Embed Size (px)
Citation preview

Family Studies of the Genetic Abnormality in Transthyretin (Prealbumin) in
Portuguese Patients with Familial Amyloidotic Polyneuropathy“ MARIA JOAO MASCARENHAS SARAIVA:
STEVEN BIRKEN, PEDRO P. COSTA, AND DEWITT S. GOODMAN
Department of Medicine College of Physicians & Surgeons of Columbia University
New York, New York 10032
Centro de Estudos de Paramiloidose Hospital de St. Antonio
Porto, Portugal
INTRODUCTION
Amyloidosis comprises a variety of disease processes characterized by the deposi- tion in tissues of fibrillar protein with a twisted @-pleated sheet configuration.’ The amyloid @-fibrils can be formed from a number of different proteins by a variety of pathogenetic mechanisms. The major protein component (called AL) associated with “primary” and with myeloma-associated amyloid is usually derived from immunoglob- ulin light chains.’,’ In other “secondary” forms of amyloidosis, and in the form associated with familial Mediterranean fever, the major fibrillar protein constituent (the AA protein) is derived from a serum acute-phase reactant, a protein designated SAA.’,’
Recently, a third protein, transthyretin (TTR, usually referred to as plasma prealbumin‘). has been related to several forms of amyloidosis, and particularly to the systemic heredofamilial amyloidoses that demonstrate autosomal dominant inheri- tance. Thus, in 1978, Costa et aL4 reported that a protein component of the amyloid fibrils that accumulate in tissues of patients with familial amyloidotic polyneuropathy (FAP), Portuguese type, is closely related to TTR. Since then, a number of investiga- tors, using both chemical and immunochemical methods,’t9 have demonstrated the association of TTR with amyloid deposits in patients with FAP from several countries
‘This work was supported by Grants HL2 1006 (SCOR). AMO5968, and HD15454. M.J.M.S. was a recipient of a scholarship from lnstituto National de Investigacso CiZntifica during part of this work.
bPermanent address: Departmento de Bioquimica, Instituto de Ciincias Biomtdicas, Universi- dade de Porto, 4000 Porto, Portugal.
‘The name transthyretin has been suggested recently by the Nomenclature Committee of the IUB and the IUPAC-IUB Joint Commission on Biochemical Nomenclature for the protein commonly called prealbumin (1981. J. Biol. Chem. 256: 12-14).
86

SARAIVA er al.: TRANSTHYRETIN VARIANT IN FAP 87
and of different ethnic origins. It was suggested that in FAP a genetic mutation may have occurred, leading to an abnormal TTR that is abnormally bound, degraded, and/or precipitated in tissues as amyloid fibrils.
Human plasma TTR has been extensively studied and characterized in the past two decades. The TTR molecule is a stable and symmetrical tetramer composed of four identical subunits, with a molecular weight of 54,980." The amino acid sequence of TTR is known," and the full three-dimensional structure of the molecule has been determined by high-resolution X-ray crystallography." TTR possesses high-affinity binding sites for both thyroxine"." and for plasma retinol-binding protein (RBP)123'' and hence plays a role in the plasma transport of both thyroid hormone and of retinol (vitamin A).
The studies reported here were undertaken in order to explore the genetic and biochemical abnormalities involved in the etiology of the Portuguese type of FAP. The studies involved detailed investigation of the properties and structure of TTR isolated from serum of patients with FAP, and of amyloid fibril protein isolated from tissues of patients who died with FAP. As described later, we have found that an abnormal TTR with a methionine for valine substitution at position 30 is deposited in tissues of FAP patients, and that this abnormal TTR circulates in relatively low concentrations in the plasma of these patients. Thus, these studies have identified a point mutation in human plasma TTR in FAP, and have shown that the mutant (variant) form of TTR is selectively deposited in tissues as the amyloid characteristic of the disease. In addition, family studies have shown that the presence of the variant TTR can be used for the diagnosis of the Portuguese variety of FAP, and for the identification of at-risk offspring in the preclinical phase of the disease.
PLASMA TTR IN PATIENTS WITH FAP
In the first phase of this project, studies were conducted that focused on plasma TTR in Portuguese patients with FAP; these studies have been reported in detail.I4 Significantly reduced (p i 0.005) levels of TTR were found in patients with FAP. Thus, the TTR levels in 24 patients with FAP (190 + 44 Kg/ml, mean * SD) were approximately two-thirds those of a group of 18 control subjects (294 64 pg/ml) from the same geographic area. In contrast, RBP levels were not reduced, nor was there an abnormality in the ratio of retinol to RBP in the patients with FAP. Hence there did not appear to be an abnormality in vitamin A transport in Portuguese patients with FAP. There did, however, appear to be abnormality of TTR metabolism, accounting for the reduced plasma levels of TTR.
TTR was isolated from pooled sera of 24 patients with FAPand was compared with normal TTR with regard to a wide range of physicochemical characteristic^.'^ FAP-TTR was indistinguishable from normal TTR with regard to a variety of variables including: (i) electrophoretic mobility; (ii) chromatographic behavior; (iii) molecular weight; (iv) stability of the TTR tetramer (resistance to dissociation); (v) immunoreactivity i n TTR radioimmunoassays using antisera prepared against both normal and FAP-TTR; (vi) ability to form a protein-protein complex with RBP and affinity for RBP as assessed by polarization of fluorescence; and (vii) overall amino acid composition. It was suggestedn4 that if an abnormal TTR is produced in this form of FAP, either it is present only in low amounts in the plasma of patients with FAP, or it is structurally almost identical to normal TTR and does not differ from normal TTR with regard to the various physical and chemical properties investigated.

88 ANNALS NEW YORK ACADEMY OF SCIENCES
AMYLOID FIBRIL PROTEIN (AF,): ISOLATION AND PARTIAL CHARACTERIZATION
Since FAP-TTR and normal TTR were indistinguishable with regard to the many physical-chemical properties examined, it became apparent that further progress towards the biochemical definition of the abnormality in FAP would require the study of the TTR-related amyloid protein from the tissues. Accordingly, studies were conducted (i) to develop improved methods for the isolation of pure and undenatured amyloid fibril protein; and (ii) to characterize the amyloid fibril protein, and compare it with plasma TTR, in a number of ways.
