Embed Size (px)
Citation preview

HN
YHMA*CCU
BgndbwMpHptopa7tDiSrtpmgiHmPsdPnt
Hcc
GASTROENTEROLOGY 2006;130:312–322
eterozygous Mutations in PMS2 Cause Hereditaryonpolyposis Colorectal Carcinoma (Lynch Syndrome)
VONNE M. C. HENDRIKS,* SHANTIE JAGMOHAN–CHANGUR,* HELEEN M. VAN DER KLIFT,*ANS MORREAU,‡ MARJO VAN PUIJENBROEK,‡ CARLI TOPS,* THEO VAN OS,§ ANJA WAGNER,¶
ARGREET G. F. M. AUSEMS,� ENCARNA GOMEZ,§ MARTIJN H. BREUNING,*NNETTE H. J. T. BRÖCKER–VRIENDS,* HANS F. A. VASEN,#,** and JUUL TH. WIJNEN*Center for Human and Clinical Genetics, ‡Department of Pathology, and #Department of Gastroenterology, Leiden University Medicalenter, Leiden; §Department of Clinical Genetics South-East Netherlands, Maastricht University Medical Center, Maastricht; ¶Department oflinical Genetics, Erasmus University Medical Center, Rotterdam; �Department of Medical Genetics, University Medical Center Utrecht,
trecht; and **Foundation for the Detection of Hereditary Tumours, Leiden, The NetherlandstgcmMH
hsMpctfles
tcttpss
bpfi
env
ackground & Aims: The role of the mismatch repairene PMS2 in hereditary nonpolyposis colorectal carci-oma (HNPCC) is not fully clarified. To date, only 7ifferent heterozygous truncating PMS2 mutations haveeen reported in HNPCC-suspected families. Our aimas to further assess the role of PMS2 in HNPCC.ethods: We performed Southern blot analysis in 112atients from MLH1-, MSH2-, and MSH6-negativeNPCC-like families. A subgroup (n � 38) of theseatients was analyzed by denaturing gradient gel elec-rophoresis (DGGE). In a second study group consistingf 775 index patients with familial colorectal cancer, weerformed immunohistochemistry using antibodiesgainst MLH1, MSH2, MSH6, and PMS2 proteins. In 8 of75 tumors, only loss of PMS2 expression was found. Inhese cases, we performed Southern blot analysis andGGE. Segregation analysis was performed in the fam-
lies with a (possibly) deleterious mutation. Results:even novel mutations were identified: 4 genomic rear-angements and 3 truncating point mutations. Three ofhese 7 families fulfill the Amsterdam II criteria. Theattern of inheritance is autosomal dominant with ailder phenotype compared with families with patho-
enic MLH1 or MSH2 mutations. Microsatellite instabil-ty and immunohistochemical analysis performed inNPCC-related tumors from proven carriers showed aicrosatellite instability high phenotype and loss ofMS2 protein expression in all tumors. Conclusions: Wehow that heterozygous truncating mutations in PMS2o play a role in a small subset of HNPCC-like families.MS2 mutation analysis is indicated in patients diag-osed with a colorectal tumor with absent staining forhe PMS2 protein.
ereditary nonpolyposis colorectal carcinoma (HNPCC)is characterized by the development of colorectal
arcinoma, endometrial carcinoma, and various other
ancers at an early age of onset and is caused by muta-ions in 1 of the 4 DNA mismatch repair (MMR)enes.1–5 In 50%–85% of the families fulfilling thelinical Amsterdam criteria for HNPCC,6,7 germlineutations have been reported in MLH1, MSH2, orSH6.8–10 Whether PMS2 plays a role in the etiology ofNPCC is uncertain.PMS2 is a MutL homologue MMR gene located on
uman chromosome 7p22 and is involved in repair ofingle base mismatches and insertion-deletion loops.3,11
ice deficient for Pms2 are prone to sarcomas and lym-homas but not intestinal adenomas and carcinomas, inontrast to mice deficient for Mlh1 or Msh2.12–14 Theseumors show microsatellite instability (MSI), which re-ects the inability of the MMR machinery to repairrrors that occur during DNA replication. These findingsuggest a role for PMS2 in carcinogenesis.
Mutation analysis of PMS2 is complicated by the facthat PMS2 is part of a family of highly homologous geneslustered on chromosome 7. There are up to 15 nonfunc-ional PMS2-related genes on the long arm resemblinghe 5= part of PMS2 and 1 nearly 100% homologousseudogene of the 3= part of the gene (exon 9 and 11–15)ituated about 700 kilobases centromeric of PMS2 on thehort arm of chromosome 7.15
So far, 7 different truncating PMS2 mutations haveeen described in heterozygous form in HNPCC-sus-ected families.3,16–18 The first patient belonged to aamily suspected of having HNPCC, but a detailed fam-ly history was not reported.3 In addition, PMS2 muta-
Abbreviations used in this paper: DGGE, denaturing gradient gellectrophoresis; HNPCC, hereditary nonpolyposis colorectal carci-oma; MMR, mismatch repair; MSI, microsatellite instability; VUCS,ariant with unknown clinical significance.
© 2006 by the American Gastroenterological Association0016-5085/06/$32.00
doi:10.1053/j.gastro.2005.10.052

tccchTiplPNmhcratebwaasftv
r
i
pTpMimePa
iiOacim7M
pm
tma
fir
Htrawrpkwngp
wopNcPcmawiisa1amidr
tsB(
February 2006 PMS2 MUTATIONS IN HNPCC 313
ions were found in 2 patients diagnosed with colorectalarcinoma at 28 and 22 years of age, respectively.16 Botholorectal carcinomas showed loss of immunohisto-hemical staining for PMS2. Only the first patientad a family history of colorectal carcinoma. Recently,runinger et al reported 3 truncating PMS2 mutations
n 6 HNPCC-suspected families. These investigatorserformed immunohistochemical analysis in 1048 se-ected colorectal carcinomas. In 16 cases (1.5%), loss ofMS2 expression in combination with MSI was found.one of the 6 families in which a truncating PMS2utation was identified fulfilled the Amsterdam criteria;
owever, 5 of the 6 families did fulfill the Bethesdariteria. No segregation analyses were performed in theeported families.17 Worthly et al described 1 HNPCC-ffected family that fulfilled the revised Amsterdam cri-eria with a PMS2 mutation segregating with the dis-ase.18 Moreover, 3 families have been described withiallelic deleterious PMS2 mutations.15,19 These familiesere characterized by the combination of brain tumors
nd colorectal adenomas and carcinomas at a young age,syndrome referred to as Turcot’s syndrome.20,21 Other
tudies in cohorts totaling about 200 HNPCC-suspectedamilies did not show any truncating PMS2 muta-ions.22–24 Therefore, these authors questioned the in-olvement of PMS2 in the pathogenesis of HNPCC.
The aim of the present study was to further define theole of PMS2 in familial colorectal carcinoma.
