Embed Size (px)
Citation preview

& Electrochemistry
Low-Band-Gap BODIPY Conjugated Copolymers for SensingVolatile Organic Compounds**
Choong Ping Sen,[a] Rekha Goswami Shrestha,[b] Lok Kumar Shrestha,[b] Katsuhiko Ariga,[b]
and Suresh Valiyaveettil*[a]
Abstract: Conjugated polymers with strong photophysical
properties are used in many applications. A homopolymer(P1) and five new low band gap copolymers based on 4,4’-difluoro-4-bora-3a,4a-diaza-s-indacene (BODIPY) and accept-
ors 3,6-dithienyldiketopyrrolopyrrole (P2), phthalimide (P3),benzotriazole (P4), 4,7-dithienyl[1,2,3]triazolo[4,5g]quinoxa-
line (P5), and 2,5-dithienylthieno[3,4-b]pyrazine (P6) wereprepared by means of Sonogashira polymerization. The char-
acterization of polymers by using 1H NMR, absorption, and
emission spectroscopy is discussed. All polymers with highmolecular weights (Mn) of 16 000 to 89 000 g mol¢1 showed
absorption maxima in the deep-red region (l= 630–760 nm)in solution and exhibited significant redshifts (up to 70 nm)
in thin films. Polymers P2, P5, and P6 showed narrow optical
band gaps of 1.38, 1.35, and 1.38 eV, respectively, which aresignificantly lower than that of P1 (1.63 eV). The HOMO andLUMO energy levels of the polymers were calculated by
using cyclic voltammetry measurements. The LUMO energylevels of BODIPY-based alternating copolymers were inde-
pendent of the acceptors ; this suggests that the majorfactor that tunes the LUMO energy levels of the polymers
could be the BODIPY core. All polymers showed selective
and reproducible detection of volatile organic solvents, suchas toluene and benzene, which could be used for develop-
ing sensors.
Introduction
4,4’-Difluoro-4-bora-3a,4a-diaza-s-indacene dyes, more com-monly known as boron dipyrromethene (BODIPY) dyes, have
attracted widespread interest owing to their optical properties,such as high molar absorption coefficient, high quantum yield,sharp absorption and emission bands, and high photostabili-ty.[1, 2] The optical properties of the BODIPY dyes can be fine-
tuned with structural modification or incorporating small sub-stituents on different positions of the BODIPY core.[3, 4] Suchstructure–property versatility of BODIPY derivatives, combinedwith synthetic accessibility, has sparked intense research onthe uses of BOIDPY in many promising applications, such as
tagging of biomolecules,[5] fluorescent chemosensors,[6] laserdyes,[7] photochromic devices,[8, 9] and light-harvesting sys-
tems.[10–15] To date, only a few reports on BODIPY-based conju-
gated polymers have been published.[16–20]
The band gap engineering of conjugated polymers byusing a donor–acceptor diad was also used for developing
BODIPY-incorporated low-band-gap conjugated copolymers.[21]
Electron deficient groups, such as phthalic amide[22–24] and ben-
zotriazole,[25–27] are attractive candidates as electron-acceptingmoieties in conjugated polymers, which show enhancement inlight absorption, better p stacking, improved interchain inter-actions, and increased solar cell performances.[28–30] Recently,
Toppare and co-workers reported triazoloquinoxaline-incorpo-rated conjugated polymers with narrow band gaps (1.00 eV)and broad absorptions between l= 700 and 1000 nm.[27]
Similarly, diketopyrrolopyrrole (DPP)-incorporated polymersshowed low band gaps,[31] low HOMO and LUMO energy
levels, broad absorptions that extended to the near-infraredregion,[32] strong p stacking,[33] high hole and electron mobili-
ties,[33–35] and high power conversion efficiencies in organic
solar cells.[32, 36]
The rapid deterioration of the environment owing to the
emission of volatile organic compounds from chemical indus-tries demands the development of sensors that could detect
such compounds rapidly and selectively upon exposure tocontaminated air. Many research groups have used a quartzcrystal microbalance (QCM)[37] to detect small molecules, for
protein immobilization, and gas sensing by measuring thechanges in frequency of the quartz crystal resonator accompa-
nied by a variation in mass changes per unit area through ad-sorption of molecules and macromolecules. Conjugated poly-
mers, such as polyaniline[38, 39] and polythiophene,[40, 41] coatedon QCM electrodes were also used for the development of
[a] C. P. Sen, Prof. Dr. S. ValiyaveettilFaculty of Science, Department of ChemistryNational University of Singapore3 Science Drive 2, Singapore 117543 (Singapore)E-mail : [email protected]
[b] Dr. R. G. Shrestha, Dr. L. K. Shrestha, Prof. Dr. K. ArigaWorld Premier International Center for MaterialsNanoarchitectonics (WPI-MANA)National Institute for Materials Science (NIMS)1-1 Namiki, Ibaraki, Tsukuba 305-0044 (Japan)
[**] BODIPY = 4,4’-difluoro-4-bora-3a,4a-diaza-s-indacene.
Supporting information for this article is available on the WWW underhttp ://dx.doi.org/10.1002/chem.201502939.
Chem. Eur. J. 2015, 21, 17344 – 17354 Ó 2015 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim17344
Full PaperDOI: 10.1002/chem.201502939

sensors by monitoring changes in mass through adsorption ofvolatile organic vapors.
Herein, we report the synthesis and characterization of low-band-gap copolymers of BODIPY with five different acceptors
incorporated into the main chain for the detection of differentorganic solvents. Polymers are designed to understand the
effect of different acceptor molecules on the overall propertiesof the polymers. A homopolymer of BODIPY (P1) and five new
alternating copolymers (P2–P6) were synthesized by means of
Sonogashira polymerization (Scheme 1). Different electron ac-
ceptors, 3,6-dithienyldiketopyrrolopyrrole (P2), phthalimide
(P3), benzotriazole (P4), 4,7-dithienyl[1,2,3]triazolo[4,5g]qui-noxaline (P5), and dithienylthieno[3,4-b]pyrazine (P6), were se-
lected for copolymerization with BODIPY. Full characterization
of the optical and electrochemical properties of all polymers isdiscussed in detail. It is conceivable that the affinity of the vol-
atile molecules depends on the structures and properties ofthe polymer backbone. Interactions of all target polymers with
a few common solvent vapors are examined by using a QCMsensor.
Results and Discussion
Synthesis and characterization
2,6-Diethynyl-BODIPY derivatives (5 a and 5 b) were synthesizedaccording to reported procedures with a few modifica-
tions.[16, 18, 19, 42] The acceptor monomers 8,[43] 10,[44] 12,[45] 16,[27]
and 19[27, 46] were synthesized and characterized by means of
NMR spectroscopy and mass spectrometry (see Figures S1–S14in the Supporting Information). The BODIPY homopolymer P1was synthesized according to a reported procedure.[47] Thetarget polymers were synthesized by means of Sonogashira
polycondensation[18, 47] in the presence of [Pd(PPh3)4] as a cata-lyst and CuI as a cocatalyst (Scheme 2), dissolved in tetrahydro-furan (THF)/diisopropylamine (DIPA) under nitrogen. The reac-
tion mixture was heated at reflux under darkness for 48 h toyield the target polymers. All crude polymers were purified by
Soxhlet extraction to yield the pure polymers (P2–P6) in goodyield (68–87 %). The purified polymers are soluble in commonorganic solvents, such as chloroform, THF, chlorobenzene, and1,2-dichlorobenzene.
