected with the wild-type AR and then subjected to increasing concentra-tions of 10-12 M belinostat (Fig. 3A-D). The re-expression of a functional wild-type AR in androgen defective PC3 and DU145 cells resulted in a significant slow-down of basal growth rate of these cells compared to parental ones in standard culture condi-tions. Belinostat significantly decreased the number of viable of PC3 and DU145 parental cells starting from the higher concentrations of 1 and 5 µM (Fig. 3A). The same tumor cells transfected and forced to express AR acquired more sensitivity to belinostat, which significantly decreased the number of viable cells starting from 0.1 µM belinostat. The same trend was clearly visible if apoptosis was used as efficacy indicator. An increment in the DNA fragmentation (Fig. 3E-H) as well as in the activity of caspase-3 , -8 and -9 (Fig. 3I-N) was observed upon treatment with belinostat in PC3 and DU145 cells trans-fected with AR when compared with parental cells.
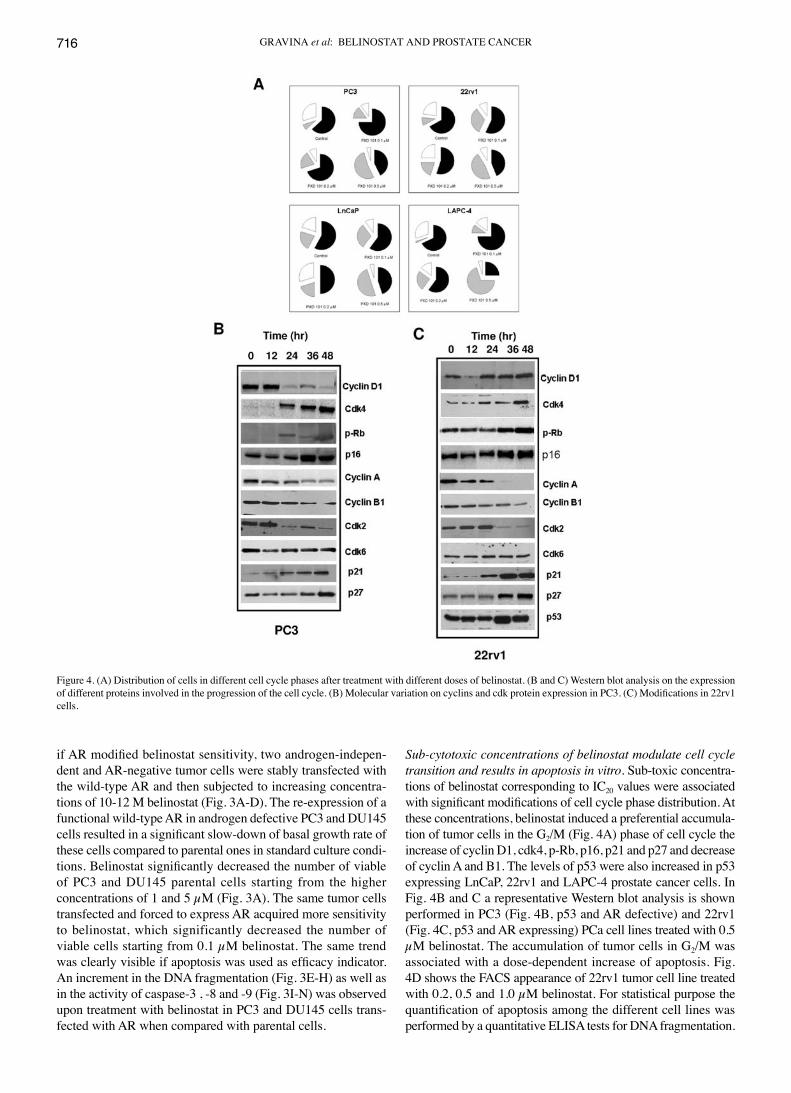
Sub-cytotoxic concentrations of belinostat modulate cell cycle transition and results in apoptosis in vitro. Sub-toxic concentra-tions of belinostat corresponding to IC20 values were associated with significant modifications of cell cycle phase distribution. At these concentrations, belinostat induced a preferential accumula-tion of tumor cells in the G2/M (Fig. 4A) phase of cell cycle the increase of cyclin D1, cdk4, p-Rb, p16, p21 and p27 and decrease of cyclin A and B1. The levels of p53 were also increased in p53 expressing LnCaP, 22rv1 and LAPC-4 prostate cancer cells. In Fig. 4B and C a representative Western blot analysis is shown performed in PC3 (Fig. 4B, p53 and AR defective) and 22rv1 (Fig. 4C, p53 and AR expressing) PCa cell lines treated with 0.5 µM belinostat. The accumulation of tumor cells in G2/M was associated with a dose-dependent increase of apoptosis. Fig. 4D shows the FACS appearance of 22rv1 tumor cell line treated with 0.2, 0.5 and 1.0 µM belinostat. For statistical purpose the quantification of apoptosis among the different cell lines was performed by a quantitative ELISA tests for DNA fragmentation.
