Embed Size (px)
Citation preview

Métodos en Ecología y Sistemática Vol. 5(2): 10-26 ISSN 1659-3049 Agosto 2010
Autor Correspondencia: [email protected]*
Gen periodo no construye filogenias dentro del complejo de especie,
Lutzomyia longipalpis (Diptera: Phlebotominae)
GABRIEL GOLCZER1, 2 & JAZZMIN ARRIVILLAGA1* 1Lab. de Genética de poblaciones (Sección Invertebrados). Lab. Ecología Molecular de insectos vectores, Universidad
Simón Bolívar, Departamento de Estudios Ambientales. Caracas, Sartenejas, Venezuela. Apartado Postal 89000. 2Postgrado en Ciencias Biológicas. Universidad Simón Bolívar, Decanato de Postgrado. Caracas, Sartenejas,
Venezuela. Apartado Postal 8900.
RESUMEN: El gen periodo (per) ha sido utilizado ampliamente en estudios de filogenia y evolución en insectos, en especial con el complejo de especie Lutzomyia longipalpis vector de la leishmaniasis visceral en el neotrópico. Dicho gen regula el ritmo de los batidos de las alas utilizado para el cortejo, lo que tiene importancia en procesos de aislamientos reproductivos. En este trabajo se utilizaron las secuencias disponibles en el GenBank del gen per del complejo L. longipalpis, para realizar análisis de métodos de distancia genética (NJ), máxima parsimonia (MP) y máxima verosimilitud (ML). En general, se obtuvieron bajos soportes estadísticos y grandes politomías en los análisis de distancia y filogenéticos (MP y ML) con todos los grupos de secuencias, lo que evidencia que el gen per no reconstruye filogenias intraespecíficas divergentes, y no es lo suficientemente variable para diferenciar especies cercanas filogenéticamente dentro del complejo. PALABRAS CLAVE: Filogenia, máxima verosimilitud, máxima parsimonia, vecino más cercano, divergencia. ABSTRACT: The period gene (per) has been used in phylogenetic and evolutionary studies on insects, especially within Lutzomyia longipalpis complex species, vector of visceral leishmaniasis on the neotropical region. This particular gene regulates the locomotor activity of the wings during courtship, influencing on reproductive isolations processes. In this work we used DNA sequences of the per gene of L. longipalpis complex available on the GenBank, to make different analyses methods: based on global similarity by genetic distance (NJ), and phylogenies based on cladistic analysis (MP) and probability methods (ML). General results based on all method criteria indicated low bootstrap values and large politomies on the phylogenetic analyses with all set of sequences, all results suggest that the per gen does not reconstruct divergent intraspecific phylogenies within the complex species, and is not enough variable to differentiate species that are near phylogenetically inside the species complex. KEY WORDS: Phylogeny, maximum likelihood, maximum parsimony, neighbor joining, divergence.
INTRODUCTION El gen periodo (per) ha sido utilizado ampliamente en estudios de filogenia y evolución en insectos. Dicho gen codifica un proteína llamada PER involucrada en procesos de ritmos circadianos y ultra diarios (Konopka y Benzer 1971), y fue identificado por primera vez en una cepa mutante de Sophophora melanogaster (=Drosophila melanogaster), cambio del nombre del género aún en discusión
por la ICZN, (Dalton 2009, 2010). La función del producto del gen per en el caso de insectos, en especial dípteros, se centra en la regulación del ritmo de las “canciones” o batidos de alas utilizados para el cortejo, con ritmos específicos para cada especie (Wheeler et al. 1991). La ocurrencia de este gen, no solo en insectos, sino también en mamíferos, sugiere un grado de conservación funcional, pero gran nivel de

Métodos en Ecología y Sistemática Vol. 5(2): ISSN 1659-3049 Agosto 2010
11
diversificación dentro de los metazoos (Sauman y Reppert 1996). Se ha demostrado su rápida tasa mutacional (Schmid y Tautz 1997), lo que ha sugerido su uso en filogenias a nivel intraespecífico de complejos de especies, de especies crípticas y de especies filogenéticamente cercanas. Por lo que, el gen per promete ser una herramienta exitosa en el estudio del aislamiento reproductivo, especialmente como marcador genético en proceso de especiación (Kyriacou y Hall 1982, Ritchie et al. 1999). El complejo de especies Lutzomyia longipalpis (Lutz y Neiva 1912), está conformado por Lutzomyia longipalpis sensu lato y Lutzomyia pseudolongipalpis (Arrivillaga y Feliciangeli 2001). Estas especies han sido incriminadas en la transmisión de la Leishmania chagasi, agente etiológico causante de la leishmaniasis (forma visceral típica y la forma no ulcerativa) en el Neotrópico (Lainson y Rangel 2005, De Lima et al. 2009). El complejo de Lutzomyia longipalpis sensu lato está conformada por cuatro especies monofiléticas determinadas por análisis cladísticos basados en secuencias de ADN mitocondrial (Arrivillaga et al. 2003), con diferente grado de aislamiento reproductivo (Lanzaro et al. 1993, Arrivillaga y Marrero 2009), representadas por las poblaciones de Brasil (especie A, no descrita formalmente), las poblaciones de América Central (especie D, no descrita formalmente), poblaciones de Colombia (especie C1, no descrita formalmente), y poblaciones de Venezuela (especie C2, no descrita formalmente, cuyas poblaciones son recolectadas en la región geográfica de los Llanos, Cordillera de la Costa y Andina). Mientras, Lutzomyia pseudolongipalpis (especie B) es la única especie descrita formalmente del complejo y es citada como una especie endémica de Venezuela, con una distribución muy restringida, en el sistema Coriano
(Arrivillaga y Feliciangeli 2001, Arrivillaga et al. 2009). Actualmente, no existen conflictos dentro de la taxonomía alfa de L. longipalpis sensu latu (término utilizado para las especies genéticas no descritas formalmente), de considerar al menos a cuatro de las especies filogenéticas, como entidades taxonómicas discretas, con base en marcadores moleculares (COI, ND4, microsatélites). Sin embargo, el polimorfismo genético evidenciado dentro de la especie filogenética A (poblaciones brasileras) han sugerido hipótesis contrastantes. Hay autores que han señalado un proceso de especiación dentro de esta especie basándose en estudios de feromonas (Maingon et al. 1993, Watts et al. 2005, Bauzer et al. 2007, Maingon et al. 2007, Araki et al. 2009), cruzamientos (Souza et al. 2008) y biología molecular con el uso del gen per, cacophony y paralytic (Oliveira et al. 2001, Bauzer et al. 2002a, Bauzer et al. 2002b, Lins et al. 2002, Boteccia et al. 2003, Lins et al. 2008, Araki et al. 2009). En contraste, otros autores han sugerido que es una unidad taxonómica simple, pero polimórfica (Lanzaro et al. 1993, Day y Ready 1999, de Azevedo et al. 2000, Uribe et al. 2001, Arrivillaga et al. 2002, Hodgkinson et al. 2002, de Queiroz Balbino et al. 2006, Pinto et al. 2010), utilizando diferentes marcadores (isoenzimas, RAPD-PCR, microsatélites, cruzamiento, citocromo b, COI, ND4), técnicas (electroforesis, PCR, SSCP, secuenciación) y métodos de análisis (polimorfismo genético, genética de poblacionales y filogenia). En el caso especifico de marcadores moleculares, la utilización del gen per en la construcción de filogenias dentro del complejo Lutzomyia longipalpis, y con énfasis en estudios intra-específicos dentro de la especie A, se inicia retrospectivamente con los estudios de Oliveira et al. (2001), quienes proponen el uso de genes reguladores de “canciones” de cortejo en L. longipalpis (y otros dípteros) como el gen

