Embed Size (px)
Citation preview

RESEARCH PAPERbph_1349 731..742
Fangchinoline inducesautophagic cell death viap53/sestrin2/AMPKsignalling in humanhepatocellular carcinomacellsNing Wang1*, Weidong Pan2*, Meifen Zhu1, Maosheng Zhang2,3,Xiaojian Hao2,4, Guangyi Liang2,5 and Yibin Feng1
1School of Chinese Medicine, Li Ka Shing Faculty of Medicine, The University of Hong Kong,
Hong Kong, 2The Key Laboratory of Chemistry for Natural Products of Guizhou Province and
Chinese Academy of Sciences, Guiyang, Guizhou Province, China, 3Zunyi Medical College,
Zunyi, Guizhou Province, China, 4State Key Laboratory of Phytochemistry and Plant Resources
in West China, Kunming Institute of Botany, Chinese Academy of Sciences, Kunming, China,
and 5Guiyang College of Traditional Chinese Medicine, Guiyang, Guizhou Province, China
CorrespondenceDr Yibin Feng, School of ChineseMedicine, Li Ka Shing Faculty ofMedicine, The University ofHong Kong, 10 Sassoon Road,Pokfulam, Hong Kong. E-mail:[email protected]; Professor GuangyiLiang, The Key Laboratory ofChemistry for Natural Productsof Guizhou Province and ChineseAcademy of Sciences, 202Shachong South Road, Guiyang,Guizhou Province, 550002,China or Guiyang College ofTraditional Chinese Medicine,Guiyang, Guizhou Province,550002, China. E-mail:guangyi_liang@126.com----------------------------------------------------------------
*Equally contributed to thisresearch.----------------------------------------------------------------
Keywordsfangchinoline; autophagy; p53;sestrin2; hepatocellularcarcinoma; AMPK----------------------------------------------------------------
Received9 February 2011Accepted25 February 2011
BACKGROUND AND PURPOSEFangchinoline is a novel anti-tumour agent with little known of its cellular and molecular mechanisms of action. Here we haveinvestigated the mode of cell death induced by fangchinoline and its underlying mechanism in two human hepatocellularcarcinoma cell lines, HepG2 and PLC/PRF/5.
EXPERIMENTAL APPROACHApoptosis and autophagy were monitored in fangchinoline-treated HepG2 and PLC/PRF/5 cells by histological methods. Thesignal transduction pathways involved in activation of autophagy were examined, using immunoblotting, real-time PCR andsiRNA techniques.
KEY RESULTSFangchinoline did not induce apoptosis in HepG2 and PLC/PRF/5 cells but triggered, dose-dependently, autophagy, analternative mode of cell death which may contribute to fangchinoline’s anti-tumour action. Nuclear translocation of p53 wasinvolved in induction of autophagy by fangchinoline, followed by selective transactivation of the autophagy-related genesestrin2 and initiation of the autophagic process. Signalling by the AMP-activated protein kinase was also involved as adownstream target of sestrin2 and induced mTOR-independent autophagic cell death in both cell lines. siRNA for Atg5 orpharmacological block of p53 abolished fangchinoline-induced autophagy and inhibition of autophagy switched cell death toapoptosis in these cells, suggesting that cell death is irreversible once autophagy is induced by fangchinoline.
CONCLUSIONS AND IMPLICATIONSFangchinoline is a highly specific agent inducing autophagic cell death in hepatocellular carcinoma cells with a novelmechanism, which elucidates the potential of fangchinoline to potentiate programmed cell death in cancer cells.
Abbreviation3-MA, 3-methylamphetamine; ACD, autophagic cell death; AMPK, AMP-activated protein kinase; AO, acridine orange;BRL, buffalo rat liver; CCD, charge-coupled device; DRAM, damage-regulated autophagy modulator; ECL,electrochemiluminescence; FBS, fetal bovine serum; GFP-LC3, green fluorescent protein-light chain 3; MDC,monodansylcadaverine; mTOR, mammalian target of rapamycin; MTT, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide; PCD, programme cell death; PCR, polymerase chain reaction; RIPA, radio-immunoprecipitation assay buffer; SCR, scramble negative control; SDS-Page, sodium dodecyl sulphate polyacrylamidegel electrophoresis
BJP British Journal ofPharmacology
DOI:10.1111/j.1476-5381.2011.01349.xwww.brjpharmacol.org
British Journal of Pharmacology (2011) 164 731–742 731© 2011 The AuthorsBritish Journal of Pharmacology © 2011 The British Pharmacological Society

IntroductionAutophagy is a mechanism of membrane-trafficking wherecytoplasmic proteins and constituents are delivered to lysos-omes for further degradation (Tang et al., 2006; 2008; Tianet al., 2006). The molecular mechanism underlying theinduction of autophagy in mammalian cells is not com-pletely described but so far more than 30 genes have beenidentified to be related to autophagy in yeast (Bommareddyet al., 2009). Autophagic vacuolization is observed in thepathogenesis of numerous neoplasms, as well as in cancerchemotherapy, indicating that the role of autophagy incancer is far from simple. Many studies reveal that autophagycould act as a survival pathway or contribute to cell deathin tumour cells treated with chemotherapeutic agents(Mizushima et al., 1998; Hanahan and Weinberg, 2000;Cheng et al., 2009). In many cases, induction of basal autoph-agy, the self-eating process, facilitates the breakdown of somelong-life cytoplasmic proteins and dysfunctional organellesto counteract the deleterious effect of endogenous metabolicstress, in response to chemotherapeutic agents and leads totumour cell survival, while excessive autophagy induced bydrugs leads to elimination of tumour cells and plays an anti-tumour role when autophagy comes to completion (Chenand Karantza-Wadsworth, 2009). The term ‘autophagic celldeath’ (ACD) is extensively employed to indicate that cellsare undergoing an alternative type of programmed cell deathsubroutine, lacking the morphological features of classicalapoptosis and manifesting with the excessive accumulationof autophagosomes in the cytoplasm (Morselli et al., 2009). Inview of this complexity, the role of autophagy must be clari-fied case by case.
Fangchinoline is a bis-benzylisoquinoline alkaloid with ofcomplex structure and has been identified as a new com-pound, sharing structural features with tetrandrine, anothercompound with a wide spectrum of anti-tumour activity invarious cancer cells. The potent anti-tumour activity of tet-randrine has been extensively reported with its proposedmechanism of inducing G1/S and G2/M arrest and stimulat-ing apoptotic cell death (Zhang et al., 2003; Meng et al., 2004;Sun et al., 2007). However, the anti-tumour activity offangchinoline and its underlying mechanism(s) remainunclear.
Here, we report the potent anti-tumour action of fangchi-noline in two hepatocellular carcinoma cell lines, HepG2 andPLC/PRF/5. Fangchinoline induced hepatoma cell death atlow concentrations (IC50 was approximately 5 mM) in bothHepG2 and PLC/PRF/5 cells, but fangchinoline used mecha-nisms different from those of its analogue, tetrandrine, topotentiate tumour cell death. Though incapable of inducingapoptosis or cell cycle arrest in tumour cells, fangchinolinewas able to initiate ACD by increasing the nuclear transloca-tion of p53 to transactivate the sestrin2 gene, a novel player inautophagy induction. Signalling by AMP-activated proteinkinase (AMPK) was then activated as downstream target ofsestrin2 and a mTOR-independent autophagy was induced.Inhibition of autophagy potentiated apoptosis when cellsweare exposed to fangchinoline. To the best of our knowl-edge, this study provides the first evidence that fangchinolinemay have potential as a chemotherapeutic agent, by inducingACD in tumour cells.
Methods
Cell line and cell cultureThe human hepatocellular carcinoma cell line HepG2 waspurchased from American Type Culture Collection (USA); thehuman hepatoma cell line PLC/PRF/5 was provided by Pro-fessor Tsao Sai-Wah. Rat normal hepatic cell line buffalo ratliver (BRL) was purchased from Sun Yat-Sen University(China). Cells were maintained in high glucose Dulbecco’sModified Eagle Medium (Invitrogen, USA) supplemented with10% fetal bovine serum (Invitrogen, USA), and incubated in ahumidified atmosphere containing 5% CO2 at 37°C.
