Embed Size (px)
Citation preview

lable at ScienceDirect
Polymer 52 (2011) 1107e1115
Contents lists avai
Polymer
journal homepage: www.elsevier .com/locate/polymer
Formation and reorganization of the mesophase of random copolymersof propylene and 1-butene
Daniela Mileva a, René Androsch a,*, Evgeny Zhuravlev b, Christoph Schick b, Bernhard Wunderlich c
aMartin-Luther-University Halle-Wittenberg, Center of Engineering Sciences, D-06099 Halle/Saale, GermanybUniversity of Rostock, Institute of Physics, D-18051 Rostock, Germanyc 200 Baltusrol Rd., Knoxville, TN 379234-37-7, USA
a r t i c l e i n f o
Article history:Received 4 December 2010Received in revised form7 January 2011Accepted 7 January 2011Available online 15 January 2011
Keywords:Poly(propyleneerane1-butene)MesophaseFast scanning chip calorimetry
* Corresponding author. Tel.: þ49 3461 46 3762; faE-mail address: [email protected] (R.
0032-3861/$ e see front matter � 2011 Elsevier Ltd.doi:10.1016/j.polymer.2011.01.021
a b s t r a c t
Fast scanning chip calorimetry (FSC) has been employed to study the kinetics of formation of themesophase of random copolymers of propylene and 1-butene from the glassy amorphous state and itsreorganization on heating. The experiments performed consistently prove a distinct decrease of the rateof mesophase formation with increasing concentration of 1-butene chain defects. The time required forisothermal mesophase formation at 300 K is of the order of 0.1 s in case of the homopolymer, while it isprolonged by one order of magnitude to 1 s in the copolymer with 11 mol-% 1-butene. Similar, cold-ordering of amorphous structure on continuous heating at 1000 K s�1 is only completed in the homo-polymer while it is almost completely suppressed in the random copolymer containing about 11 mol-%1-butene. The perfection of the mesophase and/or its reorganization into crystals is faster in thehomopolymer than in copolymers containing 1-butene. The critical heating rate for complete inhibitionof perfection and reorganization is reduced from about 40,000 K s�1 in the homopolymer to about10,000 K s�1 in copolymers. The reduced rate of mesophase formation in random copolymers ofpropylene and 1-butene is attributed to the decrease of the thermodynamic driving force for the phasetransformation.
� 2011 Elsevier Ltd. All rights reserved.
1. Introduction
Crystallization of the isotropic and quiescent melt of isotacticpolypropylene (iPP) at low supercooling leads to direct formation ofcross-hatched monoclinic lamellae which are arranged in a spher-ulitic superstructure [1e6]. Alternatively, the melt of iPP can becrystallized via intermediate formation of a metastable mesophase/conformationally disordered glass (CD glass) [7e9] at highsupercooling, and its subsequent reorganization into crystals onheating [10e15]. The crystallization via the mesophase leads toa qualitatively different semicrystalline structure than the directcrystallization from the liquid state since the crystals obtainedare of non-lamellar shape and not organized in a higher-orderspherulitic superstructure [13e18]. As a consequence, ultimateproperties of semicrystalline iPP crystallized along these differentthermodynamic pathways are largely different [19e21]. Semi-crystalline preparations of iPP containing cross-hatched lamellaeand spherulites are opaque and rather non-ductile while iPP crys-tallized via the mesophase is highly transparent and rather ductile.
x: þ49 3461 46 3891.Androsch).
All rights reserved.
The present study is related to the crystallization of iPP via themesophase, which in the following is introduced in more detail.
The conditions for the formation of the mesophase and itstransformation into crystals were quantitatively evaluated recentlyin the case of the iPP homopolymer. The mesophase forms at highsupercooling of the melt at temperatures (T) between about 330 Kand the glass transition temperature of the amorphous phase (T g) atabout 260 K [22e25]. In order to suppress the liquide crystal phasetransformation at low supercooling, the melt needs to be cooledrapidly at a rate faster than about 100K s�1 [17,24e27]. If the coolingrate is lower, then spherulitic crystallization occurs at temperatureshigher than 350 K and mesophase formation at low temperaturegets increasingly reduced with decreasing cooling rate. Isothermalanalysis of the kinetics of the liquid e mesophase transformationrevealed a half-time of the ordering process of less than a second atambient temperature [22,23]. Though the mesophase formation isfast, it can be suppressed by cooling the melt faster than 1000 K s�1
toT< T g [25]. Subsequent devitrification of the glass byheating thenallows cold-ordering/mesophase formation between T g and about350 K if the heating rate is lower than 10,000 K s�1 [28]. The liquide
mesophase transition at high supercooling is incomplete even onlong-term storage at ambient temperature, i.e., quenched prepara-tions of iPP are heterogeneous and contain about 25e50% [12,27,29]

D. Mileva et al. / Polymer 52 (2011) 1107e11151108
isometricmesophase particles of size of 5e20nm [14e18,30,31] andamorphousphase. Theamorphousphase in semimesomorphic iPP ispartially restrained and immobilized due to covalent coupling to themesophase, i.e., the amorphous phase contains mobile (MAF) andrigid amorphous fractions (RAF)with corresponding glass transitiontemperatures T g, MAF (270 K) and T g, RAF (>T g, MAF), respectively [8].The mesophase is of lower packing density than the monocliniccrystalline phase [27,32] and contains helix reversals as mostimportant conformational defect [8]. At ambient temperature, themesophase is below its own glass transition temperature (T g, Meso-
phase), i.e., the removal of helix reversals to achieve crystallineregistry of molecular stems requires heating of the mesophase toT> T g, Mesophase, with T g, Mesophase > T g, RAF. The mesophasee crystalphase transition has frequently been analyzed by temperature-resolved X-ray diffraction [10e12,15,33] and occurs in a widetemperature range starting at about 350K, being related to the onsetof helix mobility as detected by solid state NMR [34]. It has beensuggested that the mesophase e crystal phase transition is a localprocess within the ordered domains, either proceeding by helixrewinding or by a chain translation mechanism [35e39]. The mes-ophase e crystal phase transition is a thermodynamically irrevers-ible process toward equilibrium, obeys specific kinetics, and can besuppressed by heating at rates faster than about 20,000 K s�1 intothe temperature rangeof the equilibrium liquid, i.e., to temperatureshigher than the equilibrium melting temperature [28]. In this case,complete disordering/isotropization of the mesophase occurs atabout 350 K. An extensive review of the structure, stability andreorganization of the mesophase of iPP has been published recently[40].
In the present work, we focus on the evaluation of the change ofthe kinetics of mesophase formation in iPP as a result of randominsertion of 1-butene co-units into the chain. Random isotacticcopolymers of propylene and 1-butene (iPP-But) are commerciallyimportant polymeric materials, which were developed to broadenthe range of application or the processability of the iPP homopol-ymer [1,41,42]. Significant research has been performed to describestructure formation of iPP-But copolymers from solution [43], orfrom the melt at low supercooling [44e52] while studies to gaininformation about the crystallization process via intermediateformation of a metastable mesophase at high supercooling are rare.The application-related potential of iPP-But copolymers crystal-lized along this specific thermodynamic pathway has beendemonstrated [53,54], and initial investigations of the kinetics ofboth the liquid e mesophase and mesophase e crystal phasetransitions have been performed [55e57]. In detail, it has beenshown by non-isothermal fast scanning chip calorimetry (FSC) andby investigation of the structure of samples cooled at different ratethat the critical cooling rate, required to suppress crystallization atlow supercooling, slightly decreases with increasing concentrationof 1-butene in the chain [57]. This result is in agreement with theearlier observation of a decreased maximum rate of crystallizationat low supercooling [48]. Analysis of the fine structure of themesophase of iPP-But copolymers revealed that the 1-butene chaindefects are trapped in themesophase, and the concentrations of co-units in the amorphous phase and the mesophase are probablyidentical [58]. Heating of the mesophase to T > Tg, Mesophase allowsits reorganization to crystals with the co-units, initially trapped inthe mesophase, kept inside the crystals. The temperature of reor-ganization of the mesophase into crystals on slow heatingdecreases with increasing concentration of 1-butene chain defects,which has been attributed to a lowering of the glass transitiontemperature of the mesophase. Further information about thekinetics of formation and reorganization of the mesophase asa function of the copolymer composition are not available, which istherefore primary object of this work.
