Embed Size (px)
Citation preview

* CorrespScience aUniversityE-mail: d(Gon Sup†These aut
PHYTOTHERAPY RESEARCHPhytother. Res. (2015)Published online in Wiley Online Library(wileyonlinelibrary.com) DOI: 10.1002/ptr.5488
Copyright
Flavonoids of Korean Citrus aurantium L. InduceApoptosis via Intrinsic Pathway in HumanHepatoblastoma HepG2 Cells
Seung Hwan Lee1† Silvia Yumnam,1† Gyeong Eun Hong,1 Suchismita Raha,1Venu Venkatarame Gowda Saralamma,1 Ho Jeong Lee,1 Jeong Doo Heo,2 Sang Joon Lee,2Won-Sup Lee,3 Eun-Hee Kim,4 Hyeon Soo Park1* and Gon Sup Kim1
1Research Institute of Life Science, College of Veterinary Medicine (BK21 plus project), Gyeongsang National University, Gazwa,Jinju 660-701, Korea2Gyeongnam Department of Environment Toxicology and Chemistry, Toxicity Screening Research Center, Korea Institute ofToxicology, Jinju, Korea3Department of Internal Medicine, Institute of Health Sciences, Gyeongsang National University School of Medicine, GyeongnamRegional Cancer Center, Gyeongsang National University Hospital, Jinju 660-702, Korea4Department of Nursing Science, International University of Korea, Jinju 660-759, Korea
Korean Citrus aurantium L. has long been used as a medicinal herb for its anti-inflammatory, antioxidant, andanticancer properties. The present study investigates the anticancer role of flavonoids extracted from C.aurantium on human hepatoblastoma cell, HepG2. The Citrus flavonoids inhibit the proliferation of HepG2cells in a dose-dependent manner. This result was consistent with the in vivo xenograft results. Apoptosis wasdetected by cell morphology, cell cycle analysis, and immunoblot. Flavonoids decreased the level of pAkt andother downstream targets of phosphoinositide-3-kinase/Akt pathway – P-4EBP1 and P-p70S6K. The expressionsof cleaved caspase 3, Bax, and Bak were increased, while those of Bcl-2 and Bcl-xL were decreased with anincrease in the expression of Bax/Bcl-xL ratio in treated cells. Loss of mitochondrial membrane potential wasalso observed in flavonoid-treated HepG2 cells. It was also observed that the P-p38 protein level was increasedboth dose and time dependently in flavonoid-treated cells. Collectively, these results suggest that flavonoidextracted from Citrus inhibits HepG2 cell proliferation by inducing apoptosis via an intrinsic pathway. Thesefindings suggest that flavonoids extracted from C. aurantium L. are potential chemotherapeutic agents againstliver cancer. Copyright © 2015 John Wiley & Sons, Ltd.
Keywords: apoptosis; HepG2; Citrus aurantium; Akt; Bax/Bcl-xL; p-38.
INTRODUCTION
Hepatoblastoma, the most common solid tumor, is thethird leading cause of cancer-related deaths in the world(Ferlay et al., 2010). It is rare in adults but is the most com-mon malignant tumor in children. It accounts for 75% ofprimary liver tumor in childhood (Kasper et al., 2005).Because of poor prognosis, alternative therapeutic strate-gies are required for the treatment of hepatoblastoma.Natural resources have played an important role in drug
discovery. Natural products are considered safe and havelong been used as medicines in many Asian countries(Newman et al., 2003). Among these, flavonoids have beenknown for their antioxidant, antiviral, anti-inflammatory,and anticancer activity (Havsteen, 1983; Ren et al., 2003).Tea, vegetables, and fruits are themain source of flavonoids.Flavonoids like hesperidin, nobiletin, and naringin are
rich in Citrus species (Benavente-Garcia and Castillo,2008). Citrus aurantium belongs to the Rutaceae familyof the order Sapindales. The immature peel of the fruit
ondence to: Hyeon Soo Park and Gon Sup Kim, Institute of Lifend College of Veterinary Medicine, Gyeongsang National, Gazwa, Jinju, 660-701, [email protected] (Hyeon Soo Park); [email protected])hors contributed equally to this work.
© 2015 John Wiley & Sons, Ltd.
is used as a medicine for treating various diseases. It isused to treat indigestion and has demonstrated potentialas a chemotherapeutic agent. Flavonoid is one of the maincomponents of Citrus and has been reported to possessanti-inflammatory, antioxidant, and anticancer properties(Kamaraj et al., 2009; Li et al., 2007). Many have reportedthe antiproliferative effect of flavonoids on various cancercell lines (Lee et al., 2012; Luo et al., 2008).
Apoptosis is the most well-studied programmed celldeath, and activation of apoptosis is considered to be aprotective mechanism against cancer progression (Kimand Han, 2001). There are two main apoptotic pathways:the extrinsic or death receptor pathway and the intrinsicor mitochondrial pathway. It is characterized by nuclearfragmentation,membrane blebbing, chromatin condensa-tion, and apoptotic body formation (Kerr et al., 1972) andactivation of caspases is a hallmark of apoptosis (Olssonand Zhivotovsky, 2011). The main aim of the presentstudy is to study the anticancer effect of flavonoids fromCitrus aurantium (CE). In this study, we have reportedthat CE inhibits HepG2 cell proliferation by inducing ap-optosis through dephosphorylation of Akt and activatingcaspase 3 and pro-apoptotic proteins Bax and Bak whileantiapoptotic proteins Bcl-xl and Bcl-2 expressions werereduced. The involvement of p38 mitogen-activated pro-tein kinase (MAPK)was also observed in theCE-inducedapoptosis in HepG2 cells.