Cell viability assayCells were seeded in 96-well plate with density of 104 cells perwell. A series of concentrations of fangchinoline or tetran-drine (0, 10-3, 10-2, 10-1, 1, 10, 102, 103 mM) were added andfollowed by 24 h incubation. All experiments were conductedparallel with controls (0.1% DMSO). Cell viability was deter-mined by MTT assay.
Clonogenic assayClonogenic assay was conducted as described (Franken et al.,2006) with some modifications. Briefly, HepG2 and PLC/PRF/5 cells were seeded into six-well plates at a density of 103
cells per well. Twenty-four hours after seeding, cells weretreated with various concentrations of fangchinoline for 12days. Drugs were replaced when medium was refreshed.Plates were then washed with PBS, and cells were fixed in 4%paraformaldehyde and stained with 0.1% Coomassie blue(Bio-Rad) in 30% methanol and 10% acetic acid. Images werecaptured by a CCD camera.
Monodansylcadaverine & AcridineOrange stainingFor monodansylcadaverine (MDC) staining, cells were incu-bated with 0.05 mM MDC in PBS at 37°C for 10 min (Munafóand Colombo, 2001) and visualized with fluorescent micros-copy (Carl Zeiss, USA, 630 ¥ magnification, CCD camera). ForAcridine Orange (AO) staining, cells with or without treat-ment were stained by 1 mg·mL-1 AO in PBS at 37°C for 15 min(Yang et al., 2008). Then cells were washed and visualizedwith a fluorescence microscope at 630 ¥ magnification (CarlZeiss, Germany).
Quantification of GFP-LC3 punctaHepG2 cells with stable expression of GFP-LC3 were seededinto 35 mm confocal dishes and treated. The accumulation ofGFP-LC3 was examined by fluorescent microscopy at 630 ¥magnification. Autophagy was quantified by counting thepercentage of cells showing accumulation of GFP-LC3 indots. A total of 100 cells per preparation in three independentexperiments were calculated. Cells presenting several intensepunctate aggregations of GFP-LC3 were classified as showingautophagy (Shen et al., 2007).
Annexin V/PI stainingCells were seeded in six-well tissue culture plates and receiveddifferent treatments. Cells exposed to UV for 20 min followed
BJP N Wang et al.
732 British Journal of Pharmacology (2011) 164 731–742

by 6 h incubation were used as positive control. Medium wasthen discarded and cells were stained with Annexin V/ pro-pidium iodide (PI) kit (Roche, USA) according to the manu-facturer’s instructions. The stained cells were washed withPBS once and the cell cycle was analysed by flow cytometry(FC500, Beckman Coulter, USA).
Cell cycle analysisCells were seeded in six-well tissue culture plates and receiveddifferent treatments. Cells were collected by trypsinizationand fixed in 70% ethanol at 4°C overnight. Then cells werecollected and resuspended in staining buffer (50 mg·mL-1 PI inPBS supplemented with 0.1% Triton X-100) for 15 min indark at room temperature. The stained cells were washed withPBS once and the cell cycle was analysed by flow cytometry(FC500, Beckman Coulter, USA).
DNA fragmentation assayCells were harvested in lysis reagent (1% NP-40 in 20 mMEDTA, 50 mM Tris-HCl, pH 7.5) for 1 min with thoroughvortex mixing followed by centrifugation. In total, 10 mL ofproteinase K (25 mg·mL-1, Sigma, USA) was added to thesupernatant and incubated at 37°C for 2 h, 65 mL of 10 Mammonium acetate and 500 mL ice-cold ethanol were addedand thoroughly mixed. The samples were stored at -80°C for1 h to precipitate the DNA. DNA was collected by centrifuga-tion and washed with 200 mL 80% ice-cold ethanol and air-dried for 10 min. The residues were reconstituted in 30 mL ofTE buffer and the DNA fragments was separated by 2% agarosegel and visualized with a UV transilluminator (Bio-Rad, USA).
Subcellular fractionationCells were harvested in ice-cold hypotonic buffer (10 mMHEPES, 10 mM KCl, 0.1 mM EDTA, 0.4% NP-40, 0.05 mMdithiothreitol; DTT) supplemented with protease inhibitorcocktail (Roche, USA) for 5 mins followed by 14 000¥ g cen-trifugation at 4°C. Supernatant was collected as cytoplasmicfraction, and the pellet was further incubated with nuclearextraction buffer (20 mM HEPES, 400 mM NaCl, 1 mM EDTA,0.05 mM DTT) supplemented with protease inhibitor cocktail(Roche, USA) on ice for 30 min. The lysate was then centri-fuged at 14 000¥ g for 10 min at 4°C to obtain supernatant asnuclear fraction. Immunoblotting was performed to detectthe localization of p53 in cytoplasm and nuclear. Lamin B1and b-actin were used as loading controls for nuclear andcytoplasmic compartments respectively (Wei et al., 2008).
ImmunoblottingCells were lysed with radio-immunoprecipitation assay buffersupplemented with proteinase inhibitor on ice for 30 minand then centrifuged at 14 000¥ g at 4°C for 25 min. Thesupernatant was transferred and protein concentration wasdetermined by using BSA as standard. Equal amounts ofprotein were resolved by SDS-PAGE and transferred onto apolyvinylidene fluoride membrane (Biorad). Then themembrane was blocked with 5% BSA in buffer overnight at4°C. The membrane was then incubated with primary anti-bodies at 4°C overnight followed by incubation with appro-priate secondary antibodies. The immunoreactivities weredetected using electrochemiluminescence advanced kit (GE
Healthcare, UK) and visualized using a chemiluminesenceimaging system (Bio-rad, USA).
Quantitative real-time PCRTotal RNA of cells was purified by using RNeasy Mini Kit(Qiagen, Germany) following the manufacturer’s instruction.Reverse-transcription reaction was performed using Quan-tiTech Reverse Transcription Kit (QIAGEN, Germany) toprepare cDNA samples. The quantitative real-time PCR wasconducted by QuantiTect SYBR Green PCR Kit (Qiagen,Germany) with 1 mM primers for DRAM (forward:5′-TCAAATATCACCATTGATTTCTGT-3′; reverse: 5′-GCCACATACGGATGGTCATCTCTG-3′; Invitrogen, USA) or 1 mMsestrin2 primers (forward: 5′-GCATTACCTGCTGCTGCATA-3′; reverse: 5′-AAGGCCTGGATATGCTCCTT-3′; Invitrogen,USA) on LightCycler 480 real-time PCR system (Roche, USA).The expression of GAPDH was used as endogenous control(forward: 5′-GCTAGGGACGGCCTGAAG-3′; reverse: 5′-GCCCAATACGACCAAATCC-3′; Invitrogen, USA). 4 ng of cDNAwas used for each reaction.
RNA interferenceCells were transfected with siRNA against human Atg5 orAMPKa (20 nM, Santa Cruz Biotechnology Inc., USA) usingtransfection reagent Lipofectamine 2000 (Invitrogen, USA) inserum- and antibiotic-free medium for 6 h followed by 48 hincubation in normal medium.
Statistical analysisResults were analysed using Student’s t-test and are expressedas mean � SD.
MaterialsFangchinoline and tetrandrine were isolated from Radix ofStephaniae tetrandrae (Han-Fang-Ji in Chinese) by ProfessorLiang Guanyi. The purity of each chemical was more than99.0%. Propidium iodide (PI, P4170), bafilomycin A1(B1793), leupeptin (L9783), pifithrin-a (P4236) and com-pound C (P5499) and Ac-DEVD-CHO (A0385) were pur-chased from Sigma-Aldrich (USA). The pEGFP-C1 plasmidencoding human LC3 was a kind gift from Professor TamotsuYoshimori, Osaka University.