Summarizing the intention of the present work, we attempt togain further quantitative information about the kinetics of meso-phase formation and reorganization in iPP-But random copolymers.To the best of our knowledge, for random iPP-But copolymers, dataabout the kinetics of isothermal mesophase formation at ambienttemperature, the kinetics of non-isothermal mesophase formation/cold-ordering on heating, and the kinetics of reorganization ofmesophase into crystals are not available. We believe that theseinformation are necessary to further complete the knowledgeabout the various pathways of crystallization of the important classof propylene-based polymers. In addition, and perhaps moreimportantly, the data observed may allow general conclusionsabout the effect of the variation of the molecular structure/intro-duction of chain irregularities on the crystallization behavior atconditions far from thermodynamic equilibrium, if quantified bythe supercooling. The importance of such research from bothpolymer science and polymer engineering points of view has beenrecognized and resulted in organization of special seminars like the9th Lähnwitz-Seminar on Calorimetry “Transitions far from Equi-librium e Super-heating; Super-cooling” in 2006 [59], and a work-shop on “Polymer Crystallization under Conditions Relevant toProcessing” in 2010 [60]. In order to achieve the goal of the presentwork, we employed the new experimental technique of fast scan-ning chip calorimetry (FSC). First instruments were developed inthe 1990’s to be used under quasi-adiabatic conditions for fastheating up to 107 K s�1 [61,62]. Further utilization of this techniquefor research of polymer crystallization at fast cooling requiredoperation at non-adiabatic conditions, which has been achievedonly less than 10 years ago [63,64]. The particular technique of non-adiabatic FSC needed to be used since mesophase formation at highsupercooling, and cold-ordering and reorganization during heatingoccur at a time scale of few milliseconds (ms), being too fast to bemonitored by standard differential scanning calorimetry (DSC). Assuch the present study is also performed to further establish anddemonstrate the capabilities of the novel instrumental technique ofFSC for analysis of phase transitions in polymeric materials.
2. Experimental section
For the present study we used an iPP homopolymer witha mass-average molar mass and polydispersity of 373 kDa and 6.2,respectively [65]. Isotactic poly(propyleneerane1-butene) sampleswith a concentration of 6.0 and 10.9 mol-% 1-butene (iPP-But.6 andiPP-But.11) and a molar mass of 225 kDa and polydispersity of 3.1were purchased from SigmaeAldrich [66,67]. The as-receivedmaterials were compression-molded at 473 K to films of 100 mmthickness in a Collin press, using a rate of 10 Kmin�1 for cooling thefilms to ambient temperature. For subsequent FSC analyses,samples of 10e15 mm thickness and a lateral dimension of theorder of 50 mm were prepared by microscope-aided cutting. Thespecimens were placed on a thin film chip sensor XI 395 (XensorIntegration, Netherlands), with the thermal contact between thesensor and the specimen improved by Apiezon� N grease. We usedthe recently developed dual-sensor instrument setup which allowsrecording of the electrical power difference between the sampleand reference calorimeters according to the programmed tem-peratureetime profile, and subsequent conversion into anapparent sample heat capacity [68,69]. The determination of aninstrumental baseline, required to correct for temperature-dependent different losses of heat to the environment at thesample and reference sites, has been performed as recommendedin the literature [68,69]. It included the measurement of the powerdifference as a function of temperature during heating and coolingat identical rates in presence of the sample of interest, and theconstruction of a baseline based on averaging heating- and
D. Mileva et al. / Polymer 52 (2011) 1107e1115 1109
cooling-data at temperatures outside of phase transitions andinterpolation in regions of phase transitions using 2nd or 3rd orderpolynomials. While calibration of the power signal was notrequired, the measurement of temperature considering thethermal lag as a function of heating/cooling rate was checked usingthe melting temperature of indium, with the indium placed on topof a sample, to consider the thermal resistance of the latter. Inorder to achieve high cooling rate, the encapsulated measuringsystem was inserted into liquid nitrogen, and the furnace waspurged using cold, gaseous nitrogen. The mass m of the sampleswas estimated by comparing the measured total heat capacity Cp attemperatures higher than the melting temperature with specificheat capacity data cp (¼ Cp/m) available in the ATHAS data base[70]. The sample mass was of the order of 10 ng.
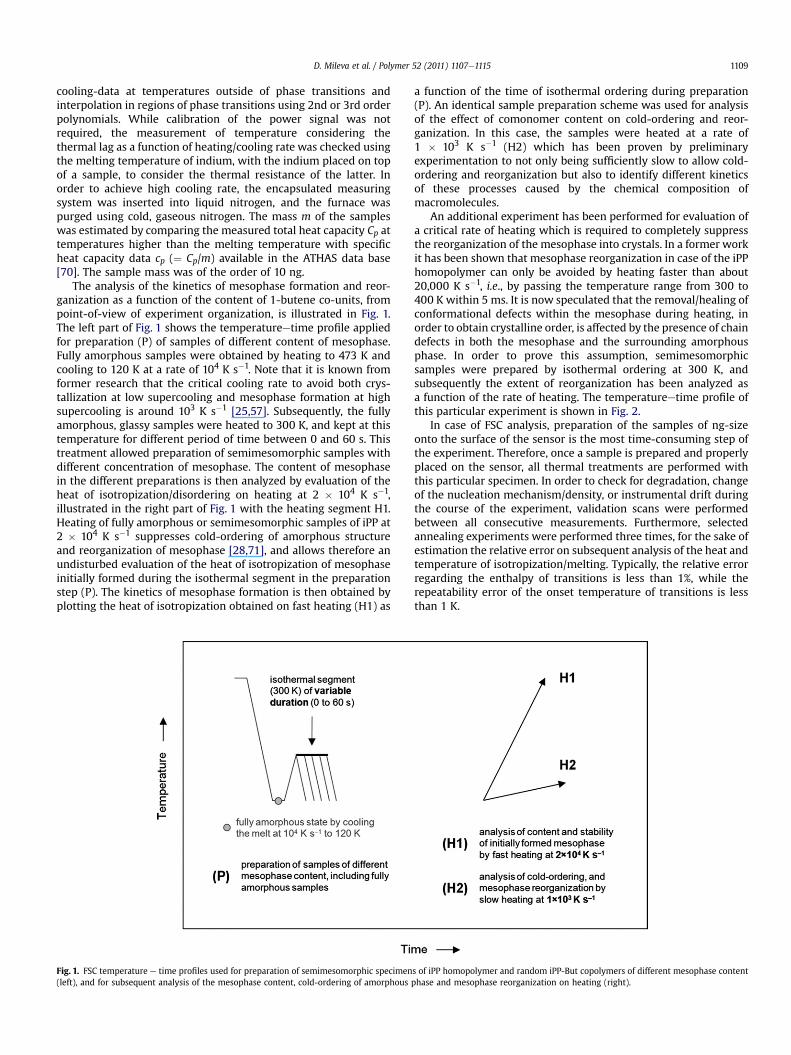
The analysis of the kinetics of mesophase formation and reor-ganization as a function of the content of 1-butene co-units, frompoint-of-view of experiment organization, is illustrated in Fig. 1.The left part of Fig. 1 shows the temperatureetime profile appliedfor preparation (P) of samples of different content of mesophase.Fully amorphous samples were obtained by heating to 473 K andcooling to 120 K at a rate of 104 K s�1. Note that it is known fromformer research that the critical cooling rate to avoid both crys-tallization at low supercooling and mesophase formation at highsupercooling is around 103 K s�1 [25,57]. Subsequently, the fullyamorphous, glassy samples were heated to 300 K, and kept at thistemperature for different period of time between 0 and 60 s. Thistreatment allowed preparation of semimesomorphic samples withdifferent concentration of mesophase. The content of mesophasein the different preparations is then analyzed by evaluation of theheat of isotropization/disordering on heating at 2 � 104 K s�1,illustrated in the right part of Fig. 1 with the heating segment H1.Heating of fully amorphous or semimesomorphic samples of iPP at2 � 104 K s�1 suppresses cold-ordering of amorphous structureand reorganization of mesophase [28,71], and allows therefore anundisturbed evaluation of the heat of isotropization of mesophaseinitially formed during the isothermal segment in the preparationstep (P). The kinetics of mesophase formation is then obtained byplotting the heat of isotropization obtained on fast heating (H1) as
Fig. 1. FSC temperature e time profiles used for preparation of semimesomorphic specimen(left), and for subsequent analysis of the mesophase content, cold-ordering of amorphous
a function of the time of isothermal ordering during preparation(P). An identical sample preparation scheme was used for analysisof the effect of comonomer content on cold-ordering and reor-ganization. In this case, the samples were heated at a rate of1 � 103 K s�1 (H2) which has been proven by preliminaryexperimentation to not only being sufficiently slow to allow cold-ordering and reorganization but also to identify different kineticsof these processes caused by the chemical composition ofmacromolecules.
