Embed Size (px)
Citation preview

This article appeared in a journal published by Elsevier. The attachedcopy is furnished to the author for internal non-commercial researchand education use, including for instruction at the authors institution
and sharing with colleagues.
Other uses, including reproduction and distribution, or selling orlicensing copies, or posting to personal, institutional or third party
websites are prohibited.
In most cases authors are permitted to post their version of thearticle (e.g. in Word or Tex form) to their personal website orinstitutional repository. Authors requiring further information
regarding Elsevier’s archiving and manuscript policies areencouraged to visit:
http://www.elsevier.com/copyright

Author's personal copy
Materials Chemistry and Physics 113 (2009) 219–226
Contents lists available at ScienceDirect
Materials Chemistry and Physics
journa l homepage: www.e lsev ier .com/ locate /matchemphys
Elaboration and characterization of fluorapatite ceramic withcontrolled porosity
K. Chaari a,∗, F. Ben Ayeda, J. Bouaziza, K. Bouzouitab
a Laboratoire de Chimie Industrielle II, Unité Céramique, Ecole Nationale d’Ingénieurs de Sfax, B.P.W, 3038 Sfax, Tunisiab Institut Supérieur des Etudes Technologiques de Sousse, B.P. 135 Sousse-Erriadh 4023, Tunisia
a r t i c l e i n f o
Article history:Received 11 April 2008Received in revised form 6 June 2008Accepted 15 July 2008
Keywords:BiomaterialSinteringFluorapatitePorosityBiomedical applications
a b s t r a c t
Porous fluorapatite ceramics were fabricated using poly vinyl butyral as a porosifier. The conditions ofspecimens heat treating were optimized. The effects of preparation conditions involving poly vinyl butyralparticle concentration, sintering time, and forming pressure (die-pressing technique) on the resultant poresize/structure as well as the pore size distribution were investigated. The experimental results showedthat the Fap ceramics with controlled pore characteristics such as pore volume fraction, pore size andpore structure are achievable. It provides the possibility to design Fap ceramics with diverse porositiessimulating that of natural bone.
© 2008 Elsevier B.V. All rights reserved.
1. Introduction
Ceramics used for the repair and reconstruction of diseased ordamaged parts of human body are termed bioceramics [1]. Withthe growing demands of bioactive materials for orthopaedic aswell as maxillofacial surgery, the utilization of calcium hydroxyap-atite (Hap, with Ca/P = 1.667) and tri-calcium phosphate (TCP, withCa/P = 1.5) as fillers, spacers, and bone graft substitutes has receivedgreat attention mainly during the past two decades, primarilybecause of their biocompatibility, bioactivity, and ostéoconductioncharacteristics with respect to host tissue [1–3].
In recent years, attention was particularly placed on the fabrica-tion of bioceramics with “porous” configuration because the porousnetwork allows the tissue to infiltrate, which further enhances theimplant-tissue attachment [4–14]. In a porous form, Hap ceram-ics can be colonized by bone tissue with the same characteristicsas peri-implanted tissues [15]. For colonization of the pores totake place, they must be larger than 50–100 �m [13] or even250–300 �m according to some researchers [16–18].
Therefore, the control of porosity within the porous ceramicsis an important subject for many investigations. Recently, a noveltechnique in the fabrication of porous ceramics has been conductedby impregnating a cellulose spongy body with inter-connectedmacropores (>150 �m) into a slurry, followed by heat treating to
∗ Corresponding author.E-mail address: [email protected] (K. Chaari).
drive off the spongy body and to densify the ceramic powder [11].A resulting ceramic with a replica spongy structure is obtained.This technique allows the fabrication of ceramic with an open-porestructure and permits the use of spongy bodies of diverse porosi-ties to simulate the natural bone structure. However, the strengthsof these replicas are relatively low and this may restrict possibleload-bearing clinical applications. More recently, Arita et al. [19]obtained porous Hap ceramic sheets by means of a tape castingtechnique with CaCO3 as a gas-forming agent. Hap ceramic sheetswith highly porous microstructure (up to 62%) were successfullydeveloped but the pore size is limited to only several micrometers.
Because of its potential for use in dental implants, Fap has beenthe subject of several studies [20–26]. In addition, the fluorapatite(Fap) and hydroxyapatite (Hap) present closely related structuresand interstate physical and chemical properties. Compared to pureHap, Fap has much higher chemical and thermal stability [27–29].This study aims at developing porous Fap ceramic with controlledporosity using poly vinyl butyral (PVB) particles as a porosifier. Theinfluences of sintering time, forming pressure and PVB particle con-tent on the resultant pore characteristics of the porous Fap ceramicwere investigated.