Figure 4. (A) Distribution of cells in different cell cycle phases after treatment with different doses of belinostat. (B and C) Western blot analysis on the expression of different proteins involved in the progression of the cell cycle. (B) Molecular variation on cyclins and cdk protein expression in PC3. (C) Modifications in 22rv1 cells.
INTERNATIONAL JOURNAL OF ONCOLOGY 40: 711-720, 2012 717
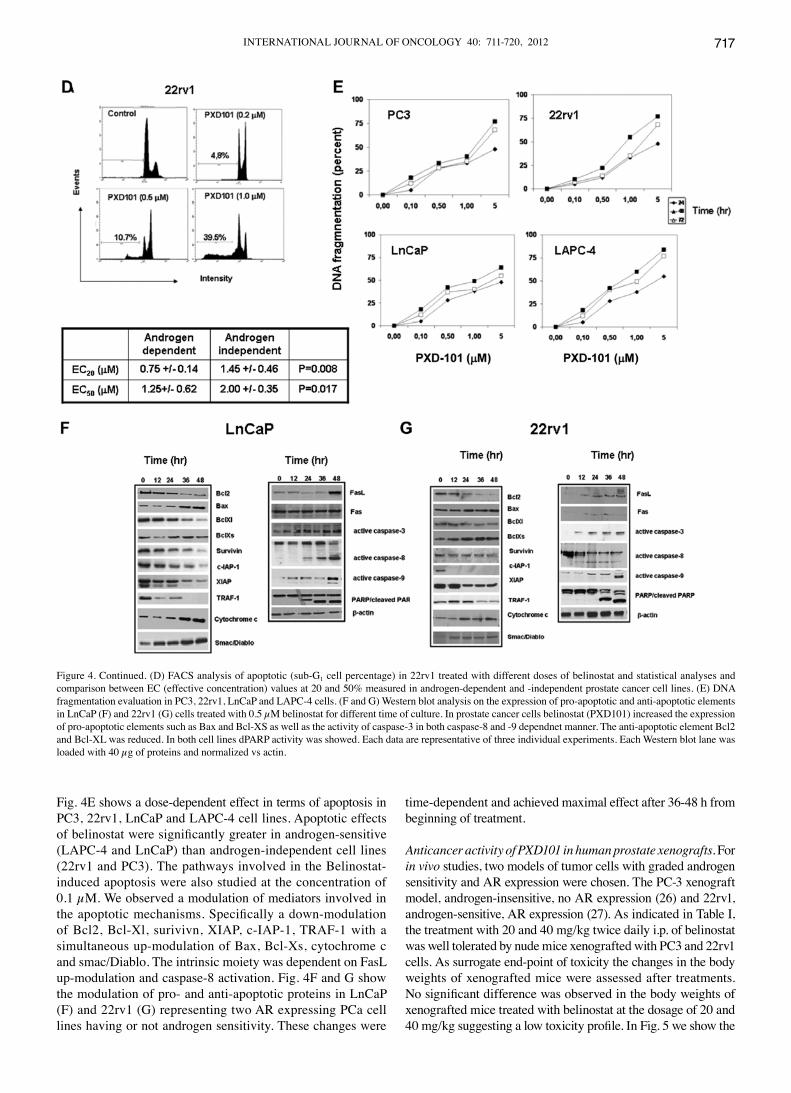
Fig. 4E shows a dose-dependent effect in terms of apoptosis in PC3, 22rv1, LnCaP and LAPC-4 cell lines. Apoptotic effects of belinostat were significantly greater in androgen-sensitive (LAPC-4 and LnCaP) than androgen-independent cell lines (22rv1 and PC3). The pathways involved in the Belinostat-induced apoptosis were also studied at the concentration of 0.1 µM. We observed a modulation of mediators involved in the apoptotic mechanisms. Specifically a down-modulation of Bcl2, Bcl-Xl, surivivn, XIAP, c-IAP-1, TRAF-1 with a simultaneous up-modulation of Bax, Bcl-Xs, cytochrome c and smac/Diablo. The intrinsic moiety was dependent on FasL up-modulation and caspase-8 activation. Fig. 4F and G show the modulation of pro- and anti-apoptotic proteins in LnCaP (F) and 22rv1 (G) representing two AR expressing PCa cell lines having or not androgen sensitivity. These changes were
time-dependent and achieved maximal effect after 36-48 h from beginning of treatment.
