Embed Size (px)
Citation preview

doi.org/10.26434/chemrxiv.7466426.v1
Hydration Structure of Sodium and Potassium Ions with DFT-MDTimothy Duignan, Gregory K. Schenter, Mirza Galib, Marcel D. Baer, Jan Wilhelm, Jürg Hutter, Mauro DelBen, Xiu Song Zhao, Christopher J. Mundy
Submitted date: 14/12/2018 • Posted date: 14/12/2018Licence: CC BY-NC-ND 4.0Citation information: Duignan, Timothy; Schenter, Gregory K.; Galib, Mirza; Baer, Marcel D.; Wilhelm, Jan;Hutter, Jürg; et al. (2018): Hydration Structure of Sodium and Potassium Ions with DFT-MD. ChemRxiv.Preprint.
The ability to reproduce the structure of water around the sodium and potassium ions as determined byexperiment is a key test of the quality of interaction potentials due to the central importance of these ions in awide range of important phenomena. Here, we simulate the Na+ and K+ ions in bulk water using the recentlydeveloped strongly constrained and appropriately normed (SCAN) functional and compare with experimentalX-ray diffraction (XRD) and X-ray adsorption fine structure (EXAFS) measurements to demonstrate that itaccurately reproduces important structural details of the hydration structure of the sodium and potassiumcations. We demonstrate that it performs substantially better than the generalized gradient approximation(GGA) based dispersion corrected revised Perdew, Burke, and Ernzerhof functional (revPBE-D3) and is evenbetter than the random phase approximation level for potassium. Both of these functionals have beendemonstrated to accurately reproduce the structure of bulk water. This improved performance compared withrevPBE-D3 is attributed to smaller fluctuations of the mean error of ion-water cluster binding energies utilizinga novel benchmark for testing functionals.
File list (1)
download fileview on ChemRxivcationstructure.pdf (356.77 KiB)

Hydration structure of sodium and potassium
ions with DFT-MD
Timothy T. Duignan,†,‡ Gregory K. Schenter,† Mirza Galib,† Marcel D. Baer,†
Jan Wilhelm,¶ Jurg Hutter,¶ Mauro Del Ben,§ Xiu Song Zhao,∗,‡ and
Christopher J. Mundy∗,†,∥
†Physical Science Division, Pacific Northwest National Laboratory, P.O. Box 999,Richland,
Washington 99352, USA
‡School of Chemical Engineering, The University of Queensland, St Lucia, Brisbane 4072,
Australia
¶Department of Chemistry, University of Zurich, CH-8057 Zurich, Switzerland.
§Computational Research Division, Lawrence Berkeley National Laboratory, Berkeley,
California 94720, USA.
∥Affiliate Professor, Department of Chemical Engineering, University of Washington,
Seattle, Washington, USA
E-mail: [email protected]; [email protected]
1

Abstract
The ability to reproduce the structure of water around the sodium and potassium
ions as determined by experiment is a key test of the quality of interaction potentials
due to the central importance of these ions in a wide range of important phenom-
ena. Here, we simulate the Na+ and K+ ions in bulk water using the recently devel-
oped strongly constrained and appropriately normed (SCAN) functional and compare
with experimental X-ray diffraction (XRD) and X-ray adsorption fine structure (EX-
AFS) measurements to demonstrate that it accurately reproduces important structural
details of the hydration structure of the sodium and potassium cations. We demon-
strate that it performs substantially better than the generalized gradient approximation
(GGA) based dispersion corrected revised Perdew, Burke, and Ernzerhof functional
(revPBE-D3) and is even better than the random phase approximation level for potas-
sium. Both of these functionals have been demonstrated to accurately reproduce the
structure of bulk water. This improved performance compared with revPBE-D3 is at-
tributed to smaller fluctuations of the mean error of ion-water cluster binding energies
utilizing a novel benchmark for testing functionals.
Introduction
The sodium and potassium ions play a central role in a large range of important industrial
and biological processes. For example, the sodium and potassium ions are considered to
be promising candidates to replace lithium ions in the next generation of energy storage
devices.1–5 Additionally, the flow of potassium and sodium ions through cell membranes is
used to control important biological processes.6 In both of these systems, an important step
is the partial desolvation of the ion as it passes through a small channel. For example,
the ions must desolvate to intercalate into electrode materials or to pass through the ion
pump in the cell membrane. This desolvation can determine the rates and mechanisms
of these processes. A recent demonstration of this point is provided by Peng et al. 7 who
2

demonstrate that interfacial sodium ion diffusion is highly sensitive to the hydration number.
It is therefore very important to build an accurate and detailed understanding of the structure
of solvent around these ions.
Many attempts have been made to simulate the properties of these ions in water.8–21
Classical forcefield molecular dynamics can reproduce a variety structural properties such
as the peak position in the radial distribution function (RDF) but the parameters for the
Lennard-Jones and polarisability interactions have to be explicitly adjusted to do so. These
models also have limited predictive power as new parameters are usually needed to model
the ion in different environments such as inside an electrode material. Hybrid quantum
mechanical/molecular mechanics (QM/MM) is an alternative approach14,21 to the problem.
However, it is unclear how large the QM region needs to be to accurately capture the full
solvent structure. Moreover, significant challenges arise associated with the treatment of the
interface between the two regions. A promising and increasingly widely used approach is
to use molecular dynamics simulations with a full quantum mechanical density functional
theory treatment of the system.22 The dispersion corrected generalized gradient approxima-
tion (GGA) functionals are by far the most widely used in the context of condensed phase
simulation due to their low computational demand. Of these functionals the revised Perdew,
Burke, and Ernzerhof functional with Grimme dispersion correction (revPBE-D3)23–25 has
been demonstrated to accurately reproduce the structure of bulk water.26 However, this
approach often demonstrate a dependence on the specific functional chosen and can fail to
quantitatively reproduce experimental measurements. For example, Galib et al. 20,27 demon-
strated that standard GGA functionals cannot reproduce the experimentally determined
water structure around the sodium cation as determined by x-ray diffraction (XRD) and
X-ray adsorption fine structure measurements (EXAFS).
It is therefore necessary to simulate these ions with quantum density functional theory
(DFT) functionals at higher rungs of the so-called “Jacobs Ladder” of accuracy.28 No signif-
icant change in peak position occurs on moving from the GGA functional PBE to the hybrid
3

