Embed Size (px)
Citation preview

B
SNB
Neurobiology of Disease 8, 405–418 (2001)doi:10.1006/nbdi.2001.0385, available online at http://www.idealibrary.com on
A
Distinct Behavioral and NeuropathologicalAbnormalities in Transgenic Mouse Modelsof HD and DRPLA
Gabriele Schilling,* Hyder A. Jinnah,† Vicky Gonzales,*Michael L. Coonfield,* Yujin Kim,‡ Jonathan D. Wood,‡
Donald L. Price,* ,†,§ Xiao-Jiang Li,¶ Nancy Jenkins,\
Neal Copeland,\ Timothy Moran,‡ Christopher A. Ross,†,‡,§ andDavid R. Borchelt* ,§
*Department of Pathology, †Department of Neurology, ‡Department of Psychiatry andehavioral Sciences, and §Department of Neuroscience, The Johns Hopkins University School
of Medicine, Baltimore, Maryland, 21205; ¶Department of Genetics, Emory Universitychool of Medicine, Atlanta, Georgia 30322; and Mouse Cancer Genetics Program,ational Cancer Institute, Frederick Cancer Research & Development Center,ldg. 539, Room 229, Frederick, Maryland 21702-1201
Received September 26, 2000; accepted January 11, 2001; published online May 3, 2001
Huntington’s disease (HD) and Dentatorubral and pallidoluysian atrophy (DRPLA) are autosomaldominant, neurodegenerative disorders caused by the expansion of polyglutamine tracts in theirrespective proteins, huntingtin and atrophin-1. We have previously generated mouse models of thesedisorders, using transgenes expressed via the prion protein promoter. Here, we report the first directcomparison of abnormalities in these models. The HD mice show abbreviated lifespans (4–6 months),hypoactivity, and mild impairment of motor skills. The DRPLA mice show severe tremors, are hyper-active, and are profoundly uncoordinated. Neuropathological analyses reveal that the distribution ofdiffuse nuclear immunolabeling and neuronal intranuclear inclusions (NII’s), in the CNS of bothmodels, was remarkably similar. Cytoplasmic aggregates of huntingtin were the major distinguishingneuropathological feature of the HD mice; mutant atrophin-1 accumulated/aggregated only in thenucleus. We suggest that the distinct behavioral and neuropathological phenotypes in these micereflect differences in the way these mutant proteins perturb neuronal function. © 2001 Academic Press
INTRODUCTION
HD and DRPLA are autosomal dominant, progres-sive disorders caused by expansions of polyglutaminetracts in their respective proteins, huntingtin (Hun-tington’ s Disease Collaborative Group, 1993) and at-rophin-1 (Koide et al., 1994; Nagafuchi et al., 1994). HDis caused by expansions of more than 36 CAG’s inhuntingtin (Rubinsztein et al., 1996), whereas DRPLAis associated with expansions of more than 49 CAG’sin atrophin-1 (Koide et al., 1994). Both diseases shareseveral features but the patterns of cell death differ(Albin et al., 1995; Ross et al., 1997a,b). In HD postmor-
0969-9961/01 $35.00Copyright © 2001 by Academic Press
ll rights of reproduction in any form reserved. 405
tem brain, caudate and putamen are the most affectedareas, whereas in DRPLA postmortem brain, cerebel-lar dentate nucleus, subthalamic nucleus, and globuspallidus display the most severe degeneration of neu-rons (Ross et al., 1997a). Postmortem brains from pa-tients with either disease show neuronal intranuclearinclusions composed of apparently aggregated poly-glutamine proteins (DiFiglia et al., 1997; Becher et al.,1998; Hayashi et al., 1998; Igarashi et al., 1998).
Neuronal intranuclear inclusions (NII’s) were firstdescribed in transgenic mice expressing the first exonof huntingtin with 115–156 CAG’s (Mangiarini et al.,1996; Davies et al., 1997). Subsequently, NII’s were

1atCNbaac
smdfod86tetmaaefmhmutata
406 Schilling et al.
A
identified in several transgenic mouse models of poly-glutamine diseases (Ordway et al., 1997; Skinner et al.,997; Reddy et al., 1998; Hodgson et al., 1999; Schilling etl., 1999a,b) and from patients with a variety of polyglu-amine disorders (DiFiglia et al., 1997; Becher et al., 1998;ummings et al., 1998; Igarashi et al., 1998). The impact ofII’s on the pathogenesis of polyglutamine diseases has
een a point for much speculation. Whether aggregatesre protective, inert, or toxic for the cells in which theyccumulate is unclear (Sisodia, 1998). In vitro, proteinsontaining long polyglutamine repeats aggregate into
b-amyloid like structures (Scherzinger et al., 1997; Scher-zinger et al., 1999), via a mechanism that may involve theformation of polar zippers (Perutz, 1999). These aggre-gates may sequester factors that regulate transcriptionand, therefore, may disrupt gene expression (Lin et al.,2000; Luthi-Carter et al., 2000).
In HD, DRPLA, and other polyglutamine diseases, theproteins encoding the mutations are widely expressed(Li et al., 1993; Margolis et al., 1996). Hence, it has beenuggested that either the toxicity of polyglutamines isodulated by adjacent protein sequences, or that subtle
ifferences in the levels of mutant gene expression con-er the selectivity of neurodegeneration. We have previ-usly described two transgenic models of polyglutamineiseases, one for HD (Schilling et al., 1999a: HD-N171-2Q) and one for DRPLA (Schilling et al., 1999b: AT-FL-5Q). In this study, we take advantage of the fact that thewo transgenic mouse models for polyglutamine dis-ases express different polyglutamine proteins under theranscriptional regulation of the same prion protein pro-
oter to directly compare the evolution of behavioralnd neuropathological abnormalities. The age of appear-nce and distribution of nuclear aggregates in both mod-ls was virtually identical. The major neuropathologicaleature that distinguishes the HD mice from the DRPLA
ice was neuronal cytoplasmic aggregates of mutantuntingtin in the HD mice. Because these two modelsanifest distinct behavioral abnormalities, despite the
se of a common transcription promoter, we concludehat aspects of the biology of these mutant proteins,part from when or where they are expressed, modulatehe manifestations of behavioral and neuropathologicalbnormalities.
MATERIALS AND METHODS
Behavioral Analysis of HD and DRPLA TransgenicMice
Six HD transgenic mice at 5 months of age andage-matched nontransgenics (or six DRPLA mice at 12
Copyright © 2001 by Academic Pressll rights of reproduction in any form reserved.
months and age-matched nontransgenics), were testedin an automated optical animal activity system (Digis-can RXYZCM, Columbus Instruments, OH) for hori-zontal activity and total distance. Mice were trans-ferred to the activity chamber two hours prior to thebeginning of the dark period (7 p.m.). Measures ofhorizontal activity and total distance were taken every120 min for 22 h with Digipro Version 1.5. The data (36data points for each genotype) were split into twogroups, light and dark period, averages were takenand plotted together with the standard deviation. Thedata from each period were compared for significanceusing a two-tailed t test.
Resident-Intruder studies. These studies focused onmale mice that were no older than 4 months of age.Older animals that had been group housed were un-reliable in these tests. Male transgenic (4) and non-transgenic (4) animals were housed singly for at least72 h before the introduction of juvenile male intruders(7 weeks). The time to the first attack, the number ofattacks, and the duration of the attacks were measuredfor 5 min. The data for HD and DRPLA transgenicmice and nontransgenics were averaged and plottedwith standard deviations and p-values from two-tailed t-tests.
Rotarod testing. Six 8-week-old naive mice fromHD-N171-82Q line 81 and AT-FL-65Q line 150 werecompared to nontransgenic mice from the same agegroup. The mice were tested on a rotarod device(Rotamex 4/8, Columbus Instruments InternationalCorporation, OH). The speed of the rod was set toincrease from 4 revolutions/min to 30 revolutions/min over a 10-min period. The interval for the mice tofall from the rod was measured in 4 trials per day overa 4-day period. At least 10 min recovery time wasallowed between trials (Clark et al., 1997). The data foreach group of animals were averaged and plottedtogether with the standard deviations.
Balance beam and wire mesh: Six HD (line 81) andDRPLA (line 150) mice were tested of each age group.The mice were transferred to the balance beam (diam-eter 5 6 mm) or inverted wire mesh (grid 5 6 3 6 mm)and the times were taken for the mice fall into a smallpan of water three times/day on three different days.Averages, standard deviation and two tailed t testswere calculated from the performances of the thirdday.