Métodos en Ecología y Sistemática Vol. 5(2): ISSN 1659-3049 Agosto 2010
12
cac y el gen per, concentrando su investigación en el gen cac, detectando polimorfismos en este, pero sin aplicar análisis bajo supuestos filogenéticos. Posteriormente, Bauzer et al. (2002b) proponen al gen per como indicador de la divergencia entre dos poblaciones simpátridas de L. longipalpis, especie A, representadas por machos con los morfotipos de una mancha y de dos manchas en los tergos abdominales. Bauzer et al. (2002a) utilizan el gen per para probar divergencia a nivel inter-poblacional, entre las poblaciones de L. longipalpis (especie A) correspondientes a las poblaciones brasileras de: Lapinha, Natal y Jacobina, señalando divergencia entre las tres poblaciones, empleando análisis de distancia genética (NJ). Adicionalmente, Bauzer et al. (2002b) proponen al gen per como indicador de la divergencia entre dos poblaciones simpátridas de L. longipalpis, especie A, representadas por machos con los morfotipos de una mancha y de dos manchas en los tergos abdominales. Sin embargo, no existen trabajos publicados en las que se hayan hecho contrastaciones directa de esta hipótesis de divergencia propuesta por Bauzer et al. (2007), que deriven de la inclusión y comparación directa entre secuencias del gen de las especie L. longipalpis sensu lato (especies: A, C1, C2 y D) y L.
pseudolongipalpis (nominada anteriormente como especie B). En este sentido, el objetivo del presente trabajo es evaluar la utilidad y la calidad del gen per en análisis filogenéticos dentro del complejo de especies L. longipalpis, utilizando el set de secuencias disponibles en el GenBank para las especies citadas, obtenidas por Bauzer et al. (2002a, 2002b) y Meneses et al. (artículo no publicado) y Araki et al. (2009). Para ello se evalúa la hipótesis de divergencia utilizando los mismos parámetros propuestos por Bauzer et al. (2002a, 2002b) usando el método de distancia del Vecino Más Cercano (NJ, por sus siglas en inglés), con la finalidad de obtener soluciones bajo los mismos criterios, y
hacer el análisis comparable. Adicionalmente, se re-analiza la data de secuencias mediante análisis cladísticos (MP, máxima parsimonia), y análisis de filogenéticos con métodos de probabilidad (Máxima Verosimilitud, MP por sus siglas en inglés).
MATERIALES Y MÉTODOS Secuencias de AD!. Se obtuvieron de la base de datos GenBank (www.ncbi.nlm.nih.gov/ Genbank/) 352 secuencias del gen per de individuos de distintas poblaciones de L. longipalpis, con un promedio de 266 pares de bases, para 15 localidades y cuatro países: Brasil, Costa Rica, Colombia y Venezuela, dentro de la distribución geográfica del complejo de especie. Dichas secuencias fueron introducidas por distintos autores (Bauzer et al. 2002a, Bauzer et al. 2002b, Araki et al. 2009) y otros investigadores Meneses et al. (secuencias introducidas en el año 2005). A continuación se nombran los códigos de acceso de las secuencias en el GenBank: AY082911-AY082957, 47 secuencias de Bauzer et al. (2002a), AF446142 - AF446207 ,65 secuencias de Bauzer et al. (2002b), EU713077 - EU713077, 156 secuencias de Araki et al. (2009) y AY560920 - AY561152, 230 secuencias de Meneses et al. (secuencias introducidas en el GenBank en el año 2005). Se utilizó como grupo externo a la especie Lutzomyia dispar (código de acceso AY071912), sin embargo, en los análisis de MP se incluyó un segundo grupo externo Lutzomyia pseudolongipalpis (secuencia de Curarigua, Edo. Lara, Venezuela código de acceso AY561046). En cada tipo de análisis dependiendo de las restricciones computacionales y de espacio gráfico para la visualización óptima de los resultados, se utilizó una batería de secuencias menor a 70 en cada análisis. Dichas secuencias fueron seleccionadas semi-aleatoriamente (selección aleatoria de secuencias dentro de un

