Embed Size (px)
Citation preview

Phytochemistry 70 (2009) 1239–1245
Contents lists available at ScienceDirect
Phytochemistry
journal homepage: www.elsevier .com/locate /phytochem
Hydroperoxy-cycloartane triterpenoids from the leaves of Markhamia lutea,a plant ingested by wild chimpanzees
Damien Lacroix a, Soizic Prado a,*, Alexandre Deville a, Sabrina Krief b, Vincent Dumontet c, John Kasenene d,Elisabeth Mouray a, Christian Bories e, Bernard Bodo a,*
a Molécules de Communication et Adaptation des Micro-organismes, Unité Associée au CNRS, Muséum National d’Histoire Naturelle, 57 Rue Cuvier, 75005 Paris, Franceb Eco-anthropologie et Ethnobiologie, MNHN-CNRS, 43 Rue Buffon, 75005 Paris, Francec Institut de Chimie des Substances Naturelles, CNRS, Avenue de la Terrasse 91198 Gif-sur-Yvette, Franced Department of Botany, Makerere University, P.O. Box 7062, Kampala, Ugandae Chimiothérapie Antiparasitaire, CNRS, Faculté de Pharmacie, Université Paris Sud, 92200 Châtenay-Malabry, France
a r t i c l e i n f o
Article history:Received 26 March 2009Received in revised form 21 June 2009Available online 11 August 2009
Keywords:CycloartaneTriterpenoid hydroperoxidesMarkhamia luteaBignoniaceaeAnti-parasitic activitiesZoopharmacognosyChimpanzees
0031-9422/$ - see front matter � 2009 Elsevier Ltd. Adoi:10.1016/j.phytochem.2009.06.020
* Corresponding authors. Tel.: +33 (0) 1 40 79 31 2E-mail addresses: [email protected] (S. Prado), bodo
a b s t r a c t
In the framework of the phytochemical investigation of plant species eaten by wild chimpanzees in theirnatural environment in Uganda, leaf samples of Markhamia lutea were selected and collected. The crudeethyl acetate extract of M. lutea leaves exhibited significant in vitro anti-parasitic activity and low cyto-toxicity against MRC5 and KB cells. Fractionation of this extract led to six cycloartane triterpenoids,musambins A–C and their 3-O-xyloside derivatives musambiosides A–C. The structures were elucidatedon the basis of spectral studies including mass spectroscopy and extensive 2D NMR. Most of the com-pounds exhibited mild anti-leishmanial and anti-trypanosomal activities.
� 2009 Elsevier Ltd. All rights reserved.
1. Introduction tane triterpenoids, musambins A–C (1–3), as well as three glyco-
CH3 COOH
R1O
CH3
HH
R2CH3
2
3
12
13
1614
4
5
6
7
8
1
9
1015
11
18
19
17
29 28
30
OHH
H
H CH3
OOH
CH3
21
2022
23
24
25
26
27
CH3H
H
H
CH3
CH2
CH3
21
2022
23 24
2526
27
OOH
H
CH3
CH
21
2022
23 24H
O1 : R1 = H ; R2 = A2 : R1 = H ; R2 = B3 : R1 = H ; R2 = C4 : R = Xyl ; R = A
HA
B
C
In continuation of a programme on bioactive compounds fromplants ingested by wild chimpanzees (Pan troglodytes schweinfur-thii) in the Kanyawara area of the Kibale National park in Ugandasince 2000 (Krief et al., 2004, 2005a,b), we have phytochemicallyinvestigated the leaves of Markhamia lutea (Benth.) K. Schum.(Bignoniaceae). This upright evergreen tree with yellow flowersand wavy thin leaves grows in South-Western Uganda and iscommonly known as ‘‘musambia” in both Rutooro and Rukiga locallanguages (Kasanene, personal communication). Traditional medi-cine uses the leaves of this species as anti-parasitic and the roots inorder to alleviate symptoms of watery and bloodness diarrhea(Kernan et al., 1998). In Kibale National Park this species is eatenby primates such as chimpanzees and black-and-white colobus(Onderdonk and Chapman, 2000; Chapman et al., 2003). Previousphytochemical researches on the roots of this plant have led tothe isolation of anti-viral phenylpropanoids (Kernan et al., 1998;Kanchanapoom et al., 2002; Sinaphet et al., 2006). However, theanti-parasitic activity of the leaves has not been yet investigated.
We now report on the isolation from the ethyl acetate extract ofthe leaves and the structural determination of three new cycloar-
ll rights reserved.
9; fax: +33 (0) 1 40 79 31 [email protected] (B. Bodo).
side derivatives, musambiosides A–C (4–6), along with theknown phaeophorbide A, b-sitosterol and pentacyclic triterpenesarjunic acid (Anjaneyulu and Rama Prasad, 1982; Conrad et al.,1998) and 2-epi-tormentic acid (Delgado et al., 1989; Hattoriet al., 1988). The biological evaluation of 1–6 pointed out anti-try-panosomal and anti-plasmodial activities.
2
CH3
2526
27
1 25 : R1 = Xyl ; R2 = B6 : R1 = Xyl ; R2 = C

1240 D. Lacroix et al. / Phytochemistry 70 (2009) 1239–1245
2. Results and discussion
2.1. Isolation
The dried and ground leaves of M. lutea were defatted withcyclohexane and then extracted with ethyl acetate at room tem-perature. From this EtOAc extract, six original cycloartane triter-penes, musambins A (1), B (2) and C (3) and their glycosidederivatives, musambiosides A (4), B (6) and C (5) were purifiedby silica gel column chromatography and C18 reversed-phase-HPLC.