2. Materials preparation
2.1. Fap powder synthesis
The Fap powder was prepared using a wet method [20,21,30]. A calcium nitratesolution (Ca(NO3)2) is slowly added using a peristaltic pump to a boiling diammo-nium phosphate ((NH4)2HPO4) and ammonium fluoride (NH4F) solution containing
0254-0584/$ – see front matter © 2008 Elsevier B.V. All rights reserved.doi:10.1016/j.matchemphys.2008.07.079

Author's personal copy
220 K. Chaari et al. / Materials Chemistry and Physics 113 (2009) 219–226
Table 1Elementary chemical analysis of Fap powder
Element Atomic number
%Theoretical %Measured
Calcium 10.00 9.98 ± 0.02Phosphorus 06.00 5.98 ± 0.02Fluor 02.00 1.97 ± 0.03Ca/P 1.667 1.668Ca/F 5.000 5.065
NH4OH to maintain the pH at 9. To adjust the pH at this value, additional concen-trated NH4OH is added when necessary. The precipitate is then filtered, washedwith hot distilled water, dried at 70 ◦C overnight and calcined at 500 ◦C for 1 h undernitrogen atmosphere. The powders obtained after sintering were examined by X-raydiffraction (Seifert XRD 3000 TT) with Cu K� radiation, Fourier transform infraredspectrometry (Perkin-Elmer FTIR 783), and scanning electron microscopy (PhillipsXL 30). The composition and Ca/P atomic ratio in raw powders were evaluated bychemical analysis. Ca and P contents are determined respectively by atomic absorp-tion spectrometry (Perkin-Elmer 5000) and colorimetric method [31]. Fluoride ionsare analysed by a specific electrode (Ingold) [32]. The specific area measurementwas performed using BET method (Micromeritrics ASPA 2010) [33].
2.2. Porous Fap ceramic preparation
The Fap and PVB powder are mixed in agate mortar. The powder mixture wasuniaxially pressed at various pressures into discs of 30 mm diameter and 7 mmthickness. Removed from the mould the specimen is a composite of ceramic andpolymer. The samples were then sintered at temperature T > 900 ◦C, after normalheating under air flue at T < 500 ◦C.
The relative density is calculated by dividing bulk density by Fap’s theoreticaldensity (3.19 g cm−3). The bulk density of the sintered body is calculated from thedimensions and weights.
The porosity of the as-sintered Fap ceramics was determined by mercuryporosimetry (Micrometric). Three to four specimens were selected to determineporosity with an error of less than 1% of the measured porosity value. The poresize and pore structure of the ceramics were examined using scanning electronmicroscopy.
Mechanical properties of porous Fap specimens were assessed using compres-sive strength testing (Brazilian test) [34,35]. The maximal rupture strength (�c) wasdetermined using the equation:
�c2F
�De(1)
where F is the tensile strength and D and e are the diameter and the thickness ofsample.
The samples were tested as received from sintering (diameter 25 mm and thick-ness 6 mm) with a crosshead speed of 2 mm min−1.
3. Results and discussion
3.1. Characteristics of synthetic Fap powder
The observation of the Fap powder microstructures synthesizedand calcined at 500 ◦C shows that it consists of fine particles but alsoof agglomerates with an important size (Fig. 1). The uncalcined andcalcined powders present a specific area close to 32 and 29 m2 g−1,respectively.
The results of the chemical analyses are consigned in Table 1.The percentages of the various elements constituting the Fap areclose to the theoretical percentages.
DTA and dilatometric measurements of Fap are summarized inTable 2. The curve DTA contains two endothermic peaks; the firstis located towards 90 ◦C, corresponding to the loss of hydrationwater. The second appears towards 1180 ◦C, is due to the formation
Fig. 1. Micrographic of synthetic Fap powder.
of a liquid phase. This is corroborated by binary eutectic betweenCaF2 and Fap [20]. Fluorite (CaF2) is an impurity in the Fap powdersynthesized [20].
From the dilatometric study, we note a beginning of sinteringtowards 715 ◦C. Beyond 1100 ◦C, we record a swelling due to theintergranular porosity formation, which minimizes the mechanicalresistance of the samples. For a temperature close to 1000 ◦C, werecorded a maximum densification [20].
The evolution of the Fap density versus the heating speed isrepresented in Table 3. The sintering temperature was then fixedat 1000 ◦C. We observe that the kinetics does not have a notoriousinfluence on the densification. Consequently, the speed of rise intemperature is fixed at 5 ◦C min−1, because the Fap was stable athigh temperature under 1100 ◦C, as noticed in previous works [36].Higher speeds are to be avoided because they can induce differ-ential constraints due to the variation in temperature between thesurface and the interior of samples.
During heat treatment, a compromise was required between thecreation of porosity with high percentages and large pores diame-ters, and a good compressive strength of the samples. Indeed, theprepared matrices can then be schematized like a network of spher-ical macrospores inter-connected in three-dimensional; the wallssurrounding them are responsible for the mechanical resistance.According to the thermal analysis and previous works, we can fixthe sintering temperature at 1000 ◦C with a rise in temperatureabout of 5 ◦C min−1 [20,36].
3.2. Fabrication of porous Fap ceramics
3.2.1. Optimization of the sintering conditionsThe temperature of the PVB elimination was evaluated by ther-
mogravimetric analysis with several heating kinetics (Fig. 2a). Theresults obtained indicate a beginning of the polymeric compounddegradation at temperatures as much lower than the heating rateis low. The kinetic study of the mass lost evolution for constant
Table 2SSA, DTA measurements, sintering domain temperature and theoretical density of Fap powder
SSA (m2 g−1) DTA measurements (endothermic peak) Sintering domain T (◦C) da
Fap (calcined at 500 ◦C) [21] 29.00 1180 ◦C (liquid phase) 715–1100 3.19
a Theoretical density.