Neurochemical Methods
Animals were killed after decapitation, their brainswere immediately removed and bisected sagittally. In

FcacpbsMCact
I
(wswtpci
dmAtmo
moaDpto(1twg
mnmamhhh
407Behavioral and Neuropathological Abnormalities in HD and DRPLA Mice
the analysis of 4-week-old animals, we homogenizedentire hemibrains. We also analyzed the forebrainsof older, severely affected, HD (at 6 months), andDRPLA (at 12 months) mice. Tissue was frozen rap-idly on dry ice, and stored at 270°C until assayed.
rozen tissues were homogenized in 10–20 vol ofhilled 50 mM sodium phosphate buffer, pH 7.0. Anliquot was then adjusted to 0.1 M sodium acetate,ooled, and centrifuged to sediment particulate matterrior to HPLC analysis. Monoamines were measuredy HPLC with electrochemical detection, with sensorset at 150, 250, 350, and 500 mV (ESA Inc., Chelmsford,
A). Exactly 10 ml of tissue extract was injected onto a18 reverse phase MD-150 column (ESA Inc.) and elutedt a flow of 0.6 ml/min for 20 min. The mobile phaseonsisted of 1.7 mM 1-tetraethylammonium and 8% ace-onitrile in 75 mM sodium phosphate buffer, pH 2.9.
mmunocytochemistry
Animals were anaesthetized in methoxyflouraneMetofane, Schering-Plough, Union, NJ) and perfused
ith 2% paraformaldehyde-lysine-sodium periodateolution through the left cardiac ventricle. The brainsere removed, postfixed in the same fix overnight,
hen transferred to phosphate buffered saline (PBS,H 7.3). To analyze the DRPLA mice, the brains wereut sagittally, processed for paraffin embedding andmmunostained (8-mm sections) with AP142 (a poly-
clonal rabbit, peptide antibody that recognizes 452–439 aa of atrophin-1) (Wood et al., 1998), at dilutions of1:10,000 or 1:25,000, following standard protocols(Becher et al., 1998). To analyze HD mice, the brainswere dissected sagittally, soaked overnight in 30%sucrose-PBS at 4°C, and frozen on crushed dry icebefore sectioning. Free-floating frozen sections (40mm) were cut on a sliding microtome (Reichert-Jung,Optische Werke AG, Vienna ) and stored in cryopro-tectant at 220°C until further use. Sections were im-munostained with either a polyclonal, rabbit GST-fusion antibody raised against an N-terminal fragmentof huntingtin, containing amino acids 1–24 and 69–92(AP360) or rabbit antibody EM48 (Li et al., 1999), bothat a dilution of 1:5,000. EM48 preferentially detectscytoplasmic aggregates of mutant huntingtin(Gutekunst et al., 1999; Li et al., 1999). Sections immu-nostained to visualize cytoplasmic aggregates fromHD mice were immunostained with EM48. HD brainsections that should visualize nuclear accumulationwere immunostained with AP360. Standard immuno-cytochemical protocols were followed (Schilling et al.,1999a,b; Borchelt et al., 1996; Becher et al., 1998). Pri-
mary antibodies were incubated overnight in 5% NGSand 0.1% Triton-X100 in PBS and detected using theElite Kit-biotinylated goat anti-rabbit secondary anti-body (Vector ABC Elite, Burlinghame, CA). Visualiza-tion of the reactivity was achieved by incubation in0.1% 39-39-diaminobenzidine tetrahydrochloride(Gibco BRL, Life Technologies, Gaithersburg, MD)and 0.01% hydrogen peroxide. Counter staining of thesections was not used to increase visibility of nuclearimmunostaining.
RESULTS
Comparison of HD and DRPLA BehavioralPhenotypes
The animals used in this study have been previouslydescribed (Schilling et al., 1999a,b). Except where in-
icated, we have focused on line 81 (of HD-N171-82Qice) for our HD mouse model and on line 150 (ofT-FL-65Q mice) for our DRPLA mouse model. These
wo lines have the earliest onset of behavioral abnor-alities that is compatible with breeding. The lifespan
f HD-line 81 mice is about 5 months (Schilling et al.,1999a), whereas the average life expectancy of our line150 DRPLA mice is about 12 months (the death curveis very broad, however) (Schilling et al., 1999b). In ourstudy, we attempted to compare animals in similarstages of disease (e.g., advanced stage: 5-month-oldHD mouse equivalent to a 9 to 12-month-old DRPLA
ouse). The HD mice express an N-terminal fragmentf huntingtin (HD-N171), containing the first 171mino acids, including the CAG repeats, whereas theRPLA mice express a full-length mutant atrophin-1rotein. The polyglutamine repeat lengths in both pro-
eins are of lengths normally associated with juvenilenset (65Q in atrophin-1 and 82Q in huntingtin)Duyao et al., 1993; Ross et al., 1997a; Hayashi et al.,998). Both lines of transgenic mice were generated inhe same strain of mice (C3H/HeJ 3 C57BL/6J),
hich should eliminate the influence of strain back-rounds on the phenotypes.The behavioral phenotypes of the HD and DRPLAice are readily distinguishable. The HD mice appear
ormal at birth, but develop unsteady gait by 2–4onths of age. HD mice in advanced stages of disease
re hypoactive relative to age-matched normal litter-ates (Figs. 1A and 1B). On measures of numbers of
orizontal photocell breaks (horizontal activity) andow these interruptions translate into locomotor be-avior (distance traveled), HD mice were hypoactive
Copyright © 2001 by Academic PressAll rights of reproduction in any form reserved.
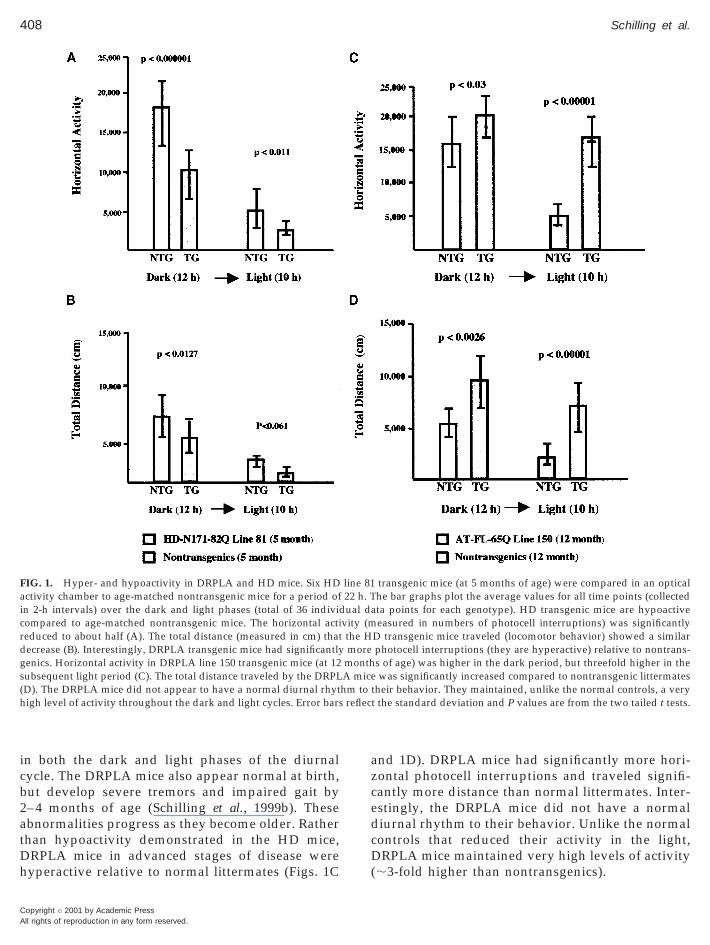
408 Schilling et al.
A
in both the dark and light phases of the diurnalcycle. The DRPLA mice also appear normal at birth,but develop severe tremors and impaired gait by2– 4 months of age (Schilling et al., 1999b). Theseabnormalities progress as they become older. Ratherthan hypoactivity demonstrated in the HD mice,DRPLA mice in advanced stages of disease werehyperactive relative to normal littermates (Figs. 1C
FIG. 1. Hyper- and hypoactivity in DRPLA and HD mice. Six HDactivity chamber to age-matched nontransgenic mice for a period ofin 2-h intervals) over the dark and light phases (total of 36 indivicompared to age-matched nontransgenic mice. The horizontal actireduced to about half (A). The total distance (measured in cm) thatdecrease (B). Interestingly, DRPLA transgenic mice had significantlygenics. Horizontal activity in DRPLA line 150 transgenic mice (at 12subsequent light period (C). The total distance traveled by the DRPL(D). The DRPLA mice did not appear to have a normal diurnal rhythigh level of activity throughout the dark and light cycles. Error bars
Copyright © 2001 by Academic Pressll rights of reproduction in any form reserved.
and 1D). DRPLA mice had significantly more hori-zontal photocell interruptions and traveled signifi-cantly more distance than normal littermates. Inter-estingly, the DRPLA mice did not have a normaldiurnal rhythm to their behavior. Unlike the normalcontrols that reduced their activity in the light,DRPLA mice maintained very high levels of activity(;3-fold higher than nontransgenics).
transgenic mice (at 5 months of age) were compared in an opticalhe bar graphs plot the average values for all time points (collectedta points for each genotype). HD transgenic mice are hypoactiveeasured in numbers of photocell interruptions) was significantly
D transgenic mice traveled (locomotor behavior) showed a similarphotocell interruptions (they are hyperactive) relative to nontrans-s of age) was higher in the dark period, but threefold higher in thewas significantly increased compared to nontransgenic littermates
their behavior. They maintained, unlike the normal controls, a veryt the standard deviation and P values are from the two tailed t tests.
line 8122 h. T
dual davity (mthe Hmore
monthA micehm to
reflec

snr
twddteo
qt(AaitCtnipScwssNspseiD
409Behavioral and Neuropathological Abnormalities in HD and DRPLA Mice
Male DRPLA mice are difficult to handle and dis-play aggressive behavior toward other mice. To deter-mine the level of aggressive behavior in the DRPLAmice, we compared them to age-matched nontrans-genics in a resident-intruder task (Table 1). DRPLAmice (at 4 months of age) attack the intruder in ap-proximately half the time compared to nontransgeniclittermates. They also demonstrate about a twofoldhigher number of average attacks, while the averagelength of the attacks appeared to be normal (Table 1).Thus, DRPLA mice display a higher level of aggres-sive behavior than the nontransgenic littermates. Thelevel of aggressive behavior in the HD mice at fourmonths of age was not significantly different fromage-matched nontransgenics (Table 1).