Received 23 June 2015Revised 28 August 2015
Accepted 15 September 2015
S. H. LEE ET AL.
MATERIALS AND METHODS
Chemicals. Dulbecco’s modified Eagles medium mediawas purchased from HyClone (Logan, UT, USA). Anti-biotics (streptomycin/penicillin) and fetal bovine serumwere obtained from Gibco (Grand Island, NY, USA).3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazoliumbromide (MTT), propidium iodide (PI), and Hoechst33342 fluorescent dye were purchased from Invitrogen(Carlsbad, CA, USA). Anti-Bcl-xL, anti-Bak, anti-pro-caspase 3, anti-pro-caspase 8, anti-pro-caspase 9,anti-poly(ADP-ribose) polymerase (PARP), Fas, andFas L antibodies were purchased from Cell SignalingTechnology (Beverly, MA, USA).
Sample preparation. Citrus aurantium L. were obtainedfrom the Citrus Genetic Resource Bank, Jeju NationalUniversity. Lyophilized fruit peel was powdered andextracted in 3L of 70% aqueous methanol for 24h usinga PT-MR 2100 apparatus (Kinematica, Lucerne,Switzerland). The extracts were combined after sevenextraction cycles and filtered using a Buchner funnel.The filtrate was concentrated to 140mL using an EyelaNVC-2100 rotary evaporator (Tokyo Rikakikai, Tokyo,Japan), and 25-mL water and 3-g NaOH were added tothe concentrate, which was extracted with 200-mL ethylacetate. After three extraction cycles, the organic layerwas neutralized with brine and dried over anhydrousMgSO4 to yield 0.6 g of flavonoid components (the ex-tract was completely dried using a vacuum evaporator).High-performance liquid chromatographic (HPLC)
analysis was performed using 1100 series LC system(Agilent Technologies, Palo Alto, CA, USA). Chro-matographic separation was carried out in a ZorbaxStable Bond Analytical SB-C18 column (4.6 × 250mm,5μm; Agilent Technologies). The binary solvent systemconsisted of 0.1% aqueous formic acid (A) andmethanol/acetonitrile (B) in a 1:1 ratio. The elution
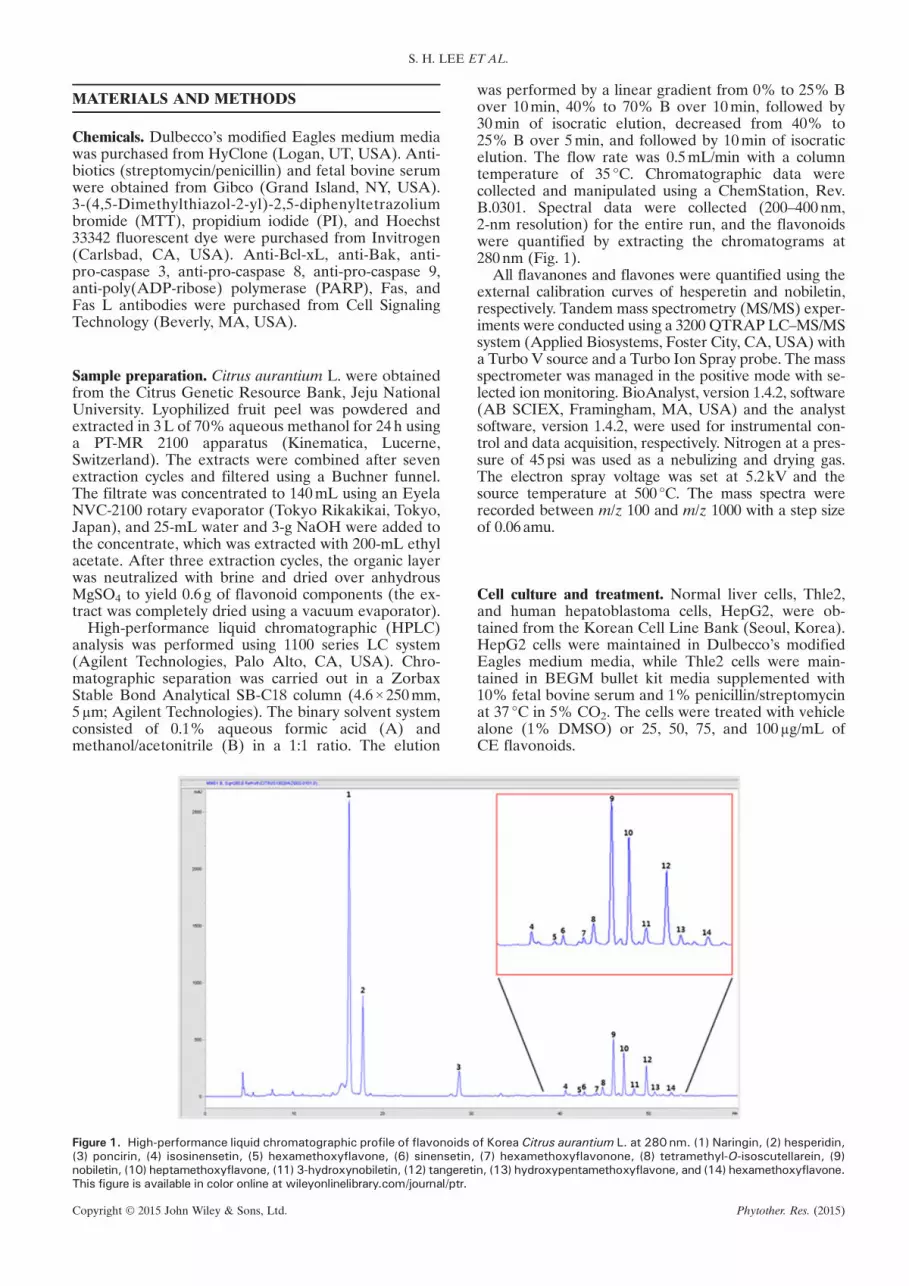
Figure 1. High-performance liquid chromatographic profile of flavonoids(3) poncirin, (4) isosinensetin, (5) hexamethoxyflavone, (6) sinensetin,nobiletin, (10) heptamethoxyflavone, (11) 3-hydroxynobiletin, (12) tangeretThis figure is available in color online at wileyonlinelibrary.com/journal/ptr.