2.2. Structure determination
Musambin A (1), was obtained as a colourless amorphous solid,optically active, ½a�22
D 78.4� (c 1.0, MeOH). Its HR-ESI-MS (negativemode) showed the quasi-molecular ion [M�H]� at m/z: 503.3372(calc. for C30H47O6: 503.3373) corresponding to the molecular for-mula C30H48O6 for 1 and indicative of seven degrees of unsatura-tion. The IR spectrum showed absorptions for carboxylic acid andhydroxyl functionalities at mmax 1701 and 3362 cm�1, respectively.The 13C J-modulated NMR spectrum (CD3OD) of 1 exhibited thethirty carbon atoms of the molecular formula, including three sp2
carbons of one carboxylic acid at dC 180.6 ppm and one mono-substituted carbon–carbon double bond at dC 114.3 (@CH2) and145.7 (>C@). Taking into account the seven degrees of unsatura-tion, 1 includes five rings. The sp3 carbons were distributed intofive methyls, 10 methylenes, seven methines including three oxy-genated ones and five quaternary carbons (Table 1). All the spectraldata favoured a triterpene structure. On the NMR spectra, the sig-nals at dC 31.1 and dH 0.70 and 0.50 forming two doublets
Table 113C NMR data for compounds (1–6) (CD3OD, 400.13 MHz, 298 K).
C No. 1 2 3 4 5 6
1 73.6 73.6 73.6 73.5 73.6 73.52 37.8 37.8 37.9 36.9 37.0 37.03 71.4 71.4 71.5 81.7 81.8 81.74 55.9 55.9 56.0 55.0 55.2 55.15 38.3 38.3 38.5 38.5 38.6 38.56 24.0 24.0 24.0 23.7 23.8 23.77 26.5 26.5 26.8 26.5 26.5 26.48 50.1 50.1 50.1 50.0 50.0 50.09 22.2 22.2 22.2 22.2 22.2 22.210 30.2 30.3 30.2 30.2 30.3 30.211 26.7 26.7 26.8 26.4 26.7 26.712 34.1 34.0 34.0 34.0 34.0 34.013 46.4 46.4 46.5 46.4 46.4 46.514 50.0 50.0 50.0 49.9 49.9 49.915 36.9 36.9 37.0 36.9 37.0 36.916 29.1 29.0 29.1 29.1 29.1 29.017 53.6 53.3 53.7 53.6 53.3 53.618 18.8 18.9 19.0 18.8 18.9 18.619 31.1 31.1 31.1 31.0 31.0 31.020 37.1 37.6 37.2 37.0 37.7 37.121 18.7 18.9 18.9 18.7 18.9 18.722 33.2 40.7 32.3 33.2 40.5 32.423 28.3 129.7 35.6 28.3 129.7 35.624 91.0 137.0 204.5 91.0 137.0 205.025 145.7 82.5 145.9 145.7 82.5 146.026 114.3 24.9 125.9 114.3 25.1 125.727 16.9 24.9 17.9 16.9 25.0 17.828 180.6 180.8 180.8 180.7 181.0 180.729 9.1 9.0 9.1 9.8 9.9 9.830 19.9 19.8 20.2 20.0 19.8 19.810 – – – 106.2 106.3 106.220 – – – 75.1 75.2 75.130 – – – 77.5 77.6 77.540 – – – 71.1 71.2 71.250 – – – 66.7 66.8 66.7
(J = 4.5 Hz) were indicative of the presence of a methylene in acyclopropyl group, suggesting a cycloartane triterpenoid structure.
Analysis of the correlations on the 1H–1H COSY spectrum al-lowed the determination of the sub-structures (a–f) which aremarked with bold bonds in Fig. 1. Especially were depicted the fol-lowing spin systems: sub-structure a (–CH(O)–CH2–CH(O)–) thetwo ending methine groups of which bearing an oxygen atom,sub-structure b (–CH2–CH2–), sub-structure c (–CH2–CH2–), sub-structure d (–CH2–CH2–CH–CH(CH3)–), sub-structure e (–CH2–CH2–) and sub-structure f (–CH(O)–C(CH3)@CH2). Assembling ofthese sub-structures was performed from the cross-peaks ob-served on the HMBC. Among the three oxygenated methane car-bons, those at C-1 (dC 73.6) and C-3 (dC 71.4) showed both cross-peaks with the protons of CH2-2 and CH-5, and in addition C-1was correlated to CH2-19 and C-3 to CH3-29 protons. The couplingconstant values, around 3.0 Hz for the proton at C-1 indicated itwas equatorial, whereas those of proton at C-3 (J = 11.9 Hz) indi-cated it was axial. The third oxidized methine at dC 91.0 ppm (C-24) was located on the side chain, because of the cross-peaks ob-served in the HMBC spectrum with the ethylenic protons (@CH2-26), the methyl at dH 1.71 ppm (CH3-27) and the methylenes atdH 1.50 ppm (CH2-23). The quaternary carbon at dC 55.9 (C-4)and the carboxyl group at dC 180.6 ppm (C-28) were both corre-lated with the methyl protons at 1.07 ppm (CH3-29) and themethine protons at dH 4.53 (H-3) and 2.58 ppm (H-5) allowing toconnect sub-structures a and b. The quaternary carbon at dC
30.2 ppm, (C-10) was correlated to CH2-19, CH-1 and CH-5. Theircoupling constants indicated that H-3 and H-5 were axial and thusa, whereas those of H-1 that it was b-equatorial. The quaternarycarbon at 22.2 pm (C-9) was correlated to protons of CH2-11,CH2-12, CH2-19 and the methine CH-8, linking sub-structure c,with sub-structures a and b. The quaternary carbons at dC 46,4(C-13) and 50.0 ppm (C-14) were both correlated with the follow-ing protons: CH3-18, CH3-30, CH2-12, CH2-15 and CH2-16, but onlythe first one gave a cross-peak with the proton of CH2-11 at dH
2.40 ppm, allowing it assignment at C-13.Concerning the side chain, the carbon at dC 91.0 ppm (CH-24)
gave HMBC correlations with the protons of CH2-26, CH3-27 andwith those of the two methylenes CH2-22 and -23. The methineat dC 37.1 ppm (C-20) was correlated with protons of CH3-21,CH-17 and CH2-16: these data allowed to define the side chainstructure and to link it at position -17 of the polycyclic moiety.Taking into account that the molecular formula involves six oxygenatoms and that the structure encloses only three oxymethinegroups in addition to a carboxyl group, it is suggested that CH-24is not linked to an alcohol group, but to a hydroperoxide group.This is confirmed by the side chain fragmentation in mass spec-trometry, leading to a fragment at m/z = 471.3086 correspondingto the departure of the hydroperoxide moiety and to the chemicalshift of the carbon >CH(O)-24 at dC 91.0 ppm, which is not that of a
OH
OH
OOH
COOH
a
b
c
d
e
f
Fig. 1. 1H–1H COSY (bold lines) and selected HMBC (H ? C) data for musambin A(1).