Author's personal copy
K. Chaari et al. / Materials Chemistry and Physics 113 (2009) 219–226 221
Table 3Relative density versus heating time of samples sintered at 1000 ◦C
Heating rate (◦C min−1) 2 5 10 20Density (%) ±1 95.6 95.7 95.8 95.9
temperatures with a speed of rise in temperature about 1 ◦C min−1
is given in Fig. 2b. We note that for treating times lower than 20 h,the PVB combustion is not total. The percentage of residual poly-mer is as much weaker than the calcination temperature is high.Indeed, for a temperature about of 340 ◦C, the PVB degradation iscarried out over 18 h, but only 70% of the PVB are eliminated. Thus,it proves to be necessary to produce a second stage at 400 ◦C toeliminate the polymer completely (Fig. 2a). Indeed, slow heating isrequired to minimize the potential for cracking due to gas pressurebuild up. So, it is necessary to proceed with precaution.
Thus, the thermal cycle must make it possible to spread out intime the gaseous emissions resulting from the polymer combustionin order not to deteriorate the material cohesion. The PVB degrada-tion optimal cycle permits the macroporosity generation withoutdamage, which can be summarized according to following ther-mal operations; in the first stage, samples were heated slowly, at1 ◦C min−1 to 340 ◦C, and held for 20 h, in the second with the samerise to 400 ◦C used to ensure complete combustion of the residualPVB. This was followed by a more rapid temperature rise 5 ◦C min−1
up to the sintering temperature to 1000 ◦C.
3.2.2. Sintering and characterization of porous Fap ceramicsAt heat treatment, we search to find a compromise between the
creation of porosity having high percentages and large pores, and agood breaking strength of the samples.
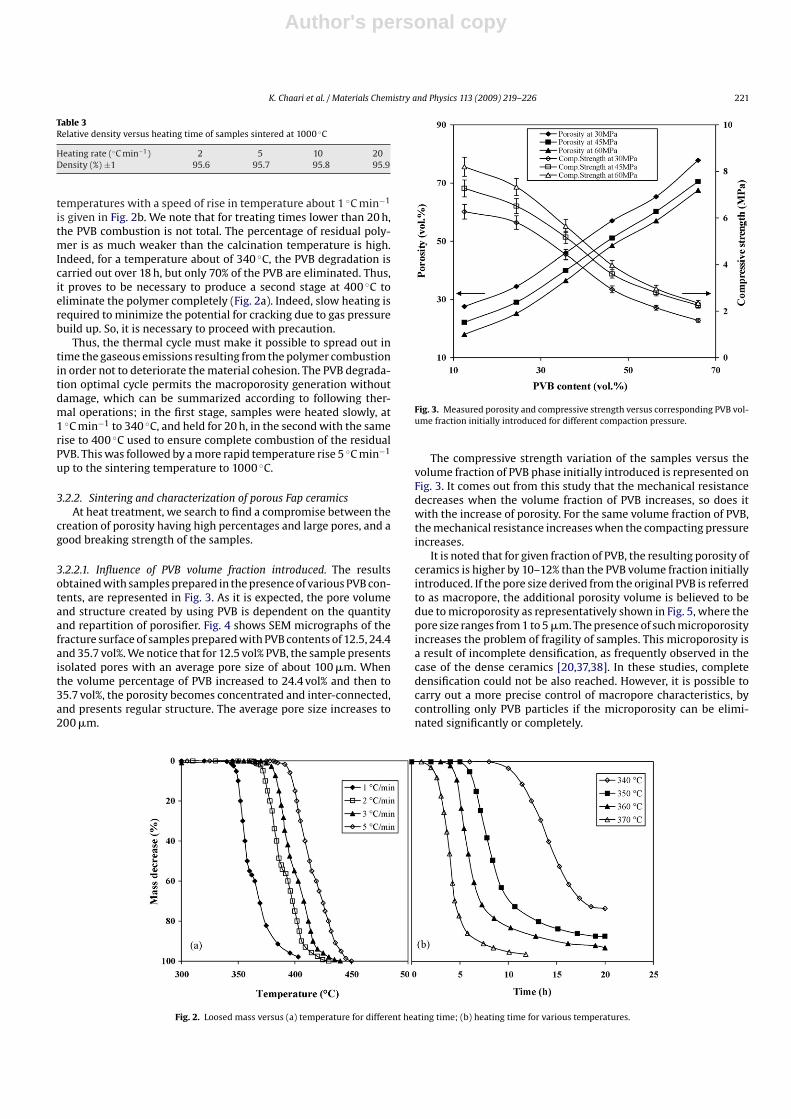
3.2.2.1. Influence of PVB volume fraction introduced. The resultsobtained with samples prepared in the presence of various PVB con-tents, are represented in Fig. 3. As it is expected, the pore volumeand structure created by using PVB is dependent on the quantityand repartition of porosifier. Fig. 4 shows SEM micrographs of thefracture surface of samples prepared with PVB contents of 12.5, 24.4and 35.7 vol%. We notice that for 12.5 vol% PVB, the sample presentsisolated pores with an average pore size of about 100 �m. Whenthe volume percentage of PVB increased to 24.4 vol% and then to35.7 vol%, the porosity becomes concentrated and inter-connected,and presents regular structure. The average pore size increases to200 �m.
