Embed Size (px)
Citation preview

Differential regulation of survivin expression and apoptosis byvitamin D3 compounds in two isogenic MCF-7 breast cancer cellsublines
Fengzhi Li*,1, Xiang Ling1, Huayi Huang1, Lisa Brattain1, Pasha Apontes1, Jianguo Wu1,Lise Binderup2, and Michael G Brattain11 Department of Pharmacology and Therapeutics, Grace Cancer Drug Center, Roswell Park CancerInstitute, Buffalo, NY, USA2 Department of Biochemistry, Leo Pharmaceutical Products, 55, Denmark
AbstractAlthough both the antiapoptotic function of survivin and vitamin D3 (VD3)-mediated cell growthinhibition and apoptosis have been extensively studied, it is not known whether survivin plays a rolein VD3 compound-mediated cell growth inhibition and apoptosis induction. Using an isogenic modelof MCF-7 breast adenocarcinoma cells (MCF-7E and MCF-7L sublines that are sensitive andresistant to VD3 compounds), we found that VD3 compounds effectively downregulated survivin inVD3-sensitive MCF-7E cells, which was associated with VD3-induced apoptosis. In contrast, VD3compounds failed to downregulate survivin in VD3-resistant MCF-7L cells, which showed resistantto VD3-induced apoptosis. However, inhibition of survivin expression by small interfering RNA(siRNA) induced cell death per se and further sensitized VD3-induced apoptosis in MCF-7L cells,indicating that the inability of these cells to respond to VD3 is due to the failure to downregulatesurvivin. Forced expression of survivin not only blocked VD3-mediated G1 cell accumulation butalso increased S and G2/M cell populations. VD3 treatment rapidly triggered the activation of p38MAPK signaling in MCF-7E cells but not in MCF- 7L cells. Moreover, inhibition of p38 activationdiminished VD3-mediated survivin inhibition and partially rescued VD3-induced cell death. Wefurther showed that VD3 increased the expression of TGFβ1 and TGFβ receptor 2, and that blockingthe function of TGFβ receptor 2 diminished VD3 compound-mediated survivin downregulation.Thus, we propose that the VD3 compound-induced growth inhibition and apoptosis induction are atleast partially dependent on survivin downregulation via VD3-induced TGFβ signaling and theactivation of p38 MAPK pathway. Targeting survivin through these pathways may lead to novelapplications for cancer therapeutics.
Keywordsvitamin D3; survivin; apoptosis; p38 MAPK; MCF-7 breast cancer cell
IntroductionA new protein family, designated inhibitors of apoptosis (IAP), was recently recognized(Deveraux and Reed, 1999; Salvesen and Duckett, 2002). All IAP family members share oneor more signature motifs termed the baculovirus IAP repeat (BIR). This zinc-binding motif
*Correspondence: F Li, Department of Pharmacology and Therapeutics, Roswell Park Cancer Institute (RPCI), Elm & Carlton Streets,Buffalo, NY 14263, USA; [email protected].
NIH Public AccessAuthor ManuscriptOncogene. Author manuscript; available in PMC 2010 February 11.
Published in final edited form as:Oncogene. 2005 February 17; 24(8): 1385. doi:10.1038/sj.onc.1208330.
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript

consists of a conserved sequence of about 70 amino acids. Eight human IAP family membershave been identified so far. They are c-IAP1, c-IAP2 (Rothe et al., 1995), XIAP (Duckett etal., 1996), NAIP (Roy et al., 1995; Liston et al., 1996), survivin (Ambrosini et al., 1997),apollon (Chen et al., 1999), ML-IAP/livin (Vucic et al., 2000; Kasof and Gomes, 2001) andILP-2 (Richter et al., 2001). Survivin is a unique member in this family as it has a number ofdistinct features that it does not share with other members. Survivin is the smallest IAP member(Ambrosini et al., 1997) and is homodimerized in solution (Chantalat et al., 2000; Muchmoreet al., 2000; Verdecia et al., 2000); it is undetectable in most normal adult tissues but highlyexpressed in human cancers (Li, 2003); expression of survivin is cell cycle-regulated with arobust increase in G2/M phase (Li et al., 1998); and survivin appears to be involved in bothinhibition of apoptosis and regulation of cell division (Li et al., 1998, 1999; Reed and Reed,1999; Li, 2003) while other IAP members are restricted to one of these two functions (Deverauxand Reed, 1999; Salvesen and Duckett, 2002).
It has been demonstrated by many studies in multiple systems that modulation of survivinexpression or function triggers programmed cell death (apoptosis) (Li, 2003). The accumulateddata from survivin studies on human cancers suggest that survivin expression in cancer isassociated with cancer progression, poor prognosis, drug resistance, and shorter patientsurvival (Li, 2003). In vitro and in vivo studies targeting survivin with antisenseoligonucleotides (Li et al., 1999;Chen et al., 2000;Olie et al., 2000;Xia et al., 2002), dominant-negative mutants (Li et al., 1998,1999;Grossman et al., 1999a,b;O’Connor et al., 2000;Mesriet al., 2001), ribozymes (Pennati et al., 2002;Choi et al., 2003), triplex DNA-formation (Shenet al., 2003) and RNA interference (Ling and Li, 2004) have shown induction of apoptosis,reduction of tumor-growth potential, and sensitization to chemotherapeutic drugs and othertherapeutic approaches (Zaffaroni and Daidone, 2002). These studies indicate that survivin isan excellent novel target for cancer therapeutics.
Vitamin D3 (VD3) compounds have been demonstrated to be effective for inhibition of cellproliferation and induction of apoptosis in vitro and in vivo in a variety of cancer cell models(Colston et al., 1992; Peehl et al., 1994; Simboli-Campbell et al., 1996; Getzenberg et al.,1997). They are currently in clinical trials for cancer therapeutics (Hansen et al., 2000; Johnsonet al., 2002). However, the mechanisms by which these compounds generate inhibition ofproliferation and induction of apoptosis in tumor cells are not fully understood. The inhibitionof cell proliferation mediated by VD3 compounds has been reported to be due to modulationof cell cycle regulators and a G1 phase arrest through induction of p21 and p27 in pancreaticand breast cancer cells (Kawa et al., 1997; Wu et al., 1997; Verlinden et al., 1998). However,another report showed that VD3-induced G1 arrest is associated with a significant decrease ofp21 and increase of p27 in squamous cell carcinoma cells (Hershberger et al., 1999). Thecontextuality of these studies in different cancer cell types suggests that inhibition ofproliferation by VD3 compounds may involve several pathways. VD3 compounds have alsobeen shown to induce apoptosis in a variety of cancers including breast cancer (James et al.,1996, 1998), colon cancer (Diaz et al., 2000), and prostate cancer (Blutt et al., 2000). Althoughthe apoptosis induced by VD3 compounds is characterized by genomic DNA fragmentation(James et al., 1998; Blutt et al., 2000), caspase activation and poly(ADP-ribose) polymerase(PARP) cleavage (Johnson et al., 2002), the mechanism by which these events happen is notvery clear. For example, it was shown that Bcl-2 expression could be positively or negativelyregulated by VD3 compounds, but not essential for apoptosis (Diaz et al., 2000). These resultssuggest that other factors or targets rather than Bcl-2 may be involved in the VD3 compound-induced growth inhibition and apoptosis.
It was reported that VD3 and its analog EB1089 inhibit the MCF-7E subline growth byinduction of transforming growth factor β receptor II (TGFβRII) (Wu et al., 1998) andTGFβ isoforms (Yang et al., 2001). In contrast, proliferation of the MCF-7L isogenic subline,
Li et al. Page 2
Oncogene. Author manuscript; available in PMC 2010 February 11.
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript

which has lost cell surface RII and is resistant to TGFβ, was not affected by these compounds(Wu et al., 1998). However, the downstream targets controlling these differential responses toVD3 compounds in these cells are not well defined. In the present study, we report that VD3-sensitive MCF-7E breast cancer cells express a low basal level of survivin that is sensitive todownregulation by VD3 compound treatment. VD3-mediated inhibition of survivin expressionin MCF-7E cells evoked apoptosis. In contrast, VD3-resistant MCF-7L breast cancer cellsexpress a high basal level of survivin that is not sensitive to inhibition by VD3 compounds,resulting in resistance to apoptosis. Our data further indicated that VD3-mediated survivinrepression is p38 MAPK activation and TGFβ signaling-dependent. Thus, our studies, for thefirst time, demonstrate that VD3-induced apoptosis is at least in part due to TGFβ and p38signaling-dependent downregulation of survivin expression. Targeting survivin through thesepathways may lead to novel applications for cancer treatment.
ResultsDifferential expression of survivin in MCF-7E and MCF-7L breast cancer cells
It has been shown that MCF-7E cells are sensitive to VD3 compounds while MCF-7L cellsare resistant to these agents (Wu et al., 1998). Accordingly, Western blot experiments revealedthat the basal level of survivin expression in MCF-7E cells is lower than that in MCF-7L cells(Figure 1a). Northern blot experiments showed that the differential expression of survivinprotein is accompanied by a differential expression of survivin mRNA (Figure 1b).
VD3 compounds downregulate survivin expression, which is essential for VD3 compound-induced apoptosis
Consistent with the fact that HL-60 leukemia cells and MCF-7E breast cancer cells are sensitiveto VD3 and its analogs. VD3 compounds inhibited survivin expression in both HL-60 andMCF-7E cells (Figure 2a and b). In contrast, VD3 compounds had no inhibitory effect onsurvivin expression in the VD3-resistant MCF-7L cells (Figure 2c). Northern blot experimentsrevealed that VD3 compounds effectively downregulated survivin mRNA in MCF-7E cellsbut not in MCF-7L cells (Figure 3a and b). In accordance with the differential modulation ofsurvivin in these cells, MCF-7E cells are sensitive to VD3-induced cell death while MCF-7Lcells are resistant to VD3 treatment (Figure 4a). However, inhibition of survivin by smallinterfering RNA (siRNA) sensitized VD3-induced cell death in MCF-7L cells (Figure 4b).This observation indicates that the inability of MCF-7L cells to respond to VD3 is due to thefailure to downregulate survivin. In addition, targeting survivin with siRNA also induced celldeath in MCF-7E cells, and a combination of siRNA with VD3 further increased cell death inMCF- 7E cells but to a lower extent than in MCF-7L cells (not shown). The induction ofapoptosis by VD3 compounds was further confirmed by examining caspaspe-9 activation andPARP (polyADP-ribose polymerase) cleavage (Figure 4c).
