Embed Size (px)
Citation preview

Episcleral Implants for Topotecan Delivery to thePosterior Segment of the Eye
Angel M. Carcaboso,1 Diego A. Chiappetta,2,3 Javier A. W. Opezzo,1 Christian Hocht,1
Adriana C. Fandino,4 J. Oscar Croxatto,5 Modesto C. Rubio,1,3 Alejandro Sosnik,2,3
David H. Abramson,6 Guillermo F. Bramuglia,1 and Guillermo L. Chantada7
PURPOSE. Intravenous or periocular topotecan has been pro-posed as new treatment modality for patients with advancedintraocular retinoblastoma, but systemic topotecan lactone ex-posure induced by both approaches may cause toxicity. Thepurpose of this study was to develop a topotecan-loaded oculardelivery system to minimize systemic exposure and achieveselective transscleral penetration.
METHODS. Biocompatible polymer implants containing low (0.3mg) or high (2.3 mg) topotecan load were manufactured andcharacterized in vitro. Adrenaline (500 �g) was coloaded toinduce local vasoconstriction in vivo in 2 of 4 animal groups.Implants were inserted into the episclera of rabbits, and topo-tecan (lactone and total) concentrations in ocular tissues andplasma were determined over a period of 48 hours.
RESULTS. In vitro, implants released 30% to 50% of the loadeddrug within 48 hours and 45% to 70% by day 10. In vivo,topotecan lactone was highly accumulated in locally exposedocular tissues (ranging from 105 to 106 ng/g in sclera andchoroid and 102 to103 ng/g in retina) over 48 hours with all theformulations studied. Low vitreous topotecan lactone levels(approximately 5 ng/mL) were found in animals receivingconcomitant local vasoconstriction and high load implants.Topotecan lactone concentrations in plasma and in contralat-eral eyes were minimal or undetectable as a marker of tissueselectivity of the proposed strategy.
CONCLUSIONS. These studies may contribute to improving theefficacy and safety of chemotherapy treatments for retinoblas-toma and may support the role of the local vasculature andtissues promoting drug clearance and local accumulation dur-ing transscleral drug delivery. (Invest Ophthalmol Vis Sci.2010;51:2126–2134) DOI:10.1167/iovs.09-4050
The systemic treatment of advanced intraocular retinoblas-toma with vitreous seeding represents a challenge because
drug penetration to the avascularized vitreous is limited by thepresence of the blood-retinal barrier.1,2 The standard of carefor these patients in most centers around the world includestumor chemoreduction with intravenously administered carbo-platin, etoposide, and vincristine followed by consolidationwith local therapy or external beam radiotherapy.3 However,the discouraging results in terms of eye preservation in thissubgroup motivated the clinical testing of novel treatmentmodalities, such as carboplatin given periocularly, which iscurrently part of the ongoing Children Oncology Group trialfor eyes with vitreous seeding.4
It is postulated that periocularly administered anti-retino-blastoma drugs may achieve better ocular penetration whileminimizing systemic drug exposure in the treatment of thesetumors.5–8 Carboplatin was the first drug to be used by theperiocular route in retinoblastoma, but its use is being recon-sidered because of local toxicity, including optic nerve atro-phy,7,9 and its limited ability to be curative.10 However, thereare few drugs other than carboplatin that have been character-ized for periocular use in this disease, and identifying othercandidates is a primary challenge for clinicians. Topotecan(TPT), a camptothecin derivative, has been studied as an alter-native for the treatment of retinoblastoma. After the report ofsingle cases from other groups, we reported encouraging ac-tivity in a series of children with extraocular disease treatedunder compassionate basis with intravenously administeredTPT.11,12 Subsequent studies in tumor-bearing animal modelsshowed that the association of TPT and carboplatin results in avery active drug combination, with potentially synergistic ac-tivity.13 However, hematopoietic toxicity limits the clinical useof these two agents concomitantly by the intravenous (IV)route.14 One way of potentially taking advantage of this com-bination and reducing its toxicity would be to give one agentby the IV route and the remaining one by the periocular route,therefore reducing the overall systemic exposure in patientswhile achieving active drug levels in the eye. The response rateto intravenously administered TPT associated with periocularcarboplatin in previously untreated patients is being investi-gated in a phase III study sponsored by St Jude Children’sResearch Hospital (Memphis, TN). In the phase III study, somepatients receive periocular carboplatin concomitantly (clinical-trials.gov identifier: NCT00186888). TPT is also being testedintravenously as second-line treatment for relapsed or resistant
From the 1Department of Pharmacology, 2The Group of Biomate-rials and Nanotechnology for Improved Medicines (BIONIMED), theDepartment of Pharmaceutical Technology, and the 3National ScienceResearch Council, Faculty of Pharmacy and Biochemistry, University ofBuenos Aires, Buenos Aires, Argentina; the Departments of 4Ophthal-mology and 7Hemato-Oncology Service, Hospital J.P. Garrahan, BuenosAires, Argentina; the 5Fundacion Oftalmologica Argentina Jorge Malb-ran, Buenos Aires, Argentina; and the 6Department of OphthalmicOncology, Memorial Sloan-Kettering Cancer Center, New York, NewYork.
Supported by the Fund for Ophthalmic Knowledge; a PostdoctoralFellowship from the Ministry of Education and Science, Spain (AMC);University of Buenos Aires Grant UBACYT B025; and the Natalie DafneFlexer Foundation.
Presented in part at the annual meeting of the Association forResearch in Vision and Ophthalmology, Fort Lauderdale, Florida, April2008.
Submitted for publication May 27, 2009; revised August 27, 2009;accepted September 15, 2009.
Disclosure: A.M. Carcaboso, None; D.A. Chiappetta, None;J.A.W. Opezzo, None; C. Hocht, None; A.C. Fandino, None; J.O.Croxatto, None; M.C. Rubio, None; A. Sosnik, None; D.H. Abram-son, None; G.F. Bramuglia, None; G.L. Chantada, None
Corresponding author: Guillermo L. Chantada, Hemato-OncologyService, Hospital J.P. Garrahan, Combate de los Pozos 1881, C1245AAL,Buenos Aires, Argentina; [email protected].
Physiology and Pharmacology
Investigative Ophthalmology & Visual Science, April 2010, Vol. 51, No. 42126 Copyright © Association for Research in Vision and Ophthalmology

retinoblastoma. Because of the potentially severe local toxicityassociated with periocular carboplatin, we studied periocularTPT in phase I trials8 to consider its clinical use alone or inassociation with IV carboplatin for the treatment of retinoblas-toma.