HCH3
HH
OH
HOOC
HH
OH
H
CH3
CH3
H
H
H
CH3 HH
O-OHH
H
HH
CH3
Fig. 2. Selected NOEs (H M H) data for musambin A (1).
D. Lacroix et al. / Phytochemistry 70 (2009) 1239–1245 1241
secondary alcohol function expected at dC 76.8 ppm, but ratherthat of a carbon bearing an hydroperoxide group which is expectedat dC 90.4 ppm (Kato et al., 1996; Cabrera and Seldes, 1995; Luoet al., 2005).
Strong NOEs observed between CH3-29 and CH2-19, betweenCH2-19 and CH-8 at dH 1.50 ppm and between this proton andthe methyl at 1.01 ppm (CH3-18), indicating these protons to beon the same side of the pentacyclic structure (Fig. 2). On the otherhand, strong NOEs were depicted between H-3 and H-5, betweenCH-17 and both CH3-30 and CH3-21, indicating they were on theother face. All the data were consistent with the structure 1a,3b-dihydroxy-24-hydroperoxy-cycloart-26-methylene-28-carboxylicacid for 1.
Musambin B (2), was obtained as a colourless amorphous solid,optically active (½a�22
D 68.4�). Its HR-ESI-MS (negative mode)showed the quasi-molecular ion [M�H]� at m/z: 503.3377 (calc.for C30H47O6: 503.3373) corresponding to the molecular formulaC30H48O6 for 2 and suggesting it was an isomer of 1. The 1H and13C NMR spectra of 2 were very similar to those of 1 and differedonly by the presence of an 1,2-disubstituted double bond at dC
129.7 (CH) and 137.0 ppm (CH) instead of an sp2 exo-methylenegroup, and by the presence of a carbon quaternary oxygenated car-bon (82.5 ppm), instead of an oxymethine group (91.0) in 1. TheCOSY spectrum indicated the same sub-structures a–d as abovedepicted for 1 (Fig. 1), but not the e and f ones. The oxygenatedquaternary carbon (C-25) was correlated in the HMBC spectrumwith the protons of two equivalent methyl singulets at dH
1.29 ppm (CH3-26 and -27) and with the two ethylenic protonsat 5.63 and 5.56 ppm (CH-23 and -24) and with the methyleneCH2-22 at 2.21 and 1.80 ppm (Tables 1 and 2). These NMR datasuggested the side chain to have the following structure: –CH(CH3)–CH2–CH@CH–C(OOH)(CH3)2, with an E-configuration ofthe disubstituted double bond (JHH = 15.9 Hz). The assignment ofthe hydroperoxide group at C-25 was deduced from the COSYand in agreement with the observed chemical shift value of thiscarbon at dC 82.5 ppm, which is characteristic of a quaternaryhydroperoxide expected at dC 82.3 ppm and differing from that ofa quaternary alcohol expected at dC 70.7 ppm (Kato et al., 1996;Cabrera and Seldes, 1995). The full assignment of the 1H and 13Cspectra of 2, resulted from complete 2D-NMR analysis. MusambinB (2) is thus a pentacyclic triterpene of the cycloartane type, isomerof 1, from which it differs only by the location on the side chain ofthe double bond and of the hydroperoxide group, and 2 has thestructure 1a,3b-dihydroxy-25-hydroperoxy-cycloart-23E-en-28-carboxylic acid.
Musambin C (3), was obtained as a colourless amorphous solid,optically active (½a�22
546 22.2�). Its HR-ESI-MS (negative mode)showed the quasi-molecular ion [M�H]� at m/z: 485.3240
[M�H]�, (calc. for C30H45O5: 485.3267) corresponding to themolecular formula C30H46O5 for 3, which differed from those of 1and 2 by the loss of two protons and an oxygen atom. The triterpe-nic polycyclic moiety was identical with those of 1 and 2 as shownby the 1D- and 2D-NMR spectra. The main difference was relatedto the side chain: a conjugated ketone carbon was depicted at dC
204.5 ppm in the 13C NMR spectrum. The spin system characteris-tic of a CH3–C@CH2 group, previously described for 1 was also ob-served (Tables 1 and 2). On the HMBC spectrum, the ketone carbongave strong cross-peaks with the following methylenes: CH2-22, -23, -26 (sp2) and CH3-27. Analysis of the 2D-NMR (COSY, HSQC andHMBC) spectra allowed total assignment of the 1H and 13C spectraof musambin C, 3 (Tables 1 and 2), and to propose the structure1a,3b-dihydroxy-cycloart-26-methylene-24-oxo-28-carboxylicacid.
The glycosidic nature of musambioside A (4) was deduced fromthe strong IR absorption at m 3387 cm�1. The HR-MS, showed thequasi-molecular ion [M�H]� at m/z: 635.3764 (calc. forC35H55O10: 635.3795) indicating the molecular formula C35H56O10
for 4. The 13C NMR depicted the 35 carbon atoms of the moleculeand indicated the presence of one –CH2OH at 66.7 ppm, one –CH(O)–OH at 106.2 ppm and three >CHOH groups at 75.1, 77.5,71.1 ppm (Table 1). Analysis of the 2D-NMR spectra for this moi-ety, suggested a cyclic pentose, and the coupling constant mea-surement of its protons (Table 3) indicated that H-20, H-30, andH-40 were axial and H-10 equatorial which was in favour of a xylosestructure. The xylose was linked to the oxygen at C-3, becausestrong correlations were depicted between the carbon C-3 at dC
81.7 ppm and proton at dH 4.30 (H-10) and reciprocally, betweenthe anomeric carbon C-10 (dC 106.2 ppm) of the sugar and H-3 atdH 4.56 ppm. All the 1D and 2D-NMR spectra indicated that thegenine triterpenic moiety was identical to musambin A (1) (Tables1–3), and thus musambioside A (4) was the 3b-D-xyloside of1a,3b-dihydroxy-24-hydroperoxy-cycloart-26-methylene-28-car-boxylic acid.