Analysis of Neurotransmitter Levels in HD andDRPLA Mice
Increases or decreases in the gross levels of motoractivity are typically associated with abnormalities ofdopaminergic neurotransmitter systems (Sahgal andSahgal, 1993) while aggressive behavior is typicallyassociated with abnormalities in the serotonergic sys-tems (Hen, 1996). To determine if the phenotypic dif-ferences between the HD and DRPLA mice might beassociated with defects in the secretion of serotonin ordopamine, we measured the levels of both, in thebrains of these mice. As shown in Table 2, the contentof these monoamines appeared normal in the brains ofboth mutants, suggesting that any disturbance in thesesystems might arise at the levels of the receptor ofthese neurotransmitters (Cha et al., 1998).
Neuropathological Comparison of HD andDRPLA Mice
There have been many reports distinguishing be-tween nuclear staining and NII’s (Skinner et al., 1997;
TABLE 1
Increased Aggressive Behavior of DRPLA Transgenic Mice
Age(months)
Time until 1stattack (s)
Average number ofattacks
NTG’s 4 150.3 6 85.2 4.1 6 2.7HD line 81 4 205.3 6 105.4 3.2 6 3.7DRPLA line 150 4 95.3 6 22.0 8.1 6 2.6
(P , 0.04) (P , 0.012)
Note. Averages were taken from three different trials with fourrandomly selected animals in each group.
Hodgson et al., 1999; Wheeler et al., 2000). In thistudy, we refer to nuclear staining as diffuse immu-olabeling that fills the nucleus, whereas NIIs willefer to larger, more discrete, aggregates.
In previous studies, we demonstrated that the dis-ribution of nuclear staining and NIIs are widespread,
hen the brains of mice with advanced stages of theiseases are examined (Schilling et al., 1999a,b). Toetermine whether the differences in behavior be-
ween HD and DRPLA mice might be due to differingvolutions of neuropathology, we examined the brainsf young, largely asymptomatic HD and DRPLA mice.By immunohistochemistry, the distribution and fre-
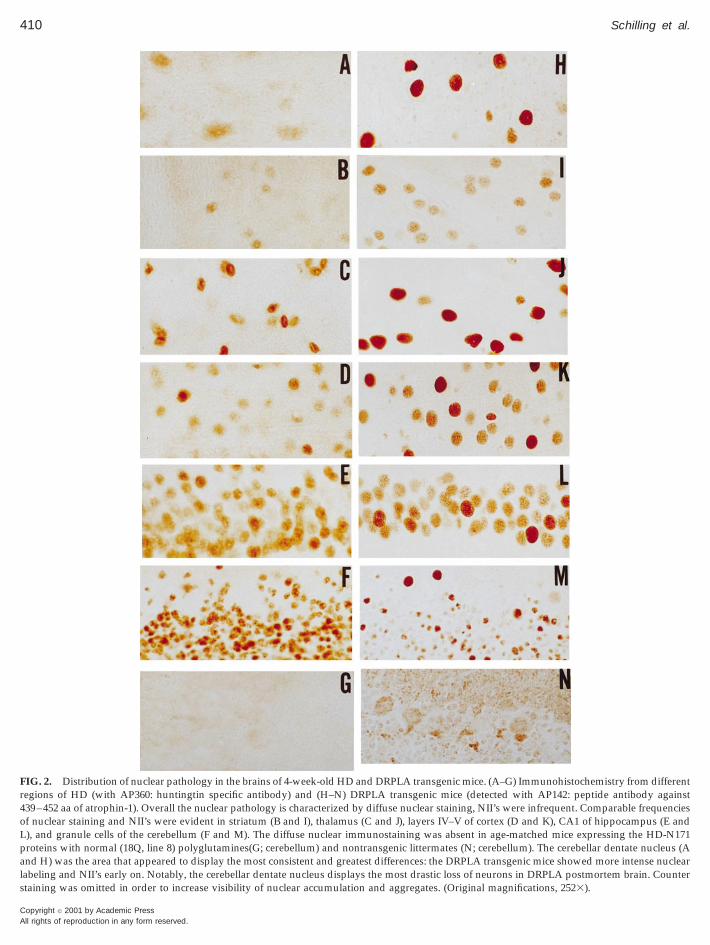
uency of nuclear staining and NIIs in DRPLA (withhe specific atrophin-1 antibody AP142) and HD miceusing the specific N-terminal huntingtin antibodyP360) appeared to be very similar at four weeks of
ge (Fig. 2). Comparable frequencies of nuclear stain-ng and aggregates were evident in striatum (B and I),halamus (C and J), layers IV–V of cortex (D and K),A1 of hippocampus (E and L), and granule cells of
he cerebellum (F and M). The diffuse nuclear immu-ostaining was absent in age-matched mice express-
ng the HD-N171 proteins with normal (18Q, line 8)olyglutamines (G; cerebellum) (also see Fig. 7 andchilling et al., 1999a) or nontransgenic littermates (N;erebellum). The cerebellar dentate nucleus (A and H)as the area that appeared to display the most con-
istent and greatest differences: the DRPLA micehowed a much more intense nuclear staining andIIs appeared very early. Cerebellar dentate nucleus
hows the most drastic loss of neurons in DRPLAostmortem brain (Ross et al., 1997b). By contrast, thetriatum, which displays the most drastic neurodegen-ration in HD postmortem brain, showed a very sim-lar pattern of diffuse nuclear staining in the HD andRPLA mice (B and I).
TABLE 2
Serotonin (5-HT) and Dopamine (DA) Levels in HDand DRPLA Transgenic Mice
Age 5-HT DA
HD 4w 7.30 6 0.51 13.90 6 0.97NTG 4w 6.55 6 0.32 14.73 6 2.95DRPLA 4w 6.43 6 0.30 12.30 6 0.44NTG 4w 6.53 6 0.78 11.92 6 0.95
HD 6m 9.64 6 0.42 27.44 6 1.68NTG 6m 10.63 6 0.53 28.25 6 3.62DRPLA 12m 8.50 6 1.68 28.29 6 6.71NTG 12m 7.51 6 0.23 24.73 6 1.36
Copyright © 2001 by Academic PressAll rights of reproduction in any form reserved.
410 Schilling et al.
A
FIG. 2. Distribution of nuclear pathology in the brains of 4-week-old HD and DRPLA transgenic mice. (A–G) Immunohistochemistry from differentregions of HD (with AP360: huntingtin specific antibody) and (H–N) DRPLA transgenic mice (detected with AP142: peptide antibody against439–452 aa of atrophin-1). Overall the nuclear pathology is characterized by diffuse nuclear staining, NII’s were infrequent. Comparable frequenciesof nuclear staining and NII’s were evident in striatum (B and I), thalamus (C and J), layers IV–V of cortex (D and K), CA1 of hippocampus (E andL), and granule cells of the cerebellum (F and M). The diffuse nuclear immunostaining was absent in age-matched mice expressing the HD-N171proteins with normal (18Q, line 8) polyglutamines(G; cerebellum) and nontransgenic littermates (N; cerebellum). The cerebellar dentate nucleus (Aand H) was the area that appeared to display the most consistent and greatest differences: the DRPLA transgenic mice showed more intense nuclearlabeling and NII’s early on. Notably, the cerebellar dentate nucleus displays the most drastic loss of neurons in DRPLA postmortem brain. Counterstaining was omitted in order to increase visibility of nuclear accumulation and aggregates. (Original magnifications, 2523).
Copyright © 2001 by Academic Pressll rights of reproduction in any form reserved.

qDi
411Behavioral and Neuropathological Abnormalities in HD and DRPLA Mice
In both models, aggregates were infrequent at fourweeks (Fig. 3). Instead, nuclei were filled with diffusenuclear staining, which might arise from aggregatesthat are small and perhaps still soluble (Fig. 3A, cortexlayers I–II in brains of 4-week-old DRPLA line 150mice with AP142 antibody). The diffuse immunostain-ing of nuclei in the cortex of 4-week-old DRPLA mice(Fig. 3A) progresses to intense nuclear staining andNIIs by 12 months of age (Fig. 3B). A similar evolutionof nuclear pathology was observed in granule cells ofDRPLA (Figs. 3C and 3D) and HD mice (Figs. 3E and3F). Overall, in both models, those neurons displayingdiffuse nuclear staining at young ages progress todisplay NIIs.