It is well known that local drug delivery to the vitreous ishampered by the innate local ocular barriers that protect theeye from potentially toxic xenobiotics.15 Confirming thispoint, in a study of the ocular pharmacokinetics of periocularTPT in a non–tumor-bearing animal model, we observed pro-fuse ocular barrier activity after the periocular administrationof TPT that limited penetration to the vitreous.16 Specifically,we found that a significant proportion of the periocularlyadministered TPT reached the vitreous through the systemiccirculation, probably because of rapid orbital clearance of thedrug. In a phase I clinical trial of periocular TPT in childrenwith relapsed or resistant intraocular retinoblastoma, we ob-served a low toxicity profile for this new treatment modalityand were able to characterize TPT plasma concentrations inthese patients.8 We found that the systemic drug exposure wassignificant, though lower than the exposure induced in thesame patients after IV administration of similar dosages. Basedon our preclinical and clinical results for periocular TPT weinferred that, though promising, this strategy may remain rel-atively inefficient in the clinical setting as a way of deliveringTPT by the transscleral route. Therefore, we aimed to design amethod for a more selective periocular administration of TPTthat would allow for the safe use of this drug alone or inassociation with carboplatin for the treatment of retinoblas-toma with vitreous seeding. Because this concern is also appli-cable to other drugs, it has motivated researchers to proposeseveral strategies to maximize drug penetration into the eyeand to achieve a prolonged exposure,15 which could be espe-cially important for S-phase specific drugs such as TPT.17 Re-cently, intense research has been focused on the design of abiocompatible polymer-based drug delivery system (DDS) torelease drugs in the orbital space, primarily for the delivery ofdrugs for the treatment of other (nontumor) ocular condi-tions.18 Based on our previous results, we hypothesized that aDDS implanted locally could saturate the adjacent eye barriersand achieve selective drug delivery to the vitreous of thetreated eye while minimizing systemic exposure. Thus, in thisstudy we developed and characterized, in vitro and in vivo, a
TPT-loaded “double-faced” polymeric DDS composed of adrug-free face designed to isolate the drug reservoir from theorbital conjunctival tissues and a drug-loaded face to concen-trate TPT in the adjacent scleral surface when inserted epis-clerally. We specifically studied the ability of the DDS, incombination with a pharmacologic strategy to constrain localblood vessels, to achieve high and long-term local drug accu-mulation in ocular tissues, which could enhance the effect ofTPT in retinoblastoma.
MATERIALS AND METHODS
Commercial TPT (Hycamtin, containing 4 mg TPT, 48 mg mannitol,and 20 mg tartaric acid) and TPT standard were donated by Glaxo-SmithKline (Buenos Aires, Argentina). At physiological pH, the activeform of TPT, a closed lactone ring (LTPT; stable at acidic pH), isreversibly hydrolyzed to an inactive carboxylate form.19
Poly(�-caprolactone) polymer (PCL; molecular weight 14,000 Da;Tm 60°C) and adrenaline were purchased from Sigma-Aldrich (BuenosAires, Argentina). The remaining reagents and chromatography sol-vents were of analytical grade.
Implant Manufacture and CharacterizationTPT implants were manufactured by a simple, fast, and reproduciblemelt-molding-compression method using a stainless steel mold (7-mmdiameter).20 A diagram of the procedure is shown in Figure 1. Depend-ing on the proportions of the matrix polymer powder and activeprinciples, different implants were manufactured. For the initial exper-iments, we developed implants with a low load (LL) of TPT. Wesubsequently explored the effect of including a vasoconstrictor (adren-aline) in the PCL matrix and increasing drug load, developing implantsnamed low load with vasoconstrictor (LLv), high load (HL), and highload with vasoconstrictor (HLv; Table 1). When included in the for-mulation, adrenaline and PCL powders were blended. Blank drug-freeimplants containing identical excipient amounts were also produced,as were pure PCL implants.
Scanning electron microscopy (SEM; JSM-35C; JEOL, Tokyo, Japan)was performed to characterize the surface and matrix structure of theimplants before and after in vitro incubations.
In Vitro Release AssayIndividual LLv and HLv implants (n � 3 each) were placed in 100-mLglass flasks containing phosphate-buffered solution (PBS; 50 mL, pH
FIGURE 1. Diagram of the melt-molding compression technique developed to produce one-side coatedTPT-loaded PCL implants. Briefly, to produce a PCL coating (drug-free layer of pure polymer), groundedPCL is introduced into the bottom of a stainless steel mold composed of a static platform and a matrix (across-section is represented). A homogeneous mixture of PCL and TPT (with hydrophilic excipients) ispoured onto the pure PCL layer, and the mold plunger is mounted (left). The filled mold is exposed to a1.7 kg/m2 pressure and heated (70°C, 1 hour) to melt the polymer and entrap the drug and excipients(middle). The system is cooled (4°C, 0.5 hour) to allow for the solidification of the implants. Implant withdimensions (right). P, pressure.
IOVS, April 2010, Vol. 51, No. 4 Implants for Topotecan Transscleral Delivery 2127

7.4) and were incubated at 37°C. After 1 hour, the complete contentof the flasks was collected and replaced with fresh PBS solution. Thissampling process was repeated daily for 10 days. The total TPT con-centration was measured in every collected sample by HPLC withfluorometric detection, as previously described.16
To characterize the long-term stability of the PCL matrix, theswelling process (absorption of fluids) and the weight loss of theimplants, blank implants and pure PCL implants (n � 3 each) wereincubated in the same conditions as for the in vitro release assay. At 1,7, 21, and 28 days, the implants were collected and carefully wiped toremove superficial liquids, and the wet weight was recorded. After adrying period of 24 hours at 37°C, the dry weight was recorded.
In Vivo Studies and Sampling Procedures
New Zealand albino rabbits (1.8–2.2 kg) used for this study werehandled according to the ARVO Statement for the Use of Animals inOphthalmic and Vision Research. Four groups of rabbits (LL, LLv, HL,HLv) were used to study ocular and plasma TPT concentrations afterthey were exposed to TPT implants. A mixture of ketamine (37.5mg/kg) and xylazine (5 mg/kg) was used to anesthetize the animals,and the DDS was implanted as shown in Figure 2.
