Embed Size (px)
Citation preview

This journal is c the Owner Societies 2013 Phys. Chem. Chem. Phys.
Cite this: DOI: 10.1039/c3cp50280e
Glycerol electro-oxidation over glassy-carbon-supported Au nanoparticles: direct influence of thecarbon support on the electrode catalytic activity
Janaina F. Gomes,*a Luiz H. S. Gasparottob and Germano Tremiliosi-Filhoa
Glycerol is at present abundantly co-produced in the biodiesel fabrication and can be used as fuel in
Direct Glycerol Fuel Cells (DGFC) for cogeneration of electricity, value-added chemicals and heat. With
this motivation, in the present work, we investigated at a fundamental level the oxidation of glycerol
over glassy carbon (GC) supported Au nanoparticles in alkaline medium using cyclic voltammetry. By
controlling the Au deposition time, we varied the GC supported Au coverage from 0.4% to 30%
maintaining a regular particle size distribution with a mean particle size of about 200 nm. An influence
of the carbon support on the activity of the GC-supported Au nanoparticles was evidenced. Results
from studies on the oxidation of glycerol and ethylene glycol on Au and Pt nanoparticles supported on
a glassy carbon, highly ordered pyrolytic graphite and dimensionally stable anode under different pH
conditions indicate that the carbon support participates actively in the oxidation of glycerol and other
alcohols. We propose that active oxygenated species are gradually formed on the glassy carbon by
potential cycling (up to the saturation of the carbon area) and these oxygenated species are additional
oxygen suppliers for the oxidation of glycerol residues adsorbed on the Au particles, following a
mechanism consisting of the synergism of two active elements: gold and carbon.
1. Introduction
Glycerol is typically used in pharmaceuticals and cosmetics indus-tries. Nowadays, it is largely produced as a co-product of the bio-diesel fabrication in the proportion of 1 kg of glycerol to 10 kg ofbiodiesel. As a consequence of the increasing interest in thisrenewable bio-fuel, a problem emerges: what to do with theadditional glycerol, which exceeds the demand of traditional tradesfor glycerol? The exceeding glycerol is partly burned in order togenerate heat that is used in biodiesel production and in manyother industrial processes. This is an environmentally correctpractice since glycerol substitutes wood and fossil fuels, such ascoal and diesel. Additionally, another fraction of the exceedingglycerol is exported to other countries, where it is used as rawmaterial in the traditional trades of glycerol. Despite some applica-tions, huge volumes of glycerol are being accumulated in manycountries. For this reason, at present, there is an opportunity fornew applications for glycerol, which is currently an abundant raw
material and presents chemical potential for the development ofnew products and processes with high value added. One of thepossibilities is the use of glycerol in Direct Glycerol Fuel Cells(DGFC)1–3 for cogeneration of electricity, chemicals and heat.
Glycerol undergoes electro-oxidation by different reaction path-ways leading to the formation of many partially oxidized products,in addition to the final product, CO2.4–11 While the production ofCO2 involves the liberation of 14 electrons per glycerol molecule,the formation of partially oxidized products involves less than this.Therefore, on one side, the formation of partial oxidation productsdecreases the faradaic efficiency of the system, but, on the otherside, interesting chemicals can be produced.
A general reaction mechanism of the electro-oxidation of glycerolon bulk Au in alkaline media can be formulated as follows:9,12,13
a Instituto de Quımica de Sao Carlos, Universidade de Sao Paulo, Post-Box 780,
13560-970 Sao Carlos, Sao Paulo, Brazil. E-mail: [email protected];
Fax: +55 16 3373 9952b Instituto de Quımica, Universidade Federal do Rio Grande do Norte, Lagoa Nova,
59072-970, Natal, RN, Brazil
Received 21st January 2013,Accepted 10th April 2013
DOI: 10.1039/c3cp50280e
www.rsc.org/pccp
PCCP
PAPER
Dow
nloa
ded
by U
NIV
ER
SID
AD
SA
O P
AU
LO
on
14/0
5/20
13 1
3:34
:35.
Pu
blis
hed
on 1
1 A
pril
2013
on
http
://pu
bs.r
sc.o
rg |
doi:1
0.10
39/C
3CP5
0280
E
View Article OnlineView Journal

Phys. Chem. Chem. Phys. This journal is c the Owner Societies 2013
Here, the adsorbed intermediates of the electro-oxidation ofglycerol on Au are not represented. Step I corresponds to theformation of 3-carbon-atom chain products, such as dihydroxy-acetone (CH2OH–CO–CH2OH), glyceric acid (CH2OH–CHOH–COOH), tartronic acid (COOH–CHOH–COOH) and mesoxalicacid (COOH–CO–COOH). These products can diffuse to thebulk of the solution or, alternatively, be further oxidizedtowards the formation of 2-carbon-atom chain products (Step II)and 1-carbon-atom chain products (Step III), for example:glycolic acid (CH2OH–COOH), glyoxylic acid (CHO–COOH),formic acid (HCOOH) and carbon dioxide (CO2).
There has been a growing interest in gold as a catalyst sinceit presents, in an alkaline environment, a higher oxidationactivity than platinum.9 This interesting behavior is attributedto the high resistance of gold toward the formation of poisoningsurface oxides.14 Although it is known that the catalytic proper-ties of materials can be further improved by employing them inthe nanometric regime,15,16 the utilization of gold nanoparticlesfor the glycerol electro-oxidation has been barely investigated.4,17
In a heterogeneous liquid phase, glycerol oxidation overgold nanoparticles supported on different carbons and oxideshas been studied and an effect of the support on the catalystproperties has been evidenced.18 In particular, under the sameexperimental conditions and with comparable gold particlesize, gold catalysts supported on carbon have shown higheractivity in the liquid phase glycerol oxidation than the magnesiasupported gold catalyst.18 Recently, the effect of the support onthe catalyst properties has also been verified in an electro-chemical environment for formic acid oxidation on Au–Pdcore–shell nanoparticles supported on ITO and Vulcan XC-72R.19
Au–Pd core–shell nanoparticles supported on Vulcan XC-72R werefound to be about 2 times more active towards formic acid electro-oxidation than Au–Pd core–shell nanoparticles supported on ITO.19
Despite the fact that the carbon support influences reactions oncarbon-supported metal particles, investigations on the role of thecarbon support in the catalyst activity are scarce.20–23 The presenceof the carbon support is known to indirectly affect the electro-chemical properties of the carbon-supported metal particles due tochanges in the metal electronic structure caused by metal–supportinteractions.20,22 However, whether the carbon support directlyinfluences the catalyst activity is still unknown.