Copyright © 2015 John Wiley & Sons, Ltd.
was performed by a linear gradient from 0% to 25% Bover 10min, 40% to 70% B over 10min, followed by30min of isocratic elution, decreased from 40% to25% B over 5min, and followed by 10min of isocraticelution. The flow rate was 0.5mL/min with a columntemperature of 35 °C. Chromatographic data werecollected and manipulated using a ChemStation, Rev.B.0301. Spectral data were collected (200–400nm,2-nm resolution) for the entire run, and the flavonoidswere quantified by extracting the chromatograms at280nm (Fig. 1).
All flavanones and flavones were quantified using theexternal calibration curves of hesperetin and nobiletin,respectively. Tandem mass spectrometry (MS/MS) exper-iments were conducted using a 3200 QTRAP LC–MS/MSsystem (Applied Biosystems, Foster City, CA, USA) witha Turbo V source and a Turbo Ion Spray probe. The massspectrometer was managed in the positive mode with se-lected ion monitoring. BioAnalyst, version 1.4.2, software(AB SCIEX, Framingham, MA, USA) and the analystsoftware, version 1.4.2, were used for instrumental con-trol and data acquisition, respectively. Nitrogen at a pres-sure of 45psi was used as a nebulizing and drying gas.The electron spray voltage was set at 5.2kV and thesource temperature at 500 °C. The mass spectra wererecorded between m/z 100 and m/z 1000 with a step sizeof 0.06amu.
Cell culture and treatment. Normal liver cells, Thle2,and human hepatoblastoma cells, HepG2, were ob-tained from the Korean Cell Line Bank (Seoul, Korea).HepG2 cells were maintained in Dulbecco’s modifiedEagles medium media, while Thle2 cells were main-tained in BEGM bullet kit media supplemented with10% fetal bovine serum and 1% penicillin/streptomycinat 37 °C in 5% CO2. The cells were treated with vehiclealone (1% DMSO) or 25, 50, 75, and 100μg/mL ofCE flavonoids.
of Korea Citrus aurantium L. at 280 nm. (1) Naringin, (2) hesperidin,(7) hexamethoxyflavonone, (8) tetramethyl-O-isoscutellarein, (9)
in, (13) hydroxypentamethoxyflavone, and (14) hexamethoxyflavone.
Phytother. Res. (2015)

CITRUS FLAVONOIDS INDUCE APOPTOSIS IN HEPG2 CELLS
Cell proliferation assay. Cell viability was determined bymeasuring the metabolism of MTT. HepG2 and Thle2cells (1 ×104 cells per well) were seeded on a 12-wellplate and were treated with various concentrations ofCE – 25, 50, 75, and 100μg/mL or vehicle alone for24h at 37 °C in 5% CO2 incubator. Five-hundred micro-liters of 0.5% (w/v) MTT solution was added to eachwell and further incubated for 3h at 37 °C. After incuba-tion, the medium was aspirated, and the formazan crys-tal was solubilized in 500μL of DMSO. After 15-minshaking, the absorbance was measured at 540nm in amicroplate reader. Cell viability was expressed as apercentage of proliferation versus the control, whichwas set as 100%.
Cell cycle distribution. Flow cytometer was used to studythe cell cycle distribution of CE-treated HepG2 cells, and1×105 HepG2 cells were seeded on a six-well plate. Afterreaching 70–80% confluence, the cells were treated with0, 25, 50, 75, and 100μg/mL of CE. After 24-h incubationat 37 °C, whole cells were harvested and fixed in 70%ethanol for 1h at 4 °C. Fixed cells were washed in 1×phosphate-buffered saline (PBS) and then further incu-bated with 1-U/mL RNase A (DNase free) and 5-μg/mLPI (Sigma-Aldrich, St. Louis, MO, USA) for 30min inthe dark. The cell cycle distribution of HepG2 cells wasanalyzed using FACS (BD Biosciences, Franklin Lakes,NJ, USA). Approximately 10,000 cells were analyzed.
Apoptotic assay. Apoptotic cells were detected using afluorescein isothiocyanate (FITC) annexin V apoptosisdetection kit 1 (BD Pharmingen, San Diego, CA,USA). Briefly, cells were treated with varying concentra-tions of CE (0, 25, 50, 75, and 100μg/mL) for 24h at 37 °C.Whole cells were harvested and washed with PBS.The washed cells were suspended in annexin V bindingbuffer containing 10-mM HEPES/NaOH, pH7.4,140-mM NaCl, and 2.5-mM CaCl2 according to themanufacturer’s protocol. The cells were stained simulta-neously with FITC-conjugated annexin Vand PI at roomtemperature (RT) for 15min in the dark prior to additionof binding buffer. The apoptotic cells were measuredusing a FACSCalibur flow cytometer. The cells weresorted into intact cells (Annexin V� PI�), early apoptoticcells (Annexin V+ PI�), late apoptotic cells (AnnexinV+ PI+), and necrotic cells (Annexin V� PI+).
Hoechst staining. For nuclear morphological analysis,cells (1×104 cells per well) were seeded onto cover glassesand treatedwith 25, 50, 75, and 100μg/mL of CEor vehiclealone. Cells were fixed with 3.5% paraformaldehyde for15min at RT. The cells were then washed three times with1× PBS and permeabilized with 1% Triton X-100 andstained with Hoechst 33342 fluorescent dye (0.5μg/mL)for 15min. Nuclear morphology was examined by fluores-cence microscopy (Leica) using a 1000× objective.