An additional experiment has been performed for evaluation ofa critical rate of heating which is required to completely suppressthe reorganization of the mesophase into crystals. In a former workit has been shown that mesophase reorganization in case of the iPPhomopolymer can only be avoided by heating faster than about20,000 K s�1, i.e., by passing the temperature range from 300 to400 K within 5 ms. It is now speculated that the removal/healing ofconformational defects within the mesophase during heating, inorder to obtain crystalline order, is affected by the presence of chaindefects in both the mesophase and the surrounding amorphousphase. In order to prove this assumption, semimesomorphicsamples were prepared by isothermal ordering at 300 K, andsubsequently the extent of reorganization has been analyzed asa function of the rate of heating. The temperatureetime profile ofthis particular experiment is shown in Fig. 2.
In case of FSC analysis, preparation of the samples of ng-sizeonto the surface of the sensor is the most time-consuming step ofthe experiment. Therefore, once a sample is prepared and properlyplaced on the sensor, all thermal treatments are performed withthis particular specimen. In order to check for degradation, changeof the nucleation mechanism/density, or instrumental drift duringthe course of the experiment, validation scans were performedbetween all consecutive measurements. Furthermore, selectedannealing experiments were performed three times, for the sake ofestimation the relative error on subsequent analysis of the heat andtemperature of isotropization/melting. Typically, the relative errorregarding the enthalpy of transitions is less than 1%, while therepeatability error of the onset temperature of transitions is lessthan 1 K.
s of iPP homopolymer and random iPP-But copolymers of different mesophase contentphase and mesophase reorganization on heating (right).
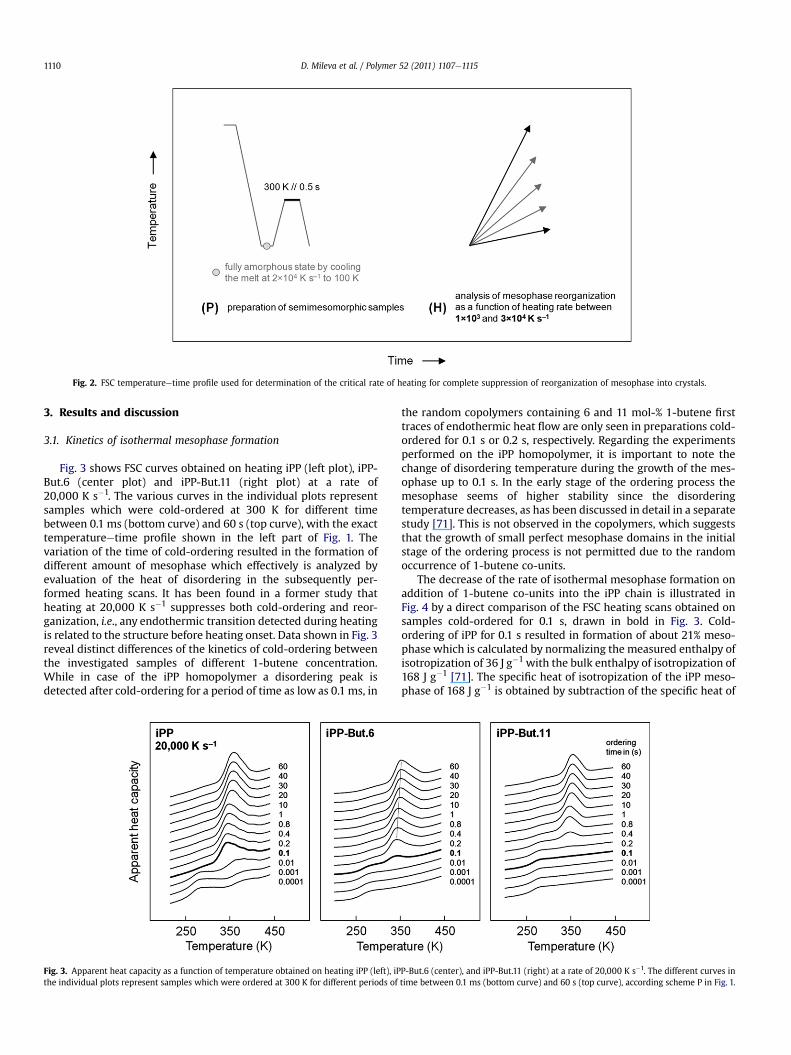
Fig. 2. FSC temperatureetime profile used for determination of the critical rate of heating for complete suppression of reorganization of mesophase into crystals.
D. Mileva et al. / Polymer 52 (2011) 1107e11151110
3. Results and discussion
3.1. Kinetics of isothermal mesophase formation
Fig. 3 shows FSC curves obtained on heating iPP (left plot), iPP-But.6 (center plot) and iPP-But.11 (right plot) at a rate of20,000 K s�1. The various curves in the individual plots representsamples which were cold-ordered at 300 K for different timebetween 0.1 ms (bottom curve) and 60 s (top curve), with the exacttemperatureetime profile shown in the left part of Fig. 1. Thevariation of the time of cold-ordering resulted in the formation ofdifferent amount of mesophase which effectively is analyzed byevaluation of the heat of disordering in the subsequently per-formed heating scans. It has been found in a former study thatheating at 20,000 K s�1 suppresses both cold-ordering and reor-ganization, i.e., any endothermic transition detected during heatingis related to the structure before heating onset. Data shown in Fig. 3reveal distinct differences of the kinetics of cold-ordering betweenthe investigated samples of different 1-butene concentration.While in case of the iPP homopolymer a disordering peak isdetected after cold-ordering for a period of time as low as 0.1 ms, in
Fig. 3. Apparent heat capacity as a function of temperature obtained on heating iPP (left), iPthe individual plots represent samples which were ordered at 300 K for different periods of
the random copolymers containing 6 and 11 mol-% 1-butene firsttraces of endothermic heat flow are only seen in preparations cold-ordered for 0.1 s or 0.2 s, respectively. Regarding the experimentsperformed on the iPP homopolymer, it is important to note thechange of disordering temperature during the growth of the mes-ophase up to 0.1 s. In the early stage of the ordering process themesophase seems of higher stability since the disorderingtemperature decreases, as has been discussed in detail in a separatestudy [71]. This is not observed in the copolymers, which suggeststhat the growth of small perfect mesophase domains in the initialstage of the ordering process is not permitted due to the randomoccurrence of 1-butene co-units.
The decrease of the rate of isothermal mesophase formation onaddition of 1-butene co-units into the iPP chain is illustrated inFig. 4 by a direct comparison of the FSC heating scans obtained onsamples cold-ordered for 0.1 s, drawn in bold in Fig. 3. Cold-ordering of iPP for 0.1 s resulted in formation of about 21% meso-phase which is calculated by normalizing the measured enthalpy ofisotropization of 36 J g�1 with the bulk enthalpy of isotropization of168 J g�1 [71]. The specific heat of isotropization of the iPP meso-phase of 168 J g�1 is obtained by subtraction of the specific heat of
P-But.6 (center), and iPP-But.11 (right) at a rate of 20,000 K s�1. The different curves intime between 0.1 ms (bottom curve) and 60 s (top curve), according scheme P in Fig. 1.