Downregulation of survivin by VD3 is also essential for VD3-mediated cell growth inhibitionUsing a survivin siRNA approach we have provided direct evidence indicating thatdownregulation of survivin is essential for VD3-induced apoptosis (Figure 4). Furtherexperiments indicated that VD3- induced survivin downregulation is also essential for VD3-mediated growth arrest. As shown in Figure 5, forced expression of survivin in MCF-7E cellsnot only blocked the accumulation of G1 cells but also increased S and G2/M cell populationsin comparison with the empty vector-transfected cell control treated with VD3. These resultsdemonstrated an essential role of survivin in VD3-mediated cell growth inhibition andapoptosis induction (Figures 4 and 5).
Li et al. Page 3
Oncogene. Author manuscript; available in PMC 2010 February 11.
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript

Modulation of survivin expression and apoptosis by growth factor deprivation in MCF-7Eand MCF-7L cells
Similar to VD3 compounds, growth factor withdrawal downregulated survivin expression inMCF-7E cells (Figure 6a) but not in MCF-7L cells (Figure 6b). Consistent with thisobservation, growth factor deprivation induced cell death in MCF-7E cells, while MCF-7Lcells showed resistance to growth factor deprivation-induced cell death (Figure 6c).
Downregulation of survivin expression by VD3 compounds and growth factor deprivation isassociated with increased p38 MAPK phosphorylation in MCF-7E cells
To delineate the potential mechanism by which VD3 compounds and growth factor withdrawaldownregulated survivin expression, MCF-7E and 7L cells were treated with VD3 or subjectedto growth factor deprivation, followed by Western blots to probe phosphorylation of p38MAPK. The results showed that both VD3 and growth factor deprivation activate p38 asreflected by the increased signal of the p38 phosphorylation (Figure 7a and b). In contrast, p38phosphorylation in MCF-7L cells was not increased by either VD3 treatment or growth factordeprivation (Figure 7c and d). This is paralleled by the lack of inhibitory effects by VD3treatment and growth factor deprivation on survivin expression in MCF-7L cells but not inMCF-7E cells (Figures 2b and 6a, b), suggesting that p38 activation may mediate inhibitionof survivin expression in MCF-7E cells. The requirement of p38 phosphorylation for survivindownregulation was further supported by the fact that inhibition of p38 activation by the p38inhibitor (PD169316) reversed survivin downregulation induced by VD3 (Figure 8a) andgrowth factor withdrawal (Figure 8b). Moreover, inhibition of p38 activation also partiallyrescued VD3-induced cell death (Figure 8a, bottom), which further confirmed the involvementof p38 activation in VD3-induced apoptosis.
Downregulation of survivin by VD3 compounds is partially TGFβ signaling-dependentIt has been demonstrated that the autocrine TGFβ induced by VD3 analogs in MCF-7E cellsis required for inhibition of cell growth and induction of apoptosis (Wu et al., 1998; Yang etal., 2001). Our data also showed that VD3 increased the expression of TGFβ1 and TGFβreceptor 2 in MCF-7E cells (Figure 9). We hypothesized that if TGFβ signaling is involved inVD3-mediated downregulation of survivin, then abrogating the function of TGFβ receptor 2should diminish the degree of VD3-mediated survivin downregulation. Consistent with thishypothesis, using a Tet-Off-controlled expression of a dominant-negative TGFβ receptor 2 tocounter against the function of endogenous wild-type TGFβ receptor 2 in MCF-7E cells(MCF7E-DNRII cells) (Ko et al., 1998b), we found that while survivin expression in theparental MCF-7E cells was not affected by doxycycline (DOX) (Figure 10a, a control), VD3compounds (EB1089) were able to activate maximal inhibitory effects on survivin expressiononly when turning the expression of a dominant-negative TGFβ receptor 2 (DNRII) off byadding DOX (Figure 10b, lane 3). However, turning expression of a dominant-negativeTGFβ receptor 2 on in the absence of DOX diminished the effect of VD3 analog, EB1089 onsurvivin downregulation (Figure 10b, lane 4). These results suggest that the VD3 compound-induced TGFβ signaling was involved (refer to the Figure 10 legend for more information).Moreover, the requirement of TGFβ and TGFβ receptor 2 signaling in downregulation ofsurvivin was further supported by the fact that TGFβ1 treatment downregulated survivinexpression in MCF- 7E cells but not in MCF-7L cells (Figure 11a and b).
DiscussionIt is known that modulation of the expression of survivin, a novel inhibitor of apoptosis (IAP)protein, is associated with cancer cell viability (Li, 2003), and that VD3 compounds, whichare currently in clinical trials, are effective for cancer cell growth inhibition and apoptosisinduction (Johnson et al., 2002). However, it is not known whether survivin is a target in VD3-
Li et al. Page 4
Oncogene. Author manuscript; available in PMC 2010 February 11.
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript

induced growth inhibition and apoptosis. In this report, we took the advantage of a uniqueisogenic model of the MCF-7 breast cancer cell lines, which consists of the VD3-sensitiveMCF-7E cells and the VD3-resistant MCF-7L cells, to investigate the role of survivin in VD3-induced cell growth inhibition and apoptosis. We demonstrated, for the first time, that survivinplays an essential role in VD3-mediated apoptosis induction (Figure 4) and cell growth arrest(Figure 5). Several lines of evidence supported our conclusion. First, VD3- resistant MCF-7Lcells express a high basal level of survivin; in contrast, VD3-sensitive MCF-7E cells expressa low basal level of survivin. Second, VD3 compounds effectively downregulate survivinexpression in VD3-sensitive MCF-7E and leukemia HL-60 cells, but these compounds haveno inhibitory effect on survivin expression in VD3-resistant MCF-7L cells. Third, VD3-induced downregulation of survivin in MCF-7E cells is associated with apoptosis; in contrast,MCF-7L cells are resistant to VD3 treatment associated with unable to downregulate survivin.Fourth, while VD3 was unable to induce cell death in MCF-7L cells (Figure 4a), these cellswere sensitized to VD3 after the inhibition of survivin expression by transfection of survivinsiRNA. Finally, forced expression of survivin not only blocked the VD3-mediated G1 cellaccumulation but also increased S and G2/M cell populations (Figure 5). To delineate thesignaling pathways involved in VD3-mediated inhibition of survivin expression in MCF-7Ecells, we provided evidence that p38 MAPK activation and TGFβ signaling appear to beinvolved in VD3-mediated survivin downregulation. Inhibition of the p38 activation bypharmacological p38 inhibitors or block of TGFβ signaling by expression of a dominant-negative TGFβ receptor 2 diminished the inhibitory effects of VD3 compounds on survivinexpression in MCF-7E cells.
Although this report has delineated a role of survivin in VD3 compound-mediated cell growthinhibition and apoptosis, a number of interesting questions arise and warrant furtherinvestigation. For example, why is survivin expression low in MCF-7E cells and high inMCF-7L cells? What is the functional relationship of p38 signaling with the TGFβ signalingin mediating survivin downregulation? Are they independent or dependent in terms ofmediating survivin downregulation? In addition, significant inhibition of survivin expressionby VD3 compounds was still obtained even if when the autocrine TGFβ signaling was blockedin the TGFβ DNRII model cells by expression of a TGFβ receptor 2 dominant-negative mutant(Figure 10b, lane 4). This observation strongly suggested that VD3 compound-mediateddownregulation of survivin is also involved in a TGFβ signal-independent pathway. It is unclearwhether the p38 activation represents the TGFβ signal-independent pathway. Nevertheless, theinvolvement of TGFβ signaling in VD3-mediated survivin downregulation was directlysupported by the fact that VD3 increased the expression of TGFβ1 and TGFβ receptor 2 (Figure9), and exogenous TGFβ1 treatment downregulated survivin expression in VD3-sensitiveMCF-7E cells but not in VD3-resistent MCF-7L cells (Figure 11).
We have shown, in this paper, that the basal level of survivin expression is higher in MCF-7Lcells than that in MCF-7E cells (Figure 1), and growth factor deprivation downregulatessurvivin expression in MCF-7E cells but not in MCF-7L cells (Figure 5). We hypothesizedthat the cell survival signaling may be higher and deregulated in MCF-7L cells in comparisonwith that in MCF-7E cells. We have examined both the Akt pathway and the Erk1/2 MAPKpathway in MCF-7E and MCF-7L cells. We found that a low level PI3K/Akt activation canbe observed with and without growth factor deprivation in MCF-7L cells. However, the Aktphosphorylation state rapidly became undetectable during growth factor deprivation inMCF-7E cells. Interestingly, we did not find significant Erk1/2 activation, with or withoutserum starvation, in both MCF-7E and MCF-7L cells.
Previous studies demonstrated that autocrine TGFβ signaling mediates VD3 compound-induced growth inhibition and apoptosis in several breast cancer cells including MCF-7E cells(Wu et al., 1998; Yang et al., 2001). TGFβ signals through two TGFβ receptors, type I and
Li et al. Page 5
Oncogene. Author manuscript; available in PMC 2010 February 11.
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript

type II receptors, which are transmembrane serine/threonine kinases, to activate Smadsignaling (Hu et al., 1998). The later interacts with certain transcription cofactors, and bindsto the TGFβ/Smad responsive element to turn on the transcription of TGFβ target genes(Padgett, 1999). Interestingly, here, we found that activation of TGFβ signaling by VD3treatment or by the addition of exogenous TGFβ1 repressed survivin expression. This findingsupports the growing evidence that TGFβ signaling through activating Smads may also mediaterepression of transcription (Attisano and Wrana, 2000). In consideration of the important roleof survivin in cancer progression, further delineation of the detailed molecular events involvedin the potential Smad-mediated repression of survivin transcription will be required and mayprovide new avenues to control survivin expression for cancer therapeutics.