DAPI and TUNEL assay. For DAPI staining, HepG2cells were seeded onto cover glasses and were treatedwith various concentrations of CE. After 24-h incubation,the cells were fixed with 37% formaldehyde and 95%
Copyright © 2015 John Wiley & Sons, Ltd.
ethanol (1:4) for 10min at RT. The cells were then washedwith PBS and stained with DAPI (4′,6-diamidino-2-phenylindole). The stained cells were examined througha fluorescence microscope.
TUNEL (Terminal deoxynucleotidyl transferase dUTPnick end labeling) staining was performed using a DNAfragmentation detection kit (QIA33; Calbiochem, SanDiego, CA,USA), according to themanufacturer’s instruc-tions. Briefly, the sections were deparaffinized and per-meabilized with proteinase K and incubated with 3%hydrogen peroxide for 5min, followed by incubation withterminal deoxynucleotidyl transferase enzyme solution for90min at 37°C. The reaction was terminated by incubationin a stop solution for 5min. The peroxidase reaction wasdeveloped with 3-3′-diaminobenzidine tetrahydrochloride(D-5905; Sigma-Aldrich). Counter staining was performedwith hematoxylin before mounting.
Mitochondrial membrane potential assay. Overnightgrown HepG2 cells incubated with vehicle (as control)or in the presence of 25, 50, 75, and 100μg/mL of CEfor 24h at 37 °C in complete media were harvestedand washed with 1× PBS. Washed cells were stainedwith 3,3′-dihexyloxacarbocyanine iodide dye and wereanalyzed by FACSCalibur flow cytometer.
Western blot. Total cell lysates were prepared using lysisbuffer (1-M Tris-HCl [pH8.0], 5-M NaCl, 1% NaN3,10% sodium dodecyl sulfate, 10% NP-40, and 0.5%C24H39NaO4). Cell lysates (50μg of protein) were sepa-rated by 12% polyacrylamide gel and then electropho-retically transferred onto a polyvinylidene membrane.After blocking in Tris-buffered saline-Tween containing5% skimmed milk powder for 30min, each membranewas incubated overnight at 4 °C with primary antibodiesagainst each target protein. After washing five times,the membranes were incubated with horseradishperoxidase-conjugated secondary antibodies, and pro-tein levels were detected using an enhanced chemilumi-nescence reagent (Anigen, Seoul, Korea). The proteinsband were quantify using IMAGEJ software, and thebands were normalized with β-actin proteins.
In vivo experiments. BALB/c nude mice (Samtaco,Osan, Korea) were used for in vivo experiments. Eachcontrol and CE-treated groups consist of three mice,and 2×106 HepG2 cells were injected to the left andright flank regions. When the tumor reaches 100mm3,25 and 50mg/kg of CE were injected three times a weekto the intraperitoneal region for 7weeks. The controlgroup provides vehicle. Tumor mass was measured bythe longest part and shortest part and calculated withthe following formula.
Tumor mass mm3� � ¼ longestð Þ=2� shortestþ 1ð Þ2
Hematoxylin and eosin staining. Xenograft tissues werefixed with 4% paraformaldehyde overnight. Then tis-sues were embedded with paraffin. Paraffin wereremoved from the embedded samples with 100%xylazine and dehydrated with different concentrations
Phytother. Res. (2015)

S. H. LEE ET AL.
of ethanol (95%, 90%, 80%, and 70%). Tissue sampleswere stained with hematoxylin for 3min and placed on0.3% acid alcohol for differentiation. The samples wererinsed with Scott’s tap water prior to exposure to eosinsolution for 3min. After staining with hematoxylinand eosin, tissue samples were dried and protectedwith a cover slide. Samples were then observed undera light microscope.
Statistical analysis. Data are expressed as the mean± standard deviation of at least three independentexperiments, and statistical analysis was carried outusing Student’s t-test, using SPSS version 10.0 forWindows (SPSS, Chicago, IL, USA). The level of statis-tical significance was set at p< 0.05.
RESULTS
Quantification and characterization of flavonoids
Flavonoids were isolated from Citrus aurantium L.using HPLC–MS/MS at the Department of Chemistry,Gyeongsang National University, by Prof. Sung ChulShin. The structures of 14 different flavonoids werecharacterized by retention time, molecular ion mass,and comparison with literature data. The HPLC chro-matogram and chemical structures are shown in Fig. 1.The mass spectral and quantification data are compiledin Table 1. The 14 flavonoids were quantified from thepeak areas of the HPLC chromatogram recorded at280nm. Quantification of these flavonoids was validatedon the basis of a representative flavonoid standardfrom the same group. Thus, flavanones (naringin,hesperidin, and poncirin) and flavones (isosinensetin,hexamethoxyflavone, sinensetin, hexamethoxyflavone,tetramethyl-O-isoscutellarein, nobiletin, heptametho-xyflavone, 3-hydroxynobiletin, tangeretin, hydroxy-pentamethoxyflavone, and hexamethoxyflavone) werequantified using the calibration curves of hesperetinand nobiletin, respectively. The recovery and precisionvalues were above 90% and below 10% for the individ-ual external standards. The calibration curves were
Table 1. Spectral data of the 14 flavonoid compounds in the Citrus aur
No. Retention time (min) MS [M+H]+
1 16.46 5812 18.02 6113 29.56 5954 41.67 3735 42.88 4036 43.40 3737 44.60 4038 45.91 3439 46.52 40310 47.50 43311 48.91 41912 50.78 37313 52.06 38914 52.72 403
Copyright © 2015 John Wiley & Sons, Ltd.
constructed by plotting the concentration of thestandard against the peak area. Plant flavonoids for whichno standards were available have been routinely quan-tified using the calibration curve of structurally relatedcompounds.