To assess the suitability of LL implants (DDS containing 0.3 mgTPT) to achieve selective TPT ocular delivery and low systemic expo-sure, a first cohort of animals received LL implants (n � 4). Afterevaluating the results showing low vitreous levels of TPT, a secondcohort (n � 5) received LLv implants. In these two groups, a terminal100-�L vitreous sample was aspirated with a 25-gauge needle in theanesthetized animals 5 hours after the insertion of the DDS, and bloodsamples were obtained through the ear vein at 1, 2, and 4 hours. TotalTPT and LTPT were determined in vitreous and plasma, as previouslydescribed.16 To study longer term (24-hour) exposure in ocular tissues,three more rabbits were similarly exposed to LLv implants, and theirplasma, vitreous, and ocular tissues were analyzed for total TPT andLTPT 24 hours later. These rabbits were anesthetized, and the implantsremoved; 30 minutes later, the rabbits were euthanatized with an IV oran intracardiac bolus injection of sodium thiopental (100 mg in 5 mLsaline). Eyes were enucleated, and the right eyes were divided in twoequal halves. One half was exposed (i.e., in contact with the DDS), and
the other half was nonexposed. Left eyes were excised in the sameway, and one of the halves was processed. All the excised tissues werewashed in ice-cold PBS, and the sclera, choroid, and retina wereimmediately dissected. The excess PBS in the tissues was absorbedwith a sponge, and tissues were weighed. To quantitatively extract TPTfrom the tissues, the tissues were exposed to two freeze-thaw cycles in100 �L distilled water, and finally 400 �L cold methanol were added.Supernatants were analyzed for total TPT and LTPT.
To further characterize the transscleral penetration process of TPTand to achieve high and long-term (24- to 48-hour) TPT concentrationsin the vitreous, animal groups HL and HLv (n � 6 and n � 8 rabbits,respectively) receiving high-load implants (DDS containing 2.3 mgTPT; Table 1) were included in the experiments. To evaluate thetransscleral release kinetics of the drug after implanting the DDS inthese two groups, two methods were used. First, to characterize theearly vitreous penetration profile of the drug released from the DDS, amicrodialysis-based multiple-sampling schedule was performed. Sec-ond, a terminal vitreous sample was aspirated 24 or 48 hours later.During the microdialysis sampling period, TPT concentrations wereassessed by means of the principle by which drugs in the targetedtissue cross the capillary-like semipermeable microdialysis probe con-tinuously perfused with a drug-free physiological fluid, until equilib-rium was reached.21 For these experiments, 2 hours after the surgicalinsertion of the DDS, in a location 3 mm away from the limbus and120o away from the implant site, the surface of the sclera was exposedby making an incision in the conjunctiva, and the microdialysis probe(Fig. 3) was inserted into the vitreous space through an incision madewith a 25-gauge needle. A second probe was inserted into the con-tralateral eye in the same location. Both probes were fixed to the sclerawith two vicryl 7-0 sutures (Fig. 3). Microdialysis membranes werethen perfused with PBS (pH 7.4) at a flow rate of 0.5 �L/min using aninfusion pump (KDS230; KD Scientific, Holliston, MA) and were left for30 to 40 minutes to equilibrate with the vitreous before sample col-lection started. Dialysates were collected every 40 minutes over aperiod of 160 minutes, covering the 160- to 320-minute time rangeafter the DDS implantation, and total TPT was analyzed. At the end ofthe sample collection, a concentrated TPT solution (500 ng/mL) wasperfused through the probe to calculate the probe recovery by the
TABLE 1. Characteristics of the TPT-Loaded PCL Implants
Coating
Matrix
Formulation(TPT load, mg) PCL (mg) PCL (mg)
Hycamtin*(mg)
TPT Powder†(mg)
Adrenaline(mg)
AddedExciplents‡
(mg)Final Weight§
(mg)
LL (0.3) 20 95 5 0 0 0 117 � 7LLv (0.3) 20 95 5 0 0.5 0 122 � 4HL (2.3) 50 45 5 2 0 0 93 � 8HLv (2.3) 50 45 5 2 0.5 0 98 � 4Blank (0) 50 45 0 0 0.5 5 94 � 5
LL, low load; LLv, low load with vasoconstrictor; HL, high load; HLv, high load with vasoconstrictor.* Hycamtin (5 mg) contains TPT (0.3 mg), mannitol (3.3 mg), and tartaric acid (1.4 mg).† TPT pure standard drug.‡ Mannitol (3.5 mg) and tartaric acid (1.5 mg).§ Mean � SD of n � 5 implants.
FIGURE 2. Surgical insertion of theDDS. (a) The implant is introducedthrough the conjunctival incision inthe superior temporal episcleral zoneof the right eye, with the uncoatedside facing the sclera. (b) The inci-sion is sutured with 5-0 sutures. (c)Final appearance of the implantedeye.
2128 Carcaboso et al. IOVS, April 2010, Vol. 51, No. 4

retrodialysis method.22 The recovery value obtained for the probes was30% � 9% and 28% � 6% (mean � SD for the HL and HLv groups,respectively), which allowed calibration of the probes to calculate theactual intravitreal TPT concentrations. At the end of the microdialysisstudy, probes were withdrawn, a 5-0 suture was used to close thesclera, and animals were allowed to recover. Either 24 or 48 hourslater, a vitreous sample (100 �L) was aspirated with a 25-gauge needleand was analyzed for total TPT and LTPT. During the experiment,plasma samples were obtained through a jugular catheter at 0.25, 0.5,1, 2, 4, 6, and 24 (or 48) hours after the insertion of the DDS. Plasmaexposure to the drug was defined as the area under the concentration-time curve (AUC), calculated as previously published.16 At the end ofthe 24- or 48-hour experiment, DDS was removed. Thirty minutes lateranimals were euthanatized, eyes were enucleated, and tissues wereprocessed as detailed for total TPT and LTPT content.
In Vivo Release Assay
To study the ability of the DDS to release the drug in vivo, LLv implantswere recovered from animals of the LLv group over a period of 7 days.
To determine the amount of TPT remaining (not released) in the DDS,the polymer was dissolved in methylene chloride (2 mL), and TPT wasextracted with distilled water (4 � 1 mL) and analyzed by HPLC. Thedifference between the initial TPT load and the unreleased TPT was theamount of drug released in vivo.
Histopathology Evaluation
The effect of HLv and blank implants on the exposed and contralateraleyes was assessed in vivo in 10 rabbit eyes. Briefly, the surgicalprocedure used in the studies was followed to expose n � 3 eyes toHLv implants and n � 3 eyes to blank implants. Control eyes contralat-eral to the implanted eyes were also studied (n � 4). After surgery, theanimals were recovered and housed normally until they were killed 48hours later, and eyes were enucleated. Histology evaluation was per-formed as previously described.16
RESULTS
Solid products with convex surfaces, 7 mm in diameter and 2.5to 3 mm in thickness, were obtained (see photograph in Fig.1). Implants were characterized by an intense yellow color onthe scleral side from TPT and a white, glossy appearance in theorbital side coated with pure PCL. According to SEM, thescleral surfaces of the implants were porous, and the orbitalsurfaces were smooth (Figs. 4a, 4b). Hydrophilic excipientsand drugs formed needle-looking crystal structures into theoriginal implant matrix, which disappeared after 10 days of thein vitro release study (Figs. 4c–e).