Amyloid fibril concentrates were obtained postmortem from the kidneys of two Portuguese patients who died with FAP. The amyloid fibrils were purified by repeated homogenization in phosphate-buffered saline solution, and then in distilled water, in a modification of the methods of Pras et al.” and Glenner et a1.,I6 as described previ~usly.~ The lyophilized amyloid fibril concentrates were solubilized and reduced with dithiothreitol in 6 M guanidine-HCI, followed by alkylation with iodoacetic acid. The alkylated preparation was dialyzed to remove guanidine and then lyophilized. The resulting preparation was soluble in a buffer of “physiological” ionic strength and pH (40 mM Tris-HCI, pH 7.4, 0.15 M NaCI), in the absence of guanidine or of detergents.
Purified amyloid fibril protein (AF,) was isolated by affinity chromatography on RBP linked to Sepharose. It has been shown that reduction and alkylation of TTR do not affect its binding properties for RBP.’* The solubilized amyloid fibril preparation was applied to an RBP-Sepharose affinity column. A major portion of the amyloid protein did not bind to the column, and was eluted directly. Another portion of the amyloid protein (approximately 20% of the total protein applied to the column) did bind to the column and was then eluted with distilled water adjusted to pH 10.4. It is known that the TTR-RBP complex is dissociated in solution of very low ionic strength and alkaline pH,I3.l7 and an identical affinity chromatography procedure has been used for the isolation of TTR from serum.” The protein that had bound to the RBP affinity column migrated as a single protein band on polyacrylamide gel electrophoresis (PAGE). This isolated protein displayed TTR immunoreactivity, and had an amino acid composition that was almost identical to that of normal TTR. Moreover, when analyzed by SDS-PAGE under dissociating conditions, this protein migrated as a single band of molecular weight of approximately 14,000 that could not be distin- guished from the TTR monomer. Thus, this protein represented a purified preparation of AF, that closely resembled plasma TTR.
Studies were also conducted on the amyloid fibril protein that did not bind to the RBP affinity column. Amino acid analysis revealed that only approximately one- fourth of the total mass of this material consisted of protein; the nature of the nonprotein component(s) is not known. The amino acid composition of the protein was, however, similar to that of TTR. Analysis of this material by SDS-gel electrophoresis showed several protein bands: major bands of molecular weights of approximately 15,000 and 32,000, and minor bands of about 42,000 and 54,000; higher-weight bands were seen as well. Immunoblotting demonstrated that all of these protein bands displayed TTR immunoreactivity. The results suggest that these protein bands all represented polymers of a TTR-related subunit, and that TTR-related protein may be the predominant, or almost the sole, protein component of the amyloid fibril.
SDS-PAGE under nondissociating conditions revealed that the purified AF, migrated almost entirely as a band of protein of molecular weight of approximately 50,000. Thus, the AF, protein appeared to form a stable tetramer under conditions where plasma TTR is normally found in tetrameric form. The ability of the purified

SARAIVA et a/.: TRANSTHYRETIN VARIANT IN FAP 89
AF, to form tetramers was confirmed by studies with labeled thyroxine that demon- strated that AF, has binding affinity for thyroxine. The binding of iodothyronine molecules by TTR requires the tetrameric quaternary structure of the protein, since the tetrameric molecule has a channel running through the center of its long axis in which the two binding sites for thyroxine are located.’’ In addition, the affinity chromatography procedure, on RBP linked to Sepharose, showed that AF, protein had binding properties for RBP very similar to those of normal TTR. Hence, the AF, protein resembled plasma TTR in forming a stable tetrameric structure, and in its binding affinities for both thyroxine and for RBP.
AF, AND I T R : COMPARATIVE STUDIES OF PRIMARY STRUCTURE
The isolation of a purified, soluble amyloid fibril protein (AF,) provided material suitable for detailed studies of its chemical structure as compared to that of TTR. Accordingly, studies were conducted to compare the structure of AF, to that of TTR which had been isolated both from normal plasma and from pooled sera of patients with FAP (FAP-TTR). The two samples of TTR were the same preparations that had been used in the detailed study of their physical-chemical characteristics summarized above. The structural studies included: (i) comparative peptide mapping by reverse- phase high-performance liquid chromatography (HPLC) after trypsin digestion; (ii) cyanogen bromide (CNBr) cleavage studies; and (iii) amino acid microsequence analysis of selected tryptic and CNBr peptides.
Comparative Tryptic Peptide Mapping
A tryptic peptide map was constructed for normal TTR by digesting the reduced and carboxymethylated protein with trypsin, and separating the resulting peptide fragments by HPLC. HPLC was carried out with Waters Associates equipment using a pBondapak C,, (0.4 x 30 cm) column, with a gradient of &SO% acetonitrile in 0.1 % phosphoric acid. The separated tryptic peptides were identified by their amino acid composition, determined by amino acid analysis, on the basis of the known amino acid sequence of TTR.”
All of the tryptic peptides were separated by HPLC under the conditions used. Peptides 4 and 10 eluted as a doublet (see below); complete separation of these peptides could be achieved, however, by rechromatography on the same column, with a trifluoroacetic acid-propanol solvent system. Several peptides were consistently found that represented combined tryptic peptides due to uncleaved lysine or arginine residues. These combined peptides included peptides 5 + 6.7 + 8, 1 1 + 12 + 13, and 12 + 13.
Tryptic peptide maps were obtained under identical conditions for samples of the purified AF, and of the normal TTR and FAP-TTR preparations. The tryptic peptide map of AF, closely resembled the peptide map of normal TTR, but with two distinct differences. First, tryptic peptide 4 (residues 22 to 34 in the TTR subunit) was absent from the AF, digest. Second, the AF, peptide map showed the presence of a peptide peak not present in the normal TTR map. This “abnormal” peptide peak eluted a t approximately 61 minutes, approximately 4 minutes after the position where normal peptide 4 would be expected to be seen. The “abnormal” peptide present in the AF, tryptic digest was collected and subjected to microsequence analysis.
FIGURE I shows some of the amino acid sequence of the human TTR subunit.” The sequence shown includes that of tryptic peptide 4 (labeled T,). The “abnormal”

90 ANNALS NEW YORK ACADEMY OF SCIENCES
peptide from the AF, digest had an amino acid composition identical to that of tryptic peptide 4, except that methionine was present and the valine content of the peptide was low. C-terminal sequence analysis by carbo~ypeptidases’~ showed the same C-terminal sequence of the final 4 residues as that of normal tryptic peptide 4. The “abnormal” peptide is hereafter referred to as peptide T:.
