Embed Size (px)
Citation preview

stp
vwafia
A
vLGLOpDTGU(k
m
Franceschetti Hereditary Recurrent Corneal Erosion
WALTER LISCH, ANTHONY J. BRON, FRANCIS L. MUNIER, DANIEL F. SCHORDERET, LEILA TIAB,CLEMENS LANGE, PARYKSHIT SAIKIA, THOMAS REINHARD, JAYNE S. WEISS, ENKEN GUNDLACH,
UWE PLEYER, CHRISTINA LISCH, AND CLAUDIA AUW-HAEDRICH
n
twoaicrpfsfwsneat
dtctisfii
itifit
● PURPOSE: To describe new affected individuals of France-chetti’s original pedigree of hereditary recurrent erosion ando classify a unique entity called Franceschetti corneal dystro-hy.
● DESIGN: Observational case series.● METHODS: Slit-lamp examination of 10 affected indi-iduals was conducted. Biomicroscopic examinationsere supplemented by peripheral corneal biopsy in 1
ffected patient with corneal haze. Tissue was processedor light and electron microscopy and immunohistochem-stry was performed. DNA analysis was carried out in 12ffected and 3 nonaffected family members.
● RESULTS: All affected individuals suffered from severeocular pain in the first decade of life, attributable to recurrentcorneal erosions. Six adult patients developed bilateral diffusesubepithelial opacifications in the central and paracentral cor-nea. The remaining 4 affected individuals had clear corneas inthe pain-free stage of the disorder. Histologic and immunohis-tochemical examination of the peripheral cornea in a singlepatient showed a subepithelial, avascular pannus. There wasnegative staining with Congo red. DNA analysis excludedmutations in the transforming growth factor beta–induced(TGFBI) gene and in the tumor-associated calcium signaltransducer 2 (TACSTD2) gene.● CONCLUSION: We have extended the pedigree of France-schetti corneal dystrophy and elaborated its natural history onthe basis of clinical examinations. A distinctive feature is theappearance of subepithelial opacities in adult life, accompaniedby a decreased frequency of recurrent erosion attacks. Itsclinical features appear to distinguish it from most other formsof dominantly inherited recurrent corneal erosion reported inthe literature. (Am J Ophthalmol 2012;153:1073–1081.© 2012 by Elsevier Inc. All rights reserved.)
Supplemental Material available at AJO.com.ccepted for publication Dec 19, 2011.From the Department of Ophthalmology, Johannes Gutenberg Uni-
ersity Mainz, Mainz, Germany (W.L.); Clinical Research Unit, Nuffieldaboratory of Ophthalmology, Oxford, United Kingdom (A.J.B.); Julesonin Eye Hospital, Department of Ophthalmology, University ofausanne, Lausanne, Switzerland (F.L.M.); Institut de Recherche enphtalmologie Sion, Sion, the University of Lausanne, and the École
olytechnique fédérale de Lausanne, Lausanne, Switzerland (F.L.M.,.F.S., L.T.); University Eye Hospital Freiburg, Freiburg, Germany (C.L.,.R., E.G., C.A.-H.); Private Ophthalmological Office Hanau, Hanau,ermany (P.S., Ch.L.); Department of Ophthalmology, Louisiana Stateniversity Health Science Center New Orleans, New Orleans, Louisiana
J.S.W.); and Department of Ophthalmology, Campus Virchow-Klini-um, Charité Universitaetsmedizin Berlin, Berlin, Germany (U.P.).Inquiries to Walter Lisch, Kurt-Blaum-Platz 8, 63450 Hanau, Ger-
sany; e-mail: [email protected]
© 2012 BY ELSEVIER INC. A0002-9394/$36.00doi:10.1016/j.ajo.2011.12.011
R ECURRENT CORNEAL EROSION IS A CONDITION OF
repeated, acute, severe eye pain attributable todefective epithelial adherence and recurrent cor-
eal epithelial separation.1 The severity of the pain reflectsthat the cornea has one of the richest sensory innervationsof the body.2 The condition is most commonly post-raumatic, following a glancing injury to the cornea, inhich case attacks are unilateral and confined to theriginal site of corneal damage. In most instances, in thebsence of infection, each attack recovers without signif-cant subepithelial corneal scarring. Less commonly, theondition is bilateral and occurs either spontaneously or inesponse to negligible trauma. In this instance a “dystro-hic” basis may be considered, although evidence of aamily history is often lacking. In recent decades, in olderubjects this presentation has been recognized as a mani-estation of epithelial basement membrane dystrophy,herein characteristic bilateral biomicroscopic features
uch as map/dot/fingerprint disorder and superficial bleb,et, and mare’s tail changes are accompanied by recurrentrosion attacks in 1 or both eyes.3,4 At least some of thesere genetically determined by a mutation affecting theransforming growth factor beta–induced (TGFBI) gene.5