The aim of the present work is to show evidence of a directeffect of the carbon support on the electro-catalytic activity ofglassy-carbon-supported Au nanoparticles toward glycerol oxi-dation. Our results indicate that the glassy carbon support inconjunction with Au nanoparticles participates actively in theglycerol oxidation.
2. Experimental
AuCl3 (30% w/w in HCl), poly(vinyl pyrrolidone) (PVP), glyceroland NaNO3 were acquired from Aldrich Co. and used withoutfurther purification. A glassy carbon (GC) cylinder (Alfa Aesar,7 mm in diameter and 5 mm high) was polished with a 2000grid silicon carbide paper, followed by 9 mm and 3 mm alumina.The GC electrode was then sonicated for 10 min in ultrapure
Milipore/Milli-Q water in order to remove alumina residues,followed by 2 voltammetric cycles in the potential rangebetween 0.05 V and 1.7 V vs. the reversible hydrogen electrode(RHE) in an O2-free 0.1 mol L�1 NaOH solution to check forremaining impurities. The electrode was then placed intoanother electrochemical cell and a meniscus was formed witha solution consisting of 7.6 mmol L�1 AuCl3 + 20 g L�1 PVP +0.1 mol L�1 NaNO3. Gold nanoparticles were produced onto theclean GC by applying a potential of 0.25 V vs. Ag/AgCl for 0.1 s,0.5 s, 5 s, 15 s, 30 s and 45 s. A platinum foil served as a counterelectrode. After each electro-deposition, the Au-modified GCelectrode was thoroughly rinsed with ultrapure water, insertedinto a conventional three-electrode cell and subjected to onevoltammetric cycle between 0.05 V to 1.6 V vs. RHE at a scanrate of 0.05 V s�1 in an O2-free 0.1 mol L�1 NaOH solution atmeniscus configuration to determine the Au electrochemicalsurface area via oxide reduction charge, considering a conver-sion factor of 386 mA cm�2 for polycrystalline Au.24 Finally,glycerol oxidation on the Au-modified GC electrodes was per-formed in an O2-free 0.1 mol L�1 glycerol + 0.1 mol L�1 NaOHsolution at 0.05 V s�1 at meniscus configuration. The referenceelectrode was the RHE and all potentials in this work arereferred to it unless otherwise stated. The electrochemicalexperiments were carried out using a 1285 Solartron potentiostat/galvanostat controlled by the CoreWare software. Field emis-sion gun microscopy (FEG) was conducted on a FEG-VP ZeissSupra 35.
3. Results and discussion3.1 Electrochemical and physical characterization of theglassy carbon supported Au particles
Fig. 1 shows a cyclic voltammogram (CV) for the GC electrode ina solution of 7.6 mmol L�1 AuCl3 + 20 g L�1 PVP + 0.1 mol L�1
NaNO3 at a scan rate of 0.05 V s�1. Gold electro-depositionbegins at around 0.5 V developing a main peak at 0.25 V and ashoulder at about 0.12 V vs. Ag/AgCl, with the latter beingattributed to the reduction of adsorbed AuCl4
�.25 During theback scan, the voltammogram displays a current loop indicativeof a nucleation controlled process and no oxidation peak wasobserved, denoting the irreversibility of gold electro-depositionon a GC substrate. The potential was then held at 0.25 V vs.Ag/AgCl for distinct deposition times and here it is illustratedfor 5 s and 45 s (independent experiments). The presence ofgold was confirmed using an EDX (not shown) and cyclicvoltammetry of the deposit in aqueous 0.1 mol L�1 NaOH (insetof Fig. 1), with the typical response of polycrystalline Au (goldoxide formation and reduction peaks).
Fig. 2 depicts the effect of the deposition time on size anddistribution of the Au nanoparticles obtained at 0.25 V vs.Ag/AgCl for distinct times of 5 s and 45 s in the 7.6 mmol L�1
AuCl3 + 20 g L�1 PVP + 0.1 mol L�1 NaNO3 electrolyte. It is clearfrom Fig. 2A (electro-deposition time = 5 s) that the sphere-shaped Au particles are in the nanometric regime, typically50–250 nm, and nicely dispersed on the surface of the GCelectrode. By increasing the electro-deposition time to 45 s in
Paper PCCP
Dow
nloa
ded
by U
NIV
ER
SID
AD
SA
O P
AU
LO
on
14/0
5/20
13 1
3:34
:35.
Pu
blis
hed
on 1
1 A
pril
2013
on
http
://pu
bs.r
sc.o
rg |
doi:1
0.10
39/C
3CP5
0280
EView Article Online
This journal is c the Owner Societies 2013 Phys. Chem. Chem. Phys.
an independent experiment (Fig. 2B), the GC substrate isextensively populated with Au, but the particle size is practicallythe same. The sequence of images presented in Fig. 2 clearlyshows the tendency of the surface to become bulk Au at longtime polarization. Similar results were obtained by Sobri andRoy26 when studying the nucleation of gold on GC electrodes. Itis important to mention that the small particles of ca. 50 nmobserved for 5 s and 45 s deposition times (Fig. 2A and B) maybe at the growing stage, but their sizes are mainly limited toabout 250 nm, as seen for longer deposition times.