Results
Fangchinoline induced death of hepatoma celllines, HepG2 and PLC/PRF/5 in vitroIn our study, a dose-dependent reduction in tumour cellviability after fangchinoline treatment for 24h was observed.The IC50 of fangchinoline was approximately 5 mM in bothHepG2 and PLC/PRF/5 cells (Figure 1A). Tetrandrine, thederivative of fangchinoline, also exhibits potent cytotoxicityto HepG2 and PLC/PRF/5 cells (Figure 1A). Sustained treat-ment, over 12 days, with fangchinoline resulted in cell deathat a lower concentration than its IC50 deterrnined after 24 h(Figure 1B); 2 mM of fangchinoline in HepG2 cells and 4 mMin PLC/PRF/5 cells were enough to potently inhibit tumour
BJPFangchinoline induces autophagy
British Journal of Pharmacology (2011) 164 731–742 733

cell growth. No drug-resistance was observed during the sus-tained treatment of fangchinoline.
Fangchinoline did not induce apoptotic celldeath in hepatoma cellsHere we used Annexin V/PI double staining to identify theinvolvement of apoptosis in fangchinoline-induced celldeath. No increase in apoptosis was observed between cellsexposed to vehicle and various doses of fangchinoline, whilecells exposed to UV showed clear apoptotic changes
(Figure 2A). Cells treated with 10 mM tetrandrine also exhib-ited apoptotic features. Cell cycle analysis showed that tet-randrine, but not fangchinoline, markedly induced G0/G1cycle arrest in both the hepatoma cell lines (Figure 2B). Con-sistent with these findings, cells exposed to UV and tetran-drine exhibited nuclear fragmentation but no DNA fragmentscould be found in hepatoma cells after treatment withfangchinoline (Figure 2C). Immunoblotting analysis revealedthat fangchinoline treatment did not increase the cleavage ofpoly-ADP ribose polymerase (PARP), a protein marker of apo-ptosis (Figure 2D). These results were further confirmed by
TeradrineA
B
Fangchinoline
110HepG2
PLC/PRF/5
HepG2
HepG2
PLC/PRF/5
PLC/PRF/5
Cel
l via
bilit
y (%
)C
ell v
iabi
lity
(%)
90
70
50
30
10
0
110
90
70
50
30
10
0
0
log (mM)
log (mM)
1 2 3 4−4 −3 −2 −1
0 1 2 3 4−4 −3 −2 −1
Figure 1The anti-tumour activity of fangchinoline in hepatocellular carcinoma cells. (A) The cytotoxicity of tetrandrine and fangchinoline. (B) Thelong-term inhibition (12 days of treatment) by fangchinoline of tumour cell survival.
BJP N Wang et al.
734 British Journal of Pharmacology (2011) 164 731–742

A
2.95% 2.79% 2.82% 0.77% 3.90% 4.87% 34.17% 3.30% 1.03%
80.81% 14.87%44.18% 16.80%93.30% 2.20%91.06% 3.82%
2.31%
92.17%
1.11% 1.65%
94.27%
B
C
D
E
ControlG0/G1:57.46%S:13.56%G2/M:26.18%
ControlG0/G1:59.40%S:11.99%G2/M:25.42%
G0/G1:56.18%S:13.37%G2/M:26.73%
2.97%
0.33% 0.76%
96.55% 2.35%
0.07%
98.53%
0.10%
0.67%
4.71% 53.66%
26.92% 14.78%
5.16% 9.92%
75.38% 9.54%
2.02%
Control
HepG2
HepG2
HepG2
HepG2
HepG2
10 10 10 10
5050 00
0
20
40
Cel
l via
bilit
y (%
)
60
80
100
UV
0
0 0 00 0 0 0
00000
0 0
00 0
0 0 000
0 00 0
5
5 5
5010
1010
10
1010
1010
+
+ +
+−
− − − − − − −−
− − − − − − −
100
100 101 102 103
FITC-A104
100 101 102 103
FITC-A
D
9717
7
178
191
264
109
122
156
0 1023
0 1023 0 1023 0 1023 0 1023
0 1023 0 1023 0 1023
EF
D
E
F
D
E
F
D
E
F
D
E
F
D
EF
D
EF
D
EF
104 100 101 102 103
FITC-A104 100 101 102 103
FITC-A104
100 101 102 103
FITC-A104 100 101 102 103
FITC-A104
100 101 102 103
FITC-A104 100 101 102 103
FITC-A104 100 101 102 103
FITC-A104 100 101 102 103
FITC-A104
101
PX
A 102
103
104
100
101
PX
A 102
103
104
100
101
PX
A 102
103
104
100
101
PX
A 102
103
104
100
101
PX
A 102
103
104
100
101
PX
A 102
103
104
100
101
PX
A 102
103
104
100
101
PX
A 102
103
104
100
101
PX
A 102
103
104
100
101
PX
A 102
103
104
PLC/PRE/5
PLC/PRE/5
PLC/PRE/5
PLC/PRE/5
PLC/PRE/5
Fangchinoline 5 μM Fangchinoline 10 μM
Fangchinoline 5 mM
G0/G1:61.94%S:12.37%G2/M:25.80%
Fangchinoline 5 mMG0/G1:60.27%S:13.49%G2/M:25.13%
Fangchinoline 10 mM
Fangchinoline (μM)Tetrandrine (μM)
UV
Fangchinoline (μM)
Fangchinoline (μM)
Ac-DEVD-CHO (μM)
Tetrandrine (μM)
G0/G1:57.92%S:12.56%G2/M:26.76%
Fangchinoline 10 mMG0/G1:74.63%S:6.91%G2/M:15.49%
Tetrandrine 10 mM
G0/G1:80.00%S:3.40%G2/M:12.63%
Tetrandrine 10 mM
Tetrandrine 10 μMUV
Figure 2Fangchinoline does not induce apoptosis in HepG2 and PLC/PRF/5 cells. (A) Cells were treated with fangchinoline or tetrandrine for 24 h or UVfor 20 min. Then cells were dually stained with Annexin V/PI and analysed by flow cytometry. (B) Cells were treated with fangchinoline ortetrandrine for 24 h and then fixed. The cell cycle was analysed with flow cytometry. (C) Cells were exposed to fangchinoline or tetrandrine for24 h or UV for 20 min. Total DNA was collected and separated on 2% agarose gel prestained with Gel-Red. (D) Cells were exposed tofangchinoline or tetrandrine for 24 h or UV for 20 min. Cells lysate was collected and protein expression was analysed by immunobloting. (E) Cellswere treated with fangchinoline for 24 h in the presence of 50 mM Ac-DEVD-CHO. Cell viability was determined by MTT assay.
BJPFangchinoline induces autophagy
British Journal of Pharmacology (2011) 164 731–742 735

the observation that fangchinoline-induced cell death couldnot be attenuated in the presence of 50 mM of Ac-DEVD-CHO, a caspase inhibitor (Figure 2E).