Anticancer activity of PXD101 in human prostate xenografts. For in vivo studies, two models of tumor cells with graded androgen sensitivity and AR expression were chosen. The PC-3 xenograft model, androgen-insensitive, no AR expression (26) and 22rv1, androgen-sensitive, AR expression (27). As indicated in Table I, the treatment with 20 and 40 mg/kg twice daily i.p. of belinostat was well tolerated by nude mice xenografted with PC3 and 22rv1 cells. As surrogate end-point of toxicity the changes in the body weights of xenografted mice were assessed after treatments. No significant difference was observed in the body weights of xenografted mice treated with belinostat at the dosage of 20 and 40 mg/kg suggesting a low toxicity profile. In Fig. 5 we show the
Figure 4. Continued. (D) FACS analysis of apoptotic (sub-G1 cell percentage) in 22rv1 treated with different doses of belinostat and statistical analyses and comparison between EC (effective concentration) values at 20 and 50% measured in androgen-dependent and -independent prostate cancer cell lines. (E) DNA fragmentation evaluation in PC3, 22rv1, LnCaP and LAPC-4 cells. (F and G) Western blot analysis on the expression of pro-apoptotic and anti-apoptotic elements in LnCaP (F) and 22rv1 (G) cells treated with 0.5 µM belinostat for different time of culture. In prostate cancer cells belinostat (PXD101) increased the expression of pro-apoptotic elements such as Bax and Bcl-XS as well as the activity of caspase-3 in both caspase-8 and -9 dependnet manner. The anti-apoptotic element Bcl2 and Bcl-XL was reduced. In both cell lines dPARP activity was showed. Each data are representative of three individual experiments. Each Western blot lane was loaded with 40 µg of proteins and normalized vs actin.
GRAVINA et al: BELINOSTAT AND PROSTATE CANCER718
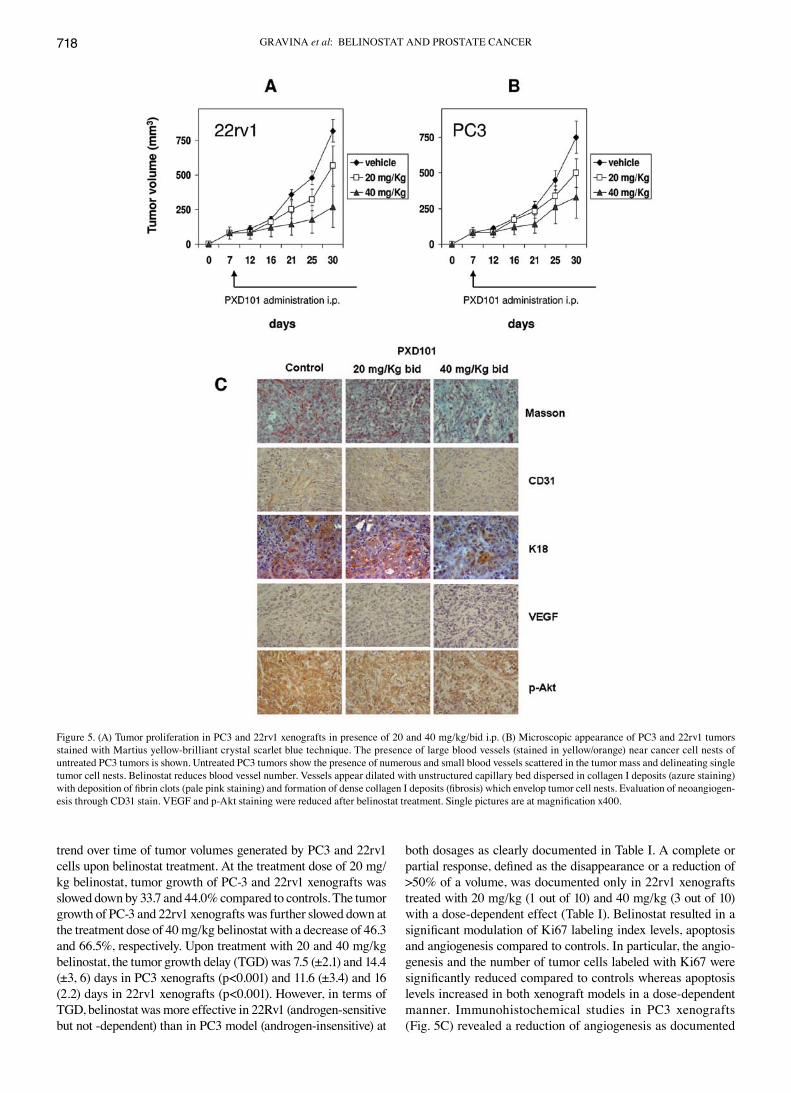