Métodos en Ecología y Sistemática Vol. 5(2): ISSN 1659-3049 Agosto 2010
13
grupo de cada localidad) utilizando un programa computacional desarrollado en lenguaje Visual Basic 2005© desarrollado por Golczer (ver Apéndice). Alineamiento. Se realizó el alineamiento múltiple de las secuencias utilizando el programa ClustalW (Li 2003) utilizando una penalización de apertura de gap de 10 y 6.66 de extensión. En el caso del análisis de parsimonia se utilizó también el programa de alineamiento MUSCLE (Edgar 2004) para evaluar si existía diferencia entre un alineamiento múltiple simple o uno basado en la construcción de arboles más parsimoniosos. Los alineamientos fueron revisados manualmente para ajustar des-alineamientos obvios (Arrivillaga et al. 2003, Kjer et al. 2006). Análisis de Distancia. El análisis de Vecino más Cercano (Neighbor Joining, NJ) fue realizado con el programa MEGA 4.0 (Molecular Evolutionary Genetic Analysis) (Tamura et al. 2007) repitiendo las especificaciones de los autores Bauzer et al. (2002a): Modelo de Kimura 2 parámetros, 1000 replicas de Bootstrap y Jacknife como método de soporte de nodos. En los análisis de NJ se utilizó a Lutzomyia dispar como grupo externo. Se realizaron varios análisis por separado donde se utilizaron: 1) todas las secuencias disponibles en el GenBank citadas en los trabajos de Bauzer et al. (2002a, 2002b), 47 y 65 secuencias respectivamente, 2) se seleccionaron semi-aleatoriamente 51 secuencias disponibles en el GenBank y citadas por Araki et al. (2009), y 3) se seleccionaron semi-aleatoriamente 48 secuencias disponibles en el GenBank introducidas por Meneses et al. en el año 2005. Finalmente, se realizo un análisis global de NJ, utilizando 2 secuencias de cada localidad (seleccionadas aleatoriamente) de cada grupo de secuencias disponibles en el GenBank pertenecientes a los autores mencionados.
ANÁLISIS FILOGENÉTICOS
Análisis Cladístico. El método de Máxima Parsimonia (MP) fue ejecutado con el programa PAUP*4.04b (Swofford 2002) y se realizaron búsquedas heurísticas de los árboles más parsimoniosos, con 100 adiciones de ramas azarosas de tipo TBR, 1000 réplicas de soporte de Bootstrap y Jacknife, y se calculó el índice de soporte de Bremer (Bremer 1994). Se incluyeron los gaps como 5ta base y se utilizó el consenso estricto de los 1000 árboles obtenidos, siguiendo la metodología de Arrivillaga et al. (2003). Los análisis de MP se hicieron con los datos separados de Bauzer et al. (2002a, 2002b), Araki et al. (2009) y Meneses et al. (introducidos en el GenBank en el año 2005) siguiendo la metodología de Arrivillaga et al. (2003) (arboles no mostrados). Luego se realizó un análisis global, se incluyo una secuencia de cada localidad seleccionada aleatoriamente (Cuadro 1). En los análisis de MP, se incluyeron 2 grupos externos, Lutzomyia dispar y Lutzomyia pseudolongipalpis. Métodos de Probabilidad. El Análisis de Máxima Verosimilitud (ML) fue ejecutado con el programa PAUP*4.04b (Swofford 2002), utilizando el modelo TIM+G de sustitución nucleotídica según el criterio de información de Akaike, (Posada y Buckley 2004) obtenido mediante el programa ModelTest 3.7 (Posada y Crandall 1998). Se realizaron búsquedas heurísticas con 50 adiciones de ramas azarosas de tipo TBR, y 1000 réplicas de soporte de Bootstrap y Jacknife. Se utilizaron las mismas secuencias reflejadas en la Cuadro 1 para hacer comparables los análisis de MP y ML.
RESULTADOS Análisis de Distancia. Al realizar los análisis de las secuencias del gen per según el procedimiento señalado por (Fig. 1A) por Bauzer et al. (2002a) y Bauzer et al. (2002b) (Fig.1B) no se encontraron diferencias en las topologías de los dendogramas. Sin embargo, se