Attacks of recurrent erosion are a well-recognized featureof the phenotypically distinct, major TGFBI corneal dys-trophies, as well as in Fuchs dystrophy;4,6 additionally,there have been a relatively small number of reports ofinherited recurrent corneal erosion occurring as a dystro-phy in its own right.7–18 All of the recurrent erosionisorders, whether traumatic or dystrophic, are presumedo be attributable to a defect in the epithelial adhesionomplex, which normally ensures a tight adhesion betweenhe basal cells of the corneal epithelium and the underly-ng basal lamina and Bowman layer. These attachmenttructures consist of the hemidesmosomes, the anchoringbrils of type VII collagen, and the basal lamina contain-ng collagens type IV and VII, laminin, and fibronectin.19
The International Committee for Classification of Cor-neal Dystrophies (IC3D) published a new classification in200820 that placed each of 25 known corneal dystrophiesnto 1 of 4 categories based on the quality of evidence forheir existence. Supportive clinical, structural, and geneticnformation was incorporated into a standard templateor each corneal dystrophy. One template, headed “Ep-thelial recurrent erosion dystrophy (ERED),” includedhe report of Franceschetti’s “Hereditary recurrent ero-
ion of the cornea”7 and the so-called “dystrophiaLL RIGHTS RESERVED. 1073

Smolandiensis” described by Hammar and associates ina family with early-onset corneal erosion and keloid-likecorneal opacifications.15,18 The study included histo-logic examination and DNA analysis in a large pedigree.Franceschetti, in 1928, described a separate large familywith early-onset recurrent corneal erosion and auto-somal dominant inheritance but, apparently, withoutother corneal changes.7 No histologic examination was
FIGURE 1. Franceschetti corneal dystrophy. Pedigree of an 8-goriginal 1928 pedigree and the newly studied family member(arrow). Solid shapes represent affected individuals. SymbolsSymbols with dots in the lower left corner represent family membeand D5S2115-D6S436 with restriction of haplotype analysis to2 [TACSTD2] on chromosome 1 and transforming growth fac
TABLE 1. Ten Affected Individuals With Franceschetti CornealErosions With and Without S
Individual Sex
Age
(y)
Visual
Acuity, OD
Visual
Acuity, OS
Reductio
Corne
Sensati
OD/O
V/22 F 101 20/200 20/200 �
VI/7 F 67 20/32 20/100 NA
VI/8 M 72 20/32 20/40 OD-,O
VI/9 F 70 20/50 20/40 �
VI/12 M 66 20/25 20/32 OD-,O
VII/4 M 44 20/20 20/20 NA
VII/14 M 37 20/40 20/32 �
VII/15 F 31 20/20 20/20 NA
VIII/1 F 12 20/20 20/20 NA
VIII/3 F 7 20/20 20/20 NA
AMD � age-related macular degeneration; NA � not available.
performed. This 6-generation pedigree included a total
AMERICAN JOURNAL OF1074
of 32 affected patients, of whom only 1, the 8-year-oldmale proband, was clinically studied with the slit lamp.Examination revealed a corneal erosion with no otherpathologic corneal findings.
The remaining 31 patients were identified as affectedbased on their ophthalmologic history alone. We have hadthe opportunity to review members of Franceschetti’soriginal family with recurrent corneal erosion syndrome,
ation family. The red line marks the border between the abovee proband V/22 is equally included in the original pedigreex represent individuals who were examined at the slit lamp.
th DNA analysis. Haplotypes are shown for D1S2890-D1S2873flanking markers (tumor-associated calcium signal transducer
eta–induced [TGFBI] on chromosome 5).
rophy (FRCD) Showing Early Onset of Innumerable Recurrentithelial Corneal Opacifation
Central Haze or
Patch-like
Opacification,
OD/OS
Cataract,
OD/OS
Cataract
Extration,
OD/OS
Penetrating
Keratoplasty,
OD/OS
AMD,
OD/OS
� – � � �
� OS� OD� � �
� � � � �
� � � � �
� � � � �
� � � � �
� � � � �
� � � � �
� � � � �
� � � � �
eners. Thwithrs withe 2tor b
Dystubep
n of
al
on,
S
S�
S�
including newly identified members. A proportion of these
OPHTHALMOLOGY JUNE 2012

FIGURE 2. Franceschetti corneal dystrophy. (Top left) Slit-lamp photograph of Patient VI/7 with diffuse, central haze of theepithelial/subepithelial layer. (Top right) Slit-lamp photograph of Patient VI/8 with diffuse, central haze of the epithelial/subepithelial layer. (Middle and Bottom) Histology of corneal biopsy specimen from Patient VI/8. (Middle left) Light microscopyshows the basal epithelium with enlarged intercellular clefts. The Bowman layer (arrow) is partially destroyed and pannus (asterisk)is found between the basal epithelium and Bowman layer (periodic acid–Schiff, �200). (Middle right) Basal epithelium (asterisk)and clefts (arrow) contain alcian blue–positive deposits (alcian blue, �400). (Bottom left) Electron microscopy shows irregularityin size and shape of the basal epithelial cells and enlarged intercellular clefts. (Bottom right) Pannus tissue with numerous fibrocytes.
FRANCESCHETTI CORNEAL DYSTROPHYVOL. 153, NO. 6 1075

mbtfG(t
um
n1T(sg(aicD
were examined clinically, and in 1 case a corneal biopsywas carried out. DNA analysis was also performed.
