Embed Size (px)
Citation preview

E.\p Dermalol IWS: 4: 199-206Prinlcil ill Denmark • All lii^hls reserved
Copyright © Munk.^gaard 1995
Experimental DermatologyISSN 0906-6705
Diminished expression of the extracellulardomain of bullous pemphigoid antigen 2(BPAG2) in the epidermal basementmembrane of patients with generalizedatrophic benign epidermolysis buUosa
Pohla-Gubo G, Lazarova Z, Giudice G J, Liebert M, Grassegger A,Hintner H, Yancey KB. Diminished expression of the extracellular domainof bullous pemphigoid antigen 2 (BPAG2) in the epidermal basementmembrane of patients with generalized atrophie benign epidermolysisbullosa.Exp Dermatol 1995: 4: 199-206. © Munksgaard, 1995
Abstract: Generalized atrophic benign epidermolysis bullosa (GABEB) isa nonlethal form of junetional epidertnolysis bullosa characterized by gen-eralized skin and mucosal blisters that heal with atrophy; other featuresinclude alopecia, nail dystrophy, large melanoeytic nevi, and autosomalrecessive inheritance. The specific aim of this study was to identify anabnormality in epidermal basement membrane adhesion molecules in wellcharacterized GABEB patients that would explain why these subjects"epidermis separates frotn their epidermal basement membrane. Cryostatsections of nonlesional skin from 8 GABEB patients in 5 different familiesas well as skin from normal volunteers (controls) were studied by in-direct immunofluorescence microscopy using rabbit antiserum directedagainst a BPAGl fusion protein or monoclonal antibodies directed againstthe extracellular domain of BPAG2 (HD18 and 233), epiligrin (PlEl),laminin 5 (GB3), types IV and VII collagen, or integrin subunits a:. «3' Pi-af,, or (34. In these studies, monoclonal antibodies HD18 and 233 showedno reactivity and diminished reactivity, respectively, to the epidermal BMof all GABEB patients. Interestingly, in one patient, the absent or dimin-ished reaetivities of monoclonal anti-BPAG2 antibodies were limited to welldemarcated portions of an otherwise intact epidermal basement mem-brane. Moreover, BPAGl, epiligrin, laminin 5, types IV and VII eollagen,and all integrin subunits under study were expressed in the same mannerin both GABEB and normal human skin. These tindings identify anabnormality in the extracellular domain of BPAG2 in the skin of GABEBpatients. BPAG2 (type XVII collagen) is a transmembrane, hemidesmo-some-associated molecule whose extracellular domain resides at the exactlevel where blisters develop in the skin of patients with GABEB. Impair-ment of this adhesion molecule may play a key role in the pathogenesisof this inherited subepidermal bullous disease.
G. Pohla-Gubo', Z. Lazarova^G. J. Giudice^, M. Lieberf*,A. Grassegger^, H. Hintner'and Kim. B. Yancey^'Department of Dermatology, Salzburg, Austria:^Dermatology Branch, National Institutes ofHealth, Bethesda, MD, USA: 'Department otDermatology, Medical College of Wisconsin,Milwaukee, Wl, USA: ^Department of Urology,IVID Anderson Cancer Center, Houston, TX, USA:^Department of Dermatology, Innsbruck, Austria
Key words: cell adhesion - hemidesmosomes -
junctional epidermolysis bullosa
Helmut Hintner, Department of Dermatology,
General Hospital Salzburg, MOIIner Hauptstrasse
48, A-5020 Salzburg, Austria
Accepted for publication 28 February 1995
Abbreviations
GABEB: generalized atrophic benign epidermo-lysis bullosaBM: basement membraneMAb: monoclonal antibody
NHS: normal human skinPBS: phosphate buffered salineFITC: fluorescein isothiocyanateBPAG: bullous pemphigoid antigenEB: epidermolysis bullosa
199

Pohla-Gubo et al.