Peptide T:was also present in small amounts in the tryptic digests of FAP-TTR. along with larger amounts (approximately four-six-fold) of the corresponding normal peptide T4. Except for this single difference, the FAP-TTR peptide map was identical to that of normal TTR.
The tryptic digests were also screened by HPLC with a different solvent system, with different pH and elution properties (a gradient of 0-30% propanol in 0.1% trifluoroacetic acid, pH 4.0). No additional differences between AF, and TTR were observed. Further evidence that the structures of AF, and TTR are probably identical except for differences associated with tryptic peptide 4 was obtained from a study
I3 20 Met-Val-Lys-Val- Leu-Asp-Ala-Val-
-CNBr 2
.................... H2N-Gly-Pro-Thr-Gly -...... .- ............................ +CNBrl-
I -CNBro-
30 I27 Arg- Gly -Ser -Pro-Ala-Ile-Asn-Val-Ala-Val-His-Val-Phe-Arg-Lys-~~~~-Glu-COOH
CNBr 2 ....4
FIGURE I. Partial amino acid sequence of the human TTR subunit.” Symbols: Half arrows pointing lo the right. sequence determined by manual Edman degradationM; halfarrowspointing to the left, sequences determined by the use of ~arboxypeptidases.’~ The nomenclature of the peptides in this figure is as follows: T,, tryptic peptide 4; CNBr 1 and 2, cyanogen bromide fragments C, and C2 from normal TTR; CNBr a and b, additional cyanogen bromide fragments (C, and C,) of amyloid fibril protein AF,. The Met residue (enclosed in a box) below Val 30 indicates the difference found between AF, and TTR from the studies described in this paper.
comparing the kinetics of trypsin digestion of AF, with those of normal TTR and FAP-TTR. The patterns of appearance of the different individual tryptic peptides were quite similar for all three proteins.
Cyanogen Bromide Cleavage Studies
Normal TTR possesses only one methionine residue, at position 13. CNBr treatment, therefore, cleaves the protein into two peptide fragments: a small peptide, residues 1-13 (called peptide C, , and labeled CNBr 1 in FIGURE I ) , and a large peptide, residues 14-127 (called peptide C2, and labeled CNBr 2 in FIGURE 1). We reasoned that if another methionine residue was present in AF, in the region of tryptic peptide 4 (residues 22-34), CNBr treatment of AF, would produce extra peptide fragments (as compared to TTR). Accordingly, samples of AF, were subjected to

SARAIVA et al.: TRANSTHYRETIN VARIANT IN FAP 91
CNBr treatment, followed by HPLC analysis and separation of the resulting peptide fragments. Samples of normal TTR and of FAP-TTR were treated and analyzed in an identical manner for comparative purposes.
Normal TTR showed an HPLC peptide pattern that consisted of a group of small peaks eluting between approximately 7 and 14 minutes, and a group of larger, overlapping peaks eluting between approximately 35 and 50 minutes. Fragment C, was identified in the early peaks, and fragment C2 in the later peaks. Both peptide fragments showed elution heterogeneity. There were no peaks observed between 15 and 30 minutes with normal TTR.
After CNBr treatment of AF,, or of FAP-TTR, the HPLC peptide maps showed two sharp peaks eluting at close to 20 minutes. These peaks were collected and subjected to amino acid analysis. Homoserine lactone was found with both peaks, indicating that they represented CNBr cleavage peptides with C-terminal methionine residues. The first of these peaks was identified as a peptide composed of residues 14-30 of the TTR subunit, but with a methionine (instead of valine) residue at its C-terminus. This peptide (called Ca, and labeled CNBr a in FIGURE 1 ) had the amino acid composition, N-terminal sequence, and C-terminal sequence appropriate for this identification (FIG. I ) . The second sharp peak was found to correspond to a peptide composed of residues 1 through 30 with a methionine at position 30 and with an uncleaved methionine at position 13. This peak is therefore referred to as peptide
If AF, (and in part FAP-TTR) contains a methionine residue a t position 30. one would also expect to find a peptide fragment corresponding to residues 3 1 through 127 after CNBr cleavage. This “extra” CNBr fragment, called peptide Cb (and labeled CNBr b in FIGURE I). was not evident in the HPLC chromatogram. We suspected, however, that this peptide was coeluting with uncleaved fragment C,. In order to verify this hypothesis, SDS-PAGE was carried out on pooled HPLC fractions corresponding to the normal peptide C, (from normal TTR), and on pooled fractions from the AF, preparation eluting at about the same position. A single peptide band, of apparent molecular weight of approximately 13,OOGI 4,000 and representing fragment C,, was seen in the normal TTR preparation. With the fractions from AF,, however, two bands were observed: one corresponding to peptide C2 and a band of apparent molecular weight of approximately 12,000. This second band corresponded to peptide Cb from AF,. Its demonstration by gel electrophoresis represented the direct demonstration of the production of the anticipated “extra” CNBr fragment, peptide cb, from AF,. Furthermore, the N-terminal sequence of Cb was identical to the sequence of normal TTR for positions 31, 32, and 33 (FIG. 1).
Taken together, these results demonstrate that the amyloid fibril protein AF, comprised a TTR variant with a methionine for valine substitution a t position 30. The variant TTR was also present in the TTR isolated from pooled sera from patients with FAP, together with larger amounts of the normal TTR. Thus, in these patients with FAP, the variant TTR was circulating in plasma along with larger amounts of normal TTR.
C, + a .
STUDIES OF I T R IN FAP KINDRED
The finding of a variant TTR in serum (and tissues) of Portuguese patients with FAP suggested that the presence of the variant TTR in plasma might be useful as a genetic marker for this type of heredofamilial amyloidosis. Accordingly, methods were developed for the effective screening of samples of serum or plasma from individual subjects that would enable us to determine whether or not the variant TTR was

92 ANNALS NEW YORK ACADEMY OF SCIENCES
present. The subjects studied included patients with FAP and their asymptomatic children. We anticipated that the detection of the presence of the variant TTR could be used for the diagnosis of the Portuguese type of FAP, for the better definition of its mode of inheritance, and for the detection of carriers of the “abnormal” protein in the preclinical phase of the disease (that is, for the prediction of the at-risk offspring of patients with FAP).