Our previous study of DRPLA mice demonstrated,that olfactory bulb does not exhibit a high frequency
FIG. 3. Evolution of nuclear pathology in the brains of HD andDRPLA transgenic models. Layers I–II of cortex display only faintdiffuse nuclear accumulation in DRPLA (line 150) mice (A) at 4weeks of age (immunostained with the specific atrophin-1 antibodyAP142). Over time, further accumulation/aggregation of polyglu-tamines leads toward the formation of more intense nuclear stainingand larger aggregates (arrow) in many of these neurons by 12months of age (B). Similarly, diffuse nuclear staining was observedin cerebellar granule cells from DRPLA (line 150) mice (C) and HD(line 81) mice (E) at 4 weeks of age (HD sections: huntingtin specificantibody AP360). In advanced stages of their phenotypes, the stain-ing develops into more intense nuclear staining and larger, nuclearaggregates (arrow) in both DRPLA (D; 12 months old) and HD (F; 5months old) mice. (Original magnification, 2523).
of nuclear staining and aggregates, as compared tocortex, despite expressing high levels of the transgene(Schilling et al., 1999b). We note a similar outcome inthe HD mice where the olfactory bulb, in mice withadvanced stages of disease, does not demonstrate dif-fuse nuclear accumulation or aggregation of mutanthuntingtin (data removed after review). Thus, neu-rons of the olfactory bulb appear to be less prone todevelop nuclear aggregates of polyglutamine pro-teins.
The prion protein promoter expresses highly in ner-vous tissue, heart/muscle, and to a lesser extent innonnervous tissues (Borchelt et al., 1996). A recentreport describing NIIs in multiple organ tissues(Sathasivam et al., 1999) led us to examine the fre-
uency of NIIs in peripheral tissues of the HD andRPLA mice. Heart, liver, stomach, skeletal muscle,
ntestine, and pancreas were immunostained for mu-
FIG. 4. Cerebellar dentate nucleus demonstrates differences in theappearance of nuclear accumulation and inclusions in HD andDRPLA transgenic mice. A lack of nuclear accumulation and inclu-sions in the neurons of the cerebellar dentate nucleus was observedin two independent HD transgenic lines: line 6 at 11 months (A) andline 81 at 5.5 months (B) (both immunostained with huntingtinspecific AP360). In contrast, nuclear accumulation and NIIs wereobserved in two independent DRPLA transgenic lines: line 79 at 11.5months (C) and line 150 at 12 months (D) (immunostained with thespecific atrophin-1 peptide antibody AP142). Cerebellar granulecells (inset B), from the same section as cerebellar dentate nucleus,demonstrated intense nuclear accumulation and NII’s in HD trans-genic mice. The inset in (A) demonstrates immunohistochemicalstaining of the same neurons in a nontransgenic mouse with AP360.Original magnification, 2523.
Copyright © 2001 by Academic PressAll rights of reproduction in any form reserved.
412 Schilling et al.
A
tant huntingtin or atrophin-1. Nuclear and cytoplas-mic aggregates were rare or absent in sections fromhearts (and all the tissues listed above) of the oldestHD (6 months) and DRPLA (19 months) mice (dataremoved after review). The absence of NII’s and cyto-plasmic aggregates in the pancreas of our HD mice(data removed after review) is consistent with previ-ous studies, which demonstrated that the resting lev-els of glucose in our HD mice do not develop overtdiabetes (Schilling et al., 1999a).
Cerebellar Pathology and Behavioral SymptomsDistinguish DRPLA Mice from HD Mice
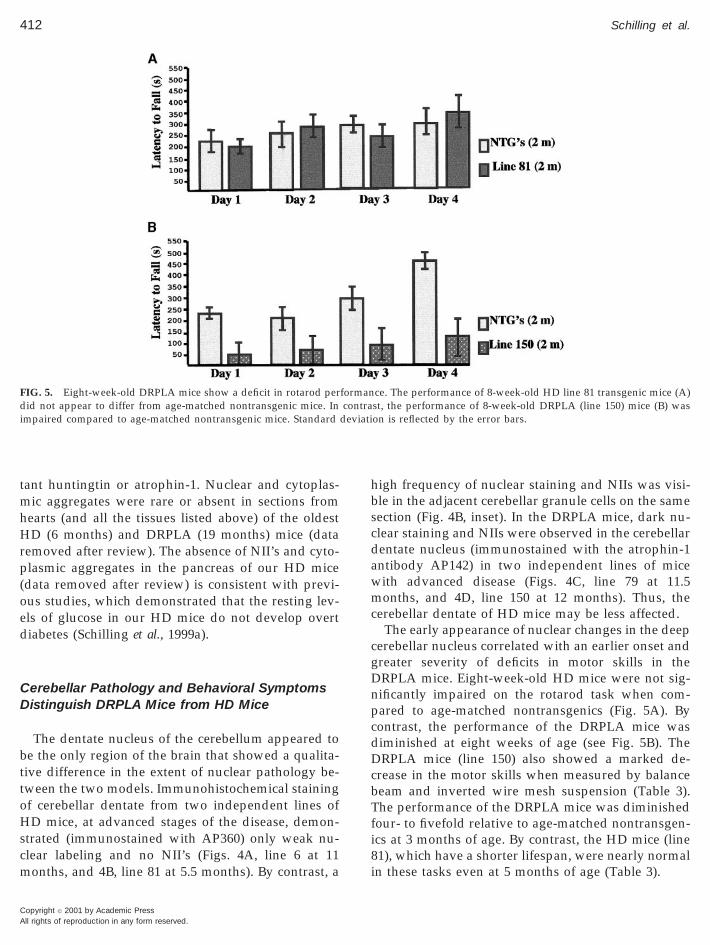
The dentate nucleus of the cerebellum appeared tobe the only region of the brain that showed a qualita-tive difference in the extent of nuclear pathology be-tween the two models. Immunohistochemical stainingof cerebellar dentate from two independent lines ofHD mice, at advanced stages of the disease, demon-strated (immunostained with AP360) only weak nu-clear labeling and no NII’s (Figs. 4A, line 6 at 11months, and 4B, line 81 at 5.5 months). By contrast, a
FIG. 5. Eight-week-old DRPLA mice show a deficit in rotarod perdid not appear to differ from age-matched nontransgenic mice. Inimpaired compared to age-matched nontransgenic mice. Standard
Copyright © 2001 by Academic Pressll rights of reproduction in any form reserved.
high frequency of nuclear staining and NIIs was visi-ble in the adjacent cerebellar granule cells on the samesection (Fig. 4B, inset). In the DRPLA mice, dark nu-clear staining and NIIs were observed in the cerebellardentate nucleus (immunostained with the atrophin-1antibody AP142) in two independent lines of micewith advanced disease (Figs. 4C, line 79 at 11.5months, and 4D, line 150 at 12 months). Thus, thecerebellar dentate of HD mice may be less affected.
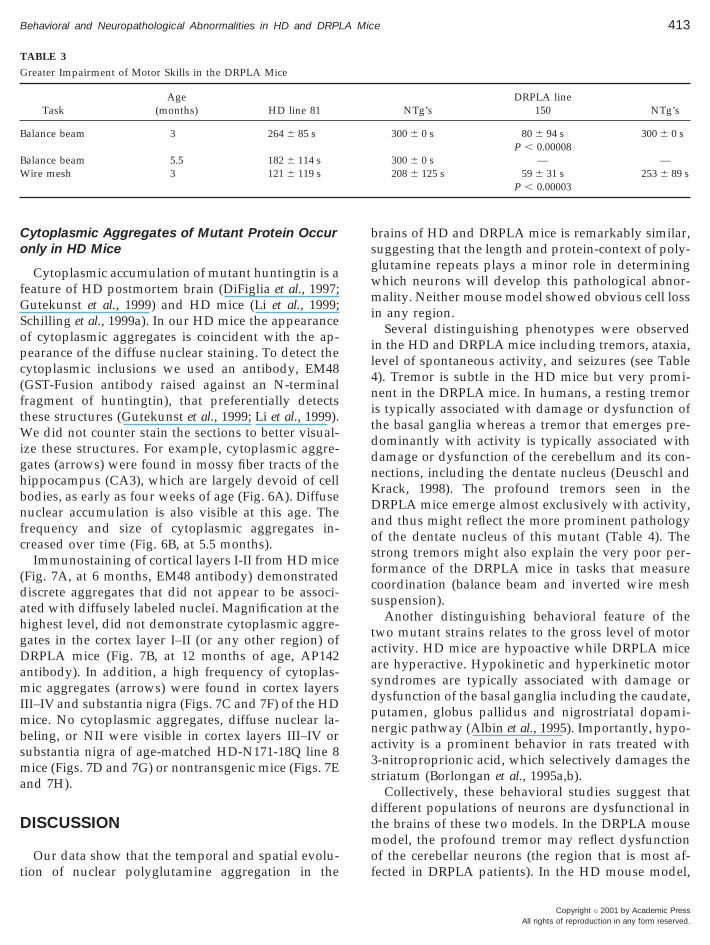
The early appearance of nuclear changes in the deepcerebellar nucleus correlated with an earlier onset andgreater severity of deficits in motor skills in theDRPLA mice. Eight-week-old HD mice were not sig-nificantly impaired on the rotarod task when com-pared to age-matched nontransgenics (Fig. 5A). Bycontrast, the performance of the DRPLA mice wasdiminished at eight weeks of age (see Fig. 5B). TheDRPLA mice (line 150) also showed a marked de-crease in the motor skills when measured by balancebeam and inverted wire mesh suspension (Table 3).The performance of the DRPLA mice was diminishedfour- to fivefold relative to age-matched nontransgen-ics at 3 months of age. By contrast, the HD mice (line81), which have a shorter lifespan, were nearly normalin these tasks even at 5 months of age (Table 3).