The HR-ESI-MS of musambioside (5) indicated the quasi-molec-ular ion [M�H]� at m/z: 635.3795 in agreement with the molecularformula C35H56O10 and suggesting it was a cycloartane triterpeneglycoside close to musambioside B (2). The 35 carbons of themolecular formula were distributed into five oxygenated carbonswith chemical shifts characteristic of the xylose moiety as aboveobserved for 4, and thirty carbons with chemical shifts very similarof the cycloartane moiety assigned for musambin B, 2 (Table 1).Analysis of the 1D- and 2D-NMR spectra of 5 confirmed the pres-ence of 1,2-disubtituted double bond at dC 129.7 (CH) and137.0 ppm (CH), allowed the structure determination of this chainidentical to that of musambin B 2 and its linkage to C-17 of thepentacyclic structure. The xylose moiety was linked to the oxygen

Table 21H NMR data for compounds (1–3) (CD3OD, 400.13 MHz, 298 K).
C No. 1 2 3
dH m (J) dH m (J) dH m (J)
1 eq 3.54 dd 3.2; 2.8 3.54 dd 3.0; 3.0 3.54 dd 3.0; 3.02 eq 1.84 ddd 13.5; 4.9; 3.2 1.82 m 1.84 max 1.79 ddd 13.5; 12.0; 2.8 1.77 m 1.80 m3ax 4.53 dd 12.0; 4.9 4.53 dd 11.9; 4.9 4.53 dd 11.9; 4.85ax 2.58 dd 12.5; 4.2 2.57 dd 12.5; 4.2 2.58 dd 12.6; 4.16a 1.30 m 1.30 m 1.31 mb 1.00 m 1.00 m 1.00 m7a 1.30 m 1.30 m 1.28 mb 1.30 m 1.30 m 1.28 m8 1.50 m 1.52 dd 11.8; 4.5 1.53 m11a 2.40 ddd 15.0; 9.0; 9.0 2.42 ddd 15.0; 8.5; 8.5 2.42 mb 1.30 m 1.25 m 1.28 m12 1.70 dd 9.0. 9.0 1.69 dd 8.5; 8.5 1.70 m15 1.30 m 1.35 m 1.35 m16a 1.91 m 1.95 m 1.96 mb 1.34 m 1.35 m 1.38 m17 1.60 m 1.61 ddd 6.5; 6.5; 6.5 1.65 m18 1.01 s 1.01 s 1.02 m19a 0.70 d 4.5 0.70 d 4.5 0.71 d 4.5b 0.50 d 4.5 0.50 d 4.5 0.51 d 4.520 1.45 m 1.45 m 1.45 m21 0.90 d 6.4 0.92 d 6.4 0.94 m22a 1.50 m 2.21 m 1.81 mb 1.00 m 1.80 m 1.28 m23a 1.50 m 5.63 ddd 15.9; 7.0; 5.0 2.74 mb 1.50 m – 2.68 m24 4.17 dd 6.6; 6.6 5.56 d 15.9 –26a 4.94 dq 1.5; 1.5 1.29 s 6.07 brsb 4.91 m – – 5.84 brs27 1.71 dd 1.5; 1.5 1.29 s 1.85 s29 1.07 s 1.07 s 1.09 s30 0.99 s 0.99 s 1.00 s
1242 D. Lacroix et al. / Phytochemistry 70 (2009) 1239–1245
at C-3, as deduced from the HMBC correlations between C-3 (dC
81.8 ppm) and H-10 (dH 4.30 ppm) and between H-3 (dH 4.56) andC-10 (dC 106.3). This linkage was confirmed by the strong NOE cor-relation in the NOESY between H-10 and H-3. Musambioside B (5)was thus the 3b-D-xyloside of 1a,3b-dihydroxy-25-hydroperoxy-cycloart-23E-en-28-carboxylic acid.
The HR-ESI-MS of musambioside 6 indicated the quasi-molecu-lar ion [M�H]� at m/z: 617.3677, (calc. for C35H53O9: 617.3690) inagreement with the molecular formula C35H54O9 for 6 and suggest-ing it was also a cycloartane triterpene glycoside, related tomusambiosides A and B (4 and 5). The 35 carbons of the molecularformula were distributed into five oxygenated carbons with chem-ical shifts characteristic of the xylose moiety as above observed for4 and 5, and thirty carbons with chemical shifts very similar of thecycloartane moiety assigned for musambin C, 3 (Table 1). Analysisof the 1D- and 2D-NMR spectra of 6 confirmed the presence of theconjugated ketone at C-24 on the side chain, allowed the structuredetermination of this chain identical to that of musambin C and itslinkage to C-17 of the pentacyclic structure. The xylose moiety wasattached the b3-oxygen of this cycloartane derivative as deducedfor compounds 4 and 5. The differences between the side chainsof 4 and 6 were the same as those between 1 and 3 (Tables 2and 3). Musambioside C (6) was thus 3b-D-xyloside of 1a,3b-dihy-droxy-cycloart-26-methylene-24-oxo-28-carboxylic acid.
Previous studies on plant hydroperoxysterols in which thehydroperoxide group was attached at C-24 on the side chain, re-port that a doubling of the signals of C-24 to C-26 was observedand this phenomena was explained by the presence of a mixtureof epimers at C-24 (Kato et al., 1996; Cabrera and Seldes, 1995;Anjaneyulu et al., 1985). This doubling of the resonance at C-24and its adjacent carbons was not observed neither for musambinsA, nor for musambioside A which is consistent with the presence ofonly one epimer at C-24 for these new compounds.