Isolation and Study of TTR from Individual Subjects
Serum samples were collected from ten Portuguese patients with FAP (five men and five women; age range, 26-45 years), and from 21 children (age range, 6-23 years) of six of the patients with FAP. The ten FAP patients all had typical clinical pictures of the disease; none of the 21 children of the FAP patients had clinical evidence of FAP. Plasmapheresis was performed in one of the FAP patients to provide a sample of plasma of 400 ml; in all other cases serum sample volumes ranged from 10 to 30 ml.
TTR was isolated from the individual small serum samples by a two-step procedure involving: (i) chromatography on Cibacron Blue F3-GA (Bio-Rad Affi-gel Blue) with a linear salt gradient in 30 m M phosphate buffer, pH 7.0; and (ii) affinity chromatog- raphy on human RBP linked to Sepharose. The RBP-Sepharose affinity column employed was the same column that had been used in the studies described earlier on the isolation of amyloid fibril protein, AF,. This two-step procedure resulted in the isolation of TTR that was estimated to be approximately 90% pure by SDS-PAGE analysis. The recoveries obtained were of the order of 16%, providing material in sufficient quantity and with a degree of purity suitable for further structural analytical studies. For the larger sample of plasma obtained by plasmapheresis, TTR was isolated by the sequence of procedures described recently14 for the isolation of TTR from pooled sera of patients with FAP.
In order to explore whether the variant (“abnormal”) TTR could be detected readily in TTR isolated from a single individual patient with FAP, portions of the TTR purified from the plasma sample from a patient with FAP were subjected to trypsin digestion or to CNBr cleavage, followed by peptide mapping by HPLC. The aim of this work was to examine the ease and clarity with which the aberrant (“abnormal”) peptides (Tf, C, and C,) could be detected in an individual patient.
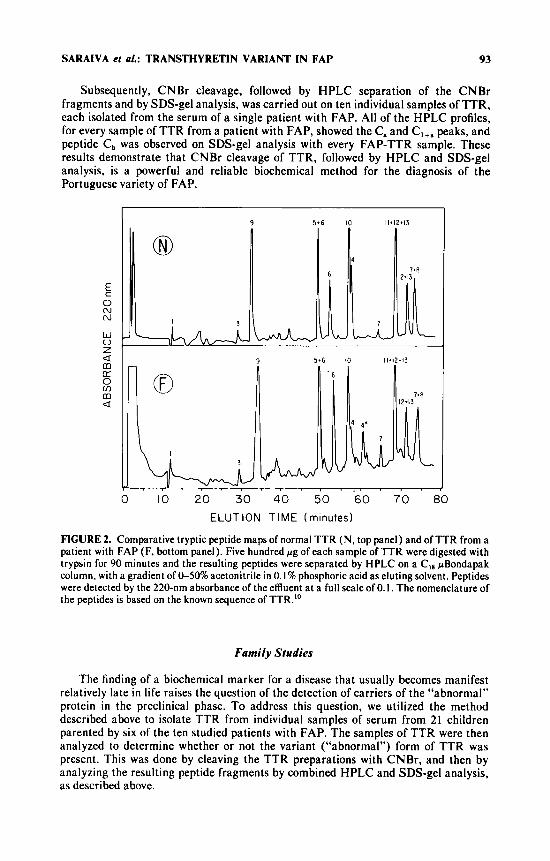
FIGURE 2 shows the HPLC tryptic peptide maps obtained for normal TTR and for TTR from the individual patient with FAP. Tryptic peptide 4 was present in relatively lower amounts in the patient’s TTR preparation, along with an approximately equal amount of tryptic peptide 4*. The latter was identified by amino acid analysis and by its having an identical elution time as peptide T: from tryptic digestions of AF, carried out in parallel in the same experiment.
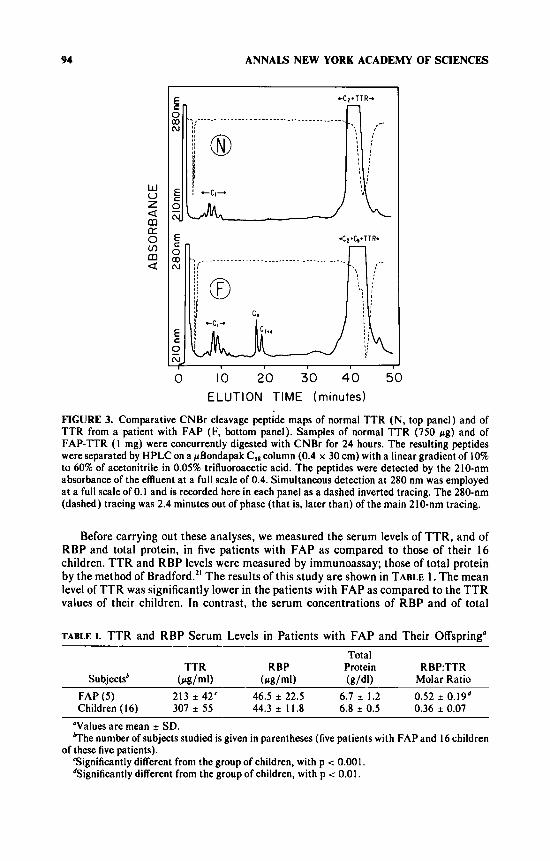
FIGURE 3 shows the comparative CNBr peptide maps obtained for normal TTR and for TTR from the plasma of the individual patient with FAP. Two sharp and closely eluting peptide peaks, not seen in normal TTR, were clearly evident in the FAP-TTR profile near 20 minutes elution time. These peptides were identified both by amino acid analysis and by their retention time, in comparison to concurrently obtained CNBr peptide maps of AF,. These two peaks represented the peptides C, and C,
The fractions where peptides C2 and Cb coeluted (as marked in the bottom panel of FIGURE 3) were collected and analyzed by SDS-PAGE, and compared with the fractions from the normal TTR preparation eluting a t the same position of the gradient. A band corresponding to peptide Cb was observed in the FAP-TTR preparation. Such a band was never observed in preparations from normal TTR.
and are so labeled in FIGURE 3.
SARAIVA er al.: TRANSTHYRETIN VARIANT IN FAP 93
Subsequently, CNBr cleavage, followed by HPLC separation of the CNBr fragments and by SDS-gel analysis, was carried out on ten individual samples of TTR, each isolated from the serum of a single patient with FAP. All of the HPLC profiles, for every sample of TTR from a patient with FAP, showed the C, and C,,, peaks, and peptide C, was observed on SDS-gel analysis with every FAP-TTR sample. These results demonstrate that CNBr cleavage of TTR, followed by HPLC and SDS-gel analysis, is a powerful and reliable biochemical method for the diagnosis of the Portuguese variety of FAP.