ce. The performance of 8-week-old HD line 81 transgenic mice (A)st, the performance of 8-week-old DRPLA (line 150) mice (B) wason is reflected by the error bars.
formancontra
deviati
GSopc(ft
413Behavioral and Neuropathological Abnormalities in HD and DRPLA Mice
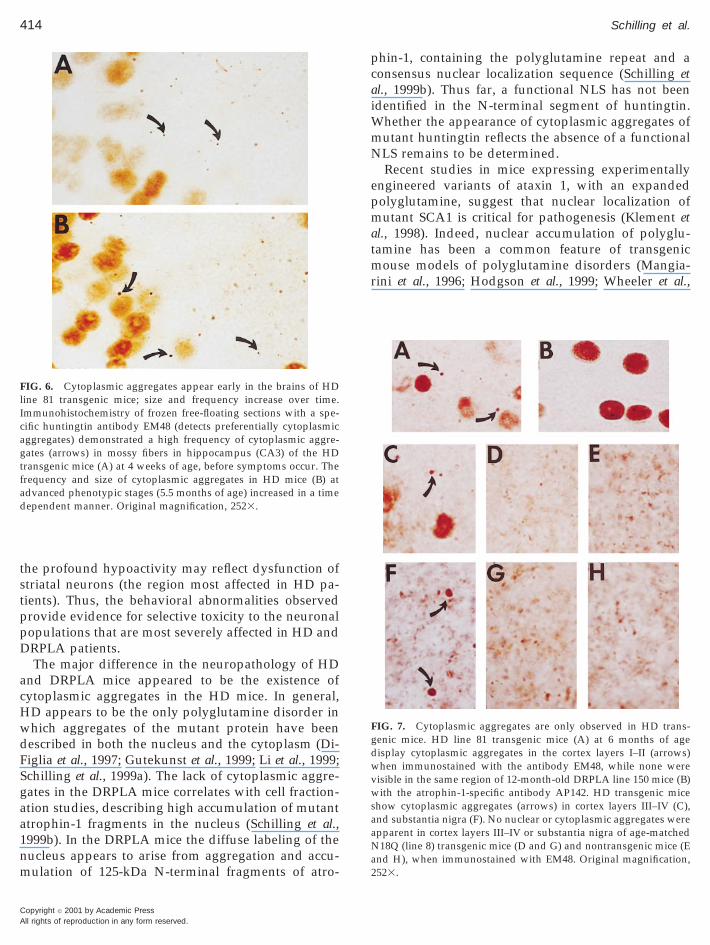
Cytoplasmic Aggregates of Mutant Protein Occuronly in HD Mice
Cytoplasmic accumulation of mutant huntingtin is afeature of HD postmortem brain (DiFiglia et al., 1997;
utekunst et al., 1999) and HD mice (Li et al., 1999;chilling et al., 1999a). In our HD mice the appearancef cytoplasmic aggregates is coincident with the ap-earance of the diffuse nuclear staining. To detect theytoplasmic inclusions we used an antibody, EM48GST-Fusion antibody raised against an N-terminalragment of huntingtin), that preferentially detectshese structures (Gutekunst et al., 1999; Li et al., 1999).
We did not counter stain the sections to better visual-ize these structures. For example, cytoplasmic aggre-gates (arrows) were found in mossy fiber tracts of thehippocampus (CA3), which are largely devoid of cellbodies, as early as four weeks of age (Fig. 6A). Diffusenuclear accumulation is also visible at this age. Thefrequency and size of cytoplasmic aggregates in-creased over time (Fig. 6B, at 5.5 months).
Immunostaining of cortical layers I-II from HD mice(Fig. 7A, at 6 months, EM48 antibody) demonstrateddiscrete aggregates that did not appear to be associ-ated with diffusely labeled nuclei. Magnification at thehighest level, did not demonstrate cytoplasmic aggre-gates in the cortex layer I–II (or any other region) ofDRPLA mice (Fig. 7B, at 12 months of age, AP142antibody). In addition, a high frequency of cytoplas-mic aggregates (arrows) were found in cortex layersIII–IV and substantia nigra (Figs. 7C and 7F) of the HDmice. No cytoplasmic aggregates, diffuse nuclear la-beling, or NII were visible in cortex layers III–IV orsubstantia nigra of age-matched HD-N171-18Q line 8mice (Figs. 7D and 7G) or nontransgenic mice (Figs. 7Eand 7H).
DISCUSSION
Our data show that the temporal and spatial evolu-tion of nuclear polyglutamine aggregation in the
TABLE 3
Greater Impairment of Motor Skills in the DRPLA Mice
TaskAge
(months) HD line 81
Balance beam 3 264 6 85 s
Balance beam 5.5 182 6 114 sWire mesh 3 121 6 119 s
brains of HD and DRPLA mice is remarkably similar,suggesting that the length and protein-context of poly-glutamine repeats plays a minor role in determiningwhich neurons will develop this pathological abnor-mality. Neither mouse model showed obvious cell lossin any region.
Several distinguishing phenotypes were observedin the HD and DRPLA mice including tremors, ataxia,level of spontaneous activity, and seizures (see Table4). Tremor is subtle in the HD mice but very promi-nent in the DRPLA mice. In humans, a resting tremoris typically associated with damage or dysfunction ofthe basal ganglia whereas a tremor that emerges pre-dominantly with activity is typically associated withdamage or dysfunction of the cerebellum and its con-nections, including the dentate nucleus (Deuschl andKrack, 1998). The profound tremors seen in theDRPLA mice emerge almost exclusively with activity,and thus might reflect the more prominent pathologyof the dentate nucleus of this mutant (Table 4). Thestrong tremors might also explain the very poor per-formance of the DRPLA mice in tasks that measurecoordination (balance beam and inverted wire meshsuspension).
Another distinguishing behavioral feature of thetwo mutant strains relates to the gross level of motoractivity. HD mice are hypoactive while DRPLA miceare hyperactive. Hypokinetic and hyperkinetic motorsyndromes are typically associated with damage ordysfunction of the basal ganglia including the caudate,putamen, globus pallidus and nigrostriatal dopami-nergic pathway (Albin et al., 1995). Importantly, hypo-activity is a prominent behavior in rats treated with3-nitroproprionic acid, which selectively damages thestriatum (Borlongan et al., 1995a,b).
Collectively, these behavioral studies suggest thatdifferent populations of neurons are dysfunctional inthe brains of these two models. In the DRPLA mousemodel, the profound tremor may reflect dysfunctionof the cerebellar neurons (the region that is most af-fected in DRPLA patients). In the HD mouse model,
NTg’sDRPLA line
150 NTg’s
300 6 0 s 80 6 94 s 300 6 0 sP , 0.00008
300 6 0 s — —208 6 125 s 59 6 31 s 253 6 89 s
P , 0.00003
Copyright © 2001 by Academic PressAll rights of reproduction in any form reserved.
Sgaa1nm
pcaiWmN
epmatmr
414 Schilling et al.
A
the profound hypoactivity may reflect dysfunction ofstriatal neurons (the region most affected in HD pa-tients). Thus, the behavioral abnormalities observedprovide evidence for selective toxicity to the neuronalpopulations that are most severely affected in HD andDRPLA patients.
The major difference in the neuropathology of HDand DRPLA mice appeared to be the existence ofcytoplasmic aggregates in the HD mice. In general,HD appears to be the only polyglutamine disorder inwhich aggregates of the mutant protein have beendescribed in both the nucleus and the cytoplasm (Di-Figlia et al., 1997; Gutekunst et al., 1999; Li et al., 1999;
chilling et al., 1999a). The lack of cytoplasmic aggre-ates in the DRPLA mice correlates with cell fraction-tion studies, describing high accumulation of mutanttrophin-1 fragments in the nucleus (Schilling et al.,999b). In the DRPLA mice the diffuse labeling of theucleus appears to arise from aggregation and accu-ulation of 125-kDa N-terminal fragments of atro-
FIG. 6. Cytoplasmic aggregates appear early in the brains of HDline 81 transgenic mice; size and frequency increase over time.Immunohistochemistry of frozen free-floating sections with a spe-cific huntingtin antibody EM48 (detects preferentially cytoplasmicaggregates) demonstrated a high frequency of cytoplasmic aggre-gates (arrows) in mossy fibers in hippocampus (CA3) of the HDtransgenic mice (A) at 4 weeks of age, before symptoms occur. Thefrequency and size of cytoplasmic aggregates in HD mice (B) atadvanced phenotypic stages (5.5 months of age) increased in a timedependent manner. Original magnification, 2523.