We also showed in this report that VD3 compound-mediated downregulation of survivinrequired p38 MAPK activation as well. It is currently unclear whether p38 activation isdownstream of TGFβ signaling or is independent of TGFβ signaling in this system. However,it was previously shown that TGFβ treatment rapidly activated p38 MAPK in human Burkittlymphoma B cells (Schrantz et al., 2001), mammary epithelial cells (Yu et al., 2002), murinehepatocytes (Yoo et al., 2003) and PC-3U prostate cancer cells (Edlund et al., 2003). Althoughthese studies showed that the activation of p38 is required for TGFβ-mediated apoptosis, thedown-stream target of p38 activation was not clear. In this report, we demonstrated thatactivation of p38 by VD3 is required for downregulation of survivin. While the underlyingmechanism for the activated p38 to down-regulate survivin is currently not clear, two possiblemechanisms may be responsible for the p38-mediated downregulation of survivin. First, theactivated p38 may phosphorylate certain transcription factor(s) responsible for survivintranscription, since VD3 compounds indeed downregulated survivin mRNA expression(Figure 3). The other possibility is that activated p38 may directly destabilize survivin proteinby phosphorylation. These possibilities remain to be investigated. Regardless of themechanism, in consistence with the finding that TGFβ-mediated apoptosis requires p38activation, our data indicated that inhibition of p38 activation blocks survivin downregulationby VD3 and partially rescued VD3-induced cell death (Figure 8a).
In addition, although we have shown in this report that VD3 compounds induced caspaseactivation and PARP cleavage (Figure 4c), it has been reported that apoptosis induced by VD3compounds could also be involved in caspase-independent pathway (Mathiasen et al., 1999;Narvaez and Welsh, 2001). Consistent with these reports, it has been shown that apoptotic celldeath triggered by downregulation of survivin could also be involved in both caspase-dependent and caspase-independent pathways (Shankar et al., 2001; Li, 2003; Liu et al.,2004).
In summary, we have demonstrated, for the first time, that survivin plays an essential role inVD3 compound-induced cell growth inhibition and apoptosis induction in breast cancer cells.Downregulation of survivin by VD3 compounds is involved in autocrine TGFβ signaling andp38 MAPK activation. Targeting survivin by these pathways may lead to novel applicationsfor cancer therapeutics.
Materials and methodsCell culture and reagents
MCF-7E cells designate the early passages (less than 200 passages) of the MCF-7 breast cancercell line while MCF-7L cells are from the later passages (more than 500 passages) of MCF-7breast cancer cells. MCF-7E cells are sensitive to growth inhibition by TGFβ or VD3 treatmentwhile MCF-7L cells are resistant to both of these agents (Ko et al., 1998a). These cells weremaintained in DMEM, supplemented with 10% fetal bovine serum (Mediatech Cellgro,Herndon, VA, USA) and penicillin (100 U/ml)/streptomycin (0.1 μg/ml) (Invitrogen, Grand
Li et al. Page 6
Oncogene. Author manuscript; available in PMC 2010 February 11.
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript

Island, NY, USA) in a humidified incubator with 5% CO2 at 37°C. The genetically modifiedMCF-7E cells (MCF7E-DNRII) (Ko et al., 1998b) were normally cultured in the same mediumabove in the presence of G418 (300–500 μg/ml) and DOX (2 μg/ml). Cells were routinelysubcultured twice weekly. VD3 compounds (1α,25(OH)2D3, EB1089 and CB1093) wereprovided from Leo Pharmaceutical Products (Ballerup, Denmark). The antibodies for survivin(FL-142), TGFβ1 (V) and TGFβ receptor 2 (L-21) were purchased from Santa Cruz (SantaCruz, CA, USA). PD169316 was purchased from CalBiochem (La Jolla, CA, USA). Phospho-p38 MAP kinase (Thr180/Tyr182) antibody and anti-p38 antibody were purchased from CellSignaling (Beverly, MA, USA). DOX and G418 were purchased from BD Biosciences (SanDiego, CA, USA). Propidium iodide (PI), TGFβ1, phosphatase inhibitor cocktail 1, monoclonalantiactin antibody and goat peroxidase- conjugated anti-rabbit IgGantibody were purchasedfrom Sigma (St Louis, MO, USA). Lipofectamine™ 2000 reagents were purchased fromInvitrogen (Carlsbad, CA, USA).
Treatment and Western blotIn all experiments, cells were treated with vitamin D3 (1α,25(OH)2D3) and/or its analogs(EB1089 and CB1093) in medium containing 10% serum. For Western blots, cells were washedwith phosphate-buffered saline (PBS) and lysed at 4°C for 30 min in PBS containing 1%Nonidet P-40, 0.5% sodium deoxycholate, 0.1% sodium dodecyl sulfate (SDS), 10 μg/mlphenylmethyl sulfonyl fluoride, and 20 μM leupeptin with or without the phosphatase inhibitorcocktail 1. Cell extracts were cleared by centrifugation at 15 000 g for 20 min at 4°. In total,50 μg total proteins from each sample were heated at 95°C for 5 min after mixing with equalvolume of 2 × SDS loading buffer. Samples were separated on 15% SDS–polyacrylamide gelelectrophoresis (SDS–PAGE) gels and electrotransferred to Immobilon-P membranes(Millipore, Bedford, MA, USA). The membrane was blocked in 5% skim milk or BSA (forphospho-specific antibodies) in TBS-T buffer (20mM Tris/HCl (pH 7.5), 0.137M NaCl, and0.05% Tween-20) at room temperature for 2–3 h. The membranes were incubated with differentprimary antibodies in TBS-T overnight at 4°C in the range of dilutions from 1: 500 to 1: 2000.After washing with TBS-T, the membrane was incubated in 5% skim milk in TBST buffercontaining a secondary antibody (1: 5000), for 45–60 min at room temperature with shaking.Proteins of interest were detected by an ECL protein detection kit (Amersham, ArlingtonHeights, IL, USA) and visualized by autoradiography after various times (20–120 s) ofexposure. For normalization of protein loading, the same membranes were stripped withstripping buffer (100mM 2-mercaptoethanol, 2% sodium dodecyl sulphate, 62.5mM Tris-HClpH 6.7) and used for Western blot with a monoclonal antibody against actin at a dilution of 1:1000 by the same procedure.
Total RNA isolation and Northern blotCells in 60 × 15mm culture dishes were washed with ice cold PBS and directly lysed in thedish with 1ml TRI REAGENT (Molecular Research Center, Cincinnati, OH, USA), The lysatewas transferred into a 1.5 ml tube and stored at room temperature for 5 min. In total, 0.2 mlchloroform was added to each tube and vortexed vigorously for 15–20 s. After incubation for2–15 min at room temperature, the samples in the tube were centrifuged at 12 000 r.p.m. for15 min at 4°C. The supernatant containing total RNAs was transferred into a new 1.5 ml RNase-free tube and total RNAs were precipitated with 0.5 ml isopropanol by centrifugation at 12 000r.p.m. for 10 min at 4°C. After washing with 75% ethanol and drying at room temperature for10 min, the total RNA pellets were resuspended in 15-μl depc-dH2O by vortexing and RNAconcentrations were determined by spectrophotometer. For Northern blot analysis, 10 μg oftotal RNAs were diluted in MOPS/formaldehyde/formamide buffer and separated on 1%formaldehyde-denatured agarose gels. The separated RNA was transferred onto Immobilon-N membrane (Millipore, Bedford, MA, USA) and crosslinked by ultraviolet (Stratagene, LaJolla, CA, USA). The membrane was then hybridized with a 32P-labeled survivin cDNA probe
Li et al. Page 7
Oncogene. Author manuscript; available in PMC 2010 February 11.
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript

in ExpressHyb hybridization solution (Clontech) for 1 h at 68°C. The expression level ofsurvivin mRNA was visualized by autoradiography after washing. The membrane was strippedin 0.1 × SSC solution and re-hybridized with a 32P-labeled glyceraldehyde 3-phosphatedehydrogenase (GAPDH) cDNA probe for an internal control.
Trypan blue exclusion staining for determination of cell viabilityCells to be counted were collected by trypsinization/centrifugation and resuspended in PBSbuffer. A small sample of the cell suspension was diluted in 0.4% (w/v) trypan blue (one samplea time since viable cells absorb trypan blue over time as well). A cover glass was centered overthe hemacytometer chambers and one chamber was filled with the cell dilution using a Pasteurpipette. Stained (dead) and unstained (viable) cells were counted in each of the four corner andcentral squares under an inverted microscope using 100× magnification, respectively. Eachcell sample was counted in this way for three times. The percentage of cell viabilities in eachsample was calculated with the formula of ‘% viability=total viable cell numbers/total cellnumbers × 100’.
Small interfering RNA (siRNA) preparationA human survivin mRNA-specific RNA oligonucleotides with 3′-TT overhangs [120GGCUGG CUU CAU CCA CUG C138TT (forward chain) and 138GCA GUG GAU GAA GCCAGC C120TT (reverse chain)] were chemically synthesized and purified by HPLC (Xeragon,Huntsville, AL, USA). Equal moles of each RNA oligonucleotide were mixed together to afinal concentration of 20 μM in annealing buffer (100mM KAc, 30mM HEPES-KOH, 2mMMgAc2, pH 7.4). After denaturation at 90°C for 1 min, the survivin siRNA mixture (designatedSRi-3) was annealed at 37°C for 60 min and stored at −80°C for transfection experiments. Ascramble RNA duplex (designated scraSRi) was also prepared as above for a negative controlin this study. The scramble sequence [5′CAGUCG CGU UUG CGA CUG GTT (forward chain)and 5′CCA GUC GCA AAC GCG ACU GTT (reverse chain)] was not present in mammaliancells by BLAST search at NCBI. The effectiveness of survivin SRi-3 on survivin inhibitionwas 75–90% and confirmed in HeLa (Ling and Li, 2004) and MCF-7 cells (Figure 4b, upperpanel).
Transfection of siRNA and cell viability analysisOne day prior to transfection, 3 × 105 cells per well were seeded in six-well plate containingculture medium without antibiotics. Cells at 40–60% confluence were transfected with thescramble (scraSRi) and survivin (SRi-3) siRNAs, respectively, as follows. Serum-free DMEM(100 μl) containing 3 μg siRNAs were mixed with 100 μl serum-free DMEM containing 9 μlLipofectamine™ 2000 reagents and held at room temperature. After the medium in a six-wellplate was replaced by serum-free DMEM (800 μl/well), the siRNA-Lipofectamine™ 2000mixture prepared above was added onto each well in the six-well plate within 20–45 min afterthe mixture was prepared. In total, 330 μl DMEM containing 30% fetal bovine serum wasadded into each well in the plate 2–4 h later. The transfected cells were treated with and withoutVD3 16 h post-transfection. Survivin expression was analysed by Western blots and cellviability was analysed microscopically 48 h after VD3 treatment by trypan blue exclusion asdescribed above.