E 52 nl
w 0 Z
(L 0 cn
a m
m a
I I I n 1 I \ . A
0 10 20 30 40 5 0 60 7 0 80
ELUTION TIME (minutes)
FIGURE 2. Comparative tryptic peptide maps of normal TTR (N, top panel) and of TTR from a patient with FAP (F, bottom panel). Five hundred pg of each sample of TTR were digested with trypsin for 90 minutes and the resulting peptides were separated by HPLC on a CI8 pBondapak column, with a gradient of &50% acetonitrile in 0. I % phosphoric acid as eluting solvent. Peptides were detected by the 220-nm absorbance of the effluent a t a full scale of 0.1. The nomenclature of the peptides is based on the known sequence of TTR.’”
Family Studies
The finding of a biochemical marker for a disease that usually becomes manifest relatively late in life raises the question of the detection of carriers of the “abnormal” protein in the preclinical phase. To address this question, we utilized the method described above to isolate TTR from individual samples of serum from 21 children parented by six of the ten studied patients with FAP. The samples of TTR were then analyzed to determine whether or not the variant (“abnormal”) form of TTR was present. This was done by cleaving the TTR preparations with CNBr, and then by analyzing the resulting peptide fragments by combined HPLC and SDS-gel analysis, as described above.
94 ANNALS NEW YORK ACADEMY OF SCIENCES
0 10 20 30 40 50 ELUTION TIME (minutes)
FIGURE 3. Comparative CNBr cleavage peptide maps of normal TTR (N, top panel) and of TTR from a patient with FAP (F, bottom panel). Samples of normal TTR (750 pg) and of FAP-TTR ( 1 mg) were concurrently digested with CNBr for 24 hours. The resulting peptides were separated by HPLC on a pBondapak C,* column (0.4 x 30 cm) with a linear gradient of 10% to 60% of acetonitrile in 0.05% trifluoroacetic acid. The peptides were detected by the 210-nm absorbance of the effluent at a full scale of 0.4. Simultaneous detection at 280 nm was employed a t a full scale of 0.1 and is recorded here in each panel as a dashed inverted tracing. The 280-nm (dashed) tracing was 2.4 minutes out of phase (that is, later than) of the main 210-nm tracing.
Before carrying out these analyses, we measured the serum levels of TTR, and of RBP and total protein, in five patients with FAP as compared to those of their 16 children. TTR and RBP levels were measured by immunoassay; those of total protein by the method of Bradford." The results of this study are shown in TABLE 1. The mean level of TTR was significantly lower in the patients with FAP as compared to the TTR values of their children. In contrast, the serum concentrations of RBP and of total
TABLE I. TTR and RBP Serum Levels in Patients with FAP and Their Offspring" Total
TTR RBP Protein RBP:TTR Subjectsb (pg/ml) (pglml) ( g / W Molar Ratio
FAP (5) 213 * 42' 46.5 * 22.5 6.7 i 1.2 0.52 * 0.19d Children (16) 307 i 55 44.3 * 11.8 6.8 * 0.5 0.36 i 0.07
"Values are mean * SD. ?he number of subjects studied is given in parentheses (five patients with FAP and 16 children
Significantly different from the group of children, with p < 0.001. dSignificantly different from the group of children, with p < 0.01.
of these five patients).
SARAIVA ef al.: TRANSTHYRETIN VARIANT IN FAP 95
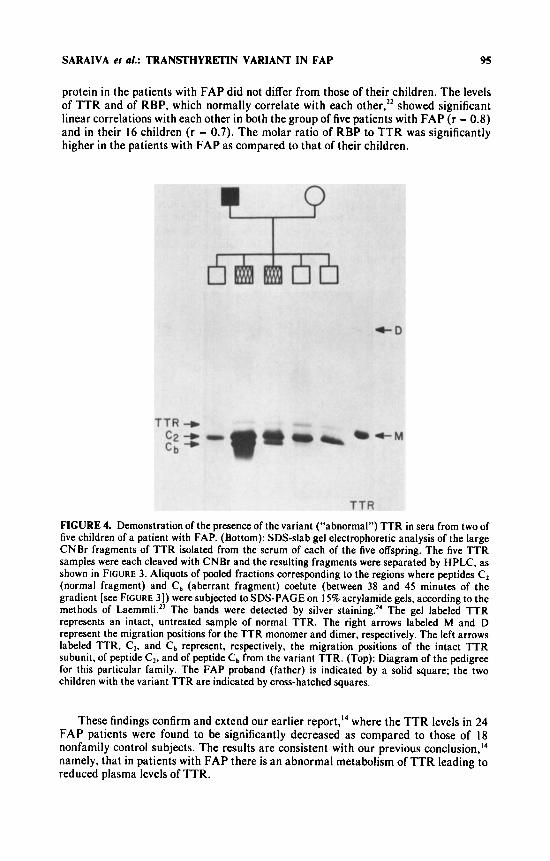
protein in the patients with FAP did not differ from those of their children. The levels of TTR and of RBP, which normally correlate with each other,22 showed significant linear correlations with each other in both the group of five patients with FAP (r = 0.8) and in their 16 children (r = 0.7). The molar ratio of RBP to TTR was significantly higher in the patients with FAP as compared to that of their children.
FIGURE 4. Demonstration of the presence of the variant (“abnormal”) TTR in sera from two of five children of a patient with FAP. (Bottom): SDS-slab gel electrophoretic analysis of the large CNBr fragments of TTR isolated from the serum of each of the five offspring. The five TTR samples were each cleaved with CNBr and the resulting fragments were separated by HPLC, as shown in FIGURE 3. Aliquots of pooled fractions corresponding to the regions where peptides Cz (normal fragment) and Cb (aberrant fragment) coelute (between 38 and 45 minutes of the gradient [see FIGURE 31) were subjected to SDS-PAGE on 15% acrylamide gels, according to the methods of LaemmkZ3 The bands were detected by silver staining.” The gel labeled TTR represents an intact, untreated sample of normal TTR. The right arrows labeled M and D represent the migration positions for the TTR monomer and dimer, respectively. The left arrows labeled TTR, Cz, and Cb represent, respectively, the migration positions of the intact TTR subunit, of peptide Cz, and of peptide Cb from the variant TTR. (Top): Diagram of the pedigree for this particular family. The FAP proband (father) is indicated by a solid square; the two children with the variant TTR are indicated by cross-hatched squares.