CE inhibits tumor growth of xenograft BALB/cnude mice
To evaluate the effect of CE, tumor growth in BALB/cnude mice was measured. Mice tumor was measuredthree times a week. CE treatment slowed tumor growth.As shown in Fig. 2a, the tumor volume was decreased inCE-treated group when compared with the control mice.This result suggests that CE inhibits tumor growth.
To study the histopathological changes in CE-treated/untreated xenograft mice, hematoxylin-and-eosin stain-ing was performed. Control xenograft tissue sampleshows that rapid tumor mass growth contains manymulti-nuclear cells and necrosis of central zone of tissue.However, CE treatment tumor tissue shows that tumormass growth was inhibited by encapsulation. Few base-ment membrane covered tumor tissue, and centraltumor region necrosis was also detected (Fig. 2b).Significant tumor mass difference was the identifiedcause of tumor mass encapsulation. Collectively, theseresults suggest that CE inhibits tumor growth in theBALB/c mice xenograft model.
Effect of CE on HepG2 cell viability and proliferation
To determine the effect of CE in vitro, cytotoxicity ofCE in HepG2 cells was studied. HepG2 cells wereseeded on 12-well plates and treated with various con-centrations (25, 50, 75, and 100μg/mL) of CE or vehiclealone for 24h, and MTT assay was carried out. Cellviability decreases dose dependently with an IC50 valueof 75-μg/mL CE (Fig. 3a). These results were consistentwith the in vivo study suggesting that CE inhibitsHepG2 cell proliferation. Furthermore, the effect ofCE on normal liver cells Thle2 was also determined. Itwas observed that CE did not have any effect on Thle2cells (Fig. 3a).
antium L
Compound Quantity ± SD (mg/kg)
Naringin 299 ± 0.5Hesperidin 210 ± 1.3Poncirin 109 ± 1.1Isosinensetin 30.4 ± 0.1Hexamethoxyflavone 15.5 ± 0.05Sinensetin 19.7 ± 0.1Hexamethoxyflavone 23.6 ± 0.07Tetramethyl-O-isoscutellarein 35.6 ± 0.1Nobiletin 201 ± 0.1Heptamethoxyflavone 169 ± 1.33-Hydroxynobiletin 28.7 ± 0.1Tangeretin 81.5 ± 0.09Hydroxypentamethoxyflavone 12.8 ± 0.04Hexamethoxyflavone 7.0 ± 0.04
Phytother. Res. (2015)

Figure 2. Citrus aurantium (CE) inhibits hepatoblastoma tumor growth in BALB/c nude mice. (a) BALB/c nude mice implanted with HepG2xenografts were treated with CE as indicated for 50 days. Tumor formation was visualized by photograph. (b) Hematoxylin-and-eosin stain-ing of CE-treated or CE-untreated tissue samples of BALB/c xenograft nude mice. This figure is available in color online at wileyonlinelibrary.com/journal/ptr.
CITRUS FLAVONOIDS INDUCE APOPTOSIS IN HEPG2 CELLS
The phosphoinositide-3-kinase/Akt/mammalian tar-get of rapamycin pathway is considered to be a centralregulator of multiple cellular processes, including me-tabolism, proliferation, and survival (Liu et al., 2009).In the present study, it was observed that the level ofpAkt was reduced in CE-treated cells dose dependently,while Akt expression remains almost the same. More-over, CE reduced the phosphorylation of downstreamtargets, 4EBP1 and p70S6K (Fig. 3b). Thus, these re-sults suggest that CE inhibits HepG2 cell proliferationbut has no effect on normal liver cells, Thle2.
Effect of CE on cell cycle progression
To determine the effect of CE on cell cycle progressionof HepG2 cells, flow cytometer analysis was performedat various concentrations of CE (25, 50, 75, and100μg/mL) for 24h. As shown in Fig. 4, CE treatmentremarkably inhibits the cell proliferation of HepG2cells. The sub-G1 phase was increased dose dependentlyfrom 5.60% in untreated control cells to 18.79% and23.74% in cells treated with 75 and 100μg/mL CE,respectively, while the G1 phase decreased dose depen-dently. These results suggest that cell death induced byCE may be apoptosis.
CE induces apoptosis in HepG2 cells
To confirm whether cell death induced by CE is apoptosisor not, annexin V–PI double staining was carried out inHepG2 cells treated with various concentrations of CE.
Copyright © 2015 John Wiley & Sons, Ltd.
It was observed that treatment with 0, 25, 50, 75, and100μg/mL of the CE for 24h progressively decreasedthe proportion of intact cells from 83.31% in untreatedcontrol cells to 57.81% in 100-μg/mL CE-treated HepG2cells (Fig. 5a). In contrast, early apoptotic cells increasedfrom 5.61% to 27.03%, and late apoptotic cells increasedfrom 7.47% to 12.58% in CE-treated cells. These resultsindicate that CE induces apoptosis in HepG2 cells.
To further confirm these results, nuclear assay wascarried out. Treated cells were stained with Hoechststain, and it was observed that the nuclei of CE-treatedcells were condensed, which is indicative of apoptosis(Fig. 5b). DAPI and TUNEL assays were also per-formed. And as shown in Fig. 5c, fragmented DNAwas observed in the CE-treated HepG2 cells. Takentogether, these results suggest that CE induces apopto-sis in HepG2 cells.