PATIENTS AND METHODS
OUR PROBAND, PATIENT V/22 OF FRANCESCHETTI’S ORIGI-
nal pedigree, allowed us to identify new affected individ-uals and extend the pedigree to 8 generations (Figure 1).Ten affected members of the family were interviewed andexamined biomicroscopically, including careful slit-lampexamination of the cornea with and without the instilla-tion of fluorescein. In 5 patients corneal sensitivity wasmeasured using a Cochet-Bonnet esthesiometer. The re-sults of these examinations are summarized in Table 1. Toassist in understanding the mechanism of the recurrenterosion attacks, a corneal biopsy was performed on PatientVI/8, a 72-year-old man, after obtaining ethical approvaland with appropriate discussion and consent. A 2 � 2-mmperilimbal biopsy was performed in the left cornea in the 1o’clock meridian, using a diamond knife to a depth of 100�m (T.R.). The following studies were performed:
● LIGHT MICROSCOPY AND IMMUNOHISTOCHEMISTRY:
One-half of the specimen was fixed in 4% formaldehydein 0.075 M phosphate buffer, dehydrated in increasingconcentrations of ethanol (70%–99%), and infiltratedwith paraffin (Merck, Darmstadt, Germany) at 60° C.Three-�m sections were cut, floated on de-ionized water at45 C, and mounted as single sections on Superfrost Plusglass slides (Menzel-Glaeser, Braunschweig, Germany).Slides were stained with hematoxylin and eosin, periodicacid–Schiff (PAS), Masson trichrome, alcian blue, andCongo red, and stained immunohistochemically with poly-clonal antibodies against claudin-7 (clone 5D10F3; Invit-rogen, Camarillo, California, USA), E-cadherin (clone4A2C7; Invitrogen), TGFBI, keratoepithelin-2, and kera-toepithelin-15, and antibodies against decorin, which has1 glycosaminoglycan chain of chondroitin/dermatansulfate(product number D 8428; Sigma, Roedermark, Germany).The TGFBI antibody to recombinant protein, includingthe full-length complementary DNA sequence (210–683)of human TGFBI, was produced in rabbits.18 Aminoeth-ylcarbazole was used as a chromogen to detect the immu-noreactions. The immunohistochemical staining of thespecimen was compared to that of a normal postmortemcornea with a fixation time of 24 hours.
● ELECTRON MICROSCOPY: The other half of the speci-en was fixed in 4% glutaraldehyde in 0.1 M phosphate
uffer (pH 7.3) for 24 hours, postfixed in 2% osmiumetroxide, processed through alcohol and propylene oxideor embedding in Araldite (Leica Microsystems, Wetzlar,ermany), and sectioned with a Reichert-Jung ultratome
Leica Microsystems). Semithin sections were stained with
oluidine blue, whereas ultrathin sections were stained withAMERICAN JOURNAL OF1076
ranyl acetate and lead citrate and examined with a trans-ission electron microscope (Zeiss, Oberkochen, Germany).
● MOLECULAR GENETIC STUDIES: Genomic deoxyribo-ucleic acid (DNA) was extracted from blood samples of5 family members and 1 spouse, and all exons of theGFBI and tumor-associated calcium signal transducer 2
TACSTD2) genes whose mutations are responsible, re-pectively, for the TGFBI-related corneal dystrophies andelatinous drop-like corneal dystrophy were sequencedF.L.M. and D.F.S.). Intron-exon boundaries were sequenceds described previously.21,22 In addition, the following flank-ng microsatellite markers were analyzed and haplotypes wereonstructed as described: D5S2115 and D5S436 (TGFBI);1S2890 and D1S230 (TACSTD2).
RESULTS
THE EXTENDED PEDIGREE IS SHOWN IN FIGURE 1. ALL AF-
fected individuals described the early onset of severeattacks of eye pain attributable to corneal erosions in 1 orthe other eye, associated with a red eye, epiphora, andphotophobia. The earliest onset was at 5 years and thelatest at 9 years. The disease demonstrated an autosomaldominant inheritance pattern with several examples ofmale-to-male transmission. Ten affected individuals (V/22, VI/7, VI/8, VI/9, VI/12, VII/4, VII/14, VII/15, VIII/1,VIII/3) and 3 nonaffected family members (VII/6, VIII/2,VIII/6) were examined at the slit lamp (Table 1) andexamples of the lesions were photographed (Figure 2, Top;Supplemental Figures 1 and 2, available at AJO.com).
A 12-year-old female patient (VIII/1) with a history ofattacks in each eye from the age of 5 suffered from ocularpain when she was examined at the slit lamp. At this timeshe showed bilateral punctate epithelial defects withoutother corneal changes. Attacks often occurred during thesecond half of the night and lasted for hours or even days.The clinical histories showed that young patients werefrequently kept home from school because of the severeocular discomfort. Although ophthalmic examination wascritical for diagnosis, healing of the epithelium prior toexamination caused some parents to incorrectly assumethat their affected children were simulating pain in orderto avoid school. The use of extended-wear contact lensesmarkedly reduced the subjective symptoms, as demon-strated in this 12-year-old girl and also in an affected adult(VII/15) who had completely clear corneas in the pain-freestage (Supplemental Figure 3, available at AJO.com). Atypical sequence of events was reported by a 42-year-oldpatient (VII/4) who also showed bilateral clear corneas inthe pain-free stage of his condition, in spite of a history ofrecurrent, early-onset epithelial erosions. His first attack ofocular pain occurred at the age of 5 years and affectedpredominantly the right eye. Severe ocular pain attacks
lasted during childhood and persisted until the age of 20.OPHTHALMOLOGY JUNE 2012

s
After age 30 the attacks occurred only 1 or 2 times peryear. The pattern of symptoms in Patient VII/4 exemplifiesthat in all other affected adults of Franceschetti’s family.The individuals VII/4, VII/15, VIII/1, and VIII/3, whowere between 7 and 44 years of age, were asymptomatic atthe time of examination and had clear corneas, withoutfluorescein staining, despite numerous previous erosiveattacks (Table 1; Supplemental Figure 3, available atAJO.com). Slit-lamp examination of Patients VI/7, VI/8,VI/9, VI/12, and VII/14, who were between 37 and 72years of age, showed bilateral central haze consisting ofdiffuse epithelial/subepithelial and patch-like opacities as-sociated with visual impairment (Table 1; Figure 2, Top;Supplemental Figures 1 and 2, available at AJO.com).Confocal microscopy of Patient VI/12 disclosed irregular,diffuse opacities at the level of the corneal epithelium(Supplemental Figure 4, available at AJO.com). Corneal
FIGURE 3. Franceschetti corneal dystrophy. Immunohistochemnegative for claudin (�400). (Top right) Normal postmortem cpecimen is negative for E-cadherin (�400). (Bottom right) Nor
sensation measured with the Cochet-Bonnet esthesiometer
FRANCESCHETTI CORNVOL. 153, NO. 6
(normal value: 5.0 cm) was reduced in Patient VI/9 toright/left 0.0 cm/2.0 cm and in Patient VII/14 to right/left1.5 cm/1.5 cm. The 101-year-old female proband (V/22),with a follow-up of 80 years, showed bilateral clear graftsafter penetrating keratoplasty performed abroad 12 yearspreviously (Table 1; Supplemental Figure 5, available atAJO.com). The residual host cornea showed diffuse sub-epithelial opacities. Unfortunately, no histologic examina-tions of the corneal buttons were performed. It was notpossible for us to get material of the specimen blocks ofPatient V/22. Family members VI/10, VI/11, VII/7, VII/9, andVII/11 were considered affected based on family history.