Implant weights and drug loads are shown in Table 1. Thedrug could be completely recovered as LTPT by solvent ex-traction after the implant was dissolved in methylene chloride,confirming the stability of the drug through the manufacturingprocess (data not shown).
In Vitro and In Vivo Release from TPT DDS
In vitro release profiles of LLv and HLv implants are presentedin Figure 5. The release rate was biphasic (faster during the first2 days and slower thereafter). During the first 2 days, 150 �gTPT was released from LLv implants and 700 �g was releasedfrom HLv implants (Figs. 5a, 5b), accounting for 50% and 30%of the total drug load, respectively. Afterward, a slower release
FIGURE 3. Diagram of the ocular microdialysis concentric probe de-signed and manufactured in the laboratory. A 10-mm-long microdialysismembrane (outer diameter [OD] 200 �m, 10,000 molecular weightcutoff; Asahi Medical Co., Japan), an inlet plastic tube (inner diameter,500 �m), and an outlet silica tube (OD 145 �m) were used to build theprobes. Epoxy adhesive (gray dots) was used to glue the componentsand to seal the end of the dialysis membrane. The probe was mountedin a nitrocellulose semirigid support, which allowed fixing the canulato the sclera with two sutures of 7-0 silk.
FIGURE 4. Scanning electron micro-graphs of LLv implants. (a) Scleralside of an intact LLv implant. (b) Or-bital side of the same implant. (c)Image of a fractured implant, show-ing the porous scleral side (top) andthe smooth orbital side (bottom). (d)Detail of the inner implant matrixshowing fine needle-like crystalsformed by the hydrophilic molecules(TPT � excipients) loaded into theimplant. (e) Detail of the matrix after10 days of incubation in PBS at 37°C;absence of crystals was observed.Original magnification: (a, b) 56�;(c) 24�; (d, e) 2000�. Scale bar: (a,b) 100 �m; (c) 1 mm; (d, e) 10 �m.
IOVS, April 2010, Vol. 51, No. 4 Implants for Topotecan Transscleral Delivery 2129

rate between days 2 and 10 was observed, reaching 70% and45% of the total loaded dose, respectively. The adrenalinerelease profile was similar (Fig. 5c). Although the instability ofadrenaline at pH 7.4 precludes obtaining accurate data, thedetection of adrenaline traces during the whole in vitro exper-iment confirms its entrapment and a sustained release trend.The shape and size of the implants was fully conserved duringthe 10 days of the in vitro study (Fig. 5d) and after the in vivostudies (Figs. 5e, 5f). Disappearance of the yellow color fromthe scleral side of the implants throughout the in vitro and invivo studies further evidenced drug release (Figs. 5d–f). In vivorelease data from eight implants are displayed in Figure 5a,overlapping the in vitro release curve, as a sign of the invitro/in vivo release correlation. The release in vivo was alsoconfirmed by the imprinting of the released TPT (yellow area)on the surface of the sclera (Fig. 5g). In vitro water absorptionby blank implants was 1.9% � 0.2% at day 1 and 4.5% � 0.6%at day 7. These values coincided with the weight loss, asdisplayed in Figure 5h (1.8% of original weight at day 1 and4.5% at day 7). Pure PCL implants showed �2% weight lossafter 28 days of incubation.
In Vivo Experiments
Figure 6a displays TPT vitreous levels 5 hours after the inser-tion of low-loaded implants (LL and LLv groups). A statisticallysignificant increase in TPT vitreous levels was found in thetreated eye compared with the contralateral eye in both groups(P � 0.001; t-test). TPT vitreous levels into the treated eyewere significantly higher for the LLv group compared with the
LL group, whereas the penetration to the contralateral eye waslow and similar in both groups. Plasma TPT levels were below4 ng/mL at 1, 2, and 4 hours and below 1 ng/mL at 24 hours.LTPT in vitreous and plasma was below the detection limit (0.5ng/mL) at all time points.
Figure 6b shows total TPT vitreous levels in the HL group,as assessed by microdialysis sampling during the 160- to 320-minute time period after the insertion of the DDS. No differ-ences between the implanted and the control eye were foundat any of the time points in this study. In animals receiving HLvimplants, however, a significantly higher penetration to thetreated eye was found compared with the contralateral eye inthe 220- to 300-minute time interval (Fig. 6c). When comparedwith the HL group, vitreous TPT levels in the HLv rabbits werehigher at 260 and 300 minutes (P � 0.05, t-test). Microdialysissampling did not allow for analyzing LTPT because lactone wasconverted to carboxylate during the 40-minute sample collec-tion in pH 7.4 buffer. Therefore, additional experiments wereperformed in rabbits with HLv implants sampled by vitreousaspiration at 5 hours; 12% � 3% LTPT was found in the vitreoustotal TPT amount (n � 3 rabbits). These experiments alsoallowed confirmation of comparable total TPT vitreous concen-tration values at 5 hours, as sampled by vitreous aspiration(13 � 6 ng/mL) and microdialysis (9 � 5 ng/mL).
TPT plasma exposure was assessed in both HL and HLvgroups for 48 hours (Fig. 6d). LTPT AUC could not be calcu-lated because LTPT was not detectable in most of the plasmasamples. In the few samples with detectable amounts of LTPT,concentrations were lower than 5 ng/mL. Total TPT plasma
FIGURE 5. Drug release profile from TPT-loaded implants. (a) In vitro cumulative release from LLv implants (closed circles; mean � SD of n � 3values) and in vivo release values calculated as the difference between the initial TPT load and the unreleased TPT in recovered implants (opencircles; individual values). (b) In vitro cumulative release from HLv implants (mean � SD of n � 3 values). (c) Adrenaline in vitro cumulative releasefrom LLv implants (mean � SD of n � 3 values). (d) LLv implants scleral side before (left) and after (right) 10 days of in vitro release. (e) Scleralside of LLv implants as preimplanted (left) or recovered from rabbits at 8 hours, 3 days, and 6 days (left to right). (f) Orbital sides of the implantsin (d). (g) Enucleated treated (left) and contralateral eyes devoid of conjunctival and muscular tissues of an albino rabbit 8 hours after the insertionof the DDS. (h) Weight loss (percentage of initial weight) of Blank implants and pure PCL implants after in vitro incubation (mean � SD of threevalues).