Métodos en Ecología y Sistemática Vol. 5(2): ISSN 1659-3049 Agosto 2010
14
señalan diferencias con los valores de bootstraps obtenidos por Bauzer et al. (2002a, 2002b), a excepción del soporte de una rama con valor igual a 64 (Fig. 1A) y una con valor igual a 51 (Fig. 1B), en nuestro análisis los valores oscilan entre 3 y 47 para los nodos mas externos. Igualmente los valores de Jacknife fueron menores a 50 en todas las ramas (a excepción de dos). En general los análisis de distancia con las secuencias del gen per presentaron bajo soporte de bootstrap en los nodos basales restándole robustez a la topología tanto basal como externa. Por lo que no se evidencia divergencia filogenética entre los morfotipos-haplotipos de una y dos manchas, representados por la nomenclatura: 1S = 1 mancha y 2S = 2 manchas (Fig. 1B), así como no se observa divergencia soportada entre las localidades de Lapinha, Natal y Jacobina (Fig. 1A). Al analizar por separado las secuencias disponibles en el genbank de Meneses et al. (Fig. 2A) y Araki et al. (2009) (Fig. 2B) se encontraron valores de Bootstrap y Jacknife menores a 50 para los nodos basales. Las secuencias de países, localidades o manchas se agrupan en clados mixtos, no evidenciándose la monofília de las secuencias de los individuos pertenecientes a una misma localidad, morfotipo, país o especie genética, por lo tanto la divergencia no se evidencia en ninguno de los análisis por grupo de secuencias. Igualmente, al analizar la matriz de secuencias completa reflejadas en la Cuadro 1, con los datos de Bauzer et al. (2002a, 2002b), Araki et al. (Araki et al. 2009), y Meneses et al. (Fig. 3) se obtuvieron valores de Bootstrap y Jacknife menores a 50 en los nodos basales del árbol NJ. Tampoco en este caso se encontraron clados monofiléticos formados por las secuencias de localidades cercanas, mismos países o mismo morfotipo, observándose que no existe estructura divergente bajo este análisis. Lo anterior, evidencia que para todos los grupos de secuencias no existe divergencia entre las
especies propuestas, ni agrupación por tipo de feromona, morfotipo de macha, genética, que sugiera aislamiento reproductivo. Análisis Cladístico. Al construir los árboles filogenéticos de máxima parsimonia con cada grupo de secuencias, se encontró en cada caso una politomía dura (Braun y Kimball 2001), dado a los valores de Bootstrap y Jacknife menores a 50, para la mayoría de las ramas independiente del tipo de alineamiento utilizado, grupo de secuencias, soporte de rama y método de trabajo (Fig. 4). En cada caso, las secuencia obtenidas de ejemplares provenientes de Curarigua (L. pseudolongipalpis) se encuentra dentro de la politomía, compartiendo un clado con L. longipalpis sensu lato (que incluye las cuatro especies no descritas). Las soluciones no indican la existencia de grupos divergentes, que evidencien grupos monofiléticos dentro del complejo de especies, basado en las secuencia del gen analizado. Al obtener árboles de parsimonia en politomía sin diferenciación de grupos, bajos valores de bootstrap (representados con el colapso de las ramas) y bajos valores de CI y RI, se demuestra la poca información filogenética que aporta el gen per, basado en el gran número de homoplasias, y pocas sinapomorfías.
Análisis de Máxima Verosimilitud. Al realizar los árboles filogenéticos utilizando el método de Máxima Verosimilitud, se obtienen resultados muy similares en cuanto a topología y soporte a los análisis de MP y NJ (árboles no mostrados). El resultado evidencia una politomía dura, y no se observa ninguna estructura en la topología (Fig. 5).
Métodos en Ecología y Sistemática Vol. 5(2): ISSN 1659-3049 Agosto 2010
15
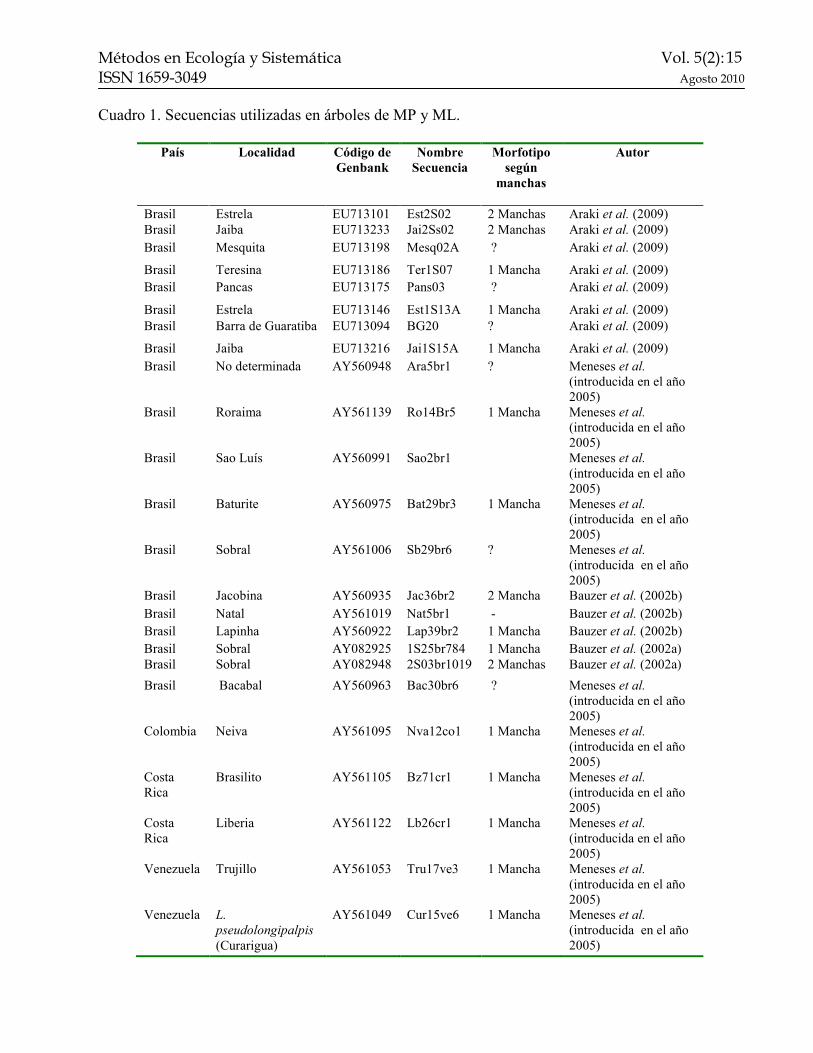
Cuadro 1. Secuencias utilizadas en árboles de MP y ML.
País Localidad Código de
Genbank
!ombre
Secuencia
Morfotipo
según
manchas
Autor
Brasil Estrela EU713101 Est2S02 2 Manchas Araki et al. (2009) Brasil Jaiba EU713233 Jai2Ss02 2 Manchas Araki et al. (2009) Brasil Mesquita EU713198 Mesq02A ? Araki et al. (2009)
Brasil Teresina EU713186 Ter1S07 1 Mancha Araki et al. (2009) Brasil Pancas EU713175 Pans03 ? Araki et al. (2009)
Brasil Estrela EU713146 Est1S13A 1 Mancha Araki et al. (2009) Brasil Barra de Guaratiba EU713094 BG20 ? Araki et al. (2009)
Brasil Jaiba EU713216 Jai1S15A 1 Mancha Araki et al. (2009) Brasil No determinada AY560948 Ara5br1 ? Meneses et al.
(introducida en el año 2005)
Brasil Roraima AY561139 Ro14Br5 1 Mancha Meneses et al. (introducida en el año 2005)
Brasil Sao Luís AY560991 Sao2br1 Meneses et al. (introducida en el año 2005)
Brasil Baturite AY560975 Bat29br3 1 Mancha Meneses et al. (introducida en el año 2005)
Brasil Sobral AY561006 Sb29br6 ? Meneses et al. (introducida en el año 2005)
Brasil Jacobina AY560935 Jac36br2 2 Mancha Bauzer et al. (2002b) Brasil Natal AY561019 Nat5br1 - Bauzer et al. (2002b) Brasil Lapinha AY560922 Lap39br2 1 Mancha Bauzer et al. (2002b) Brasil Sobral AY082925 1S25br784 1 Mancha Bauzer et al. (2002a) Brasil Sobral AY082948 2S03br1019 2 Manchas Bauzer et al. (2002a)
Brasil Bacabal AY560963 Bac30br6 ? Meneses et al. (introducida en el año 2005)
Colombia Neiva AY561095 Nva12co1 1 Mancha Meneses et al. (introducida en el año 2005)
Costa Rica
Brasilito AY561105 Bz71cr1 1 Mancha Meneses et al. (introducida en el año 2005)
Costa Rica
Liberia AY561122 Lb26cr1 1 Mancha Meneses et al. (introducida en el año 2005)
Venezuela Trujillo AY561053 Tru17ve3 1 Mancha Meneses et al. (introducida en el año 2005)
Venezuela L.
pseudolongipalpis
(Curarigua)
AY561049 Cur15ve6 1 Mancha Meneses et al. (introducida en el año 2005)