2.3. Biological activity
The anti-parasitic activities of the extracts of M. lutea leaves andpurified compounds were evaluated for their ability to inhibit thegrowth of Plasmodium falciparum, Trypanosoma brucei brucei andLeishmania donovani and also for their cytotoxicity against KBand MRC5 cells (Table 4). The crude ethyl acetate extract exhibitedthe most significant anti-parasitic activity in vitro and low cytotox-icity against human MRC5 and KB cells. Thus, this extract was spe-cifically active against T. brucei brucei (EC50: 1.9 lg/ml) and P.falciparum (IC50: 10.2 lg/ml) and poorly active against L. donovani(EC50: 42.0 lg/ml). For the six compounds isolated by fractionationfrom this EtOAc extract, weak activity or no activity was observedagainst L. donovani and against P. falciparum. The keto analoguemusambin C (3) was the best growth inhibitor of P. falciparum withIC50 10.2 lg/ml. All the compounds showed very weak or no cyto-toxicity against KB and MRC5 cells. The more interesting resultswere obtained for the anti-trypanosomal activity: musambin B(2) which had an EC50 of 1.9 lg/ml was almost as active as the ref-erence drugs [1.0 lg/ml for bis(aminoethylthio)-4-melaminophe-narsine and 1.4 lg/ml for pentamidine]. In addition, the lowcytotoxicity of musambin B (2) together with its relatively goodselectivity index (SI > 25) are promising. The two other aglycones,musambins A (1) and C (3) showed significative, but weaker activ-ity with EC50 of 7.8 and 15.6 lg/ml, respectively. As generally ob-served, the activity of their xylosides were weaker. These resultsindicated that the anti-parasitic activities observed for musambinsA–C explained the activity of the EtOAc extract from which theyhave been isolated.
The phytochemical investigation of M. lutea led to the isolationof six new cycloartane triterpenoids and one of them, musambin B(2) showed a potent anti-trypanosomal activity. Further structure–activity relationship studies will be carried out in order to identify
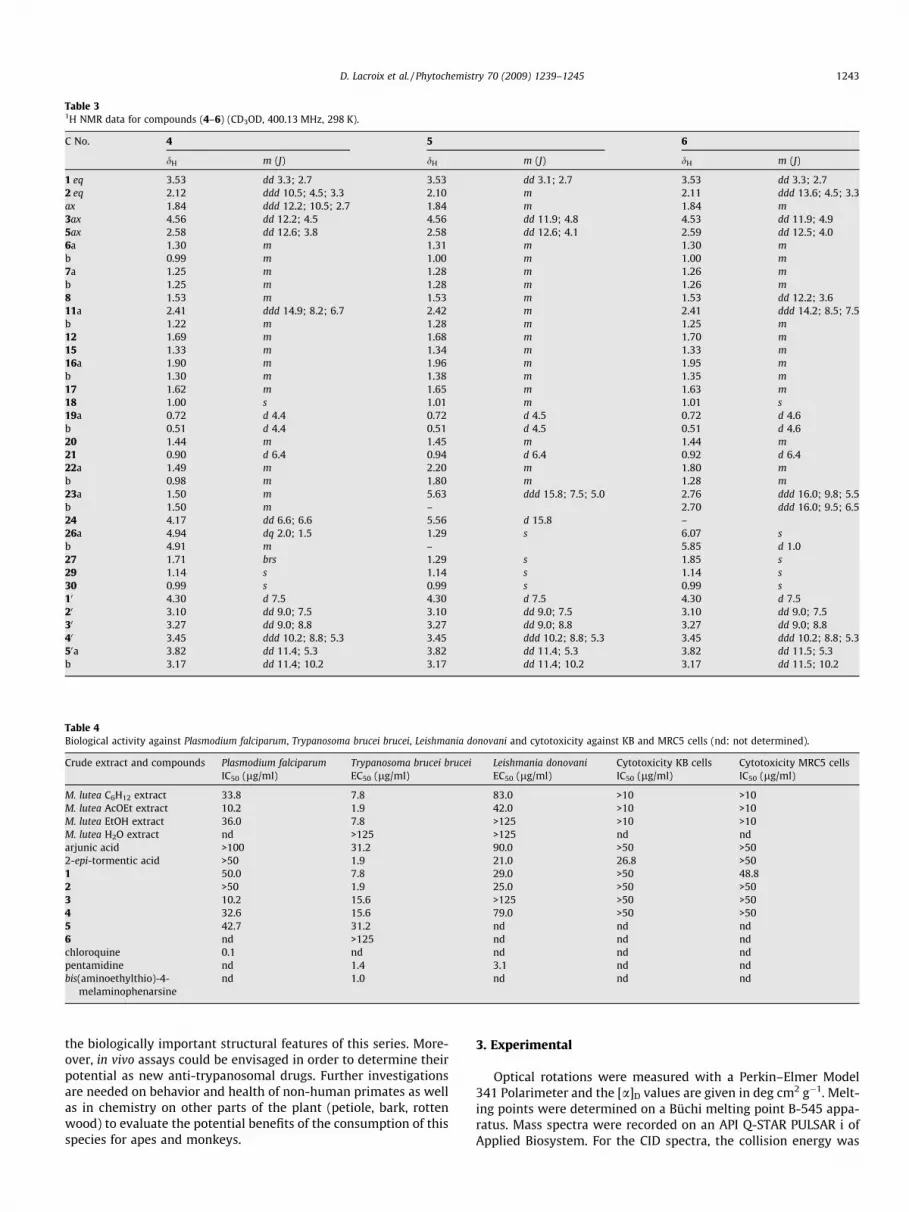
Table 31H NMR data for compounds (4–6) (CD3OD, 400.13 MHz, 298 K).