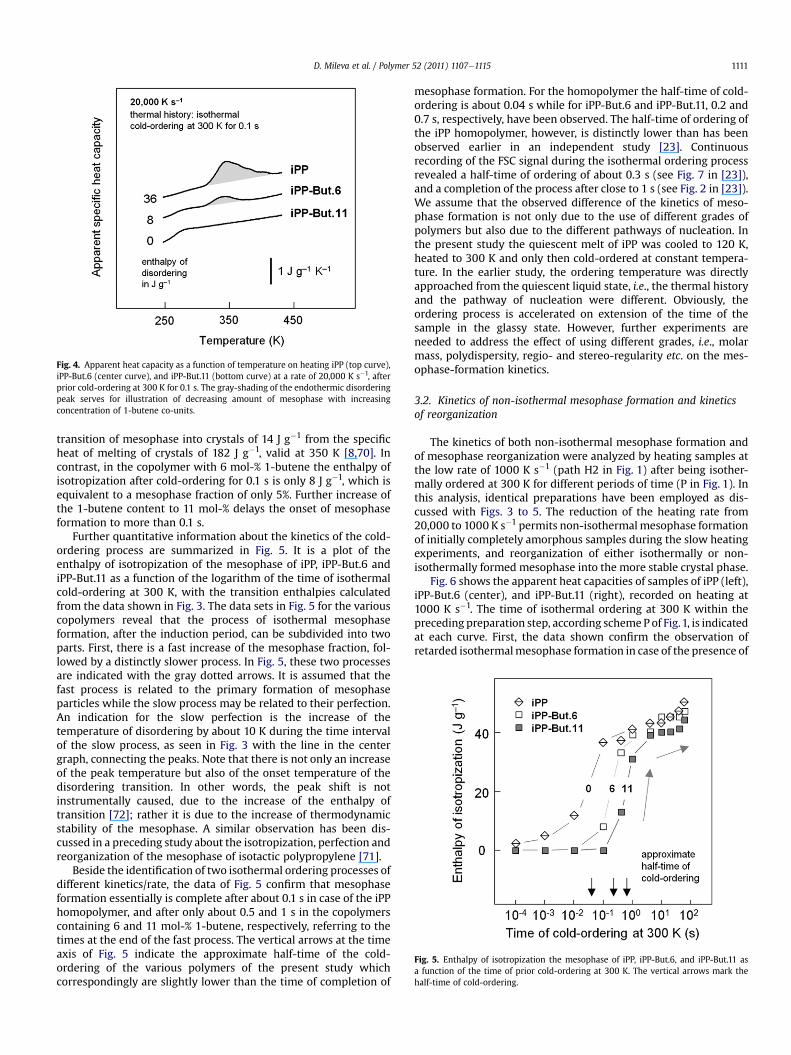
Fig. 4. Apparent heat capacity as a function of temperature on heating iPP (top curve),iPP-But.6 (center curve), and iPP-But.11 (bottom curve) at a rate of 20,000 K s�1, afterprior cold-ordering at 300 K for 0.1 s. The gray-shading of the endothermic disorderingpeak serves for illustration of decreasing amount of mesophase with increasingconcentration of 1-butene co-units.
Fig. 5. Enthalpy of isotropization the mesophase of iPP, iPP-But.6, and iPP-But.11 asa function of the time of prior cold-ordering at 300 K. The vertical arrows mark thehalf-time of cold-ordering.
D. Mileva et al. / Polymer 52 (2011) 1107e1115 1111
transition of mesophase into crystals of 14 J g�1 from the specificheat of melting of crystals of 182 J g�1, valid at 350 K [8,70]. Incontrast, in the copolymer with 6 mol-% 1-butene the enthalpy ofisotropization after cold-ordering for 0.1 s is only 8 J g�1, which isequivalent to a mesophase fraction of only 5%. Further increase ofthe 1-butene content to 11 mol-% delays the onset of mesophaseformation to more than 0.1 s.
Further quantitative information about the kinetics of the cold-ordering process are summarized in Fig. 5. It is a plot of theenthalpy of isotropization of the mesophase of iPP, iPP-But.6 andiPP-But.11 as a function of the logarithm of the time of isothermalcold-ordering at 300 K, with the transition enthalpies calculatedfrom the data shown in Fig. 3. The data sets in Fig. 5 for the variouscopolymers reveal that the process of isothermal mesophaseformation, after the induction period, can be subdivided into twoparts. First, there is a fast increase of the mesophase fraction, fol-lowed by a distinctly slower process. In Fig. 5, these two processesare indicated with the gray dotted arrows. It is assumed that thefast process is related to the primary formation of mesophaseparticles while the slow process may be related to their perfection.An indication for the slow perfection is the increase of thetemperature of disordering by about 10 K during the time intervalof the slow process, as seen in Fig. 3 with the line in the centergraph, connecting the peaks. Note that there is not only an increaseof the peak temperature but also of the onset temperature of thedisordering transition. In other words, the peak shift is notinstrumentally caused, due to the increase of the enthalpy oftransition [72]; rather it is due to the increase of thermodynamicstability of the mesophase. A similar observation has been dis-cussed in a preceding study about the isotropization, perfection andreorganization of the mesophase of isotactic polypropylene [71].
Beside the identification of two isothermal ordering processes ofdifferent kinetics/rate, the data of Fig. 5 confirm that mesophaseformation essentially is complete after about 0.1 s in case of the iPPhomopolymer, and after only about 0.5 and 1 s in the copolymerscontaining 6 and 11 mol-% 1-butene, respectively, referring to thetimes at the end of the fast process. The vertical arrows at the timeaxis of Fig. 5 indicate the approximate half-time of the cold-ordering of the various polymers of the present study whichcorrespondingly are slightly lower than the time of completion of
mesophase formation. For the homopolymer the half-time of cold-ordering is about 0.04 s while for iPP-But.6 and iPP-But.11, 0.2 and0.7 s, respectively, have been observed. The half-time of ordering ofthe iPP homopolymer, however, is distinctly lower than has beenobserved earlier in an independent study [23]. Continuousrecording of the FSC signal during the isothermal ordering processrevealed a half-time of ordering of about 0.3 s (see Fig. 7 in [23]),and a completion of the process after close to 1 s (see Fig. 2 in [23]).We assume that the observed difference of the kinetics of meso-phase formation is not only due to the use of different grades ofpolymers but also due to the different pathways of nucleation. Inthe present study the quiescent melt of iPP was cooled to 120 K,heated to 300 K and only then cold-ordered at constant tempera-ture. In the earlier study, the ordering temperature was directlyapproached from the quiescent liquid state, i.e., the thermal historyand the pathway of nucleation were different. Obviously, theordering process is accelerated on extension of the time of thesample in the glassy state. However, further experiments areneeded to address the effect of using different grades, i.e., molarmass, polydispersity, regio- and stereo-regularity etc. on the mes-ophase-formation kinetics.
3.2. Kinetics of non-isothermal mesophase formation and kineticsof reorganization
The kinetics of both non-isothermal mesophase formation andof mesophase reorganization were analyzed by heating samples atthe low rate of 1000 K s�1 (path H2 in Fig. 1) after being isother-mally ordered at 300 K for different periods of time (P in Fig. 1). Inthis analysis, identical preparations have been employed as dis-cussed with Figs. 3 to 5. The reduction of the heating rate from20,000 to 1000 K s�1 permits non-isothermalmesophase formationof initially completely amorphous samples during the slow heatingexperiments, and reorganization of either isothermally or non-isothermally formed mesophase into the more stable crystal phase.