Fig. 3. Measured porosity and compressive strength versus corresponding PVB vol-ume fraction initially introduced for different compaction pressure.
The compressive strength variation of the samples versus thevolume fraction of PVB phase initially introduced is represented onFig. 3. It comes out from this study that the mechanical resistancedecreases when the volume fraction of PVB increases, so does itwith the increase of porosity. For the same volume fraction of PVB,the mechanical resistance increases when the compacting pressureincreases.
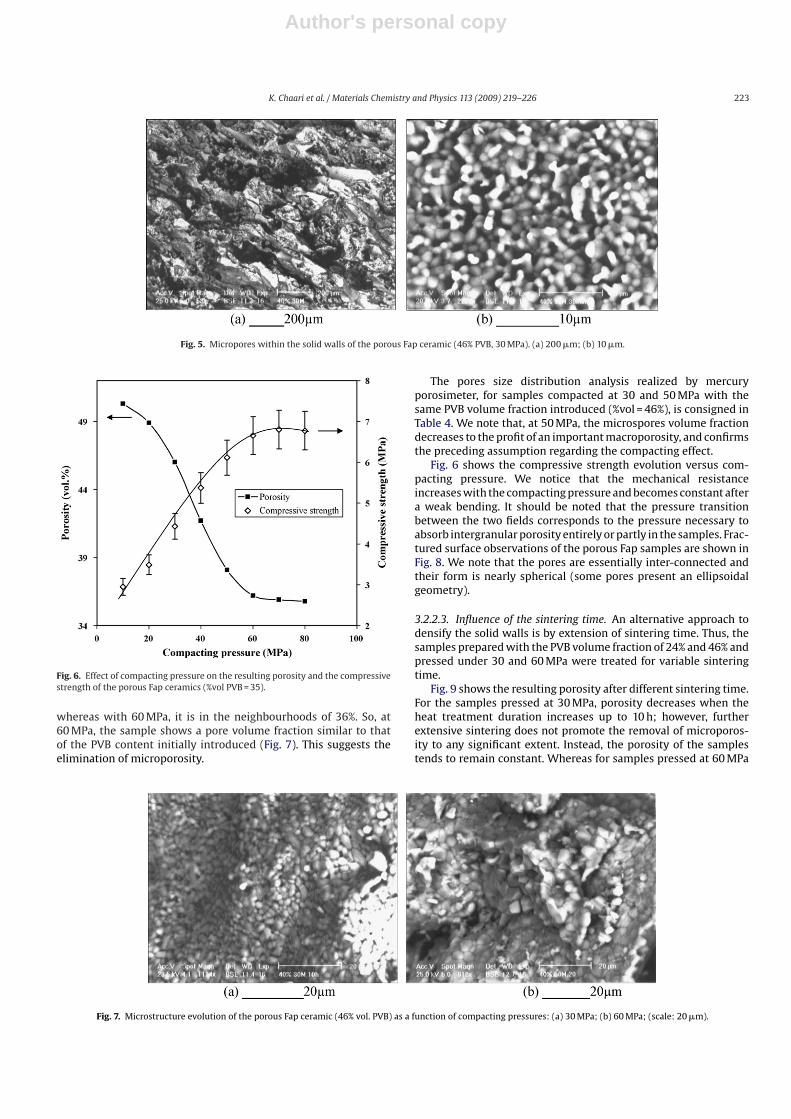
It is noted that for given fraction of PVB, the resulting porosity ofceramics is higher by 10–12% than the PVB volume fraction initiallyintroduced. If the pore size derived from the original PVB is referredto as macropore, the additional porosity volume is believed to bedue to microporosity as representatively shown in Fig. 5, where thepore size ranges from 1 to 5 �m. The presence of such microporosityincreases the problem of fragility of samples. This microporosity isa result of incomplete densification, as frequently observed in thecase of the dense ceramics [20,37,38]. In these studies, completedensification could not be also reached. However, it is possible tocarry out a more precise control of macropore characteristics, bycontrolling only PVB particles if the microporosity can be elimi-nated significantly or completely.
Fig. 2. Loosed mass versus (a) temperature for different heating time; (b) heating time for various temperatures.

Author's personal copy
222 K. Chaari et al. / Materials Chemistry and Physics 113 (2009) 219–226
Fig. 4. Macropores evolution with PVB volume fraction initially introduced in the porous Fap ceramic: (a) 12.5 vol% PVB, scale: 1 mm, 15×; (b) 12.5 vol% PVB, scale: 200 �m,75×; (c) 24.4 vol% PVB, scale: 1 mm, 15×; (d) 24.4 vol% PVB, scale: 200 �m, 75×; (e) 35.7 vol% PVB, scale: 1 mm, 15× (f) 35.7 vol% PVB, scale: 200 �m, 75×.