2130 Carcaboso et al. IOVS, April 2010, Vol. 51, No. 4
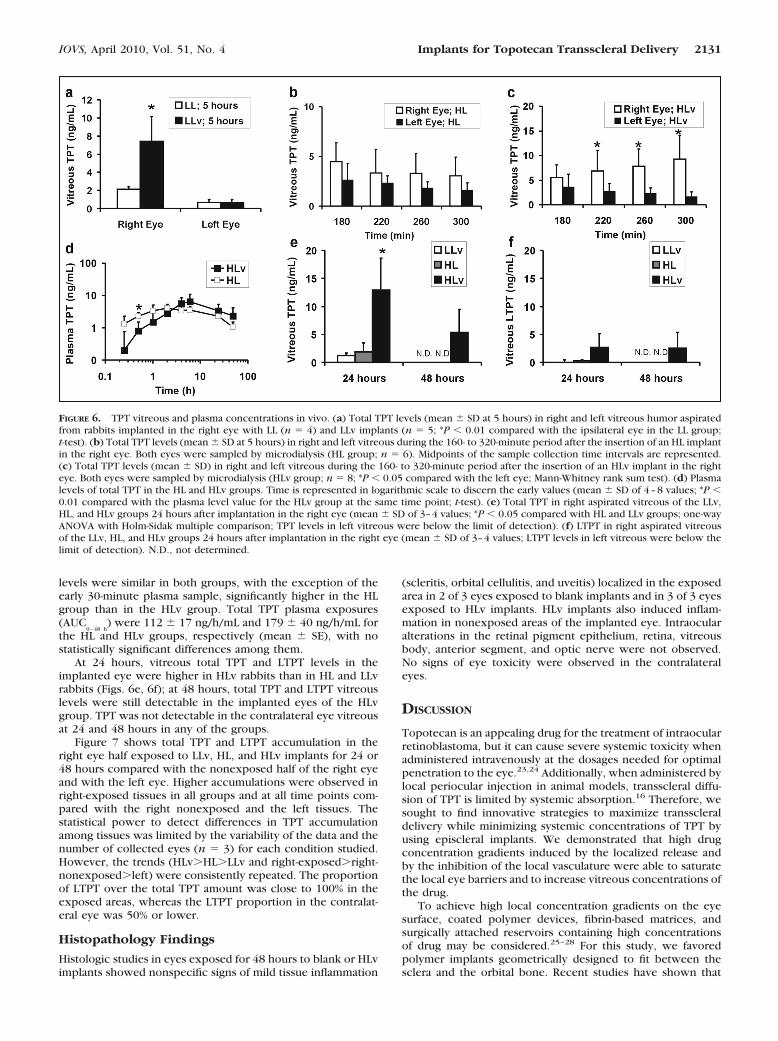
levels were similar in both groups, with the exception of theearly 30-minute plasma sample, significantly higher in the HLgroup than in the HLv group. Total TPT plasma exposures(AUC
0–48 h) were 112 � 17 ng/h/mL and 179 � 40 ng/h/mL for
the HL and HLv groups, respectively (mean � SE), with nostatistically significant differences among them.
At 24 hours, vitreous total TPT and LTPT levels in theimplanted eye were higher in HLv rabbits than in HL and LLvrabbits (Figs. 6e, 6f); at 48 hours, total TPT and LTPT vitreouslevels were still detectable in the implanted eyes of the HLvgroup. TPT was not detectable in the contralateral eye vitreousat 24 and 48 hours in any of the groups.
Figure 7 shows total TPT and LTPT accumulation in theright eye half exposed to LLv, HL, and HLv implants for 24 or48 hours compared with the nonexposed half of the right eyeand with the left eye. Higher accumulations were observed inright-exposed tissues in all groups and at all time points com-pared with the right nonexposed and the left tissues. Thestatistical power to detect differences in TPT accumulationamong tissues was limited by the variability of the data and thenumber of collected eyes (n � 3) for each condition studied.However, the trends (HLv�HL�LLv and right-exposed�right-nonexposed�left) were consistently repeated. The proportionof LTPT over the total TPT amount was close to 100% in theexposed areas, whereas the LTPT proportion in the contralat-eral eye was 50% or lower.
Histopathology Findings
Histologic studies in eyes exposed for 48 hours to blank or HLvimplants showed nonspecific signs of mild tissue inflammation
(scleritis, orbital cellulitis, and uveitis) localized in the exposedarea in 2 of 3 eyes exposed to blank implants and in 3 of 3 eyesexposed to HLv implants. HLv implants also induced inflam-mation in nonexposed areas of the implanted eye. Intraocularalterations in the retinal pigment epithelium, retina, vitreousbody, anterior segment, and optic nerve were not observed.No signs of eye toxicity were observed in the contralateraleyes.
DISCUSSION
Topotecan is an appealing drug for the treatment of intraocularretinoblastoma, but it can cause severe systemic toxicity whenadministered intravenously at the dosages needed for optimalpenetration to the eye.23,24 Additionally, when administered bylocal periocular injection in animal models, transscleral diffu-sion of TPT is limited by systemic absorption.16 Therefore, wesought to find innovative strategies to maximize transscleraldelivery while minimizing systemic concentrations of TPT byusing episcleral implants. We demonstrated that high drugconcentration gradients induced by the localized release andby the inhibition of the local vasculature were able to saturatethe local eye barriers and to increase vitreous concentrations ofthe drug.
To achieve high local concentration gradients on the eyesurface, coated polymer devices, fibrin-based matrices, andsurgically attached reservoirs containing high concentrationsof drug may be considered.25–28 For this study, we favoredpolymer implants geometrically designed to fit between thesclera and the orbital bone. Recent studies have shown that
FIGURE 6. TPT vitreous and plasma concentrations in vivo. (a) Total TPT levels (mean � SD at 5 hours) in right and left vitreous humor aspiratedfrom rabbits implanted in the right eye with LL (n � 4) and LLv implants (n � 5; *P � 0.01 compared with the ipsilateral eye in the LL group;t-test). (b) Total TPT levels (mean � SD at 5 hours) in right and left vitreous during the 160- to 320-minute period after the insertion of an HL implantin the right eye. Both eyes were sampled by microdialysis (HL group; n � 6). Midpoints of the sample collection time intervals are represented.(c) Total TPT levels (mean � SD) in right and left vitreous during the 160- to 320-minute period after the insertion of an HLv implant in the righteye. Both eyes were sampled by microdialysis (HLv group; n � 8; *P � 0.05 compared with the left eye; Mann-Whitney rank sum test). (d) Plasmalevels of total TPT in the HL and HLv groups. Time is represented in logarithmic scale to discern the early values (mean � SD of 4–8 values; *P �0.01 compared with the plasma level value for the HLv group at the same time point; t-test). (e) Total TPT in right aspirated vitreous of the LLv,HL, and HLv groups 24 hours after implantation in the right eye (mean � SD of 3–4 values; *P � 0.05 compared with HL and LLv groups; one-wayANOVA with Holm-Sidak multiple comparison; TPT levels in left vitreous were below the limit of detection). (f) LTPT in right aspirated vitreousof the LLv, HL, and HLv groups 24 hours after implantation in the right eye (mean � SD of 3–4 values; LTPT levels in left vitreous were below thelimit of detection). N.D., not determined.