Métodos en Ecología y Sistemática Vol. 5(2): ISSN 1659-3049 Agosto 2010
16
Figura 1. (A) Dendograma construido según los métodos de Bauzer et al. (2002a). Si se representa el grupo externo L. dispar. (B) Dendograma construido según los criterios propuestos en la metodología de Bauzer et al. (2002b), con 1000 replicas de bootstrap. Los soportes de las ramas basales representan los valores de Bootstrap/Jacknife, y los soportes de los nodos mas interno representan solo valores de Botstrap.

Métodos en Ecología y Sistemática Vol. 5(2): ISSN 1659-3049 Agosto 2010
17
Figura 2. (A) Dendograma construido según el método de Bauzer et al. (2002b), con un valor de bootstrap de 1000 replicas a partir de 48 secuencias seleccionadas al azar de Meneses et al. (B) Dendograma construido según el método de Bauzer et al. (2002b), con un valor de bootstrap de 1000 replicas a partir de 51 secuencias seleccionadas al azar de Araki et al. (2009). Los soportes de las ramas basales representan los valores de Bootstrap/Jacknife, y los soportes de los nodos mas interno representan solo valores de Botstrap.

Métodos en Ecología y Sistemática Vol. 5(2): ISSN 1659-3049 Agosto 2010
18
Figura 3. Dendograma construido según el método de Bauzer et al. (2002b), con un valor de bootstrap de 1000 replicas a partir de 51 secuencias seleccionadas al azar de Bauzer et al. (2002a, 2002b), Araki et al. (2009) y Meneses et al. Los soportes de las ramas basales representan los valores de Bootstrap/Jacknife, y los soportes de los nodos mas interno representan solo valores de Botstrap.

Métodos en Ecología y Sistemática Vol. 5(2): ISSN 1659-3049 Agosto 2010
19
Figura 4. Consenso estricto de los 1000 más parsimoniosos encontrados en 1000 replicas de bootstrap con 50 búsquedas heurísticas, de una secuencia por localidad (ver Cuadro 1) a partir de los datos de Bauzer et al. (2002a, 2002b), Araki et al. (2009) y Meneses et al. Longitud del árbol 136, CI= 0.5809 y RI= 0.4191. Los soportes de las ramas representan valores de Bootstrap/Soporte de Bremer/Jacknife (excepto el nodo basal= B/J).

Métodos en Ecología y Sistemática Vol. 5(2): ISSN 1659-3049 Agosto 2010
20
Figura 5. Consenso estricto del árbol de Máxima Verosimilitud (ML) en 1000 replicas de bootstrap con 100 búsquedas heurísticas, de una secuencia por localidad a partir de secuencias de Bauzer et al. (2002a, 2002b), Araki et al. (2009) y Meneses et al. (ver Cuadro 1). Los soportes de las ramas internas representan valores de Bootstrap /Jacknife (en el caso del soporte de Jacknife sólo se muestran los valores superiores a 50).