3.2 Glycerol electro-oxidation over glassy-carbon-supportedAu particles in alkaline medium
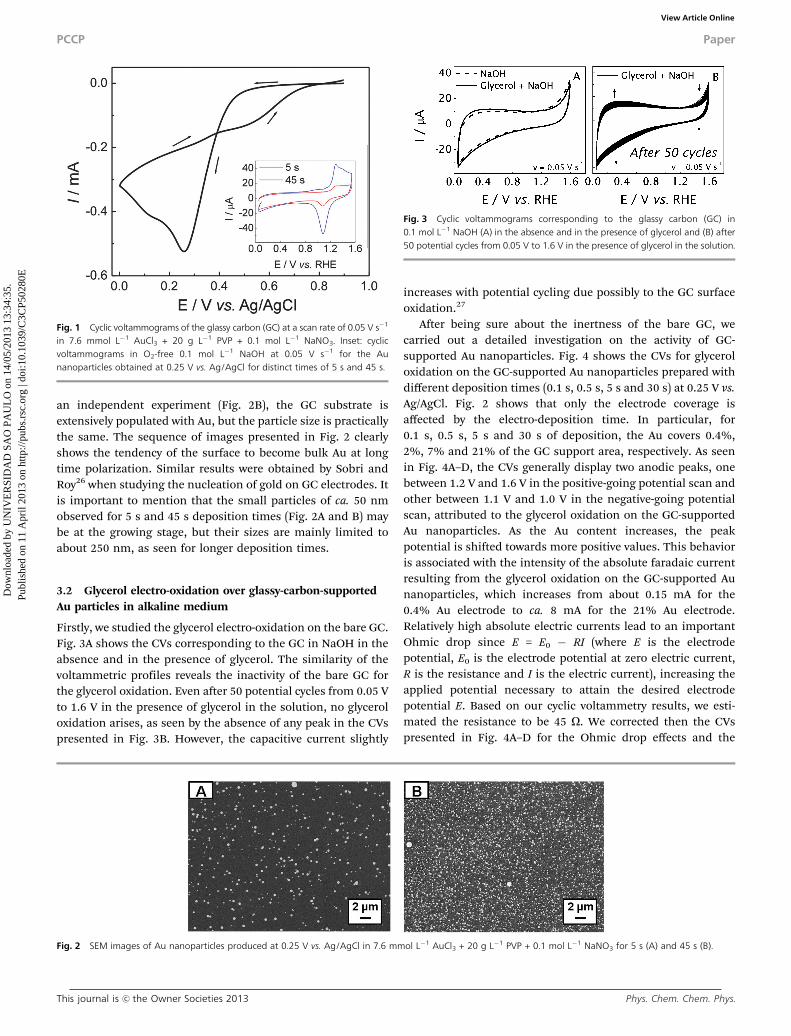
Firstly, we studied the glycerol electro-oxidation on the bare GC.Fig. 3A shows the CVs corresponding to the GC in NaOH in theabsence and in the presence of glycerol. The similarity of thevoltammetric profiles reveals the inactivity of the bare GC forthe glycerol oxidation. Even after 50 potential cycles from 0.05 Vto 1.6 V in the presence of glycerol in the solution, no glyceroloxidation arises, as seen by the absence of any peak in the CVspresented in Fig. 3B. However, the capacitive current slightly
increases with potential cycling due possibly to the GC surfaceoxidation.27
After being sure about the inertness of the bare GC, wecarried out a detailed investigation on the activity of GC-supported Au nanoparticles. Fig. 4 shows the CVs for glyceroloxidation on the GC-supported Au nanoparticles prepared withdifferent deposition times (0.1 s, 0.5 s, 5 s and 30 s) at 0.25 V vs.Ag/AgCl. Fig. 2 shows that only the electrode coverage isaffected by the electro-deposition time. In particular, for0.1 s, 0.5 s, 5 s and 30 s of deposition, the Au covers 0.4%,2%, 7% and 21% of the GC support area, respectively. As seenin Fig. 4A–D, the CVs generally display two anodic peaks, onebetween 1.2 V and 1.6 V in the positive-going potential scan andother between 1.1 V and 1.0 V in the negative-going potentialscan, attributed to the glycerol oxidation on the GC-supportedAu nanoparticles. As the Au content increases, the peakpotential is shifted towards more positive values. This behavioris associated with the intensity of the absolute faradaic currentresulting from the glycerol oxidation on the GC-supported Aunanoparticles, which increases from about 0.15 mA for the0.4% Au electrode to ca. 8 mA for the 21% Au electrode.Relatively high absolute electric currents lead to an importantOhmic drop since E = E0 � RI (where E is the electrodepotential, E0 is the electrode potential at zero electric current,R is the resistance and I is the electric current), increasing theapplied potential necessary to attain the desired electrodepotential E. Based on our cyclic voltammetry results, we esti-mated the resistance to be 45 O. We corrected then the CVspresented in Fig. 4A–D for the Ohmic drop effects and the
Fig. 1 Cyclic voltammograms of the glassy carbon (GC) at a scan rate of 0.05 V s�1
in 7.6 mmol L�1 AuCl3 + 20 g L�1 PVP + 0.1 mol L�1 NaNO3. Inset: cyclicvoltammograms in O2-free 0.1 mol L�1 NaOH at 0.05 V s�1 for the Aunanoparticles obtained at 0.25 V vs. Ag/AgCl for distinct times of 5 s and 45 s.
Fig. 2 SEM images of Au nanoparticles produced at 0.25 V vs. Ag/AgCl in 7.6 mmol L�1 AuCl3 + 20 g L�1 PVP + 0.1 mol L�1 NaNO3 for 5 s (A) and 45 s (B).
Fig. 3 Cyclic voltammograms corresponding to the glassy carbon (GC) in0.1 mol L�1 NaOH (A) in the absence and in the presence of glycerol and (B) after50 potential cycles from 0.05 V to 1.6 V in the presence of glycerol in the solution.
PCCP Paper
Dow
nloa
ded
by U
NIV
ER
SID
AD
SA
O P
AU
LO
on
14/0
5/20
13 1
3:34
:35.
Pu
blis
hed
on 1
1 A
pril
2013
on
http
://pu
bs.r
sc.o
rg |
doi:1
0.10
39/C
3CP5
0280
EView Article Online

Phys. Chem. Chem. Phys. This journal is c the Owner Societies 2013
corresponding corrected CVs are presented in Fig. 4E–H. Pos-sibly we worked with a small overcompensation of the resistant,seen by the atypical shape of the back peak of the CVspresented in Fig. 4G and H. By comparing the corrected CVswith the non-corrected CVs, it is evident that glycerol oxidationon electrodes with 7% and 21% Au coverage was significantlyaffected by Ohmic drop effects. Therefore, in the potentialregion corresponding to glycerol oxidation, when we apply0.8–1.6 V, it may actually correspond to a lower electrodepotential vs. RHE. In particular, at the maximum current, theapplied potential of 1.6 V may correspond to the electrodepotential of 1.2 V after the Ohmic drop correction.
Following we will analyze the voltammetric response (notcorrected for the Ohmic drop effects) of glycerol oxidation onGC-supported Au particles with potential cycling.