These findings confirm and extend our earlier report,14 where the TTR levels in 24 FAP patients were found to be significantly decreased as compared to those of 18 nonfamily control subjects. The results are consistent with our previous conclu~ion,’~ namely, that in patients with FAP there is an abnormal metabolism of TTR leading to reduced plasma levels of TTR.
96 ANNALS NEW YORK ACADEMY OF SCIENCES
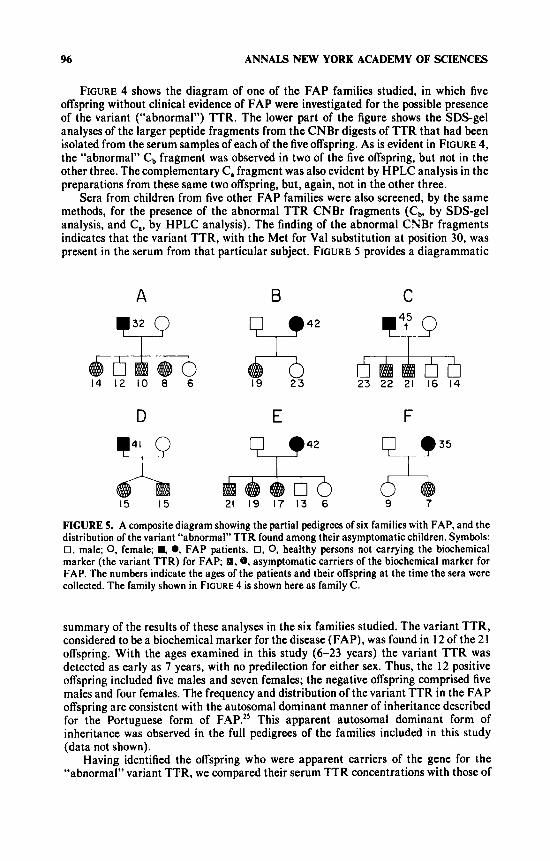
FIGURE 4 shows the diagram of one of the FAP families studied, in which five offspring without clinical evidence of FAP were investigated for the possible presence of the variant (“abnormal”) TTR. The lower part of the figure shows the SDS-gel analyses of the larger peptide fragments from the CNBr digests of TTR that had been isolated from the serum samples of each of the five offspring. As is evident in FIGURE 4, the “abnormal” Cb fragment was observed in two of the five offspring, but not in the other three. The complementary C, fragment was also evident by HPLC analysis in the preparations from these same two offspring, but, again, not in the other three.
Sera from children from five other FAP families were also screened, by the same methods, for the presence of the abnormal TTR CNBr fragments (Cb, by SDS-gel analysis, and C,, by HPLC analysis). The finding of the abnormal CNBr fragments indicates that the variant TTR, with the Met for Val substitution at position 30, was present in the serum from that particular subject. FIGURE 5 provides a diagrammatic
A B 42
14 12 10 8 6
D E
15 15 21 19 17 13 6
C
v 23 22 21 16 14
9 7
FIGURE 5. A composite diagram showing the partial pedigrees of six families with FAP, and the distribution of the variant “abnormal” TTR found among their asymptomatic children. Symbols: 0, male; 0, female; ., 0, FAP patients. 0, 0, healthy persons not carrying the biochemical marker (the variant TTR) for FAP; 8.8, asymptomatic carriers of the biochemical marker for FAP. The numbers indicate the ages of the patients and their offspring at the time the sera were collected. The family shown in FIGURE 4 is shown here as family C.
summary of the results of these analyses in the six families studied. The variant TTR, considered to be a biochemical marker for the disease (FAP), was found in 12 of the 21 offspring. With the ages examined in this study (6-23 years) the variant TTR was detected as early as 7 years, with no predilection for either sex. Thus, the 12 positive offspring included five males and seven females; the negative offspring comprised five males and four females. The frequency and distribution of the variant TTR in the FAP offspring are consistent with the autosomal dominant manner of inheritance described for the Portuguese form of FAP.” This apparent autosomal dominant form of inheritance was observed in the full pedigrees of the families included in this study (data not shown).
Having identified the offspring who were apparent carriers of the gene for the “abnormal” variant TTR. we compared their serum TTR concentrations with those of

SARAIVA er a/.: TRANSTHYRETIN VARIANT IN FAP 91
their apparently normal siblings. The mean (*SD) TTR level observed for the offspring carriers was 325 42 Fg/ml, and that for their noncarrier sibs was 283 * 62 pg/ml. The difference between the two groups is not statistically significant. Thus, in the Portuguese type of FAP, TTR levels cannot be used as a predictive marker in the preclinical phase of the disease.
DISCUSSION
The studies described in this paper have demonstrated that the amyloid fibril protein, AF,, purified from the tissues of two Portuguese patients who died with FAP, comprised a TTR variant with a methionine for valine substitution at position 30. In the development of this work, the structural difference between AF, and TTR was first localized to a single tryptic peptide (peptide T4) by comparative HPLC mapping of tryptic peptides. The nature of the difference was then identified by CNBr cleavage studies and by amino acid microsequence analyses of aberrant CNBr and tryptic peptides. The variant TTR was also found to be present in the TTR isolated from FAP pooled sera, and in TTR isolated from individual serum samples from ten patients with FAP. Thus, in these patients with FAP, the variant TTR was circulating in plasma along with larger amounts of normal TTR.
The AF, studied here was isolated by a novel procedure involving affinity chromatography of a solubilized fibril preparation on RBP linked to Sepharose. isolated AF, resembled plasma TTR in forming a stable tetrameric structure, and in its binding affinities for both RBP and thyroxine. It appears that the Met for Val substitution at position 30 in AF, did not substantially alter any of those aspects of the structure of TTR that are involved in the subunit-subunit interactions that form the tetrameric molecule, or in the interactions of TTR with RBP or thyroxine.