Métodos en Ecología y Sistemática Vol. 5(2): ISSN 1659-3049 Agosto 2010
21
DISCUSIÓN En el presente estudio, hemos analizado todas las secuencias almacenadas en el GenBank del gen per para el complejo de especies L. longipalpis, reconstruyendo los dendogramas previamente publicados (Bauzer et al. 2002a, Bauzer et al. 2002b) y (Araki et al. 2009), no publicados (Meneses et al.) y utilizando otros métodos para comparar los resultados obtenidos. Los análisis de distancia aunque muestran topologías resueltas en ciertos grupos, presentan valores de bootstrap que no son mayores a 50 en los nodos basales, lo que le resta soporte estadístico a la topología y por consiguiente a la hipótesis de divergencia entre poblaciones brasileras planteada por Bauzer et al. (2002a, 2002b, 2007), que atribuye a las poblaciones de Brasil (especie A) el estatus de complejo de especies. Lo anterior, no es respaldado por los resultados obtenidos con las filogenias del gen per, adicionalmente es incongruente con otros resultados obtenidos con otros marcadores moleculares (Uribe et al. 2001, Arrivillaga et al. 2003), donde la especie A es un clado monofilético, conformado por todas las poblaciones de Brasil, independientemente de su morfotipo según manchas, o feromonas. La inclusión de la secuencia de L.
pseudolongipalpis dentro del grupo de todas las secuencias del complejo L. longipalpis demuestra la limitación del gen per de reconstruir la divergencia entre especies hermanas, y a su vez no refleja la divergencia entre las poblaciones (especies dentro de L. longipalpis sensu lato), a diferencia de otros marcadores moleculares como citocromo oxidasa I (Arrivillaga et al. 2002), ND4 (Uribe et al., 2001) y microsatélites: LIST6-002, LIST6-004, LIST6-006, LIST6-012, y LIST6-029 (Watts et al. 2005). Se ha evidenciado que el método de construcción de los árboles filogenéticos o dendogramas puede variar el resultado con el mismo grupo de secuencias (Khuner y
Felsenstein 1994), por ende al analizar los datos con métodos de máxima parsimonia o máxima verosimilitud se pudieran reconstruir topologías resueltas y con un mejor soporte, en contraste con los métodos de distancia. En este particular, utilizando análisis de MP, ML, NJ, todos los análisis son concordantes con base en la interpretación de no divergencia. Lo anterior comprueba que el análisis utilizado no influye en este caso en los resultados, como indican Khuner y Felsenstein (1994). Adicionalmente, se observó correlación en la interpretación biológica derivada del mismo análisis independientemente del tipo de soporte de rama usado (Bootstrap, Jacknife, Bremer), dado a las características de la data molecular, gran número de secuencias y caracteres con alto número de homoplasias y bajo número de caracteres sinapomórficos (Müller 2005). Sin embargo, el uso del gen per ha permitido obtener topologías resueltas empleando estos métodos NJ, MP, ML, obteniéndose grupos monofiléticos bien soportados en el estudio de otros grupos de especies (Wang y Hey 1996, Barr et al. 2005). Dentro del género Lutzomyia a nivel supraespecífico, el gen per fue utilizado en construcciones de arboles de distancia con varias especies, obteniéndose topologías resueltas y soportadas con valores altos de bootstrap (Mazzoni et al. 2002). Sin embargo, esto no ocurre al comparar grupos de secuencias de individuos de la misma especie, de especies cercanas o complejo de especies, siendo este el último caso la situación de L. longipalpis sensu lato y L. pseudolongipalpis.
La inhabilidad de reconstruir filogenias intraespecíficas divergentes a partir de especies cercanas implica que el gen per, aunque se presuma que se encuentra involucrado en procesos de especiación (Oliveira et al. 2001), no es un marcador molecular para procesos de divergencia reciente, tan o más confiable como genes mitocondriales anteriormente utilizados

Métodos en Ecología y Sistemática Vol. 5(2): ISSN 1659-3049 Agosto 2010
22
en el género Lutzomyia, ya que el gen no describe de forma adecuada la filogenia de la especie o del complejo de especies (Edwards 2009). La confiabilidad yace en el supuesto de que el gen permite diferenciar individuos o linajes a nivel de género y reconstruye una aproximación de la historia evolutiva de las especies o poblaciones (Ishikawa et al. 1999, Uribe et al. 2001, Arrivillaga et al. 2002, Testa et al. 2002).
REFERENCIAS Araki AS, Vigoder FM, Bauzer LG, Ferreira GE,
Souza !A, Araujo IB, Hamilton JG, Brazil RP, Peixoto AA. 2009. Molecular and behavioral differentiation among Brazilian populations of Lutzomyia longipalpis (Diptera: Psychodidae: Phlebotominae). PLoS Negl Trop Dis 3(1): 365. Arrivillaga J, Feliciangeli D. 2001. Lutzomyia pseudolongipalpis: The First New Species Within the longipalpis (Diptera: Psychodidae: Phlebotominae) Complex from La Rinconada, Curarigua, Lara State, Venezuela. Journal of Medical Entomology 38(6): 783-790. Arrivillaga J, Marrero R. 2009. Two new populations of Lutzomyia pseudolongipalpis Arrivillaga & Feliciangeli (Diptera: Phlebotominae) vector of visceral leishmaniasis in Venezuela. Neotropical Entomology 38(4): 556-559. Arrivillaga J, Mutebi J, Piñango H, !orris D,
Alexander B, Feliciangeli M, Lanzaro G. 2003. The Taxonomic Status of Genetically Divergent Populations of Lutzomyia longipalpis (Diptera: Psychodidae) Based on the Distribution of Mitochondrial and Isozyme Variation. Journal of Medical Entomology 40(5): 615-627. Arrivillaga J, !orris D, Feliciangeli MD, Lanzaro G. 2002. Phylogeography of the neotropical sand fly Lutzomyia longipalpis inferred from mitochondrial DNA sequences. Infection, Genetics and Evolution 2: 83-95. Arrivillaga J, Salerno P, Rangel Y. 2009. Aislamiento reproductivo asimétrico entre Lutzomyia pseudolongipalpis y Lutzomyia
longipalpis (especie C2), vectores neotropicales de leishmaniasis visceral (Diptera: Pshychodidae). Biología Tropical 57: 22-31. Barr !, Cui L, McPheron B. 2005. Molecular Systematics of Nuclear Gene period in Genus Anastrepha (Tephritidae). Anals of the Entomological Society of America 98(2): 173-180. Bauzer L, Gesto J, Souza !, Ward R, Hamilton J, Kyriacou C, Peixoto A. 2002a. Molecular Divergence in the period Gene Between Two Putative Sympatric Species of the Lutzomyia longipalpis Complex. Molecular Biology and Evolution 19(9): 1624-1627. Bauzer L, Souza !, Maingon R, Peixoto A. 2007. Lutzomyia longipalpis in Brazil: a complex or a single species? A mini-review. Memórias do Instituto Oswaldo Cruz 102(1): 1-12. Bauzer L, Souza !, Ward R, Kyriacou C, Peixoto A. 2002b. The period gene and genetic differentiation between three Brazilian populations of Lutzomyia longipalpis. Insect Molecular Ecology 11(4): 315-323. Braun E, Kimball R. 2001. Polytomies, the power of Phylogenetic inference, and the Stochastic Nature of Molecular Evolution: a comment on Walsh et al. (1999). Evolution 55(6): 1261–1263. Bremer K. 1994. Branch Support and Tree Stability. Cladistics 10: 295-304. Dalton R. 2009. A fly by any other name: Drosophila experts argue over reclassification proposal. Nature 457: 368. Dalton R. 2010. What's in a name? Fly world is abuzz: Proposed reorganization of Drosophila fruitfly genus might throw out its most celebrated member. Nature 464: 825. Day J, Ready P. 1999. Relative abundance, isolation and structure of phlebotomine microsatellites. Insect Molecular Ecology 8(4): 575-580.