trend over time of tumor volumes generated by PC3 and 22rv1 cells upon belinostat treatment. At the treatment dose of 20 mg/kg belinostat, tumor growth of PC-3 and 22rv1 xenografts was slowed down by 33.7 and 44.0% compared to controls. The tumor growth of PC-3 and 22rv1 xenografts was further slowed down at the treatment dose of 40 mg/kg belinostat with a decrease of 46.3 and 66.5%, respectively. Upon treatment with 20 and 40 mg/kg belinostat, the tumor growth delay (TGD) was 7.5 (±2.1) and 14.4 (±3, 6) days in PC3 xenografts (p<0.001) and 11.6 (±3.4) and 16 (2.2) days in 22rv1 xenografts (p<0.001). However, in terms of TGD, belinostat was more effective in 22Rv1 (androgen-sensitive but not -dependent) than in PC3 model (androgen-insensitive) at
both dosages as clearly documented in Table I. A complete or partial response, defined as the disappearance or a reduction of >50% of a volume, was documented only in 22rv1 xenografts treated with 20 mg/kg (1 out of 10) and 40 mg/kg (3 out of 10) with a dose-dependent effect (Table I). Belinostat resulted in a significant modulation of Ki67 labeling index levels, apoptosis and angiogenesis compared to controls. In particular, the angio-genesis and the number of tumor cells labeled with Ki67 were significantly reduced compared to controls whereas apoptosis levels increased in both xenograft models in a dose-dependent manner. Immunohistochemical studies in PC3 xenografts (Fig. 5C) revealed a reduction of angiogenesis as documented
Figure 5. (A) Tumor proliferation in PC3 and 22rv1 xenografts in presence of 20 and 40 mg/kg/bid i.p. (B) Microscopic appearance of PC3 and 22rv1 tumors stained with Martius yellow-brilliant crystal scarlet blue technique. The presence of large blood vessels (stained in yellow/orange) near cancer cell nests of untreated PC3 tumors is shown. Untreated PC3 tumors show the presence of numerous and small blood vessels scattered in the tumor mass and delineating single tumor cell nests. Belinostat reduces blood vessel number. Vessels appear dilated with unstructured capillary bed dispersed in collagen I deposits (azure staining) with deposition of fibrin clots (pale pink staining) and formation of dense collagen I deposits (fibrosis) which envelop tumor cell nests. Evaluation of neoangiogen-esis through CD31 stain. VEGF and p-Akt staining were reduced after belinostat treatment. Single pictures are at magnification x400.