Patients and MethodsMutation analysis for PMS2 mutations was performed
n 2 groups of patients.The first group comprised 112 patients from families sus-
ected of HNPCC, 44 of which fulfill the Amsterdam criteria.his group of 112 patients originates from a group of 184atients in which mutation analysis of MLH1, MSH2, andSH6 has been performed previously.25,26 The 112 patients,
n whom no germline mutation in one of the previouslyentioned MMR genes was found, were analyzed with South-
rn blot hybridization for large genomic rearrangements inMS2. In a subgroup of these families (the first 38 of 112), welso performed scanning for point mutations in PMS2.
The second group was selected from a tissue microarraymmunohistochemistry study comprising 775 different (famil-al) cases of colorectal carcinoma and endometrial carcinoma.27
f these 775 cases, 406 fulfilled the revised Bethesda criteriand 76 did not.28 Of the 406 cases fulfilling the Bethesdariteria, 88 fulfilled the Amsterdam II criteria. Of the remain-ng 293 cases suspected of having HNPCC, no detailed infor-ation on family history was available. Tumors from 8 of these
75 patients showed loss of PMS2 expression and retained
LH1, MSH2, and MSH6 expression. In these 8 patients, we herformed Southern blot analysis followed by a search for pointutations.When a genomic rearrangement or a truncating point mu-
ation was detected, DNA and tumor material from familyembers were collected for mutation analysis, MSI analysis,
nd immunohistochemistry studies.In total, 95 spouses from the MMR mutation–positive
amilies were used as controls to test whether variants occurredn the “healthy” population. All controls and cases with colo-ectal carcinoma are of Dutch descent.
Southern Blot Analysis
Southern blot analysis was performed with MunI andindIII digests, followed by hybridization with 2 complemen-
ary DNA probes encompassing PMS2 exon 6–11 and 11–15,espectively. These probes detect fragments containing PMS2nd the pseudogene homologous with the 3= part of PMS2,hich can be distinguished from each other by differences in
estriction fragment size. Rearrangements occurring in the 5=art of the PMS2 will be detected by alterations of the 12.8-ilobase-long exon 1–7 containing MunI restriction fragmenthen hybridized with the exon 6–11 probe. The numerousearly identical copies of exon 1–5 present in the humanenome do not allow the use of a probe derived from the 5=art of the gene.29
Point Mutation Analysis
All exons, including their intron-exon boundaries,ith the exception of exon 2, were analyzed by direct nucle-tide sequencing on an ABI 3730 automated sequencer (Ap-lied Biosystems, Foster City, CA) using published protocols.ewly designed primers and specifically developed polymerase
hain reaction protocols for a DNA-specific amplification ofMS2 are shown in Table 1. Primers for the specific amplifi-ation of exons 1, 3–5, 9, and 11–13 of PMS2 were designedaking use of the few nucleotides divergent between PMS2
nd its pseudogenes. The most 3= nucleotide of the primersas specific for PMS2 and specific for its pseudogenes, allow-
ng amplification of only PMS2-specific sequences. The spec-ficity of the amplicons could be checked by at least 1 PMS2-pecific nucleotide not present in its homologues. Thispproach was possible for all exons with the exception of exons4 and 15 because the only nucleotide different between PMS2nd its paralogue genes represents a single nucleotide poly-orphism in both sequences. We scanned exon 2 by denatur-
ng gradient gel electrophoresis (DGGE) using the primersescribed by Nicolaides et al11 modified by elongation of theeverse primer with a GC clamp of 55 nucleotides.
MSI Analysis
MSI analysis was performed on paired DNA fromumor and normal tissue using the Bethesda panel of micro-atellite markers D2S123, D5S346, D17S250, BAT25, andAT26,30 supplemented by 3 mononucleotide markers
BAT40, MSH3, and MSH6).31 Tumors were regarded as MSI
igh if at least 30% of the markers from the Bethesda panel
sm
4scwCtLPsDGapii
(n
nfPPst(osTc
T
1
3
4
4
6
7
8
9
1
1
1
1
1
1
1
NaA4(a li ann
314 HENDRIKS ET AL GASTROENTEROLOGY Vol. 130, No. 2
howed instability, MSI low if �30% showed instability, oricrosatellite stable if none of the markers showed instability.
Immunohistochemistry
Immunohistochemical staining was performed on-�m whole slides of formalin-fixed, paraffin-embedded tis-ues or on tissue microarray sections, which were mounted onoated slides using a tape transfer system.32 Slides were stainedith antibodies against MLH1 (initially with clone 14, 1:75,albiogen, Cambridge, MA; later supplemented and substi-
uted by clone G168-728, 1:50, BD Biosciences, Franklinakes, NJ), MSH2 (clone GB-12, 1:100; Oncogene Researchroducts, San Diego, CA), MSH6 (clone 44, 1:400; BD Bio-ciences), and PMS2 (clone A16-4, 1:50; BD Biosciences) in aAKO Techmate 500� automated tissue stainer (DAKO,lostrup, Denmark) using standard protocols and procedures
s indicated by the manufacturer. Staining patterns of MMRroteins were evaluated using normal epithelial, stromal, ornflammatory cells or the centers of lymphoid follicles as
able 1. Primers and Polymerase Chain Reaction Conditions
Exon Polymerase chain reaction primers BufferM
(mm
1F:CCG CGC TCC TAC CTG CAC GT C1R:ACC GGA AGA CTG CGA GCC CC3F:ACT GAT AGC ATG GGT CCG A3R:CAG AGC TGA AAG AGA G3F:ACT GAT AGC ATG GGT CCG A4R:AGA AGC TAG AAG TTG AGA TG5F:GAT GAC TCC TCA ACC A A5R:TCC AGA CAG GAT CTT AT6F:GTA ACT TGA GCT GTG TAA TTC C6R:ATC ACT AGA GCA ATA AGA GG7F:AAA GAA AAC AGA TTA AGT CC A7R:AAA GAC ACG AAA CTA TTA GC8F:GCT TCA CTC TGG AAT CCT GCC C A8R:AGC TCC ACG TAA ACT GCC TA9F:TGC TGG TTG GTT AGA ATA GA A9R:ATT CCA GTC ATA GCA GAG C