Light microscopy of biopsy from Patient VI/8 (Figure2, Middle) showed an irregular basal epithelium withenlarged intercellular spaces. Alcian blue–positive de-posits were present both intracellularly and intercellu-larly. The Bowman layer was partially destroyed and an
of Patient VI/8 after corneal biopsy. (Top left) The specimen isa as control is positive for claudin (�400). (Bottom left) Theostmortem cornea as control is positive for E-cadherin (�400).
istryorne
mal p
avascular connective tissue pannus was present between
EAL DYSTROPHY 1077

TABLE 2. Epithelial Recurrent Erosion Dystrophya
Study Autosomal Dominant Inheritance
Early Ocular Pain Attributable to Corneal
Erosion
Reduction of
Ocular Pain in
Adult
Corneal Erosion Without Other
Corneal Changes
Late Corneal Haze and
Patch-like Opacities Histology
DNA
Analysis
Franceschetti
19287
� 6 generations � 1 8-year-old boy – � – – –
Wales 19558 � 6 generations � 1 8-year-old and 1 9-year-old � � � � �
Holt 19569 � (?) 3 generations; 3 female
subjects; no father-son
inheritance
� between the age of 4 and 6 years � � � � �
Remler 195910 �(?) 11-year-old brotherb and
12-year-old sister
� � � � � �
Legrand 196311 �(?) mother and 2 daughters � ? � � � � �
Shindo 196812 �(?) 4 generations; no father-son
inheritance
�? in 1 54-year-old female subject
onset from age 20
� � in 1 patient with punctate
opacities of cornea
� � �
Remler 198313 � (?) 40-year-old fatherb and 9-
year-old daughter
� � � � � �
Feder 199314 �(?) 3 generations; no father-son
inheritance
6 affected individuals between 5
and 14 years
� � due to family record � � �
Hammar 200815
and 201018
� 6 generations; 21 affected � during the first 5 years of life � � � youngest individual 7
years old
� �
Hammar 200916 � 7 generations; 84 affected � between the age of 4 and 7 years � � � youngest individual
16 years old
� �
Vincent 200917 � 3 generations; 6 affected � average age of 6 years � � � � �
Current study � 8 generations; 10 new affected � between the age of 5 and 9 years � � between 37 and 79
years
� �
a� indicates reported; �indicates not reported.bThe same patient.
AM
ERIC
AN
JOU
RN
AL
OF
OPH
THA
LMO
LOG
Y1078
JUN
E2012

aStiBDThmcTDppmrgcamb
ntdWcrapp
ocbBesbna7Adssr
icypdrFamdcl
cbiws
the basal epithelium and Bowman layer. Congo redstaining was negative.
Electron microscopy (Figure 2, Bottom; SupplementalFigures 6 and 7, available at AJO.com) showed irregularityin size and shape of the basal epithelial cells and, as alreadyfound in conventional histology, enlarged intercellularclefts containing deposits, which corresponded to thealcian blue–positive ones. There was also pannus tissuecontaining numerous fibrocytes.
● IMMUNOHISTOCHEMISTRY: In the specimen studiedhere, there was a lack of expression of both the tightjunctional protein claudin 7 and E-cadherin, a componentof the desmosome (Figure 3, Top and Bottom). There waspositive staining for keratoepithelin-2 and keratoepithe-lin-15 in basal epithelium and basal membrane (Supple-mental Figures 8 and 9, available at AJO.com), more thanin the normal postmortem cornea, where only slightstaining was seen in few basal epithelial cells. Decorinexpression appears to be enhanced in the basal epitheliallayer compared to the normal postmortem cornea (Sup-plemental Figures 10 and 11, available at AJO.com).
● DNA AND LINKAGE ANALYSIS: Sequencing of all exonsnd the promoter of TGFBI gene did not reveal any variant.imilarly, sequencing of the TACSTD2 gene was also nega-ive. In order to exclude the presence of large rearrangementsn both genes, we performed haplotype analysis (Figure 1).oth genes were excluded by haplotype analysis. Markers1S2890 and D1S230 define a 5-Mb interval aroundACSTD2. The nonaffected female patient, VII/6, wasomozygous for the D1S2890 213-150 haplotype that wasost prevalent among affected individuals. D1S230 is ex-
luded in Patients VII/4, VII/14, VIII/1, and VIII/3. ForGFBI, exclusion was similarly suggested using marker5S2115 located at 0.6 Mb proximal to TGFBI. The affectedatients VII/7 and VII/9 did not share the 171-allele that wasresent in the other affected family members. D5S436, aore distal marker, was also analyzed and showed even more
ecombinations. Simulation analysis with the current pedi-ree showed that the max logarithm of the odds score thatan be expected is 2.6, mostly because many spouses were notvailable. Unfortunately, to recruit more individuals wasore complicated than estimated, and therefore we have not
een able to start a whole genome linkage analysis.