functional PBE0,17 indicating that hybrid functionals are unlikely to solve this issue. To this
end, the strongly constrained and appropriately normed (SCAN) meta-GGA functional has
recently been developed. The additional step on Jacobs Ladder has the advantage that the
computational costs are comparable to standard GGA functionals. SCAN has been devel-
oped to satisfy 17 known constraints that a general exchange-correlation functional should
satisfy. It can accurately reproduce binding energies and structures of a variety of molecules
without empirical dispersion corrections.29 Furthermore, Chen et al. 30 and Zheng et al. 31
have used this functional to simulate the structure of bulk liquid water determining a den-
sity of 1.05 g/cm−1 and showing good agreement with experimental oxygen-oxygen RDFs
at room temperature when simulated at an elevated temperature of 330 K. This functional
has also recently been applied to calculate the potential of mean force of the NaCl dimer
in water.32 Additionally, the random phase approximation (RPA) approximation to electron
correlation has recently been implemented for condensed phase calculations.33,34 This level
of theory represents the highest (fifth) rung of the Jacobs ladder and provides an accurate
description of bulk liquid water.34 The RPA method comes with additional computational
costs over standard GGA functionals due to the explicit treatment of electron correlation
and the requirement of larger basis sets. Nevertheless, it is important to quantitatively de-
termine whether these more sophisticated functionals can overcome the limitations of the
GGA based description of ion solvation.
Structural analysis
One of the main the differences between this study and the studies of Chen et al. 30 and Zheng
et al. 31 is the simulation at 300 K rather than the 330K used and using newly optimized
pseudo potentials for the oxygen atom. Herein, we use the same pseudo potentials as was
used in Chen et al. 30 and Zheng et al. 31 for hydrogen. The resulting oxygen-oxygen RDFs
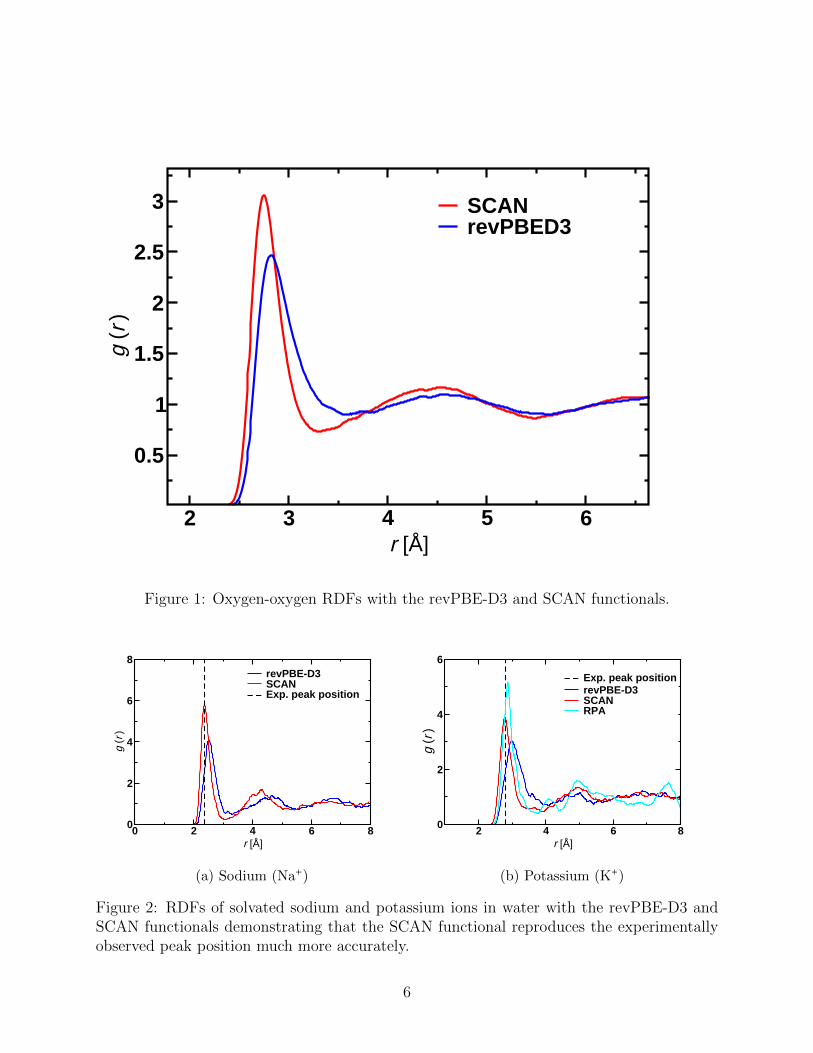
are shown in Figure 1 for both revPBE-D3 and SCAN. The revPBE-D3 functional has been
shown to give near perfect agreement with the experimentally determined RDF.26,35 The
4

structure is significantly more enhanced for the SCAN functional resembling36 the structure
using the GGA of Becke37 and Lee, Yang and Parr38 (BLYP) in addition to the dispersion
correction (D2) put forth by Grimme.39 The origin of the discrepancy in comparison with
previous SCAN studies30,31 is not immediately apparent. The structural effects associated
with quantum nuclear effects have been demonstrated to be quite small and so likely cannot
explain the discrepancy.40,41 Nevertheless, the slightly over-structured behaviour of SCAN is
consistent with the work of Yao 42 who also simulated at 300 K with CP2K43 and CPMD44
demonstrating good agreement between the two basis set approaches, namely gaussian func-
tions and plane-waves. Simulation of 20 ps utilizing SCAN water in a slab configuration
at 300 K using the protocol discussed herein is consistent with the density of 1.05 g/cm3
calculated by Chen et al..30 These results presented here suggest some caution in the use
of SCAN to describe pure water may be appropriate as its RDF and density appear to be
comparable to BLYP-D2 at 300 K in terms of agreement with experimental measurements.26
Table 1: RDF peak positions for different DFT functionals compared with ex-periment demonstrating that the SCAN functional provides closer agreement tothat predicted from X-ray diffraction and EXAFS.20 Units are in A, error is ±0.05 A.
revPBE-D3 SCAN RPA ExperimentNa+ 2.51 2.36 - 2.38K+ 2.98 2.78 2.86 2.79
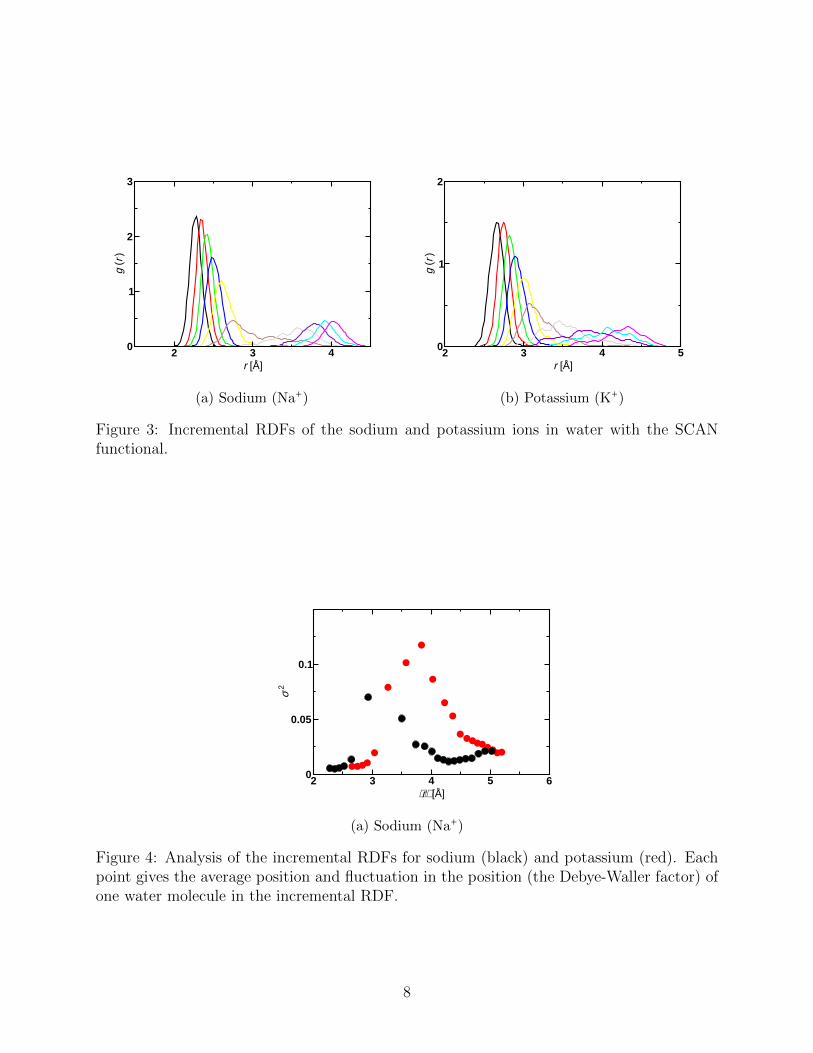
Figure 2 depicts the RDF for the sodium and potassium ions utilizing both revPBE-D3
and SCAN functionals. Significant differences are clear. As shown in Table 1, the experi-
mental peak position is reproduced much more accurately with the SCAN functional than
with the GGA. The inability to reproduce this peak position has been problematic with
other GGA functionals such as BLYP-D2.20 To the best of our knowledge this is the first
demonstration that the SCAN functional can significantly outperform simpler GGA func-
tionals in reproducing bulk structural properties in the condensed phase. As previously
stated, previous research has demonstrated that SCAN can accurately reproduce bulk wa-
ter structure.30,31 This is also true for the revPBE-D3 GGA functional.26,35 Similarly, the
5
2 3 4 5 6r [Å]
0.5
1
1.5
2
2.5
3
g (r
)
SCANrevPBED3
Figure 1: Oxygen-oxygen RDFs with the revPBE-D3 and SCAN functionals.
0 2 4 6 8r [Å]
0
2
4
6
8
g (r
)
revPBE-D3SCANExp. peak position
(a) Sodium (Na+)
2 4 6 8r [Å]
0
2
4
6
g (r
)
Exp. peak positionrevPBE-D3SCANRPA
(b) Potassium (K+)
Figure 2: RDFs of solvated sodium and potassium ions in water with the revPBE-D3 andSCAN functionals demonstrating that the SCAN functional reproduces the experimentallyobserved peak position much more accurately.
6