The molecular weights of all polymers were calculated byusing gel permeation chromatography (GPC) with THF as theeluent and polystyrene as standards. As expected, the 1H NMR
signals of the polymers were broader than those of the mono-mers. All signals in the 1H NMR spectra of the monomers and
polymers are comparable, except for the disappearance of theethynyl proton signal at d= 3.31 ppm (see Figure S15–S20 in
the Supporting Information). The estimated number-averaged
molecular weights (Mn) of the polymers ranged from 1.6 Õ 104
to 8.9 Õ 104 g mol¢1 with a polydispersity index (PDI) of less
than 2 (Table 1). The incorporation of branched alkyl chains onthe monomers led to the formation of soluble high-molecular-
weight polymers.
Thermal properties of polymers
The thermal stability of the polymers was evaluated by means
of TGA at a heating rate of 10 8C min¢1 under a nitrogen at-mosphere. All samples were dried in a vacuum oven at 50 8C
to remove traces of solvents before analysis. From the data,polymer P2 has the highest decomposition temperature
(431 8C), whereas P6 has the lowest (381 8C; Figure 1 and
Table 1). In comparison, the decomposition temperatures ofP1, P3, P4, and P5 were 390, 404, 415, and 418 8C, respectively,
(Table 1), which implies high thermal stability. The phase transi-tion of the polymers was studied by means of differential scan-
ning calorimetry (DSC) at a heating rate of 10 8C min¢1 undera nitrogen atmosphere. No clear phase transition can be ob-
served (see Figures S21–S26 in the Supporting Information),which indicates a rigid polymer backbone.[16, 18]
Photophysical properties of the BODIPY-based conjugatedpolymers
The absorption properties of the BODIPY-based conjugated
polymers (P1–P6) were measured in dilute chloroform and as
thin films. The 2,6-diethynyl-BODIPY core possesses a character-istic So!S1 (p to p*) transition at l�540 nm and a So!S2 (p
to p*) transition at l�350–450 nm.[16] Significant redshifts inthe absorption maximum were observed for all polymers (P1–
P6) relative to that of the BODIPY monomer due to extendedconjugation along the polymer backbone. The absorption max-
Scheme 1. Chemical structures of BODIPY polymers P1–P6.
Chem. Eur. J. 2015, 21, 17344 – 17354 www.chemeurj.org Ó 2015 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim17345
Full Paper

imum for BODIPY homopolymerP1 was measured in chloroform
at l = 665 nm and in a thin filmat l= 725 nm (Figure 2 A). The
observed second absorptionmaximum at l= 720 nm in
chloroform with a comparableextinction coefficient to that of
l= 665 nm is accounted for the
aggregated form of P1. Whena poor solvent, hexane, was
added to the solution of P1 inchloroform, the intensity of the
absorption at l= 665 nm wasquenched, whereas the intensityat l= 720 nm was enhanced.
The observed changes for allpolymers in chloroform before
and after the addition of hexaneare shown in Figure 2.
The absorption maxima of P3and P4 in chloroform are l= 610
and 630 nm, respectively, where-
as the absorption maximum ofP4 is more redshifted (20 nm)
than that of P3. The emissionmaxima of P3 and P4 are l=
640 and 650 nm, respectively, indilute chloroform. The absorp-
tion maxima of P3 and P4 in the
solid state are l= 680 and 700 nm, respectively, as a resultfrom polymer aggregation. No significant differences were
found in the solid-state optical band gap of polymers by re-placing the phthalimide moiety in P3 with the benzotriazole
group in P4. However, when DPP is copolymerized withBODIPY to form P2, the absorption maximum in chloroform
and the thin film are significantly redshifted to l= 760 and
780 nm, respectively, and the emission maximum to l=
800 nm. The optical band gaps of P2 in chloroform and in the
thin film are estimated to be 1.51 and 1.38 eV. These observa-tions are consistent with the fact that DPP is a stronger accept-
or, which leads to a polymer with a low band gap.[31, 32]
The absorption maxima of P5 in chloroform and in the thin
film are l= 690 and 730 nm, respectively, and the optical bandgap in the thin film is 1.35 eV. Meanwhile, the optical bandgap for P5 is much lower than those of other polymers be-
cause the combination of quinoxaline and benzotriazole intoone acceptor unit provides a highly electron-deficient acceptor
unit. The absorption maximum has shifted to l= 625 nm inchloroform and l= 710 nm in P6. The large shift in absorption
maximum (85 nm) of P6 from solution to the solid state indi-
cates an efficient p–p transition and strong interaction be-tween polymer chains in P6 relative to that of P5 (40 nm). This
might be due to a high electron density of thiophene, whichresults in stronger electrostatic interactions between polymer
chains. The optical band gap for P6 is 1.38 eV. It is observedthat P2, P5, and P6 have a broad absorption range that covers
Scheme 2. Synthesis of BODIPY-based conjugated polymers P1–P6.
Table 1. Molecular weights of BODIPY-based conjugated polymers.
Polymer Mn[a]
[g mol¢1]Mw
[a]
[g mol¢1]PDI[a] Tdec
[b]
[8C]
P1 75 600 109 560 1.45 390P2 89 760 142 100 1.58 431P3 58 310 116 210 1.99 404P4 68 120 123 500 1.81 415P5 16 860 25 530 1.51 418P6 44 060 65 570 1.49 381
[a] Mn, Mw, and PDIs were determined by GPC by using polystyrene asa standard in THF. [b] The decomposition temperature was determined atthe midpoint of the thermogravimetric analysis (TGA) weight loss curve.
Figure 1. TGA results for BODIPY-based conjugated polymers under a nitro-gen atmosphere at a heating rate of 10 8C min¢1. Most polymers showeddegradation temperatures in the range of 310–430 8C.
Chem. Eur. J. 2015, 21, 17344 – 17354 www.chemeurj.org Ó 2015 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim17346
Full Paper

the visible to near-infraredregion. In particular, polymer P2has the highest extinction coeffi-cient of all polymers, which
makes it a potential polymer fororganic photovoltaic (OPV) appli-
cations. Polymers P2 and P5showed larger Stokes shifts than
those of P1, P3, P4, and P6,
which indicated that the laterpolymers had rigid conforma-
tions in the solution state. Thefluorescence quantum yields of
P1–P6 (Table 2) were determinedin chloroform by using fluores-cein (0.1 m NaOH, quantum yield
of 0.85) as a reference.[18, 48] Poly-mers P1, P3, and P4 showed
quantum yields of 18.3, 19.0,and 15.4 %, respectively. In con-
trast, smaller fluorescence quan-tum yields were found in P2, P5,
and P6 with 3.8, 2.9, and 2.1 %,
respectively, owing to the heavysulfur atom effect from thio-
phene.[18, 47, 49] Moreover, solventeffects on the polymer were ex-
amined by taking absorptionspectra in toluene (see Fig-
ure S27 in the Supporting Infor-
mation). Polymers P2 (20 nm)and P5 (15 nm) showed the larg-
est redshifts of lmax, whereasthose of P1 (6 nm), P3 (10 nm),
and P4 (5 nm) were smaller. Thissuggests that the DPP and tria-
zoloquinoxaline moieties in P2and P5 have a stronger affinitytowards toluene.