Fangchinoline induces autophagy inhepatoma cells HepG2 and PLC/PRF/5In our study, dose-dependent increases of MDC and AO stain-ing in hepatoma cells exposed to fangchinoline showed theaccumulation of autophagic vacuoles in acidic compartmentsenriched in lipids (Bampton et al., 2005), which may indicatean autophagy induction by fangchinoline (Figure 3A,B).Induction of autophagy by fangchinoline was confirmed bythe immunoblot analysis on expression of LC3-II, a biomar-ker of autophagy (Figure 3C). In contrast, tetrandrine was notable to induce autophagy at 10 mM, at which dose it exhibitspotent pro-apoptotic action (Figure 3A–C). To furtherdescribe the role of autophagy in the anti-tumour action offangchinoline, we examined if fangchinoline could alsoinduce autophagy in normal hepatic cells. No induction ofautophagy was observed in BRL cells with 5 or 10 mM fangchi-noline treatment, concentrations which were capable ofkilling tumour cells (Figure 3C). Moreover, to further eluci-date that exposure to fangchinoline could induce autophagy,HepG2 cells stably expressing GFP-LC3 were treated withfangchinoline for 24 h and significant increase of green fluo-rescent signal was observed, indicating accumulation of GFP-LC3 protein in cells exposed to fangchinoline. Thisaccumulation of GFP-LC3 was blocked by siRNA for the Atg5gene (Figure 3D).Similar results were observed in the quanti-fication of GFP-LC3 cell localisation, showing that fangchi-noline intervention increases the punctate nature of theautophagosomes and that this inductive action was blockedby Atg5 gene silencing (Figure 3D). To monitor the dynamicsof fangchinoline-induced autophagy, we exposed HepG2cells to different doses of fangchinoline in the presence ofbafilomycin A1 (32 nM) or leupeptin (50 mM). Addition ofbafilomycin A1 blocked the fusion process of autophagosomeand lysosome, and we observed a significant decrease ofLC3-I, the 18 kDa cytosolic constituent, after fangchinolinetreatment while the lipid membrane form of LC3-II remainedthe same (Figure 3E). However, the presence of the lysosomeinhibitor leupeptin, which blocks the lysosomal degradationof autophagic vacuoles, revealed no effect on fangchinoline-induced autophagy in HepG2 cells (Figure 3E). Different pat-terns of fangchinoline-induced autophagy in cells in thepresence of bafilomycin A1 or leupeptin indicates thatfangchinoline initiates autophagy by slowing the off-rateprocess (lysosomal degradation) of autophagy rather thanincreasing the on-rate process (autophagosome formation).
Fangchinoline induces translocation of p53 inhepatoma cellsThe human tumour suppressor protein p53, which transacti-vates many of pro-apoptotic and cell cycle arresting-relatedgenes, is well recognized for its ability to induce apoptosis. Asa transcriptional factor, a novel function of p53 in regulatingautophagy has been identified by recent studies. The pool ofcytoplasmic p53 may repress autophagy ina transcription-dependent manner whereas translocation of p53 into the
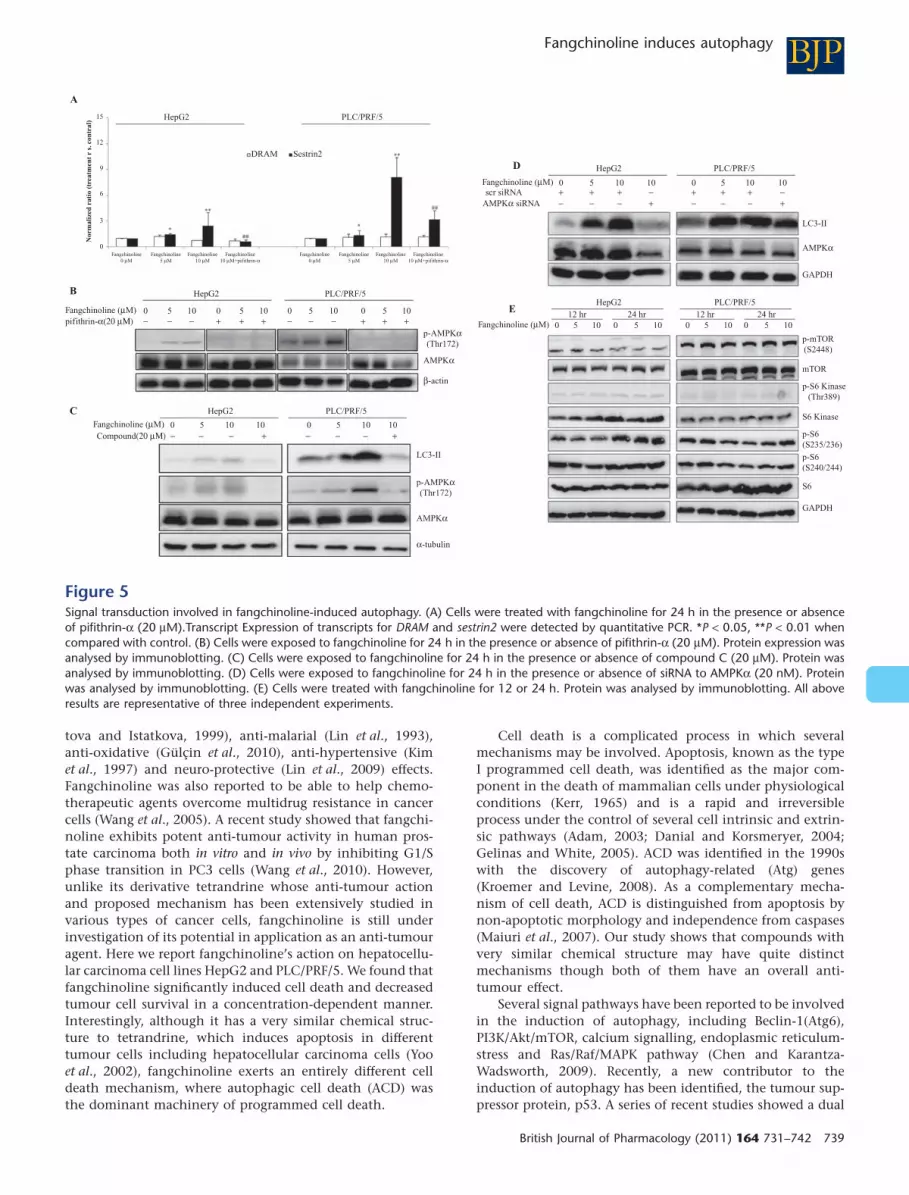
nucleus induces autophagy via activating the transcription ofspecific genes (Maiuri et al., 2009). In our study, no significantdecrease of the total level of p53 protein in cells treated withfangchinoline could be observed. However, a concentration-dependent increase of nuclear import of p53 could be foundin both HepG2 and PLC/PRF/5 cells exposed to fangchinoline(Figure 4A), indicating that fangchinoline may act as a modu-lator to increase the p53 nuclear localization without alteringits total expression level. The autophagy-inducing action offangchinoline was completely blocked by co-treatment withpifithrin-a (20 mM), suggesting a central role of p53 infangchinoline-stimulated autophagy in hepatoma cells(Figure 4B). Although the exact mechanism of pifithrin-a ininhibiting p53 transcription activity remains unclear, initialstudy suggested that pifithrin-a may modulate the nuclearimport or export of p53, or may decrease the stability ofnuclear p53 (Waters et al., 2010). Our results suggest that thetranscriptional activity of p53 is required for the initiation ofautophagy in hepatoma cell exposed to fangchinoline.
Involvement of senstrin2 but not DRAM infangchinoline-induced autophagy inhepatoma cellsWe examined the conventional signal transduction pathwayswhich may activate autophagy in human cancer cells. As weobserved, fangchinoline has little effect on Beclin-1, p38MAPK, p42/p44 MAPK or Akt signalling pathway (Figure 4C).Autophagy induction by p53 nuclear translocations includedthe up-regulation of DRAM and sestrin2 in p53-sufficient butnot in p53-deficient cells (Maiuri et al., 2009). Determined byquantitative real-time PCR, increased sestrin2 transcript butnot that of DRAM could be observed in cells treated withfangchinoline. Consistent with this, adding the p53 inhibi-tor, pifithrin-a, completely blocked the transactivation ofsestrin2, suggesting that nuclear import of p53 may berequired for the transcriptional activation of sestrin2(Figure 5A). Recent studies found that removal of sestrin2could suppress autophagy induced by various stimulators,where activation of AMPK signalling and inhibition of mTORare involved as the key mechanisms (Budanov and Karin,2008; D’Amelio and Cecconi, 2009). To further examine theeffect of p53 translocation on the AMPK/mTOR signallingpathway, cells were exposed to fangchinoline in the presenceor absence of pifithrin-a. We observed that the AMPK signal-ling could be activated by treatment of fangchinoline in theabsence of pifithrin-a. However, this stimulatory effect offangchinoline on AMPK pathway as well, was completelydown-regulated (Figure 5B). The induction of autophagy wasattenuated in the presence of an AMPK inhibitor or siRNA toAMPKa (Figure 5C,D). These results indicate that AMPK acti-vation may be necessary for fangchinoline-induced autoph-agy. It has been reported that mTOR inhibition follows as adownstream target of AMPK activation. Controversially, wefound that fangchinoline could induce autophagy via theactivation of the AMPK pathway without inhibiting eitherphosphorylation of mTOR or of its substrates p70 S6 kinaseand S6 ribosomal protein (Figure 5E). This reveals that abypassing mechanism of AMPK signalling targets to autoph-agy activation independent of mTOR suppression, may beinvolved in fangchinoline’s action.