Métodos en Ecología y Sistemática Vol. 5(2): ISSN 1659-3049 Agosto 2010
23
de Azevedo A, Monteiro F, Cabello P, Souza !, Goreti M, Rangel E. 2000. Studies on Populations of Lutzomyia longipalpis (Lutz & Neiva 1912) (Diptera: Psychodidae: Phlebotominae) in Brazil. Memórias do Instituto Oswaldo Cruz 95(3): 305-322. De Lima H, Rodriguez !, Feliciangeli MD,
Barrios MA, Sosa A, Agrela I, Sanchez E, Lopez O. 2009. Cutaneous leishmaniasis due to Leishmania chagasi/L. infantum in an endemic area of Guarico State, Venezuela. Royal Society of Tropical Medicine and Hygiene 103(7): 721-726. de Queiroz Balbino V, Coutinho-Abreu I, Sonoda
I, Melo M, de Andrade P, de Castro J, Rebêlo J, Carvalho S, Ramalho-Ortigão M. 2006. Genetic structure of natural populations of the sand fly Lutzomyia longipalpis (Diptera: Psychodidae) from the Brazilian northeastern region. Acta Tropica 98(1): 15-24. Edgar RC. 2004. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Research 32(5): 1792-1797. Edwards SV. 2009. Is a new and general theory of molecular systematics emerging? Evolution 63(1): 1-19. Hodgkinson V, Birungi J, Haghpanah M, Joshi S, Munstermann L. 2002. Rapid Identification of Mitochondrial Cytochrome B Haplotypes by Single Strand Conformation Polymorphism in Lutzomyia longipalpis (Diptera: Psychodidae) Populations. Journal of Medical Entomology 39(4): 689-694. Ishikawa E, Ready P, de Souza A, Day J, Rangel E, Davies C, Shaw J. 1999. A Mitochondrial DNA Phylogeny Indicates Close Relationships between Populations of Lutzomyia whitmani (Diptera: Psychodidae, Phlebotominae) from the Rain-forest Regions of Amazônia and Northeast Brazil. Memórias do Instituto Oswaldo Cruz 94(3): 339-345. Khuner M, Felsenstein J. 1994. A Simulation Comparision of Phylogeny Algorithms under Equal and Unequal Evolutionary Rates. Molecular Biology and Evolution 11(3): 459-468.
Kjer K, Gillespie J, Ober K. 2006. Structural Homology in Ribosomal RNA, and a Deliberation on POY. Arthropod Systematics & Phylogeny 64(2): 159-164. Konopka RJ, Benzer S. 1971. Clock mutants of Drosophila melanogaster. Proceedings of the National Academy of Science 68(9): 2112-2116. Kyriacou C, Hall J. 1982. The function of courtship song rhythms in Drosophila. Animal Behavior 30(1): 784-801. Lainson R, Rangel E. 2005. Lutzomyia longipalpis and the eco-epidemiology of American visceral leishmaniasis, with particular reference to Brazil - A Review. Memórias do Instituto Oswaldo Cruz 100(8): 811-827. Lanzaro G, Ostrovska K, Herrero M, Lawyer P, Warburg A. 1993. Lutzomyia longipalpis is a species complex: genetic divergence and interspecific hybrid sterility among three populations. American Journal of Tropical Medicine and Hygiene 48(1): 839-847. Li K. 2003. ClustalW-MPI: ClustalW analysis using distributed and parallel computing. . Bioinformatics 19(12): 1585-1586. Lins R, Oliveira S, Souza !, de Queiroz R,
Justiniano S, Ward R, Kyriacou C, Peixoto A. 2002. Molecular evolution of the cacophony IVS6 region in sandflies. Insect Molecular Ecology 11(2): 117-122. Lins RM, Souza !A, Peixoto AA. 2008. Genetic divergence between two sympatric species of the Lutzomyia longipalpis complex in the paralytic gene, a locus associated with insecticide resistance and lovesong production. Memórias do Instituto Oswaldo Cruz 103(7): 736-740. Maingon R, Feliciangeli D, Ward R, Chance M,
Adamson R, Rodriguez !, Convit J, Petralanda I, Hernandez A, Segovia M. 1993. Molecular approaches applied to the epidemiology of leishmaniasis in Venezuela. Archives Institute Pasteur Tunis 70(3-4): 309-324.