Electrochemical properties ofBODIPY-based conjugated poly-mers
Cyclic voltammetry (CV) wasused to evaluate the redox be-
havior of the polymers. A solu-tion of polymer in chloroform
(1 mg mL¢1) was drop-cast ona 2 mm diameter platinum disk
electrode to form a thin film.
The cyclic voltammograms wererecorded in a 0.1 m solution of
tetrabutylammonium hexafluoro-phosphate in anhydrous acetoni-
trile at a scan rate of 100 mV s¢1
and a standard calomel elec-
Figure 2. A) Absorption spectra of polymers in chloroform (&), a 1:1 mixture of chloroform/hexane (*), and thinfilms (~). B) Solution-state photoluminescence spectra of polymers in chloroform (&) and a 1:1 mixture of chloro-form/hexane (*). Polymer thin films were prepared by drop-casting a solution of polymer onto quartz plates, fol-lowed by slow evaporation at room temperature.
Chem. Eur. J. 2015, 21, 17344 – 17354 www.chemeurj.org Ó 2015 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim17347
Full Paper

trode (SCE) as a reference electrode, which was calibrated withthe ferrocene/ferrocenium ion (Fc/Fc+) redox couple (Figure 3).
The oxidation and reduction potentials were determined fromthe onset potentials of oxidation and reduction sweeps. The
corresponding HOMO and LUMO energy levels of the polymerswere estimated by assuming the absolute HOMO energy level
of ferrocene to be ¢4.8 eV relative to the vacuum level. All
electrochemical data of the polymers are summarized inTable 3 for comparison. CV traces of BODIPY homopolymer P1show two irreversible peaks: an oxidation at + 1.03 V and a re-duction at ¢1.71 V. The latter is the characteristic reduction
peak of the BODIPY core. The HOMO and LUMO energy levelsof P1 are ¢5.32 and ¢3.65 eV, respectively, and the electro-
chemical band gap is 1.67 eV. Both P3 and P4 show similar oxi-
dation and reduction patterns to that of P1. In addition, theHOMO energy levels of P3 and P4 are ¢5.36 and ¢5.32 eV, re-
spectively, whereas the LUMO energy levels of both polymersare ¢3.68 eV. Because the HOMO and LUMO energy levels in
P1, P3, and P4 are similar, the incorporation of benzotriazoleor phthalimide units as acceptors did not significantly perturb
the energy levels of the copolymers. The electrochemical band
gaps of P1, P3, and P4 are 1.67, 1.68, and 1.64 eV, respectively,which are in good agreement with the thin-film optical band
gaps.Polymer P2 shows a quasi-reversible oxidation at + 0.75 V
and an irreversible oxidation at + 1.00 V. The quasi-reversibleoxidation can be assigned to the oxidation of DPP and
a second oxidation peak arises from oxidation of the BODIPYcore. However, there is only one irreversible reduction peakobserved at ¢1.78 V for P2. The corresponding HOMO andLUMO energy levels of P2 are ¢5.15 and ¢3.62 eV, respective-ly. In comparison, polymer P2, with the DPP unit, has relatively
high-lying HOMO energy levels and similar LUMO energylevels to those of P1. The electrochemical band gap of P2 is
1.54 eV, which is lower than those of P1, P3, and P4. This is ex-pected because the DPP unit is a strong acceptor, as revealedfrom results of photophysical studies. On the other hand, cou-
pling of the BODIPY unit with P3 or P4 has shown minimumchanges in LUMO–HOMO energy levels and band gaps of the
polymers. This indicates that the electron affinities of bothphthalimide and benzotriazole units are comparable.
Polymer P5 shows two irreversible oxidation peaks at + 0.72and at + 0.97 V. The first peak corresponds to the oxidation of
quinoxaline and the second peak is from the oxidation of theBODIPY core. On the CV reduction sweep, polymer P5 shows
a different reduction pattern from those of P1–P4. Three re-duction peaks at ¢1.35, ¢1.48, and ¢1.85 V were observed,
and accounted for characteristic reduction peaks of the qui-
noxaline unit in P5.[27] The corresponding HOMO and LUMOenergy levels are at ¢5.21 and ¢3.69 eV, respectively, and the
calculated electrochemical band gap is 1.52 eV. Similarly, poly-mer P6 shows two irreversible oxidation peaks at + 0.63 and
+ 1.04 V, and an irreversible reduction peak at ¢1.55 V. TheHOMO and LUMO energy levels of P6 calculated from the oxi-
dation and reduction onsets are ¢5.14 and ¢3.61 eV, respec-
tively. By attaching dithenyl[1,2,3]triazolo[4,5g]quinoxaline (15)or di(thiophen-2-yl)thieno[3,4-b]pyrazine (18) to the BODIPY on
the polymer backbone, the HOMO energy levels were destabi-lized by 0.11–0.18 V, compared with that of P1. The LUMO
energy levels of P5 and P6 are similar to that of P1. Also, allpolymers (P1–P6) show similar values in LUMO energy levels
of around 3.61–3.69 eV, which implies that the LUMO energy
levels of BODIPY-based copolymers are not significantly affect-ed by the nature of acceptors on the polymer backbone.[20]
Theoretical calculations on BODIPY polymers
DFT calculations were performed on the monomers and model
dimers in the gas phase with corresponding repeat units togain a better understanding of the electrochemical propertiesof polymers P1–P6. The frontier orbital energy levels were esti-mated from the optimized geometry by using the Gaussi-an 09[50] program at the B3LYP/6-311g(d,p) level. HOMO and
LUMO surface plots of model compounds are compared inFigure 4. In the case of P3 and P4, the HOMO wave function is
delocalized on both monomers, whereas the LUMO wave func-
tion is exclusively found on the BODIPY moiety. Thus, polymersP3 and P4 are expected to show similar optical and electro-
chemical properties to those of P1. This is reflected from boththeoretical and experimental studies that P1, P3, and P4 exhib-
it similar frontier orbitals energy levels and band gaps(Table 4).
Table 2. Summary of the photophysical properties of BODIPY-based conjugated polymers (P1–P6).
Polymer Absorption Fluorescence[a]
CHCl3 Thin film CHCl3 Toluene CHCl3
lmax [nm] e[b] [cm2 mg¢1] lonset[c] [nm] Eg
[e]opt soln [eV] lmax
[d] [nm] lonset[c] [nm] Eg
[e]opt film [eV] lem [nm] lem [nm] Ff
[f] [%]
P1 665 78 735 1.69 725 760 1.63 680 685 18.3P2 760 105 820 1.51 780 895 1.38 800 805 3.8P3 610 70 715 1.74 680 735 1.69 640 645 19.0P4 630 75 715 1.73 700 740 1.68 650 650 15.4P5 690 40 840 1.48 730 920 1.35 750 750 2.9P6 625 50 825 1.51 710 895 1.38 650 655 2.1
[a] No detectable emission was observed from thin films of the polymers, owing to strong quenching effects. The concentrations of P1, P2, P3, P4, P5, andP6 were 3.0, 2.0, 3.6, 4.8, 1.8, and 4.0 mg mL¢1, respectively. [b] The extinction coefficient, e, was calculated by dividing absorbance by concentration[mg cm¢3] and cuvette path length (1 cm). [c] lonset was calculated from the intersection of the tangent lines drawn to the lowest energy absorption edgeto the baseline. [d] lmax was measured for the polymer thin film on a quartz plate. [e] Eg = 1240/lonset. [f] Fluorescence quantum yields of the polymers weredetermined in chloroform by using fluorescein (0.1 m NaOH, quantum yield of 0.85) as a reference.