Survivin expression vector transfection and cell cycle analysisFor the transfection of expression vectors, pEGFPc1 control vector and pEGFPc1-survivinexpression vector were transfected into MCF-7E cells same as above. The transfected cellswere treated with VD3 16 h post-transfection and analysed by PI staining and cell flowcytometry through gating the transfected green cells 24 h after VD3 treatment.
Li et al. Page 8
Oncogene. Author manuscript; available in PMC 2010 February 11.
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript

AcknowledgmentsThis work was sponsored in part by foundation grants from Wendy Will Case Cancer Fund Inc. (Chicago, IL), ElsaU Pardee Foundation (Midland, Michigan) and Concern Foundation (Beverly Hill, CA) to FL, by NIH R01 grants toMGB (CA72001 and CA38173) and to FL (CA109481), and by shared resources of NIH Cancer Center Support Grant(CA16056) to Roswell Park Cancer Institute.
Abbreviations
IAP inhibitor of apoptosis
BIR baculovirus IAP repeat
VD3 vitamin D3
PARP poly (ADP-ribose) polymerase
TGFβ RII transforming growth factor receptor II
TGFβ DNR II dominant-negative TGFβ receptor 2
siRNA small interfering RNA
DOX doxycycline
SDS-PAGE sodium dodecyl sulfate-polyacrylamide gel electrophoresis
GAPDH glyceraldehyde 3-phosphate dehydrogenase
PI propidium iodide
s.d standard deviation
ReferencesAmbrosini G, Adida C, Altieri DC. Nat Med 1997;3:917–921. [PubMed: 9256286]Attisano L, Wrana JL. Curr Opin Cell Biol 2000;12:235–243. [PubMed: 10712925]Blutt SE, Polek TC, Stewart LV, Kattan MW, Weigel NL. Cancer Res 2000;60:779–782. [PubMed:
10706079]Chantalat L, Skoufias DA, Kleman JP, Jung B, Dideberg O, Margolis RL. Mol Cell 2000;6:183–189.
[PubMed: 10949039]Chen J, Wu W, Tahir SK, Kroeger PE, Rosenberg SH, Cowsert LM, Bennett F, Krajewski S, Krajewska
M, Welsh K, Reed JC, Ng SC. Neoplasia 2000;2:235–241. [PubMed: 10935509]Chen Z, Naito M, Hori S, Mashima T, Yamori T, Tsuruo T. Biochem Biophys Res Commun
1999;264:847–854. [PubMed: 10544019]Choi KS, Lee TH, Jung MH. Cancer Gene Ther 2003;10:87–95. [PubMed: 12536196]Colston KW, Chander SK, Mackay AG, Coombes RC. Biochem Pharmacol 1992;44:693–702. [PubMed:
1324683]Deveraux QL, Reed JC. Genes Dev 1999;13:239–252. [PubMed: 9990849]Diaz GD, Paraskeva C, Thomas MG, Binderup L, Hague A. Cancer Res 2000;60:2304–2312. [PubMed:
10786699]Duckett CS, Nava VE, Gedrich RW, Clem RJ, Van Dongen JL, Gilfillan MC, Shiels H, Hardwick JM,
Thompson CB. EMBO J 1996;15:2685–2694. [PubMed: 8654366]Edlund S, Bu S, Schuster N, Aspenstrom P, Heuchel R, Heldin NE, ten Dijke P, Heldin CH, Landstrom
M. Mol Biol Cell 2003;14:529–544. [PubMed: 12589052]Getzenberg RH, Light BW, Lapco PE, Konety BR, Nangia AK, Acierno JS, Dhir R, Shurin Z, Day RS,
Trump DL, Johnson CS. Urology 1997;50:999–1006. [PubMed: 9426741]Grossman D, McNiff JM, Li F, Altieri DC. J Invest Dermatol 1999a;113:1076–1081. [PubMed:
10594755]
Li et al. Page 9
Oncogene. Author manuscript; available in PMC 2010 February 11.
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript

Grossman D, McNiff JM, Li F, Altieri DC. Lab Invest 1999b;79:1121–1126. [PubMed: 10496530]Hansen CM, Hamberg KJ, Binderup E, Binderup L. Curr Pharm Des 2000;6:803–828. [PubMed:
10828309]Hershberger PA, Modzelewski RA, Shurin ZR, Rueger RM, Trump DL, Johnson CS. Cancer Res
1999;59:2644–2649. [PubMed: 10363987]Hu PP, Datto MB, Wang XF. Endocr Rev 1998;19:349–363. [PubMed: 9626558]James SY, Mackay AG, Colston KW. J Steroid Biochem Mol Biol 1996;58:395–401. [PubMed: 8903423]James SY, Mercer E, Brady M, Binderup L, Colston KW. Br J Pharmacol 1998;125:953–962. [PubMed:
9846632]Johnson CS, Hershberger PA, Trump DL. Cancer Metastasis Rev 2002;21:147–158. [PubMed:
12465754]Kasof GM, Gomes BC. J Biol Chem 2001;276:3238–3246. [PubMed: 11024045]Kawa S, Nikaido T, Aoki Y, Zhai Y, Kumagai T, Furihata K, Fujii S, Kiyosawa K. Br J Cancer
1997;76:884–889. [PubMed: 9328147]Ko Y, Banerji SS, Liu Y, Li W, Liang J, Soule HD, Pauley RJ, Willson JK, Zborowska E, Brattain MG.
J Cell Physiol 1998a;176:424–434. [PubMed: 9648930]Ko Y, Koli KM, Banerji SS, Li W, Zborowska E, Willson JKV, Brattain MG, Arteaga CL. Int J Oncol
1998b;12:87–94. [PubMed: 9454891]Li F. J Cell Physiol 2003;197:8–29. [PubMed: 12942537]Li F, Ackermann EJ, Bennett CF, Rothermel AL, Plescia J, Tognin S, Villa A, Marchisio PC, Altieri DC.
Nat Cell Biol 1999;1:461–466. [PubMed: 10587640]Li F, Ambrosini G, Chu EY, Plescia J, Tognin S, Marchisio PC, Altieri DC. Nature 1998;396:580–584.
[PubMed: 9859993]Ling X, Li F. Bio Tech 2004;36:450–460.Liston P, Roy N, Tamai K, Lefebvre C, Baird S, Cherton-Horvat G, Farahani R, McLean M, Ikeda JE,
MacKenzie A, Korneluk RG. Nature 1996;379:349–353. [PubMed: 8552191]Liu T, Brouha B, Grossman D. Oncogene 2004;23:39–48. [PubMed: 14712209]Mathiasen IS, Lademann U, Jaattela M. Cancer Res 1999;59:4848–4856. [PubMed: 10519395]Mesri M, Wall NR, Li J, Kim RW, Altieri DC. J Clin Invest 2001;108:981–990. [PubMed: 11581299]Muchmore SW, Chen J, Jakob C, Zakula D, Matayoshi ED, Wu W, Zhang H, Li F, Ng SC, Altieri DC.
Mol Cell 2000;6:173–182. [PubMed: 10949038]Narvaez CJ, Welsh J. J Biol Chem 2001;276:9101–9107. [PubMed: 11053435]O’Connor DS, Grossman D, Plescia J, Li F, Zhang H, Villa A, Tognin S, Marchisio PC, Altieri DC. Proc
Natl Acad Sci USA 2000;97:13103–13107. [PubMed: 11069302]Olie RA, Simoes-Wust AP, Baumann B, Leech SH, Fabbro D, Stahel RA, Zangemeister-Wittke U.
Cancer Res 2000;60:2805–2809. [PubMed: 10850418]Padgett RW. Cancer Metastasis Rev 1999;18:247–259. [PubMed: 10728987]Peehl DM, Skowronski RJ, Leung GK, Wong ST, Stamey TA, Feldman D. Cancer Res 1994;54:805–
810. [PubMed: 7508338]Pennati M, Colella G, Folini M, Citti L, Daidone MG, Zaffaroni N. J Clin Invest 2002;109:285–286.
[PubMed: 11805141]Reed JC, Reed SI. Nat Cell Biol 1999;1:E199–E200. [PubMed: 10587656]Richter BW, Mir SS, Eiben LJ, Lewis J, Reffey SB, Frattini A, Tian L, Frank S, Youle RJ, Nelson DL,
Notarangelo LD, Vezzoni P, Fearnhead HO, Duckett CS. Mol Cell Biol 2001;21:4292–4301.[PubMed: 11390657]
Rothe M, Pan MG, Henzel WJ, Ayres TM, Goeddel DV. Cell 1995;83:1243–1252. [PubMed: 8548810]Roy N, Mahadevan MS, McLean M, Shutler G, Yaraghi Z, Farahani R, Baird S, Besner-Johnston A,
Lefebvre C, Kang X, Salih M, Aubry H, Tamai K, Guan X, Ioannou P, Crawford TO, de Jong PJ,Surh L, Ikeda J-E, Korneluk RG, MacKenzie A. Cell 1995;80:167–178. [PubMed: 7813013]
Salvesen GS, Duckett CS. Nat Rev Mol Cell Biol 2002;3:401–410. [PubMed: 12042762]Schrantz N, Bourgeade MF, Mouhamad S, Leca G, Sharma S, Vazquez A. Mol Biol Cell 2001;12:3139–
3151. [PubMed: 11598198]
Li et al. Page 10
Oncogene. Author manuscript; available in PMC 2010 February 11.