IOVS, April 2010, Vol. 51, No. 4 Implants for Topotecan Transscleral Delivery 2131

polyethylene devices tightly sutured to the sclera enhance drugdiffusion through the indented region of the eye by maximiz-ing contact with the ocular surface.27 The thickness and con-vex surface design of our DDS allowed the indentation of thesclera and prevented local migration of the implant without theneed for sutures (Fig. 4g). PCL polymer was used in the implantmatrix given its proven biocompatibility and its versatility toentrap hydrophilic substances to be released with a controlledand predictable pattern.20,29 In addition, the slow biodegrad-ability of PCL allowed the size, shape, and hardness of theimplants to be retained throughout the in vivo studies, facili-tating easy surgical removal from the implantation site at thecompletion of treatment or in the case of acute local toxicity.
Because TPT exerts most of its antiretinoblastoma effect inthe first 15 minutes of cellular exposure,13 we designed im-plants that initially release high amounts of drug to saturate thelocal barriers and to achieve high local concentrations of thedrug. Long-lasting release at a slower rate achieved with ourDDS might have been important in recruiting more retinoblas-toma cells at the S-phase of the cycle. In fact, the use ofprotracted administration schedules of intravenously adminis-tered TPT resulted in improved antitumor activity in otherpediatric malignancies with a drug sensitivity profile compara-ble to that of retinoblastoma.30 Preliminary in vitro experi-ments showed very slow TPT release rate from implants loadedwith pure TPT and no hydrophilic substances (data notshown). We have previously demonstrated that the release ofhydrophilic drugs from PCL implants can be correlated with
the loaded amount of hydrophilic substance in the PCL ma-trix.20 Therefore, we hypothesized that the hydrophilic addi-tive substances (excipients) of the commercial TPT, whenhomogeneously incorporated in the slow degradation PCL de-vice, would form pores allowing the absorption of fluids andthe release of the drug at the desired release rate. This pointwas confirmed after in vitro incubation by observing the extentof fluid absorption and mass loss of the implants loaded withhydrophilic excipients and the disappearance of the originalcrystals in the DDS matrix (Fig. 4). To allow comparison withour previous data of TPT vitreous and plasma exposure inrabbits receiving 1 mg/eye (periocular) or 1 mg/rabbit (IV) ofTPT solution,16 the amount of drug loaded in HL and HLvimplants was selected to release a total dose of approximately1 mg in 48 hours, as tested in vitro (Fig. 5).
The function of the drug released in vitro could not bestudied because most of the drug in solution was quicklyconverted to the inactive carboxylate at pH 7.4 and 37°C.However, we inferred that the stability of the lactone TPT forminto the implant might have been favored, both in vitro and invivo, by the acid excipients (tartaric acid) coloaded in the DDS,which could have provided an acid microenvironment in thepolymer matrix. This rationale was supported by the observa-tion that only LTPT was detected in the implants recoveredfrom the rabbits included in the in vivo release study (Fig. 5a).Biodegradable polymers based on poly-D,L-lactide-co-glycolideacid have been reported to stabilize camptothecin lactonesbecause of a hydrolytic process that produces carboxylic acid
FIGURE 7. TPT accumulation (nano-gram of TPT per gram of tissue) inthe right eye half exposed to LLv, HL,and HLv implants for 24 or 48 hourscompared with the right nonex-posed eye half and the left eye. RE,right-exposed half; RNE, right-nonex-posed half; L, left eye. Total TPT(a–c) and proportion of LTPT overtotal TPT (d–f) were assessed in ret-ina (a, d), choroid (b, e), and sclera(c, f). Mean � SD of n � 3 values arerepresented. #, LTPT under limit ofdetection.
2132 Carcaboso et al. IOVS, April 2010, Vol. 51, No. 4

functional groups, generating a low pH inside the deliverysystems.31 These FDA-approved materials could be an alterna-tive to the also approved PCL polymer we test here; however,the low cost of PCL could be important for the eventualapplication of similar devices in developing countries, whereadvanced retinoblastoma is more prevalent.32
Our initial in vivo experiments with LL formulations led toselective TPT delivery to the exposed eye and very low plasmaexposure. However, these experiments showed relatively lowTPT levels in the treated vitreous. Levels in the range of 10ng/mL are needed for antitumor activity in retinoblastoma, andthey were not achieved by these implants.13 In our previousstudy, we were able to show in postmortem experiments thatthe sclera is permeable to TPT, but the drug failed to reach asignificant transscleral penetration in vivo, probably because arapid clearance from the orbit by the regional circulation.16
Therefore, we hypothesized that vasoconstriction could im-prove the transscleral flow of TPT; hence, we developed ourLLv implants, finding a significant increase in TPT vitreousconcentrations compared with the LL implants. However, vit-reous TPT concentrations at 24 hours were almost undetect-able with low-loaded formulations. Therefore, we increasedthe TPT load in the implants and studied high loaded formula-tions (HL and HLv). Surprisingly, even though the TPT load washigher, HL implants did not reach vitreous TPT levels achievedat 5 hours with the LLv approach, highlighting the criticaleffect of local vasoconstriction. A remarkable increase wasobserved in local drug penetration with HLv implants com-pared with HL formulations, further supporting the relevanceof a strategy to inhibit local vessels to reduce local clearance ofthe drug, as previously reported with �-adrenergic selectiveinhibitors such as oxymetazoline.33
Plasma total TPT exposure in rabbits receiving HL and HLvimplants was approximately three times lower than in rabbitsreceiving 1 mg drug either periocularly or intravenously.16
Most interestingly, LTPT levels were low or undetectable in theimplanted rabbits, whereas 30% and 40% of total TPT exposurein plasma was lactone in the rabbits that received periocularadministration and IV administration, respectively.16 There-fore, episcleral implants significantly decreased systemic expo-sure to the potentially toxic lactone form of TPT. The signifi-cantly lower systemic TPT levels during the earlier time pointsusing HLv implants (Fig. 5d) likely resulted from the inhibitionof systemic absorption of the drug by vasoconstriction.