3.2.2.2. Influence of the compacting pressure. To try to control poros-ity, we have to study the effect of compacting pressure variation.The volume fraction of PVB phase initially introduced is fixed at 35%(vol%).
Fig. 6 shows that the pore volume decreases slowly when thecompacting pressure increases up to 30 MPa, then falls very quickly
beyond this value. For pressures higher than 60 MPa, porosityremains constant. This is reasonable because the mixture (Fap pow-ders + PVB particles) is expected to be consolidated more denselyat higher compacting pressure and this promotes the removal ofmicroporosity within the solid walls. Under these conditions, for acompacting pressure of 30 MPa, the pores volume is about of 46%,
Table 4Pore size distribution of the sintered porous ceramics at two compacting pressures
Compacting pressure (MPa) Total porosity (%vol) Porosity >5 �m macroporosity (%) ±1 Porosity <5 �m microporosity (%) ±0.5
30 57.0 32.6 24.450 49.2 37.3 11.9
The volume fraction of PVB phase initially introduced is fixed at 46% vol. PVB.
Author's personal copy
K. Chaari et al. / Materials Chemistry and Physics 113 (2009) 219–226 223
Fig. 5. Micropores within the solid walls of the porous Fap ceramic (46% PVB, 30 MPa). (a) 200 �m; (b) 10 �m.
Fig. 6. Effect of compacting pressure on the resulting porosity and the compressivestrength of the porous Fap ceramics (%vol PVB = 35).
whereas with 60 MPa, it is in the neighbourhoods of 36%. So, at60 MPa, the sample shows a pore volume fraction similar to thatof the PVB content initially introduced (Fig. 7). This suggests theelimination of microporosity.
The pores size distribution analysis realized by mercuryporosimeter, for samples compacted at 30 and 50 MPa with thesame PVB volume fraction introduced (%vol = 46%), is consigned inTable 4. We note that, at 50 MPa, the microspores volume fractiondecreases to the profit of an important macroporosity, and confirmsthe preceding assumption regarding the compacting effect.
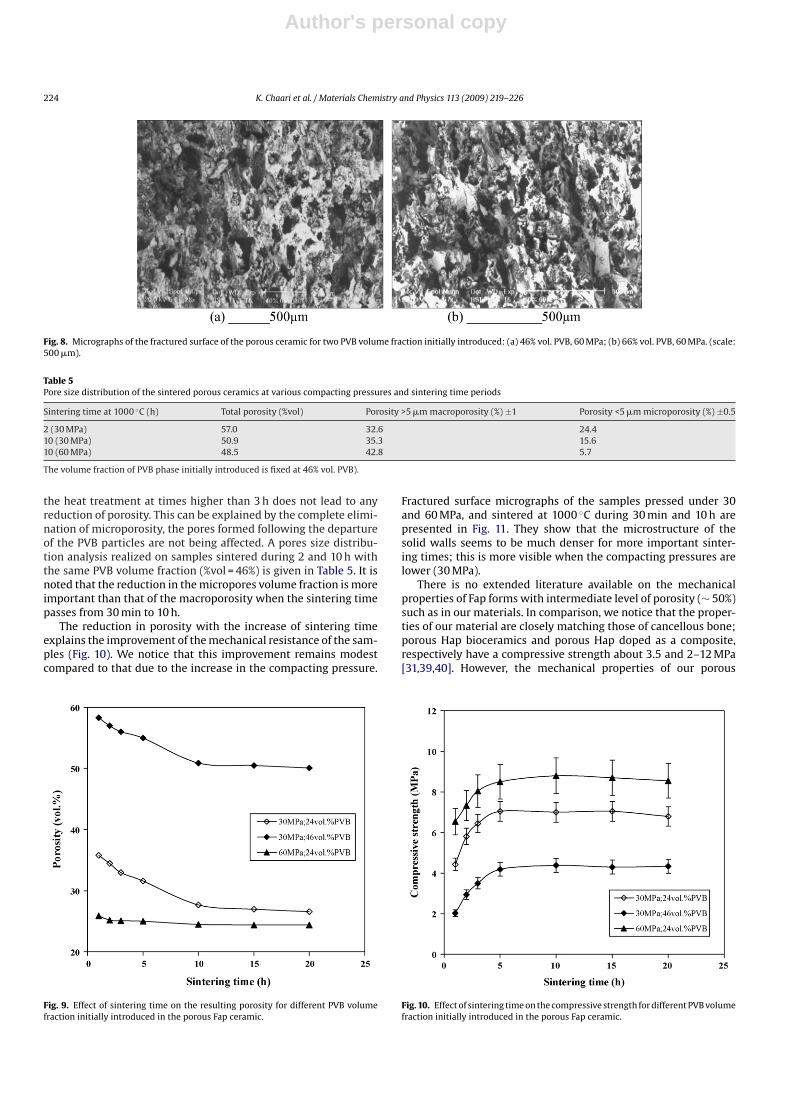
Fig. 6 shows the compressive strength evolution versus com-pacting pressure. We notice that the mechanical resistanceincreases with the compacting pressure and becomes constant aftera weak bending. It should be noted that the pressure transitionbetween the two fields corresponds to the pressure necessary toabsorb intergranular porosity entirely or partly in the samples. Frac-tured surface observations of the porous Fap samples are shown inFig. 8. We note that the pores are essentially inter-connected andtheir form is nearly spherical (some pores present an ellipsoidalgeometry).