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript

Shankar SL, Mani S, O’Guin KN, Kandimalla ER, Agrawal S, Shafit-Zagardo B. J Neurochem2001;79:426–436. [PubMed: 11677271]
Shen C, Buck A, Polat B, Schmid-Kotsas A, Matuschek C, Gross HJ, Bachem M, Reske SN. CancerGene Ther 2003;10:403–410. [PubMed: 12719710]
Simboli-Campbell M, Narvaez CJ, Tenniswood M, Welsh J. J Steroid Biochem Mol Biol 1996;58:367–376. [PubMed: 8903420]
Verdecia MA, Huang H, Dutil E, Kaiser DA, Hunter T, Noel JP. Nat Struct Biol 2000;7:602–608.[PubMed: 10876248]
Verlinden L, Verstuyf A, Convents R, Marcelis S, Van Camp M, Bouillon R. Mol Cell Endocrinol1998;142:57–65. [PubMed: 9783903]
Vucic D, Stennicke HR, Pisabarro MT, Salvesen GS, Dixit VM. Curr Biol 2000;10:1359–1366. [PubMed:11084335]
Wu G, Fan RS, Li W, Ko TC, Brattain MG. Oncogene 1997;15:1555–1563. [PubMed: 9380407]Wu G, Fan RS, Li W, Srinivas V, Brattain MG. J Biol Chem 1998;273:7749–7756. [PubMed: 9516484]Xia C, Xu Z, Yuan X, Uematsu K, You L, Li K, Li L, McCormick F, Jablons DM. Mol Cancer Ther
2002;1:687–694. [PubMed: 12479365]Yang L, Yang J, Venkateswarlu S, Ko T, Brattain MG. J Cell Physiol 2001;188:383–393. [PubMed:
11473365]Yoo J, Ghiassi M, Jirmanova L, Balliet AG, Hoffman B, Fornace AJ Jr, Liebermann DA, Bottinger EP,
Roberts AB. J Biol Chem 2003;278:43001–43007. [PubMed: 12933797]Yu L, Hebert MC, Zhang YE. EMBO J 2002;21:3749–3759. [PubMed: 12110587]Zaffaroni N, Daidone MG. Drug Resist Update 2002;5:65–72.
Li et al. Page 11
Oncogene. Author manuscript; available in PMC 2010 February 11.
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
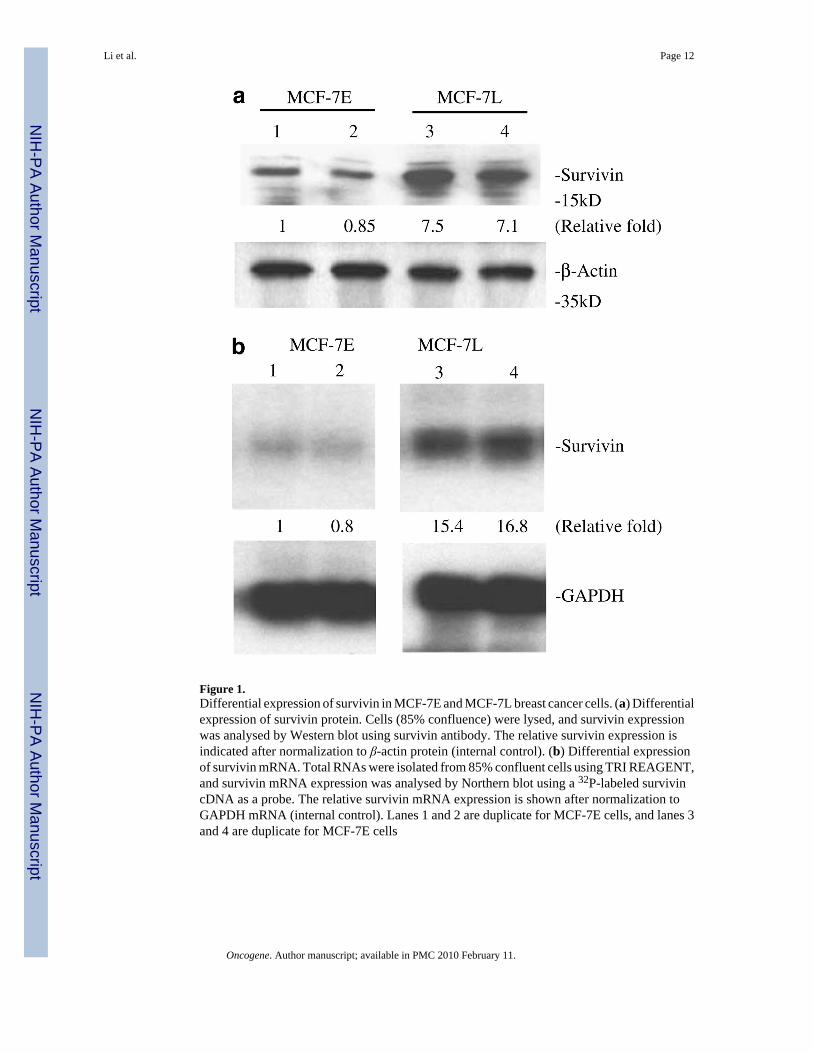
Figure 1.Differential expression of survivin in MCF-7E and MCF-7L breast cancer cells. (a) Differentialexpression of survivin protein. Cells (85% confluence) were lysed, and survivin expressionwas analysed by Western blot using survivin antibody. The relative survivin expression isindicated after normalization to β-actin protein (internal control). (b) Differential expressionof survivin mRNA. Total RNAs were isolated from 85% confluent cells using TRI REAGENT,and survivin mRNA expression was analysed by Northern blot using a 32P-labeled survivincDNA as a probe. The relative survivin mRNA expression is shown after normalization toGAPDH mRNA (internal control). Lanes 1 and 2 are duplicate for MCF-7E cells, and lanes 3and 4 are duplicate for MCF-7E cells
Li et al. Page 12
Oncogene. Author manuscript; available in PMC 2010 February 11.
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
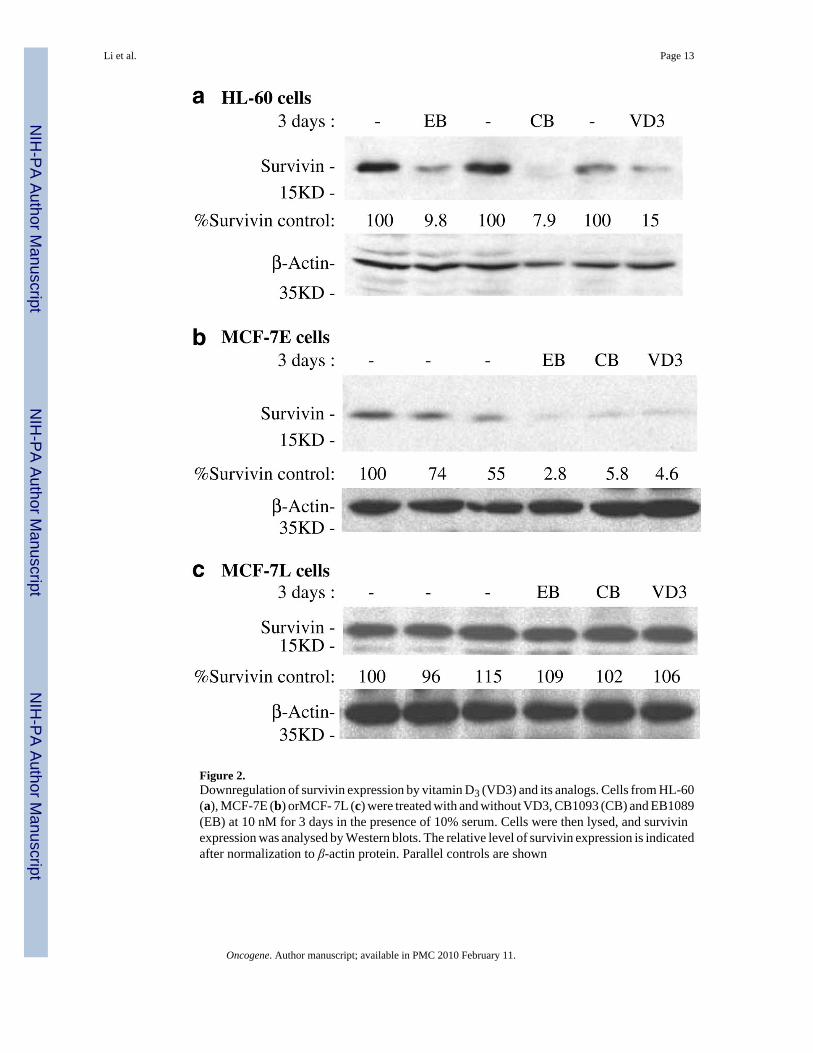
Figure 2.Downregulation of survivin expression by vitamin D3 (VD3) and its analogs. Cells from HL-60(a), MCF-7E (b) orMCF- 7L (c) were treated with and without VD3, CB1093 (CB) and EB1089(EB) at 10 nM for 3 days in the presence of 10% serum. Cells were then lysed, and survivinexpression was analysed by Western blots. The relative level of survivin expression is indicatedafter normalization to β-actin protein. Parallel controls are shown
Li et al. Page 13
Oncogene. Author manuscript; available in PMC 2010 February 11.
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
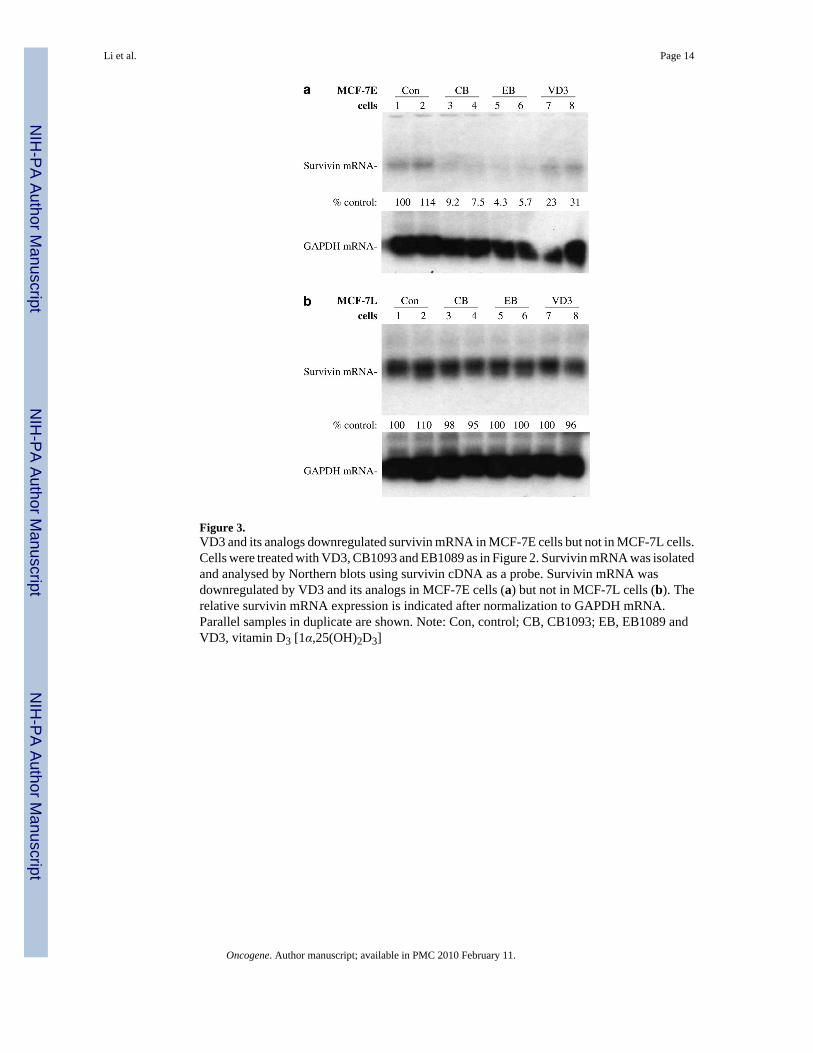
Figure 3.VD3 and its analogs downregulated survivin mRNA in MCF-7E cells but not in MCF-7L cells.Cells were treated with VD3, CB1093 and EB1089 as in Figure 2. Survivin mRNA was isolatedand analysed by Northern blots using survivin cDNA as a probe. Survivin mRNA wasdownregulated by VD3 and its analogs in MCF-7E cells (a) but not in MCF-7L cells (b). Therelative survivin mRNA expression is indicated after normalization to GAPDH mRNA.Parallel samples in duplicate are shown. Note: Con, control; CB, CB1093; EB, EB1089 andVD3, vitamin D3 [1α,25(OH)2D3]
Li et al. Page 14
Oncogene. Author manuscript; available in PMC 2010 February 11.