C No. 4 5 6
dH m (J) dH m (J) dH m (J)
1 eq 3.53 dd 3.3; 2.7 3.53 dd 3.1; 2.7 3.53 dd 3.3; 2.72 eq 2.12 ddd 10.5; 4.5; 3.3 2.10 m 2.11 ddd 13.6; 4.5; 3.3ax 1.84 ddd 12.2; 10.5; 2.7 1.84 m 1.84 m3ax 4.56 dd 12.2; 4.5 4.56 dd 11.9; 4.8 4.53 dd 11.9; 4.95ax 2.58 dd 12.6; 3.8 2.58 dd 12.6; 4.1 2.59 dd 12.5; 4.06a 1.30 m 1.31 m 1.30 mb 0.99 m 1.00 m 1.00 m7a 1.25 m 1.28 m 1.26 mb 1.25 m 1.28 m 1.26 m8 1.53 m 1.53 m 1.53 dd 12.2; 3.611a 2.41 ddd 14.9; 8.2; 6.7 2.42 m 2.41 ddd 14.2; 8.5; 7.5b 1.22 m 1.28 m 1.25 m12 1.69 m 1.68 m 1.70 m15 1.33 m 1.34 m 1.33 m16a 1.90 m 1.96 m 1.95 mb 1.30 m 1.38 m 1.35 m17 1.62 m 1.65 m 1.63 m18 1.00 s 1.01 m 1.01 s19a 0.72 d 4.4 0.72 d 4.5 0.72 d 4.6b 0.51 d 4.4 0.51 d 4.5 0.51 d 4.620 1.44 m 1.45 m 1.44 m21 0.90 d 6.4 0.94 d 6.4 0.92 d 6.422a 1.49 m 2.20 m 1.80 mb 0.98 m 1.80 m 1.28 m23a 1.50 m 5.63 ddd 15.8; 7.5; 5.0 2.76 ddd 16.0; 9.8; 5.5b 1.50 m – 2.70 ddd 16.0; 9.5; 6.524 4.17 dd 6.6; 6.6 5.56 d 15.8 –26a 4.94 dq 2.0; 1.5 1.29 s 6.07 sb 4.91 m – 5.85 d 1.027 1.71 brs 1.29 s 1.85 s29 1.14 s 1.14 s 1.14 s30 0.99 s 0.99 s 0.99 s10 4.30 d 7.5 4.30 d 7.5 4.30 d 7.520 3.10 dd 9.0; 7.5 3.10 dd 9.0; 7.5 3.10 dd 9.0; 7.530 3.27 dd 9.0; 8.8 3.27 dd 9.0; 8.8 3.27 dd 9.0; 8.840 3.45 ddd 10.2; 8.8; 5.3 3.45 ddd 10.2; 8.8; 5.3 3.45 ddd 10.2; 8.8; 5.350a 3.82 dd 11.4; 5.3 3.82 dd 11.4; 5.3 3.82 dd 11.5; 5.3b 3.17 dd 11.4; 10.2 3.17 dd 11.4; 10.2 3.17 dd 11.5; 10.2
Table 4Biological activity against Plasmodium falciparum, Trypanosoma brucei brucei, Leishmania donovani and cytotoxicity against KB and MRC5 cells (nd: not determined).
Crude extract and compounds Plasmodium falciparumIC50 (lg/ml)
Trypanosoma brucei bruceiEC50 (lg/ml)
Leishmania donovaniEC50 (lg/ml)
Cytotoxicity KB cellsIC50 (lg/ml)
Cytotoxicity MRC5 cellsIC50 (lg/ml)
M. lutea C6H12 extract 33.8 7.8 83.0 >10 >10M. lutea AcOEt extract 10.2 1.9 42.0 >10 >10M. lutea EtOH extract 36.0 7.8 >125 >10 >10M. lutea H2O extract nd >125 >125 nd ndarjunic acid >100 31.2 90.0 >50 >502-epi-tormentic acid >50 1.9 21.0 26.8 >501 50.0 7.8 29.0 >50 48.82 >50 1.9 25.0 >50 >503 10.2 15.6 >125 >50 >504 32.6 15.6 79.0 >50 >505 42.7 31.2 nd nd nd6 nd >125 nd nd ndchloroquine 0.1 nd nd nd ndpentamidine nd 1.4 3.1 nd ndbis(aminoethylthio)-4-
melaminophenarsinend 1.0 nd nd nd
D. Lacroix et al. / Phytochemistry 70 (2009) 1239–1245 1243
the biologically important structural features of this series. More-over, in vivo assays could be envisaged in order to determine theirpotential as new anti-trypanosomal drugs. Further investigationsare needed on behavior and health of non-human primates as wellas in chemistry on other parts of the plant (petiole, bark, rottenwood) to evaluate the potential benefits of the consumption of thisspecies for apes and monkeys.
3. Experimental
Optical rotations were measured with a Perkin–Elmer Model341 Polarimeter and the [a]D values are given in deg cm2 g�1. Melt-ing points were determined on a Büchi melting point B-545 appa-ratus. Mass spectra were recorded on an API Q-STAR PULSAR i ofApplied Biosystem. For the CID spectra, the collision energy was

1244 D. Lacroix et al. / Phytochemistry 70 (2009) 1239–1245
40 eV and the collision gas was nitrogen. 13C NMR spectra were re-corded on an AC 300 BRUKER spectrometer operating at 75.47 MHz(for 13C). 1H and 2D-NMR spectra were recorded on an Avance 400BRUKER spectrometer operating at 400.13 MHz. For HMBC experi-ments the delay (1/2J) was 70 ms and for the NOESY experimentsthe mixing time was 150 ms.
3.1. Plant material
Leaves of M. lutea (Benth.) K. Schum., syn. Markhamia platycalyx(Baker) Sprague and Dolichandrone platycalyx Baker (Bignoniaceae),were collected at Kanyawara Kibale National Park (Uganda) in June2003. Voucher specimens (UFC-0012) were deposited in the Her-barium of the National Museum of Natural History (Paris) and inthe Herbarium of the Makerere University (Uganda).
3.2. Extraction and isolation
The dried and powdered leaves of M. lutea (1.18 kg) weremacerated two times with cyclohexane (2.5 l) for 24 h and thecombined extracts yielded an oil (3.2 g) which was discarded.The leaves were then extracted 3� with AcOEt (5 l) for 24 h atroom temperature to give after evaporation of the solvent underreduced pressure the AcOEt extract (57.0 g). Then, 20 g of this ex-tract were submitted to MPLC chromatography on 60 H Merck Sigel (650 g) to yield 33 fractions eluted successively with cyclohex-ane/CH2Cl2 gradient (from 8/2 to pure CH2Cl2) to yield fractions F-1to F-10, then with a CH2Cl2/MeOH gradient from 10/0 to 8/2 toyield fractions F-11 to F-31 and then successively with AcOEt toyield fraction F-32 and MeOH to yield fraction F-33. From fractionF-13, b-sitosterol was isolated by crystallisation (140 mg).
Fraction F-18 (528 mg) was chromatographed on silica gelcolumn chromatography with a CH2Cl2/MeOH gradient from 1/0to 0/1 to give three fractions, and the middle fraction further chro-matographed on silica gel column chromatography eluted with acyclohexane/AcOEt gradient from 1/0 to 0/1 to give three fractions:Fraction F-3 was composed of pure 2-epi-tormentic acid (160 mg).