Na-Cl potential of mean force (PMF) has been computed with SCAN, however relatively
small differences are observed when compared to other DFT functionals and there are no
comparisons of the outcomes of the aforementioned PMF with experimental results to demon-
strate an improvement. It has already been demonstrated that the PMF calculated with the
BLYP-D2 functional leads to reasonably good agreement with experimental osmotic/activity
coefficients.45
Previous studies have determined that solvation structure of the potassium ion as one
of the most difficult cases to reproduce using GGA functionals.16 Given the good overall
agreement of the solvation structure of the sodium ion with SCAN, it gives rise to the
question of what the level of performance will be for wavefunction methods that include
electron correlation effects. To this end, we have also simulated the potassium ion treating
electron correlation at the level of the random-phase approximation (RPA).34 Due to the
large computational costs of this functional we have not yet attempted the simulation of
the sodium ion with this method. Figure 2 suggests that the RPA functional performs
substantially better than the GGA in comparison with the experimental peak position.
Although the K–O distance is somewhat larger than the experimental value, this may be
due to a lack of statistics. Nevertheless, given that we used the converged GGA simulation
to generate the initial configuration it is clear that the RPA is producing results more in
line with EXAFS experiment. Interestingly the RPA is also more structured than both the
GGA or the meta-GGA. Given the excellent agreement of RPA results for water under bulk
homogeneous conditions,46 overall the performance of the RPA is satisfactory and hints that
properly converged correlated wavefunction methods may well out-perform lower rungs of
Jacob’s Ladder but at a large computational overhead.
A key property of interest is the hydration number of the ions. However, this quantity is
not well defined for ions with water molecules that cross between hydration layers. The best
way to understand the ion hydration and its model dependencies is through examination
of the incremental RDFs that separates the distribution into contributions from the closest
7
2 3 4r [Å]
0
1
2
3
g (r
)
(a) Sodium (Na+)
2 3 4 5r [Å]
0
1
2
g (r
)
(b) Potassium (K+)
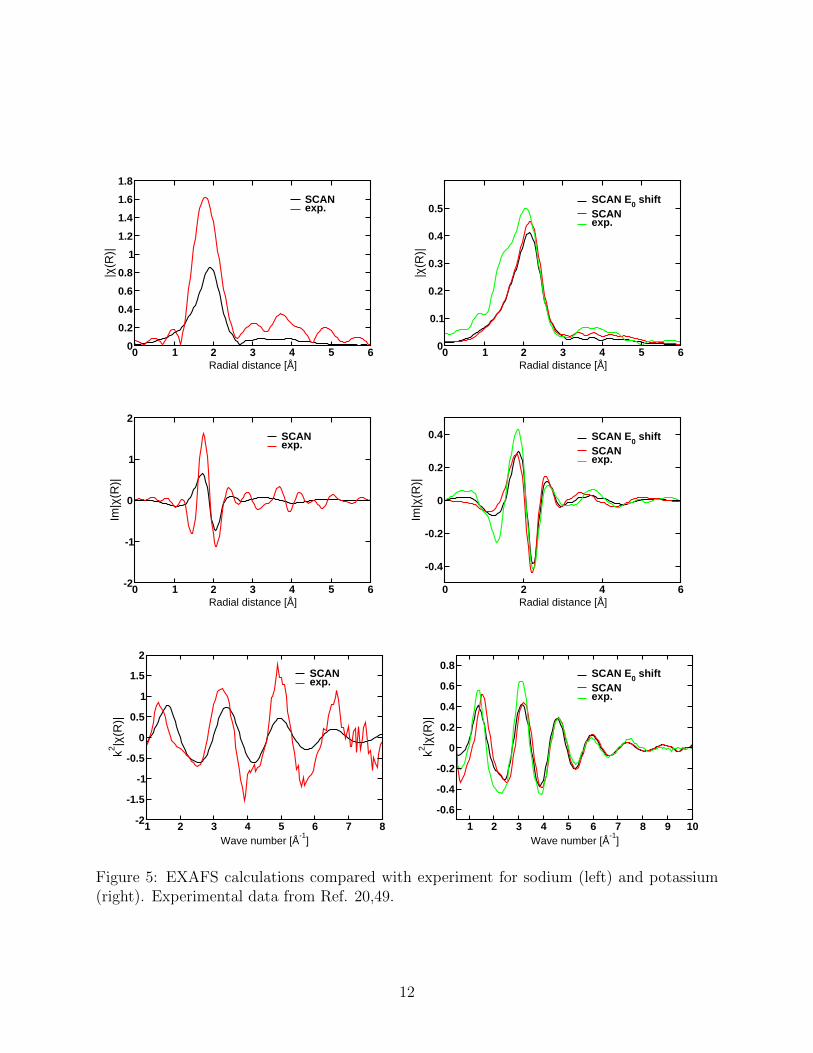
Figure 3: Incremental RDFs of the sodium and potassium ions in water with the SCANfunctional.
2 3 4 5 6⟨r⟩ [Å]
0
0.05
0.1
σ2
(a) Sodium (Na+)
Figure 4: Analysis of the incremental RDFs for sodium (black) and potassium (red). Eachpoint gives the average position and fluctuation in the position (the Debye-Waller factor) ofone water molecule in the incremental RDF.
8