Fig. 6 shows the apparent heat capacities of samples of iPP (left),iPP-But.6 (center), and iPP-But.11 (right), recorded on heating at1000 K s�1. The time of isothermal ordering at 300 K within thepreceding preparation step, according scheme P of Fig.1, is indicatedat each curve. First, the data shown confirm the observation ofretarded isothermalmesophase formation in case of the presence of
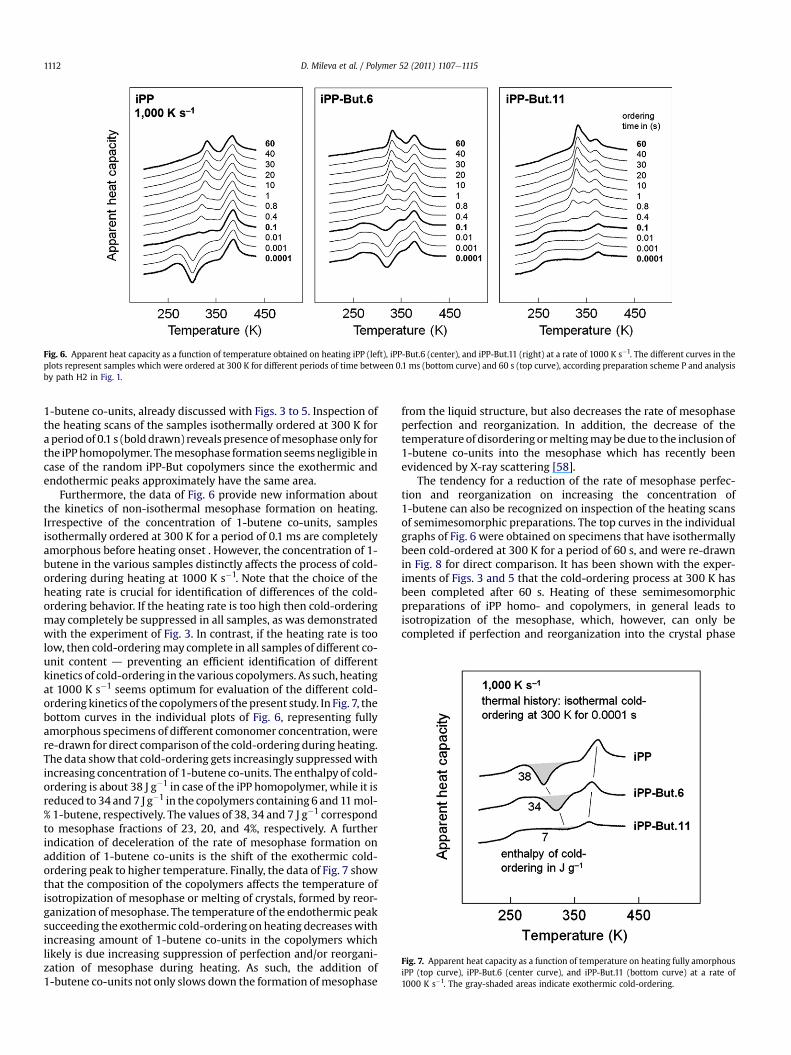
Fig. 6. Apparent heat capacity as a function of temperature obtained on heating iPP (left), iPP-But.6 (center), and iPP-But.11 (right) at a rate of 1000 K s�1. The different curves in theplots represent samples which were ordered at 300 K for different periods of time between 0.1 ms (bottom curve) and 60 s (top curve), according preparation scheme P and analysisby path H2 in Fig. 1.
Fig. 7. Apparent heat capacity as a function of temperature on heating fully amorphousiPP (top curve), iPP-But.6 (center curve), and iPP-But.11 (bottom curve) at a rate of1000 K s�1. The gray-shaded areas indicate exothermic cold-ordering.
D. Mileva et al. / Polymer 52 (2011) 1107e11151112
1-butene co-units, already discussed with Figs. 3 to 5. Inspection ofthe heating scans of the samples isothermally ordered at 300 K fora periodof 0.1 s (bolddrawn) reveals presence ofmesophase only forthe iPPhomopolymer. Themesophase formation seemsnegligible incase of the random iPP-But copolymers since the exothermic andendothermic peaks approximately have the same area.
Furthermore, the data of Fig. 6 provide new information aboutthe kinetics of non-isothermal mesophase formation on heating.Irrespective of the concentration of 1-butene co-units, samplesisothermally ordered at 300 K for a period of 0.1 ms are completelyamorphous before heating onset . However, the concentration of 1-butene in the various samples distinctly affects the process of cold-ordering during heating at 1000 K s�1. Note that the choice of theheating rate is crucial for identification of differences of the cold-ordering behavior. If the heating rate is too high then cold-orderingmay completely be suppressed in all samples, as was demonstratedwith the experiment of Fig. 3. In contrast, if the heating rate is toolow, then cold-orderingmay complete in all samples of different co-unit content d preventing an efficient identification of differentkinetics of cold-ordering in the various copolymers. As such, heatingat 1000 K s�1 seems optimum for evaluation of the different cold-ordering kinetics of the copolymers of the present study. In Fig. 7, thebottom curves in the individual plots of Fig. 6, representing fullyamorphous specimens of different comonomer concentration, werere-drawn for direct comparison of the cold-ordering during heating.The data show that cold-ordering gets increasingly suppressedwithincreasing concentration of 1-butene co-units. The enthalpy of cold-ordering is about 38 J g�1 in case of the iPP homopolymer, while it isreduced to 34 and 7 J g�1 in the copolymers containing 6 and 11mol-% 1-butene, respectively. The values of 38, 34 and 7 J g�1 correspondto mesophase fractions of 23, 20, and 4%, respectively. A furtherindication of deceleration of the rate of mesophase formation onaddition of 1-butene co-units is the shift of the exothermic cold-ordering peak to higher temperature. Finally, the data of Fig. 7 showthat the composition of the copolymers affects the temperature ofisotropization of mesophase or melting of crystals, formed by reor-ganization ofmesophase. The temperature of the endothermic peaksucceeding the exothermic cold-ordering on heating decreaseswithincreasing amount of 1-butene co-units in the copolymers whichlikely is due increasing suppression of perfection and/or reorgani-zation of mesophase during heating. As such, the addition of1-butene co-units not only slows down the formation ofmesophase
from the liquid structure, but also decreases the rate of mesophaseperfection and reorganization. In addition, the decrease of thetemperatureof disorderingormeltingmaybedue to the inclusion of1-butene co-units into the mesophase which has recently beenevidenced by X-ray scattering [58].
The tendency for a reduction of the rate of mesophase perfec-tion and reorganization on increasing the concentration of1-butene can also be recognized on inspection of the heating scansof semimesomorphic preparations. The top curves in the individualgraphs of Fig. 6 were obtained on specimens that have isothermallybeen cold-ordered at 300 K for a period of 60 s, and were re-drawnin Fig. 8 for direct comparison. It has been shown with the exper-iments of Figs. 3 and 5 that the cold-ordering process at 300 K hasbeen completed after 60 s. Heating of these semimesomorphicpreparations of iPP homo- and copolymers, in general leads toisotropization of the mesophase, which, however, can only becompleted if perfection and reorganization into the crystal phase
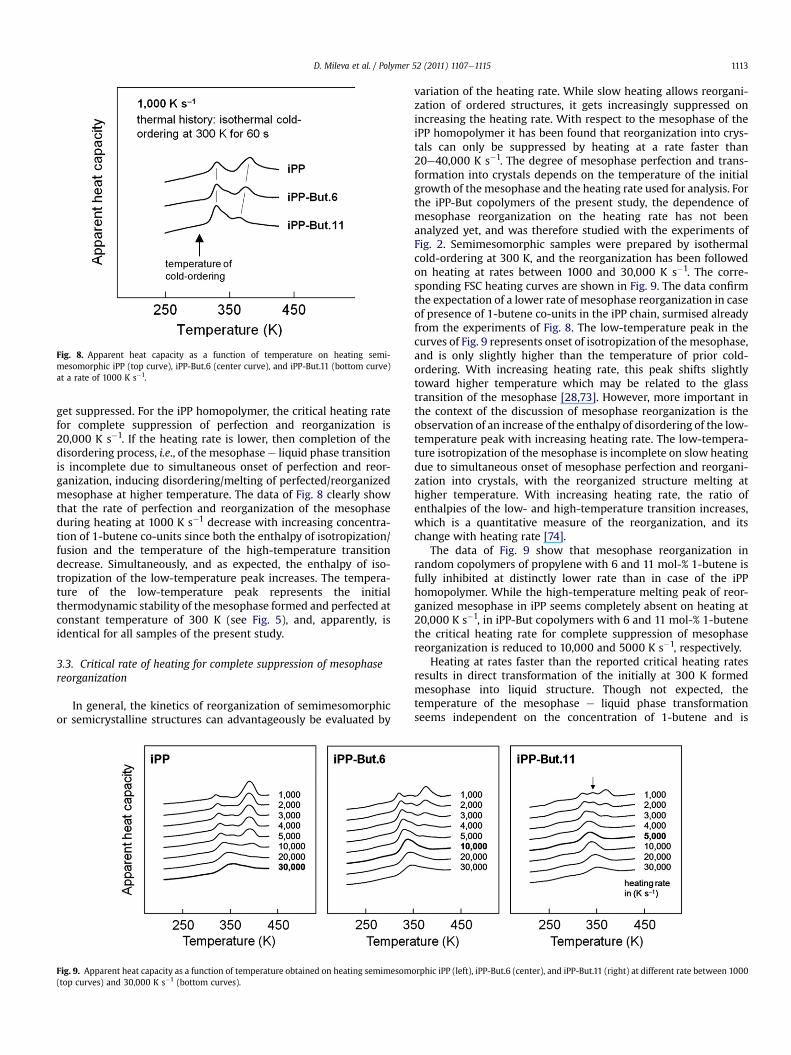
Fig. 8. Apparent heat capacity as a function of temperature on heating semi-mesomorphic iPP (top curve), iPP-But.6 (center curve), and iPP-But.11 (bottom curve)at a rate of 1000 K s�1.