DISCUSSION
ALL PATIENTS REPORTED ATTACKS OF SEVERE EYE PAIN
attributable to erosions of the corneal epithelium since earlychildhood. Because, in the past, our knowledge of the clinicalphenotype of Franceschetti’s hereditary recurrent cornealerosion has been based on the examination of a single8-year-old affected member of the pedigree, it has been
assumed that the natural history was one of recurrent attacks aFRANCESCHETTI CORNVOL. 153, NO. 6
without consequent subepithelial scarring or opacity.7 Weow show for the first time that diffuse and patchy subepi-helial corneal opacities develop in patients in their fourthecade and older, particularly affecting the central cornea.e found that most affected elderly individuals had these
orneal opacities and that they were associated with aeduction in best-corrected visual acuity, to between 20/32nd 20/100 (Table 1). We assume, but cannot confirm, thatenetrating keratoplasties carried out in Patient V/22 wereerformed because of the visual effect of such opacities.
Histopathologic examination of corneal tissue from 1lder patient, from a peripheral region affected by superfi-ial corneal haze, revealed a connective tissue pannus lyingetween the epithelial basal lamina and a fragmentedowman layer. The early onset of recurrent epithelialrosion without any signs of corneal opacification coulduggest a hereditary disturbance of the adhesion complexetween basal epithelium and Bowman layer. Our immu-ohistochemical examination of the epithelium disclosedlack of expression of the tight junctional protein claudinand of E-cadherin, a component of the desmosome.lthough these proteins might play an etiologic role in the
evelopment of dystrophy-induced recurrent corneal ero-ion attributable to a loosening of the cell formation, iteems more likely that their deficiency reflects a secondaryesponse to multiple erosive attacks.
On the basis of the ultrastructural findings in 1 patient,t is reasonable to postulate that, in the Franceschettiorneal dystrophy, repeated erosions, occurring over manyears, stimulate invasion by activated keratocytes, whichenetrate through the fragmented Bowman layer and layown a connective tissue pannus.23 The question arises, ineviewing the clinical features and natural history ofranceschetti dystrophy, whether it can be adjudged to beunique dystrophy in its own right, with sufficient infor-ation to distinguish it from other superficial corneal
ystrophies. In our opinion, the clinical phenotype can belearly differentiated from most other well-known epithe-ial and Bowman layer corneal dystrophies.20 Certain other
inherited corneal dystrophies have been described thatresemble the dystrophy described here, in having earlypainful recurrent corneal erosion attacks of early onset andevidence of subepithelial corneal opacification.8–18 Thesedeserve further mention. We have also summarized thecurrent knowledge about these rare forms of cornealdystrophy in tabular form (Table 2). The solitary report ofsubepithelial mucinous corneal dystrophy showed similarclinical symptoms and signs to those of our family pre-sented here, with the early onset of recurrent erosions andof central, subepithelial opacities and haze.14 Histologi-ally, eosinophilic, periodic acid–Schiff-positive, alcianlue–positive, hyaluronidase-sensitive material was presentn a subepithelial band anterior to the Bowman layer,hich was focally replaced by collagenous scar. The
ubepithelial band stained positively with antibodies
gainst chondroitin 4 sulfate. This publication from 1993EAL DYSTROPHY 1079

haaipkad
ooediorphbcrtccgdbsTtp
fifpdarbfioagctscddSeeaZapmasmfct
did not present a DNA analysis.14 In the cornea of ourpatient, studied histologically, there were only smallamounts of alcian blue–positive material intracellularlyand within the basal intercellular clefts. Partial Bowmanlayer destruction was present in addition to a pannusbetween epithelium and Bowman layer. Although we didnot use the full range of antibodies employed by Feder andassociates,14 the light microscopic features, despite en-
anced decorin expression in the basal epithelial layer,rgue against the subepithelial mucinous dystrophy. In thebsence of evidence for a TGFBI gene mutation, wenterpreted the keratoepithelin-2 and keratoepithelin-15ositivity as a nonspecific pathologic response. Nonspecificeratoepithelin positivity has been described in secondarymyloid deposits of corneas with Fuchs endothelial cornealystrophy and subsequent bullous keratopathy.24
Hammar and associates have presented 2 Swedish fam-ilies with the early onset of recurrent corneal erosions anddevelopment of subepithelial fibrosis.15,16,18 They namedthe disorders after the areas of Sweden where the 2 familieslive: dystrophia Smolandiensis (after the county Småland)and dystrophia Helsinglandica (after the county Helsing-land). The authors regard these dystrophies as unique.