Introduction
Genertilized atrophic benign epidermolysis bullosa(GABEB) is a form of junctional EB that is in-herited in an autosomal recessive manner (1, 2),GABEB is characterized by widespread and re-peated bullous and erosive lesions of the skin andmucous membranes that typically develop follow-ing minor trauma. Such lesions are present in thesepatients at birth, making it difficult to distinguishthese individuals from those with other forms ofjunctional EB such as the potentially life-threaten-ing Herhtz variant. Unlike the latter group, pa-tients with GABEB typically survive childhoodand develop fewer lesions with age. Moreover, as aconsequence of their disease, these patients acquirea characteristic phenotype consisting of atrophicscars at sites of prior blisters, onychodystrophy,onycholysis, atrophic alopecia, defects of dentalenamel, severe caries, loss of teeth, and the devel-opment of GABEB nevi (1,2), The latter are nevo-cellular nevi that commonly develop in early child-hood; interestingly, they often match the con-figuration of former blisters and may become quitelarge (3),
Light microscopy of lesional skin from patientswith GABEB demonstrates a "subepidermal"split. By electron microscopy or antigen mapping(4) the plane of cleavage has been shown to- bewithin the lamina lucida between the plasma mem-brane of basal keratinocytes and the lamina densa.This junctional blistering is thought to be causedby a defect in hemidesmosomes that are rudimen-tary or absent in patients with this disease (5), Todefine a possible structural defect in these patients'epidermal basement membrane (BM), we havestudied biopsies of clinically normal appearingskin from eight ptitients with GABEB using apanel of antibodies directed against adhesion mol-ecules in basal keratinocytes and epidermal BM(6-15),
Material and methods
Patients
8 patients with GABEB who have been character-ized in detail were subjects for this investigation (1,3), The clinical features in patients 1 through 5 areunchanged from prior descriptions (1, 3); theseadults continue to develop blisters and their seque-lae at sites of minor trauma. Patient no, 6 (case no,8 in ref, (1)) is currently 17 years old and manifeststhe characteristic phenotype of other GABEB pa-tients in this series (i,e,, atrophic scars at sites ofprior blisters, alopecia, and onychodystrophy). Pa-tient no, 7 (case in ref, (3)) is now 16 years of ageand has also developed the same phenotype. All
patients in this group have GABEB nevi. Morpho-logically, these nevi manifest as dermal-shagreen-like plaques (patient nos, 1 through 6) or dark,polycyclic pigmented lesions (patients 7 and 8),These nevi are considered a hallmark of this par-ticular cohort of GABEB patients. All patientswere biopsied on clinically normal appearing skinof the upper and/or lower inner arm for these im-munofluorescence mapping studies. Normal hu-man skin (NHS) was used as control.
Reagents
Antibodies used to assess the expression of variousadhesion molecules in human epidermal BM arelisted in Table 1, The following additional infor-mation is provided about certain key reagents.Rabbit anti-BPAGl antiserum was provided byDr, John R, Stanley (Dermatology Branch, NIH,Bethesda, MD), This antiserum was raised againstFPl, a previously decribed bacterial fusion proteincontaining peptide sequence corresponding to1179 bp of BPAGl cDNA; this region of the mol-ecule extends from the coiled-coiled rod domain(5' end) to the noncoiled region just before the Brepeat (3' end) (15), This antiserum immunopreci-pitates BPAGl from metabolically radiolabeledhuman keratinocytes and binds hemidesmosomesin basal keratinocytes by immunoelectron micro-scopy (15), Two murine monoclonal antibodies(MAbs) directed against the extracellular domainof BPAG2 were employed in this study; both dem-onstrate strong reactivity to normal epidermal BMwith minimal background by indirect immunoflu-orescence microscopy, MAb HDI8 is specificallydirected against the BPAG2 fusion protein GST'-NAl, a bacterial recombinant that corresponds to
Table 1. First-step antibodies and control reagents
Antigen
integrin subunit a3integrin subunit (3,integrin subinut aeintegrin subunit P4integrin subunit a^BPAG1 (BP230)BPAG2 (BP180)BPAG2 (BP180)
epiligrinlaminin 5type iV coilagentype VII collagenhuman IgGi
Antibody
mouse MAbmouse Mabrat MAbmouse MAbmouse MAbrabbit antiserum^"'mouse MAb 233mouse MAb HD18
mouse MAb P1E1mouse MAb GB3mouse MAbmouse MAbmouse MAb
Diiution
1:201:201:201:201:201:201:201:20
1:101:101:201:201:10
Source
Chemicon (Temecuia, CA)ChemioonChemiconChemiconChemiconDr J.R. StanieyDr. K. OwaribeDrs. Liebert and Giudice(manuscript inpreparation)Dr W. CarterSera-Lab (GB)Dakopatts (Denmark)ChemiconSouthern(Birmingham, AL)
Rabbit antiserum directed against a BPAG1 fusion protein.
200

Diminished BPAG2 in GABEB patients
Table 2. Adhesion molecules in the skin of patients with GABEB
Antibody
anti-integrin subunit a^anti-integrin subunit Pianti-integrin subunit a6anti-integrin subunit P4anti-integrin subunit a2anti-BPAG1anti-epiligrin (P1E1)anti-laminin 5 (GB3)anti-fype VII collagenanti-BPAG2 (MAb 233)anti-BPAG2 (MAb HD18)
Pt. no. 155/M
(case no. 1in ref. (1))
+a)
-1-
-t-
++++++
_t l )
Pt. no. 252/M
(case no. 2in ref. (1))
-1-
++++++++
+/-—
Pt. no. 353/F
(case no. 3in ref. (1))
+-t-
++-1-
++++
+ &+/-'='
Patient no./age
Pt. no. 448/F
(case no. 4in ref. (1))
+-1-
-1-
-t-
+++++
+/-
(years) sex (M/F)
Pt. no. 540/M
(case no. 5in ref. (1))
+
+-1-
-1-
-1-
-1-
-1-
++
+1-
Pt. no. 617/M
(case no. 8in ref. (1))
-1-
-1-
-1-
++++4-
++/-
Pt. no. 7
16/F(case inref. (3))
+
++-1-
++++-1-
+/-
Pt. no. 84/F
+
++++++++
+/-
' + indicates staining intensity equal to tbat seen in normal human skin.' + / - indicates markedly reduced staining intensity.