Fig. 5A and B show CVs for glycerol oxidation on the electrodewith 2% Au coverage (prepared with 0.5 s Au deposition time).When the meniscus is initially formed (black-line CV of Fig. 5A),a gradual electrode activation takes place upon potential cycling,as seen by increasing current density, up to the stabilization aftersome cycles.† After refreshing the solution at the electrode/solution interface by bubbling nitrogen in the bulk solution
and, in addition, by breaking/reassembling the meniscus, anincrease of the current density is observed (red-line CVs). Theseresults indicate that under initial experimental conditions someof the reaction products of the glycerol oxidation, less reactivethan glycerol itself, accumulate at the electrode interface par-tially inhibiting further glycerol oxidation. By refreshing thesolution at the electrode/solution interface, the relatively lowreactive species are quickly expelled from the interfacial regionliberating Au sites for oxidizing fresh glycerol. The oxidation ratecontinues to increase with potential cycling until reachingstabilization of the cyclic voltammogram after some cycles. Afterthe 4th solution refreshing (Fig. 5B), the electrode starts to beslowly deactivated by potential cycling.
Based on these results, it seems that two different effectsinfluence the glycerol oxidation rate over the GC-supported Aunanoparticles with potential cycling:� effect (1) is associated to the gradual and finite formation
of surface active species, which play an important role in thealcohol electro-catalysis;� effect (2) is related to the accumulation of inactive or hardly
active glycerol oxidation products at the electrode/solution interface.Effects (1) and (2) contribute positively (up to a limited extent)
and negatively to the overall faradaic current, respectively.One of the possible reasons for the electrode activation
[above-mentioned as effect (1)] could be due to electrode ageingby potential cycling. The ageing process results in surfacereconstruction with an increase of the mean particle size anda decrease of the surface active area and roughness.29,30 Thedriving force for the electrode ageing is the low stability ofsmall particles.30 However, in the present work, electrodeageing by potential cycling can be ruled out by several observa-tions: (i) the cyclic voltammograms of the GC-supported Aunanoparticles in NaOH taken just before and after the glyceroloxidation did not present any significant change concerningthe charges of the Au oxide reduction peak, as seen in Fig. 5C;(ii) as shown later in Fig. 12, species contributing to theelectrode activation are removed by abundantly rinsing theelectrode with water between two consecutive measurements,as seen by the electrode re-activation at the beginning of eachmeasurement. If Au particles had grown by potential cycling, noreversibility of the Au particle size would be expected by rinsingthe electrode with water; (iii) SEM images taken immediatelybefore (Fig. 6A) and after (Fig. 6B) the glycerol oxidation onGC-supported Au nanoparticles are quite similar, providingdirect evidence that no substantial change of the Au particlesize distribution occurred after potential cycling. The absence ofthe ageing process may be explained by the stability of thepresent GC-supported Au nanoparticles, which range between50 nm to 250 nm (as shown in Fig. 2). The reason why theelectrode is activated by potential cycling will be discussed later.
Fig. 7 presents the effect of potential cycling on the glyceroloxidation on the GC-supported Au electrode with 0.4% to 21%Au coverage. Fig. 7A and B shows electrode deactivationby potential cycling. Thus, effect (2) prevails over effect (1).This performance may be associated with lower real potentialsdue to Ohmic drop effects. The CVs presented in Fig. 7C–E
Fig. 4 First CVs in O2-free 0.1 mol L�1 glycerol + 0.1 mol L�1 NaOH at 0.05 V s�1
for glycerol oxidation on the GC-supported Au nanoparticles prepared withdifferent deposition times, specifically: 0.1 s (A), 0.5 s (B), 5 s (C) and 30 s (D).In (E–H) the CVs presented in (A–D), respectively, are corrected for the Ohmicdrop effect.
† It is very important to emphasize that the same electrode activation was alsoobserved for Au nanoparticles deposited onto HOPG without PVP and, inaddition, using a GC counter electrode instead of platinum. These resultsindicate that the effect of electrode activation by potential cycling is due neitherto PVP residue elimination nor to contamination from dissolution of the Ptcounter electrode.28
Paper PCCP
Dow
nloa
ded
by U
NIV
ER
SID
AD
SA
O P
AU
LO
on
14/0
5/20
13 1
3:34
:35.
Pu
blis
hed
on 1
1 A
pril
2013
on
http
://pu
bs.r
sc.o
rg |
doi:1
0.10
39/C
3CP5
0280
EView Article Online

This journal is c the Owner Societies 2013 Phys. Chem. Chem. Phys.
show electrode activation by potential cycling. Therefore, effect(1) predominates over effect (2). This behavior may be related tothe formation of oxygenated species on the GC surface at highpotentials. Fig. 7 also reveals that, after activation by potentialcycling, the electrode with 0.4% Au coverage (Fig. 7D) oxidizesglycerol as well as the electrode with more than 15 times its Aucoverage (Fig. 7B), as seen by quite similar current densities.After polishing the electrode with 0.4% Au coverage (Fig. 7E) avery low amount of gold probably remained. In fact, no trace ofAu is seen in the CV corresponding to the polished electrode inpure electrolytic solution (inset of Fig. 7E), which was obtainedimmediately before potential cycling in the presence of glycerol.Surprisingly, despite the very low amount of gold, the activationof the electrode by potential cycling in the presence of glycerolis still observed (Fig. 7E). However, no important change
(with respect to the characteristics of Au) in the CV of theelectrode in pure electrolyte was observed after the potentialcycles in the presence of glycerol (inset of Fig. 7E). The activa-tion of the electrode by potential sweeping in the presence ofglycerol was observed not only in potentiodynamic experi-ments, but also in potentiostatic ones, as seen in Fig. 8, byincreasing current intensity as a function of successive sets ofpotential steps. Furthermore, both effects (1) and (2) seem to beassociated with the presence of the carbon support, since thecyclic voltammograms of a bulk gold electrode in contact withglycerol are stable with potential cycling independently of theupper potential limit, as shown in Fig. 9B–E.
Based on the previous discussion, we propose that thecarbon support (in conjunction with Au particles) plays a rolein the oxidation of glycerol in alkaline medium, mainly related
Fig. 6 SEM images of Au nanoparticles produced at 0.25 V vs. Ag/AgCl in 7.6 mmol L�1 AuCl3 + 20 g L�1 PVP + 0.1 mol L�1 NaNO3 for 5 s deposition time takenbefore (A) and after (B) glycerol electro-oxidation on them.