BJP N Wang et al.
736 British Journal of Pharmacology (2011) 164 731–742

AFangchinoline 0 μM
HepG2
HepG2
HepG2
MD
C s
tain
ing
posi
tive
cel
l (%
)O
A s
tain
ing
posi
tive
cel
l (%
)
Pun
ctat
ion
per
100
cell
PLC/PRF/5
PLC/PRF/5
0
20
40
60
80
100
0
20
40
60
80
100
0
20
40
60
80
100
HepG2
HepG2
HepG2
LC3-II
LC3-I
Baflomycin A1(32 nM)
0 0
+ + ++ + +
05 5 510 10 10
0 5 10 0 5 10 0 5 10 0 5 10 0 5 10
LC3-II
α-tubulin
β-actin
β-actin
LC3-II
Atg5
Atg5siRNA
scrsiRNA
α-tubulin
PLC/PRF/5
PLC/PRF/5 HepG2 PLC/PRF/5BRL
Fangchinoline 0 μM
Fangchinoline (μM)
Fangchinoline (μM)
Leupeptin (50 μM)
Fangchinoline0 μM
Fangchinoline5 μM
Fangchinoline10 μM
Terandrine10 μM
Fangchinoline0 μM
Fangchinoline5 μM
Fangchinoline10 μM
Terandrine10 μM
Terandrine10 μM
Fangchinoline0 μM
Fangchinoline5 μM
Fangchinoline10 μM
Fangchinoline5 μM+scr
siRNA
Fangchinoline5 μM+Atg5
siRNA
Fangchinoline 5 μM
Fangchinoline 5 μM
Fangchinoline 10 μM
Fangchinoline 10 μM
Tetrandrine 10 μM
Tetrandrine 10 μM
Tetrandrine (μM)
B
C
D
E
Figure 3Fangchinoline induces autophagy in HepG2 and PLC/PRF/5 cells. (A) Cells were exposed to fangchinoline or tetrandrine for 24 h. Then cells werestained by monodansylcadaverine (MDC) and visualized by fluorescence microscopy (630 ¥ magnification). Percentage of cells with positivestaining was calculated. **P < 0.01 significantly different from control. (B) Cells were exposed to fangchinoline or tetrandrine for 24 h. Then cellswere stained with acridine orange (AO) and visualized by fluorescence microscopy (630¥). Percentage of cells with positive staining wascalculated. **P < 0.01 significantly different from control. (C) Cells were treated with fangchinoline or tetrandrine for 24 h and target protein wasanalysed by immunoblotting. **P < 0.01 significantly different from control. (D) Cells stably expressing GFP-LC3 fusion protein were exposed tofangchinoline or tetrandrine for 24 h. The induction of autophagy was blocked by introducing siRNA to Atg5 into cells and a scrambled negativecontrol (scr) siRNA was used as negative control. Percentage of cells with autophagic punctate fluorescence was determined. **P < 0.01significantly different from control. ##P < 0.01, cells with siRNA to Atg5 significantly different from cells with scr siRNA. (E) Cells were exposed tofangchinoline for 24 h in the presence or absence of bafilomycin A1 (32 nM) or leupeptin (50 mM). Cell lysate was collected and protein expressionwas analysed by immunobloting. All of the results are representative of three independent experiments and are shown as mean + SD.
BJPFangchinoline induces autophagy
British Journal of Pharmacology (2011) 164 731–742 737

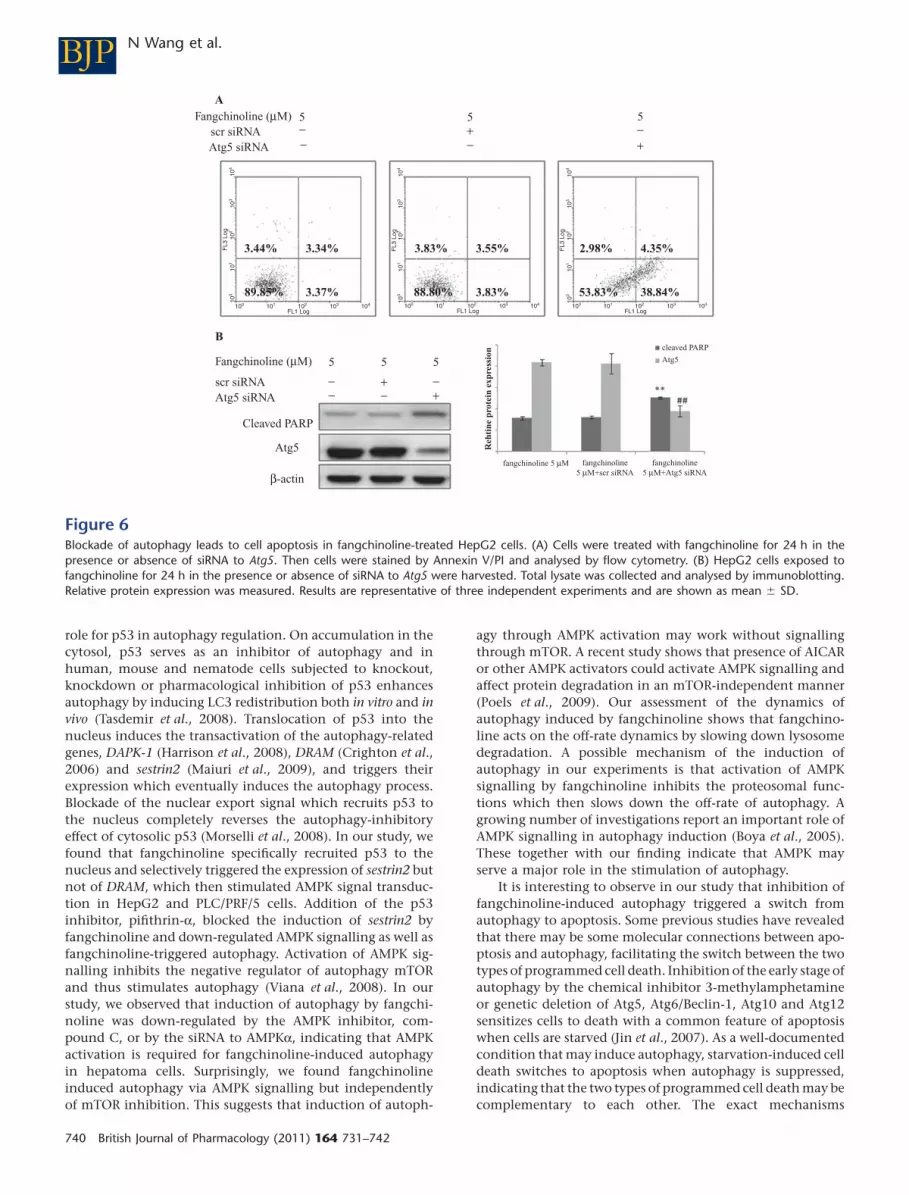
Inhibition of autophagy switches it toapoptosis in hepatoma cells withfangchinoline interventionBecause autophagy is a novel property of fangchinoline ininducing tumour cell death, we further examined thatwhether inhibition of autophagy in fangchinoline-treatedcells resulted in cell survival. Interestingly, by inhibitingautophagy through genetic inhibition of Atg5, we found thatapoptosis was initiated in HepG2 cells exposed to fangchino-line (Figure 6A). This observation was further confirmed byimmunoblotting, where cleavage of PARP, a marker of cell
undergoing apoptosis (Oliver et al., 1998), was significantlyincreased when autophagy was inhibited in HepG2 cellsexposed to fangchinoline (Figure 6B). Our results indicatedthat apoptosis may be an alternative route to cell deathinduced by fangchinoline when ACD is inhibited.