Chem. Eur. J. 2015, 21, 17344 – 17354 www.chemeurj.org Ó 2015 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim17348
Full Paper
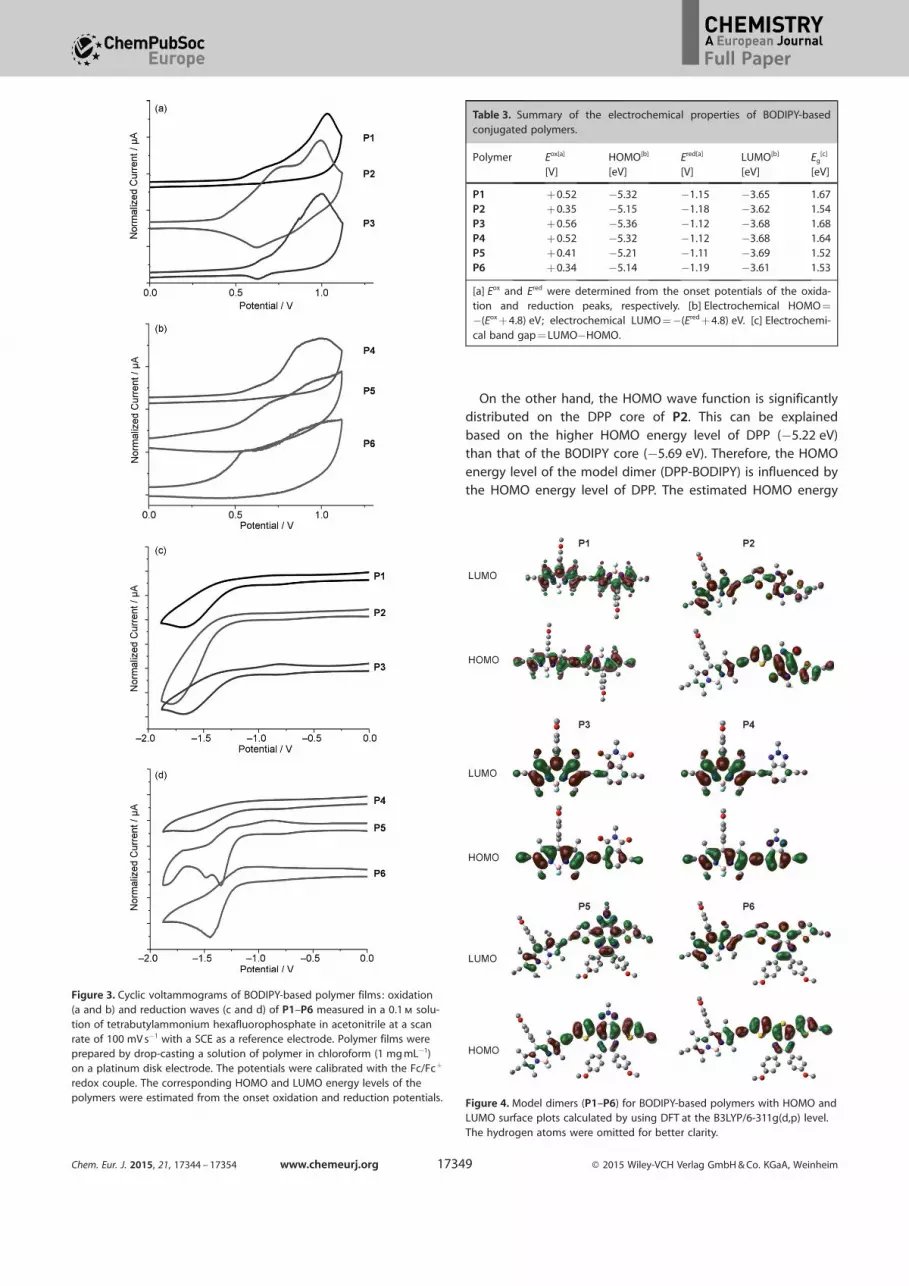
On the other hand, the HOMO wave function is significantlydistributed on the DPP core of P2. This can be explained
based on the higher HOMO energy level of DPP (¢5.22 eV)than that of the BODIPY core (¢5.69 eV). Therefore, the HOMO
energy level of the model dimer (DPP-BODIPY) is influenced bythe HOMO energy level of DPP. The estimated HOMO energy
Figure 3. Cyclic voltammograms of BODIPY-based polymer films: oxidation(a and b) and reduction waves (c and d) of P1–P6 measured in a 0.1 m solu-tion of tetrabutylammonium hexafluorophosphate in acetonitrile at a scanrate of 100 mV s¢1 with a SCE as a reference electrode. Polymer films wereprepared by drop-casting a solution of polymer in chloroform (1 mg mL¢1)on a platinum disk electrode. The potentials were calibrated with the Fc/Fc+
redox couple. The corresponding HOMO and LUMO energy levels of thepolymers were estimated from the onset oxidation and reduction potentials.
Table 3. Summary of the electrochemical properties of BODIPY-basedconjugated polymers.
Polymer Eox[a]
[V]HOMO[b]
[eV]Ered[a]
[V]LUMO[b]
[eV]Eg
[c]
[eV]
P1 + 0.52 ¢5.32 ¢1.15 ¢3.65 1.67P2 + 0.35 ¢5.15 ¢1.18 ¢3.62 1.54P3 + 0.56 ¢5.36 ¢1.12 ¢3.68 1.68P4 + 0.52 ¢5.32 ¢1.12 ¢3.68 1.64P5 + 0.41 ¢5.21 ¢1.11 ¢3.69 1.52P6 + 0.34 ¢5.14 ¢1.19 ¢3.61 1.53
[a] Eox and Ered were determined from the onset potentials of the oxida-tion and reduction peaks, respectively. [b] Electrochemical HOMO =
¢(Eox + 4.8) eV; electrochemical LUMO =¢(Ered + 4.8) eV. [c] Electrochemi-cal band gap = LUMO¢HOMO.
Figure 4. Model dimers (P1–P6) for BODIPY-based polymers with HOMO andLUMO surface plots calculated by using DFT at the B3LYP/6-311g(d,p) level.The hydrogen atoms were omitted for better clarity.
Chem. Eur. J. 2015, 21, 17344 – 17354 www.chemeurj.org Ó 2015 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim17349
Full Paper

level is relatively higher than that in P1. Conversely, the LUMO
wave function of the model dimer is delocalized on the back-bone and no significant differences in LUMO energy levels be-
tween P2 and P1 were observed based on theoretical calcula-tions and CV results.
Surface morphology of polymers
The surface morphology of the polymer films was examined
by means of field-emission (FE) SEM. Dilute solutions of poly-mers in THF, toluene, chloroform, and chlorobenzene were
drop-cast on a glass substrate at room temperature to obtainsmooth and featureless thin films. In the case of dilute solu-
Table 4. Summary of calculated HOMO and LUMO energy levels of monomers and model dimers of P1–P6 by using DFT at the B3LYP/6-311g(d,p) level.