water molecules. Figure 3 provides this quantity for the sodium and potassium ions using the
SCAN functional. Additional incremental RDFs for the other functionals are provided in the
SI. Figure 4 provides an analysis of the average position and spread of each water molecule
for both ions. There are substantial similarities between the two ions. For instance, it is clear
that both ions have five relatively tightly bound water molecules and a more diffuse sixth
water molecule that occupies the first hydration shell but can often be in the second shell.20
In contrast water molecules beyond the first shell show qualitatively different behaviour for
the two ions. The seventh water molecule for sodium almost always occupies the second
hydration shell and is more localised than the sixth. With potassium the seventh and eight
water molecules bridge the first and second hydration shells blurring the distinction between
hydration layers and are more diffuse than the sixth water molecule. This is consistent with
potassium being classified more as a chaotrope in contrast with the kosomotrope status of
the sodium ion.47 The hydration number defined as the integral of the RDF at the minima
gives values of 5.6±0.2 and 7.5±0.8 with the SCAN functional. The large uncertainty in the
potassium ion coordination number is due to the broad and flat minimum of the RDF and
is the result of the fact that this ion does not have a well defined first hydration layer. These
results are consistent with XAFS and XRD data which gives a sodium hydration number of
5.4 to 5.9.20
EXAFS analysis
Given a detailed description of molecular interactions, an associated ensemble of configu-
rations can be generated. From this ensemble, collective properties and responses of the
system can be determined. In particular, it is possible to generate the X-ray absorption fine
structure signal which is a unique spectral signature associated with the generated ensemble.
(In this work we concentrate on the ”extended” region of the signal, referred to as EXAFS.)
This provides an effective benchmark of our description of molecular interactions, giving us
confidence in its ability to generate accurate ensembles and be used to predict phenomena
9

that are not directly measurable.
EXAFS is an effective probe of the local solvent structure about a solute photo-electron
source. The signal is most sensitive to the solute - nearest solvent distance and its fluctuations
which are measured by the Debye-Waller factor of the solute-solvent vibration, σ2. In Fig. 4,
the lowest five distance with the smallest σ2 contribute the most to the signal. Taking
these and assuming independent single electron scattering events reproduces the qualitative
structure of the signal. EXAFS is sensitive to the balance between solute-solvent and solvent-
solvent interactions that determine the collective solvent structure about the solvent.
In order to connect to experimental measurement, we take configurations from a canonical
ensemble corresponding to the density and temperature of the measurement, determined
from the SCAN description of molecular interaction. For each configuration, we generate an
EXAFS signal using the feff9 code developed by Rehr and coworkers.48 We take the mean
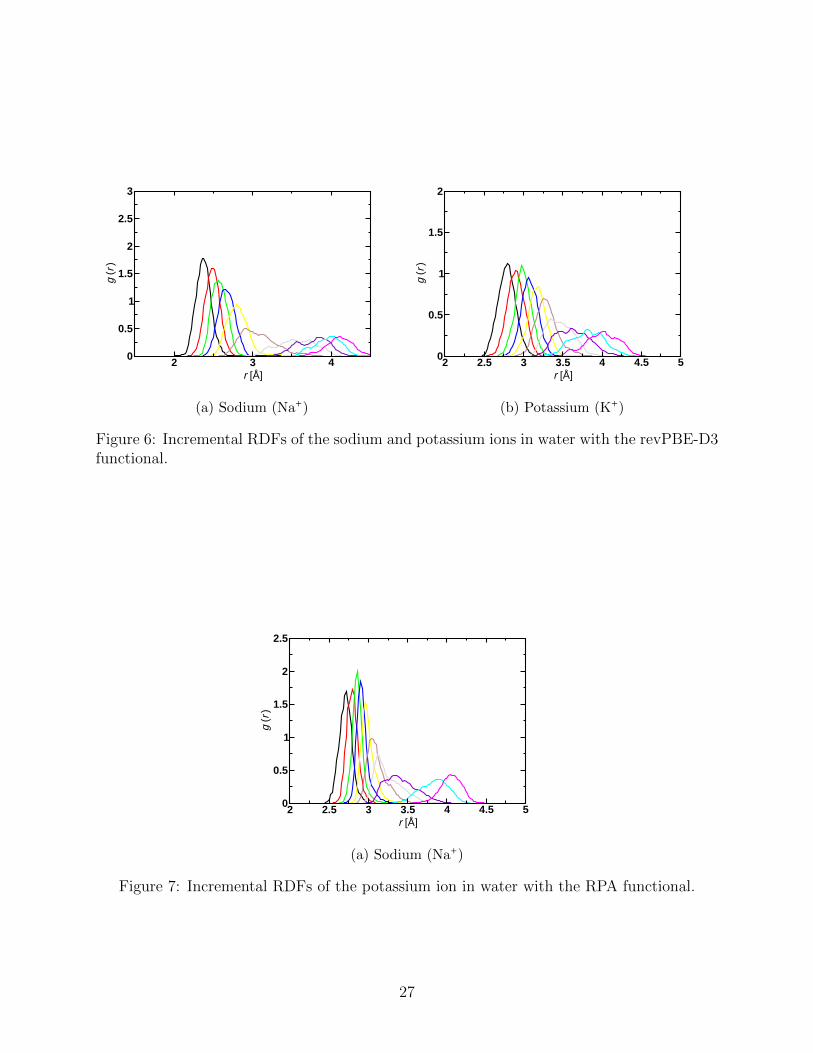
of these configurations to generate a signal corresponding to the ensemble. In Figure 5 we
compare the k2 weighted fine structure, k2 χ(k), vs. k (in A−1), generated in this manner
compared to experimental measurement.20,49
For the case of K+, we recover unprecedented agreement with the measured signal. In
∣χ(r)∣ we recover both the main peak position and width at 2A, but in addition, we recover the
multiple scattering contributions between 3 A and 5 A. The k2χ(k) agreement in amplitude
and frequency from 3 A to 9 A gives us confidence in the consistency between measurement
and simulation.
In comparing measured and simulated signals, for the case of Na+, it is still a challenge
to recover the large amplitude of the measured EXAFS signal. The position of the peak
of ∣χ(r)∣ and the interference pattern of Im(χ(r)) appear to match the measured signal.
In addition, the effect Debye-Waller factor (width of χ(r)) appears to be recovered. The
frequency of the oscillations of k2χ(k) are recovered. The ion-water distance and fluctuations
appear to be recovered by the simulation. It is important to note that for Na+ the EXAFS
data is less reliable as for a low-Z atom the fluorescence self-absorption correction is difficult
10

to apply quantitatively.
For Na+ the mean square variation in the distance obtained from the full width at half
maximum of the Gaussian fit can be determined accurately from experimental XRD to be
0.020 A2. This agrees perfectly with the value determined from the simulation data of SCAN.
Discussion and Error Analysis
It is important to understand why this improvement in the structural properties is observed
with the SCAN functional. Obviously the fundamental answer lies in the more accurate
description of exchange and correlation. However, there should also be a discernible im-
provement in the energetics of the system, i.e., the energies computed with SCAN should
more closely agree with high level correlated quantum mechanical calculations than energies
computed with revPBE-D3. Unfortunately, there are various choices for how to compare the
energies for these systems and there is no systematic analysis of which approach is best. If a
reliable method could be identified that did not require high level calculations on very large
systems it would provide a useful tool and serve as a simple and computationally feasible
diagnostic test to determine which DFT functional will be best suited to reproduce struc-
tural details. Currently the only options for this approach rely on qualitative arguments
or to perform the full, computationally demanding simulations. It is not uncommon for
DFT functionals at similar levels of the theory to predict substantially different structural
details.26,32 This is also a challenge in the area of classical force field development where
force field dependence remains a significant issue.
A standard method used in the development of forcefields is to fit the parameters to
reproduce ion-water binding energies calculated with a high-level of quantum mechanical
theory.11,50–52 Similarly, it is common to use ion-water binding energies to benchmark and
compare different levels of DFT theory.51,52 Normally it is the absolute binding energy of a
11
0 1 2 3 4 5 6Radial distance [Å]
0
0.2
0.4
0.6
0.81
1.2
1.4
1.6
1.8
|χ(R
)|
SCAN exp.
0 1 2 3 4 5 6Radial distance [Å]
0
0.1
0.2
0.3
0.4
0.5
|χ(R
)|
SCAN E0 shiftSCANexp.
0 1 2 3 4 5 6Radial distance [Å]
-2
-1
0
1
2
Im|χ
(R)|
SCANexp.
0 2 4 6Radial distance [Å]
-0.4
-0.2
0
0.2
0.4
Im|χ
(R)|
SCAN E0 shiftSCANexp.
1 2 3 4 5 6 7 8Wave number [Å-1]
-2
-1.5
-1
-0.5
0
0.5
1
1.5
2
k2 |χ(R
)|
SCAN exp.
1 2 3 4 5 6 7 8 9 10Wave number [Å-1]
-0.6
-0.4
-0.2
0
0.2
0.4
0.6
0.8
k2 |χ(R
)|
SCAN E0 shiftSCANexp.
Figure 5: EXAFS calculations compared with experiment for sodium (left) and potassium(right). Experimental data from Ref. 20,49.
12