^ Patient no. 3 demonstrates segments of epidermal basement membrane in which BPAG2 stains with the same intensity as that in normal human skin as well
as segments where staining is either markedly diminisbed (MAb 233) or completely absent (HD18). See text for details.
' - indicates no staining.
amino acids 542-665 and 865-890 of this molecule(ref, (6) and Drs, Liebert and Giudice, manuscriptin preparation), MAb 233 reacts with the collagen-ase sensitive ectodomain of BPAG2 as describedpreviously (13),
Imniunofluorescenee tnieroseopy studies
Indirect immunofluorescence microscopy was per-formed as previously described (16), In brief, 5 \xmcryostat sections of NHS or patient skin wereplaced on glass slides and incubated with the first-step antibodies listed in Table 1 for 30 min at roomtemperature, Eollowing three 5-min washes withphosphate buffered saline (PBS), the appropriatesecond-step antibodies were added to skin sectionsfor a 2nd 30-min incubation; slides were washedagain, mounted, and independently evaluated bytwo investigators. Second-step antibodies used inthis study included: fluorescein isothiocyanate(FITC)-conjugated sheep F(ab')2 anti-mouse IgGand IgM (GRUB, Kaumberg, Austria), dilution1:40; EITC-goat E(ab')2 anti-mouse IgG (Tago,Inc, Burlingame, CA), dilution 1:40; EITC-goatE(ab')2 anti-rat IgG (Tago, Inc), dilution 1:80; andEITC-goat F(ab')2 anti-rabbit IgG (Tago, Inc), di-lution 1:80, Controls included the replacement ofthe first-step antibodies by an irrelevant controlMAb (specifically, murine monoclonal anti-humanIgGi [Southern Biotechnology Associates, Bir-mingham, AL], dilution 1:10), or normal rabbitserum (in the case of rabbit anti-buUous pemphig-oid antigen 1 [BPAGl]), All first-step antibodies
(and controls) were used at high concentrations toassure detection of target epitopes.
Results