D. Mileva et al. / Polymer 52 (2011) 1107e1115 1113
get suppressed. For the iPP homopolymer, the critical heating ratefor complete suppression of perfection and reorganization is20,000 K s�1. If the heating rate is lower, then completion of thedisordering process, i.e., of the mesophasee liquid phase transitionis incomplete due to simultaneous onset of perfection and reor-ganization, inducing disordering/melting of perfected/reorganizedmesophase at higher temperature. The data of Fig. 8 clearly showthat the rate of perfection and reorganization of the mesophaseduring heating at 1000 K s�1 decrease with increasing concentra-tion of 1-butene co-units since both the enthalpy of isotropization/fusion and the temperature of the high-temperature transitiondecrease. Simultaneously, and as expected, the enthalpy of iso-tropization of the low-temperature peak increases. The tempera-ture of the low-temperature peak represents the initialthermodynamic stability of themesophase formed and perfected atconstant temperature of 300 K (see Fig. 5), and, apparently, isidentical for all samples of the present study.
3.3. Critical rate of heating for complete suppression of mesophasereorganization
In general, the kinetics of reorganization of semimesomorphicor semicrystalline structures can advantageously be evaluated by
Fig. 9. Apparent heat capacity as a function of temperature obtained on heating semimesom(top curves) and 30,000 K s�1 (bottom curves).
variation of the heating rate. While slow heating allows reorgani-zation of ordered structures, it gets increasingly suppressed onincreasing the heating rate. With respect to the mesophase of theiPP homopolymer it has been found that reorganization into crys-tals can only be suppressed by heating at a rate faster than20e40,000 K s�1. The degree of mesophase perfection and trans-formation into crystals depends on the temperature of the initialgrowth of the mesophase and the heating rate used for analysis. Forthe iPP-But copolymers of the present study, the dependence ofmesophase reorganization on the heating rate has not beenanalyzed yet, and was therefore studied with the experiments ofFig. 2. Semimesomorphic samples were prepared by isothermalcold-ordering at 300 K, and the reorganization has been followedon heating at rates between 1000 and 30,000 K s�1. The corre-sponding FSC heating curves are shown in Fig. 9. The data confirmthe expectation of a lower rate of mesophase reorganization in caseof presence of 1-butene co-units in the iPP chain, surmised alreadyfrom the experiments of Fig. 8. The low-temperature peak in thecurves of Fig. 9 represents onset of isotropization of the mesophase,and is only slightly higher than the temperature of prior cold-ordering. With increasing heating rate, this peak shifts slightlytoward higher temperature which may be related to the glasstransition of the mesophase [28,73]. However, more important inthe context of the discussion of mesophase reorganization is theobservation of an increase of the enthalpy of disordering of the low-temperature peak with increasing heating rate. The low-tempera-ture isotropization of the mesophase is incomplete on slow heatingdue to simultaneous onset of mesophase perfection and reorgani-zation into crystals, with the reorganized structure melting athigher temperature. With increasing heating rate, the ratio ofenthalpies of the low- and high-temperature transition increases,which is a quantitative measure of the reorganization, and itschange with heating rate [74].
The data of Fig. 9 show that mesophase reorganization inrandom copolymers of propylene with 6 and 11 mol-% 1-butene isfully inhibited at distinctly lower rate than in case of the iPPhomopolymer. While the high-temperature melting peak of reor-ganized mesophase in iPP seems completely absent on heating at20,000 K s�1, in iPP-But copolymers with 6 and 11 mol-% 1-butenethe critical heating rate for complete suppression of mesophasereorganization is reduced to 10,000 and 5000 K s�1, respectively.
Heating at rates faster than the reported critical heating ratesresults in direct transformation of the initially at 300 K formedmesophase into liquid structure. Though not expected, thetemperature of the mesophase e liquid phase transformationseems independent on the concentration of 1-butene and is
orphic iPP (left), iPP-But.6 (center), and iPP-But.11 (right) at different rate between 1000

D. Mileva et al. / Polymer 52 (2011) 1107e11151114
approximately 350 K on heating at 30,000 K s�1. Initially weexpected a decrease not only of the critical rate of heating forcomplete suppression of reorganization but also of the temperatureof the mesophase e liquid phase transformation, based on theknowledge of incorporation of 1-butene co-units into the meso-phase, [58] decreasing their thermodynamic stability. However,assessing the thermodynamic stability of the mesophase asa function of comonomer concentration, as defined by intersectionof the free-enthalpy e temperature curves, would require analysisof the equilibrium transition temperature. In the present study, themesophase in all samples of different co-unit concentration wasformed at identical temperature of 300 K, though differentrespective supercooling below the equilibrium melting tempera-ture. As such it is not surprising that the disordering temperature inall copolymers is unchanged.
Finally, careful viewing of the data of Fig. 9 suggests annealing/perfection of mesophase, as is indicated by detection of a smallendothermic peak on heating at low rate, following the low-temperature isotropization peak of initially formedmesophase (seearrow in the right plot). On fast heating, the annealing/perfection ofmesophase apparently is reduced, and is then only recognized asa shoulder. The observation of a discrete annealing peak oncontinuous heating may display the maximum degree of perfectionof themesophase, ormay be due to interruption of the annealing bythe shallow mesophase e crystal transition exotherm, precedingthe final melting peak.
4. Conclusions
In the present study, the effect of presence of 1-butene co-unitsin the iPP macromolecule on the kinetics of structure formation athigh supercooling and the kinetics of temperature-triggered reor-ganization has been evaluated. It has been shown that mesophaseformation at high supercooling is distinctly slower in random iPP-But copolymers than in the iPP homopolymer, with the decrease ofthe rate of both isothermal and non-isothermal mesophaseformation being dependent on the concentration of 1-butene chaindefects (see Figs. 4, 5 and 7). The finding of a decreased rate ofordering at high supercooling is in line with independent obser-vation of a lowered rate of crystallization at low supercooling. Thedecrease of both the rate of crystallization at low supercooling andthe rate of mesophase formation at high supercooling, however,cannot primarily be attributed to the selection of moleculesegments free of constitutional defects at the growth front of theordered phase. X-ray analyses revealed incorporation of 1-buteneco-units into the crystalline phase and into the mesophase,apparently without partitioning between ordered and amorphousphases [58]. It is therefore suggested that the decrease of the (gross)rate of crystallization and rate of mesophase formation in iPP-Butcopolymers, compared to the iPP homopolymer, is mainly due tothe decrease of the thermodynamic driving force, i.e., the differenceof the bulk free enthalpies of parent and daughter phases of theordering process. In random copolymers, the free enthalpy of theliquid phase is lower than that of the homopolymer at identicaltemperature (otherwise, there would be phase separation), whichreduces the free-enthalpy difference between liquid and orderedphases and the equilibrium melting temperature [75]. For iPP-based random copolymers, the decrease of the equilibriummeltingtemperature has been discussed and seems experimentally proven[55,76e78], and the link between the rate of phase transformationand the free-enthalpy difference between parent and daughterphases is well documented with the TurnbulleFisher equation [79].The advance of the present work, however, seems the proof thatheterogeneous nucleation evident on crystallization at low super-cooling, and homogeneous nucleation, assumed to be the dominant
nucleation mechanism on mesophase formation at high super-cooling, obviously obey identical rules regarding the relationshipbetween the thermodynamic driving force and the rate of phasetransformation. Despite this result is expected from the classicalnucleation and crystallization theory [80,81], experimentalevidence is rare since homogenous nucleation in bulk samplesbarely can be assessed regarding its kinetics with standardanalytics. With the present work, the experimental tool of fastscanning chip calorimetry has been proven to be outstandingbeneficial for quantitative analysis of the kinetics of homogeneousnucleation/crystallization at high supercooling of iPP-based mate-rials, finally providing valuable data for further establishment ofcorrelations between the chemical architecture of macromoleculesand structure formation from the quiescent liquid state.