cluster that is used. This is defined as:
EBind2 = EC+(H2O)n −EC+ − nE(H2O) (1)
Where C+ is the cation. Often only very small clusters are considered and the structures
of the clusters are minimum energy structures or artificial perturbations about it. This is
not the best choice, as the minimum energy structure of an ion-water dimer for instance will
be very different to the typical structures that water molecules take around ions under the
conditions of bulk solvation. Therefore, it is preferable to extract from simulation clusters
of the closest n waters about the ion. Larger water clusters than dimers and trimers will
be more relevant as they will incorporate the desired many-body effects. The definition of
the binding energy given above is not ideal for large cluster sizes, because the error will be
dominated by the error in the water-water interactions. If we are interested in improving
the description of the water structure specifically around the ion a better choice may be:
EBind1 = EC+(H2O)n −EC+ −E(H2O)n (2)
where the E(H2O)n is computed by simply removing the ion and recomputing the energy. This
expression gives the binding energy of the ion to the water not the water-water interaction.
This is also a very important quantity as it is the definition of the energy used in the potential
distribution theorem to determine the solvation free energy of the ion.53
When comparing with multiple structures it is customary to calculate the RMSD in this
binding energy between the DFT functional and a higher level of theory such as MP2. This
is given by:
RMSD =√
1
N∑N
(EMP2Bind1 −EDFT
Bind1)2
(3)
where the sum is over a N frames extracted from the simulation. It should be noted that this
is not the optimal choice if the goal is to accurately reproduce structural properties of the
13

ion in water. The possibility to have large average errors in the total binding energy and still
accurately reproduce the structural details produces a confounding picture for researchers
that are solely concerned with the accuracy of any particular method. This is because the
water structure is actually determined by (mean) forces rather than energies and thus a large
error in the total binding energy will not affect the structural properties if there is not a
large variation in binding energies from structure to structure. Assuming the ions are never
isolated in vacuum, the average error in the total binding energy will be irrelevant in terms
of the structural details. An alternative way of seeing this is that the relative probability
of two states is given by the ratio of the Boltzmann factors which should not be altered by
large constant error terms. That is:
e−βE1+Eerror
e−βE2+Eerror= e−βE1
e−βE2(4)
We suggest that a more effective quantity to examine is the standard deviation of the mean
signed error. This is given by:
SD-ME =√
1
N − 1∑N
((EMP2Bind1 −EDFT
Bind1) − ⟨EMP2Bind1 −EDFT
Bind1⟩)2
(5)
The angle brackets term is the average error in the DFT binding energy compared with
MP2 over the N structures sampled. This expression captures the variation of the error from
structure to structure. If this quantity is zero throughout the entire simulation then the
structural details should be perfectly reproduced regardless of the whether the total binding
energy is accurate or not. Standard basis sets used in condensed phase simulation are
demonstrably far from the complete basis set limit and running simulations at the complete
basis set limit is not computationally feasible. Basis set superposition error is known to be
a substantial issue in clusters and thus is not reasonable to expect DFT-MD methods to
reproduce absolute binding free energies in any case.
When we compute the standard deviation of the mean signed error using the EBind1
14

quantity given above we can see that the error going from revPBE-D3 to SCAN is reduced
by a factor of 7 (5.2 kJmol−1 to 0.7 kJmol−1) for Na(H2O)8 clusters and by a factor of 5
(7.8 kJmol−1 to 1.6 kJmol−1) for K(H2O)8 clusters. The resulting variation with the SCAN
functional is reduced below both thermal noise (2.4 kJmol−1) and “chemical accuracy” (4.2
kJmol−1 ) explaining the satisfactory experimental agreement of structural properties. A
very similar result is also seen for 32 water molecule clusters although a smaller basis set
for the reference MP2 calculations must be used. This reduction is not seen for the RMSD
error where it can actually be slightly larger for SCAN than revPBE-D3. We did not provide
an analysis for the RPA functional due to large memory requirements required to perform
these calculations in the large box sizes used to approximate a non-periodic system. The
supplementary information (SI) provides a detailed comparison of the different methods of
error estimation. It is true that many properties, such as single ion solvation free energies,
are sensitive to the accuracy of the absolute binding free energy not just the variation in
the error. However, once accurate structures are obtained, standard perturbative methods
can be used to estimate absolute binding energies in situations where this quantity is of
importance. See Ref. 53 for an example of this.
In summary, we have demonstrated that the newly developed SCAN functional can ac-
curately reproduce key structural properties of water around the sodium and potassium ions
that simple GGA functionals fail to reproduce. Additionally these properties cannot be ac-
curately reproduced in reasonable computational times using high level RPA functional. We
have argued that the fluctuation of the mean signed error of the ion-water binding energy
should be used to assess the quality of a DFT functional or forcefield and that this quantity
is consistent with the improved performance of the SCAN functional over revPBE-D3.
15

Computational Details
Ab initio Simulations
For the revPBE and SCAN functionals, Born-Oppenheimer ab initio molecular dynamics
simulations within the NVT (at 300 K) ensemble using periodic boundary conditions are
performed within the CP2K simulation suite (http:www.cp2k.org) containing the QuickStep
module for the DFT calculations.54 The D3 dispersion correction due to Grimme25 was used
for revPBE. A 0.5 fs time step was used. We used a double ζ basis set that has been optimized
for the condensed phase55 in conjunction with GTH pseudopotentials56 using a 400 Ry cutoff
for the auxiliary plane wave basis for the revPBE-D3 simulations and a 1200 Ry cutoff for the
SCAN simulations.32,57 The pseudopotentials for the oxygen atom were reoptimized for the
SCAN functional using the ATOM code of CP2K. A Nose-Hoover thermostat was attached
to every degree of freedom to ensure equilibration.58 The energies were accumulated for ≈
12 ps after 3 ps of equilibration. The sodium and potassium simulations for revPBE-D3 and
SCAN consisted of one sodium ion in a box of water molecules of dimensions 14.33 A3.
For the RPA simulations of potassium the procedure outlined in Ref. 3333,34 was used.
The simulation box consisted of 63 water molecules and a single potassium ion with dimen-
sions of 12.423A3. Basis sets of correlation consistent triple zeta quality, were used including
new basis sets for the potassium. The cutoff is set to 800 Ry. Core electrons are replaced by
pseudopotentials that have been parametrized for the PBE functional. The Kohn-Sham PBE
orbitals were used as input for the RPA calculation. The Monte Carlo (MC) simulations have
been performed employing the same setup as given in Ref. 46, with T = 295 K and p = 1 bar.
The MC efficiency is improved with the presampling of moves, the approximated potential is
calculated using the revPBE-D3 functional. The initial configuration has been equilibrated
with a 15 ps NVT-MD run at the experimental density using the revPBE-D3 level. The
statistics from the MC simulations were accumulated for 6100 frames after equilibration for
500.
16