The antibodies employed in this study were se-lected to evaluate the expression of adhesion mol-ecules in basal keratinocytes and the lamina lucida,lamina densa, and the sublamina densa region ofhuman epidermal BM, In normal human skin,integrin subunits a2, a3, and Pi were presentaround the periphery of basal keratinocytes, Inte-grin subunit a^ was faintly expressed on the per-iphery of basal and immediately suprabasal kera-tinocytes, Integrin subunits af, and P4, BPAGl,BPAG2, epiligrin, laminin 5, type IV collagen, andtype VIl collagen were all present in a bright andcontinuous linear pattern in BM of normal humanskin using reagents at the concentrations specifiedin Table 1,
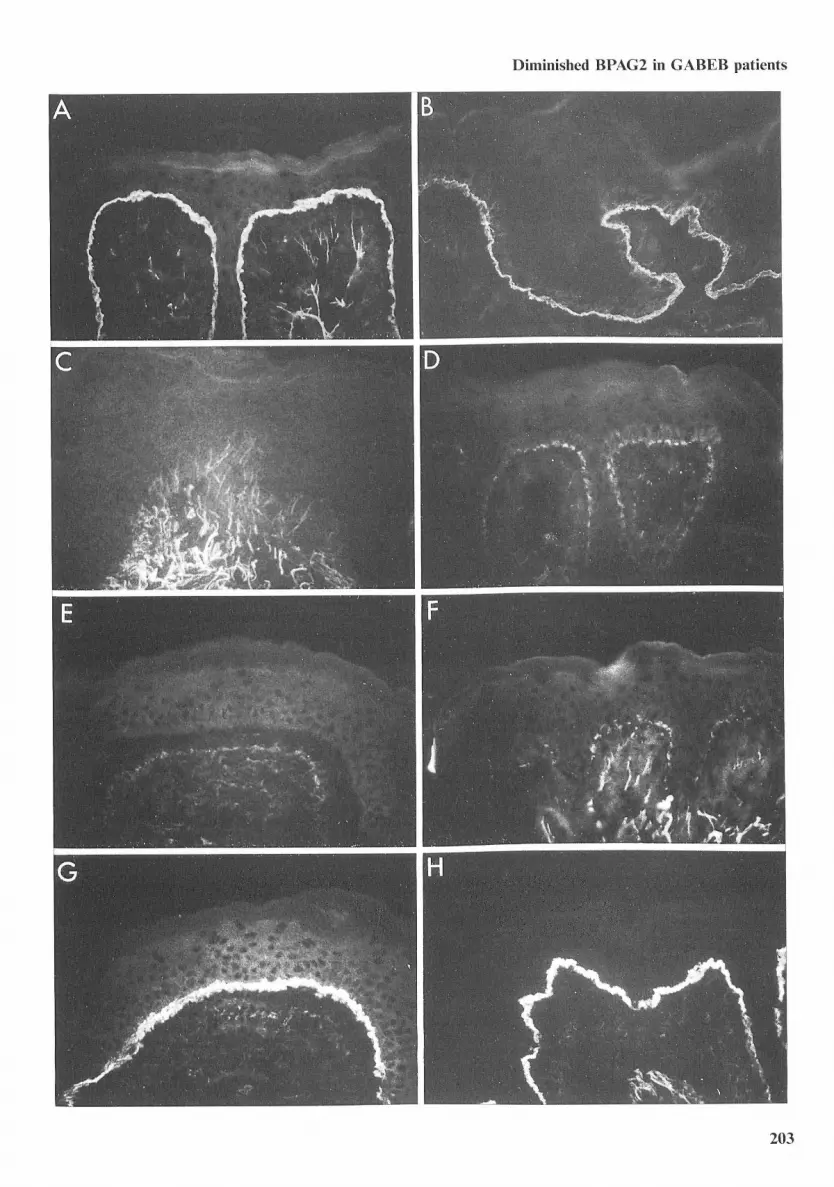
BPAGl, epiligrin, laminin 5, types IV and VIIcollagen, and integrin subunits ao, a^. Pi, a^, andp4 were expressed in the skin of all GABEB pa-tients with the same intensity and distribution asthat found in normal human skin (Table 2), Incontrast, MAbs HD 18 and 233 directed againstthe extracellular domain of BPAG2 showed no re-activity or markedly diminished reactivity, respec-tively, to the epidermal BM of skin samples fromall patients with GABEB (Eig, 1, Table 2), Interest-ingly, in one patient (specifically patient 3, a sisterof 3 other patients in this study), the absent ordiminished reactivities of monoclonal anti-BPAG2
201

Pohla-Gubo et al.
antibodies were limited to well demarcated por-tions of epidermal BM (Eig, 2), Interveningstretches of epidermal BM in this patient's skinshowed BPAG2 reactivity that was indistinguish-able from that found in normal human skin (Eig,2), Careful studies of sequential sections of patient3's skin demonstrated that areas of epidermal BMdevoid of BPAG2 contained normal amounts oflaminin 5, epiligrin, and type VII collagen - find-ings indicating that the lack of BPAG2 reactivitywas not due to a frank interruption of the epider-mal BM or an artifact secondary to tissue pro-cessing (Eig, 2), This unique pattern was observedin two separate biopsies from this patient. In allother GABEB patients, the absent or diminishedreactivity of BPAG2 was seen throughout their en-tire epidermal BM; this impairment was docu-mented in a total of eleven biopsies from theseeight patients.
When first-step antibodies were replaced by anirrelevant antibody (or serum) for control pur-poses, there was no reactivity in skin sections fromany GABEB patients or normal volunteers.