Acknowledgments
The authors gratefully acknowledge financial support by theDeutsche Forschungsgemeinschaft (DFG). EZ acknowledges finan-cial support by a European Union funded Marie Curie ESTfellowship.
References
[1] Pasquini N. Polypropylene handbook. Munich: Carl Hanser Verlag; 2005.[2] Natta G, Corradini P. Structure and properties of isotactic polypropylene.
Nuovo Cimento Suppl 1960;15:40e51.[3] Hock CW. Morphology of polypropylene crystallized from the melt. J Polym
Sci Part A 1966;4:227e42.[4] Binsbergen FL, De Lange BGM. Morphology of polypropylene crystallized from
the melt. Polymer 1968;9:23e40.[5] Norton DR, Keller A. The spherulitic and lamellar morphology of melt-crys-
tallized isotactic polypropylene. Polymer 1985;26:704e16.[6] Olley RH, Bassett DC. On the development of polypropylene spherulites.
Polymer 1989;30:399e409.[7] Wunderlich B, Grebowicz J. Thermotropic mesophases and mesophase tran-
sitions of linear, flexible macromolecules. Adv Polym Sci 1984;60:1e59.[8] Grebowicz J, Lau SF, Wunderlich B. The thermal properties of polypropylene.
J Polym Sci Symp 1984;71:19e37.[9] Wu ZQ, Dann VL, Cheng SZD, Wunderlich B. Fast DSC applied to the crystal-
lization of polypropylene. J Therm Anal 1988;34:105e14.[10] Zannetti R, Celotti G, Fichera A, Francesconi R. The structural effects of
annealing time and temperature on the paracrystalecrystal transition inisotactic polypropylene. Makromol Chem 1969;128:137e42.
[11] O’Kane WJ, Young RJ, Ryan AJ, Bras W, Derbyshire GE, Mant GR. SimultaneousSAXS/WAXS and d.s.c. analysis of the melting and recrystallization behaviourof quenched polypropylene. Polymer 1994;35:1352e8.
[12] Konishi T, Nishida K, Kanaya T. Crystallization of isotactic polypropylene fromprequenched mesomorphic phase. Macromolecules 2006;39:8035e40.
[13] Grubb DT, Yoon DY. Morphology of quenched and annelaed isotatic poly-propylene. Polym Commun 1986;27:84e8.
[14] Hsu CC, Geil PH, Miyaji H, Asai K. Structure and properties of polypropylenecrystallized from the glassy state. J Polym Sci Polym Phys 1986;24:2379e401.
[15] Wang ZG, Hsiao BS, Srinivas S, Brown GM, Tsou AH, Cheng SZD, et al. Phasetransformation in quenched mesomorphic isotactic polypropylene. Polymer2001;42:7561e6.
[16] Ogawa T, Miyaji H, Asai K. Nodular structure of polypropylene. J Phys Soc JpnLett 1985;54:3668e70.
[17] Zia Q, Androsch R, Radusch HJ, Piccarolo S. Morphology, reorganization, andstability of mesomorphic nanocrystals in isotactic polypropylene. Polymer2006;47:8163e72.
[18] Zia Q, Radusch HJ, Androsch R. Direct analysis of nodular crystals in isotacticpolypropylene by atomic force microscopy, and its correlation with calori-metric data. Polymer 2007;48:3504e11.
[19] Gezovich DM, Geil PH. Deformation and aging of quenched polypropylene.Polym Eng Sci 1968;8:210e5.
[20] Zia Q, Androsch R, Radusch HJ. Effect of structure at the micrometer andnanometer length scales on the light transmission of isotactic polypropylene.J Appl Polym Sci 2010;117:1013e20.
[21] Zia Q, Radusch HJ, Androsch R. Deformation behavior of isotactic poly-propylene crystallized via a mesophase. Polym Bull 2009;63:755e71.
[22] Silvestre C, Cimmino S, Duraccio D, Schick C. Isothermal crystallization ofisotactic poly(propylene) studied by superfast calorimetry. Macromol RapComm 2007;28:875e81.
[23] De Santis F, Adamovsky S, Titomanlio G, Schick C. Isothermal nanocalorimetryof isotactic polypropylene. Macromolecules 2007;40:9026e31.

D. Mileva et al. / Polymer 52 (2011) 1107e1115 1115
[24] Gradys A, Sajkiewicz P, Minakov AA, Adamovsky S, Schick C, Hashimoto T,et al. Crystallization of polypropylene at various cooling rates. Mat Sci Eng2005;A413eA414:442e6.
[25] De Santis F, Adamovsky S, Titomanlio G, Schick C. Scanning nanocalorimetryat high cooling rate of isotactic polypropylene. Macromolecules2006;39:2562e7.
[26] Piccarolo S. Morphological changes in isotactic polypropylene as a function ofcooling rate. J Macromol Sci Phys 1992;B31:501e11.
[27] Piccarolo S, Alessi S, Brucato V, Titomanlio G. Crystallization behaviour at highcooling rates of two polypropylenes. In: Dosier M, editor. Crystallization ofpolymers. Dordrecht: Kluwer; 1993. p. 475e80.
[28] Mileva D, Androsch R, Zhuravlev E, Schick C. The temperature of melting ofthe mesophase of isotactic polypropylene. Macromolecules 2009;42:7275e8.
[29] Farrow GJ. Measurement of the smectic content in undrawn polypropylenefilaments. J Appl Polym Sci 1965;9:1227e32.
[30] Zia Q, Androsch R, Radusch HJ, Ingolic E. Crystal morphology of rapidly cooledisotactic polypropylene: a comparative study by TEM and AFM. Polym Bull2008;60:791e8.
[31] Zia Q, Androsch R. Effect of atomic force microscope tip geometry on theevaluation of the crystal size of semicrystalline polymers. Meas Sci Technol2009;20. 097003. p. 4.
[32] Natta G. Progress in stereospecific polymerization. Makromol Chem1960;35:94e131.
[33] Androsch R, Wunderlich B. Reversible crystallization and melting at the lateralsurface of isotactic polypropylene crystals. Macromolecules 2001;34:5950e60.
[34] Schaefer D, Spiess HW, Suter UW, Fleming WW. Two-dimensional solid-stateNMR studies of ultraslow chain motion: glass transition in atactic poly(propylene) versus helical jumps in isotactic poly(propylene). Macromole-cules 1990;23:3431e9.
[35] De Rosa C. Temperature dependence of intramolecular disorder in the high-temperature phase of poly(tetrafluoroethylene) (phase I). Macromolecules1988;21:1174e6.
[36] Watanabe J, Okamoto S, Satoh K, Sakajiri K, Furuya H, Abe A. Reversible helix-helix transition of poly(b-phenylpropyl L-aspartate) involving a screw-senseinversion in the solid state. Macromolecules 1996;29:7084e8.
[37] Lotz B, Mathieu C, Thierry A, Lovinger AJ, Rosa C, De Ruiz de Ballesteros O, et al.Chirality constraints in crystal�crystal transformations: isotactic poly(1-butene) versus syndiotactic polypropylene. Macromolecules 1998;31:9253e7.
[38] Wang C, Hsieh TC, Cheng YW. Solution-electrospun isotactic polypropylenefibers: processing and microstructure development during stepwise anneal-ing. Macromolecules 2010;43:9022e9.