CE mediates intrinsic apoptotic pathway in HepG2 cells
Mitochondria play an essential role in the propagation ofapoptosis, and mitochondrial dysfunction usually triggersspecific cellular signaling to induce apoptosis. Increasingevidence suggests that the disruption of mitochondrial in-tegrity is a critical step in cells undergoing apoptosis anda decreasing mitochondrial membrane potential is associ-ated with mitochondrial dysfunction. Therefore, loss ofmitochondrial membrane potential is an important eventduring the mitochondrial-mediated apoptosis. To studythe effect of CE on mitochondrial membrane potential,HepG2 cells untreated or treated with CE for 24h werestained with 3,3′-dihexyloxacarbocyanine iodide dye,
Phytother. Res. (2015)

Figure 3. Effect of Citrus aurantium (CE) on the viability and proliferation of HepG2 cells. (a) HepG2 and Thle2 cells were treated with variousconcentrations of the flavonoids for 24 h. Cell viability was then determined by a 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bro-mide assay. Cell viability is represented as the percentage relative absorbance compared with the controls. (b) HepG2 cells were treated withvarious concentrations of CE, and protein expressions of Akt, P-4E-BP1, and P-p70S6K were determined by western blot.
Figure 4. Effect of Citrus aurantium (CE) on cell cycle distribution in HepG2 cells. Cells treated with vehicle (dimethyl sulfoxide) or indicatedconcentration of CE were collected and assessed by propidium iodide staining. Data represent the mean ± SD of three replicate independentexperiments. The asterisk (*) indicates a significant difference from the control group (*p<0.05). This figure is available in color online atwileyonlinelibrary.com/journal/ptr.
S. H. LEE ET AL.
Copyright © 2015 John Wiley & Sons, Ltd. Phytother. Res. (2015)

Figure 5. Citrus aurantium (CE) induces apoptosis in HepG2 cells. (a) HepG2 cells were treated with indicated concentration of CE for 24 h.Apoptosis was assessed by annexin V–PI double staining. (b) Hoechst 33258 staining of HepG2 cells treated with or without flavonoids CEfor 24 h. Fragmented or condensed nuclei could be observed at 1000× magnification in the flavonoids-treated group as indicated by thearrows. (c) DAPI and TUNEL assays of treated or untreated cells were carried out. Fragmented DNA was observed in CE-treated cells. Thisfigure is available in color online at wileyonlinelibrary.com/journal/ptr.
CITRUS FLAVONOIDS INDUCE APOPTOSIS IN HEPG2 CELLS
and change of fluorescent intensity was assessed by flowcytometer. It was observed that CE treatment signifi-cantly reduces the mitochondrial membrane potential ofHepG2 cells (Fig. 6a).In addition, mitochondrial-related protein expression
was checked by immunoblot. It was observed that theBax/Bcl-xL ratio was significantly increased as the ex-pressions of antiapoptotic proteins Bcl-2 and Bcl-xLwere decreased, while pro-apoptotic proteins Bak andBax expressions were increased dose dependently inCE-treated HepG2 cells (Fig. 6b). Furthermore, asshown in Fig. 7, treatment of HepG2 cells with variousconcentrations of CE (25, 50, 75, and 100μg/mL) for24h resulted in a significant decrease of pro-caspases3, 8, and 9, while active caspases 3 and 9 were signifi-cantly increased, which further leads to the cleavage ofPARP, but protein expression of active caspase 8 wasdecreased in the CE-treated cells. Both Fas and Fas Lprotein expressions were also significantly reduced inCE-treated HepG2 cells. Taken together, these resultssuggest that CE-induced apoptosis in HepG2 cells ismediated through the intrinsic pathway.
Involvement of p38 in CE induced apoptosis
p38 MAPK plays an important role in the regulation ofapoptosis. In order to study the involvement of p38 inCE-induced apoptosis, the protein expression of p38 in
Copyright © 2015 John Wiley & Sons, Ltd.
cells treated with various concentrations of CE wasdetermined by western blot. As shown in Fig. 8, it wasobserved that the levels of p38 were decreased dose de-pendently in the treated cells when compared with theuntreated cells, while the levels of phosphorylated p38were increased in the treated cells. When HepG2 cellswere treated with 100μg/mL of CE for different timeperiods, the protein expression of Pp38 also increasedtime dependently. But CE has no effect on other MAPKfamily proteins – ERK and JNK (results not shown).These results suggest the involvement of p38 in CE-induced apoptosis in HepG2 cells.
DISCUSSION
Plants have a long history in treatment of various dis-eases, and over 60% of the currently used anticancerdrugs are derived from natural resources (Balunasand Kinghorn, 2005). The Citrus species has a promis-ing role in cancer therapy. Many phytochemicals likeflavonoids are richly present in Citrus. In the presentstudy, flavonoids extracted from Citrus aurantium L.have been used to determine its anticancer effect onHepG2 cells.
Fourteen flavonoids have been isolated from C.aurantium. The major flavonoids (based on quantity) werenobiletin, naringin, and hesperidin. These flavonoids have
Phytother. Res. (2015)

Figure 6. Citrus aurantium (CE) induces apoptosis through the mitochondrial pathway in HepG2 cells. (a) Mitochondrial membrane potentialloss induced by CE. CE-treated/untreated cells for 24 h were stained with 3,3′-dihexyloxacarbocyanine iodide stains, and change in fluores-cent intensity was determined by flow cytometer. (b) HepG2 cells were treated with indicated concentrations of CE, and protein expressionsof pro-apoptotic and antiapoptotic proteins Bax, Bak, Bcl, and Bcl-xL were determined by western blot. Data represent the mean ± SD ofthree replicate independent experiments. The asterisk (*) indicates a significant difference from the control group (*p<0.05). This figureis available in color online at wileyonlinelibrary.com/journal/ptr.