0 10F:TGA GAC GCT GTC TGA AAA TA B10R:GCT TTA GAA GCT GTT TGT AC
1-5= 11aF:CCA GTC CTG AAC TCC TAG CC D11aR:CCC TGG GGA GCT GGC CGC AT
1-3= 11bF:TGG AGA AGG ACT CGG GGC AC B11bR:CCG ATC CCA GGC TCT CCC A
2 12F:TAG CCT GGG TGA CAG AAC GA B12R:AGG TCT TGC TGT GTT GTC CG
3 13F:TGT GAC ACT TAG CTG AGT AG B13R:TTG GCC TCC CAG AGT GCT GG
4 14F:AAC GTG TTT GTC AAG TCA TG A14R:TGA ACT TGG CTC ACT GCA AG
5 15F:TGT CCA GCC TTA TTT TAG AG A15R:ATC TCA GGT GAT CCG
OTE. Standard thermocycling conditions on a PTC-200 thermocyclernneal temperature, 45 seconds at 72°C, with addition of 10% glyce, Amplitaq buffer (Applied Biosystems); B, AmplitaqGold buffer (App.5 mmol/L MgCl2, 0.5 mmol/L deoxynucleoside triphosphate, 0.NH4)2SO4, 67 mmol/L Tris-HCl, pH 8.8, 1.5 mmol/L MgCl2, 7 �mol/Seven cycli; RAMP �1°C/cycli, starting at 63°C, followed by 33 cyc
nternal controls. Stained slides were scored as either positive b
showing nuclear staining in at least some tumor cells) oregative (with a positive internal control).27
Results
Southern Blot Analysis
In a cohort of 120 patients with colorectal carci-oma from (suspected) HNPCC-affected families, 4 dif-erent genomic rearrangements have been identified inMS2. In family 1 (NL225), we identified a complexMS2 rearrangement: a deletion of the complete codingequences of PMS2 in combination with a duplication ofhe PMS2 pseudogene encompassing exons 9 and 11–15Table 2 and Figure 1). In the DNA of the index patientf family 2 (NLB 54.597), a deletion including theequences of exon 10 was found (Table 2 and Figure 2).29
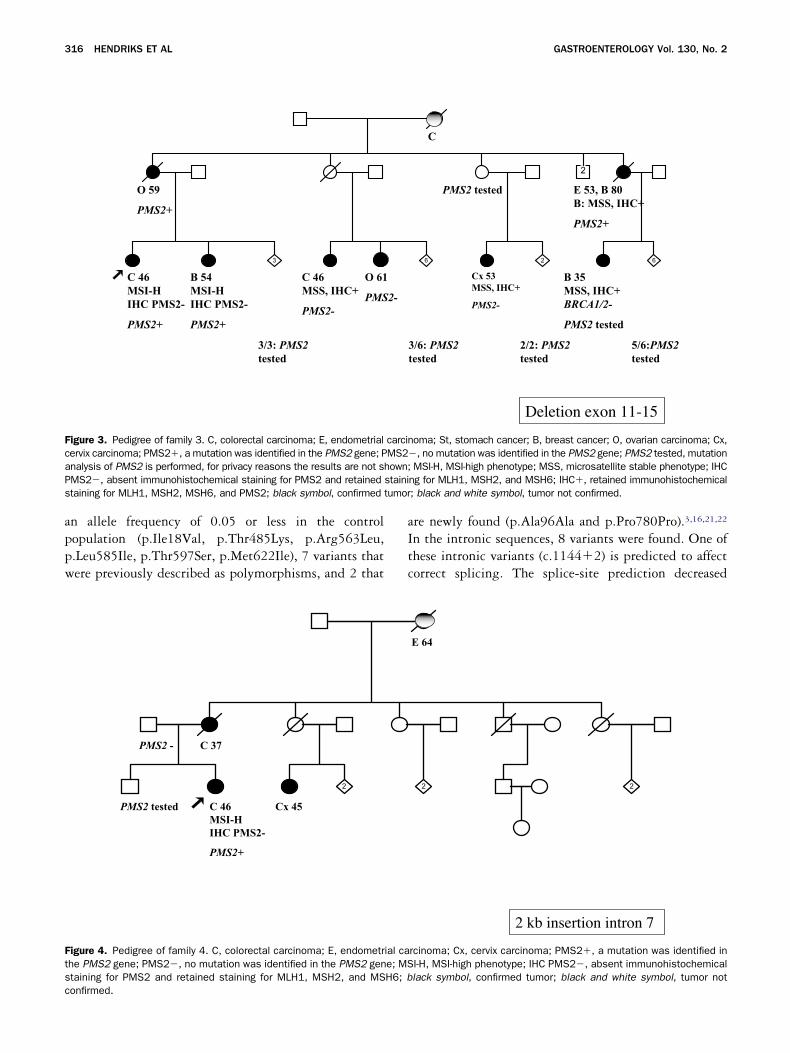
he third deletion, found in family 3 (NLB 51.355),onsists of exon 11–15 (Figure 3). In family 4, a 2-kilo-
d for Point Mutation Analysis
)Anneal temperature
(°C) Sequencing primer
62 1F
55 3F
60 TTG AGT AAC ACT GCT TTG GGA AAT
55 ACT CCT CAA CCA TTT AGA TC
55 6R
55 7R
55 8R
55 9F
52 10F
a ATT TTG AAC ATT GAA CTG TG
62 AGG AGA AAC GCT TGA ACC CG
62 GAT CCT CCT GCC TTG GCC TC
58 TGC TGG GAT TAC AGA CGT GA
60 CTG AGA CCT TCC TCG ACT GC
56 GAA GAA TGC TTC TGA AGA G
seconds at 94°C; 35 cycles of 30 seconds at 94°C, 45 seconds atnd 2.5 mmol/L Mg2�.iosystems); C, 45 mmol/L Tris-HCl, pH 8.3, 11 mmol/L (NH4)2SO4,g/�L bovine serum albumin; 4.4 �mol/L EDTA; D, 16.6 mmol/LTA, 50 mmol/L �-mercaptoethanol.eal temperature at 55°C.
Use
g2�
ol/L
1.5
1.5
3.5
1.5
; 180rol alied B11 �L ED
ase insertion close to the 3= splice site of intron 7 of

PWnep
hT
s�wM
wttkpv(
T
1
23
4
a
Wa
FiMf
Fttttafa
February 2006 PMS2 MUTATIONS IN HNPCC 315
MS2 was found (NLB 52.093) (Table 2 and Figure 4).e performed RNA analysis to determine the pathoge-
icity of this insertion, and we observed monoallelicxpression of a polymorphism present in exon 11 of theatient DNA, indicating that only 1 allele is expressed.Southern blot analysis was extended to affected and
ealthy family members from whom DNA was available.he results are shown in Figures 1–4.
Point Mutation Analysis
We performed DGGE analysis of exon 2 andequencing of all other exons of PMS2 in a subgroup (n
38) of the cohort of 112 patients and in 8 patientsith loss of PMS2 expression and retained MLH1,SH2, and MSH6 expression in their tumors. In total,
able 2. Genomic Rearrangements in PMS2
Family no. Genomic rearrangement
NL 225 Deletion exon 1–15 (� whole PMS2) andduplication of 3=-related gene (c.1-?_2589�?del)a
NLB 54.597 Deletion exon 10 (c.989-?_1144�?del)NLB 52.093 2-kilobase insertion in intron 7
(g.46227�?_47919-?ins)NLB 51.355 Deletion exon 11–15 (c.1145-
?_2589�?del)
Mutation nomenclature according to http://www.hgvs.org/mutnomen/.e used accession numbers AC005995.3 and U14658 for the genomicnd complementary DNA sequences, respectively.44
PMS2 tested PM
PMS2 tested
PMS2 -
C 26 (4 carcinomas) and >14 adenomas MSI-H, IHC PMS2-
PMS2+
igure 1. Pedigree of family 1. C, colorectal carcinoma; PMS2�, adentified in the PMS2 gene; PMS2 tested, mutation analysis of PMSI-high phenotype; MSS, microsatellite stable phenotype; IHC PMS2
or MLH1, MSH2, and MSH6; IHC�, retained immunohistochemical staining
e identified 30 variants: 3 protein-truncating muta-ions (all detected in the 8 patients who were selected onhe basis of immunohistochemistry), 6 variants of un-nown clinical significance (VUCS), and 21 polymor-hisms (Table 3). In the PMS2 coding sequence, 17ariants were detected: 2 truncating mutationsp.Arg628Stop and c.856delG, p.286fs), 6 VUCS with
3
Deletion exon 1-15
PMS2 tested
sted Adenocarcarcinoma 65 MSS, IHC+
ation was identified in the PMS2 gene; PMS2�, no mutation wasperformed, for privacy reasons the results are not shown; MSI-H,
bsent immunohistochemical staining for PMS2 and retained staining
2
Deletion exon 10PMS2 tested PMS2 tested PMS2 tested
St Br C
B 51O 55 PMS2+
C 59,67,71, E 81 MSI-H, IHC PMS2-
PMS2+
igure 2. Pedigree of family 2. C, colorectal carcinoma; E, endome-rial carcinoma; St, stomach cancer; O, ovarian carcinoma; Br, brainumor; PMS2�, a mutation was identified in the PMS2 gene; PMS2ested, mutation analysis of PMS2 is performed, for privacy reasonshe results are not shown; MSI-H, MSI-high phenotype; IHC PMS2�,bsent immunohistochemical staining for PMS2 and positive stainingor MLH1, MSH2, and MSH6; black symbol, confirmed tumor; blacknd white symbol, tumor not confirmed.