[39] Androsch R. In situ atomic force microscopy of the mesomorphic-monoclinicphase transition in isotactic polypropylene. Macromolecules 2008;41:533e5.
[40] Androsch R, Di Lorenzo ML, Schick C, Wunderlich B. Mesophases in poly-ethylene, polypropylene and poly(1-butene). Polymer 2010;51:4639e62.
[41] De Rosa C, Auriemma F, Ruiz de Ballesteros O, Resconi L, Camurati I. Tailoringthe physical properties of isotactic polypropylene through incorporation ofcomonomers and the precise control of stereo and regioregularity by metal-locene catalysts. Chem Mater 2007;19:5122e30.
[42] Maier C, Calafut T. Polypropylene: the definite user’s guide and data book.Norwich: Plastics design library by William Andrew; 1998.
[43] Cavallo C, Martuscelli E, Pracella M. Properties of solution grown crystals ofisotactic propylene/butene-1 copolymers. Polymer 1977;18:42e8.
[44] Turner-Jones A. Cocrystallization in copolymers of a-olefins IIdButene-1copolymers and polybutene type II/I crystal phase transition. Polymer1966;7:23e59.
[45] Gou Q, Li H, Yu Z, Chen E, Zhang Y, Yan S. Crystallization behavior ofa propylene-1-butene random copolymer in its a and g modifications. ColloidPolym Sci 2007;285:1149e55.
[46] Marega C, Marigo A, Saini R, Ferrari P. The influence of thermal treatment andprocessing on the structure and morphology of poly(propylene-ran-1-butene)copolymers. Polym Int 2001;50:442e8.
[47] De Rosa C, Auriemma F, Ruiz de Ballesteros O, Resconi L, Camurati I. Crys-tallization behavior of isotactic propyleneeethylene and propyleneebutenecopolymers: effect of comonomers versus stereodefects on crystallizationproperties of isotactic polypropylene. Macromolecules 2007;40:6600e16.
[48] Jeon K, Palza H, Quijada R, Alamo RG. Effect of comonomer type on thecrystallization kinetics and crystalline structure of random isotactic propylene1-alkene copolymers. Polymer 2009;50:832e44.
[49] Hosier IL, Alamo RG, Lin JS. Lamellar morphology of random metallocenepropylene copolymers studied by atomic force microscopy. Polymer2004;45:3441e55.
[50] Hosier IL, Alamo RG, Esteso P, Isasi JR, Mandelkern L. Formation of the a and gpolymorphs in random metalloceneepropylene copolymers. Effect ofconcentration and type of comonomer. Macromolecules 2003;36:5623e36.
[51] Hosoda S, Hori H, Yada K, Nakahara S, Tsuji M. Degree of comonomer inclu-sion into lamella crystal for propylene/olefin copolymers. Polymer2002;43:7451e60.
[52] Abiru T, Mizuno A, Weigand F. Microstructural characterization of propylene-butene-1 copolymer using temperature rising elution fractionation. J ApplPolym Sci 1998;68:1493e501.
[53] Mileva D, Androsch R, Radusch HJ. Effect of structure on light transmission inisotactic polypropylene and random propylene-1-butene copolymers. PolymBull 2009;62:561e71.
[54] Mileva D, Zia Q, Androsch R. Tensile properties of random copolymers ofpropylene with ethylene and 1-butene: effect of crystallinity and crystal habit.Polym Bull 2010;65:623e34.
[55] Mileva D, Androsch R, Radusch HJ. Effect of cooling rate on melt-crystalliza-tion of random propylene-ethylene and propylene-1-butene copolymers.Polym Bull 2008;61:643e54.
[56] Mileva D, Zia Q, Androsch R, Radusch HJ, Piccarolo S. Mesophase formation inpoly(propylene-ran-1-butene) by rapid cooling. Polymer 2009;50:5482e9.
[57] Mileva D, Androsch R, Zhuravlev E, Schick C. Critical rate of cooling forcomplete suppression of crystallization of random copolymers of propylenewith ethylene and 1-butene. Thermochim Acta 2009;492:67e72.
[58] Mileva D, Androsch R, Funari SS, Wunderlich B. X-ray study of crystallizationof random copolymers of propylene and 1-butene via a mesophase. Polymer2010;51:5212e20.
[59] 9th Lähnwitz seminar on calorimetry “Transitions far from Equilibrium eSuper-heating; Super-cooling”, May 29 e June 1, 2006, Rostock, Germany(organized by Schick C.) see also Thermochim Acta; 2009, 492 (1e2).
[60] Workshop on “Polymer Crystallization under Conditions Relevant to Pro-cessing”, May 27e28, 2010, Genova, Italy (organized by Alfonso GC, PetersGWM).
[61] Efremov MY, Olson EA, Zhang M, Schiettekatte F, Zhang ZS, Allen LH. Ultra-sensitive, fast, thin-film differential scanning calorimeter. Rev Sci Inst2004;75:179e91.
[62] Allen LH, Ramanath G, Lai SL, Ma Z, Lee S, Allman DDJ, et al. 1,000,000 C/s thinfilm electrical heater: in situ resistivity measurements of Al and Ti/Si thinfilms during ultra rapid thermal annealing. Appl Phys Lett 1994;64:417e9.
[63] Adamovsky SA, Minakov AA, Schick C. Scanning microcalorimetry at highcooling rate. Thermochim Acta 2003;403:55e63.
[64] Minakov AA, Schick C. Ultrafast thermal processing and nanocalorimetry atheating and cooling rates up to 1 MK/s. Rev Sci Inst 2007;78:073902e10.
[65] Private communication, Basell Bayreuth Chemie GmbH, 2001.[66] Sigma Aldrich, product information.[67] Private communication, Linz, Borealis, 2008.[68] Zhuravlev E, Schick C. Fast scanning power compensated differential scanning
nano-calorimeter: 1. The device. Thermochim Acta 2010;505:11e3.[69] Zhuravlev E, Schick C. Fast scanning power compensated differential scanning
nano-calorimeter: 2. Heat capacity analysis. Thermochim Acta 2010;505:14e21.[70] Advanced thermal analysis system ATHAS, with its data bank available in the
internet at http://athas.prz.rzeszow.pl.[71] Mileva D, Androsch R, Zhuravlev E, Schick C, Wunderlich B. Isotropization,
perfection and reorganization of the mesophase of isotactic polypropylene.Thermochim Acta; 2010, in press.
[72] Hemminger WF, Cammenga HK. Methoden der Thermischen Analyse. Berlin:Springer; 1989.
[73] Minakov AA, Wurm A, Schick C. Superheating in linear polymers studied byultrafast nanocalorimetry. Eur Phys J E 2007;23:43e53.
[74] Minakov AA, Mordvintsev DA, Schick C. Melting and reorganization of poly(ethylene terephthalate) on fast heating (1,000 K/s). Polymer 2004;45:3755e63.
[75] Flory PJ. Theory of crystallization in copolymers. Trans Faraday Soc1955;51:848e57.
[76] Mezghani K, Phillips PJ. g-phase in propylene copolymers at atmosphericpressure. Polymer 1995;36:2407e11.
[77] Alamo RG, Ghosal A, Chatterjee J, Thompson KL. Linear growth rates ofrandom propylene ethylene copolymers. The changeover from g dominatedgrowth to mixed (a þ g) polymorphic growth. Polymer 2005;46:8774e89.
[78] Jeon K, Chiari YL, Alamo RG. Maximum rate of crystallization and morphologyof random propylene ethylene copolymers as a function of comonomercontent up to 21 mol%. Macromolecules 2008;41:95e108.
[79] Turnbull D, Fisher JC. Rate of nucleation in condensed systems. J Chem Phys1949;17:71e3.
[80] Hoffmann JD, Davis GT, Lauritzen JI. The rate of crystallization of linearpolymers with chain folding. In: Hannay HB, editor. Crystalline and non-crystalline solids. Treatise on solid state chemistry, vol. 3. New York: PlenumPress; 1976.
[81] Wunderlich B. Macromolecular physics. In: Crystal nucleation, growth,annealing, vol 2. New York: Academic Press; 1976.





