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
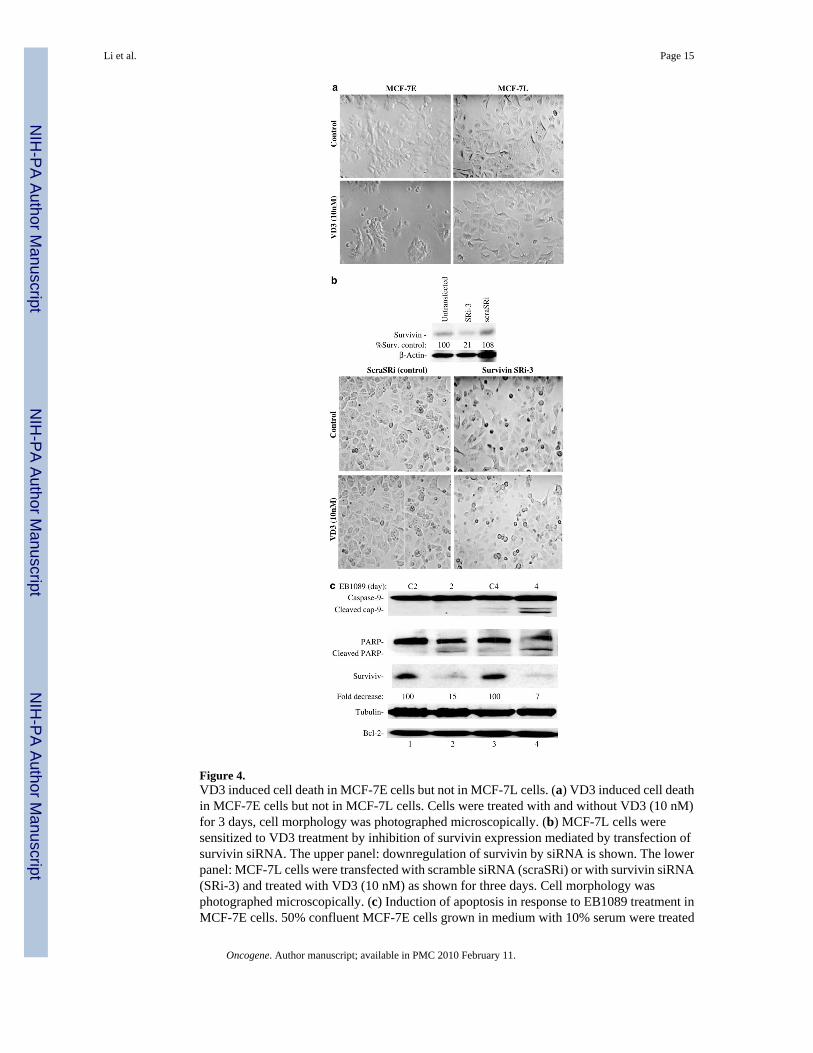
Figure 4.VD3 induced cell death in MCF-7E cells but not in MCF-7L cells. (a) VD3 induced cell deathin MCF-7E cells but not in MCF-7L cells. Cells were treated with and without VD3 (10 nM)for 3 days, cell morphology was photographed microscopically. (b) MCF-7L cells weresensitized to VD3 treatment by inhibition of survivin expression mediated by transfection ofsurvivin siRNA. The upper panel: downregulation of survivin by siRNA is shown. The lowerpanel: MCF-7L cells were transfected with scramble siRNA (scraSRi) or with survivin siRNA(SRi-3) and treated with VD3 (10 nM) as shown for three days. Cell morphology wasphotographed microscopically. (c) Induction of apoptosis in response to EB1089 treatment inMCF-7E cells. 50% confluent MCF-7E cells grown in medium with 10% serum were treated
Li et al. Page 15
Oncogene. Author manuscript; available in PMC 2010 February 11.
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript

with (lanes 2 and 4) or without (lanes 1 and 3) VD3 analog EB1089 for 2 and 4 days as indicated.Cells were lysed for analysis of PARP cleavage, caspase-9 activation, and expression ofsurvivin and Bcl-2 by Western blots. Tubulin is an internal control for total protein loading
Li et al. Page 16
Oncogene. Author manuscript; available in PMC 2010 February 11.
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
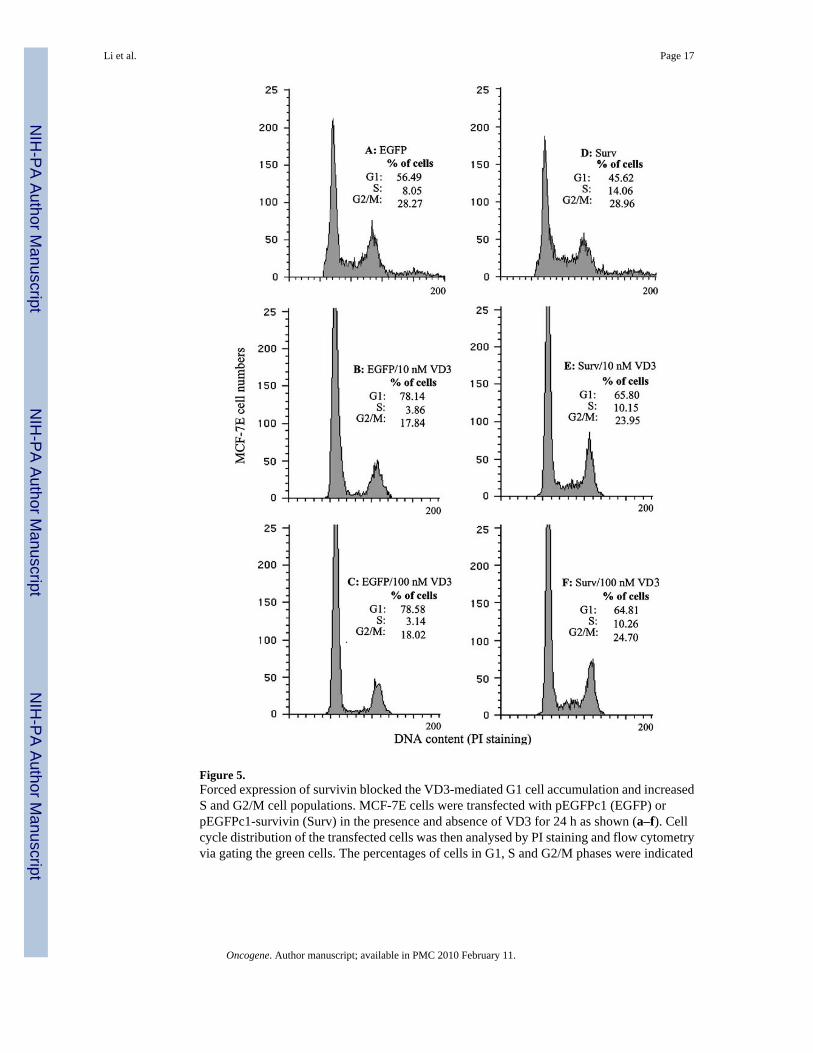
Figure 5.Forced expression of survivin blocked the VD3-mediated G1 cell accumulation and increasedS and G2/M cell populations. MCF-7E cells were transfected with pEGFPc1 (EGFP) orpEGFPc1-survivin (Surv) in the presence and absence of VD3 for 24 h as shown (a–f). Cellcycle distribution of the transfected cells was then analysed by PI staining and flow cytometryvia gating the green cells. The percentages of cells in G1, S and G2/M phases were indicated
Li et al. Page 17
Oncogene. Author manuscript; available in PMC 2010 February 11.