S2 te
mutS2 is�, a
for MLH1, MSH2, MSH6, and PMS2; black symbol, confirmed tumor.
appw
aItc
FcaP stainis umor
Ftsc
316 HENDRIKS ET AL GASTROENTEROLOGY Vol. 130, No. 2
n allele frequency of 0.05 or less in the controlopulation (p.Ile18Val, p.Thr485Lys, p.Arg563Leu,.Leu585Ile, p.Thr597Ser, p.Met622Ile), 7 variants thatere previously described as polymorphisms, and 2 that
3
O 59
PMS2+
O 61
PMS
C 46 MSS, IHC+
PMS2-
3/3: PMS2tested
B 54MSI-HIHC PMS2-
PMS2+
C 46MSI-HIHC PMS2-
PMS2+
igure 3. Pedigree of family 3. C, colorectal carcinoma; E, endometrialervix carcinoma; PMS2�, a mutation was identified in the PMS2 gene; Pnalysis of PMS2 is performed, for privacy reasons the results are not shMS2�, absent immunohistochemical staining for PMS2 and retainedtaining for MLH1, MSH2, MSH6, and PMS2; black symbol, confirmed t
2
PMS2 - C 37
Cx 45PMS2 tested C 46MSI-HIHC PMS2-
PMS2+
igure 4. Pedigree of family 4. C, colorectal carcinoma; E, endometrhe PMS2 gene; PMS2�, no mutation was identified in the PMS2 gentaining for PMS2 and retained staining for MLH1, MSH2, and MS
onfirmed.re newly found (p.Ala96Ala and p.Pro780Pro).3,16,21,22
n the intronic sequences, 8 variants were found. One ofhese intronic variants (c.1144�2) is predicted to affectorrect splicing. The splice-site prediction decreased
2
6 2 6
C
PMS2 tested E 53, B 80B: MSS, IHC+
PMS2+
B 35MSS, IHC+ BRCA1/2-
PMS2 tested
5/6:PMS2tested
2/2: PMS2tested
Cx 53MSS, IHC+
PMS2-
3/6: PMS2tested
Deletion exon 11-15
oma; St, stomach cancer; B, breast cancer; O, ovarian carcinoma; Cx,, no mutation was identified in the PMS2 gene; PMS2 tested, mutation
MSI-H, MSI-high phenotype; MSS, microsatellite stable phenotype; IHCng for MLH1, MSH2, and MSH6; IHC�, retained immunohistochemical; black and white symbol, tumor not confirmed.
2 2
2 kb insertion intron 7
E 64
rcinoma; Cx, cervix carcinoma; PMS2�, a mutation was identified inI-H, MSI-high phenotype; IHC PMS2�, absent immunohistochemicallack symbol, confirmed tumor; black and white symbol, tumor not
2-
carcinMS2�own;
ial cae; MSH6; b

f(idnt(3tt
(at
atw5(
agaai
T
NNa
b
c
d
e
f dele
February 2006 PMS2 MUTATIONS IN HNPCC 317
rom 1.00 to 0.00 by introduction of the mutationhttp://www.fruitfly.org/seq_tools/splice.html). The other 7ntronic variants seemed to be neutral variants because theyid not occur directly in the splice consensus sequences, didot predict any change in splicing using the splice predic-ion software, and were found in high frequencies. In thesupposed) PMS2 promoter region, we detected 5 variants:
polymorphisms, 1 that did not show cosegregation withhe disease in the family, and 1 that was only found in 1 ofhe controls.
The 3 families with predicted deleterious mutationsFigures 5–7) and families with VUCS (Table 3) werenalyzed more extensively. Mutation analysis was ex-
able 3. PMS2 Variants Found by Point Mutation Analysis
Exon VariantsGroup with negative
PMS2 staining (n � 8)
1 c.1–340 g�tc.1–323 g�t 5/8c.1–195 t�cc.1–154 c�g 5/8c.1–101 a�g
2 c.52A�G, p.1lel8Vala
c.59G�A, p.Arg20Gln3 c.250�108a�gb
4 c.288C�A, p.Ala96Ala 1/86 c.537�17a�gb 1/87 c.780G�C, p.Ser260Ser 1/88 c.856delG, Asp286, fsX Family 7 (see Figure 7)9 c.988�78g�ac
10 c.1144�2 t>a, r.spl Family 5 (see Figure 5)11 c.1408C�T, p.Pro470Ser 3/8
c.1454C�A, p.Thr485Lysc.1621A�G, p.Lys541Glu 2/8c.1688G�T,
p.Arg563Leud1/8
c.1753C�A, p.Leu585Ilee 1/8c.1789A�T, p.Thr597Serc
c.1866G�A, p.Met622Ilef 1/8c.1882C>T, p.Arg628X Family 6 (see Figure 6)c.2006�6g�ab
12 c.2007�4g�ab 2/8c.2007�7c�tb
13 c.2007�117t�cb
14 c.2322A�G,p.Asn775Ser
c.2338C�T, p.Pro780Pro15 c.2466C�T, p.Leu822Leu
c.2570G�C, p.Gly857Asp
OTE. Mutations printed in bold text are truncating.D, not done.Conserved codon.Splice-site prediction software did not predict a deleterious effect oIn family NA19, the variant is inherited from a spouse.Identified in both affected individuals from family 7 (NLB 60.067) wVariant identified in patient with Arg628Stop, both inherited from thDescribed to cause aberrant interaction with MLH142 and a possible
ended to family members from whom DNA was avail- H
ble to determine cosegregation between the variant andhe disease. In families 5–7, the respective mutationsere indeed shown to segregate with the disease (Figures–7). This was not the case in the families with VUCSTable 3).