3.2.2.3. Influence of the sintering time. An alternative approach todensify the solid walls is by extension of sintering time. Thus, thesamples prepared with the PVB volume fraction of 24% and 46% andpressed under 30 and 60 MPa were treated for variable sinteringtime.
Fig. 9 shows the resulting porosity after different sintering time.For the samples pressed at 30 MPa, porosity decreases when theheat treatment duration increases up to 10 h; however, furtherextensive sintering does not promote the removal of microporos-ity to any significant extent. Instead, the porosity of the samplestends to remain constant. Whereas for samples pressed at 60 MPa
Fig. 7. Microstructure evolution of the porous Fap ceramic (46% vol. PVB) as a function of compacting pressures: (a) 30 MPa; (b) 60 MPa; (scale: 20 �m).
Author's personal copy
224 K. Chaari et al. / Materials Chemistry and Physics 113 (2009) 219–226
Fig. 8. Micrographs of the fractured surface of the porous ceramic for two PVB volume fraction initially introduced: (a) 46% vol. PVB, 60 MPa; (b) 66% vol. PVB, 60 MPa. (scale:500 �m).
Table 5Pore size distribution of the sintered porous ceramics at various compacting pressures and sintering time periods
Sintering time at 1000 ◦C (h) Total porosity (%vol) Porosity >5 �m macroporosity (%) ±1 Porosity <5 �m microporosity (%) ±0.5
2 (30 MPa) 57.0 32.6 24.410 (30 MPa) 50.9 35.3 15.610 (60 MPa) 48.5 42.8 5.7
The volume fraction of PVB phase initially introduced is fixed at 46% vol. PVB).
the heat treatment at times higher than 3 h does not lead to anyreduction of porosity. This can be explained by the complete elimi-nation of microporosity, the pores formed following the departureof the PVB particles are not being affected. A pores size distribu-tion analysis realized on samples sintered during 2 and 10 h withthe same PVB volume fraction (%vol = 46%) is given in Table 5. It isnoted that the reduction in the micropores volume fraction is moreimportant than that of the macroporosity when the sintering timepasses from 30 min to 10 h.
The reduction in porosity with the increase of sintering timeexplains the improvement of the mechanical resistance of the sam-ples (Fig. 10). We notice that this improvement remains modestcompared to that due to the increase in the compacting pressure.
Fig. 9. Effect of sintering time on the resulting porosity for different PVB volumefraction initially introduced in the porous Fap ceramic.
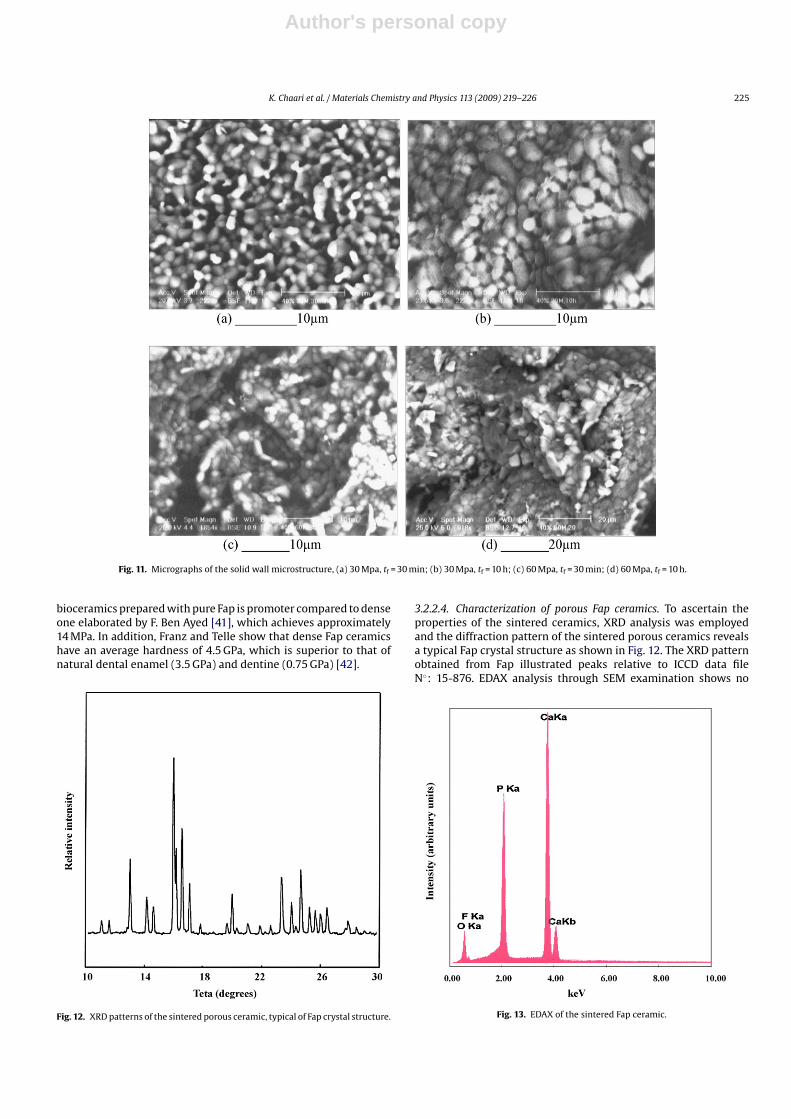
Fractured surface micrographs of the samples pressed under 30and 60 MPa, and sintered at 1000 ◦C during 30 min and 10 h arepresented in Fig. 11. They show that the microstructure of thesolid walls seems to be much denser for more important sinter-ing times; this is more visible when the compacting pressures arelower (30 MPa).