INTERNATIONAL JOURNAL OF ONCOLOGY 40: 711-720, 2012 719
by CD31 Masson's trichrome staining. The immunostaining for VEGF was also sensitively reduced after belinostat treatment as well as K18 and p-Akt.
Discussion
The increase in HDAC and DNMT expression could represent an active role of HDACs and DNMTs in prostate cancer or a cancer epi-phenomenon. It has been reported previously that hydroxamic acid-based inhibitors such as belinostat, are potent inducers of differentiation and/or apoptosis in transformed cells in culture including prostate cancer cells (10,28,29). Here we confirm that Belinostat, a member of hydroxamic acid-based inhibitors, suppresses, in vitro and in vivo, the growth of a wide panel of prostate tumor cells with graded androgen dependence at micro-molar concentrations. Here we demonstrate that belinostat can suppress the growth of both androgen-dependent and -insensitive prostate cancer cells in culture at micromolar concentrations, and that belinostat suppress tumor growth to a higher extent in nude mice bearing human androgen-dependent 22rv1 prostate tumors than androgen-independent PC-3 prostate tumors. This strongly indicates a functional relevance of increased HDACs in prostate cancer rather than cancer epi-phenomenon. In addition, HDACs regulate expression of many genes known to participate in cancer invasion and metastasis, such as extracellular matrix (ECM), metastasis-associated proteins (MTAs), metastasis suppressor gene KAI1 and NF-κB, and AR function partici-pating in hormone refractory development (30-35).
In the present study, accumulation of acetylated histones was demonstrated in all prostate cell models treated with belinostat. Nevertheless, the growth-suppressive and apoptotic activity of these agents appears to be confined to transformed cells. Treatment of non-tumor prostate epithelial cells (BPH-1) with belinostat caused no growth inhibition at doses that suppress the growth of tumor cell lines, although both the normal and trans-formed cells showed similar increases in histone acetylation. This evidence suggests that belinostat may have a differential effect on tumor and non-tumor tissues. Additionally, we demon-strated that androgen-sensitive tumor models are more sensitive
to the antitumor effects of belinostat in vitro and in vivo. Finally, among the androgen-independent models we showed that the presence of AR is a marker of belinostat efficacy since the trans-fection of AR-negative tumor cells with this nuclear receptor greatly increased the sensitivity to belinostat. It has been also demonstrated that histone deacetylases are required for androgen receptor function in hormone-sensitive and castrate-resistant prostate cancer and we demonstrated that analogously to SAHA, belinostat show additive effects with castration or bicalutamide in castration-resistant prostate cancer models (Gravina et al, 22nd EORTC-NCI-AACR Symposium on Molecular targets and Cancer Therapeutics, Berlin, 2010). In addition belinostat was able to up-modulate the PTEN protein levels in PTEN positive prostate cancer cells (36) and therefore to slow down the Akt activity which is able to modulate both the androgen-dependent and -independent AR activation (1). The slow-down of cell proliferation was associated with a significant increase of P21 and P27 and with the preferential accumulation of tumor cells in the G2/M phase of the cell cycle. The cell cycle block was followed by apoptosis, as described for other HDAC inhibi-tors, and was associated to both intrinsic and extrinsic apoptotic program (37). Apoptosis induction by belinostat was manifested at higher concentrations and was associated with the expression of many genes related to this event. In the belinostat-treated tumor cells, expression of Bax and caspase-3 increased with a contextual decrease of the anti-apoptotic gene Bcl-2. However doses less than IC20 efficiently inhibited the growth of prostate cancer tumor cell in vitro although this effect was more evident at longer times. In vivo administration of 20 or 40 mg/kg/bid i.p., belinostat was well tolerated by nude mice and tumor growth was significantly suppressed by 33.7 and 44.0%, and by 46.3 and 66.5%, after treatment with 20 and 40 mg/kg belinostat bid, respectively, in PC-3 and 22rv1 xenografts. A complete or partial remission of the tumor was observed in 22rv1 xenografts treated with 20 mg/kg/bid (1 out 10) and 40 mg/kg/bid (3 out 10).