MSI Analysis and Immunohistochemistry
Tumor material was collected from the availableffected family members from the families with aenomic rearrangement or a truncating mutation for MSInalysis and immunohistochemistry studies. The resultsre shown in Figures 1–7. Immunohistochemical stain-ng of MLH1, MSH2, MSH6, and PMS2 of all available
Subset (n � 38) fromthe group of 112
cases, allelefrequency
Control population(n � 95),
allele frequency Cosegregation
0.01 0 No0.21 0.280.07 0.050.13 0.180 0.010.05 0 No0.06 0.100.01 ND0.01 ND0.43 ND0.12 ND0 ND Yes0.22 ND0 ND Yes0.43 ND0.07 0.05 No0.16 ND0.01 0.01 No
0 00.03 0.01 No0.03 0.02 No0 ND Yes0.07 ND0.20 ND0.09 ND0.07 ND0.27 ND
0.24 ND0.25 ND0.28 ND
splicing machinery.
truncating PMS2 mutation (Figure 7).ther of the index patient.tion at protein level.16
n the
ith ae mo
NPCC-related tumors from carriers of heterozygous

tPeaioomcsn
mmiutauoawhybtota
c8aw5d(a
AM
P
Ftigtaibc
CM
Fttnnaib
FtPmrafa
318 HENDRIKS ET AL GASTROENTEROLOGY Vol. 130, No. 2
runcating PMS2 mutations showed complete loss ofMS2 and presence of the other 3 MMR proteins. Anxample of the immunohistochemical staining pattern incolorectal tumor from a carrier of a truncating mutation
n PMS2 is shown in Figure 8. The complete inactivationf PMS2 in the tumors also leads to MSI, which webserved in all HNPCC-related tumors from the provenutation carriers. Tumors diagnosed in nonmutation
arriers showed retained immunohistochemical expres-ion of PMS2 and no MSI, indicating that these carci-omas arose through a different tumorigenic pathway.
3
c.1144+2
E 52
E 37
PMS2+
denoma 73SS, IHC+
MS2-
C 62MSI-HIHC PMS2-
B 45, E 52BRCA1/2-
PMS2+
igure 5. Pedigree of family 5. C, colorectal carcinoma; E, endome-rial carcinoma; B, breast cancer; PMS2�, a mutation was identifiedn the PMS2 gene; PMS2�, no mutation was identified in the PMS2ene; MSI-H, MSI-high phenotype; MSS, microsatellite stable pheno-ype; IHC PMS2�, absent immunohistochemical staining for PMS2nd retained staining for MLH1, MSH2, and MSH6; IHC�, retained
mmunohistochemical staining for MLH1, MSH2, MSH6, and PMS2;lack symbol, confirmed tumor; black and white symbol, tumor notonfirmed.
5
6
p.Arg628Stop
Br 64,SS, IHC+
C 55, B 71B: MSS, IHC+
PMS2+
C 42MSI-H, IHC PMS2-
PMS2+
Adenoma 42
PMS2+
igure 6. Pedigree of family 6. C, colorectal carcinoma; E, endome-rial carcinoma; St, stomach cancer; B, breast cancer; Br, brainumor; PMS2�, a mutation was identified in the PMS2 gene; PMS2�,o mutation was identified in the PMS2 gene; MSI-H, MSI-high phe-otype; IHC PMS2�, absent immunohistochemical staining for PMS2nd retained staining for MLH1, MSH2, and MSH6; IHC�, retainedmmunohistochemical staining for MLH1, MSH2, MSH6, and PMS2;
tlack symbol, confirmed tumor.
Clinical Phenotype of Families With aDeleterious PMS2 Mutation
Clinical data of the 7 families with a truncatingutation are summarized in Figures 1–7. The results ofutation analysis of affected individuals are also shown
n the figures. In families 1–4, healthy at-risk individ-als were tested as well. However, for privacy reasons,hese results are not shown in the figures. Of 33 affectednd healthy first-degree relatives from families 1–4 whonderwent DNA analysis, 18 carriers were identified, 7f whom were affected with an HNPCC-related tumornd 1 who was affected with an MSI-high breast cancerith loss of PMS2 expression. The mean age of the 10ealthy carriers was 53 years, with an age range of 35–90ears. There was no clear difference in tumor spectrumetween the families with a genomic rearrangement andhose with a truncating point mutation. Although only 3f the 7 families fulfilled the Amsterdam criteria, in 6 ofhe 7 families, the pattern of inheritance appears to beutosomal dominant.
The most common cancer in the proven mutationarriers was colorectal carcinoma, which was observed inindividuals. The mean age at diagnosis was 52 years
nd ranged from 26 to 80 years. Endometrial carcinomaas found in 5 patients, with a mean age at diagnosis of4 years and a range of 37–81 years. Breast cancer wasiagnosed in 5 proven carriers at a mean age of 57 yearsrange, 35–80 years). Four of these breast tumors werenalyzed for MSI and immunohistochemistry. One of the
5
6
c.856delG, p286fs
C 70 C 80MSI-H, IHC PMS2-
PMS2+
E 47MSI-H, IHC PMS2-
PMS2+
igure 7. Pedigree of family 7. C, colorectal carcinoma; E, endome-rial carcinoma; PMS2�, a mutation was identified in the PMS2 gene;MS2�, no mutation was identified in the PMS2 gene; PMS2 tested,utation analysis of PMS2 is performed, for privacy reasons the
esults are not shown; MSI-H, MSI-high phenotype; IHC PMS2�,bsent immunohistochemical staining for PMS2 and retained stainingor MLH1, MSH2, and MSH6; black symbol, confirmed tumor; blacknd white symbol, tumor not confirmed.
umors showed an MSI-high phenotype and loss of PMS2

ea
wfwgmw
abhgBdfacustTnt
HdoifTtpPgHclwdiH
wwfpiwnohipoT
Ftpr(s
February 2006 PMS2 MUTATIONS IN HNPCC 319
xpression. Ovarian carcinoma was found in 2 cases, at 55nd 59 years of age, respectively.
The index patient in family 1 (Figure 1) was diagnosedith 4 colorectal carcinomas at age 26 years and was
ound to have more than 14 adenomas. Because no MSIas found in normal tissue, the presence of a biallelicermline mutation was unlikely. To exclude involve-ent of MYH and APC, mutation analysis of these genes
igure 8. Immunohistochemical staining of tissue microarray sec-ions from 875 cases of familial colorectal cancer including (sus-ected) HNPCC. (A) Abrogation of PMS2 in carcinoma nuclei, withetained staining of stromal nuclei in a case with a mutation in PMS2.B) MLH1 staining is present in tumor nuclei of this case. (C) Retainedtaining of tumor nuclei for PMS2 in a case without a PMS2 mutation.
as performed. No mutations were found. f
Family 3 (Figure 3) included 3 cases of breast cancernd 1 case of ovarian carcinoma. The combination ofreast cancer and ovarian cancer in this family could alsoave been caused by a mutation in one of the BRCAenes. Therefore, mutation analysis of the BRCA1 andRCA2 genes was performed in the individual whoeveloped breast cancer at age 35 years. No mutation wasound. This family also contains 3 individuals who wereffected with a carcinoma but were proven not to bearriers of the PMS2 deletion: the first of these individ-als developed a cervix carcinoma at age 35 years, theecond developed ovarian carcinoma at age 61 years, andhe third developed colorectal carcinoma at age 46 years.he colorectal tumor showed a microsatellite stable phe-otype and positive staining for PMS2. In conclusion,hese tumors likely represent phenocopies.