Métodos en Ecología y Sistemática Vol. 5(2): ISSN 1659-3049 Agosto 2010
24
Maingon R, Ward R, Hamilton J, Bauzer L, Peixoto A. 2007. The Lutzomyia longipalpis species complex: does population substructure matter to Leishmania transmission? Trends in Parasitology 24(1): 12-17. Mazzoni C, Gomes C, Souza !, de Queiroz R,
Justiniano S, Ward R, Kyriacou C, Peixoto A. 2002. Molecular Evolution of the period Gene in Sandflies. Journal of Molecular Evolution 55(1): 553-562. Müller K. 2005. The efficiency of different search strategies in estimating parsimony jackknife, bootstrap, and Bremer support. BMC Evolutionary Biology 5: 58-67. Oliveira S, Bottechia M, Bauzer L, Souza !, Ward R, Kyriacou C, Peixoto A. 2001. Courtship Song Genes and Speciation in Sand Flies. Memórias do Instituto Oswaldo Cruz 96(3): 403-405. Pinto IS, Filho JD, Santos CB, Falqueto A, Leite YL 2010. Phylogenetic relationships among species of Lutzomyia, subgenus Lutzomyia (Diptera: Psychodidae). Journal of Medical Entomology 47(1): 16-21. Posada D, Buckley TR. 2004. Model selection and model averaging in phylogenetics: advantages of akaike information criterion and bayesian approaches over likelihood ratio tests. Systems Biology 53(5): 793-808. Posada D, Crandall KA. 1998. MODELTEST: testing the model of DNA substitution. Bioinformatics 14(9): 817-818. Ritchie MG, Halsey EJ, Gleason JM. 1999. Drosophila song as a species-specific mating signal and the behavioural importance of Kyriacou & Hall cycles in D. melanogaster song. Animal Behavior 58(3): 649-657. Sauman I, Reppert SM. 1996. Circadian clock neurons in the silkmoth Antheraea pernyi: novel mechanisms of Period protein regulation. Neuron 17(5): 889-900.
Schmid KJ, Tautz D. 1997. A screen for fast evolving genes from Drosophila. Proceedings of the National Academy of Science 94(18): 9746-9750. Souza !A, Andrade-Coelho CA, Vigoder FM, Ward RD, Peixoto AA. 2008. Reproductive isolation between sympatric and allopatric Brazilian populations of Lutzomyia longipalpis s.l. (Diptera: Psychodidae). Memórias do Instituto Oswaldo Cruz 103(2): 216-219. Swofford DL. 2002. PAUP*. Phylogenetics analysis using parsimony* and other methods. Sinauer Associates, Sunderland, Mass. Tamura K, Dudley J, !ei M, Kumar S. 2007. MEGA4 Molecular Evolutionary Genetics Analysis (MEGA) software version 4.0. Molecular Biology and Evolution 241: 596-1599. Testa J, Montoya-Lerma J, Cadena H, Oviedo M, Ready P. 2002. Molecular identification of vectors of Leishmania in Colombia: mitochondrial introgression in the Lutzomyia townsendi series. Acta Tropical 84(3): 205-218. Uribe S, Lehmann T, Rowton E, Vélez I, Porter C. 2001. Speciation and Population Structure in the Morphospecies Lutzomyia longipalpis (Lutz & Neiva) as Derived from the Mitochondrial ND4 Gene. Molecular Phylogenetics and Evolution 18(1): 84-93. Wang R, Hey J. 1996. The Speciation history of Drosophila pseudoobscura and Close Relatives: Inferences from DNA Sequence Variation at the Period Locus. Genetics 144(1): 1113-1126. Watts PC, Hamilton JG, Ward RD, !oyes HA,
Souza !A, Kemp SJ, Feliciangeli MD, Brazil R, Maingon RD. 2005. Male sex pheromones and the phylogeographic structure of the Lutzomyia
longipalpis species complex (Diptera: Psychodidae) from Brazil and Venezuela. American Journal of Tropical Medicine and Hygiene 73(4): 734-743. Wheeler DA, Kyriacou CP, Greenacre ML, Yu
Q, Rutila JE, Rosbash M, Hall JC. 1991. Molecular transfer of a species-specific behavior

Métodos en Ecología y Sistemática Vol. 5(2): ISSN 1659-3049 Agosto 2010
25
from Drosophila simulans to Drosophila
melanogaster. Science 251(4997): 1082-1085. Recibido: 11 Junio, 2010. Aceptado: 27 Julio, 2010.

Métodos en Ecología y Sistemática Vol. 5(2): ISSN 1659-3049 Agosto 2010
26
APENDICE Ejemplo de Código en lenguaje Visual Basic 2005© de selección semi-aleatoria: Selección aleatoria de secuencias dentro de un grupo de cada localidad). Desarrollado por Golczer, G. Public Class Form1 Public CantSec As Integer = Public ArregloDatos(0 To CantSec) As Dato Public DatosM(0 To (CantSec / 2)) As Dato Public matriz12s(0 To 3, 0 To 12) Public i As Integer Public j As Integer Public k As Integer Public l As Integer Private Sub Form1_Load(ByVal sender As System.Object, ByVal e As System.EventArgs) Handles MyBase.Load matriz12s(0, 1) = 1 matriz12s(1, 1) = "TRC277F-12SR" matriz12s(2, 1) = 0 matriz12s(3, 1) = "ATTTTAGTCTATTCAGAGGAATTTGTTTTATAATCGATATTCCCCGTTATACCTTACTTATTTTATTTAATTTGTATATCGTTGTCATTAGAATATTTTATAAGAAAAATAATTTTCTGAAATTACATAAAATTTTATGTCAAATCAAGGTGCAGTATATATTTAAGTAAAAATGAATTACATTAATATTTAATTAAATGAATTAAAGATTGAAATATCTTTATGAAGGTGGATTTGATAGTAAATTTTAGAA" (SE REPITE CON TANTAS SECUENCIAS SE QUIERAN SELECCIONAR) End Sub Private Sub Button1_Click(ByVal sender As System.Object, ByVal e As System.EventArgs) Handles Button1.Click i = 0 For j = 1 To 12 For k = 1 To 12 If j = j = False And matriz12s(2, j) = 0 = True And matriz12s(2, k) = 0 = True Then If matriz12s(3, j) = matriz12s(3, k) = True Then
matrizHap12s(i) = matriz12s(3, j) matriz12s(2, j) = 1 matriz12s(2, k) = 1 l = i i = l + 1 End If End If Next Next For j = 0 To i Debug.Print(matrizHap12s(i)) Next End Sub End Class





