The proportion of LTPT over total TPT 5 hours after theinsertion of HLv implants in the implanted eye vitreous was12%. After 24 and 48 hours, the proportion increased to 20%and 40%, respectively, as shown in Figures 6e and 6f. Thisresult highlights the capacity of the local barriers to impedelocal drug penetration even at high concentration gradientsand supports the saturation of the barriers over time withcontinuously released drug. Our findings should be translatedwith caution to the clinical situation because barriers might bealtered in eyes affected by retinoblastoma, and our non–tumor-bearing animal model was therefore limited.34 In addition, therabbit eye, though fairly similar to the human eye in size andvolume proportion between the intraocular compartments,may account for additional model limitations because of its lackof internal retinal vessels, high peripheral choroidal flow,35 orabsence of orbital fat,36 among other factors. Interestingly, thescleral thickness reported for rabbit eyes (0.44 mm),37 thoughnarrower than in adult humans (0.61 mm),38 is similar to thatreported in a 9-day-old infant (0.40 mm),38 which would sup-port the use of rabbit eyes for our studies aiming at an earlychildhood disease. The scleral permeability of several drugs hasbeen studied and correlated with their physicochemical prop-erties, showing that molecular size is the most relevant factorlimiting the transscleral transit of drugs in vitro.39,40 Molecules
in the size range of the TPT free base (MWt 421 Da) have beenfound to permeate human and rabbit scleras.41,42 However, itis likely that the sclera plays a minor role in limiting local drugdelivery compared with more specialized tissues, such as thechoroid and the Bruch’s membrane.43
Finally, we provide evidence that the active form of thedrug, LTPT, is accumulated in the sclera and choroid exposedto the implants. These tissues seem to play an active role intransscleral drug delivery, acting as a powerful sink that pre-cludes the penetration of LTPT to the retina and the vitreous,either redirecting the drug to the systemic circulation or accu-mulating it in their own cells. We hypothesize that the drug istaken up by the transport proteins present in ocular tissues.44
Experimental data support the differential affinity of drug trans-porters for lactone and carboxylic forms of drugs,45 whichwould explain why carboxylate TPT is predominantly found inthe vitreous exposed to the implants, whereas LTPT is en-trapped in the ocular barriers.
To conclude, we have developed a technological strategy toenhance and sustain the penetration of TPT to the posteriorsegment of treated eyes and to reduce potentially toxic sys-temic drug exposure. The present research also contributes tothe understanding of the active role of the ocular tissues,sclera, and choroid in the clearance of TPT during its transientpath toward the intraocular compartments. Our observationsmay help design pharmacologic and technological strategies toovercome the barrier activity of ocular tissues during trans-scleral drug penetration, which may ultimately contribute toimproving current retinoblastoma treatments.
References
1. Cunha-Vaz JG. The blood-retinal barriers system: basic conceptsand clinical evaluation. Exp Eye Res. 2004;78:715–721.
2. Wilson TW, Chan HS, Moselhy GM, Heydt DD Jr, Frey CM, GallieBL. Penetration of chemotherapy into vitreous is increased bycryotherapy and cyclosporine in rabbits. Arch Ophthalmol. 1996;114:1390–1395.
3. Shields JA, Shields CL, Meadows AT. Chemoreduction in the man-agement of retinoblastoma. Am J Ophthalmol. 2005;140:505–506.
4. Chintagumpala M, Chevez-Barrios P, Paysse EA, Plon SE, HurwitzR. Retinoblastoma: review of current management. Oncologist.2007;12:1237–1246.
5. Mendelsohn ME, Abramson DH, Madden T, Tong W, Tran HT,Dunkel IJ. Intraocular concentrations of chemotherapeutic agentsafter systemic or local administration. Arch Ophthalmol. 1998;116:1209–1212.
6. Dunkel IJ, Lee TC, Shi W, et al. A phase II trial of carboplatin forintraocular retinoblastoma. Pediatr Blood Cancer. 2007;49:643–648.
7. Abramson DH, Frank CM, Dunkel IJ. A phase I/II study of subcon-junctival carboplatin for intraocular retinoblastoma. Ophthalmol-ogy. 1999;106:1947–1950.
8. Chantada GL, Fandino AC, Carcaboso AM, et al. A phase I study ofperiocular topotecan in children with intraocular retinoblastoma.Invest Ophthalmol Vis Sci. 2009;50:1492–1496.
9. Schmack I, Hubbard GB, Kang SJ, Aaberg TM Jr, Grossniklaus HE.Ischemic necrosis and atrophy of the optic nerve after periocularcarboplatin injection for intraocular retinoblastoma. Am J Oph-thalmol. 2006;142:310–315.
10. Abramson DH. Periocular chemotherapy for retinoblastoma: suc-cess with problems? Arch Ophthalmol. 2005;123:128–129; authorreply 129.
11. Frangoul H, Ames MM, Mosher RB, et al. Phase I study of topotecanadministered as a 21-day continuous infusion in children withrecurrent solid tumors: a report from the Children’s Cancer Group.Clin Cancer Res. 1999;5:3956–3962.
12. Chantada GL, Fandino AC, Casak SJ, Mato G, Manzitti J, Schvartz-man E. Activity of topotecan in retinoblastoma. Ophthalmic Genet.2004;25:37–43.
IOVS, April 2010, Vol. 51, No. 4 Implants for Topotecan Transscleral Delivery 2133

13. Laurie NA, Gray JK, Zhang J, et al. Topotecan combination che-motherapy in two new rodent models of retinoblastoma. ClinCancer Res. 2005;11:7569–7578.
14. Athale UH, Stewart C, Kuttesch JF, et al. Phase I study of combi-nation topotecan and carboplatin in pediatric solid tumors. J ClinOncol. 2002;20:88–95.
15. Hughes PM, Olejnik O, Chang-Lin JE, Wilson CG. Topical andsystemic drug delivery to the posterior segments. Adv Drug DelivRev. 2005;57:2010–2032.
16. Carcaboso AM, Bramuglia GF, Chantada GL, et al. Topotecan vit-reous levels after periocular or intravenous delivery in rabbits: analternative for retinoblastoma chemotherapy. Invest OphthalmolVis Sci. 2007;48:3761–3767.