Discussion
Until now, only 7 patients suspected of havingNPCC with heterozygous PMS2 mutations have been
escribed in the literature. Unfortunately, informationn family history and segregation analysis was very lim-ted.3,16–18 Other studies in about 200 HNPCC-suspectedamilies did not show any truncating PMS2 mutations.22–24
hese findings explain why many investigators questionedhe role of PMS2 in the pathogenesis of HNPCC. In theresent study, 120 families were analyzed for mutations inMS2. We identified 7 PMS2 mutations, including 4enomic rearrangements and 3 point mutations. AllNPCC-associated tumors that were identified in the
arriers of these mutations were MSI high and showedoss of immunostaining of PMS2. The tumor spectrumas similar to that seen in families associated withefects in other MMR genes. Our findings stronglyndicate that heterozygous PMS2 mutations causeNPCC.Before offering presymptomatic testing in families
ith PMS2 mutations, the clinical phenotype associatedith such mutations should be known. Six of the 7
amilies with a truncating PMS2 mutation showed aattern of inheritance compatible with autosomal dom-nant inheritance. The most frequently observed canceras colorectal carcinoma, followed by endometrial carci-oma and ovarian carcinoma. The mean age of diagnosisf colorectal carcinoma was approximately 7–8 yearsigher than the mean age of diagnosis observed in fam-lies associated with MLH1 and MSH2 mutations. In arevious study, we also reported a higher mean age ofnset of cancer in families with an MSH6 mutation.33
he majority of families with PMS2 mutations (like
amilies with MSH6 mutations) is small and does not
cpefbftaM
wcmmioomeldhdrPanfndi
iltstSPe
fepoTfcmiqH
wtip
HmHpaee
eiahfPp
iacMr
pm
320 HENDRIKS ET AL GASTROENTEROLOGY Vol. 130, No. 2
omply with the Amsterdam criteria. The attenuatedhenotype of families with PMS2 mutations may bexplained by the fact that in the absence of PMS2, aunctional MLH1 and MLH3 heterodimeric protein cane formed that is able to repair DNA mismatches. Inamilies with MLH1 or MSH2 gene defects, no alterna-ive heterodimers can be created, with as a consequencecomplete inactivation of the MMR system with massiveSI and a higher risk of cancer.34
In the literature, several families have been describedith biallelic PMS2 mutations.15,19,35 These families are
haracterized by the combined occurrence of brain tu-ors and the development of multiple colorectal adeno-as and carcinomas. In such families, the pattern of
nheritance seems to be recessive. Carriers of homozygousr compound heterozygous mutations in MLH1, MSH2,r MSH6 also present with childhood onset of cancer,ost frequently hematologic cancers and to a lesser
xtent colorectal carcinoma. In some of the cases, café auait spots have been described.36–40 In our families, weid not find patients with homozygous or compoundeterozygous PMS2 mutations. One of the families weescribe, family 1 (NL225), could fit with an autosomalecessive inheritance pattern. In carriers of a homozygousMS2 mutation, MSI in normal tissue would be expectednd has indeed been reported by De Rosa et al.19 We didot find MSI in normal tissue of the patient from thisamily, although we did not use the same dilution tech-ique De Rosa et al used. The other 6 families witheleterious PMS2 mutations fit with an autosomal dom-nant inheritance pattern.
Several groups have not been able to identify truncat-ng mutations in PMS2 in HNPCC (suspected) fami-ies.22–24 Mutations could have remained undetected dueo many pseudogenes that complicate mutation analy-is.11,15,16 We used the few nucleotides divergent be-ween PMS2 and its pseudogenes to select digests forouthern analysis and to design primers specific forMS2. With this approach, we were able to analyze allxons except exons 14 and 15.
In addition to the 7 truncating PMS2 mutations, weound 6 VUCS. Five of these variants are clustered inxon 11 of PMS2. The carboxyl terminus of the PMS2rotein is known to interact with the carboxyl terminusf MLH1 to form a functional heterodimeric protein.41
his interaction is not only essential for a proper MMRunction but also for the stability of the proteins. Re-ently, Yuan et al showed that single nucleotide poly-orphisms in the 3= end of PMS2 could diminish the
nteractions between PMS2 and MLH142 and conse-uently influence the stability of the PMS2 protein.
owever, in the families with exon 11 VUCS, in whiche tested several affected individuals, no cosegregation ofhe respective mutations with the disease was found,ndicating that these mutations are not likely to beathogenic.Mutations in PMS2 appear to be a rare cause ofNPCC. In the present study, we identified 4 PMS2utations (2.2%) in 184 families suspected of havingNPCC. Hampel et al performed MSI analysis in a
opulation-based series of 1066 colorectal carcinomasnd identified 2 PMS2 mutations (0.18%).43 Truningert al reported 6 PMS2 mutations (0.57%) in an uns-lected series of 1048 colorectal carcinoma cases.17
In conclusion, with the identification of 7 novel het-rozygous disease-causing mutations in the PMS2 genen HNPCC (suspected) families, and the finding of MSInd loss of expression of the PMS2 protein by immuno-istochemistry in all HNPCC-related tumors from af-ected carriers, we showed the role for the MMR geneMS2 in tumorigenesis in a subset of HNPCC (sus-ected) families.The families associated with PMS2 mutations were, as
n MSH6 mutation–associated families, characterized byutosomal dominant inheritance and a higher age ofancer diagnosis compared with families with MLH1 andSH2 mutations. More studies are needed to calculate
eliable penetrance figures.Immunohistochemical analysis, including the PMS2
rotein, is the first step in identifying individuals forutation analysis of PMS2.
References1. Fishel R, Lescoe MK, Rao MR, Copeland NG, Jenkins NA, Garber
J, Kane M, Kolodner R. The human mutator gene homolog MSH2and its association with hereditary nonpolyposis colon cancer.Cell 1993;75:1027–1038.
2. Bronner CE, Baker SM, Morrison PT, Warren G, Smith LG, LescoeMK, Kane M, Earabino C, Lipford J, Lindblom A. Mutation in theDNA mismatch repair gene homologue hMLH1 is associated withhereditary non-polyposis colon cancer. Nature 1994;368:258–261.
3. Nicolaides NC, Papadopoulos N, Liu B, Wei YF, Carter KC, RubenSM, Rosen CA, Haseltine WA, Fleischmann RD, Fraser CM. Mu-tations of two PMS homologues in hereditary nonpolyposis coloncancer. Nature 1994;371:75–80.
4. Miyaki M, Konishi M, Tanaka K, Kikuchi-Yanoshita R, Muraoka M,Yasuno M, Igari T, Koike M, Chiba M, Mori T. Germline mutationof MSH6 as the cause of hereditary nonpolyposis colorectalcancer. Nat Genet 1997;17:271–272.
5. Akiyama Y, Sato H, Yamada T, Nagasaki H, Tsuchiya A, Abe R,Yuasa Y. Germ-line mutation of the hMSH6/GTBP gene in anatypical hereditary nonpolyposis colorectal cancer kindred. Can-cer Res 1997;57:3920–3923.