Fig. 5 CVs in O2-free 0.1 mol L�1 glycerol + 0.1 mol L�1 NaOH at 0.05 V s�1 for glycerol oxidation on the GC-supported Au nanoparticles prepared with 0.5 sdeposition time (A) and (B). In (A) and (B), ‘‘refreshing’’ corresponds to solution refreshing at the electrode/solution interface by bubbling nitrogen in the bulk solutionand, in addition, by breaking/remaking the meniscus. (C) CVs in O2-free 0.1 mol L�1 NaOH at 0.05 V s�1 before and after potential cycling in the presence of glycerol inthe solution.
PCCP Paper
Dow
nloa
ded
by U
NIV
ER
SID
AD
SA
O P
AU
LO
on
14/0
5/20
13 1
3:34
:35.
Pu
blis
hed
on 1
1 A
pril
2013
on
http
://pu
bs.r
sc.o
rg |
doi:1
0.10
39/C
3CP5
0280
EView Article Online

Phys. Chem. Chem. Phys. This journal is c the Owner Societies 2013
to the glycerol oxidation rate, not with the overpotential of theoxidation reaction. Active species contributing to effect (1) arepossibly formed on the carbon surface at high potentials duringthe positive-going potential scan. These species may be addi-tional oxygen suppliers to the oxidation of the adsorbatesresulting from the glycerol adsorption on Au particles duringthe following negative- and positive-going potential scans.
In this way, the overpotential of the oxidation reaction islimited by the dissociative adsorption of glycerol on the Aunanoparticles. The layer of oxygenated species may be graduallyformed up to the saturation of the exposed carbon area. Sinceimportant Ohmic drop effects were observed for glycerol oxida-tion on GC-supported Au electrodes with 21% (A) and 7% (B) Aucoverage, the formation of oxygenated species on the carbonsupport may be hindered and this could explain why effect (2)prevailed over effect (1) on these electrodes.
We have also investigated the glycerol oxidation over poly-crystalline Pt and GC-supported Pt particles in order to check
Fig. 7 CVs in O2-free 0.1 mol L�1 glycerol + 0.1 mol L�1 NaOH at 0.05 V s�1 for glycerol oxidation on the GC-supported Au electrodes with 21% (A), 7% (B), 2% (C),0.4% (D) Au coverage and the mechanically polished GC-supported Au electrode with 0.4% Au coverage (E), inset: CV in O2-free 0.1 mol L�1 NaOH at 0.05 V s�1 beforeand after potential cycling in the presence of glycerol in the solution.
Fig. 8 Cronoamperograms of O2-free 0.1 mol L�1 glycerol + 0.1 mol L�1 NaOHon the mechanically polished GC-supported Au electrode, taken at 1.3 V vs. RHE.Potential was stepped from 0.05 V (t = 5 s) to 1.3 V (t = 30 s) to 1.6 V (t = 5 s) vs.RHE. The black, red, green, blue and cyano curves correspond to the first, second,third, fourth, and fifth set of potential steps, respectively.
Fig. 9 CVs of a polycrystalline bulk Au electrode in O2-free 0.1 mol L�1 NaOH inthe absence (A) and presence (B–E) of 0.1 mol L�1 glycerol, at 0.05 V s�1.
Paper PCCP
Dow
nloa
ded
by U
NIV
ER
SID
AD
SA
O P
AU
LO
on
14/0
5/20
13 1
3:34
:35.
Pu
blis
hed
on 1
1 A
pril
2013
on
http
://pu
bs.r
sc.o
rg |
doi:1
0.10
39/C
3CP5
0280
EView Article Online

This journal is c the Owner Societies 2013 Phys. Chem. Chem. Phys.
whether the carbon support effects were specific for Au. Resultsof this investigation are shown in Fig. 10.
As observed for bulk gold, the CVs corresponding to theelectro-oxidation of glycerol over a bulk platinum electrode inalkaline medium are also stable with potential cycling(Fig. 10A). In opposition, Fig. 10B and C show that the CVs ofglycerol electro-oxidation over GC-supported Pt particles inalkaline medium vary with potential cycling. Finally, as seenfor GC-supported gold particles, the GC-supported Pt particleselectrode is also activated even with very low amount of Pt onthe support (Fig. 10C). This additional evidence reinforces thatthe carbon support (in conjunction with Au or Pt particles) plays
a role in the glycerol oxidation in alkaline medium. Moreover,the effects associated with the carbon support are not specific forcarbon supported Au particles, but it seems to be generallyapplicable for carbon-supported active metal particles.
Pt was not only used to confirm whether the observed effectis specific to Au, but also to verify whether it is specific to thealkaline media, since Au is hardly active in acidic media.31 Asshown in Fig. 11, the effects correlated with the carbon supportare also present for the glycerol oxidation under acidic solutionconditions, but much less pronounced than in alkaline medium.It is known that both alcohol reactivity and metal activity areinfluenced by the pH.9,12,14,32,33 In alkaline media, gold
Fig. 10 CVs of a polycrystalline bulk Pt electrode in O2-free 0.1 mol L�1 glycerol + 0.1 mol L�1 NaOH at 0.05 V s�1 (A), inset: CV in O2-free 0.1 mol L�1 NaOH at 0.05 V s�1;CVs in O2-free 0.1 mol L�1 glycerol + 0.1 mol L�1 NaOH at 0.05 V s�1 for glycerol oxidation on (B) GC-supported Pt nanoparticles prepared with 20 s deposition time andon (C) GC-supported Pt nanoparticles prepared with 20 s deposition time after mechanical polishing. In (B), ‘‘refreshing’’ corresponds to solution refreshing at theelectrode/solution interface by bubbling nitrogen in the bulk solution and, in addition, by breaking/remaking the meniscus.
Fig. 11 (A) CVs at 0.05 V s�1 of a polycrystalline bulk Pt electrode in O2-free 0.1 mol L�1 HClO4 in the absence (dashed line) and presence (solid line) of 0.1 mol L�1
glycerol and (B) CVs at 0.05 V s�1 of GC-supported Pt nanoparticles prepared with 5 s deposition time in O2-free 0.1 mol L�1 HClO4 in the absence (black line) andpresence (red and green lines) of 0.1 mol L�1 glycerol; In (B), ‘‘refreshing’’ corresponds to solution refreshing at the electrode/solution interface by bubbling nitrogenin the bulk solution and, in addition, by breaking/remaking the meniscus.