Structure HOMO [eV] LUMO [eV] Structure HOMO [eV] LUMO [eV]
P1 ¢5.69 ¢2.85 ¢5.55 ¢2.85
P2 ¢5.22 ¢2.67 ¢5.11 ¢3.03
P3 ¢7.24 ¢2.19 ¢5.69 ¢2.95
P4 ¢6.37 ¢1.49 ¢5.47 ¢2.89
P5 ¢5.00 ¢2.73 ¢4.91 ¢2.98
P6 ¢5.00 ¢2.62 ¢4.91 ¢2.94
Chem. Eur. J. 2015, 21, 17344 – 17354 www.chemeurj.org Ó 2015 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim17350
Full Paper

tions in chloroform, polymer thin films with honeycomb struc-tures, with a hole diameter of 0.2 to 1.0 mm, were formed
upon slow evaporation of chloroform (Figure 5). All polymerfilms cast from chloroform gave similar surface morphologies.
In a separate experiment, polymers were precipitated froma solution in chloroform by adding appropriate amounts of
methanol, and the resulting suspension was drop-cast on theglass substrate. All polymers showed random-shaped precipi-
tates with no particular morphologies (see Figure S28 in the
Supporting Information).
Studies on solvent vapor adsorption of polymer films
The gas-sensing properties of P1–P6 were examined by usinga QCM method. Figure 6 a and b shows typical examples of
the time dependencies of frequency shifts for the QCM elec-
trode prepared by using P2 and P5, respectively, upon expo-sure to different solvent vapors (water, methanol, acetone,
benzene, and toluene). Frequency shifts are very rapid uponexposure of the polymer film to solvent molecules and largely
depend on the nature of the solvent molecules. For instance,the frequency shift caused due to water adsorption (7 Hz) for
the P2 electrode is extremely low, which indicates no interac-
tions between polymers and water. The frequency shiftscaused by methanol (224 Hz) and acetone adsorption (542 Hz)
are much higher than that of water adsorption, but lower thanthose of aromatic solvent vapors, such as benzene (1667 Hz)
and toluene (2579 Hz); this shows the higher selectivity of P2towards aromatic solvents. Polymer P5 electrode also dis-
played similar vapor adsorption properties. Frequency shiftscaused by water, methanol, acetone, benzene, and toluene
were approximately 1, 147, 675, 1776, and 2785 Hz, respective-ly. The selectivity of sensing decreases in the following order:
toluene>benzene>acetone>methanol>water. The rest ofthe polymers (P1, P3, P4, and P6) also followed similar selectiv-
ity trends (see Figure S29 in the Supporting Information). Thehigher selectivity towards aromatic solvent vapors could be
due to strong p–p interactions between polymers and aromat-
ic solvent molecules.[51] In particular, polymers P2 and P5showed higher toluene adsorption than other polymers. Thiscould be caused by stronger p interactions from electron-defi-cient DPP and triazoloquinoxaline with aromatic solvents,which is consistent with absorption studies that showed thelargest redshifts of lmax for P2 and P5 from toluene. Figure 6 c
demonstrates that sensing of solvent vapors could be repeti-
tively performed by alternate exposure and removal of toluenevapors to the polymer (e.g. , P2) electrode. Reproducibility data
for P1 and P6 upon exposure of toluene and benzene vaporsare shown in Figure S30 in the Supporting Information. A sum-
mary of frequency shifts for all electrodes upon exposure ofdifferent solvent vapors is presented in Figure 6 d.
Conclusion
We have successfully synthesized five novel BODIPY-based al-
ternating copolymers with P2, P3, P4, P5, and P6 as acceptorsby means of palladium-catalyzed Sonogashira polymerization.
All copolymers showed absorption maxima at longer wave-
lengths than that of the BODIPY monomer. In particular, poly-mer P2 exhibited a low optical band gap and the highest ex-
tinction coefficient, with an absorption maximum in the deep-red region, of all other copolymers. The HOMO and LUMO
energy levels of all copolymers were estimated from cyclic vol-tammograms. All copolymers showed good thermal stability
up to 360 8C. Honeycomb morphology was observed for all
polymers thin films obtained by drop-casting a solution inchloroform on a glass plate under ambient conditions. The
gas-sensing capability of the polymers was studied by usinga QCM. These polymers with strong p interactions showed
strong affinity towards benzene and toluene vapors with highreproducibility.
Experimental Section
Instrumentation
1H and 13C NMR spectra were recorded on Bruker Avance AV300(300 MHz) and Bruker Avance AV500 (500 MHz) spectrometers byusing CDCl3 or CD2Cl2 as the solvent. The chemical shifts were re-ported in ppm and referenced to the residual solvent signal : s =singlet, d = doublet, t = triplet, m = multiplet, and br = broad. EI-MSresults were obtained on a Finnigan TSQ7000 spectrometer.MALDI-TOF mass spectra were measured on a Bruker Autoflex IIITOF/TOF instrument equipped with reflectron analyzers. GPC spec-tra for polymers were measured on a Waters e2695 alliance systemequipped with Waters 2414 refractive index detector. THF wasused as the eluent, with a flow rate of 0.3 mL min¢1 at 40 8C, and
Figure 5. FESEM micrographs of the polymer films prepared by drop-castingsolutions of polymers in chloroform on a precleaned glass substrate. Chloro-form was allowed to evaporate slowly at room temperature. All polymersshowed honeycomb structures with micron-sized diameters.
Chem. Eur. J. 2015, 21, 17344 – 17354 www.chemeurj.org Ó 2015 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim17351
Full Paper

polystyrene standards were used for calibration. UV/Vis spectrawere measured on a UV-1800 Shimadzu UV/Vis spectrophotometerwith an optical filter calibrated at a bandwidth of 1 nm. The emis-sion spectra were measured on a RF-5301PC Shimadzu spectro-fluorophotometer. The cyclic voltammograms were recorded witha computer-controlled CHI electrochemical analyzer at a constantscan rate of 100 mV s¢1. The potentials were calibrated by usingthe ferrocene/ferrocenium ion redox couple as an internal refer-ence. The onset of oxidation (Eox
onset) and reduction (Eredonset) were
used to estimate HOMO (EHOMO) and LUMO (ELUMO) energy levels ofpolymers by using the equation EHOMO =¢(4.8 + Eox
onset) and ELUMO =¢(4.8 + Ered
onset). TGA and DSC data were recorded under a nitrogenatmosphere on TA Instruments 2960 and TA instrument 2920 devi-ces. SEM images was recorded on a JEOL JSM-6701F field-emissionscanning electron microscope.
Thin films of polymers were prepared as follows: Polymer (1.0 mg)was dissolved in organic solvents (toluene, THF, chlorobenzene,and chloroform; analytical grade; 1.0 mL) and the undissolvedpolymer was filtered prior to drop-casting. The polymer solutionwas drop-cast on a glass substrate, and solvent was allowed toevaporate under ambient conditions. The film was dried in vacuumprior to FESEM examination.
Materials
All chemicals and reagents were purchased from commercial sour-ces (Sigma Aldrich, Alfa Aesar, and Merck) and used without furtherpurification. Reactions were monitored by TLC carried out on silica-gel plates. Preparative separations were performed by columnchromatography on silica gel, grade 60 (0.040–0.063 mm) fromMerck.
General procedure for Sonogashira polymerization
Both monomers, [Pd(PPh3)4] catalyst, and CuI cocatalyst wereadded to a two-necked round-bottomed flask under nitrogen. A2:1 mixture of THF and DIPA was degassed with nitrogen for15 min and transferred into the reaction flask under a nitrogen at-mosphere. The reaction mixture was stirred at 65 8C for 48 h underdarkness. Excess solvent was removed under reduced pressure,polymers were precipitated in excess methanol, and collected byfiltration. The crude polymers were purified by Soxhlet extractionby using various organic solvents. Methanol was used to removethe unreacted monomers and catalyst. Acetone and hexane wereused to remove the low-molecular-weight oligomers, and hotchloroform was used to extract the target polymers. The chloro-form fraction was concentrated and reprecipitated from methanolto yield the target polymers.