6. Vasen HF, Mecklin JP, Khan PM, Lynch HT. The InternationalCollaborative Group on Hereditary Non-Polyposis Colorectal Can-cer (ICG-HNPCC). Dis Colon Rectum 1991;34:424–425.
7. Vasen HF, Watson P, Mecklin JP, Lynch HT. New clinical criteria
for hereditary nonpolyposis colorectal cancer (HNPCC, Lynch syn-
1
1
1
1
1
1
1
1
1
1
2
2
2
2
2
2
2
2
2
2
3
3
3
3
February 2006 PMS2 MUTATIONS IN HNPCC 321
drome) proposed by the International Collaborative Group onHNPCC. Gastroenterology 1999;116:1453–1456.
8. Liu B, Parsons R, Papadopoulos N, Nicolaides NC, Lynch HT,Watson P, Jass JR, Dunlop M, Wyllie A, Peltomaki P, de laChapelle A, Hamilton SR, Vogelstein B, Kinzler KW. Analysis ofmismatch repair genes in hereditary non-polyposis colorectal can-cer patients. Nat Med 1996;2:169–174.
9. Wijnen J, Khan PM, Vasen H, van der Klift H, Mulder A, Leeuwen-Cornelisse I, Bakker B, Losekoot M, Moller P, Fodde R. Hereditarynonpolyposis colorectal cancer families not complying with theAmsterdam criteria show extremely low frequency of mismatch-repair-gene mutations. Am J Hum Genet 1997;61:329–335.
0. Wagner A, Barrows A, Wijnen JT, van der Klift H, Franken PF,Verkuijlen P, Nakagawa H, Geugien M, Jaghmohan-Changur S,Breukel C, Meijers-Heijboer H, Morreau H, van Puijenbroek M,Burn J, Coronel S, Kinarski Y, Okimoto R, Watson P, Lynch JF, dela Chapelle A, Lynch HT, Fodde R. Molecular analysis of heredi-tary nonpolyposis colorectal cancer in the United States: highmutation detection rate among clinically selected families andcharacterization of an American founder genomic deletion of theMSH2 gene. Am J Hum Genet 2003;72:1088–1100.
1. Nicolaides NC, Carter KC, Shell BK, Papadopoulos N, VogelsteinB, Kinzler KW. Genomic organization of the human PMS2 genefamily. Genomics 1995;30:195–206.
2. Baker SM, Bronner CE, Zhang L, Plug AW, Robatzek M, Warren G,Elliott EA, Yu J, Ashley T, Arnheim N. Male mice defective in theDNA mismatch repair gene PMS2 exhibit abnormal chromosomesynapsis in meiosis. Cell 1995;82:309–319.
3. Reitmair AH, Cai JC, Bjerknes M, Redston M, Cheng H, Pind MT,Hay K, Mitri A, Bapat BV, Mak TW, Gallinger S. MSH2 deficiencycontributes to accelerated APC-mediated intestinal tumorigene-sis. Cancer Res 1996;56:2922–2926.
4. Prolla TA, Baker SM, Harris AC, Tsao JL, Yao X, Bronner CE,Zheng B, Gordon M, Reneker J, Arnheim N, Shibata D, Bradley A,Liskay RM. Tumour susceptibility and spontaneous mutation inmice deficient in Mlh1, Pms1 and Pms2 DNA mismatch repair.Nat Genet 1998;18:276–279.
5. De Vos M, Hayward BE, Picton S, Sheridan E, Bonthron DT. NovelPMS2 pseudogenes can conceal recessive mutations causing adistinctive childhood cancer syndrome. Am J Hum Genet 2004;74:954–964.
6. Nakagawa H, Lockman JC, Frankel WL, Hampel H, Steenblock K,Burgart LJ, Thibodeau SN, de la Chapelle A. Mismatch repairgene PMS2: disease-causing germline mutations are frequent inpatients whose tumors stain negative for PMS2 protein, butparalogous genes obscure mutation detection and interpretation.Cancer Res 2004;64:4721–4727.
7. Truninger K, Menigatti M, Luz, J, Russell A, Haider R, GebbersJ-O, Bannwart F, Yurtsever H, Neuweiler J, Riehle H-M, CattaruzzaM, Heinimann K, Schär, P, Jiricny J, Marra G. Immunohistochem-ical analysis reveals high frequency of PMS2 defects in colorectalcancer. Gastroenterology 2005;128:1160–1171.
8. Worthly DL, Walsh MD, Barker M, Ruszkiewicz A, Bennett G,Phillips K, Suthers G. Familial mutations in PMS2 can causeautosomal dominant hereditary nonpolyposis colorectal cancer.Gastroenterology 2005;128:1431–1436.
9. De Rosa M, Fasano C, Panariello L, Scarano MI, Belli G, IannelliA, Ciciliano F, Izzo P. Evidence for a recessive inheritance ofTurcot’s syndrome caused by compound heterozygous mutationswithin the PMS2 gene. Oncogene 2000;19:1719–1723.
0. Miyaki M, Nishio J, Konishi M, Kikuchi-Yanoshita R, Tanaka K,Muraoka M, Nagato M, Chong JM, Koike M, Terada T, KawaharaY, Fukutome A, Tomiyama J, Chuganji Y, Momoi M, Utsunomiya J.Drastic genetic instability of tumors and normal tissues in Turcot
syndrome. Oncogene 1997;15:2877–2881.1. Turcot J, Despres JP, St Pierre F. Malignant tumors of the centralnervous system associated with familial polyposis of the colon:report of two cases. Dis Colon Rectum 1959;2:465–468.
2. Viel A, Novella E, Genuardi M, Capozzi E, Fornasarig M, PedroniM, Santarosa M, De Leon MP, Della PL, Anti M, Boiocchi M. Lackof PMS2 gene-truncating mutations in patients with hereditarycolorectal cancer. Int J Oncol 1998;13:565–569.
3. Wang Q, Lasset C, Desseigne F, Saurin JC, Maugard C, NavarroC, Ruano E, Descos L, Trillet-Lenoir V, Bosset JF, Puisieux A.Prevalence of germline mutations of hMLH1, hMSH2, hPMS1,hPMS2, and hMSH6 genes in 75 French kindreds with nonpol-yposis colorectal cancer. Hum Genet 1999;105:79–85.
4. Liu T, Yan H, Kuismanen S, Percesepe A, Bisgaard ML, PedroniM, Benatti P, Kinzler KW, Vogelstein B, Ponz DL, Peltomaki P,Lindblom A. The role of hPMS1 and hPMS2 in predisposing tocolorectal cancer. Cancer Res 2001;61:7798–7802.
5. Wijnen JT, Vasen HF, Khan PM, Zwinderman AH, van der Klift H,Mulder A, Tops C, Moller P, Fodde R. Clinical findings with impli-cations for genetic testing in families with clustering of colorectalcancer. N Engl J Med 1998;339:511–518.
6. Wijnen J, de Leeuw W, Vasen H, van der Klift H, Moller P,Stormorken A, Meijers-Heijboer H, Lindhout D, Menko F, VossenS, Moslein G, Tops C, Brocker-Vriends A, Wu Y, Hofstra R, Sij-mons R, Cornelisse C, Morreau H, Fodde R. Familial endometrialcancer in female carriers of MSH6 germline mutations. Nat Genet1999;23:142–144.