Fraction F-20 (371.00 mg) was chromatographed on a Sepha-dex LH-20 column with MeOH as eluent to give six sub-fractionsf-1 to f-6. Sub-fraction f-5 was submitted to HPLC semi-prepara-tive chromatography on RP-18 Si gel eluted with an acetonitrile/water gradient from 50/50 to 95/5 (detection 220 nm, 3 ml/min).Arjunic acid as pure compound (15 mg – Rt: 13 min) was col-lected. Fraction F-21 (309.0 mg) was chromatographed on aSephadex LH-20 column with MeOH as eluent to give pure phae-ophorbide (5 mg).
Fraction F-29 (753 mg) was chromatographed on a SephadexLH-20 column with MeOH as eluent to give three fractions. Theresulting fraction 2 was first chromatographed on Si gel with acyclohexane/AcOEt gradient from 50/50 to 0/100 to yield 3 sub-fractions. Sub-fraction 2 was purified by HPLC (RP-18 Si gel, detec-tion 220 nm, 3 ml/min) to yield three pure compounds: musambinA (1) (200 mg; Rt: 19.0 min), musambin B (2) (100 mg; Rt:20.0 min) and musambin C (3) (20 mg; Rt: 22.0 min).
Fraction F-32 (340 mg) was chromatographed (MPLC) on a RP-18 Silica gel column, eluted with a MeOH/H2O gradient from 1/9to 10/0, to yield 10 sub-fractions. Sub-fraction 8 was purified byHPLC (RP-18 Si gel, detection 220 nm, 3 ml/min) eluted with anacetonitrile/H2O gradient to give pure musambioside A (4)(20.0 mg, Rt: 14.0 min), musambioside B (5) (5 mg, Rt: 15.0 min),musambioside C (6) (3 mg, Rt: 20.0 min).
Musambin A (1), C30H48O6; amorphous colourless solid; ½a�22D
78.4� (c 1.0, MeOH). ½a�22546 92.4� (c 1.0, MeOH). HR-MS, m/z:
503.3372 [M�H]�, (calc. for C30H47O6: 503.3373). IR (KBr) mmax
(cm�1): 3362, 2922, 2853, 1701. 1H and 13C NMR see Tables 1and 2.
Musambin B (2), C30H48O6; amorphous colourless solid; ½a�22D
68.4� (c 1.0, MeOH). ½a�22546 79.6� (c 1.0, MeOH). HR-MS, m/z:
503.3377 [M�H]�, (calc. for C30H47O6: 503.3373). IR (KBr) mmax
(cm�1): 3352, 2943, 2874, 1703, 1659. 1H and 13C NMR see Tables1 and 2.
Musambin C (3), C30H46O5; amorphous colourless solid; ½a�22546
22.2� (c 0.2, MeOH). HR-MS, m/z: 485.3240 [M�H]�, (calc. forC30H45O5: 485.3267). IR (KBr) mmax (cm�1): 3385, 2930, 2874,1676. 1H and 13C NMR see Tables 1 and 2.
Musambioside A (4), C35H56O10; amorphous colourless solid;½a�22
546 32.0� (c 0.6, MeOH). HR-MS, m/z: 635.3764 [M�H]�, (calcd.for C35H55O10: 635.3795). IR (KBr) mmax (cm�1): 3387, 2941, 2880,1703, 1678.1H and 13C NMR see Tables 1 and 2.
Musambioside B (5), C35H56O10; amorphous colourless solid;½a�22
546 38.0� (c 0.3, MeOH); HR-MS, m/z: 635.3795 [M�H]�, (calcd.for C35H55O10: 635.3795). IR (KBr) mmax (cm�1): 3399, 2933, 2872,1705. 1H and 13C NMR see Tables 1 and 2.
Musambioside C (6), C35H54O9; amorphous colourless solid;½a�22
546 33.0� (c 0.3, MeOH). HR-MS, m/z: 617.3677 [M�H]�, (calc.for C35H53O9: 617.3690). 1H and 13C NMR see Tables 1 and 2.
2-epi-tormentic acid: C30H48O5; amorphous colourless solid;13C NMR (CD3OD, 100 MHz) d ppm: 45.5 (C-1), 72.2 (C-2), 79.6(C-3), 39.2 (C-4), 56.8 (C-5), 19.4 (C-6), 34.3 (C-7), 41.2 (C-8),49.0 (C-9), 38.0 (C-10), 24.8 (C-11), 129.6 (C-12), 140.0 (C-13),42.8 (C-14), 29.5 (C-15), 26.7 (C-16), 49.0 (C-17), 55.1 (C-18),76.6 (C-19), 43.1 (C-20), 27.3 (C-21), 39.0 (C-22), 30.3 (C-23),17.9 (C-24), 16.9 (C-25), 17.6 (C-26), 24.9 (C-27), 182.2 (C-28),27.1 (C-29), 16.6 (C-30); 1H NMR (CD3OD, 400 MHz) d ppm (m, J,Hz): 2.05 (1H, dd, 14.5, 3.5, H-1 eq), 1.16 (1H, dd, 14.5, 3.5, H-1ax), 4.01 (1H, ddd, 3.5, 3.5, 3.5, H-2), 3.15 (1H, d, 3.5, H-3), 0.88(1H, dd, 10.8, 2.0, H-5), 1.56 (2H, m, H-6), 1.56 (1H, m, H-7a),1.44 (1H, m, H-7b), 1.65 (1H, m, H-9), 2.05 (2H, m, H-11), 5.30(1H, dd, 3.7, 3.7, H-12), 1.82 (1H, m, H-15a), 1.02 (1H, m, H-15b),2.57 (1H, ddd, 13.3, 13.3, 4.7, H-16a), 1.52 (1H, m, H-16b), 2.50(1H, brs, H-18), 1.35 (1H, m, H-20), 1.75 (1H, m, H-21a), 1.25 (1H,m, H-21b), 1.75 (1H, m, H-22a), 1.65 (1H, m, H-22b), 1.02 (3H, s,H-23), 1.02 (3H, s, H-24), 1.26 (3H, s, H-25), 0.81 (3H, s, H-26),1.33 (3H, s, H-27), 1.20 (3H, s, H-29), 0.93 (3H, d, 6.7, H-30).