PCCP Paper
Dow
nloa
ded
by U
NIV
ER
SID
AD
SA
O P
AU
LO
on
14/0
5/20
13 1
3:34
:35.
Pu
blis
hed
on 1
1 A
pril
2013
on
http
://pu
bs.r
sc.o
rg |
doi:1
0.10
39/C
3CP5
0280
EView Article Online

Phys. Chem. Chem. Phys. This journal is c the Owner Societies 2013
interacts much more with glycerol and other alcohols than inacidic medium. It has been shown that the glycerol oxidationreaction on gold is significantly favored with increasing thepH.9,12,32 This positive effect of the high pH has also beenobserved for Pt.9,12,32 Under acidic conditions, however, onlyplatinum exhibits catalytic activity, while Au completely lacks itscatalytic properties. One of the explanations for the distinctbehavior observed for Au and Pt in alkaline medium is that theybecome covered by OH� species above the zero-charge potentialand, through the formation of hydrogen bridges, Au–OH andPt–OH interact with alcohol molecules that can be further oxi-dized.34 In addition to that, the acid–base equilibrium involvingthe alcoholic group of alcohols may also contribute to the highcatalytic activities in alkaline media. At high pH, alcohol moleculesdeprotonate and the deprotonated species (negatively charged) arebelieved to be the main reactant in alkaline media.12
The electro-oxidation of ethylene glycol in alkaline mediumover GC-supported Au nanoparticles was also investigated inorder to make sure whether the influence of the carbon supportis specific to glycerol or not. Results of this study, which arepresented in Fig. 12, reveal that the behavior of the electrodewith 0.4% Au coverage in ethylene glycol is similar to thatobserved in glycerol. Then, we affirm that the carbon support(in conjunction with metal particles) influences not only theelectro-oxidation of glycerol, but also ethylene glycol and pos-sibly other alcohols.
Another important aspect of the results presented in Fig. 12is that by abundantly rinsing the electrode with water between
two consecutive measurements, species contributing to theelectrode activation are removed, as seen by the electrodere-activation at the beginning of each measurement, shown inFig. 12B–D. This performance is completely different comparedwith that observed when solution at the electrode/solutioninterface was refreshed by bubbling nitrogen in the bulksolution and, in addition, by breaking/remaking the meniscus.Unlike rinsing the electrode with water, solution refreshingdoes not remove species contributing to the electrode activa-tion by potential cycling in the presence of alcohols. Therefore,based on these results, we infer that the species contributing tothe electrode activation are weakly bonded to the carbonsupport. These species may be formed at early stages of carbonoxidation, most likely C–OH.35
In addition to preparing the Au nanoparticles throughelectro-deposition, the GC-supported Au was also prepared bysputtering a 1 nm thick Au layer on the glassy carbon support.In this technique, argon ions are accelerated against a ‘‘target’’,which is the metal source. In the collision, metal atoms areejected from the ‘‘target’’ and physically deposited onto asubstrate. The thickness of the metallic deposit is controlledby the mass of the deposited metal layer through the metaldensity. The sputtered Au was then applied for the glycerolelectro-oxidation in alkaline medium, as shown in Fig. 13.
As seen in Fig. 13A, the electrode prepared by the sputteringmethod deactivates with potential cycling in the presence ofglycerol in alkaline medium. This behavior is comparable tothat observed for the GC-supported Au electrode with 21% Au
Fig. 12 CVs at 0.05 V s�1 of a GC-supported Au electrode with 0.4% Au coverage in (A and C) O2-free 0.1 mol L�1 ethylene glycol + 0.1 mol L�1 NaOH and in (B and D)O2-free 0.1 mol L�1 glycerol + 0.1 mol L�1 NaOH. In (A) and (B), ‘‘refreshing’’ corresponds to solution refreshing at the electrode/solution interface by bubbling nitrogen inthe bulk solution and, in addition, by breaking/remaking the meniscus. Note: (A–D) are consecutive measurements and between (A) and (B), (B) and (C),(C) and (D), the GC-supported Au nanoparticle electrode was abundantly rinsed with Milli-Q water.
Paper PCCP
Dow
nloa
ded
by U
NIV
ER
SID
AD
SA
O P
AU
LO
on
14/0
5/20
13 1
3:34
:35.
Pu
blis
hed
on 1
1 A
pril
2013
on
http
://pu
bs.r
sc.o
rg |
doi:1
0.10
39/C
3CP5
0280
EView Article Online

This journal is c the Owner Societies 2013 Phys. Chem. Chem. Phys.
coverage under the same experimental conditions (Fig. 7A). Onthe other hand, the response of the mechanically polishedGC-supported Au nanoparticles prepared by the sputteringmethod is particular, as seen in Fig. 13B. After polishing, unlikethe electrode prepared by gold electro-deposition (Fig. 7E), theelectrode prepared by the sputtering method does notre-activate with potential cycling. The sputtered gold must havebeen completely removed from the GC surface, accounting forthe lack of activity towards glycerol oxidation. Differently, theelectro-deposited Au nanoparticles might also be located in thenanostructure of the carbon support. Upon polishing, Auparticles on the surface of the carbon support may be removed,but some amount of gold may be enclosed into the GCmolecular structure. In conjunction, gold particles (in themolecular structure of the carbon support) and the carbonsupport show activation with potential cycling. These resultsdemonstrate that both gold particles and the carbon supportmay play complementary roles in the electro-oxidation ofalcohols, possibly through a mechanism consisting of twoactive elements, in which Au adsorbs oxygenated species andalcohol molecules and the carbon support acts as an additionaloxygen source for the oxidation of alcohol residues adsorbed onAu since its surface is partially oxidized.
A commercial Ti/Ru0.3Ti0.7O2 dimensionally stable anode(DSA) was also employed as a support for the Au nanoparticles.DSA-supported Au nanoparticles were prepared with 0.1 s Auelectro-deposition time and the resulting electrode was appliedto the glycerol oxidation in alkaline medium. The results of thisstudy are presented in Fig. 14.
DSA is hardly active towards the glycerol oxidation in alkalinemedium, as seen by the similarity between the CVs in thepresence and absence of glycerol (Fig. 14A). Some glyceroloxidation on the DSA is evidenced only above 1.2 V (red line inFig. 14A). In the presence of gold particles, however, the DSA-supported Au nanoparticles present relatively high activity to theglycerol oxidation (Fig. 14B). Additionally, comparable to theGC-supported Au nanoparticles prepared with 0.1 s deposition time,the DSA-supported Au nanoparticles prepared with 0.1 s depositiontime also show electrode activation with potential cycling.