Polymer P1: Compounds 3 b (0.15 g, 0.16 mmol) and 5 b (0.12 g,0.16 mmol) were added to a two-necked round-bottomed flask,followed by [Pd(PPh3)4] (0.009 g, 5 mol %) and CuI (0.003 g,10 mol %), under a nitrogen atmosphere. A degassed mixture ofTHF (30 mL) and DIPA (15 mL) was added by means of a syringe todissolve both monomers and catalysts. The reaction mixture washeated to 65 8C for 48 h under darkness. Purification of polymer
Figure 6. a) QCM frequency shifts of the P2 electrode upon exposure to dif-ferent solvent vapors (methanol, acetone, benzene, and toluene). b) The cor-responding QCM frequency shifts of the P5 electrode. c) Reproducibility testfor the P2 electrode upon exposure and removal of toluene vapors as a typi-cal example. d) Summary of frequency shifts of P1, P2, P3, P4, P5, and P6electrodes upon exposure to water, methanol, acetone, benzene, and tolu-ene vapors.
Chem. Eur. J. 2015, 21, 17344 – 17354 www.chemeurj.org Ó 2015 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim17352
Full Paper

was carried out according to the general procedure. The chloro-form fraction was concentrated under reduced pressure and pre-cipitated in methanol to yield a dark-black polymer (0.17 g, 76 %).1H NMR (300 MHz, CDCl3, 25 8C): d= 7.10 (br s, 2 H), 6.99 (br s, 2 H),3.87 (br s, 2 H), 2.63 (br s, 6 H), 1.53–1.27 (br s, 47 H), 0.88 ppm (br s,6 H); IR (KBr): n= 3437, 2926, 2850, 2203, 2147, 1615, 1521, 1473,1436, 1391, 1363, 1315, 1248, 1230, 1175, 1093, 1005, 835, 765,704, 583, 555, 528 cm¢1; elemental analysis calcd (%) forC45H67BF2N2O: C 77.12, H 9.64, N 4.00; found: C 74.31, H 8.49, N3.78; GPC (THF, polystyrene standard): Mn : 75 600 g mol¢1; Mw:109 560 g mol¢1; PDI: 1.45.
Polymer P2 : Compounds 5 a (0.10 g, 0.2 mmol) and 8 (0.22 g,0.2 mmol) were added to a two-necked round-bottomed flask, fol-lowed by [Pd(PPh3)4] (0.001 g, 5 mol %) and CuI (0.004 g, 10 mol %),under a nitrogen atmosphere. A degassed mixture of THF (40 mL)and DIPA (20 mL) was added by means of a syringe to dissolveboth monomers and catalysts. The reaction mixture was heated to65 8C for 48 h under darkness. Purification of the polymer was car-ried out according to the general procedure. The chloroform frac-tion was concentrated under reduced pressure and precipitated inmethanol to yield a deep dark-blue polymer (0.22 g, 78 %). 1H NMR(300 MHz, CDCl3, 25 8C): d= 8.84 (br s, 2 H), 7.16 (br s, 2 H), 7.04 (br s,4 H), 3.93 (br s, 6 H), 2.66 (br s, 6 H), 1.85–1.21 (br s, 97 H), 0.86 ppm(br s, 18 H); IR (KBr): n= 3437, 2926, 2848, 2190, 1668, 1606, 1526,1465, 1412, 1364, 1311, 1246, 1214, 1177, 1133, 1088, 1011, 829,813, 768, 732, 703, 582, 570, 537 cm¢1; elemental analysis calcd (%)for C93H137BF2N4O3S2 : C 75.88, H 9.38, N 3.81, S 4.36; found: C74.46, H 9.02, N 3.64, S 3.73; GPC (THF, polystyrene standard): Mn :89 760 g mol¢1; Mw: 142 100 g mol¢1; PDI: 1.58.
Polymer P3 : Compounds 5 b (0.10 g, 0.14 mmol) and 10 (0.06 g,0.14 mmol) were added to a two-necked round-bottomed flask,followed by [Pd(PPh3)4] (0.008 g, 5 mol %) and CuI (0.003 g,10 mol %), under a nitrogen atmosphere. A degassed mixture ofTHF (30 mL) and DIPA (15 mL) was added by means of a syringe todissolve both monomers and catalysts. The reaction mixture washeated to 65 8C for 48 h under darkness. Purification of the poly-mer was carried out according to the general procedure. Thechloroform fraction was concentrated under reduced pressure andprecipitated in methanol to yield a dark-black polymer (0.096 g,69 %). 1H NMR (300 MHz, CDCl3, 25 8C): d= 7.67 (br s, 2 H), 7.13 (br s,2 H), 7.03 (br s, 2 H), 3.88 (br s, 2 H), 3.54 (br s, 2 H), 2.65 (br s, 6 H),1.82–1.28 (br s, 56 H), 0.87–0.85 ppm (br s, 12 H); IR (KBr): n= 3451,2923, 2852, 2206, 2141, 1770, 1713, 1607, 1521, 1468, 1440, 1398,1365, 1315, 1281, 1246, 1185, 1083, 1009, 948, 834, 766, 706, 582,536 cm¢1; elemental analysis calcd (%) for C63H86BF2N3O3 : C 77.04,H 8.83, N 4.28; found: C 70.82, H 8.18, N 3.91; GPC (THF, polystyr-ene standard): Mn : 58 310 g mol¢1; Mw: 116 210 g mol¢1; PDI: 1.99.
Polymer P4 : Compounds 5 b (0.10 g, 0.14 mmol) and 12 (0.065 g,0.14 mmol) were added to a two-necked round-bottomed flask,followed by [Pd(PPh3)4] (0.0084 g, 5 mol %) and CuI (0.003 g,10 mol %), under a nitrogen atmosphere. A degassed mixture ofTHF (30 mL) and DIPA (15 mL) was added by means of a syringe todissolve both monomers and catalysts. The reaction mixture washeated to 65 8C for 48 h under darkness. Purification of the poly-mer was carried out according to the general procedure. Thechloroform fraction was concentrated under reduced pressure andprecipitated in methanol to yield a dark-black polymer (0.10 g,68 %). 1H NMR (300 MHz, CDCl3, 25 8C): d= 7.64–7.46 (br s, 2 H), 7.14(br s, 2 H), 7.03 (br s, 2 H), 4.68 (d, br, 2 H), 3.89 (br s, 2 H), 2.66 (br s,6 H), 2.10–1.26 (br s, 67 H), 0.86 ppm (br s, 9 H); IR (KBr): n= 3439,
2921, 2849, 2201, 2140, 1608, 1524, 1467, 1390, 1365, 1311, 1246,1191, 1089, 1009, 950, 832, 763, 705, 580, 535 cm¢1; elemental anal-ysis calcd (%) for C66H94BF2N5O: C 77.27, H 9.38, N 6.93; found: C74.52, H 8.90, N 5.51; GPC (THF, polystyrene standard): Mn :68 120 g mol¢1; Mw: 123 500 g mol¢1; PDI: 1.81.