The structure of the TTR subunit is dominated by eight @-strands (labeled A to H), into which about 50% of the amino acids are organized as two 4-stranded @-sheets (DAGH and CBEF).” The monomers are linked into stable dimers by further @-sheet interactions, and the dimers are assembled into complete tetrameric molecules. Valine 30 of TTR is located on @-strand Band is packed into the subunit core located between the two @-sheets. it is perhaps surprising that the substitution of a Met for Val a t this position did not have more significant effects on the structure, and hence on the physical-chemical properties, of the variant TTR.
Pras et al. have reported recentlyz6 that the amyloid fibril protein from tissues of a deceased Jewish patient with FAP consisted largely of a variant TTR subunit with a glycine for threonine substitution a t position 49, along with variant TTR fragments 1-48 and 49-127. Threonine 49 of TTR is located on @-strand C of the core of the subunit, close to valine 30. In fact, threonine 49 is one of the amino acids whose side chain is in contact with that of valine 30 in the three-dimensional structure of the protein.’’ These findings raise the possibility that these residues are part of a structural domain which, if altered, is critically involved in the pathogenesis of FAP. Such a domain might be important in some unknown aspect of TTR metabolism or function.
As reported here, we have developed an effective procedure that can be used to determine whether or not the variant TTR is present (to any significant extent) in the plasma of any particular individual subject. This procedure involves, first, the isolation of TTR from rather small samples of serum (1CL20 ml) with a degree of purity suitable for structural analysis. Isolation of TTR is achieved by a relatively simple and rapid two-step procedure, with the second step consisting of affinity chromatography on RBP linked to Sepharose. The isolated TTR is then cleaved with CNBr and analyzed

98 ANNALS NEW YORK ACADEMY OF SCIENCES
by combined HPLC and SDS-gel analysis to determine whether or not the aberrant CNBr peptides (C, and C,) are present. The results presented here indicate that this biochemical procedure can be used both for the definitive diagnosis of FAP in affected patients, and also as a presymptomatic biochemical test for the presence of the genetic abnormality that leads to the development of the clinical disease.
The fact that the same TTR variant found in amyloid fibril deposits in tissues was also detected in the sera of FAP patients when analyzed individually, and that segregation for the “abnormal” TTR was observed on family analysis, indicates that the presence of the variant TTR (with Met substituted for Val 30) is highly likely to be a direct and true genetic marker of the disease. The observed distribution of the mutant TTR within affected families (FIGURE 5 ) is consistent with the autosomal dominant mode of inheritance of FAP.2S Moreover, in all FAP patients, and in their affected offspring, a mixture of both normal and variant TTR was found in the circulation. Thus, the FAP patients appeared to be heterozygous for the variant, mutant form of TTR, a feature also consistent with an autosomal dominant trait.
The detailed mechanisms whereby the alteration in the primary structure of TTR leads to the pathogenesis of FAP remain to be determined. The amino acid substitution reported here is, however, most probably the specific underlying biochemical cause of the disease. It is likely that the Met for Val substitution represents a point mutation within the coding sequences of the TTR structural gene. Although the codon for Val 30 of TTR is not known, one possible codon for Val is GUG, which could yield Met by a single nucleotide change (GUG - AUG). Patients with FAP appear to have one gene for normal TTR, and one for the variant TTR; these genes are likely to represent co-dominant alleles a t a single gene locus, since both genes are expressed in affected subjects.
As reported previ~usly’~ and here, plasma TTR levels are reduced in patients with FAP. In contrast, plasma TTR levels were not reduced in asymptomatic FAP offspring who were carriers of the mutant, variant TTR. These findings suggest that the accumulation of the variant TTR in tissues may occur gradually over many years, perhaps over the lifetime of the patient with FAP. It is possible that only when sufficient variant TTR has deposited in tissues to permit its observation as amyloid does sufficient perturbation of TTR metabolism occur to lead to a reduced plasma level. On the basis of alterations found in the nerve fibers of some asymptomatic children, it has been suggested that a neuropathic process precedes the formation of amyloid in the Portuguese type of FAP.” Information is needed about the mechanism of amyloid formation in FAP, and about the factors that affect the synthesis and degradation rates of the amyloid-forming protein.
Definitive proof that the presence of the variant TTR can be used as a preclinical biochemical marker of the disease will require longitudinal studies in families to determine whether offspring of patients with FAP and who have the variant TTR will develop the clinical disease, whereas offspring without the variant TTR remain healthy. From the information available, it is likely that this will indeed be the case. Ultimately, the availability of a predictive, biochemical marker of the disease would be of value in assuring persons without the variant TTR that they are not a t risk of developing FAP, and in attempts a t prevention or early treatment of the disease in affected individuals.
SUMMARY
Amyloid deposits in several heredofamilial forms of amyloidosis are chemically related to transthyretin (TTR, the protein usually referred to as prealbumin). A genetically abnormal TTR may be involved. Studies were conducted on TTR isolated

SARAIVA et a/.: TRANSTHYRETIN VARIANT IN FAP 99
from sera of patients with familial amyloidotic polyneuropathy (FAP), and on amyloid fibril protein (AF,) isolated from tissues of two Portuguese patients who died with FAP. AF,, purified by affinity chromatography on retinol-binding protein (RBP), resembled plasma TTR in forming a stable tetrameric structure, and in its binding affinities for both thyroxine and RBP. Purified AF, was found to comprise a TTR variant with a methionine for valine substitution at position 30. This conclusion was based upon studies that included: (i) comparative peptide mapping by reverse-phase high-performance liquid chromatography after trypsin digestion; (ii) cyanogen bro- mide (CNBr) cleavage studies; and (iii) amino acid microsequence analysis of selected tryptic and CNBr peptides. The variant TTR was also found to be present in serum samples from FAP patients, along with larger amounts of normal TTR. An effective, small-scale procedure was developed to determine whether or not the variant TTR was present in the plasma of an individual subject. This procedure involved isolation of TTR by affinity chromatography on RBP, followed by CNBr cleavage, and analysis for the presence of specific aberrant CNBr peptides. Studies with six kindreds, including 21 asymptomatic children of 6 patients with FAP, showed that the "abnormal" TTR can be detected and used as a preclinical marker of the disease in affected children of patients with FAP. I t is likely that the variant TTR represents a point mutation within the TTR structural gene, and that the normal and mutant genes act as co-dominant alleles a t a single locus in FAP. The distribution of the mutant TTR within the six families was consistent with the autosomal dominant mode of inheritance of FAP. The mutant TTR apparently selectively deposits in tissues as the amyloid characteristic of the disease.