In conclusion the evidence here reported indicates that belinostat results in a cytotoxic and pro-apoptotic activity preferentially in androgen-sensitive preclinical models and this activity is influenced by AR and this supports further studies
Table I. Antitumor activity of PXD101 in 22rv1 and PC3 xenograft.
Treatment Dose Weight Tumor TGD PI Apoptosis Vessels Complete/ Drug mg/kg of mice weight (days) Ki67% (%) partial Gr ± SD (mg ± SD) response
PC3 Saline 25.0±1 585±235 37.3±4.5 <2 30±4.3 0/10 Belinostat 20 mg/kg, bid 24.4±1.7 388±180 7.5 24.4±2.7 12.4±2.6 20.4±2.7 0/10 Belinostat 40 mg/kg, bid 23.0±2.3 314±117 14.4 14.7±1.3 25.6±3.1 10.4±2.3 0/10 P=0.100 P=0.008 P<0.001 P<0.001 P<0.001 P=1.000
22rv1 Saline 25.3±1.8 652±200 45.5±6.5 <2 38.5±5 0/100 Belinostat 20 mg/kg, bid 24.1±2 365±133 19.6 18.8±6.5 15.4±3.0 11.4±0.8 1/10 Belinostat 40 mg/kg, bid 24.0±1.4 218±125 16.0 11.7±0.5 28.4±3.0 7.5±0.8 3/10 P=0.200 P<0.001 P<0.001 P<0.001 P<0.001 P=0.133

GRAVINA et al: BELINOSTAT AND PROSTATE CANCER720
to evaluate the effects of belinostat with hormone manipulation therapies such as castration or treatments with the anti-androgen bicalutamide.
References
1. Welsbie DS, Xu J, Chen Y, Borsu L, Scher HI, Rosen N and Sawyers CL: Histone deacetylases are required for androgen receptor function in hormone-sensitive and castrate-resistant prostate cancer. Cancer Res 69: 958-966, 2009.
2. Cress WD and Seto E: Histone deacetylases, transcriptional control, and cancer. J Cell Physiol 184: 1-16, 2000.
3. Wang L, Zou X, Berger AD, Twiss C, Peng Y, Li Y, Chiu J, Guo H, Satagopan J, Wilton A, Gerald W, Basch R, Wang Z, Osman I and Lee P: Increased expression of histone deacetylaces (HDACs) and inhibition of prostate cancer growth and invasion by HDAC inhibitor SAHA. Am J Transl Res 1: 62-71, 2009.
4. Patra SK, Patra A and Dahiya R: Histone deacetylase and DNA methyltransferase in human prostate cancer. Biochem Biophys Res Commun 287: 705-713, 2001.
5. Halkidou K, Gaughan L, Cook S, Leung HY, Neal DE and Robson CN: Upregulation and nuclear recruitment of HDAC1 in hormone refractory prostate cancer. Prostate 59: 177-189, 2004.
6. Halkidou K, Cook S, Leung HY, Neal DE and Robson CN: Nuclear accumulation of histone deacetylase 4 (HDAC4) coincides with the loss of androgen sensitivity in hormone refractory cancer of the prostate. Eur Urol 45: 382-389, 2004.
7. Fischle W, Dequiedt F, Hendzel MJ, Guenther MG, Lazar MA, Voelter W and Verdin E: Enzymatic activity associated with class II HDACs is dependent on a multiprotein complex containing HDAC3 and SMRT/N-CoR. Mol Cell 9: 45-57, 2002.
8. Khan N, Jeffers M, Kumar S, Hackett C, Boldog F, Khramtsov N, Qian X, Mills E, Berghs SC, Carey N, Finn PW, Collins LS, Tumber A, Ritchie JW, Jensen PB, Lichenstein HS and Sehested M: Determination of the class and isoform selectivity of small-molecule histone deacetylase inhibitors. Biochem J 409: 581-589, 2008.
9. Plumb JA, Finn PW, Williams RJ, Bandara MJ, Romero MR, Watkins CJ, La Thangue NB and Brown R: Pharmacodynamic response and inhibition of growth of human tumor xenografts by the novel histone deacetylase inhibitor PXD101. Mol Cancer Ther 2: 721-728, 2003.