In dystrophia Smolandiensis, a large family with auto-somal dominant inheritance was presented in 6 genera-tions.15 A total of 28 individuals were evaluated bypthalmologic examination. By the age of 5, the majorityf the affected individuals suffered from recurrent cornealrosions. The erosive symptoms usually lasted from 1 to 7ays. The attacks generally declined in frequency andntensity with age. A total of 52% of the patients devel-ped central keloid-like corneal opacities. Nine patientseceived corneal grafts. Corneal tissue examined afterenetrating keratoplasty showed stromal amyloid depositsistologically.18 In our patient, histologic examination of aiopsy specimen including Bowman layer and superficialorneal stroma disclosed no amyloid deposits in theseegions. The DNA analysis of the Swedish family excludedhe following genes: TGFBI, gelsolin, keratin 3 and 12, andollagen type VIII alpha 2.15 Our DNA analysis of Fran-eschetti’s family excluded the TGFBI and TACSTD2enes. Once mutations have been identified for this disor-er, an appropriate update in the IC3D classification cane based both on the clinical pattern of findings and on aupposedly independent genetic cause for this dystrophy.he 2 dystrophies share a common clinical onset but differ
o some extent in their subsequent clinical course. The
resence of raised, keloid-like corneal lesions and theAMERICAN JOURNAL OF1080
nding of amyloid deposits in histologic specimens wouldavor a diagnosis of dystrophia Smolandiensis. In dystro-hia Helsinglandica, a 7-generation family with autosomalominant inheritance was described.16 By the age of 7,lmost all of the affected individuals suffered from recur-ent corneal erosions. Erosive symptoms usually lasted foretween 1 and 10 days. The attacks generally declined inrequency and intensity from the late 20s, but all examinedndividuals had developed subepithelial fibrosis by the agef 37. No histologic results were presented. The DNAnalysis of this Swedish family excluded the followingenes: TGFBI, keratin 3 and12, gelsolin, decorin, andollagen type VIII alpha 2. Currently we have no certaintyhat dystrophia Helsinglandica is distinct from France-chetti corneal dystrophy. The absence of a subepithelialorneal opacity after the age of 40 years would make theiagnosis of dystrophia Helsinglandica less likely, but theifference in severity of corneal opacifications in the 2wedish families and in Franceschetti’s family could bexplained by the presence of polymorphisms and a differ-nce in expressivity of a common gene. Vincent andssociates17 reported a 3-generation family from Newealand demonstrating autosomal dominant inheritancend presenting with frequent, recurrent corneal erosions,rimarily during the first decade. Six affected familyembers showed discrete grayish-white oval-round or
nnular opacities at the level of the Bowman layer and theuperficial anterior stroma. Unfortunately, histologic infor-ation was not available. The reported features in this
amily do not exclude the diagnosis of Franceschettiorneal dystrophy. Other families presented in the litera-ure8–13 showed recurrent corneal erosions in the first
decade without histologic or genetic examination (Table2). In the recent IC3D classification20 various forms ofcorneal dystrophy, manifesting recurrent corneal erosion,were bundled together under the heading “epithelial re-current erosion dystrophy (ERED).” This included dystro-phia Smolandiensis and the Franceschetti dystrophy. Wesuggest that it is now possible to regard the Franceschetticorneal dystrophy as a dystrophy in its own right, incategory 3, that is, with a well-characterized clinicalphenotype but not yet mapped to a particular chromosome(Appendix). The typical history of Franceschetti cornealdystrophy is of a dominant disorder with recurrent epithe-lial erosions presenting in the first decade, declining infrequency and severity at a later age, and associated with acentral, disk-like haze of the superficial cornea from middle
age. Keloid opacities do not occur.ALL AUTHORS HAVE COMPLETED AND SUBMITTED THE ICMJE FORM FOR DISCLOSURE OF POTENTIAL CONFLICTS OFInterest. The genetic examinations of this study were supported by grant #320030-127558 from the Swiss National Science Foundation, Bern,Switzerland (F.L.M. & D.F.S.). The authors indicate no financial conflict of interest. Involved in design and conduct of study (W.L., A.J.B., F.M., D.F.S.,L.T., P.S., J.S.W., C.A.-H.); collection of data (W.L., C.L., P.S., D.F.S., L.T., E.G., U.P., Ch.L., C.A.-H.); management, analysis, and interpretationof data (W.L., A.J.B., F.M., D.F.S., L.T., T.R., C.A.-H.); and preparation, review, and approval of the manuscript (W.L., A.J.B., F.M., D.F.S., L.T., P.S.,J.S.W., Ch.L., C.A.-H.). This retrospective study was approved by the Institutional Review Board of the University Eye Hospital Freiburg. The studywas conducted in adherence to the tenets of the Declaration of Helsinki. Informed consent was obtained from all patients.
OPHTHALMOLOGY JUNE 2012

1
1
1
1
1
1
1
1
1
1
2
2
2
2
2
REFERENCES
1. McDermott ML. The corneal epithelium. In: Podos SM,Yanoff M, eds. Textbook of Ophthalmology. Vol. 8. ExternalDiseases: Cornea, Conjunctiva, Sclera, Eyelids, LacrimalSystem. Chandler JW, Sugar J, Edelhauser HF, associateeditors. London: Mosby; 1994:4.1–4.12.
2. Rosza AJ, Beuerman RW. Density and organization of freenerve endings in the corneal epithelium of the rabbit. Pain1982;14(2):105–120.
3. Reidy JJ, Paulus MP, Gona S. Recurrent erosions of the cornea:epidemiology and treatment. Cornea 2000;19(6):767–771.
4. Bron AJ, Aldave AJ, Akhtar S, Stewart HS, Nischal KK.Inherited dystrophies and developmental anomalies of thecornea. In: Williams WTEJ, ed. Duane’s Clinical Ophthalmol-ogy. Philadelphia: Lippincott Williams & Wilkins; 2006:1–213.
5. Boutboul S, Black GCM, Moore JE, et al. A subset of patientswith epithelial basement membrane corneal dystrophy havemutations in TGFBI/BIGH3. Hum Mutat 2006;27(6):553–557.