Copyright © 2001 by Academic Pressll rights of reproduction in any form reserved.
hin-1, containing the polyglutamine repeat and aonsensus nuclear localization sequence (Schilling etl., 1999b). Thus far, a functional NLS has not beendentified in the N-terminal segment of huntingtin.
hether the appearance of cytoplasmic aggregates ofutant huntingtin reflects the absence of a functionalLS remains to be determined.Recent studies in mice expressing experimentally
ngineered variants of ataxin 1, with an expandedolyglutamine, suggest that nuclear localization ofutant SCA1 is critical for pathogenesis (Klement et
l., 1998). Indeed, nuclear accumulation of polyglu-amine has been a common feature of transgenic
ouse models of polyglutamine disorders (Mangia-ini et al., 1996; Hodgson et al., 1999; Wheeler et al.,
FIG. 7. Cytoplasmic aggregates are only observed in HD trans-genic mice. HD line 81 transgenic mice (A) at 6 months of agedisplay cytoplasmic aggregates in the cortex layers I–II (arrows)when immunostained with the antibody EM48, while none werevisible in the same region of 12-month-old DRPLA line 150 mice (B)with the atrophin-1-specific antibody AP142. HD transgenic miceshow cytoplasmic aggregates (arrows) in cortex layers III–IV (C),and substantia nigra (F). No nuclear or cytoplasmic aggregates wereapparent in cortex layers III–IV or substantia nigra of age-matchedN18Q (line 8) transgenic mice (D and G) and nontransgenic mice (Eand H), when immunostained with EM48. Original magnification,2523.

2sm(m
ofIpgTbspnHDdioot
snerThwo(edwotwrTtd
C
op
415Behavioral and Neuropathological Abnormalities in HD and DRPLA Mice
2000) and experimental transgenic models (Ordway etal., 1997). Importantly, the tendency of mutant hun-tingtin to aggregate in the cytoplasm did not signifi-cantly influence the temporal or spatial distribution ofnuclear pathology in the HD mice. Hence, it does notappear that some neurons are protected from nuclearabnormalities because mutant huntingtin is seques-tered into cytoplasmic aggregates. Whether the differ-ences in the behavioral phenotypes of HD and DRPLAmice are caused by cytoplasmic aggregates remains tobe established. In SCA6, aggregates of ataxin-6 withexpanded polyglutamines are found primarily in thecytoplasm (Ishikawa et al., 1999a,b) and only cytoplas-mic aggregates were found in neurons of transgenicmice expressing mutant ataxin-2 (Huynh et al., 2000).Thus, these, and other studies (Li et al., 1999, 2000),suggest that cytoplasmic aggregates of mutant hun-tingtin may be toxic and thus the differences in phe-notypes between the HD and DRPLA mice may result,in part, from impact of cytoplasmic huntingtin aggre-gates on neuronal function.
Alternatively, it is possible that part, or all, of thephenotypic differences observed in the HD andDRPLA mice are manifestations of events occurring inthe nucleus. It is possible that differences in the rep-ertoire of protein-protein interactions between hun-tingtin and atrophin-1 govern the specificity of toxic-ity. Several proteins interact with the N-terminus ofhuntingtin, including a number of transcription fac-tors (Wanker et al., 1997; Boutell et al., 1999; Kazantsevet al., 1999; Steffan et al., 2000), while atrophin-1 has
TABLE 4
Phenotypes of HD and DRPLA Mouse Mode
HD line 81 DRPLA line 150
Body weight Reduced ReducedNucl. accumulation Widespread WidespreadNucl. aggregation Widespread WidespreadEarly death 6 months 12–15 monthsTremors Subtle (4 months) Strong (6 weeks)Balance beam Not aff. (12 weeks) Affected (12 weeks)Motor deficits ;12 weeks ;4 weeksAggressive No YesSpont. activity Hypoactive at 5
monthsHyperactive at 12
monthsSeizures No YesDCN pathology Subtle SevereCytopl. aggregation Yes No
Note. Data on the lifespans, body weights, distribution of nuclearpathology, onset of tremors, motor deficits, and seizures are de-scribed in Schilling et al. (1999a,b).
been shown to interact with a different repertoire ofproteins, including proteins with WW-domains andproteins that regulate transcription (Wood et al., 1998,000). In favor of this theory is a recent study demon-trating changes in gene expression in transgenicouse models of SCAI (Lin et al., 2000) and HD
Luthi-Carter et al., 2000). Thus, polyglutamine toxicityay be the result of altered gene expression.Previous studies of the HD exon-1 mice have dem-
nstrated that many different cell types are capable oforming polyglutamine NII’s (Sathasivam et al., 1999).n the exon-1 HD mice, the appearance of NII in theancreatic islet cells correlates with the appearance oflucose intolerance and diabetes (Hurlbert et al., 1999).he prion protein vector expresses relatively highly inrain and heart (Borchelt et al., 1996), with other tis-ues showing much lower levels of expression. Ourrevious study of DRPLA mice detected infrequentuclear aggregates in heart (Schilling et al., 1999b).owever, the present study of hearts in our HD andRPLA mice, and pancreas in the HD mice, failed toemonstrate diffuse nuclear labeling or NIIs, suggest-
ng that nuclear pathology is rare or absent in thesergan systems. Thus, the behavioral phenotypes ofur transgenic mice are probably caused by dysfunc-ion of the CNS.
Our analysis of behavioral abnormalities demon-trated that our HD and DRPLA mice demonstrate aumber of distinct features. In both cases, we havengineered the mice to express mutant proteins withepeat lengths that are consistent with a juvenile onset.o identify pathological correlates to the behavior, weave extended our previous study of pathology,hich demonstrated that most neurons in the CNS of
ur terminally ill HD and DRPLA mice develop NII’sSchilling et al., 1999a,b), to examine animals at mucharlier stages of disease. We find little evidence ofifferences in the level and extent of nuclear changes,ith virtually every type of neuron showing evidence
f nuclear accumulation of the respective mutant pro-ein at about the same age. This finding is consistent
ith the notion that these two models express theespective proteins at approximately equivalent doses.hus, we are inclined to believe that the differences in
he phenotypes of these mice do not arise from highlyisparate levels of mutant protein expression.
onclusions
Comparing behavioral and pathological phenotypesf mice expressing different transgenes harboringolyglutamine expansions has been complicated by
Copyright © 2001 by Academic PressAll rights of reproduction in any form reserved.

B
B
B
C
C
C
D
D
D
D
G
H
HH
H
416 Schilling et al.
A
the fact that prior studies have used different trans-gene promoters (with different developmental pat-terns of expression) or different strains of mice. Ourstudy is the first to examine the evolution of neuro-pathological changes in mice, of the same strain, ex-pressing two different polyglutamine proteins underthe influence of the same promoter. In this setting wecan expect that the developmental pattern of tran-scription for the two genes will be identical. Previousstudies of transgene expression with the PrP vectorhave demonstrated that the pattern of expression isnot significantly influenced by transgene integration(Borchelt et al., 1996). Our study of the early pathologyin our models demonstrates that, with the exception ofthe olfactory bulb and the dentate of the cerebellum,all neuronal populations appear to be equally prone toaccumulate mutant huntingtin fragments and mutantatrophin-1 in the nucleus. The differences in the be-havior of HD and DRPLA mice suggest that the sim-ple accumulation of a polyglutamine moiety in thenucleus does not lead to a uniform diminution inneuronal function. Instead, it appears that some otheraspect of these mutant proteins (repeat length, loca-tion of aggregates, or protein context) modulateswhether and how neurons will be affected and henceproduces the distinctive behavioral phenotypes of theHD and DRPLA mice.
ACKNOWLEDGMENTS
This work was supported by the National Institute of HealthGrants (NS34172, NS16375, and NS38144), The Huntington’s Dis-ease Society of America Coalition for the Cure, Hereditary DiseaseFoundation, by the DeVelbiss Fund and a bequest from the estate ofSar and Brita Levitan. We are especially grateful to Debbie Swing forhelp in making the transgenic animals.
REFERENCES
Albin, R. L., Young, A. B., & Penney, J. B. (1995) The functionalanatomy of disorders of the basal banglia. Trends Neurosci. 18,63–64.
Becher, M. W., Kotzuk, J. A., Sharp, A. H., Davies, S. W., Bates, G. P.,Price, D. L., & Ross C. A. (1998) Intranuclear neuronal inclusionsin Huntington’s disease and dentatorubral and pallidoluysianatrophy: Correlation between the density of inclusions and IT15CAG triplet repeat length. Neurobiol. Dis. 4, 387–397.