Polymer P5 : Compounds 5 b (0.066 g, 0.09 mmol) and 16 (0.106 g,0.09 mmol) were added to a two-necked round-bottomed flask,followed by [Pd(PPh3)4] (0.0053 g, 5 mol %) and CuI (0.002 g,10 mol %), under a nitrogen atmosphere. A degassed mixture ofTHF (30 mL) and DIPA (15 mL) was added by means of a syringe todissolve both monomers and catalysts. The reaction mixture washeated to 65 8C for 48 h under darkness. Purification of the poly-mer was carried out according to the general procedure. Thechloroform fraction was concentrated under reduced pressure andprecipitated in methanol to yield a dark-green polymer (0.13 g,87 %). 1H NMR (300 MHz, CDCl3, 25 8C): d= 8.81 (br s, 2 H), 7.76 (br s,4 H), 7.57 (br s, 2 H), 7.06 (br s, 2 H), 6.93 (br s, 6 H), 4.86 (br s, 2 H),3.92 (br s, 6 H), 2.78 (br s, 6 H), 2.22-1.27 (br s, 85 H), 0.95 ppm (br s,21 H); IR (KBr): n= 3443, 2921, 2851, 2186, 2137, 1606, 1525, 1464,1426, 1389, 1366, 1305, 1244, 1176, 1121, 1079, 1009, 981, 828,804, 766, 729, 703, 675, 584, 538 cm¢1; elemental analysis calcd (%)for C103H138BF2N7O3S2 : C 75.66, H 8.51, N 6.00, S 3.92; found: C74.73, H 7.83, N 5.56, S 3.54; GPC (THF, polystyrene standard): Mn :16 860 g mol¢1; Mw: 25 500 g mol¢1; PDI: 1.51.
Polymer P6 : Compounds 5 b (0.07 g, 0.09 mmol) and 19 (0.08 g,0.09 mmol) were added to a two-necked round-bottomed flask,followed by [Pd(PPh3)4] (0.005 g, 5 mol %) and CuI (0.002 g,10 mol %), under a nitrogen atmosphere. A degassed mixture ofTHF (30 mL) and DIPA (15 mL) was added by means of a syringe todissolve both monomers and catalysts. The reaction mixture washeated to 65 8C for 48 h under darkness. Purification of the poly-mer was carried out according to the general procedure. Thechloroform fraction was concentrated under reduced pressure andprecipitated in methanol to yield a dark-green polymer (0.08 g,69 %). 1H NMR (300 MHz, CDCl3, 25 8C): d= 7.48 (br s, 4 H), 7.47 (br s,2 H), 7.17 (br s, 2 H), 7.03 (br s, 4 H), 6.88 (br s, 4 H), 3.88 (br s, 6 H),2.73 (br s, 6 H), 1.86–1.26 (br s, 65 H), 0.92 ppm (br s, 18 H); IR (KBr):n= 3437, 2921, 2851, 2192, 2137, 1606, 1521, 1465, 1389, 1362,1311, 1246, 1184, 1080, 1007, 832, 764, 706, 609, 582, 536 cm¢1; el-emental analysis calcd (%) for C89H113BF2N4O3S3 : C 74.65, H 7.95, N3.91, S 6.72; found: C 72.82, H 7.88, N 3.64, S 5.81; GPC (THF, poly-styrene standard): Mn : 44 060 g mol¢1; Mw: 65 570 g mol¢1; PDI: 1.49.
Sample preparations for properties studies
Vapor sensing by QCM : In this study, a resonance frequency of9 MHz (AT-cut) was used. The frequency of the QCM electrode wasmeasured during adsorption and was recorded when it hadbecome stable, for example, the QCM frequency in air was stablewithin �2 Hz in 1 h. The polymers (P1–P6, 4.0 mg each) were dis-persed in isopropanol (2 mg mL¢1). The mixtures were then ultraso-nicated for 30 min in a bath sonicator and 3 mL of this dispersionwas drop-cast on the QCM electrodes. The electrode was dried at60 8C in vacuum for 24 h before measurements. The prepared QCMelectrode was then applied to the QCM instrument and exposedto the solvents in a sealed container to prevent the escape ofvapors during the adsorption measurements. Between measure-ments, the electrode was exposed to air to desorb the solventvapor. The recovery of the initial frequency value was taken as anindication of complete desorption. Experiments were carried out at25 8C.
Chem. Eur. J. 2015, 21, 17344 – 17354 www.chemeurj.org Ó 2015 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim17353
Full Paper

When the surface of a quartz crystal electrode is coated with poly-mer, which is exposed to different solvent vapors, changes in massoccurs. Such change in mass (m’ in g cm¢2) can be measured bythe oscillating frequency of the electrode. The frequency change(Df) corresponds to the sample amount loaded on the QCM elec-trode, and can be calculated from the Sauerbrey equation [Eq. (1)] .
Df ¼ ð 2f 20ffiffiffiffiffiffiffiffiffiffi
1QmQp Þm ð1Þ
in which fo (Hz) is the natural frequency of the quartz crystal, 1Q isthe quartz density (2.649 g cm¢3), and mQ is the shear modulus(2.947 Õ 1011 g cm¢1 s¢2).
Acknowledgements
We thank the National University of Singapore for funding andthe Department of Chemistry for technical support.
Keywords: electrochemistry · polymers · sensors ·supramolecular chemistry · volatile organic vapors
[1] R. Ziessel, G. Ulrich, A. Harriman, New J. Chem. 2007, 31, 496 – 501.[2] G. Ulrich, R. Ziessel, A. Harriman, Angew. Chem. Int. Ed. 2008, 47, 1184 –
1201; Angew. Chem. 2008, 120, 1202 – 1219.[3] A. Loudet, K. Burgess, Chem. Rev. 2007, 107, 4891 – 4932.[4] Y. Hayashi, N. Obata, M. Tamaru, S. Yamaguchi, Y. Matsuo, A. Saeki, S.
Seki, Y. Kureishi, S. Saito, H. Shinokubo, Org. Lett. 2012, 14, 866 – 869.[5] J. Li, I. Kim, E. Roche, D. Beeman, A. Lynch, C. Ding, Z. Ma, Bioorg. Med.
Chem. Lett. 2006, 16, 794 – 797.[6] N. Boens, V. Leen, W. Dehaen, Chem. Soc. Rev. 2012, 41, 1130 – 1172.[7] S. Mula, A. Ray, M. Banerjee, T. Chaudhuri, K. Dasgupta, S. Chattopad-
hyay, J. Org. Chem. 2008, 73, 2146 – 2154.[8] M. Tomasulo, E. Deniz, R. Alvarado, F. Raymo, J. Phys. Chem. C 2008, 112,
8038 – 8045.[9] E. Deniz, S. Ray, M. Tomasulo, S. Impellizzeri, S. Sortino, F. Raymo, J.
Phys. Chem. A 2010, 114, 11567 – 11575.[10] S. Kolemen, O. Bozdemir, Y. Cakmak, G. Barin, S. Erten-Ela, M. Marszalek,
J. Yum, S. Zakeeruddin, M. Nazeeruddin, M. Gratzel, E. Akkaya, Chem.Sci. 2011, 2, 949 – 954.
[11] S. Erten-Ela, M. Yilmaz, B. Icli, Y. Dede, S. Icli, E. Akkaya, Org. Lett. 2008,10, 3299 – 3302.
[12] B. Kim, B. Ma, V. Donuru, H. Liu, J. Frechet, Chem. Commun. 2010, 46,4148 – 4150.