Ruthenium and titanium oxides, which are elements of theDSA plate, are known to be active species in the CO and alcoholoxidation over Pt or Au-based catalysts.36–38 In particular,Iwasita et al.36 proposed that methanol is oxidized over PtRuelectrodes through a bifunctional mechanism, in which theadsorption of methanol occurs preferentially on platinum, butalso on ruthenium, and the formation of oxygenated species
Fig. 13 CVs in O2-free 0.1 mol L�1 glycerol + 0.1 mol L�1 NaOH at 0.05 V s�1 on (A) an electrode prepared by sputtering a 1 nm thick Au layer on the GC and (B) aftermechanically polishing the sputtered electrode. In (A), ‘‘refreshing’’ corresponds to solution refreshing at the electrode/solution interface by bubbling nitrogen in thebulk solution and, in addition, by breaking/remaking the meniscus.
Fig. 14 (A) CVs in O2-free 0.1 mol L�1 NaOH at 0.05 V s�1 over the DSA in the absence (black line) and presence (red line) of 0.1 mol L�1 glycerol and (B) CVs inO2-free 0.1 mol L�1 glycerol + 0.1 mol L�1 NaOH over DSA supported Au nanoparticles prepared with 0.1 s deposition time; inset: CVs in O2-free 0.1 mol L�1 NaOH overDSA supported Au nanoparticles prepared with 0.1 s deposition time; scan rate = 0.05 V s�1.
PCCP Paper
Dow
nloa
ded
by U
NIV
ER
SID
AD
SA
O P
AU
LO
on
14/0
5/20
13 1
3:34
:35.
Pu
blis
hed
on 1
1 A
pril
2013
on
http
://pu
bs.r
sc.o
rg |
doi:1
0.10
39/C
3CP5
0280
EView Article Online

Phys. Chem. Chem. Phys. This journal is c the Owner Societies 2013
occurs on the latter at lower potentials than on platinum.Ruthenium oxides act as the oxygen supplier, providing oxygenatoms to the oxidation of methanol residues adsorbed on Ptand Ru, such as CO. Therefore, in the presence of the secondelement, the onset potential is shifted toward lower overpoten-tials. In the present work, we believe that ruthenium andtitanium oxides may also act as oxygen suppliers for theoxidation of glycerol residues adsorbed on the DSA-supportedAu nanoparticles electrode. By potential cycling, the electrodemay be activated due to the continuous formation of activeoxides on the DSA. Unlike the bifunctional mechanism, theonset potential of the glycerol oxidation on DSA-supported Aunanoparticles is not shifted toward lower overpotentials. Thebeginning of the oxidation reaction may be limited by thedissociative adsorption of glycerol on the Au nanoparticles.
Based on this indirect evidence, we propose that oxygenatedspecies gradually formed on the surface of the glassy carbon bypotential cycling (up to the saturation of the carbon area)directly influence the glycerol electro-oxidation, acting as addi-tional oxygen suppliers to the oxidation of glycerol residuesadsorbed on the Au particles. Alternatively, we cannot discardthe possibility of the CO (e.g. from carbon support oxidation athigh potentials) promoting effect as that observed by Koper andco-workers for methanol electro-oxidation on the COad–Au(111)surface.39
The mechanism we suggest consists of two active elements:Au and carbon. In this mechanism, Au particles and oxygenatedspecies formed on carbon play complementary roles:
CH2OH–CHOH–CH2OH + Au(H2O) -
Au(CH2OH–CHOH–CH2OH)ads + H2O (1)
Au(H2O) - Au–OH + H+ + e� (2)
C(H2O) - C–OH + H+ + e� (3)
Au(CH2OH–CHOH–CH2OH)ads + Au–OH + C–OH - Au
+ C + products + ne� (4)
where Au is a gold site, and C is a carbon site.In steps (2) and (3) it is proposed that oxygenated species are
formed on Au and carbon by water splitting, but other possi-bilities cannot be excluded (e.g., from OH� in solution).
Glassy carbon is know to be active towards the oxygenreduction reaction (ORR).40 Since the mechanism of this reac-tion involves interaction between oxygen molecules and thecarbon surface, it is reasonable to propose that the carbonsurface may also interact with oxygen from water or OH�.
We suggest that the formation of glycerol electro-oxidationproducts involving extra oxygen atoms (with respect to theoriginal glycerol molecule) may be more favored on theGC-supported Au particles than on bulk Au due to the availabilityof additional oxygenated species formed on the carbon support.Further FTIR spectroscopy and/or liquid chromatography experi-ments would be necessary to confirm this hypothesis.
4. Conclusions
The oxidation of glycerol over glassy carbon-supported Aunanoparticles in alkaline medium was investigated using cyclicvoltammetry. The support coverage with gold nanoparticles wasvaried from less than 0.4% up to 30% of gold, maintainingregular particle size distribution. Scanning electron microscopyanalyses show that the mean particle size under our workingconditions was about 200 nm. We verified that there is an effectof the carbon support on the activity of the GC-supported Aunanoparticles. Results from studies about the oxidation ofglycerol and ethylene glycol on Au and Pt nanoparticles sup-ported on the glassy carbon (GC), highly ordered pyrolyticgraphite (HOPG) and dimensionally stable anode (DSA) underdifferent pH conditions, indicate that the carbon supportparticipates actively in the oxidation of glycerol and otheralcohols. We propose that active oxygenated species are graduallyformed on the glassy carbon surface by potential cycling (up tothe saturation of the surface layer) and these oxygenated specieson carbon are additional oxygen suppliers to the oxidation ofglycerol residues adsorbed on the Au particles, following amechanism consisting of two active elements: gold and carbon.
Acknowledgements
The authors thank CNPq and FAPESP for the overall support ofthis research. We thank Jonas Garcia Ferreira Jr. and Dr MauroRoberto Fernandes for assistance with experiments during theinitial stages of this work.