Borchelt, D. R., Davis, J., Fischer, M., Lee, M. K., Slunt, H. H.,Ratovitsky, T., Regard, J., Copeland, N. G., Jenkins, N. A., Sisodia,S. S., & Price, D. L. (1996) A vector for expressing foreign genes inthe brains and hearts of transgenic mice. Genet. Anal. 13, 159–163.
orlongan, C. V., Koutouzis, T. K., Freeman, T. B., Cahill, D. W., &Sanberg, P. R. (1995a) Behavioral pathology induced by repeated
Copyright © 2001 by Academic Pressll rights of reproduction in any form reserved.
systemic injections of 3-nitroproprionic acid mimics the motoricsymptoms of huntington’s disease. Brain Res. 697, 254–257.
orlongan, C. V., Koutouzis, T. K., Randall, T. S., Freeman, T. B.,Cahill, D. W., & Sanberg, P. R. (1995b) Systemic 3-nitroproprionicacid: Behavioral deficits and striatla damage in adult rats. BrainRes. Bull. 36, 549–556.
outell, J. M., Thomas, P., Neal, J. W., Weston, V. J., Duce, J., Harper,P. S., & Jones, A. L. (1999) Aberrant interactions of transcriptionalrepressor proteins with the Huntington’s disease gene product,huntingtin. Hum. Mol Genet. 8, 1647–1655.
ha, J. H., Kosinski, C. M., Kerner, J. A., Alsdorf, S. A., Mangiarini,L., Davies, S. W., Penney, J. B., Bates, G. P., & Young, A. B. (1998)Altered brain neurotransmitter receptors in transgenic mice ex-pressing a portion of an abnormal human huntington diseasegene. Proc. Natl. Acad. Sci. USA 95, 6480–6485.
lark, H. B., Burright, E. N., Yunis, W. S., Larson, S., Wilcox, C.,Hartman, B., Matilla, A., Zoghbi, H. Y., & Orr, H. T. (1997)Purkinje cell expression of a mutant allele of SCA1 in transgenicmice leads to disparate effects on motor behaviors, followed by aprogressive cerebellar dysfunction and histological alterations.J. Neurosci. 17, 7385–7395.
ummings, C. J., Mancini, M. A., Antalffy, B., DeFranco, D. B., Orr,H. T., & Zoghbi, H. Y. (1998) Chaperone suppression of aggrega-tion and altered subcellular proteasome localization imply pro-tein misfolding in SCA1. Nat. Genet. 19, 148–154.avies, S. W., Turmaine, M., Cozens, B. A., DiFiglia, M., Sharp,A. H., Ross, C. A., Scherzinger, E., Wanker, E. E., Mangiarini, L.,& Bates, G. P. (1997) Formation of neuronal intranuclear inclu-sions underlies the neurological dysfunction in mice transgenicfor the HD mutation. Cell 90, 537–548.euschl, G., & Krack, P. (1998) Tremors: Differential diagnosis,neurophysiology, and pharmacology. In Parkinson’s Disease andMovement Disorders. Williams & Wilkins, Baltimore, MD.iFiglia, M., Sapp, E., Chase, K. O., Davies, S. W., Bates, G. P.,Vonsattel, J. P., & Aronin, N. (1997) Aggregation of huntingtin inneuronal intranuclear inclusions and dystrophic neurites in brain.Science 277, 1990–1993.uyao, M., Ambrose, C., Myers, R., Novelletto, A., Persichetti, F.,Frontali, M., Folstein, S., Ross, C., Franz , M., Abbott, M., et al.(1993) Trinucleotide repeat length instability and age of onset inHuntington’s disease. Nat. Genet. 4, 387–392.utekunst, C. A., Li, S. H., Yi, H., Mulroy, J. S., Kuemmerle, S.,Jones, R., Rye, D., Ferrante, R. J., Hersch, S. M., & Li, X. J. (1999)Nuclear and neuropil aggregates in Huntington’s disease: Rela-tionship to neuropathology. J. Neurosci. 2522–2534.ayashi, Y., Kakita, A., Yamada, M., Koide, R., Igarashi, S., Takano,H., Ikeuchi, T., Wakabayashi, K., Egawa, S., Tsuji, S., & Takahashi,H. (1998) Hereditary dentatorubral-pallidoluysian atrophy: De-tection of widespread ubiquitinated neuronal and glial intranu-clear inclusions in the brain. Acta Neuropathol. (Berlin) 96, 547–552.en, R. (1996) Mean genes. Neuron 16, 17–21.odgson, J. G., Agopyan, N., Gutekunst, C. A., Leavitt, B. R.,LePiane, F., Singaraja, R., Smith, D. J., Bissada, N., McCutcheon,K., Nasir, J., Jamot, L., Li, X. J., Stevens, M. E., Rosemond, E.,Roder, J. C., Phillips, A. G., Rubin, E. M., Hersch, S. M., & Hayden,M. R. (1999) A YAC mouse model for Huntington’s disease withfull-length mutant huntingtin, cytoplasmic toxicity, and selectivestriatal neurodegeneration. Neuron 23, 181–192.untington’ s Disease Collaborative Group. (1993) A novel genecontaining a trinucleotide repeat that is expanded and unstable inHuntington’s disease chromosomes. Cell 72, 971–983.

I
I
I
K
K
K
L
L
L
L
L
M
M
N
O
P
R
R
R
R
S
S
S
S
S
417Behavioral and Neuropathological Abnormalities in HD and DRPLA Mice
Hurlbert, M. S., Zhou, W., Wasmeier, C., Kaddis, F. G., Hutton, J. C.,& Freed, C. R. (1999) Mice transgenic for an expanded CAGrepeat in the Huntington’s disease gene develop diabetes. Diabetes48, 649–651.
Huynh, D. P., Figueroa, K., Hoang, N., & Pulst, S. M. (2000) Nuclearlocalization or inclusion body formation of ataxin-2 are not nec-essary for SCA2 pathogenesis in mouse or human. Nat. Genet. 26,44–50.
garashi, S., Koide, R., Shimohata, T., Yamada, M., Hayashi, Y.,Takano, H., Date, H., Oyake, M., Sato, T., Sato, A., Egawa, S.,Ikeuchi, T. H., Nakano, R., Tanaka, K., Hozumi, I., Inuzuka, T.,Takahash,H., & Tsuji, S. (1998) Suppression of aggregate forma-tion and apoptosis by transglutaminase inhibitors in cells express-ing truncated DRPLA protein with an expanded polyglutaminestretch. Nat. Genet. 18, 111–117.
shikawa, K., Fujigasaki, H., Saegusa, H., Ohwada, K., Fujita, T.,Iwamoto, H., Komatsuzaki, Y., Toru, S., Toriyama, H., Watanabe,M., Ohkoshi, N., Shoji, S., Kanazawa, I., Tanabe, T., & MizusawaH. (1999a) Abundant expression and cytoplasmic aggregations of[alpha]1A voltage-dependent calcium channel protein associatedwith neurodegeneration in spinocerebellar ataxia type 6. Hum.Mol. Genet. 8, 1185–1193.
shikawa, K., Watanabe, M., Yoshizawa, K., Fujita, T., Iwamoto, H.,Yoshizawa, T., Harada, K., Nakamagoe, K., Komatsuzaki, Y.,Satoh, A., Doi, M., Ogata, T., Kanazawa, I., Shoji, S., & Mizusawa,H. (1999b) Clinical, neuropathological, and molecular study intwo families with spinocerebellar ataxia type 6 (SCA6). J. Neurol.Neurosurg. Psychiatry. 67, 86–89.
azantsev, A., Preisinger, E., Dranovsky, A., Goldgaber, D., Hous-man, D. (1999) Insoluble detergent-resistant aggregates form be-tween pathological and nonpathological lengths of polyglutaminein mammalian cells. Proc. Natl. Acad. Sci. USA 96, 11404–11409.
lement, I. A., Skinner, P. J., Kaytor, M. D., Yi, H., Hersch, S. M.,Clark, H. B., Zoghbi, H. Y., & Orr, H. T. (1998) Ataxin-1 nuclearlocalization and aggregation: Role in polyglutamine-induced dis-ease in SCA1 transgenic mice. Cell 95, 41–53.
oide R., Ikeuchi T., Onodera O., Tanaka H., Igarashi S., Endo K.,Takahashi H., Kondo R., Ishikawa A., Hayashi T., et al. (1994)Unstable expansion of CAG repeat in hereditary dentatorubral-pallidoluysian atrophy (DRPLA). Nat. Genet. 6, 9–13.
i, H., Li, S. H., Cheng, A. L., Mangiarini, L., Bates, G. P., & Li, X. J.(1999) Ultrastructural localization and progressive formation ofneuropil aggregates in Huntington’s disease transgenic mice.Hum. Mol. Genet. 8, 1227–1236.
i, H., Li, S. H., Johnston, H., Shelbourne, P. F., & Li, X. J. (2000)Amino-terminal fragments of mutant huntingtin show selectiveaccumulation in striatal neurons and synaptic toxicity. Nat. Genet.25, 385–389.