ORCA59 was used to calculate the cluster energies at the MP2 level of theory. Clusters
of 8 and 32 water molecules were used in the cluster correction calculation with 50 frames
extracted from the 15ps trajectory. The aug-cc-pVxZ basis set was used for the oxygen
and hydrogen atoms.60 Similarly, the cc-pCVxZ basis set was used for the sodium ion61 and
the cc-pwCVxZ basis set for the potassium ion.62 x was either D or Q. Frozen cores were
used for the MP2 calculations. RI-MP2 was used for the 32 water cluster calculations using
automatically generated auxiliary basis functions. For the revPBE-D3 and SCAN cluster
energy calculations CP2K was used with the periodicity none option and a larger cell size
to remove any box size dependence. Otherwise the same parameters, basis sets etc. as the
simulation were used.
EXAFS
The measurements have correction of the multi-electron features. (see Ref. 49). In addition,
a shift in E0 is universally applied to the ensemble, adjusted to fit the first peak at 2A−1 In
Figure 5 we Fourier transform the χ(k) to construct χ(r). This is applied in a consistent
manner to both the measurement and the simulation. χ(r) consists of a distribution of scat-
tering distances that is distorted by the interaction of a photoelectron with the underlying
charge density. This interaction is accounted for in the multiple scattering code, feff. Ex-
perimental measurement of these systems is challenging due to issues associated with Multi
Electron Electron Scattering. In the current work, we carve out clusters of the nearest 10
water molecules to the cation to use as input for EXAFS calculation using default settings of
the FEFF9 code. For the K+ and Na+ systems every 10th configuration was chosen to make
up the ensemble, this gives 3055 configurations for K+ and 3029 configurations for Na+. The
ensemble averaged χ(k) signal is recovered from an average of configurations. Next we take
the Fourier transform of the k2 χ(k) signal to generate the ∣χ(R)∣ and Im[χ(R)] signals.
When compared to simple anions and doubly charged cations, it is has been a challenge
to find the right combination of molecular interaction and statistical mechanical sampling to
17

reproduce the measured EXAFS signals for Na+ and K+ aqueous solvation. It is a challenge
to find a balance between the strong cation-water interaction while maintaining the integrity
of the collective solvent structure, solvent-solvent interaction in the presence of the cation
and disrupting the hydrogen-bonding network, while also describing the long-range, bulk
structure and response of water. The use of a single molecular framework to consistently
describe each of these molecular responses remains a challenge.
Acknowledgements
This research used resources of the National Energy Research Scientific Computing Center,
a DOE Office of Science User Facility supported by the Office of Science of the U.S. Depart-
ment of Energy under Contract No. DE-AC02-05CH11231 in addition to Pacific Northwest
National Laboratory (PNNL) Institutional Computing resources. TTD, MG, GKS and CJM
were supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy
Sciences, Division of Chemical Sciences, Geosciences, and Biosciences. MD Baer was sup-
ported by MS3 (Materials Synthesis and Simulation Across Scales) Initiative, a Laboratory
Directed Research and Development Program PNNL. PNNL is a multiprogram national
laboratory operated by Battelle for the U.S. Department of Energy. JW and JH are sup-
port by The National Centre of Competence in Research (NCCR) Materials Revolution:
Computational Design and Discovery of Novel Materials (MARVEL) of the Swiss National
Science Foundation (SNSF). XSZ and TTD acknowledge the Australian Research Council
(ARC) funding via project number FL170100101. MDB is supported by the Center for
Computational Study of Excited-State Phenomena in Energy Materials (C2SEPEM) and
by the SciDAC Program on Excited State Phenomena in Energy Materials at the Lawrence
Berkeley National Laboratory, which is funded by the U.S. Department of Energy, Office of
Science, Basic Energy Sciences, Materials Sciences and Engineering Division under Contract
No. DE-AC02-05CH11231, as part of the Computational Materials Sciences Program.
18

References
(1) Kubota, K.; Komaba, S. ReviewPractical Issues and Future Perspective for Na-Ion
Batteries. J. Electrochem. Soc. 2015, 162, A2538–A2550.
(2) Wessells, C. D.; Peddada, S. V.; Huggins, R. A.; Cui, Y. Nickel hexacyanoferrate
nanoparticle electrodes for aqueous sodium and potassium ion batteries. Nano Lett.
2011, 11, 5421–5425.
(3) Liu, Y.; Fan, F.; Wang, J.; Liu, Y.; Chen, H.; Jungjohann, K. L.; Xu, Y.; Zhu, Y.; Bi-
gio, D.; Zhu, T. et al. In situ transmission electron microscopy study of electrochemical
sodiation and potassiation of carbon nanofibers. Nano Lett. 2014, 14, 3445–3452.
(4) Jian, Z.; Xing, Z.; Bommier, C.; Li, Z.; Ji, X. Hard Carbon Microspheres: Potassium-
Ion Anode Versus Sodium-Ion Anode. Adv. Energy Mater. 2016, 6, 1501874.
(5) Avall, G.; Mindemark, J.; Brandell, D.; Johansson, P. Sodium-Ion Battery Electrolytes:
Modeling and Simulations. Adv. Energy Mater. 2018, 8, 1703036.
(6) Clausen, M. J. V.; Poulsen, H. Met. Cell. Met. Ions Life Sci. vol 12.; Springer, Dor-
drecht, 2013; pp 41–67.
(7) Peng, J.; Cao, D.; He, Z.; Guo, J.; Hapala, P.; Ma, R.; Cheng, B.; Chen, J.; Xie, W. J.;
Li, X.-Z. et al. The effect of hydration number on the interfacial transport of sodium
ions. Nature 2018, 557, 701–705.
(8) Dang, L. X.; Rice, J. E.; Caldwell, J.; Kollman, P. A. Ion Solvation in Polarizable
Water: Molecular Dynamics Simulations. J. Am. Chem. Soc. 1991, 113, 2481–2486.
(9) Chang, T.-M.; Dang, L. X. Detailed Study of Potassium Solvation Using Molecular
Dynamics Techniques. J. Phys. Chem. B 1999, 103, 4714–4720.
(10) White, J. A.; Schwegler, E.; Galli, G.; Gygi, F. The solvation of Na+ in water: First-
principles simulations. J. Chem. Phys. 2000, 113, 4668.
19