Discussion
Fangchinoline was reported to exert several pharmacologicalactions in vitro and in vivo, including anti-inflammatory (Hris-
HepG2 HepG2
HepG2
Beclin-1
p-Akt
p-p38
p38
p-Erk1/2
Erk1/2
Akt
p-GSK3B
GSK3B
p53
Rel
atin
epr
otei
n ex
pres
sion
Rel
atin
epr
otei
n ex
pres
sion
p53
Lamin B1
Lamin B1
β-actin
β-actin
α-tubulin
Cytosol
cytosol
cytosol
nuclear
nuclear
fangchinoline0 μM
fangchinoline2.5 μM
fangchinoline5 μM
fangchinoline10 μM
fangchinoline0 μM
fangchinoline 0 μM fangchinoline 10 μM fangchinoline 10 μM+pifithrin-α
Fangchinoline (μM) 0 5 10 0 5 10
fangchinoline2.5 μM
fangchinoline5 μM
fangchinoline10 μM
Nuclear
0
20
40
60
Pun
ctat
ion
of %
cel
ls 80
100
0 2.5 5 10 0 2.5 5 10Fangchinoline (μM)
A
B C
PLC/PRF/5
PLC/PRF/5
PLC/PRF/5
Figure 4Fangchinoline induced translocation of p53 in HepG2 and PLC/PRF/5 cells. (A) Cells exposed to various doses of fangchinoline was harvested andcytosolic protein and nuclear protein were isolated using different extraction buffer and analysed by immunoblotting. *P < 0.05, **P < 0.01significantly different from control. (B) HepG2 cells stably expressing GFP-LC3 fusion protein were exposed to fangchinoline for 24 h in thepresence or absence of pifithrin-a (20 mM) and then visualized (630 ¥ magnification). Percentage of cells with autophagic punctate fluorescencewas determined. P < 0.01 significantly different from control. ##P < 0.01 significantly different between the presence and absence of pifithrin-a(20 mM). All above results are representative of three independent experiments and are shown as mean + SD. (C) HepG2 and PLC cells was treatedwith fangchinoline for 24 h and the total protein was collected. Protein expression was determined by immunoblotting.
BJP N Wang et al.
738 British Journal of Pharmacology (2011) 164 731–742
tova and Istatkova, 1999), anti-malarial (Lin et al., 1993),anti-oxidative (Gülçin et al., 2010), anti-hypertensive (Kimet al., 1997) and neuro-protective (Lin et al., 2009) effects.Fangchinoline was also reported to be able to help chemo-therapeutic agents overcome multidrug resistance in cancercells (Wang et al., 2005). A recent study showed that fangchi-noline exhibits potent anti-tumour activity in human pros-tate carcinoma both in vitro and in vivo by inhibiting G1/Sphase transition in PC3 cells (Wang et al., 2010). However,unlike its derivative tetrandrine whose anti-tumour actionand proposed mechanism has been extensively studied invarious types of cancer cells, fangchinoline is still underinvestigation of its potential in application as an anti-tumouragent. Here we report fangchinoline’s action on hepatocellu-lar carcinoma cell lines HepG2 and PLC/PRF/5. We found thatfangchinoline significantly induced cell death and decreasedtumour cell survival in a concentration-dependent manner.Interestingly, although it has a very similar chemical struc-ture to tetrandrine, which induces apoptosis in differenttumour cells including hepatocellular carcinoma cells (Yooet al., 2002), fangchinoline exerts an entirely different celldeath mechanism, where autophagic cell death (ACD) wasthe dominant machinery of programmed cell death.
Cell death is a complicated process in which severalmechanisms may be involved. Apoptosis, known as the typeI programmed cell death, was identified as the major com-ponent in the death of mammalian cells under physiologicalconditions (Kerr, 1965) and is a rapid and irreversibleprocess under the control of several cell intrinsic and extrin-sic pathways (Adam, 2003; Danial and Korsmeryer, 2004;Gelinas and White, 2005). ACD was identified in the 1990swith the discovery of autophagy-related (Atg) genes(Kroemer and Levine, 2008). As a complementary mecha-nism of cell death, ACD is distinguished from apoptosis bynon-apoptotic morphology and independence from caspases(Maiuri et al., 2007). Our study shows that compounds withvery similar chemical structure may have quite distinctmechanisms though both of them have an overall anti-tumour effect.
Several signal pathways have been reported to be involvedin the induction of autophagy, including Beclin-1(Atg6),PI3K/Akt/mTOR, calcium signalling, endoplasmic reticulum-stress and Ras/Raf/MAPK pathway (Chen and Karantza-Wadsworth, 2009). Recently, a new contributor to theinduction of autophagy has been identified, the tumour sup-pressor protein, p53. A series of recent studies showed a dual
HepG2
HepG2
scr siRNAAMPKα siRNA
HepG2
LC3-II
LC3-II
GAPDH
GAPDH
p-mTOR(S2448)
p-S6 Kinase(Thr389)
p-S6(S235/236)
p-S6(S240/244)
S6
S6 Kinase
mTOR
p-AMPKα(Thr172)
p-AMPKα(Thr172)
AMPKα
AMPKα
AMPKα
β-actin
α-tubulin
HepG2
A
B
D
E
C
0
0
0
5−
− − − − − −
− − − − −+ +
+ +
+ + + +
+ + + + + +++
−−−− −−−
−
5
510
1010 0 5 1010
100 5 100 5 100
5 10 100
5 100 5 100 5 100 5 100
5 10 100
3
6
Nor
mal
ized
rat
io (
trea
tmen
t r
s. c
ontr
al)
9
12
15
DRAM Sestrin2
PLC/PRF/5
PLC/PRF/5
HepG212 hr 24 hr 12 hr 24 hr
PLC/PRF/5PLC/PRF/5
PLC/PRF/5
Fangchinoline0 μM
Fangchinoline (μM)
Fangchinoline (μM)
Fangchinoline (μM)
Fangchinoline (μM)
Fangchinoline5 μM
Fangchinoline10 μM
Fangchinoline10 μM+pifithrin-α
Fangchinoline0 μM
Fangchinoline5 μM
Fangchinoline10 μM
Fangchinoline10 μM+pifithrin-α
pifithrin-α(20 μΜ)
Compound(20 μΜ)
Figure 5Signal transduction involved in fangchinoline-induced autophagy. (A) Cells were treated with fangchinoline for 24 h in the presence or absenceof pifithrin-a (20 mM).Transcript Expression of transcripts for DRAM and sestrin2 were detected by quantitative PCR. *P < 0.05, **P < 0.01 whencompared with control. (B) Cells were exposed to fangchinoline for 24 h in the presence or absence of pifithrin-a (20 mM). Protein expression wasanalysed by immunoblotting. (C) Cells were exposed to fangchinoline for 24 h in the presence or absence of compound C (20 mM). Protein wasanalysed by immunoblotting. (D) Cells were exposed to fangchinoline for 24 h in the presence or absence of siRNA to AMPKa (20 nM). Proteinwas analysed by immunoblotting. (E) Cells were treated with fangchinoline for 12 or 24 h. Protein was analysed by immunoblotting. All aboveresults are representative of three independent experiments.