S. H. LEE ET AL.
been reported to be involved in cell cycle arrest, apoptosis,and antiproliferation of many cancer cell lines (Lee et al.,2010; Li et al., 2013). In the present study, it was observedthat CE induces cell death of HepG2 cells with an IC50value of 75-μg/mL CE. These results were consistent withthe in vivo study where 25 and 50mg/kg of CE reducedthe tumor growth in a xenograft BALB/c nude micemodel. The sub-G1 cells were increased significantly inthe CE-treated cells. The phosphoinositide-3-kinase/Aktsignaling pathway plays a central role in the control of cellgrowth, proliferation, metabolism, survival, and angiogen-esis (Fasolo and Sessa, 2012; Sheppard et al., 2012). Aktis usually overexpressed in cancer cells. Thus, drugstargeting Akt are considered to be a potential chemother-apeutics approach (Osaki et al., 2004; Yap et al., 2008). Inthe present study, it was observed that phosphorylation ofAkt and downstream targets 4EBP1 and p70S6K weredownregulated in CE-treated cells when compared withthe untreated cells. These results suggest that CE inhibitsHepG2 cell proliferation and induces cell death.Apoptosis is a programmed cell death morphologi-
cally characterized by cell shrinkage and condensationof chromatin leading to pyknosis (Kerr et al., 1972).There are two main apoptotic pathways: the extrinsicor death receptor pathway and the intrinsic or mito-chondrial pathway. The extrinsic pathway is mediated
Copyright © 2015 John Wiley & Sons, Ltd.
by death receptors, which are members of the tumor ne-crosis factor receptor gene superfamily (Locksley et al.,2001), while the intrinsic signaling pathway is mediatedby a diverse array of non-receptor-mediated stimuli thatproduce intracellular signals that act directly on targetswithin the cell. These stimuli cause changes in the innermitochondrial membrane, resulting in loss of mitochon-drial membrane potential and release of pro-apoptoticproteins. Thus, the intrinsic apoptotic pathway is amitochondrial-initiated event (Saelens et al., 2004).Caspases play an important role in both these pathways(Elmore, 2007). In the extrinsic pathway, the death re-ceptors activate the initiator caspase 8, which in turn ac-tivates caspases 3 and 7 directly or either activates Bidprotein, which translocates to the mitochondria and re-leases cytochrome c (Hengartner, 2000). While in the in-trinsic pathway, cellular stress causes the release ofcytochrome c from the mitochondrial inner membraneto the cytosol, Bcl-2 family members (Bax, Bak, andBid) may release cytochrome c, which translocates intothe mitochondria. Apoptosomes, caspase-activatingcomplexes, are then formed, which activates initiatorcaspase 9. Caspase 9 subsequently activates caspases 7and 3 ( Slee et al., 2001). These executioner caspases willfurther cleave PARP, cytokeratins, and other nuclearproteins, which causes morphological and biochemical
Phytother. Res. (2015)

Figure 7. Citrus aurantium (CE) induces caspase-dependent intrinsic apoptosis in HepG2 cells. HepG2 cells were treated with indicated con-centrations of CE, and protein expressions of caspases 3, 8, and 9; poly(ADP-ribose) polymerase (PARP); Fas; and Fas L were determined bywestern blot. Data represent the mean ± SD of three replicate independent experiments. The asterisk (*) indicates a significant differencefrom the control group (*p<0.05).
Figure 8. Regulation of p38 mitogen-activated protein kinase by Citrus aurantium (CE). HepG2 cells were treated with indicated concentra-tion of CE or with 100 μg/mL for the indicated time, and p38 and P-p38 protein expressions were determined by western blot.
CITRUS FLAVONOIDS INDUCE APOPTOSIS IN HEPG2 CELLS
changes in the cells (Slee et al., 2001). In the presentstudy, the protein expressions of pro-caspases 3, 8, and9 were reduced, which consequently increases the activ-ity of caspase 3 in CE-treated cells. This activationfurther leads to PARP cleavage. In addition, the expres-sions of pro-apoptotic proteins Bax and Bak were in-creased, while that of antiapoptotic proteins Bcl-2 andBcl-xL was reduced in the CE-treated cells. It was alsoobserved that CE treatment induces loss of mitochon-drial potential in HepG2 cells. Furthermore, condensed
Copyright © 2015 John Wiley & Sons, Ltd.
nuclei and fragmented DNA were observed in CE-treated HepG2 cells. As caspase 3 is the key execu-tioner caspase, and cleavage of PARP is a biochemicalhallmark of apoptosis, the increase in Bax/Bcl-xLratio, and loss of mitochondrial membrane potentialsuggests that CE induces mitochondrial-mediated apo-ptosis pathway in HepG2 cells.
p38 MAPK acts as tumor suppressor, and its activationaugments apoptosis (Deacon et al., 2003). In many cancercells, it has been reported that activation of p38 promotes
Phytother. Res. (2015)

S. H. LEE ET AL.
apoptosis (Chiu et al., 2008; Shin et al., 2009; Song et al.,2012). Similarly, in the present study, it was observed thatp38 is activated in CE-treated cells, suggesting the involve-ment of p38 in CE-induced apoptosis in HepG2 cells.In conclusion, flavonoids extracted from Citrus
aurantium L. inhibit the proliferation of HepG2 cellsand induce apoptosis by dephosphorylating Akt and af-fecting the level of mitochondrial proteins. The presentstudy highlights the potential use of flavonoids of C.aurantium against hepatoblastoma. Thus, Citrus flavo-noids inducing apoptosis via the intrinsic pathway inHepG2 cells may provide a potential therapeutic rolein the treatment of liver cancer.
Copyright © 2015 John Wiley & Sons, Ltd.
Acknowledgements
This work was supported by a grant from the National ResearchFoundation (NRF) of Korea funded by the Ministry of Science,ICT & Future Planning (nos. 2012M3A9B8019303 and 2012R1A2A2A06045015) and National R&D Program for Cancer Control,Ministry for Health, Welfare and Family Affairs, Republic of Korea(no. 0820050).
Conflict of Interest
The authors have declared that there is no conflict of interest.
REFERENCES
Balunas MJ, Kinghorn AD. 2005. Drug discovery from medicinalplants. Life Sci 78: 431–441.
Benavente-Garcia O, Castillo J. 2008. Update on uses and proper-ties of citrus flavonoids: new findings in anticancer, cardiovas-cular, and anti-inflammatory activity. J Agric Food Chem 56:6185–6205.