10. Qian X, La Rochelle WJ, Ara G, Wu F, Petersen KD, Thougaard A, Sehested M, Lichenstein HS and Jeffers M: Activity of PXD101, a histone deacetylase inhibitor, in preclinical ovarian cancer studies. Mol Cancer Ther 5: 2086-2095, 2006.
11. Duan J, Friedman J, Nottingham L, Chen Z, Ara G and van Waes C: Nuclear factor-κB p65 small interfering RNA or proteasome inhibitor bortezomib sensitizes head and neck squamous cell carcinomas to classic histone deacetylase inhibitors and novel histone deacetylase inhibitor PXD101. Mol Cancer Ther 6: 37-50, 2007.
12. Tumber A, Collins LS, Petersen KD, Thougaard A, Christiansen SJ, Dejligbjerg M, Jensen PB, Sehested M and Ritchie JW: The histone deacetylase inhibitor PXD101 synergises with 5-fluorouracil to inhibit colon cancer growth in vitro and in vivo. Cancer Chemother Pharmacol 60: 275-283, 2007.
13. Prince HM, Bishton MJ and Harrison SJ: Clinical studies of histone deacetylase inhibitors. Clin Cancer Res 15: 3958-3969, 2009.
14. Tepper CG, Boucher DL, Ryan PE, et al: Characterization of a novel androgen receptor mutation in a relapsed CWR22 prostate cancer xenograft and cell line. Cancer Res 62: 6606-6614, 2002.
15. Craft N, Shostak Y, Carey M, et al: A mechanism for hormone-independent prostate cancer through modulation of androgen receptor signaling by the HER-2/neu tyrosine kinase. Nat Med 5: 280-286, 1999.
16. Karan D, Kelly DL, Rizzino A, Lin MF and Batra SK: Expression profile of differentially-regulated genes during progression of androgen-independent growth in human prostate cancer cells. Carcinogenesis 23: 967-972, 2002.
17. Lin DL, Tarnowski CP, Zhang J, Dai J, Rohn E, Patel AH, Morris MD and Keller ET: Bone metastatic LNCaP-derivative C4-2B prostate cancer cell line mineralizes in vitro. Prostate 47: 212-221, 2001.
18. Zhao H, Dupont J, Yakar S, et al: PTEN inhibits cell proliferation and induces apoptosis by downregulating cell surface IGF-IR expression in prostate cancer cells. Oncogene 23: 786-794, 2004.
19. Bonaccorsi L, Carloni V, Muratori M, et al: Androgen receptor expression in prostate carcinoma cells suppresses alpha6beta4 integrin-mediated invasive phenotype. Endocrinology 141: 3172-3176, 2000.
20. Scaccianoce E, Festuccia C, Dondi D, et al: Characterization of prostate cancer DU145 cells expressing the recombinant androgen receptor. Oncol Res 14: 101-112, 2003.
21. Korenchuk S, Lehr JE, MClean L, Lee YG, Whitney S, Vessella R, Lin DL and Pienta KJ: VCaP, a cell-based model system of human prostate cancer. In Vivo 15: 163-168, 2001.
22. Pienta KJ, Abate-Shen C, Agus DB, Attar RM, Chung LW, Greenberg NM, Hahn WC, Isaacs JT, Navone NM, Peehl DM, Simons JW, Solit DB, Soule HR, van Dyke TA, Weber MJ, Wu L and Vessella RL: The current state of preclinical prostate cancer animal models. Prostate 68: 629-639, 2008.
23. Prewett MC, Hooper AT, Bassi R, Ellis LM, Waksal HW and Hicklin DJ: Enhanced antitumor activity of anti-epidermal growth factor receptor monoclonal antibody IMC-C225 in combination with irinotecan (CPT-11) against human colorectal tumor xeno-grafts. Clin Cancer Res 8: 994-1003, 2002.
24. Bruzzese F, Di Gennaro E, Avallone A, Pepe S, Arra C, Caraglia M, Tagliaferri P and Budillon A: Synergistic antitumor activity of epidermal growth factor receptor tyrosine kinase inhibitor gefitinib and IFN-alpha in head and neck cancer cells in vitro and in vivo. Clin Cancer Res 12: 617-625, 2006.