ACKNOWLEDGMENTS
We are grateful to Dr. R. Canfield for advice and discussions during the course of this work, and to Mr. C. Barreda for performing some of the amino acid analyses.
REFERENCES
1 . 2.
3.
4.
5 .
6. 7. 8. 9.
10.
1 1 .
12. 13.
14.
GLENNER, G. 1980. N. Engl. J . Med. 302: 1283-1292. GLENNER, G., W. TERRY, M. HARADA, C. ISERSKY & D. PAGE. 1971. Science 172 1 1 5 0 -
LEVIN, M., E. C. FRANKLIN, B. FRANGIONE & M. PRAS. 1972. J. Clin. Invest. 51: 2773-
COSTA, P. P., A. S. FIGUEIRA & F. R . BRAVO. 1978. Proc. Natl. Acad. Sci. USA
TAWARA, S.. S. ARAKI, K. TOSHIMORI. H. NAKAGAWA & S. OHTAKI. 1981. J. Lab. Clin.
BENSON, M. D. 1981. J. Clin. Invest. 67: 1035-1041. SKINNER, M. & A. COHEN. 1981. Biochem. Biophys. Res. Commun. 99 1326-1332. DALAKIS, M. C. & W. K. ENGEL. 1981. Arch. Neurol. 38 42W22. PRAS, M.. E. C. FRANKLIN, F. PRELLI & B. FRANGIONE. 1981. J. Exp. Med. 154989-
KANDA. Y., D. S. GOODMAN, R. E. CANFIELD & F. J . MORGAN. 1974. J . Biol. Chem.
BLAKE, C. C. F., J. J. GEISOW, S. J. OATLEY, B. RERAT & C. RERAT. 1978. J. Mol. Biol.
RAT, A.,T.SHIRATORI & D. S.GOODMAN. 1970. J . Biol. Chem. 245: 1903-1912. V A N JAARSVELD, P. P., H. EDELHOCH, D. S. GOODMAN & J. ROBEINS. 1973. J. Biol. Chem.
SARAIVA, M. J. M., P. P. COSTA & D. S. GOODMAN. 1983. J . Lab. Clin. Med.
1151.
2780.
7 5 4499-4503.
Med. 98 8 1 1-822.
993.
2 4 9 6796-6805.
121: 339-356.
248: 46984705.
102 590-603.

ANNALS NEW YORK ACADEMY OF SCIENCES 100
15.
16.
17. 18.
19. 20. 21. 22. 23. 24. 25.
26.
27.
PRAS. M., M. SCHUBERT, D. ZUCKER-FRANKLIN, A. RIMON & E. C. FRANKLIN. 1968. J.
GLENNER, G. G., P. CUATRECASAS, C. ISERSKY, H. A. BLADEN & E. D. EANES. 1969. J.
PETERSON, P. A. 1971. J. Biol. Chem. 246: 44-49. NAVAB, M.. A. K. MALLIA, Y. KANDA & D. S. GOODMAN. 1977. J. Biol. Chem.
AMBLER, R. 1972. Methods Enzymol. 2 5 262-272. LEVY, W. P. 1981. Methods Enzymol. 79: 27-31. BRADFORD, M. 1976. Anal. Biochem. 7 2 248-254. SMITH, F. R. & D. S. GOODMAN. 1971. J. Clin. Invest. 50: 2426-2436. LAEMMLI, U. K. 1970. Nature (London) 227: 680-685. OAKLEY, B., D. R. KIRSCH & N. R. MORRIS. 1980. Anal. Biochem. 105 361-363. GLENNER, G. G., T. R. IGNACZACK & D. L. PAGE. 1978. I n The Metabolic Basis of
Inherited Disease, 4th ed. J.B. Stanbury, J.B. Wyngaarden & D.S. Fredrickson, Eds.: 1308-1339. McGraw-Hill. New York. N.Y.
PRAS, M., F. PRELLI, E. C. FRANKLIN & B. FRANGIONE. 1983. Proc. Natl. Acad. Sci. USA
CARVALHO, J., A. COIMBRA & C. ANDRADE. 1976. Brain 99: 1-10,
Clin. Invest. 47: 924-933.
Histochem. Cytochem. 17: 769-780.
252: 51W5106.
8 0 539-542.
DISCUSSION
DR. A. KAPPAS (Rockefeller University, New York, N.Y.): What is the relation of genetics to the predisposition to other forms of amyloidosis besides this clearly hereditary type?
DR. D. GOODMAN: There is a series of genetic familial amyloidoses that have minor clinical variations from each other and that go under the names of Type I, Type 11, Type Ill and Type IV. These are all candidates for TTR-related amyloidoses. The other more common clinical varieties of amyloidosis are not genetically determined. These would include, amyloidosis associated with multiple myeloma, with chronic inflammation or other primary forms. Familial Mediterranean fever is associated with amyloidoses, but a different protein is involved.
Three proteins have been identified that can lead to amyloidoses. They all have a great deal of @ structure and it is the /3 structure that makes a protein potentially amyloidogenic and able to form this kind of fibril. We think that the method of isolating the fibril protein and getting a very pure protein will potentially enable Dr. Blake" to do X-ray crystal structure studies to see what is different about a protein with a methionine substitution for valine a t position 30. Maybe that will give insight into why the variant TTR forms polymers and fibrils.
Dr. Blake's initial thoughts were that this amino acid substitution should cause a significant perturbation in the structure, and yet it does not appear to do so. The variant TTR (AF,) forms stable tetramers. AF, can bind thyroxine and it can bind retinol-binding protein. Why it is neuropathic is not known, but TTR has something to do with the nervous system. TTR is present in high concentrations in cerebrospinal fluid, which is something that needs further research.
DR. G. D. LAWRENCE (Mount Sinai School of Medicine, New York, N.Y.): Since you know what these aberrations are, Dr. Goodman, what kind of an approach can you take for either prophylactic or therapeutic treatment of patients with this disease?
GOODMAN: At the moment there is no specific plan. Clearly, if one could develop an approach for prenatal diagnosis there is obvious action that could be taken regarding family counseling and abortion. TTR turns over very rapidly so that exchange transfusions would not be very useful, but one can start thinking in those directions. Otherwise I do not have any hypotheses for therapeutic approaches at this time.





