There is no extended literature available on the mechanicalproperties of Fap forms with intermediate level of porosity (∼ 50%)such as in our materials. In comparison, we notice that the proper-ties of our material are closely matching those of cancellous bone;porous Hap bioceramics and porous Hap doped as a composite,respectively have a compressive strength about 3.5 and 2–12 MPa[31,39,40]. However, the mechanical properties of our porous
Fig. 10. Effect of sintering time on the compressive strength for different PVB volumefraction initially introduced in the porous Fap ceramic.
Author's personal copy
K. Chaari et al. / Materials Chemistry and Physics 113 (2009) 219–226 225
Fig. 11. Micrographs of the solid wall microstructure, (a) 30 Mpa, tf = 30 min; (b) 30 Mpa, tf = 10 h; (c) 60 Mpa, tf = 30 min; (d) 60 Mpa, tf = 10 h.
bioceramics prepared with pure Fap is promoter compared to denseone elaborated by F. Ben Ayed [41], which achieves approximately14 MPa. In addition, Franz and Telle show that dense Fap ceramicshave an average hardness of 4.5 GPa, which is superior to that ofnatural dental enamel (3.5 GPa) and dentine (0.75 GPa) [42].
Fig. 12. XRD patterns of the sintered porous ceramic, typical of Fap crystal structure.
3.2.2.4. Characterization of porous Fap ceramics. To ascertain theproperties of the sintered ceramics, XRD analysis was employedand the diffraction pattern of the sintered porous ceramics revealsa typical Fap crystal structure as shown in Fig. 12. The XRD patternobtained from Fap illustrated peaks relative to ICCD data fileN◦: 15-876. EDAX analysis through SEM examination shows no
Fig. 13. EDAX of the sintered Fap ceramic.

Author's personal copy
226 K. Chaari et al. / Materials Chemistry and Physics 113 (2009) 219–226
impurity other than Ca or P elements present (Fig. 13). Therewere no other crystalline phases to be traced during heating. Thisresult is logical because at our sintering temperature fluorapatiteremains steady [20,21]
4. Conclusion
In this study of the production of Fap with controlled porositywe have shown that:
(i) The control of porosity values in the final, sintered Fap sam-ples was found to be attained by essentially changing the PVBamounts, forming pressure and sintering time.
(ii) Slow heating is required to minimize the potential for crackingdue to gas pressure build up.
(iii) The pore sizes in our Fap bioceramics were typically distributedin the range 50–300 �m. In addition the pores were inter-connected.
(iv) The compressive strength tests performed on cylinder piecesof such samples yielded fracture strengths over the range of4–9 MPa.
(v) The sintered specimens exhibited a pure form of Fap charac-terized patterns; there were no other crystalline phases to betraced during heating.
This technique of porous bioceramic manufacturing may easilybe used in other ceramic phases and materials, and therefore, hasa promising potential for future applications. In order to improvethe mechanical properties of these porous Fap bioceramics, manyreinforcements, including ceramic particles and carbon fibers arecurrently underway.
References
[1] K. DeGroot, Biomaterials 1 (1980) 47.[2] M. Jarcho, Clin. Orthop. Rel. Res. 157 (1981) 259.[3] C.J. Damien, J.R. Parsons, J. Appl. Biomater. 2 (1990) 187.[4] R.E. Holmes, Plast. Reconstr. Surg. 63 (1979) 626.[5] R.E. Holmes, V. Mooney, R. Bucholz, A. Tencer, Clin. Orthop. Rel. Res. 188 (1984)
252.[6] E.W. White, E.C. Shors, Dental Clin. N. Am. 30 (1986) 49.[7] E.C. Shors, E.W. White, G. Kopchok, Mater. Res. Soc. Symp. Proc. 110 (1989) 211.[8] H. Ohgushi, K. Okumura, T. Yoshikawa, J. Biomed. Mater. Res. 26 (1992) 885.