7. de Jong AE, van Puijenbroek M, Hendriks Y, Tops C, Wijnen J,Ausems MG, Meijers-Heijboer H, Wagner A, van Os TA, Brocker-Vriends AH, Vasen HF, Morreau H. Microsatellite instability, im-munohistochemistry, and additional PMS2 staining in suspectedhereditary nonpolyposis colorectal cancer. Clin Cancer Res2004;10:972–980.
8. Umar A, Boland CR, Terdiman JP, Syngal S, de la Chapelle A,Rüschhoff J, Fishel R, Lindor NM, Burgart LJ, Hamelin R, HamiltonSR, Hiatt RA, Jass J, Lindblom A, Lynch HT, Peltomaki P, RamseySD, Rodriguez-Bigas MA, Vasen HFA, Hawk ET, Barrett JC, Freed-man AN, Srivastava S. Revised Bethesda guidelines for heredi-tary nonpolyposis colorectal cancer (Lynch syndrome) and micro-satellite instability. J Natl Cancer Inst 2004;96:261–268.
9. van der Klift H, Wijnen J, Wagner A, Verkuijlen P, Tops C, OtwayR, Kohonen-Corish M, Vasen H, Oliani C, Barana D, Moller P,Delozier-Blanchet C, Hutter P, Foulkes W, Lynch H, Burn J,Moslein G, Fodde R. Molecular characterization of the spectrumof genomic deletions in the mismatch repair genes MSH2, MLH1,MSH6, and PMS2 responsible for hereditary nonpolyposis colo-rectal cancer (HNPCC). Genes Chromosomes Cancer 2005;44:123–138.
0. Boland CR, Thibodeau SN, Hamilton SR, Sidransky D, EshlemanJR, Burt RW, Meltzer SJ, Rodriguez-Bigas MA, Fodde R, RanzaniGN, Srivastava S. A National Cancer Institute Workshop on Mi-crosatellite Instability for cancer detection and familial predispo-sition: development of international criteria for the determinationof microsatellite instability in colorectal cancer. Cancer Res1998;58:5248–5257.
1. Hendriks Y, Franken P, Dierssen JW, de Leeuw W, Wijnen J, DreefE, Tops C, Breuning M, Brocker-Vriends A, Vasen H, Fodde R,Morreau H. Conventional and tissue microarray immunohisto-chemical expression analysis of mismatch repair in hereditarycolorectal tumors. Am J Pathol 2003;162:469–477.
2. Bubendorf L, Nocito A, Moch H, Sauter G. Tissue microarray(TMA) technology: miniaturized pathology archives for high-throughput in situ studies. J Pathol 2001;195:72–79.
3. Hendriks YM, Wagner A, Morreau H, Menko F, Stormorken A,Quehenberger F, Sandkuijl L, Moller P, Genuardi M, Van Hou-welingen H, Tops C, van Puijenbroek M, Verkuijlen P, Kenter G,Van Mil A, Meijers-Heijboer H, Tan GB, Breuning MH, Fodde R,
Wijnen JT, Brocker-Vriends AH, Vasen H. Cancer risk in hereditary
3
3
3
3
3
3
4
4
4
4
4
pPy
322 HENDRIKS ET AL GASTROENTEROLOGY Vol. 130, No. 2
nonpolyposis colorectal cancer due to MSH6 mutations: impacton counseling and surveillance. Gastroenterology 2004;127:17–25.
4. Peltomaki P. Deficient DNA mismatch repair: a common etiologicfactor for colon cancer. Hum Mol Genet 2001;10:735–740.
5. Hamilton SR, Liu B, Parsons RE, Papadopoulos N, Jen J, PowellSM, Krush AJ, Berk T, Cohen Z, Tetu B. The molecular basis ofTurcot’s syndrome. N Engl J Med 1995;332:839–847.
6. Wang Q, Lasset C, Desseigne F, Frappaz D, Bergeron C, NavarroC, Ruano E, Puisieux A. Neurofibromatosis and early onset ofcancers in hMLH1-deficient children. Cancer Res 1999;59:294–297.
7. Ricciardone MD, Özçelik T, Cevher B, Özdag H, Tuncer M, GürgeyA, Uzunalimoglu Ö, Çetinkaya, H, Tanyeli A, Erken E, Öztürk M.Human MLH1 deficiency predisposes to hematological malig-nancy and neurofibromatosis type 1. Cancer Res 1999;59:290–293.
8. Whiteside D, McLeod R, Graham G, Steckley JL, Booth K, Som-erville MJ, Andrew SE. A homozygous germ-line mutation in thehuman MSH2 gene predisposes to hematological malignancyand multiple café-au-lait spots. Cancer Res 2002;62:359–362.
9. Gallinger S, Aronson M, Shayan K, Ratcliffe EM, Gerstle JT,Parkin PC, Rothenmund H, Croitoru M, Baumann E, Durie PR,Weksberg R, Pollett A, Riddell RH, Ngan BY, Cutz E, Lagarde AE,Chan HS. Gastrointestinal cancers and neurofibromatosis type 1features in children with a germline homozygous MLH1 mutation.Gastroenterology 2004;126:576–585.
0. Menko FH, Kaspers GL, Meijer GA, Claes K, Hagen JM, Gille JJP.
A homozygous MSH6 mutation in a child with café-au-lait spots, toligodendroglioma and rectal cancer. Fam Cancer 2004;3:123–127.
1. Guerrette S, Acharya S, Fishel R. The interaction of the humanMutL homologues in hereditary nonpolyposis colon cancer. J BiolChem 1999;274:6336–6341.
2. Yuan ZQ, Gottlieb B, Beitel LK, Wong N, Gordon PH, Wang Q,Puisieux A, Foulkes WD, Trifiro M. Polymorphisms and HNPCC:PMS2-MLH1 protein interactions diminished by single nucleotidepolymorphisms. Hum Mutat 2002;19:108–113.
3. Hampel H, Frankel WL, Martin E, Arnold M, Khanduja K, KeublerP, Nakagawa H, Sotamaa K, Prior TW, Westman J, Panescu J, FixD, Lockman J, Comeras I, de la Chapelle A. Screening for theLynch syndrome (hereditary nonpolyposis colorectal cancer).N Engl J Med 2005;352:1851–1860.
4. den Dunnen JT, Antonarakis SE. Mutation nomenclature exten-sions and suggestions to describe complex mutations: a discus-sion. Hum Mutat 2000;15:7–12.
Received July 13, 2005. Accepted October 19, 2005.Address requests for reprints to: Yvonne M. C. Hendriks, MD, De-
artment of Clinical Genetics, K5-R, Leiden University Medical Center,O Box 9600, 2300 RC Leiden, The Netherlands. e-mail:[email protected]; fax: (31)-71-5266749.Supported by ZonMw (grant 9607.0136.1).Y.M.C.H., S.J.-C., and H.M.V. contributed equally to this work.The authors thank the families that have so kindly participated in
his study.





