BJPFangchinoline induces autophagy
British Journal of Pharmacology (2011) 164 731–742 739
role for p53 in autophagy regulation. On accumulation in thecytosol, p53 serves as an inhibitor of autophagy and inhuman, mouse and nematode cells subjected to knockout,knockdown or pharmacological inhibition of p53 enhancesautophagy by inducing LC3 redistribution both in vitro and invivo (Tasdemir et al., 2008). Translocation of p53 into thenucleus induces the transactivation of the autophagy-relatedgenes, DAPK-1 (Harrison et al., 2008), DRAM (Crighton et al.,2006) and sestrin2 (Maiuri et al., 2009), and triggers theirexpression which eventually induces the autophagy process.Blockade of the nuclear export signal which recruits p53 tothe nucleus completely reverses the autophagy-inhibitoryeffect of cytosolic p53 (Morselli et al., 2008). In our study, wefound that fangchinoline specifically recruited p53 to thenucleus and selectively triggered the expression of sestrin2 butnot of DRAM, which then stimulated AMPK signal transduc-tion in HepG2 and PLC/PRF/5 cells. Addition of the p53inhibitor, pifithrin-a, blocked the induction of sestrin2 byfangchinoline and down-regulated AMPK signalling as well asfangchinoline-triggered autophagy. Activation of AMPK sig-nalling inhibits the negative regulator of autophagy mTORand thus stimulates autophagy (Viana et al., 2008). In ourstudy, we observed that induction of autophagy by fangchi-noline was down-regulated by the AMPK inhibitor, com-pound C, or by the siRNA to AMPKa, indicating that AMPKactivation is required for fangchinoline-induced autophagyin hepatoma cells. Surprisingly, we found fangchinolineinduced autophagy via AMPK signalling but independentlyof mTOR inhibition. This suggests that induction of autoph-
agy through AMPK activation may work without signallingthrough mTOR. A recent study shows that presence of AICARor other AMPK activators could activate AMPK signalling andaffect protein degradation in an mTOR-independent manner(Poels et al., 2009). Our assessment of the dynamics ofautophagy induced by fangchinoline shows that fangchino-line acts on the off-rate dynamics by slowing down lysosomedegradation. A possible mechanism of the induction ofautophagy in our experiments is that activation of AMPKsignalling by fangchinoline inhibits the proteosomal func-tions which then slows down the off-rate of autophagy. Agrowing number of investigations report an important role ofAMPK signalling in autophagy induction (Boya et al., 2005).These together with our finding indicate that AMPK mayserve a major role in the stimulation of autophagy.
It is interesting to observe in our study that inhibition offangchinoline-induced autophagy triggered a switch fromautophagy to apoptosis. Some previous studies have revealedthat there may be some molecular connections between apo-ptosis and autophagy, facilitating the switch between the twotypes of programmed cell death. Inhibition of the early stage ofautophagy by the chemical inhibitor 3-methylamphetamineor genetic deletion of Atg5, Atg6/Beclin-1, Atg10 and Atg12sensitizes cells to death with a common feature of apoptosiswhen cells are starved (Jin et al., 2007). As a well-documentedcondition that may induce autophagy, starvation-induced celldeath switches to apoptosis when autophagy is suppressed,indicating that the two types of programmed cell death may becomplementary to each other. The exact mechanisms
A
B
Reh
tine
pro
tein
exp
ress
ion
Fangchinoline (μM)
Fangchinoline (μM)
fangchinoline 5 μM fangchinoline5 μM+scr siRNA
fangchinoline5 μM+Atg5 siRNA
scr siRNAAtg5 siRNA
scr siRNAAtg5 siRNA
Cleaved PARP
Atg5
β-actin
5
5 5 5cleaved PARP
Atg5
−
−−
−−
−−
−+
++
+5 5
100 101
FL1 Log
3.44% 3.34%
89.85% 3.37%
3.83%
3.83%
2.98% 4.35%
53.83% 38.84%
3.55%
88.80%
FL3
Log
102 103 104 100 101
FL1 Log102 103 104 100 101
FL1 Log102 103 104
100
101
102
103
104
FL3
Log
100
101
102
103
104
FL3
Log
100
101
102
103
104
Figure 6Blockade of autophagy leads to cell apoptosis in fangchinoline-treated HepG2 cells. (A) Cells were treated with fangchinoline for 24 h in thepresence or absence of siRNA to Atg5. Then cells were stained by Annexin V/PI and analysed by flow cytometry. (B) HepG2 cells exposed tofangchinoline for 24 h in the presence or absence of siRNA to Atg5 were harvested. Total lysate was collected and analysed by immunoblotting.Relative protein expression was measured. Results are representative of three independent experiments and are shown as mean � SD.
BJP N Wang et al.
740 British Journal of Pharmacology (2011) 164 731–742

involved in fangchinoline-induced apoptosis when ACD isinhibited is under investigation.
In conclusion, the mode of cell death induced by fangchi-noline in HepG2 and PLC/PRF/5 hepatocellular carcinomacells has been investigated. We found that fangchinoline didnot induce apoptosis in HepG2 and PLC/PRF/5 cells. Instead,an excessive autophagy was triggered by fangchinoline,concentration-dependently, initiating an alternative mode ofcell death which may contribute to fangchinoline’s anti-tumour action. Nuclear localization of p53 in tumour cellsexposed to fangchinoline selectively transactivated theautophagy-related sestrin2 and stimulated AMPK signalling toinitiate the autophagic process. Blockade of AMPK signallingattenuated autophagy induction in fangchinoline-treatedcells. Genetic inhibition of Atg5 blocked induction of autoph-agy fangchinoline and switched it to apoptosis, in hepatocel-lular carcinoma cells. Our results shed light on the potentialof fangchinoline as a tumour therapy agent with a novelmechanism in mediating cancer cell death.
Acknowledgements
The study was financially supported by grants from theresearch council of the University of Hong Kong (ProjectCodes: 10400413 and 10400699), Government-MatchingGrant Scheme (fourth Phase, Project Code: 20740314) andthe Science and Technology Department of Guizhou Prov-ince, China (Project Code: QKHYSCN [2009]4010 andQKHRCTD[2008]4008). The authors are grateful to thesupport of Professors Yung-Chi Cheng, Sai-Wah Tsao, YaoTong and Allan SY Lau. The authors would like to expressthanks to Ms Oi Yee Chow, Ms Cindy Lee, Mr Keith Wong andMr Freddy Tsang for their technical support. We also thankProfessor Tamotsu Yoshimori for kindly providing GFP-LC3plasmid.
Conflict of interest
The authors have declared no conflict of interest.
ReferencesAdam JM (2003). Ways of dying: multiple pathways to apoptosis.Gene Dev 17: 2481–2495.
Bampton ET, Goemans CG, Niranjan D, Mizushima N,Tolkovsky AM (2005). The dynamics of autophagy visualized in livecells: from autophagosome formation to fusion withendo/lysosomes. Autophagy 1: 23–36.
Bommareddy A, Hahm ER, Xiao D, Powolny AA, Fisher AL, Jiang Yet al. (2009). Atg5 regulates phenethyl isothiocyanate-inducedautophagic and apoptotic cell death in human prostate cancer cells.Cancer Res 69: 3704–3712.
Boya P, González-Polo RA, Casares N, Perfettini JL, Dessen P,Larochette N et al. (2005). Inhibition of macroautophagy triggersapoptosis. Mol Cell Biol 25: 1025–1040.
Budanov AV, Karin M (2008). p53 target genes sestrin1 and sestrin2connect genotoxic stress and mTOR signaling. Cell 134: 451–460.
Chen N, Karantza-Wadsworth V (2009). Role and regulation ofautophagy in cancer. Biochim Biophys Acta 1793: 1516–1523.
Cheng Y, Qiu F, Ikejima T (2009). Molecular mechanisms oforidonin-induced apoptosis and autophagy in murine fibrosarcomaL929 cells. Autophagy 5: 430–431.
Crighton D, Wilkinson S, O’Prey J, Syed N, Smith P, Harrison PRet al. (2006). DRAM, a p53-induced modulator of autophagy, iscritical for apoptosis. Cell 126: 121–134.
D’Amelio M, Cecconi F (2009). A novel player in the p53-mediatedautophagy: Sestrin2. Cell Cycle 8: 1467.
Danial NN, Korsmeryer SJ (2004). Cell death: critical control points.Cell 116: 205–219.
Franken NA, Rodermond HM, Stap J, Haveman J, van Bree C(2006). Clonogenic assay of cells in vitro. Nat Protoc 1: 2315–2319.
Gelinas C, White E (2005). BH3-only proteins in control: specificityregulates MCL-1 and BAK-mediated apoptosis. Genes Dev 19:1263–1268.