Discussion
Epidermolysis bullosa (EB) is the term applied toa group of inherited mechanobuUous disorderscharacterized by fragility of the skin and mucousmembranes (17), In the past few years disease genecandidates and distinct mutations have been iden-tified in patients with three different types of EB,In patients with EB simplex, point mutations havebeen identified in genes encoding keratins 5 and14, the predominant keratins in basal keratinocytes(18-22), Such mutations are thought to causealterations in the intermediate filament cytoskel-eton of basal keratinocytes that result in intraepid-ermal blisters in patients with this disease (23), Inthe dystrophie forms of EB, linkage analysis ofaffected kindreds identified type VII collagen as aleading disease gene candidate (24), Subsequentstudies of the gene encoding this collagen in pa-tients with dominant or recessive forms of dys-trophie EB identified specific mutations that areheld responsible for impaired anchoring fibril for-
mation and consequent blisters in the sublaminadensa area (24-27), In patients with Herlitz's junc-tional EB, immunofluorescence microscopy studiesfound absent or tnarkedly diminished expressionof a matrix protein normally present in the laminalucida of human epidermal BM (9, 11, 28, 29),This matrix protein has recently been identified aslaminin 5, a laminin isofortn that is associatedwith anchoring filaments and located at the exactultrastruetural site where blisters form in patientswith Herlitz's disease (7, 14), Pursuing the hypo-thesis that this alteration plays a key role in thepathogenesis of this form of junctional EB, a re-cent study has linked the Herlitz's disease pheno-type in 4 families to the gene encoding a specificlaminin 5 subunit polypeptide (i, e,, laminin sub-unit 72) (30), Moreover, specific mutations havebeen identified in the laminin 72 gene of some pa-tients with Herlitz's disease (30, 31), These muta-tions are thought to cause impairments in thestructure and function of anchoring filaments thatresult in blisters in the lamina lucida of these pa-tients' epidermal BM, Interestingly, disease linkageto laminin 5 subunit 72 has been excluded in otherHerlitz's disease families indicating that an abnor-tnality in another laminin 5 subunit or some otheradhesion molecule is responsible for the same dis-ease phenotype in these individuals (30, 32),Studies attetnpting to link such cases of Herlitz'sjunctional EB to a defect in a specific gene are cur-rently in progress,
GABEB is a variant form of junctiontil EB (1,2), Though adults with this disease have a charac-teristic phenotype, neonates with GABEB areoften indistinguishable from patients with themore serious and often life-threatening Herlitz'sjunctional EB, In this study, we have demonstratedthat two different tnonoclonal antibodies directeda'gainst the extracellular domain of BPAG2 showeither no reactivity or diminished reactivity to theepidermal BM of patients with GABEB, More-over, we have also demonstrated that (unlike pa-tients with Herhtz's disease) GABEB patients ex-press normal amounts of laminin 5 in their epider-mal BM, Other disease candidates (specifically,BPAGl, epiligrin, integrin subunits a,, pi, a ,, and
Fig. I. tndirect immunotluorescence microscopy of cryostat sections of normal tuiman skin (A and B) or clinically normal appearingskin from patients witti GABEB (C thirougti H) using MAbs HDI8 (A, C, and E) and 233 (B, D, and F) directed against tlieextracellular domain of BPAG2 as well as monoclonal anti-laminin 5 antibody GB3 (G) and monoclonal anti-epiligrin antibodyPI El (H). All first-step antibodies were used at high concentrations to assure detection of target epitopes. Bright continuous stainingof BPAG2 was noted in the epidermal BM of normal human skin (A and B). In contrast, there was no evidence of HD18 bindingto the epidermal BM of patients with GABEB (C and E) and markedly diminished staining of GABEB patients" skin with MAb233 (D and F). Antibodies directed against laminin 5 (G) and epiligrin (H) as well as BPAGl, type IV collagen, type Vlt collagen,and integrin subunits a2, aj . Pi, a ,, and P4 (data not shown) were normally expressed in the skin of all GABEB patients.
202
Diminished BPAG2 in GABEB patients
203

Pohla-Gubo et al.
Fig. 2. Indirect immunofluorescence microscopy of cryostat sections of clinically normal appearing skin from GABEB patientnumber 3 (A through D). In this patient, the lack of reactivity of monoclonal anti-BPAG2 HD18 (A through C) as well as themarkedly diminished reactivity of MAb 233 (data not shown) was limited to well demarcated portions of epidermal BM; interveningstretches of epidermal BM show staining that is indistinguishable from that found in normal human skin. Studies of sequentialsections of this patient's skin show that sites devoid of BPAG2 contain normal amounts of laminin 5 (D) as well as epiligrin andtype VII collagen (data not shown). This same pattern was observed in 2 separate biopsies from this patient.