Chiu SJ, Chao JI, Lee YJ, Hsu TS. 2008. Regulation of gamma-H2AX and securin contribute to apoptosis by oxaliplatin via ap38 mitogen-activated protein kinase-dependent pathway inhuman colorectal cancer cells. Toxicol Lett 179: 63–70.
Deacon K, Mistry P, Chernoff J, Blank JL, Patel R. 2003. p38Mitogen-activated protein kinase mediates cell death and p21-activated kinase mediates cell survival during chemotherapeuticdrug-induced mitotic arrest. Mol Biol Cell 14: 2071–2087.
Elmore S. 2007. Apoptosis: a review of programmed cell death.Toxicol Pathol 35: 495–516.
Fasolo A, Sessa C. 2012. Targeting mTOR pathways in humanmalignancies. Curr Pharm Des 18: 2766–2777.
Ferlay J, Shin HR, Bray F, Forman D, Mathers C, Parkin DM. 2010.Estimates of worldwide burden of cancer in 2008:GLOBOCAN 2008. Int J Cancer 127: 2893–2917.
Havsteen B. 1983. Flavonoids, a class of natural products of highpharmacological potency. Biochem Pharmacol 32: 1141–1148.
Hengartner MO. 2000. The biochemistry of apoptosis. Nature407: 770–776.
Kamaraj S, Ramakrishnan G, Anandakumar P, Jagan S, Devaki T.2009. Antioxidant and anticancer efficacy of hesperidin inbenzo(a)pyrene induced lung carcinogenesis in mice. InvestNew Drugs 27: 214–222.
Kasper HU, Longerich T, Stippel DL, Kern MA, Drebber U,Schirmacher P. 2005. Mixed hepatoblastoma in an adult. ArchPathol Lab Med 129: 234–237.
Kerr JF, Wyllie AH, Currie AR. 1972. Apoptosis: a basic biologicalphenomenon with wide-ranging implications in tissue kinetics.Br J Cancer 26: 239–257.
Kim SO, Han J. 2001. Pan-caspase inhibitor zVAD enhances celldeath in RAW246.7macrophages. J EndotoxinRes7: 292–296.
Lee CJ, Wilson L, Jordan MA, Nguyen V, Tang J, Smiyun G. 2010.Hesperidin suppressed proliferations of both human breastcancer and androgen-dependent prostate cancer cells.Phytother Res 24(Suppl 1): S15–S19.
Lee DH, Park KI, Park HS, et al. 2012. Flavonoids Isolated fromKorea Citrus aurantium L. induce G2/M phase arrest and apo-ptosis in human gastric cancer AGS cells. Evid Based Comple-ment Alternat Med 2012: 515901.
Li Y, Fang H, Xu W. 2007. Recent advance in the research offlavonoids as anticancer agents. Mini Rev Med Chem 7:663–678.
Li H, Yang B, Huang J, et al. 2013. Naringin inhibits growth poten-tial of human triple-negative breast cancer cells by targetingbeta-catenin signaling pathway. Toxicol Lett 220: 219–228.
Liu P, Cheng H, Roberts TM, Zhao JJ. 2009. Targeting thephosphoinositide 3-kinase pathway in cancer. Nat Rev DrugDiscov 8: 627–644.
Locksley RM, Killeen N, Lenardo MJ. 2001. The TNF and TNFreceptor superfamilies: integrating mammalian biology. Cell104: 487–501.
Luo G, Guan X, Zhou L. 2008. Apoptotic effect of citrus fruitextract nobiletin on lung cancer cell line A549 in vitro andin vivo. Cancer Biol Ther 7: 966–973.
Newman DJ, Cragg GM, Snader KM. 2003. Natural products assources of new drugs over the period 1981–2002. J Nat Prod66: 1022–1037.
Olsson M, Zhivotovsky B. 2011. Caspases and cancer. Cell DeathDiffer 18: 1441–1449.
Osaki M, Oshimura M, Ito H. 2004. PI3K-Akt pathway: itsfunctions and alterations in human cancer. Apoptosis 9:667–676.
Ren W, Qiao Z, Wang H, Zhu L, Zhang L. 2003. Flavonoids: prom-ising anticancer agents. Med Res Rev 23: 519–534.
Saelens X, Festjens N, Vande Walle L, van Gurp M, van Loo G,Vandenabeele P. 2004. Toxic proteins released frommitochon-dria in cell death. Oncogene 23: 2861–2874.
Sheppard K, Kinross KM, Solomon B, Pearson RB, Phillips WA.2012. Targeting PI3 kinase/AKT/mTOR signaling in cancer.Crit Rev Oncog 17: 69–95.
Shin DY, Lee WS, Lu JN, et al. 2009. Induction of apoptosis inhuman colon cancer HCT-116 cells by anthocyanins throughsuppression of Akt and activation of p38-MAPK. Int J Oncol35: 1499–1504.
Slee EA, Adrain C, Martin SJ. 2001. Executioner caspase-3, -6,and -7 perform distinct, non-redundant roles during the demo-lition phase of apoptosis. J Biol Chem 276: 7320–7326.
Song H, Wohltmann M, Tan M, Bao S, Ladenson JH, Turk J. 2012.Group VIA PLA2 (iPLA2beta) is activated upstream of p38mitogen-activated protein kinase (MAPK) in pancreatic isletbeta-cell signaling. J Biol Chem 287: 5528–5541.
Yap TA, Garrett MD, Walton MI, Raynaud F, de Bono JS, WorkmanP. 2008. Targeting the PI3K-AKT-mTOR pathway: progress,pitfalls, and promises. Curr Opin Pharmacol 8: 393–412.
Phytother. Res. (2015)





