25. Gravina G, Marampon F, Petini F, Biordi L, Sherris D, Jannini EA, Tombolini V and Festuccia C: The TORC1/TORC2 inhibitor, Palomid 529, reduces tumor growth and sensitizes to docetaxel and cisplatin in aggressive and hormone refractory prostate cancer cells. Endocr Relat Cancer (In press).
26. Tai S, Sun Y, Squires JM, Zhang H, Oh WK, Liang CZ and Huang J: PC3 is a cell line characteristic of prostatic small cell carcinoma. Prostate (In press).
27. Yokomizo A, Shiota M, Kashiwagi E, Kuroiwa K, Tatsugami K, Inokuchi J, Takeuchi A and Naito S: Statins reduce the androgen sensitivity and cell proliferation by decreasing the androgen receptor protein in prostate cancer cells. Prostate 71: 298-304, 2011.
28. Rephaeli A, Blank-Porat D, Tarasenko N, Entin-Meer M, Levovich I, Cutts SM, Phillips DR, Malik Z and Nudelman A: In vivo and in vitro antitumor activity of butyroyloxymethyl-diethyl phosphate (AN-7), a histone deacetylase inhibitor, in human prostate cancer. Int J Cancer 116: 226-235, 2005.
29. Xia Q, Sung J, Chowdhury W, Chen CL, Höti N, Shabbeer S, Carducci M and Rodriguez R: Chronic administration of valproic acid inhibits prostate cancer cell growth in vitro and in vivo. Cancer Res 66: 7237-7244, 2006.
30. Kumar R, Wang RA and Bagheri-Yarmand R: Emerging roles of MTA family members in human cancers. Semin Oncol 30: 30-37, 2003.
31. Kim JH, Kim B, Cai L, Choi HJ, Ohgi KA, Tran C, Chen C, Chung CH, Huber O, Rose DW, Sawyers CL, Rosenfeld MG and Baek SH: Transcriptional regulation of a metastasis suppressor gene by Tip60 and beta-catenin complexes. Nature 434: 921-926, 2005.
32. Takada Y, Gillenwater A, Ichikawa H and Aggarwal BB: Suberoylanilide hydroxamic acid potentiates apoptosis, inhibits invasion, and abolishes osteoclastogenesis by suppressing nuclear factor-kappaB activation. J Biol Chem 281: 5612-5622, 2006.
33. Walton TJ, Li G, Seth R, McArdle SE, Bishop MC and Rees RC: DNA demethylation and histone deacetylation inhibition co-operate to re-express estrogen receptor beta and induce apoptosis in prostate cancer cell-lines. Prostate 68: 210-222, 2008.
34. Qian DZ, Wei YF, Wang X, Kato Y, Cheng L and Pili R: Antitumor activity of the histone deacetylase inhibitor MS-275 in prostate cancer models. Prostate 67: 1182-1193, 2007.
35. Hernandez M, Shao Q, Yang XJ, Luh SP, Kandouz M, Batist G, Laird DW and Alaoui-Jamali MA: A histone deacetylation-dependent mechanism for transcriptional repression of the gap junction gene cx43 in prostate cancer cells. Prostate 66: 1151-1161, 2006.
36. Gravina GL, Biordi L, Martella F, Flati V, Ricevuto E, Ficorella C, Tombolini V and Festuccia C: Epigenetic modulation of PTEN expression during antiandrogenic therapies in human prostate cancer. Int J Oncol 35: 1133-1139, 2009.
37. Häcker S, Dittrich A, Mohr A, Schweitzer T, Rutkowski S, Krauss J, Debatin KM and Fulda S: Histone deacetylase inhibitors cooperate with IFN-gamma to restore caspase-8 expression and overcome TRAIL resistance in cancers with silencing of caspase-8. Oncogene 28: 3097-3110, 2009.














![Evaluation of androgen-induced effects on the uptake of [18F]FDG, [11C]choline and [11C]acetate in an androgen-sensitive and androgen-independent prostate cancer xenograft model](https://img.pdfslide.net/doc/110x75/6361861c2fd302ae64094544/evaluation-of-androgen-induced-effects-on-the-uptake-of-18ffdg-11ccholine-and.jpg)