P4 as well as other BM proteins) were also found tobe expressed nortnally in these patients' epidermalBM, Impaired expression of BPAG2 in GABEBpatients is of particular interest sinee prior ultra-structural studies of these patients' skin haveshown absent or minitnal numbers of hemidesmo-somes - the basal keratinocyte adhesion unit inwhich BPAG2 is localized. Moreover, BPAG2 is atransmembrane adhesion molecule whose extra-cellular domain is localized to the same level whereblisters develop in patients with GABEB (33, 34),In recent preliminary studies, Jonkman et al haveused yet another monoclonal antibody directedagainst the extracellular domain of BPAG2 todemonstrate impaired expression of this adhesionmolecule in the epidermal BM of 1 patient withGABEB (J Invest Dermatol 1994: 102:609, abstr,).Interestingly, these investigators found thatBPAG2 was expressed normally in two patients
204
with Herlitz's disease and one individual with an-other junetional EB variant. Similarly, we havefound that monoclonal antibodies HD18 and 233detect BPAG2 in the epidertnal BM of two patientswith EB simplex (one with localized disease (i,e,,Weber-Cockayne) and one with generalized disease(i,e,, Koebner), data not shown). In sum, findingsin these two studies suggest that impaired ex-pression of BPAG2 tnay be specific to patients withGABEB,
The correlates discussed above suggest thatBPAG2 is a relevant disease candidate for patientswith GABEB, While it is tetnpting to suggest thatimpaired expression of BPAG2 is the pritnary de-fect in patients with this inherited bullous disease,additional studies are required to detnonstrate thatthis alteration is not secondary to some other ab-normality. Potential primary defects that might ex-plain secondary loss of BPAG2 epitopes in the skin

Diminished BPAG2 in GABEB patients
of GABEB patients iticlude impairment in the syn-thesis or processing of this molecule, overexpres-sion of a protease that destroys or alters this pro-tein, or aberrant expression of another matrix ele-ment that perturbs or tnasks normal expression ofBPAG2 epitopes. Moreover, the diminished ex-pression of BPAG2 epitopes observed in these in-direct immunofiuoi"escence microscopy studiesdoes not tiecessarily indicate absetiee of this pro-tein in the skin of patients with GABEB, Certainlyfindings with MAb 233 as well as a polyclotial anti-serum against the ectodomain of BPAG2 (Pohla-Gubo et al, J Invest Dermtaol 1994: 102:562,abstr,) indicate that some BPAG2 epitopes arepresent in the skin of GABEB patients. Nonethe-less, partial loss of critical BPAG2 detertninants asa consequence of sotne other primary abnortnalitycould still account for blister fortnation in patientswith GABEB, In brief, fvn'ther studies are requiredto detertnine if these alterations are the cause orthe result of this inherited disease.
Other adhesion tnolecules within the lamina lu-cida of epidermal BM lepresent logical diseasecandidates for patients with GABEB, One suchcandidate is uncein (i,e,, 19-DEJ-l), Uticein is ahemidesmosotne-anchoring filament associatedcotnplex that has been shown to be defectively ex-pressed in the epidermal BM of a large number ofpatients with various fortns of junctional EB (aswell as sotne patients with recessive dystrophie EB)(35), The significance of itnpaired expression of un-cein in the skiti of junctional EB patients (just asour findings of impaired expression of BPAG2 inpatients with GABEB) awaits clarification at themolecular level. It is plausible that such studiesnicty identify mutations in diffet"ent proteins thatgive rise to similar phenotypes as well as mutationsat different sites withiti the same gene that resultin disease phenotypes of varying severity. Withinthis context, the fact that GABEB patients in thecurrent study are located in a comtnon geographicregion increases the likelihood that their defect isshared (and potentially unique).
In one of our patients, the impaired expressionof BPAG2 was restricted to segmental portions ofotherwise intact epidermal BM - an unusual find-ing that was confirtned in two separate biopsies ofnormal appearing skin from this patient. Clin-ically, this patient was indistinguishable from otherGABEB patients in this group, including three sib-lings who all demonstrated impaired expression ofBPAG2 throughout their epidermal BM, The sig-nificance of the segtnental impairtnent in BPAG2expression in this patient is unknown. Theoreticalexplanations include a form of tnosaicistn causedby a reverse or back mutatioti in sotnatic cells thatresult in sectorial expression of this protein in epi-
dermal BM, Alternatively, if there are different re-cessive tnutations on each allele of this individual,an utiequal crossover event during sotnatic mitosis(e,g,, a chromatid or partial chroniatid exchange)tnight allow reconstitutioti of a normal allele in apopulation of cells that could subsequently expressthis protein normally. It will be interesting to seeif this pattern etnerges as other patients with GAB-EB are similarly studied. In any event, the nonreac-tivity or ditninished reactivity of tnonoclonal anti-bodies directed against the extracellular domain ofBPAG2 as well as the normal expression of latninin5 in the epidermal BM of piatients with GABEBare findings that should facilitate the early distinc-tion of newborns with this form of junctional EBfrom those with Herlitz's disease. Moreover, thesefindings represent another example (36-38) wherean adhesion tnolecule in human epidertnal BM (inthis case BPAG2) is defectively expressed in theskin of patients with an inherited bullous disease(i,e,, GABEB) and targeted by autoantibodies inpatients with acquired autoimmune bullous dis-eases (i,e,, bullous petnphigoid, herpes gestationis).Such abnortnalities attest to the itnportance ofBPAG2 in maintaining adhesion of the epidermisto the epidertnal BM, Euture studies ofthe BPAG2gene in patients with GABEB should further ourunderstanding of epidertnal biology in health anddisease.
References
1. Hintner H, Wolff K. Generalized atrophic benign epiderm-olysis bullosa. Arch Dermatol 1982: 118; 375-384.
2. Paller AS, Fine JD, Kaplan S, Pearson RW. The generalizedatrophic benign form of junctional epidermolysis bullosa.Arch Dermatol t986; 122; 704-7t0.