References
1 R. L. Arechederra, B. L. Treu and S. D. Minteer, J. PowerSources, 2007, 173, 156–161.
2 A. Ilie, M. Simoes, S. Baranton, C. Coutanceau andS. Martemianov, J. Power Sources, 2011, 196, 4965–4971.
3 Z. Zhang, L. Xin and W. Li, Appl. Catal., B, 2012, 119, 40–48.4 M. Simoes, S. Baranton and C. Coutanceau, Appl. Catal., B,
2010, 93, 354–362.5 M. Avramov-Ivic, J. M. Leger, B. Beden, F. Hahn and
C. Lamy, J. Electroanal. Chem., 1993, 351, 285–297.6 L. Roquet, E. M. Belgsir, J. M. Leger and C. Lamy, Electrochim.
Acta, 1994, 39, 2387–2394.7 G. Yildiz and F. Kadirgan, J. Electrochem. Soc., 1994, 141,
725–730.8 Y. Kwon and M. T. M. Koper, Anal. Chem., 2010, 82, 5420–5424.9 J. Gomes and G. Tremiliosi-Filho, Electrocatalysis, 2011, 2,
96–105.10 D. Z. Jeffery and G. A. Camara, Electrochem. Commun., 2010,
12, 1129–1132.11 J. F. Gomes, F. B. C. de Paula, L. H. S. Gasparotto and
G. Tremiliosi-Filho, Electrochim. Acta, 2012, 76, 88–93.12 Y. Kwon, K. J. P. Schouten and M. T. M. Koper,
ChemCatChem, 2011, 3, 1176–1185.13 C. Martins, M. J. Giz and G. A. Camara, Electrochim. Acta,
2011, 56, 4549–4553.
Paper PCCP
Dow
nloa
ded
by U
NIV
ER
SID
AD
SA
O P
AU
LO
on
14/0
5/20
13 1
3:34
:35.
Pu
blis
hed
on 1
1 A
pril
2013
on
http
://pu
bs.r
sc.o
rg |
doi:1
0.10
39/C
3CP5
0280
EView Article Online

This journal is c the Owner Societies 2013 Phys. Chem. Chem. Phys.
14 Y. Kwon, S. C. S. Lai, P. Rodriguez and M. T. M. Koper, J. Am.Chem. Soc., 2011, 133, 6914.
15 M. C. Daniel and D. Astruc, Chem. Rev., 2004, 104, 293–346.16 M. Haruta and M. Date, Appl. Catal., A, 2001, 222,
427–437.17 C. P. Fu, H. H. Zhou, D. Xie, L. Sun, Y. F. Yin, J. H. Chen and
Y. F. Kuang, Colloid Polym. Sci., 2010, 288, 1097–1103.18 S. Demirel-Gulen, M. Lucas and P. Claus, Catal. Today, 2005,
102–103, 166–172.19 V. Celorrio, M. G. M. de Oca, D. Plana, R. Moliner,
M. J. Lazaro and D. J. Fermin, J. Phys. Chem. C, 2012, 116,6275–6282.
20 E. Antolini, Appl. Catal., B, 2009, 88, 1–24.21 L. Calvillo, V. Celorrio, R. Moliner and M. J. Lazaro, Mater.
Chem. Phys., 2011, 127, 335–341.22 L. Calvillo, M. Gangeri, S. Perathoner, G. Centi, R. Moliner
and M. J. Lazaro, Int. J. Hydrogen Energy, 2011, 36,9805–9814.
23 V. Celorrio, M. G. M. de Oca, D. Plana, R. Moliner, D. J.Fermin and M. J. Lazaro, Int. J. Hydrogen Energy, 2012, 37,7152–7160.
24 G. Tremiliosi-Filho, L. H. Dall’Antonia and G. Jerkiewicz,J. Electroanal. Chem., 2005, 578, 1–8.
25 F. M. El-Cheick, F. A. Rashwan, H. A. Mahmoud andM. El-Rouby, J. Solid State Electrochem., 2010, 14, 1425–1443.
26 S. Sobri and S. Roy, J. Eng. Sci. Technol., 2008, 3, 62.27 J. Maruyama and I. Abe, Electrochim. Acta, 2001, 46,
3381–3386.
28 A. I. Yanson, P. Rodriguez, N. Garcia-Araez, R. V. Mom,F. D. Tichelaar and M. T. M. Koper, Angew. Chem., Int. Ed.,2011, 50, 6346–6350.
29 C. Grolleau, C. Coutanceau, F. Pierre and J. M. Leger,Electrochim. Acta, 2008, 53, 7157–7165.
30 S. N. Pron’kin, O. A. Petrii, G. A. Tsirlina and D. J. Schiffrin,J. Electroanal. Chem., 2000, 480, 112–119.
31 J. F. Gomes and G. Tremiliosi-Filho, Electrocatalysis, 2011, 2,96–105.
32 A. Kahyaoglu, B. Beden and C. Lamy, Electrochim. Acta,1984, 29, 1489–1492.
33 S. C. S. Lai, S. E. F. Kleijn, F. T. Z. Ozturk, V. C. V. Vellinga,J. Koning, P. Rodriguez and M. T. M. Koper, Catal. Today,2010, 154, 92–104.
34 P. Ocon, C. Alonso, R. Celdran and J. Gonzalez-Velasco,J. Electroanal. Chem., 1986, 206, 179–196.
35 B. Avasarala, R. Moore and P. Haldar, Electrochim. Acta,2010, 55, 4765–4771.
36 T. Iwasita, H. Hoster, A. John-Anacker, W. F. Lin andW. Vielstich, Langmuir, 2000, 16, 522–529.
37 G. A. Camara, R. B. de Lima and T. Iwasita, J. Electroanal.Chem., 2005, 585, 128–131.
38 B. E. Hayden, D. Pletcher and J. P. Suchsland, Angew. Chem.,Int. Ed., 2007, 46, 3530–3532.
39 P. Rodriguez, Y. Kwon and M. T. M. Koper, Nat. Chem.,2012, 4, 177–182.
40 A. Garcia, L. S. Gasparotto, J. Gomes and G. Tremiliosi-Filho,Electrocatalysis, 2012, 3, 147–152.
PCCP Paper
Dow
nloa
ded
by U
NIV
ER
SID
AD
SA
O P
AU
LO
on
14/0
5/20
13 1
3:34
:35.
Pu
blis
hed
on 1
1 A
pril
2013
on
http
://pu
bs.r
sc.o
rg |
doi:1
0.10
39/C
3CP5
0280
EView Article Online





