[9] C.P.A.T. Klein, K. DeGroot, C. Weiqun, L. Yubao, Z. Xingdong, Biomaterials 15(1994) 31.
[10] M. Fabbri, G.C. Celotti, A. Ravaglioli, Biomaterials 15 (1994) 474.[11] J.C. Le Huec, T. Schaeverbeke, D. Clement, J. Faber, A. LeRebeller, Biomaterials
16 (1995) 113.[12] M. Fabbri, G.C. Celotti, A. Ravaglioli, Biomaterials 16 (1995) 225.[13] D.M. Liu, J. Mater. Sci. Lett. 15 (1996) 419.[14] D.M. Liu, Biomaterials 1996 (17) (1955).[15] N. Passuti, G. Daculsi, J.M. Rogez, S. Martin, J.V. Bainvel, Clin. Orthoped. 148
(1989) 169.[16] J.J. Klawiter, J.G. Bagwell, A.M. Weinstein, B.W. Sauer, J.R. Pruitt, J. Biomed. Mater.
Res. 10 (1976) 311.[17] P.S. Eggli, W. MuÈller, R.K. Schenk, Clin. Orthoped. 232 (1987) 127.[18] G. Daculsi, N. Passuti, Biomaterials 11 (1990) 86.[19] I.H. Arita, V.M. Castano, D.S. Wilkinson, J. Mater. Sci. Mater. Med. 6 (1995) 19.[20] F. Ben Ayed, J. Bouaziz, K. Bouzouita, J. Eur. Ceram. Soc. 20 (8) (2000) 1069.[21] F. Ben Ayed, J. Bouaziz, K. Bouzouita, J. Alloys. Compd. 322 (1–2) (2001) 238.[22] F. Ben Ayed, J. Bouaziz, I. Khattech, K. Bouzouita, Ann. Chim. Sci. Mater. 26 (6)
(2001) 75.[23] K. Chaari, S. Baklouti, J. Bouaziz, K. Bouzouita, Ann. Chim. Sci. Matér. 29 (4)
(2004) 1.[24] K. Chaari, J. Bouaziz, K. Bouzouita, J. Colloid Interface Sci. 285 (2) (2005) 469.[25] F. Ben Ayed, J. Bouaziz, C. R. Physique 8 (1) (2007) 101.[26] F. Ben Ayed, J. Bouaziz, J. Eur. Ceram. Soc. 28 (10) (2008) 1995.[27] H.W. Kim, Y.J. Noh, Y.H. Koh, H.E. Kim, J. Am. Ceram. Soc. 86 (12) (2003) 2019.[28] H.W. Kim, Y.M. Kong, C.J. Bae, Y.J. Noh, H.E. Kim, Biomaterials 25 (2004)
2919.[29] H.W. Kim, H.E. Kim, J.C. Knowles, Biomaterials 25 (2004) 3351.[30] J.C. Heughebaert, Contribution à l’étude de l’évolution des orthophosphates de
calcium précipités en orthophosphates apatitiques, Thèse, INP, Toulouse (1977).[31] A. Gee, V.R. Deitz, Anal. Chem. 25 (1953) 1320.[32] J. Bassett, R.C. Denney, G.H. Jeffery, J. Mendhamn, Vogel’s TextBook of Quanti-
tative Inorganic Analysis, Longman, London (1978) 589.[33] S. Brunauer, P.H. Emmet, J. Teller, J. Amer. Chem. Soc. 60 (1938) 310.[34] ISRM, Suggested methods for determining tensile strength of rock materials,
Int. J. Rock Mech. Min. Sci. Geomech. Abstr. 15 (1978) 99.[35] ASTM C496, Standard test method for splitting tensile strength of cylindri-
cal concrete specimens Annual Book of ASTM, Standards, vol. 0.042, ASTM,Philadelphia, 1984, p. 336.
[36] E. Fidancevska, G. Ruseska, J. Bossert, Y.-M. Lin, A.R. Boccaccini, Mater.Chem.Phys. 103 (2007) 95.
[37] A. Royer, J.C. Viguie, M. Heughebaert, J.C. Heughebaert, J. Mater. Sci.: Mater.Med. 4 (1993) 76.
[38] A.C. Tas, F. Korkusuz, M. Timuein, N. Akkas, J. Mater. Sci.: Mater. Med. 8 (1997)91.
[39] R. Van Audekerche, M. Martens, in: G.W. Hasting, P. Ducheyne (Eds.), Mechan-ical Properties of Cancellous Bone Natural and Living Biomaterials, CRC Press,Boca Raton, FL, 1984, p. 88.
[40] A. Odgaard, F. Linde, J. Biomechanics 8 (1991) 691.[41] F. Ben Ayed, J. Bouaziz, K. Bouzouita, Ann. Chim. Sci. Mater. 31 (4) (2006) 393.[42] E.D. Franz, R. Telle, Reaction hot pressing fluorapatite for dental implants, in:
P. Vincenzini (Ed.), High Tech. Ceram., Elsevier Science Publishers B.V., Amster-dam, 1987, p. 31.





