(11) Carrillo-Tripp, M.; Saint-Martin, H.; Ortega-Blake, I. A comparative study of the hy-
dration of Na+ and K+ with refined polarizable model potentials. J. Chem. Phys. 2003,
118, 7062–7073.
(12) Varma, S.; Rempe, S. B. Coordination numbers of alkali metal ions in aqueous solutions.
Biophys. Chem. 2006, 124, 192–199.
(13) Azam, S. S.; Hofer, T. S.; Randolf, B. R.; Rode, B. M. Hydration of Sodium (I) and
Potassium (I) Revisited : A Comparative QM / MM and QMCF MD Simulation Study
of Weakly Hydrated Ions. J. Phys. Chem. 2009, 113, 1827–1834.
(14) Rowley, C. N. The Solvation Structure of Na+ and K+ in Liquid Water Determined
from High Level ab Initio Molecular Dynamics Simulations. J. Chem. Theory Comput.
2012, 8, 3526–3535.
(15) Bankura, A.; Carnevale, V.; Klein, M. L. Hydration structure of salt solutions from ab
initio molecular dynamics. J. Chem. Phys. 2013, 138, 014501.
(16) Bankura, A.; Carnevale, V.; Klein, M. L. Hydration Structure of Na+ and K+ from Ab
Initio Molecular Dynamics Based on Modern Density Functional Theory. Mol. Phys.
2014, 112, 1448–1456.
(17) Gaiduk, A. P.; Zhang, C.; Gygi, F.; Galli, G. Structural and electronic properties of
aqueous NaCl solutions from ab initio molecular dynamics simulations with hybrid
density functionals. Chem. Phys. Lett. 2014, 604, 89–96.
(18) Soniat, M.; Rogers, D. M.; Rempe, S. B. Dispersion- and Exchange-Corrected Density
Functional Theory for Sodium Ion Hydration. J. Chem. Theory Comput. 2015, 11,
2958–2967.
(19) Ikeda, T.; Boero, M. Role of van der Waals corrections in first principles simulations of
alkali metal ions in aqueous solutions. J. Chem. Phys. 2015, 143, 194510.
20

(20) Galib, M.; Baer, M. D.; Skinner, L. B.; Mundy, C. J.; Huthwelker, T.; Schenter, G. K.;
Benmore, C. J.; Govind, N.; Fulton, J. L. Revisiting the hydration structure of aqueous
Na+. J. Chem. Phys. 2017, 146, 084504.
(21) Hofer, T. S.; Hunenberger, P. H. Absolute proton hydration free energy, surface po-
tential of water, and redox potential of the hydrogen electrode from first principles:
QM/MM MD free-energy simulations of sodium and potassium hydration. J. Chem.
Phys. 2018, 148, 222814.
(22) Pham, T. A. Ab initio simulations of liquid electrolytes for energy conversion and
storage. Int. J. Quantum Chem. 2018, e25795.
(23) Perdew, J. P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made
Simple. Phys. Rev. Lett. 1996, 77, 3865–3868.
(24) Zhang, Y.; Yang, W. Comment on Generalized Gradient Approximation Made Simple.
Phys. Rev. Lett. 1998, 80, 890–890.
(25) Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A Consistent and Accurate Ab Ini-
tio Parametrization of Density Functional Dispersion Correction (DFT-D) for the 94
Elements H-Pu. J. Chem. Phys. 2010, 132, 154104.
(26) Galib, M.; Duignan, T. T.; Misteli, Y.; Baer, M. D.; Schenter, G. K.; Hutter, J.;
Mundy, C. J. Mass Density Fluctuations in Quantum and Classical descriptions of
Liquid Water. J. Chem. Phys. 2017, 146, 244501.
(27) Galib, M.; Schenter, G. K.; Mundy, C. J.; Govind, N.; Fulton, J. L. Unraveling the
spectral signatures of solvent ordering in K-edge XANES of aqueous Na+. J. Chem.
Phys. 2018, 149, 124503.
(28) Perdew, J. P.; Ruzsinszky, A.; Tao, J.; Staroverov, V. N.; Scuseria, G. E.; Csonka, G. I.
21

Prescription for the design and selection of density functional approximations: More
constraint satisfaction with fewer fits. J. Chem. Phys. 2005, 123, 062201.
(29) Sun, J.; Ruzsinszky, A.; Perdew, J. Strongly Constrained and Appropriately Normed
Semilocal Density Functional. Phys. Rev. Lett. 2015, 115, 036402.
(30) Chen, M.; Ko, H.-y.; Remsing, R. C.; Calegari, M. F.; Santra, B.; Sun, Z. Ab initio
theory and modeling of water. Proc. Natl. Acad. Sci. 2017, 24–26.
(31) Zheng, L.; Chen, M.; Sun, Z.; Ko, H.-Y.; Santra, B.; Dhuvad, P.; Wu, X. Structural,
Electronic, and Dynamical Properties of Liquid Water by ab initio Molecular Dynamics
based on SCAN Functional within the Canonical Ensemble. J. Chem. Phys. 2018, 148,
164505.
(32) Yao, Y.; Kanai, Y. Free Energy Profile of NaCl in Water: First-Principles Molecu-
lar Dynamics with SCAN and ωB97X-V Exchange-Correlation Functionals. J. Chem.
Theory Comput. 2018, 14, 884–893.
(33) Del Ben, M.; Hutter, J.; VandeVondele, J. Probing the structural and dynamical prop-
erties of liquid water with models including non-local electron correlation. J. Chem.
Phys. 2015, 143, 054506.
(34) Del Ben, M.; Schutt, O.; Wentz, T.; Messmer, P.; Hutter, J.; VandeVondele, J. Enabling
simulation at the fifth rung of DFT: Large scale RPA calculations with excellent time
to solution. Comput. Phys. Commun. 2015, 187, 120–129.
(35) Marsalek, O.; Markland, T. E. Quantum dynamics and spectroscopy of ab initio liquid
water: the interplay of nuclear and electronic quantum effects. J. Phys. Chem. Lett.
2017, 8, 1545–1551.
(36) Baer, M. D.; Mundy, C. J.; McGrath, M. J.; Kuo, I.-F. W.; Siepmann, J. I.; Tobias, D. J.
22

Re-examining the properties of the aqueous vaporliquid interface using dispersion cor-
rected density functional theory. J. Chem. Phys. 2011, 135, 124712.
(37) Becke, A. D. Density-Functional Exchange-Energy Approximation with Correct
Asymptotic Behavior. Phys. Rev. A 1988, 38, 3098–3100.
(38) Lee, C.; Yang, W.; Parr, R. G. Development of the Colle-Salvetti Correlation-Energy
Formula into a Functional of the Electron Density. Phys. Rev. B 1988, 37, 785–789.
(39) Grimme, S. Accurate Description of van der Waals Complexes by Density Functional
Theory Including Empirical Corrections. J. Comput. Chem. 2004, 25, 1463–1473.
(40) Medders, G. R.; Babin, V.; Paesani, F. Development of a First-Principles Water Poten-
tial with Flexible Monomers. III. Liquid Phase Properties. J. Chem. Theory Comput.
2014, 10, 2906–2910.
(41) Ceriotti, M.; Fang, W.; Kusalik, P. G.; McKenzie, R. H.; Michaelides, A.;
Morales, M. A.; Markland, T. E. Nuclear Quantum Effects in Water and Aqueous
Systems: Experiment, Theory, and Current Challenges. Chem. Rev. 2016, 116, 7529–
7550.
(42) Yao, Y. Advancing Molecular Dynamics Simulations of Aqueous Ionic Solutions. Ph.D.
thesis, University of North Carolina, 2018.
(43) Hutter, J.; Iannuzzi, M.; Schiffmann, F.; VandeVondele, J. ¡Scp¿Cp2K:¡/Scp¿ Atomistic
Simulations of Condensed Matter Systems. Wiley Interdiscip. Rev. Comput. Mol. Sci.
2014, 4, 15–25.
(44) Car, R.; Parrinello, M. Unified Approach for Molecular Dynamics and Density-
Functional Theory R. Phys. Rev. Lett. 1985, 55, 2471–2474.
(45) Duignan, T. T.; Baer, M. D.; Mundy, C. J. Ions Interacting in Solution: Moving from
Intrinsic to Collective Properties. Curr. Opin. Colloid Interface Sci. 2016, 23, 58–65.
23