i, S. H., Schilling, G., Li, X. J., Margolis, R. L., Stine, O. C., Wagster,M. V., Abbott, V. H., Franz, M. L., & Ranen, N. G. (1993) Hun-tington’s disease gene (IT15) is widely expressed in human andrat tissues. Neuron 11, 985–993.
in, X., Antalffy, B., Kang, D., Orr, H. T., & Zoghbi, H. Y. (2000)Polyglutamine expansion down-regulates specific neuronal genesbefore pathologic changes in SCA1. Nat. Neurosci. 3, 157–163.
uthi-Carter, R., Strand, A., Peters, N. L., Solano, S. M., Holling-sworth, Z. R., Menon, A. S., Frey, A. S., Spektor, B. S., Penney,E. B., Schilling, G., Ross, C. A., Borchelt, D. R., Tapscott, S. J.,Young, A. B., Cha, J. H., & Olson, J. M. (2000) Decreased expres-sion of striatal signaling genes in a mouse model of Huntington’sdisease. Hum. Mol. Genet. 9, 1259–1271.
angiarini, L., Sathasivam, K., Seller, M., Cozens, B., Harper, A.,Hetherington, C., Lawton, M., Trottier, Y., Lehrach, H., Davies,S. W., & Bates, G. P. (1996) Exon 1 of the HD gene with anexpanded CAG repeat is sufficient to cause a progressive neuro-logical phenotype in transgenic mice. Cell 87, 493–506.argolis, R. L., Li, S. H., Young, W. S., Wagster, M. V., Stine, O. C.,Kidwai, A. S., Ashworth, R. G., & Ross, C. A. (1996) DRPLA gene(atrophin-1) sequence and mRNA expression in human brain.Brain Res. Mol. Brain Res. 36, 219–226.agafuchi, S., Yanagisawa, H., Ohsaki, E., Shirayama, T., Tadokoro,K., Inoue, T., & Yamada, M. (1994) Structure and expression of thegene responsible for the triplet repeat disorder, dentatorubral andpallidoluysian atrophy (DRPLA). Nat. Genet. 8, 177–182.rdway, J. M., Tallaksen-Greene, S., Gutekunst, C. A., Bernstein,E. M., Cearley, J. A., Wiener, H. W., Dure, L. S., Lindsey. R.,Hersch, S. M., Jope, R. S., Albin, R. L., & Detloff, P. J. (1997)Ectopically expressed CAG repeats cause intranuclear inclusionsand a progressive late onset neurological phenotype in the mouse.Cell 91, 753–763.
erutz, M. F. (1999) Glutamine repeats and neurodegenerative dis-eases: Molecular aspects. Trends Biochem. Sci. 24, 58–63.
eddy, P. H., Williams, M., Charles, V., Garrett, L., Pike-Buchanan,L., Whetsell, W. O., Jr., Miller, G., & Tagle, D. A. (1998) Behav-ioural abnormalities and selective neuronal loss in HD transgenicmice expressing mutated full-length HD cDNA. Nat. Genet. 20,198–202.
oss, C. A., Becher, M. W., Colomer, V., Engelender, S., Wood, J. D.,& Sharp, A. H. (1997a) Huntington’s disease and dentatorubral-pallidoluysian atrophy: Proteins, pathogenesis and pathology.Brain Pathol. 7, 1003–1016.
oss, C. A., Margolis, R. L., Rosenblatt, N. G., Becher, M. W., &Aylward, E. (1997b) Huntington’s disease and the related disor-der, dentatorubral-pallidoluysian atrophy (DRPLA). Mol. Med. 76,305–338.
ubinsztein, D. C., Leggo, J., Coles, R., Almqvist, E., Biancalana, V.,Cassiman, J. J., Chotai, K., Connarty, M., Crauford, D., Curtis, A.,Curtis, D., Davidson, M. J., Differ, A. M., Dode, C., Dodge, A.,Frontali, M., Ranen, N. G., Stine, O. C., Sherr, M., Abbott, M. H.,Franz, M. L., Graham, C. A., Harper, P. S., Hedreen, J. C., Hayden,M. R., et al. (1996) Phenotypic characterization of individuals with30–40 CAG repeats in the Huntington disease (HD) gene revealsHD cases with 36 repeats and apparently normal elderly individ-uals with 36–39 repeats. Am. J. Hum. Genet. 59, 16–22.
ahgal, A., & Sahgal, A. (1993) Behavioral neuroscience: A practicalapproach. Oxford Univ. Press, New York.
athasivam, K., Hobbs, C., Turmaine, M., Mangiarini, L., Mahal, A.,Bertaux, F., Wanker, E. E., Doherty, P., Davies, S. W., & Bates,G. P. (1999) Formation of polyglutamine inclusions in non-CNStissue. Hum. Mol. Genet. 8, 813–822.
cherzinger, E., Lurz, R., Turmaine, M., Mangiarini, L., Hollenbach,B., Hasenbank, R., Bates, G. P., Davies, S. W., Lehrach, H., &Wanker, E. E. (1997) Huntingtin-encoded polyglutamine expan-sions form amyloid-like protein aggregates in vitro and in vivo.Cell 90, 549–558.
cherzinger, E., Sittler, A., Schweiger, K., Heiser, V., Lurz, R.,Hasenbank, R., Bates, G. P., Lehrach, H., & Wanker E. E. (1999)Self-assembly of polyglutamine-containing huntingtin fragmentsinto amyloid-like fibrils: Implications for Huntington’s diseasepathology. Proc. Natl. Acad. Sci. USA 96, 4604–4609.
chilling, G., Becher, M. W., Sharp, A. H., Jinnah, H. A., Duan, K.,Kotzuk, J. A., Slunt, H. H., Ratovitski, T., Cooper, J. K., Jenkins,N. A., Copeland, N. G., Price, D. L., Ros, C. A., & Borchelt, D. R.
Copyright © 2001 by Academic PressAll rights of reproduction in any form reserved.

S
S
S
S
W
W
W
418 Schilling et al.
A
(1999a) Intranuclear inclusions and neuritic aggregates in trans-genic mice expressing a mutant N-terminal fragment of hunting-tin. Hum. Mol. Genet. 8, 397–407.
chilling, G., Wood, J. D., Duan, K., Slunt, H. H., Gonzales, V.,Yamada, M., Cooper, J. K., Margolis, R. L., Jenkins, N. A., Cope-land, N. G., Takahashi, H., Tsuji S., Price, D. L., Borchelt, D. R., &Ross, C. A. (1999b) Nuclear accumulation of truncated atrophin-1fragments in a transgenic mouse model of DRPLA. Neuron 24,275–286.
isodia, S. S. (1998) Nuclear inclusions in glutamine repeat disor-ders: are they pernicious, coincidental, or beneficial?. Cell 95, 1–4.
kinner, P. J., Koshy, B. T., Cummings, C. J., Klement, I. A., Helin,K., Servadio, A., Zoghbi, H. Y., & Orr, H. T. (1997) Ataxin-1 withan expanded glutamine tract alters nuclear matrix-associatedstructures. Nature 389, 971–974.
teffan, J. S., Kazantsev, A., Spasic-Boskovic, O., Greenwald, M.,Zhu, Y. Z., Gohler, H., Wanker, E. E., Bates, G. P., Housman, D. E.,& Thompson, L. M. (2000) The Huntington’s disease proteininteracts with p53 and CREB-binding protein and represses tran-scription. Proc. Natl. Acad. Sci. USA 97, 6763–6768.
Copyright © 2001 by Academic Pressll rights of reproduction in any form reserved.
Wanker, E. E., Rovira, C., Scherzinger, E., Hasenbank, R., Walter, S.,Tait, D., Colicelli, J., & Lehrach, H. (1997) HIP-I: A huntingtininteracting protein isolated by the yeast two-hybrid system. Hum.Mol. Genet. 6, 487–495.heeler, V. C., White, J. K., Gutekunst, C. A., Vrbanac, V., Weaver,M., Li, X. J., Li, S. H., Yi, H., Vonsattel, J. P., Gusella, J. F., Hersch,S., Auerbach, W., Joyner, A. L., & MacDonald, M. E. (2000) Longglutamine tracts cause nuclear localization of a novel form ofhuntingtin in medium spiny striatal neurons in Hdh(Q92) andHdh(Q111) knock-in mice. Hum. Mol. Genet. 9, 503–513.ood, J. D., Yuan, J., Margolis, R. L., Colomer, V., Duan, K., Kushi,J., Kaminsky, Z., Kleiderlein, J. J., Sharp, A. H., & Ross, C. A.(1998) Atrophin-1, the DRPLA gene product, interacts with twofamilies of WW domain-containing proteins. Mol. Cell. Neurosci.11, 149–160.ood, J. D., Nucifora, F. C., Duan, K., Zhang, C., Wang, J., Kim, Y.,Schilling, G., Sacchi, N., Liu, J. M., & Ross, C. A. (2000) Atro-phin-1, the DRPLA gene product, interacts with ETO/MTG-8 inthe nuclear matrix and represses transcription. J. Cell. Biol. 3,939–948.





