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
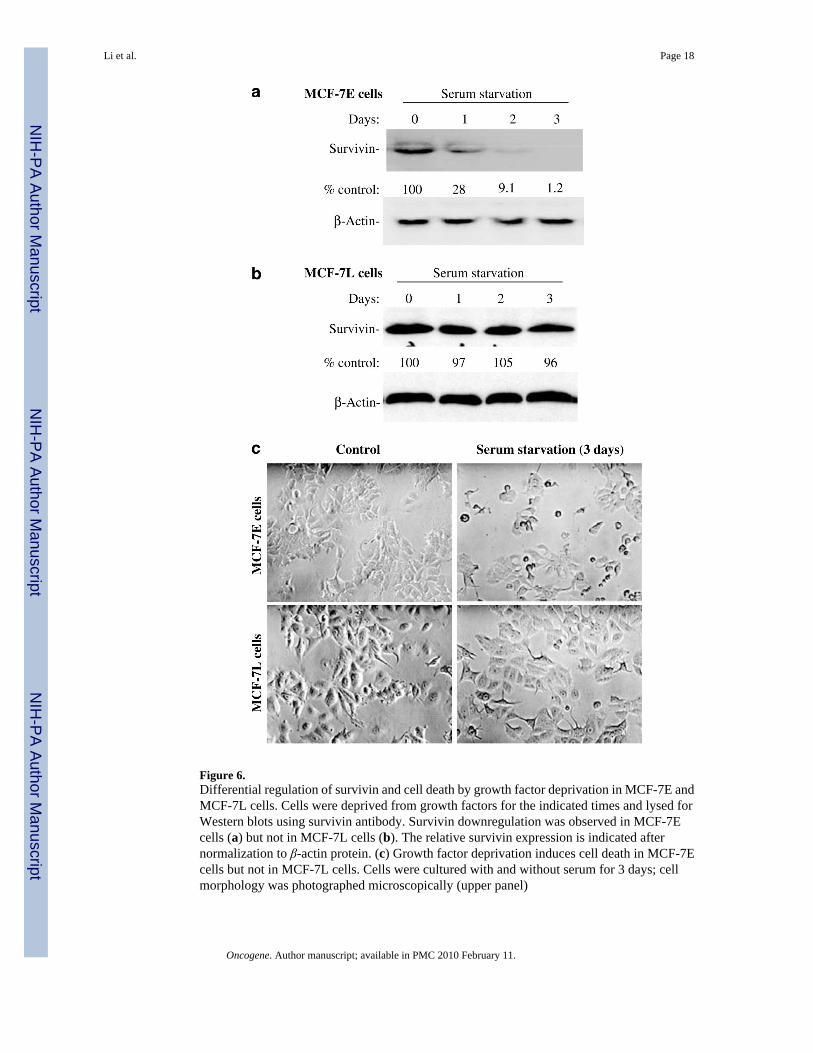
Figure 6.Differential regulation of survivin and cell death by growth factor deprivation in MCF-7E andMCF-7L cells. Cells were deprived from growth factors for the indicated times and lysed forWestern blots using survivin antibody. Survivin downregulation was observed in MCF-7Ecells (a) but not in MCF-7L cells (b). The relative survivin expression is indicated afternormalization to β-actin protein. (c) Growth factor deprivation induces cell death in MCF-7Ecells but not in MCF-7L cells. Cells were cultured with and without serum for 3 days; cellmorphology was photographed microscopically (upper panel)
Li et al. Page 18
Oncogene. Author manuscript; available in PMC 2010 February 11.
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
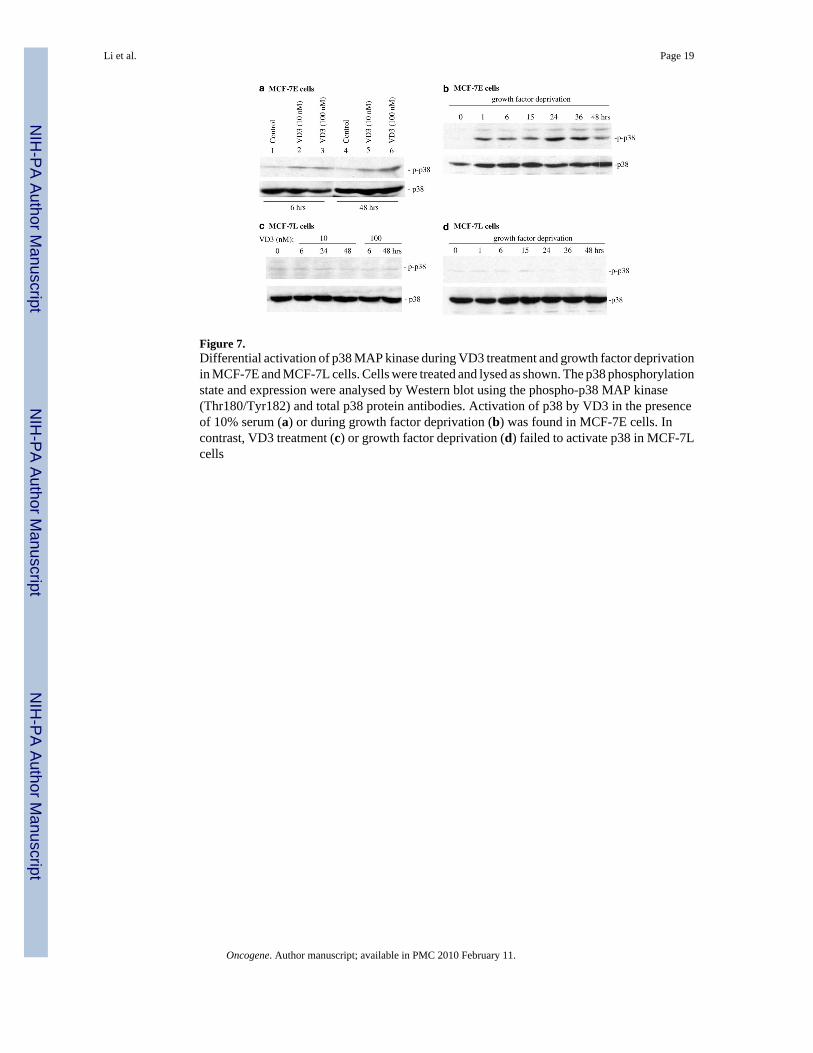
Figure 7.Differential activation of p38 MAP kinase during VD3 treatment and growth factor deprivationin MCF-7E and MCF-7L cells. Cells were treated and lysed as shown. The p38 phosphorylationstate and expression were analysed by Western blot using the phospho-p38 MAP kinase(Thr180/Tyr182) and total p38 protein antibodies. Activation of p38 by VD3 in the presenceof 10% serum (a) or during growth factor deprivation (b) was found in MCF-7E cells. Incontrast, VD3 treatment (c) or growth factor deprivation (d) failed to activate p38 in MCF-7Lcells
Li et al. Page 19
Oncogene. Author manuscript; available in PMC 2010 February 11.
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
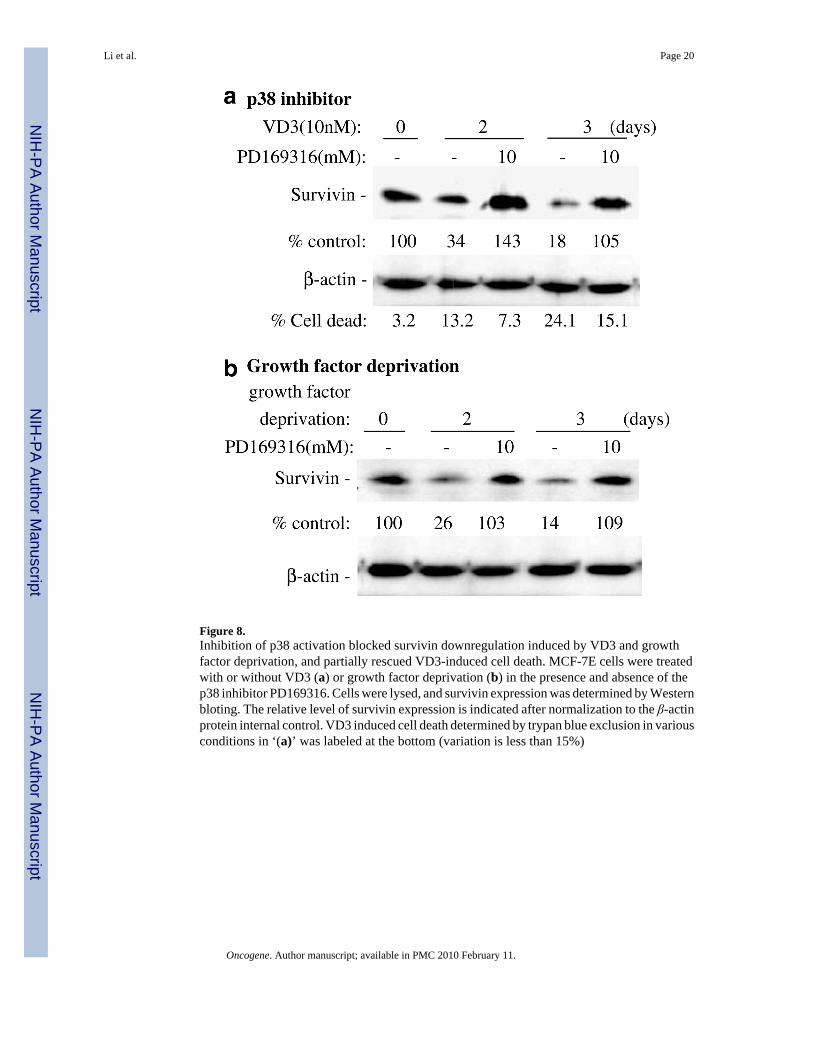
Figure 8.Inhibition of p38 activation blocked survivin downregulation induced by VD3 and growthfactor deprivation, and partially rescued VD3-induced cell death. MCF-7E cells were treatedwith or without VD3 (a) or growth factor deprivation (b) in the presence and absence of thep38 inhibitor PD169316. Cells were lysed, and survivin expression was determined by Westernbloting. The relative level of survivin expression is indicated after normalization to the β-actinprotein internal control. VD3 induced cell death determined by trypan blue exclusion in variousconditions in ‘(a)’ was labeled at the bottom (variation is less than 15%)
Li et al. Page 20
Oncogene. Author manuscript; available in PMC 2010 February 11.