6. Bron AJ, Burgess SEP. Inherited recurrent corneal erosion.Trans Ophthalmol Soc UK 1981(2):239–243.
7. Franceschetti A. Hereditäre rezidivierende Erosion derHornhaut. Z Augenheilk 1928;66(4):309–316.
8. Wales HJ. A family history of corneal erosions. TransOphthalmol Soc NZ 1955;8:77–78.
9. Holt LB. Corneal dystrophies: three dominant heredity types inPiedmont North Carolina. NC Med J 1956;17(5):225–227.
0. Remler O. Uber familiäres Auftreten von rezidivierenderHornhauterosion und ihre therapeutische Beeinflussung.Klin Monatsbl Augenheilkd 1959;135(4):263–270.
1. Legrand J. Dystrophie épithéliale cornéenne récidivantefamiliale. Bull Soc Ophtalmol FR 1963(5):384–387.
2. Shindo S. Familial recurrent corneal erosion. Nippon GankaGakkai Zasshi 1968;72(7):998–1004.
3. Remler O. Beitrag zur hereditären rezidivierenden Horn-hauterosion. Klin Mbl Augenheilkd 1983;183(1):59.
4. Feder RS, Jay M, Yue BYJT, Stock EL, O’Grady RB, Roth SI.Subepithelial mucinous corneal dystrophy. Clinical and patholog-ical correlations. Arch Ophthalmol 1993;111(8):1106–1114.
5. Hammar B, Björck E, Lagerstedt K, Dellby A, Fragerholm P.A new corneal disease with recurrent erosive episodes andautosomal-dominant inheritance. Acta Ophthalmol Scand2008;86(7):758–763.
6. Hammar B, Björck E, Lind H, Lagerstedt K, Dellby A,Fragerholm P. Dystrophia Helsinglandica: a new type ofhereditary corneal recurrent erosions with late subepithelialfibrosis. Acta Ophthalmol Scand 2009;87(6):659–665.
7. Vincent A, Markie DM, De Karolyi B, et al. Exclusion ofknown corneal dystrophy genes in an autosomal dominantpedigree of a unique anterior membrane corneal dystrophy.Mol Vis 2009;15:1700–1708.
8. Hammar B, Lagali N, Ek S, Seregard S, Dellby A, Fragerholm P.Dystrophia Smolandiensis: a novel morphological picture of recur-rent corneal erosions. Acta Ophthalmol Scand 2010;88(4):394–400.
9. Gibson IK, Inatomi T. Extracellular matrix and growth factors incorneal wound healing. Curr Opin Ophthalmol 1995;6(4):3–10.
0. Weiss JS, Moeller HU, Lisch W, et al. The IC3D classification
of the corneal dystrophies. Cornea 2008;27(Suppl 2): S1–42.FRANCESCHETTI CORNVOL. 153, NO. 6
1. Streeten B, Qi Y, Klintworth GK, Eagle RC, Jr, Strauss JA,Bennett K. Immunolocalization of �ig-h3 protein in 5q31linked corneal dystrophies and normal corneas. Arch Oph-thalmol 1999;117(1):67–75.
2. Munier FL, Korvatska E, Djemai A, et al. Kerato-epithelinmutations in four 5q31-linked corneal dystrophies. NatGenet 1997;15(3):247–251.
3. Mc Tigue JW. The human cornea: a light and electronmicroscopic study of the cornea and its alterations in variousdystrophies. Trans Am Ophthalmol Soc 1967;65:591–660.
4. Suesskind D, Auw-Haedrich C, Schorderet DF, Munier FL,Loeffler KU. Keratoepithelin in secondary corneal amyloidosis.Graefes Arch Clin Exp Ophthalmol 2006;244(6):725–731.
APPENDIX. Proposed Revision of the IC3D Classificationof the Corneal Dystrophies20 Incorporating Franceschetti
Corneal Dystrophy (FRCD) as a Distinct Clinical Entity
MIM None
Genetic locus Unknown
Gene Exclusion of TGFBI and TACSTD2
Inheritance Autosomal dominant
Onset Early childhood
Signs Inconspicuous cornea during pain-
free interval
Epithelial corneal erosion
Central, subepithelial diffuse opacity
Central, subepithelial diffuse opacity
� erosion
Symptoms Severe ocular painful attacks
attributable to recurrent epithelial
erosions during the whole life, but
above all in childhood
The attacks often start during the
night
Visual impairment, depending on the
presence of central opacification
Course Slow reduction of ocular pain attacks
in the advanced age
Slow progression of central opacities
Light microscopy Irregular basal epithelium with
intracellular and intercellular Alcian
blue–positive deposits
Partial destruction of Bowman layer
Pannus between basal epithelium
and Bowman layer
Negative Congo red staining
Electron microscopy Presumably “dystrophic”
mitochondria in between 2 basal
epithelial cells
Immunohistochemistry Segmental reduced expression of
the tight junction protein claudin
and E-cadherin
Confocal microscopy Accumulation of pathologic material
at the level of Bowman layer
Category 3
MIM � Mendelian inheritance in man.
EAL DYSTROPHY 1081

Biosketch
Walter Lisch, MD, lectured topics in ophthalmology with the thesis “Hereditary vitreoretinal degenerations” at theUniversity of Tuebingen, Germany. In 1989, he became Professor of Ophthalmology at the University of Tuebingen.Dr Lisch headed the Department of Ophthalmology, Klinikum Hanau, from 1991 until 2008. He is currently scientificcollaborator of the Department of Ophthalmology, Johannes Gutenberg University Mainz. Dr Lisch has published morethan 200 scientific papers and is a member of many international and national ophthalmological societies (seewww.whonamedit.com).