3. Grubauer G, Hintner H, Klein G, Fritseh P. Erworbene,flachige Riesen-Nitvuszellnavi bei generalisierter, atrophisi-erender, benigner Epidertnolysis bullosa. Hautarzt 1989;40; 523-526.
4. Hinttier H, Stingl G, Schuler G, Fritseh P, Stanley J, KatzS, Wolff K. Immunotluorescence mapping of antigenic de-terminants within the dermal epiderrnal junction in mech-atiobullous diseases. ,1 Invest Dertnatol 1981; 76; 113-118.
5. Tidman M,I, Eady RAJ. Hemidesmosome heterogeneity injunctionat epidermolysis bullosa revealed by morphometricanalysis. J Invest Dermatol 1986; 86; 51-56.
6. Giudice GJ, Emery DJ, Zelickson BD, Anhalt GJ, Lui Z,Diaz EA. Bullous pemphigoid and herpes gestationis auto-antibodies recognize a common non-coUagenous site on theBP180 ectodomain. J Immunol 1993; 15l" 5742-5750,
7. Rouselle P. Lunstrutn GP, Keene DR, Burgeson RE. Kal-inin: an epithelium-speeilic basement membrane adhesionmolecule that is a component of anchoring filaments. J CellBiol 199t: tt4; 567-576.
8. Carter WG, Ryan MC, Gahr PJ. Epiligrin, a new adhesionligand for integrin a^p, in epithelial basement membranes.Cell 1991; 65; 599-610.
9. Verrando P, Blanchet-Bardon C, Pisani A, Thotnas L, Cam-bazard F, Eady RAJ, Schotield O, Ortonne JP. Monoclonalantibody GB3 defines a widespread defect of several base-
205

Pohla-Gubo et al.
ment tnembranes and a keratinocyte dysfunetion in pa-tients with lethal junctional epidertnolysis bullosa. Eab In-vest 1991: 64: 85-92.
10. Verrando P, Hsi BL, Yeh CJ, Pisani A, Serieys N, OrtonneJP. Monoclonal antibody GB3, a new probe for the studyof human basement metnbranes and hemidesmosomes. ExpCell Res 1987: 170: 116-128.
11. Verrando P, Scholfield O, Ishida-Yamamoto A, AberdamD, Partouche O, Eady RAJ, Ortonne JP Nieein (BM-600)in junctional epidermolysis bullosa: polyelonal antibodiesprovide new clues for pathogenic fole. J Invest Dermatol1993: 101: 738-743.
12. Jonktnan MF, De Jong MCJM, Heeres K, Sonnenberg A.Expression of integrin a(,P4 in junetional epidermolysis bul-losa. J Invest Dermatol 1992: 99; 489-496.
13. Nishizawa Y, Uetnatsu J, Owaribe K. HD4, a 180 kDa bull-ous pemphigoid antigen, is a major transmetnbrane glyco-protein ofthe hemidesmosome. J Bioehem 1993; 113; 493-501.
14. Marinkovich MP, Verrando P, Keene DR, Meneguzzi G,Lunstrutn GP, Ortonne JP, Burgeson RE. Basement mem-brane proteins kalinin and nieein are structurally and im-munologically identical. Lab Invest 1993; 69; 295-299.
15. Tanaka T, Korman N, Shimizu H, Eady RAJ, Kalus-Kov-tun V, Cehrs K, Stanley JR. Production of rabbit antibodiesagainst carboxy-terminal epitopes encoded by bullous pem-phigoid cDNA. J Invest Dermatol 1990; 94; 617-623.
t6, Dotnloge-Hultsch N, Bisalbutra R Gammon WR, YanceyKB. Direct immunofluorescence microscopy of 1 tnol/L so-dium chloride-treated patient skin. J Atn Acad Dermatol1991; 24; 946-951,
17. Fine JD, Bauer EA, Briggaman RA, Carter DM, EadyRAJ, Esterly NB, Holbrook KA, Hurwitz S, Johnson L,Lin A, Pearson R, Sybert VP. Revised clinical and labora-tory criteria for subtypes of inherited epidermolysis bullo-sa. J Am Acad Dermatol 1991; 24; It9-135.
18. Bonifas JM, Rothman AL, Epstein EH. Epidermolysis bul-losa simplex; evidence in two families for keratin gene ab-normalities. Science 1991; 254; 1202-1205.
19. Coulombe PA, Hutton ME, Letai A, Hebert A, Paller AS,Fuchs E. Point mutations in human keratin 14 genes ofepidermolysis bullosa simplex patients; genetic and func-tional analyses. Cell 1991; 66; 1301-1311.
20. Eane EB, Rugg EL, Navsaria H, Leigh IM, HeagertyARM, Ishida-Yamamoto A, Eady RAJ. A mutation in theconserved helix termination peptide of keratin 5 in heredi-tary skin blistering. Nature 1992; 356; 244-246.