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
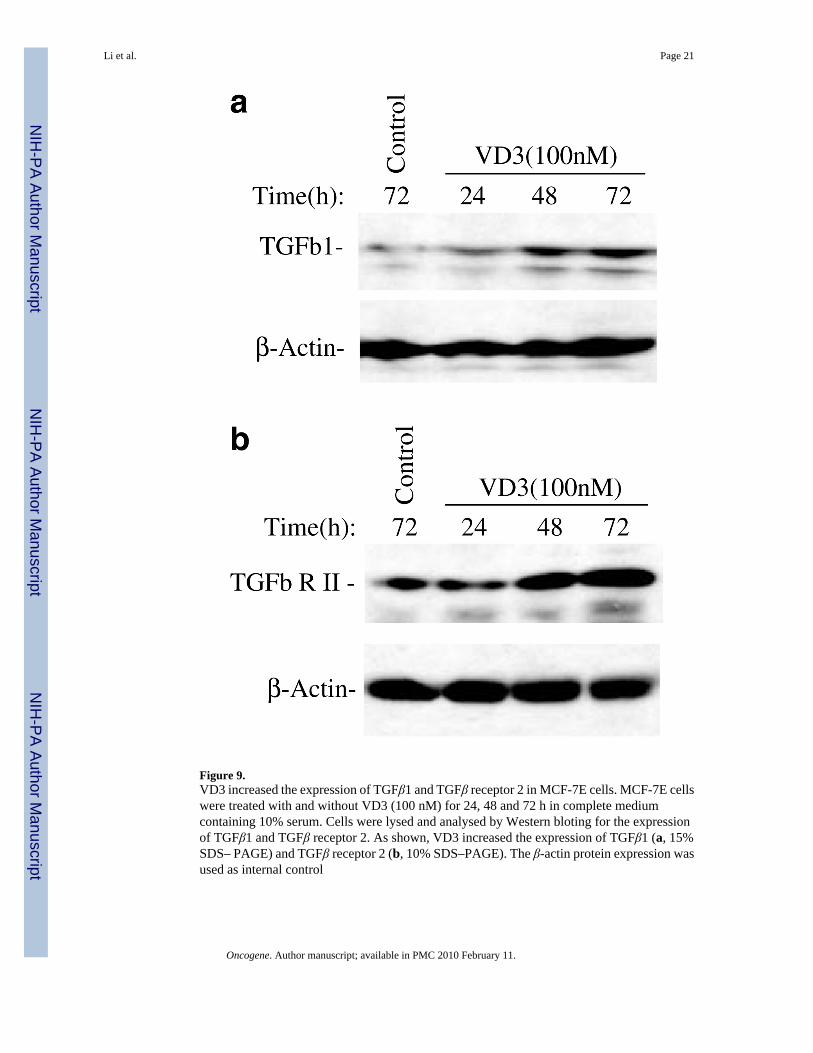
Figure 9.VD3 increased the expression of TGFβ1 and TGFβ receptor 2 in MCF-7E cells. MCF-7E cellswere treated with and without VD3 (100 nM) for 24, 48 and 72 h in complete mediumcontaining 10% serum. Cells were lysed and analysed by Western bloting for the expressionof TGFβ1 and TGFβ receptor 2. As shown, VD3 increased the expression of TGFβ1 (a, 15%SDS– PAGE) and TGFβ receptor 2 (b, 10% SDS–PAGE). The β-actin protein expression wasused as internal control
Li et al. Page 21
Oncogene. Author manuscript; available in PMC 2010 February 11.
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
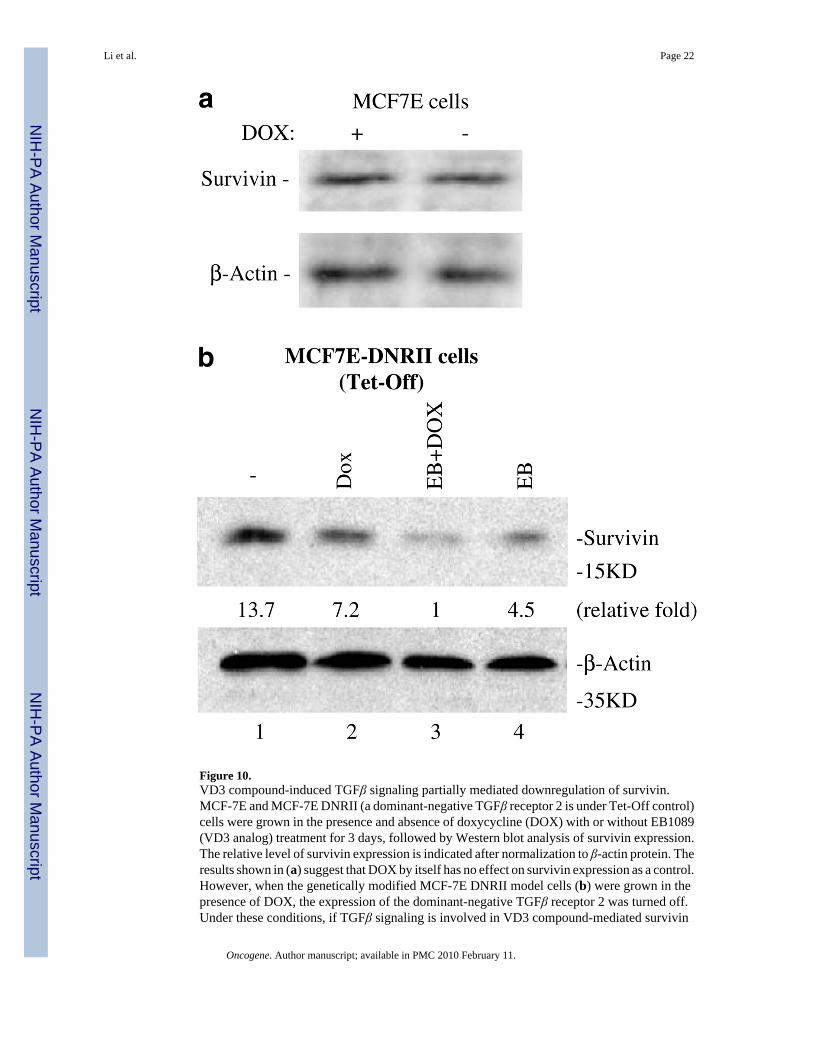
Figure 10.VD3 compound-induced TGFβ signaling partially mediated downregulation of survivin.MCF-7E and MCF-7E DNRII (a dominant-negative TGFβ receptor 2 is under Tet-Off control)cells were grown in the presence and absence of doxycycline (DOX) with or without EB1089(VD3 analog) treatment for 3 days, followed by Western blot analysis of survivin expression.The relative level of survivin expression is indicated after normalization to β-actin protein. Theresults shown in (a) suggest that DOX by itself has no effect on survivin expression as a control.However, when the genetically modified MCF-7E DNRII model cells (b) were grown in thepresence of DOX, the expression of the dominant-negative TGFβ receptor 2 was turned off.Under these conditions, if TGFβ signaling is involved in VD3 compound-mediated survivin
Li et al. Page 22
Oncogene. Author manuscript; available in PMC 2010 February 11.
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript

downregulation, a maximal downregulation of survivin by EB1089 should be obtained (Lane3). In contrast, in the absence of DOX, the expression of the dominant-negative TGFβ receptor2 would be turned on to counter against the function of endogenous wildtype TGFβ receptor2. Under these conditions, EB1089-mediated downregulation of survivin would be diminishedif TGFβ signaling were involved (lane 4 in comparison with lane 3). In addition, using thegenetically modified MCF-7E cells, the different expression of survivin in lane 2 versus lane1 suggests that there is an inherent autocrine TGFβ signaling in MCF-7E cells
Li et al. Page 23
Oncogene. Author manuscript; available in PMC 2010 February 11.
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
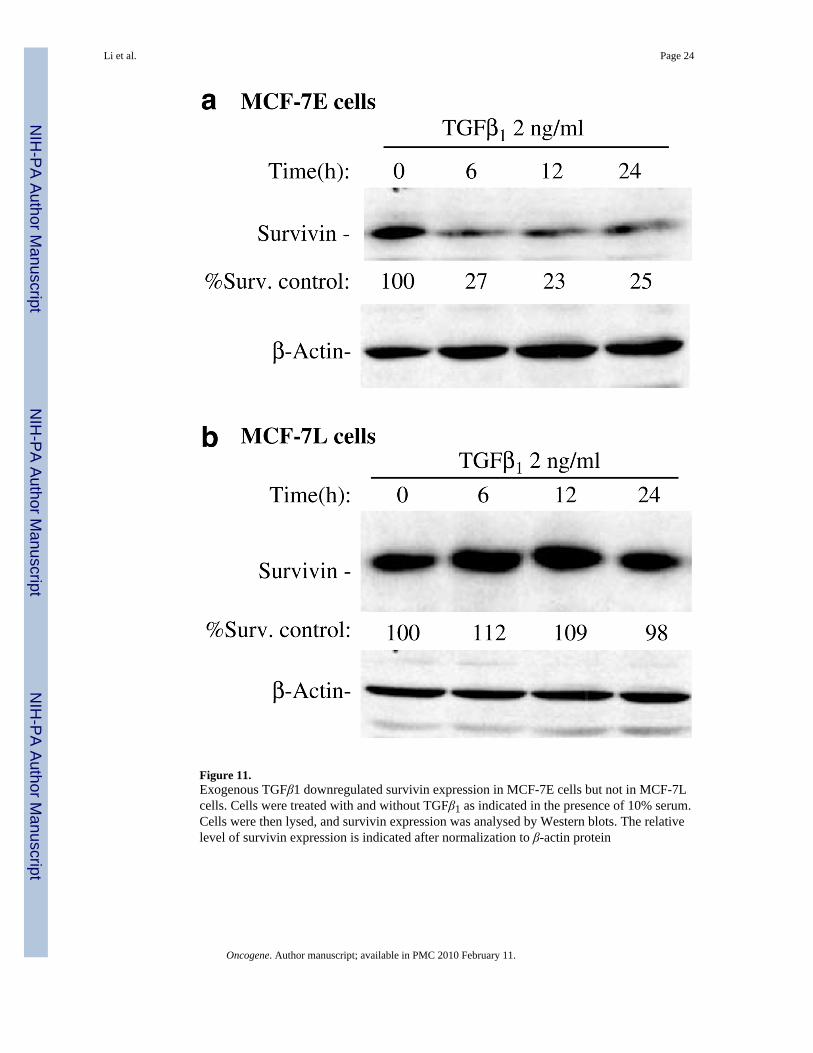
Figure 11.Exogenous TGFβ1 downregulated survivin expression in MCF-7E cells but not in MCF-7Lcells. Cells were treated with and without TGFβ1 as indicated in the presence of 10% serum.Cells were then lysed, and survivin expression was analysed by Western blots. The relativelevel of survivin expression is indicated after normalization to β-actin protein
Li et al. Page 24
Oncogene. Author manuscript; available in PMC 2010 February 11.
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript


























![Influence of cell cycle on responses of MCF7 cells to benzo[a]pyrene](https://img.pdfslide.net/doc/110x75/6313ece23ed465f0570ae658/influence-of-cell-cycle-on-responses-of-mcf7-cells-to-benzoapyrene.jpg)