FRANCESCHETTI CORNEAL DYSTROPHYVOL. 153, NO. 6 1081.e1

SUPPLEMENTAL FIGURE 2. Franceschetti corneal dystro-phy: slit-lamp photograph of Patient VII/14 with diffuse,central haze of the epithelial/subepithelial layer.
SUPPLEMENTAL FIGURE 1. Franceschetti corneal dystro-phy: slit-lamp photograph of Patient VI/12 with central/para-central patch-like opacities of the epithelial/subepithelial layer.
AMERICAN JOURNAL OF1081.e2
SUPPLEMENTAL FIGURE 3. Franceschetti corneal dystro-phy: slit-lamp photograph of 32-year-old female patient (VII/15) without corneal opacification in spite of early onset ofrecurrent corneal erosions.
SUPPLEMENTAL FIGURE 4. Franceschetti corneal dystro-phy: confocal microscopy of Patient VI/12 showing irregulardiffuse opacities at the level of epithelial wing cells.
OPHTHALMOLOGY JUNE 2012
FRANCESCHETTI CORNVOL. 153, NO. 6
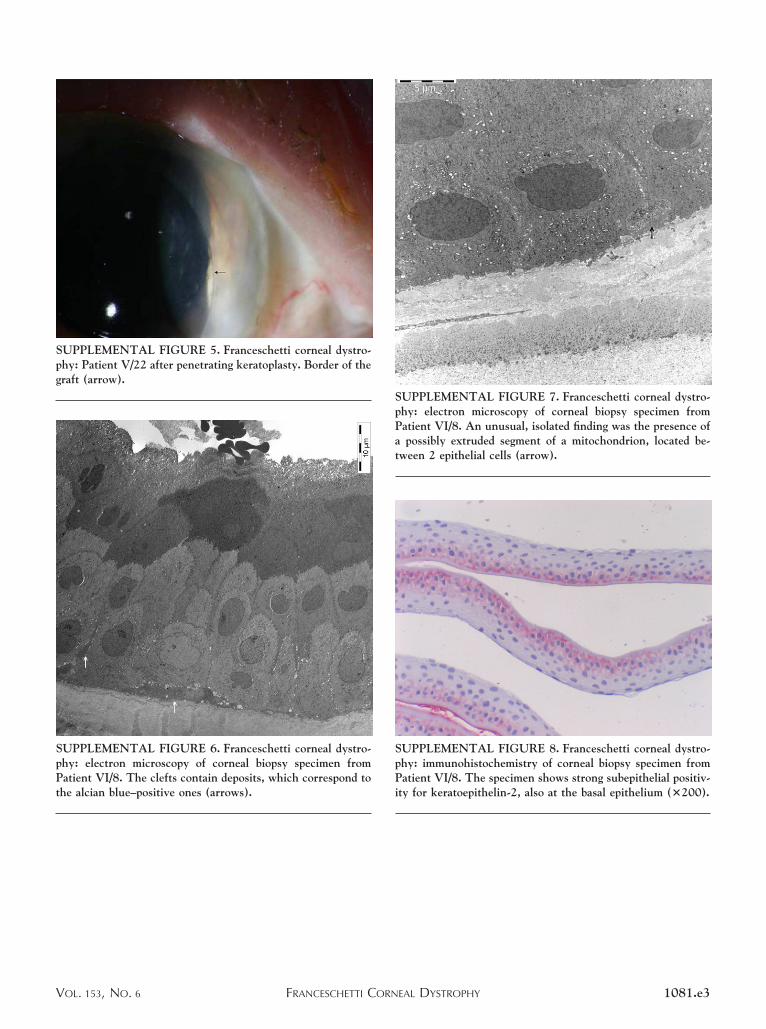
SUPPLEMENTAL FIGURE 8. Franceschetti corneal dystro-phy: immunohistochemistry of corneal biopsy specimen fromPatient VI/8. The specimen shows strong subepithelial positiv-ity for keratoepithelin-2, also at the basal epithelium (�200).
SUPPLEMENTAL FIGURE 5. Franceschetti corneal dystro-phy: Patient V/22 after penetrating keratoplasty. Border of thegraft (arrow).
SUPPLEMENTAL FIGURE 6. Franceschetti corneal dystro-phy: electron microscopy of corneal biopsy specimen fromPatient VI/8. The clefts contain deposits, which correspond tothe alcian blue–positive ones (arrows).
SUPPLEMENTAL FIGURE 7. Franceschetti corneal dystro-phy: electron microscopy of corneal biopsy specimen fromPatient VI/8. An unusual, isolated finding was the presence ofa possibly extruded segment of a mitochondrion, located be-tween 2 epithelial cells (arrow).
EAL DYSTROPHY 1081.e3

SUPPLEMENTAL FIGURE 9. Franceschetti corneal dystro-phy: immunohistochemistry of corneal biopsy specimen fromPatient VI/8. The specimen shows moderate subepithelialpositivity for keratoepithelin-15 (�200).
SUPPLEMENTAL FIGURE 10. Franceschetti corneal dystro-phy: immunohistochemistry of corneal biopsy specimen fromPatient VI/8. Decorin seems to be enhanced in the basalepithelial layer (�200).
AMERICAN JOURNAL OF1081.e4
SUPPLEMENTAL FIGURE 11. Franceschetti corneal dystro-phy: normal postmortem cornea shows strong decorin back-ground staining of the stroma.
OPHTHALMOLOGY JUNE 2012





