[13] T. Rousseau, A. Cravino, E. Ripaud, P. Leriche, S. Rihn, A. De Nicola, R.Ziessel, J. Roncali, Chem. Commun. 2010, 46, 5082 – 5084.
[14] T. Bura, N. Leclerc, S. Fall, P. Leveque, T. Heiser, P. Retailleau, S. Rihn, A.Mirloup, R. Ziessel, J. Am. Chem. Soc. 2012, 134, 17404 – 17407.
[15] H. Lin, W. Huang, Y. Chen, H. Chou, C. Hsu, J. Lin, H. Lin, Chem.Commun. 2012, 48, 8913 – 8915.
[16] V. Donuru, G. Vegesna, S. Velayudham, G. Meng, H. Liu, J. Polym. Sci.Part A J. Polym. Sci. Part A: Polym. Chem. 2009, 47, 5354 – 5366.
[17] F. Alemdaroglu, S. Alexander, D. Ji, D. Prusty, M. Borsch, A. Herrmann,Macromolecules 2009, 42, 6529 – 6536.
[18] V. Donuru, G. Vegesna, S. Velayudham, S. Green, H. Liu, Chem. Mater.2009, 21, 2130 – 2138.
[19] B. Popere, A. Della Pelle, S. Thayumanavan, Macromolecules 2011, 44,4767 – 4776.
[20] B. Popere, A. Della Pelle, A. Poe, G. Balaji, S. Thayumanavan, Chem. Sci.2012, 3, 3093 – 3102.
[21] E. Havinga, W. Tenhoeve, H. Wynberg, Polym. Bull. 1992, 29, 119 – 126.[22] X. Guo, F. Kim, S. Jenekhe, M. Watson, J. Am. Chem. Soc. 2009, 131,
7206 – 7207.
[23] H. Xin, X. Guo, F. Kim, G. Ren, M. Watson, S. Jenekhe, J. Mater. Chem.2009, 19, 5303 – 5310.
[24] J. Lee, K. Song, J. Ku, T. Sung, D. Moon, Sol. Energy Mater. Sol. Cells2011, 95, 3377 – 3384.
[25] A. Tanimoto, T. Yamamoto, Macromolecules 2006, 39, 3546 – 3552.[26] A. Balan, D. Baran, G. Gunbas, A. Durmus, F. Ozyurt, L. Toppare, Chem.
Commun. 2009, 6768 – 6770.[27] S. Ozdemir, M. Sendur, G. Oktem, O. Dogan, L. Toppare, J. Mater. Chem.
2012, 22, 4687 – 4694.[28] A. Ballantyne, L. Chen, J. Dane, T. Hammant, F. Braun, M. Heeney, W.
Duffy, I. McCulloch, D. Bradley, J. Nelson, Adv. Funct. Mater. 2008, 18,2373 – 2380.
[29] T. Chu, J. Lu, S. Beaupre, Y. Zhang, J. Pouliot, J. Zhou, A. Najari, M. Le-clerc, Y. Tao, Adv. Funct. Mater. 2012, 22, 2345 – 2351.
[30] J. Intemann, K. Yao, H. Yip, Y. Xu, Y. Li, P. Liang, F. Ding, X. Li, A. Jen,Chem. Mater. 2013, 25, 3188 – 3195.
[31] Y. Zou, D. Gendron, R. Neagu-Plesu, M. Leclerc, Macromolecules 2009,42, 6361 – 6365.
[32] S. Qu, H. Tian, Chem. Commun. 2012, 48, 3039 – 3051.[33] B. Lim, J. Yeo, D. Khim, D. Kim, Macromol. Rapid Commun. 2011, 32,
1551 – 1556.[34] P. Sonar, S. Singh, Y. Li, M. Soh, A. Dodabalapur, Adv. Mater. 2010, 22,
5409 – 5413.[35] T. Nelson, T. Young, J. Liu, S. Mishra, J. Belot, C. Balliet, A. Javier, T. Kowa-
lewski, R. McCullough, Adv. Mater. 2010, 22, 4617 – 4621.[36] J. Jung, F. Liu, T. Russell, W. Jo, Energy Environ. Sci. 2012, 5, 6857 – 6861.[37] S. M. Chang, Y. Iwasaki, M. Suzuki, E. Tamiya, I. Karube, H. Muramatsu,
Anal. Chim. Acta 1991, 249, 323 – 329.[38] Z. K. Chen, S. C. Ng, S. F. Y. Li, L. Zhong, L. G. Xu, H. S. O. Chan, Synth.
Met. 1997, 87, 201 – 204.[39] A. Talaie, J. A. Romagnoli, Synth. Met. 1996, 82, 231 – 235.[40] S. R. Kim, S. A. Choi, J. D. Kim, K. J. Kim, C. Lee, S. B. Rhee, Synth. Met.
1995, 71, 2027 – 2028.[41] P. Si, J. Mortensen, A. Kornolov, J. Denborg, P. J. Moller, Anal. Chim. Acta
2007, 597, 223 – 230.[42] G. Meng, S. Velayudham, A. Smith, R. Luck, H. Liu, Macromolecules 2009,
42, 1995 – 2001.[43] L. Huo, J. Hou, H. Chen, S. Zhang, Y. Jiang, T. Chen, Y. Yang, Macromole-
cules 2009, 42, 6564 – 6571.[44] J. Chen, M. Shi, X. Hu, M. Wang, H. Chen, Polymer 2010, 51, 2897 – 2902.[45] T. Tam, H. Tan, W. Ye, S. Mhaisalkar, A. Grimsdale, Org. Lett. 2012, 14,
532 – 535.[46] S. Tarkuc, E. K. Unver, Y. A. Udum, C. Tanyeli, L. Toppare, Electrochim.
Acta 2010, 55, 7254 – 7258.[47] V. R. Donuru, S. L. Zhu, S. Green, H. Y. Liu, Polymer 2010, 51, 5359 – 5368.[48] Y. Gabe, Y. Urano, K. Kikuchi, H. Kojima, T. Nagano, J. Am. Chem. Soc.
2004, 126, 3357 – 3367.[49] S. P. G. Costa, E. Oliveira, C. Lodeiro, M. M. M. Raposo, Tetrahedron Lett.
2008, 49, 5258 – 5261.[50] M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R.
Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Na-katsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G.Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Ha-segawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven,J. A. Montgomery, Jr. , J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E.Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Ra-ghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N.Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo,J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R.Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakr-zewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Dan-iels, ©. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski, and D. J. Fox,Gaussian 09, revision A.02; Gaussian, Inc. , Wallingford, CT, 2009.
[51] L. K. Shrestha, R. G. Shrestha, Y. Yamauchi, J. P. Hill, T. Nishimura, K. Miya-zawa, T. Kawai, S. Okada, K. Wakabayashi, K. Ariga, Angew. Chem. Int. Ed.2015, 54, 951 – 955; Angew. Chem. 2015, 127, 965 – 969.
Received: July 27, 2015Published online on October 14, 2015
Chem. Eur. J. 2015, 21, 17344 – 17354 www.chemeurj.org Ó 2015 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim17354
Full Paper












![Electrochemistry of Cu[sub 3]N with Lithium](https://img.pdfslide.net/doc/110x75/634c896ac64cf0e3560cac9a/electrochemistry-of-cusub-3n-with-lithium.jpg)
