17. Houghton PJ, Cheshire PJ, Myers L, Stewart CF, Synold TW,Houghton JA. Evaluation of 9-dimethylaminomethyl-10-hydroxy-camptothecin against xenografts derived from adult and childhoodsolid tumors. Cancer Chemother Pharmacol. 1992;31:229–239.
18. Gaudana R, Jwala J, Boddu SH, Mitra AK. Recent perspectives inocular drug delivery. Pharm Res. 2009;26:1197–1216.
19. Burke TG, Bom D. Camptothecin design and delivery approachesfor elevating anti-topoisomerase I activities in vivo. Ann N Y AcadSci. 2000;922:36–45.
20. Carcaboso AM, Chiappetta DA, Hocht C, et al. In vitro/in vivocharacterization of melt-molded gabapentin-loaded poly(epsilon-caprolactone) implants for sustained release in animal studies. EurJ Pharm Biopharm. 2008;70:666–673.
21. Rittenhouse KD, Pollack GM. Microdialysis and drug delivery tothe eye. Adv Drug Deliv Rev. 2000;45:229–241.
22. de Lange EC, de Boer AG, Breimer DD. Methodological issues inmicrodialysis sampling for pharmacokinetic studies. Adv DrugDeliv Rev. 2000;45:125–148.
23. Nitschke R, Parkhurst J, Sullivan J, Harris MB, Bernstein M, Pratt C.Topotecan in pediatric patients with recurrent and progressivesolid tumors: a Pediatric Oncology Group phase II study. J PediatrHematol Oncol. 1998;20:315–318.
24. Leger F, Loos WJ, Bugat R, et al. Mechanism-based models fortopotecan-induced neutropenia. Clin Pharmacol Ther. 2004;76:567–578.
25. Sasaki H, Nagano T, Sakanaka K, et al. One-side-coated insert as aunique ophthalmic drug delivery system. J Control Release. 2003;92:241–247.
26. Wang G, Tucker IG, Roberts MS, Hirst LW. In vitro and in vivoevaluation in rabbits of a controlled release 5-fluorouracil subcon-junctival implant based on poly(D,L-lactide-co-glycolide). PharmRes. 1996;13:1059–1064.
27. Pontes de Carvalho RA, Krausse ML, Murphree AL, Schmitt EE,Campochiaro PA, Maumenee IH. Delivery from episcleral exo-plants. Invest Ophthalmol Vis Sci. 2006;47:4532–4539.
28. Tsui JY, Dalgard C, Van Quill KR, et al. Subconjunctival topotecanin fibrin sealant in the treatment of transgenic murine retinoblas-toma. Invest Ophthalmol Vis Sci. 2008;49:490–496.
29. Sun H, Mei L, Song C, Cui X, Wang P. The in vivo degradation,absorption and excretion of PCL-based implant. Biomaterials.2006;27:1735–1740.
30. Santana VM, Furman WL, Billups CA, et al. Improved response inhigh-risk neuroblastoma with protracted topotecan administrationusing a pharmacokinetically guided dosing approach. J Clin Oncol.2005;23:4039–4047.
31. Shenderova A, Burke TG, Schwendeman SP. The acidic microcli-mate in poly(lactide-co-glycolide) microspheres stabilizes campto-thecins. Pharm Res. 1999;16:241–248.
32. Rodriguez-Galindo C, Wilson MW, Chantada G, et al. Retinoblastoma:one world, one vision. Pediatrics. 2008;122:e763–770.
33. Miller DJ, Li SK, Tuitupou AL, et al. Passive and oxymetazoline-enhanced delivery with a lens device: pharmacokinetics and efficacystudies with rabbits. J Ocul Pharmacol Ther. 2008;24:385–391.
34. Abramson DH, Frank CM, Chantada GL, et al. Intraocular carbo-platin concentrations following intravenous administration for hu-man intraocular retinoblastoma. Ophthalmic Genet. 1999;20:31–36.
35. Nork TM, Kim CB, Shanmuganayagam D, Van Lysel MS, Ver HoeveJN, Folts JD. Measurement of regional choroidal blood flow inrabbits and monkeys using fluorescent microspheres. Arch Oph-thalmol. 2006;124:860–868.
36. Davis FA. The anatomy and histology of the eye and orbit of therabbit. Trans Am Ophthalmol Soc. 1929;27:400 402–441.
37. Boubriak OA, Urban JP, Akhtar S, Meek KM, Bron AJ. The effect ofhydration and matrix composition on solute diffusion in rabbitsclera. Exp Eye Res. 2000;71:503–514.
38. Olsen TW, Edelhauser HF, Lim JI, Geroski DH. Human scleralpermeability: effects of age, cryotherapy, transscleral diode laser,and surgical thinning. Invest Ophthalmol Vis Sci. 1995;36:1893–1903.
39. Prausnitz MR, Noonan JS. Permeability of cornea, sclera, andconjunctiva: a literature analysis for drug delivery to the eye.J Pharm Sci. 1998;87:1479–1488.
40. Ahmed I, Gokhale RD, Shah MV, Patton TF. Physicochemicaldeterminants of drug diffusion across the conjunctiva, sclera, andcornea. J Pharm Sci. 1987;76:583–586.
41. Cruysberg LP, Franklin AJ, Sanders J, et al. Effective transscleraldelivery of two retinal anti-angiogenic molecules: carboxyamido-triazole (CAI) and 2-methoxyestradiol (2ME2). Retina. 2005;25:1022–1031.
42. Okabe K, Kimura H, Okabe J, et al. Effect of benzalkonium chlo-ride on transscleral drug delivery. Invest Ophthalmol Vis Sci.2005;46:703–708.
43. Cheruvu NP, Kompella UB. Bovine and porcine transscleral solutetransport: influence of lipophilicity and the choroid-Bruch’s layer.Invest Ophthalmol Vis Sci. 2006;47:4513–4522.
44. Zhang T, Xiang CD, Gale D, Carreiro S, Wu EY, Zhang EY. Drugtransporter and cytochrome P450 mRNA expression in humanocular barriers: implications for ocular drug disposition. DrugMetab Dispos. 2008;36:1300–1307.
45. Chen C, Mireles RJ, Campbell SD, et al. Differential interaction of3-hydroxy-3-methylglutaryl-coa reductase inhibitors with ABCB1,ABCC2, and OATP1B1. Drug Metab Dispos. 2005;33:537–546.
2134 Carcaboso et al. IOVS, April 2010, Vol. 51, No. 4





