21. Hovnanian A, Pollack E, Hilal L, Rochat A, Prost C, Bar-randon Y, Goossens M, A missense mutation in the roddomain of keratin 14 associated with recessive epidermo-lysis bullosa simplex. Nature Genet 1993; 3; 327-332.
22. Chan YM, Yu QC, Fine JD, Fuchs E. The genetic basis ofWeber-Cockayne epidermolysis bullosa simplex. Proe NatlAead Sci USA 1993: 90: 7414-74t8.
23. Epstein EH, Jr. Molecular genetics of epidertnolysis bullo-sa. Science 1992: 256; 799-804.
24. Hovnanian A, Christiano A, Uitto J. The molecular gen-etics of dystrophie epidermolysis bullosa. Arch Dermatol1993; 129; 1566-t570.
25. Christiano AM, Greenspan,DS, Hoffmann GG, Zhang X,Tamai Y, Lin AN, Dietz HC, Hovnanian A, Uitto J. Amissense mutation in type VII collagen in recessive dys-trophie epidermolysis bullosa. Nature Genet 1939; 4; 62-66.
26. Hilal L, Roehat A, Duquesnoy P, Blanchet-Batdon C,Wechsler J, Martin D, Christiano AM, Barrandon Y, UittoJ, Goossens M, Hovnanian A. A hotnozygous insertion-deletion in the type VII collagen gene (COL7A1) inHallopeau-Siemens dystrophie epidermolysis bullosa. Na-ture Genet 1993; 5; 287-293.
27. Christiano A, Ryynanen M, Uitto J. Dominant dystrophieepidermolysis bullosa results from a glycine substitution inthe triple helical dotnain of the type VII collagen gene(COL7At). J Itivest Dermatol 1994; 102-559.
28. Heagerty AHM, Kennedy AR, Eady RAJ, Hsi B-L, Ver-rando P Yeh C-J, Ortonne J-P. GB3 tnonoclonal antibodyfor diagnosis of junctional epidertnolysis bullosa. Lancett986; 8485; 860.
29. Meneguzzi G, Marinkovich MP Aberdam D, Burgeson RE,Ortonne J-P. Abnormal expression of kalinin in the Her-litz's JEB epithelial basement membrane. Exp Dertnatol1992; 1; 221-229,
30. Aberdam D, Galliano MF, Vailly J, Pulkkinen L, BonifasJ, Christiano AM, Tryggvason K, Uitto J, Epstein EH, Or-tonne JP, Meneguzzi G. Herlitz's junctional epidermolysisbullosa is linked to mutations in the gene (LAMC2) for the72 subttnit of nieein/kalinin (LAMININ-5). Nature Genet1994; 6; 299-304.
31. Pulkkinen L, Christiano AM, Airenne T, Kaakana H,Tryggvason K, Uitto J. Mutations in the Y2 chain gene(LAMC2) of kalinin/laminin 5 in the junetional forms ofepidermolysis bullosa. Nature Genet 1994; 6; 293-298.
32. Baudoin C, Miquet C, Blanchet-Bardon C, Gatnbini C,Meneguzzi G. Herlitz junetional epidermolysis bullosakeratinoeytes display heterogeneous defects of nieein/kal-inin gene expression. J. Clin Invest 1994; 93; 862-869.
33. Diaz L, Ratrie H, Saunders WS, Futamura S, SquiqueraHL, Anhalt GJ, Giudiee GJ. Isolation of a human epider-mal cDNA corresponding to the 180-kD autoantigen tec-ognized by bullous petnphigoid and herpes gestationis sera.J Clin Invest 1990; 86; 1088-1094.
34. Giudice GJ, Emwery DJ, Diaz LA. Cloning and pritnarystructural analysis of the bullous pemphigoid autoantigen,BPl80. J Invest Dermatol 1992; 99; 243-250.
35. Fine JD, Horiguchi Y, Couehman JR. t9-DEJ-t, a hemi-desmosotne-anehoring filament cotnplex-assoeiated mono-clonal antibody. Areh Dermatol 1989; 125; 520-523.
36. Domloge-Hultsch N, Gatnmon WR, Briggaman RA, GilSG, Carter WG, Yancey KB. Epiligrin, the tnajor hutnankeratinocyte integrin ligand, is a target in both an acquiredautoimmune disease and an inherited subepidertnal blis-tering skin disease. J Clin Invest 1992; 90; 1628-1633.
37. Yaneey KB. Adhesion molecules; interactions of keratino-cytes with epidermal basement tnembrane. Prog Dermatol1994: 28: l-t2.
38. Yancey KB. From bedside to bench and back: The diag-nosis and biology of bullous diseases. Arch Dermatol 1994:130; 983-987.
206




