Gülçin I, Elias R, Gepdiremen A, Chea A, Topal F (2010).Antioxidant activity of bisbenzylisoquinoline alkaloids fromStephania rotunda: cepharanthine and fangchinoline. J EnzymeInhib Med Chem 25: 44–53.
Hanahan D, Weinberg RA (2000). The hallmarks of cancer. Cell100: 57–70.
Harrison B, Kraus M, Burch L, Stevens C, Craig A, Gordon-Weeks Pet al. (2008). DAPK-1 binding to a linear peptide motif in MAP1Bstimulates autophagy and membrane blebbing. J Biol Chem 283:9999–10014.
Hristova M, Istatkova R (1999). Complement-mediatedantiinflammatory effect of bisbenzylisoquinoline alkaloidfangchinoline. Phytomedicine 6: 357–362.
Jin S, DiPaola RS, Mathew R, White E (2007). Metabolic catastropheas a means to cancer cell death. J Cell Sci 120: 379–383.
Kerr JF (1965). A histochemical study of hypertrophy and ischaemicinjury of rat liver with special reference to changes in lysosomes. JPathol Bacteriol 90: 419–435.
Kim HS, Zhang YH, Oh KW, Ahn HY (1997). Vasodilating andhypotensive effects of fangchinoline and tetrandrine on the rataorta and the stroke-prone spontaneously hypertensive rat. JEthnopharmacol 58: 117–123.
Kroemer G, Levine B (2008). Autophagic cell death: the story of amisnomer. Nat Rev Mol Cell Biol 9: 1004–1010.
Lin LZ, Shieh HL, Angerhofer CK, Pezzuto JM, Cordell GA, Xue Let al. (1993). Cytotoxic and antimalarial bisbenzylisoquinolinealkaloids from Cyclea barbata. J Nat Prod 56: 22–29.
Lin TY, Lu CW, Tien LT, Chuang SH, Wang YR, Chang WH et al.(2009). Fangchinoline inhibits glutamate release from rat cerebralcortex nerve terminals (synaptosomes). Neurochem Int 54:506–512.
Maiuri MC, Zalckvar E, Kimchi A, Kroemer G (2007). Self-eatingand self-killing: crosstalk between autophagy and apoptosis. NatRev Mol Cell Biol 8: 741–752.
Maiuri MC, Malik SA, Morselli E, Kepp O, Criollo A, Mouchel PLet al. (2009). Stimulation of autophagy by the p53 target geneSestrin2. Cell Cycle 8: 1571–1576.
BJPFangchinoline induces autophagy
British Journal of Pharmacology (2011) 164 731–742 741

Meng LH, Zhang H, Hayward L, Takemura H, Shao RG, Pommier Y(2004). Tetrandrine induces early G1 arrest in human coloncarcinoma cells by down-regulating the activity and inducing thedegradation of G1-S-specific cyclin-dependent kinases and byinducing p53 and p21Cip1. Cancer Res 64: 9086–9092.
Mizushima N, Sugita H, Yoshimori T, Ohsumi Y (1998). A newprotein conjugation system in human. The counterpart of the yeastApg12p conjugation system essential for autophagy. J Biol Chem273: 33889–33892.
Morselli E, Tasdemir E, Maiuri MC, Galluzzi L, Kepp O, Criollo Aet al. (2008). Mutant p53 protein localized in the cytoplasm inhibitsautophagy. Cell Cycle 7: 3056–3061.
Morselli E, Galluzzi L, Kepp O, Vicencio JM, Criollo A, Maiuri MCet al. (2009). Anti- and pro-tumor functions of autophagy. BiochimBiophys Acta 1793: 1524–1532.
Munafó DB, Colombo MI (2001). A novel assay to study autophagy:regulation of autophagosome vacuole size by amino aciddeprivation. J Cell Sci 114: 3619–3629.
Oliver FJ, de la Rubia G, Rolli V, Ruiz-Ruiz MC, de Murcia G,Murcia JM (1998). Importance of poly(ADP-ribose) polymerase andits cleavage in apoptosis. Lesson from an uncleavable mutant. J BiolChem 273: 33533–33539.
Poels J, Spasic MR, Callaerts P, Norga KK (2009). Expanding rolesfor AMP-activated protein kinase in neuronal survival andautophagy. Bioessays 31: 944–952.
Shen S, Kepp O, Martins I, Vitale I, Souquère S, Castedo M et al.(2007). Defective autophagy associated with LC3 puncta inepothilone-resistant cancer cells. Cell Cycle 9: 377–383.
Sun X, Xu R, Deng Y, Cheng H, Ma·J, Ji J et al. (2007). Effects oftetrandrine on apoptosis and radiosensitivity of nasopharyngealcarcinoma cell line CNE. Acta Biochim Biophys Sin 39: 869–878.
Tang G, Xu Z, Goldman JE (2006). Synergistic effects of theSAPK/JNK and the proteasome pathway on glial fibrillary acidicprotein (GFAP) accumulation in Alexander disease. J Bio Chem 281:38634–38643.
Tang G, Yue Z, Talloczy Z, Hagemann T, Cho W, Messing A et al.(2008). Autophagy induced by Alexander disease-mutant GFAPaccumulation is regulated by p38/MAPK and mTOR signalingpathways. Hum Mol Genet 17: 1540–1555.
Tasdemir E, Maiuri MC, Galluzzi L, Vitale I, Djavaheri-Mergny M,D’Amelio M et al. (2008). Regulation of autophagy by cytoplasmicp53. Nat Cell Biol 10: 676–687.
Tian R, Gregor M, Wiche G, Goldman JE (2006). Plectin regulatesthe organization of glial fibrillary acidic protein in Alexanderdisease. Am J Pathol 168: 888–897.
Viana R, Aguado C, Esteban I, Moreno D, Viollet B, Knecht E et al.(2008). Role of AMP-activated protein kinase in autophagy andproteasome function. Biochem Biophys Res Commun 369:964–968.
Wang FP, Wang L, Yang JS, Nomura M, Miyamoto K (2005).Reversal of P-glycoprotein-dependent resistance to vinblastine bynewly synthesized bisbenzylisoquinoline alkaloids in mouseleukemia P388 cells. Biol Pharm Bull 28: 1979–1982.
Wang CD, Huang JG, Gao X, Li Y, Zhou SY, Yan X et al. (2010).Fangchinoline induced G1/S arrest by modulating expression ofp27, PCNA, and cyclin D in human prostate carcinoma cancer PC3cells and tumor xenograft. Biosci Biotechnol Biochem 74: 488–493.
Waters FJ, Shavlakadze T, McIldowie MJ, Piggott MJ, Grounds MD(2010). Use of pifithrin to inhibit p53-mediated signalling of TNFin dystrophic muscles of mdx mice. Mol Cell Biochem 337:119–131.
Wei S, Yang HC, Chuang HC, Yang J, Kulp SK, Lu PJ et al. (2008).A novel mechanism by which thiazolidinediones facilitate theproteasomal degradation of cyclin D1 in cancer cells. J Biol Chem283: 26759–26770.
Yang C, Kaushal V, Shah SV, Kaushal GP (2008). Autophagy isassociated with apoptosis in cisplatin injury to renal tubularepithelial cells. Am J Physiol Renal Physiol 294: F777–F787.
Yoo SM, Oh SH, Lee SJ, Lee BW, Ko WG, Moon CK et al. (2002).Inhibition of proliferation and induction of apoptosis bytetrandrine in HepG2 cells. J Ethnopharmacol 81: 225–229.
Zhang YH, Fang LH, Ku BS (2003). Fangchinoline inhibits rat aorticvascular smooth muscle cell proliferation and cell cycle progressionthrough inhibition of ERK1/2 activation and c-fos expression.Biochem Pharmacol 66: 1853–1860.
BJP N Wang et al.
742 British Journal of Pharmacology (2011) 164 731–742





