(46) Del Ben, M.; Schonherr, M.; Hutter, J.; VandeVondele, J. Bulk liquid water at ambient
temperature and pressure from MP2 theory. J. Phys. Chem. Lett. 2013, 4, 3753–3759.
(47) Kunz, W. Specific Ion Effects in Colloidal and Biological Systems. Curr. Opin. Colloid
Interface Sci. 2010, 15, 34–39.
(48) Rehr, J. J.; Kas, J. J.; Vila, F. D.; Prange, M. P.; Jorissen, K. Parameter-free calcula-
tions of X-ray spectra with FEFF9. Phys. Chem. Chem. Phys. 2010, 12, 5503–5513.
(49) Glezakou, V. A.; Chen, Y.; Fulton, J. L.; Schenter, G. K.; Dang, L. X. Electronic struc-
ture, statistical mechanical simulations, and EXAFS spectroscopy of aqueous potas-
sium. Theor. Chem. Acc. 2006, 115, 86–99.
(50) Lamoureux, G.; Roux, B. Absolute Hydration Free Energy Scale for Alkali and Halide
Ions Established from Simulations with a Polarizable Force Field. J. Phys. Chem. B
2006, 110, 3308–3322.
(51) Arismendi-Arrieta, D. J.; Riera, M.; Bajaj, P.; Prosmiti, R.; Paesani, F. The i-TTM
Model for Ab Initio-Based Ion-Water Interaction Potentials. 1. Halide-Water Potential
Energy Functions . J. Phys. Chem. B 2016, 120, 1822–1832.
(52) Riera, M.; Gotz, A. W.; Paesani, F. The i-TTM model for ab initio-based ionwater
interaction potentials. II. Alkali metal ionwater potential energy functions. Phys. Chem.
Chem. Phys. 2016, 18, 30334–30343.
(53) Duignan, T. T.; Baer, M. D.; Schenter, G. K.; Mundy, C. J. Real single ion solvation
free energies with quantum mechanical simulation. Chem. Sci. 2017, 8, 6131 – 6140.
(54) VandeVondele, J.; Krack, M.; Mohamed, F.; Parrinello, M.; Chassaing, T.; Hutter, J.
Quickstep: Fast and Accurate Density Functional Calculations using a Mixed Gaussian
and Plane Waves Approach. Comput. Phys. Commun. 2005, 167, 103–128.
24

(55) VandeVondele, J.; Hutter, J. Gaussian Basis Sets for Accurate Calculations on Molec-
ular Systems in Gas and Condensed Phases. J. Chem. Phys. 2007, 127, 114105.
(56) Goedecker, S.; Teter, M.; Hutter, J. Separable Dual-Space Gaussian Pseudopotentials.
Phys. Rev. B 1996, 54, 1703–1710.
(57) Miceli, G.; Hutter, J.; Pasquarello, A. Liquid Water through Density-Functional Molec-
ular Dynamics: Plane-Wave vs Atomic-Orbital Basis Sets. J. Chem. Theory Comput.
2016, 12, 3456–3462.
(58) Martyna, G. J.; Klein, M. L.; Tuckerman, M. Nose–Hoover Chains: The Canonical
Ensemble via Continuous Dynamics. J. Chem. Phys. 1992, 97, 2635–2643.
(59) Neese, F. The ORCA program system. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2012,
2, 73–78.
(60) Dunning, Jr., T. H. Gaussian Basis Sets for use in Correlated Molecular Calculations. I.
The Atoms Boron through Neon and Hydrogen. J. Chem. Phys. 1989, 90, 1007–1023.
(61) Woon, D. E.; Dunning, Jr., T. H. Gaussian Basis Sets for use in Correlated Molecular
Calculations. IV. Calculation of Static Electrical Response Properties. J. Chem. Phys.
1994, 100, 2975–2988.
(62) Peterson, K. Private communication. 2017.
Supplementary Information
Cluster binding energies
The ion cluster binding energies are defined as:
EBind1 = EC+(H2O)n −EC+ −E(H2O)n (6)
25

EBind2 = EC+(H2O)n −EC+ − nE(H2O) (7)
The difference is that EBind2 includes the contribution from water-water interactions as well
as the ion-water binding. It is clear that the minimal SD-ME (1) error for SCAN compared
Table 2: Mean unsigned error in the cluster binding energies and fluctuationsin the mean error. The (1) and (2) indicates whether EBind1 or EBind2 was used.RMSD indicates the root mean squared deviation error estimate, whereas SDME indicates the standard deviation in the mean signed error. The DZ or QZin brackets indicates whether double zeta or quadruple zeta basis sets were usedfor the MP2 level calculation. Units are in kJmol−1.
RMSD(1) SD-ME(1) RMSD(2) SD-ME (2)Na+(H2O)8(revPBE-D3)(DZ) 5.8 5.8 9.4 7.8
Na+(H2O)8(SCAN)(DZ) 9.0 2.2 7.7 4.6Na+(H2O)8(revPBE-D3)(QZ) 8.1 5.2 17.5 4.1
Na+(H2O)8(SCAN)(QZ) 11.0 0.7 24.6 2.9Na+(H2O)32(revPBE-D3)(DZ) 16.3 5.5 81.7 11.5
Na+(H2O)32(SCAN(DZ) 9.3 1.9 23.3 8.4K+(H2O)8(revPBE-D3)(DZ) 10.0 6.3 8.5 7.2
K+(H2O)8(SCAN)(DZ) 9.7 1.9 7.7 4.0K+(H2O)8(revPBE-D3)(QZ) 8.3 7.8 13.2 10.1
K+(H2O)8(SCAN) (QZ) 2.0 1.6 11.9 2.8K+(H2O)32(revPBE-D3)(DZ) 24.4 6.8 54.3 12.2
K+(H2O)32(SCAN)(DZ) 6.9 2.1 20.3 8.1
with revPBE-D3 explains why SCAN has such good structural performance. The RMSD
error for the Na+(H2O)8(QZ) case actually goes up from revPBE-D3 to SCAN demonstrating
that this is not a good estimate of the error if reproducing structural properties is the aim.
It is clear that the error using the double zeta level of theory for the MP2 calculations
shows a less dramatic shift which is not surprising considering there will be significant error
associated with the smaller basis set. 32 water molecule clusters were also computed showing
that the standard deviation in the error does not change significantly as you go to the larger
cluster sizes.
Incremental RDFs
Here we provide incremental RDFs for the other functionals studied.
26
2 3 4r [Å]
0
0.5
1
1.5
2
2.5
3
g (r
)
(a) Sodium (Na+)
2 2.5 3 3.5 4 4.5 5r [Å]
0
0.5
1
1.5
2
g (r
)(b) Potassium (K+)
Figure 6: Incremental RDFs of the sodium and potassium ions in water with the revPBE-D3functional.
2 2.5 3 3.5 4 4.5 5r [Å]
0
0.5
1
1.5
2
2.5
g (r
)
(a) Sodium (Na+)
Figure 7: Incremental RDFs of the potassium ion in water with the RPA functional.
27

Graphical TOC Entry
0 2 4 6 8r [Å]
0
2
4
6
8
g (r
)
revPBE-D3SCANExp. peak position
28

download fileview on ChemRxivcationstructure.pdf (356.77 KiB)
















![MD-272_273 Full Manual [English]](https://img.pdfslide.net/doc/110x75/6329927f0b4264421f0069b9/md-272273-full-manual-english.jpg)






![Carte drept constitutional.[conspecte md]](https://img.pdfslide.net/doc/110x75/6319ccbebc8291e22e0f468f/carte-drept-constitutionalconspecte-md.jpg)