3.3. Biological evaluations
3.3.1. In vitro anti-plasmodial activityThe anti-plasmodial activity was evaluated against the chloro-
quine-resistant FcB1/Colombia strain of P. falciparum. The testwas performed using the method of Desjardins et al. (1979).Extracts and pure compounds were diluted in DMSO at 20 lg/mlwith culture medium to the expected concentrations in 96-wellmicroplates. Asynchronous parasite cultures were then added (1%parasitemia and 1% final hematocrite), and plates were maintainedfor 24 h at 37 �C in a candle jar. [3H]-hypoxanthine (0.5 lCi) wassubsequently added to each well, and parasites were maintainedfor an additional 24 h. After freezing and thawing, the cells wereharvested from each well onto glass fibber filters, and the dried fil-ters were counted in a scintillation spectrometer. The growth inhi-bition for each well was determined by comparison of theradioactivity incorporated into the treated culture with that inthe control culture maintained on the same plate. The concentra-tion causing 50% inhibition (IC50) was obtained from the drug con-centration–response curve. The DMSO concentration neverexceeded 0.1% and did not inhibit the parasite growth.
3.3.2. In vitro anti-leishmanial assayL. donovani (MHOM/ET/L82/LV9) promastigotes were kindly
provided by Croft, from the WHO collection at the London Schoolof Hygiene and Tropical Medicine. Assays were performed as pre-viously described by M’Bongo et al. (1997) and Okpekon et al.

D. Lacroix et al. / Phytochemistry 70 (2009) 1239–1245 1245
(2004). Briefly, promastigotes were grown at 27 �C in HEPES(25 mM) buffered RPMI 1640 medium containing 10% FCS and50 lg/ml gentamycin. Assays were performed in 96-well microti-tre plates. Drugs were serially diluted in culture medium (100 ll/well); 100 ll of parasites from a logarithmic phase culture(1.75 � 106 promastigotes/ml) were then added to each well andplates were maintained at 27 �C under a 5% CO2 atmosphere. Theviability of parasites was evaluated by the MTT colorimetric meth-od. The anti-leishmanial activity was expressed as the EC50 after a72 h incubation period. The initial concentration for screening was15 lg/ml. Pentamidine was used as a reference drug.
3.3.3. In vitro anti-trypanosomal assayThe anti-parasitic activity was assessed using the method de-
scribed by Loiseau et al. (2000). Briefly, the bloodstream forms ofT. brucei brucei GVR strain were maintained in vitro for 24 h inthe dark at 37 �C in a 5% CO2 atmosphere, in Minimum EssentialMedium (Gibco-BRL) including 25 mM HEPES and Earle’s salts,and supplemented with 2 mM L-glutamine, 1 g of additional glu-cose per liter, 10 ml of minimum essential medium non-essentialamino acids (100�; Gibco-BRL) per liter, 0.2 mM 2-mercap-toethanol, 2 mM sodium pyruvate, 0.1 mM hypoxanthine,0.016 mM thymidine, 15% heat-inactivated horse serum (Gibco-BRL) and 50 lg gentamycin per milliliter. The 96-well plates werefilled as in the leishmanicidal assay, except that the parasitesadded were 2 � 105 trypomastigotes from the blood of a mousecollected aseptically. The anti-trypanosomal activity was ex-pressed as the EC50 after a 24 h incubation period. Pentamidinediisothionate (Pentacarinat�) and bis(aminoethylthio)-4-melamin-ophenarsine dihydrochloride were as used the reference drug.
3.3.4. CytotoxicityThe human tumor cell lines KB (mouth epidermoid carcinoma)
and the human diploid embryonic lung cells MRC5 were seededinto 96-well microplates at 2000 cells per well. The cytotoxicityassays were performed according to published procedures (Pierreet al., 1991; Tempete et al., 1995).
Acknowledgments
The French ‘‘Ministère de la Recherche” is acknowledged for afellowship for one of us (DL), the ‘‘Yves Rocher” company for itsgenerous contribution to this project and the region Ile-de-Francefor the funding of the 400 MHz NMR and the ESI-TOF mass spec-trometers. The National Academy of Medecine is also gratefullyacknowledged for the attribution of the prize Hugues Gounellede Pontavel to DL. We wish to thank Mr L. Dubost for the massspectra and Prof. Ph. Grellier for its helps. SK wishes to thankICSN-CNRS for granting the fieldwork related to the plants collec-tion. This work was done within the framework of the ‘‘Collabora-tive phytochemical research using selected flora and fauna species
in Uganda” (CNRS, MNHN, MU and UWA) program and the authorsare grateful to the Uganda Wildlife Authority (UWA) and to theUganda National Council for Science and Technology (UNCST) forallowing access and fieldwork in the Kibale National Park.
References
Anjaneyulu, A.S.R., Rama Prasad, A.V., 1982. Chemical examination of the roots ofTerminalia arjuna – the structure of arjunoside III and arjunoside IV, two newtriterpenoid glycosides. Phytochemistry 21, 2057–2060.
Anjaneyulu, V., Rao, G.S., Connolly, J.D., 1985. Occurrence of 24-epimers of cycloart-25-ene-3b, 24-diols in the stem of Euphorbia trigona. Phytochemistry 24, 1610–1612.
Cabrera, G.M., Seldes, A.M., 1995. Hydroperoxycycloartanes from Tillandsiarecurvata. J. Nat. Prod. 58, 1920–1924.
Chapman, C.A., Chapman, L.J., Rode, K.D., Hauck, E.M., McDowell, L.R., 2003.Variation in the nutritional value of prilate foods: among trees, time periodsand areas. Int. J. Primatol. 24, 317–333.
Conrad, J., Vogler, B., Klaiber, I., Roos, G., Walter, U., Kraus, W., 1998. Two triterpenesesters from Terminalia macroptera bark. Phytochemistry 48, 647–650.
Delgado, G., Hernandez, J., Pereda-Miranda, R., 1989. Triterpenoids from Cunilalathryfolia. Phytochemistry 28, 1483–1485.
Desjardins, R.E., Candfield, C.J., Haynes, J.D., Chulay, J.D., 1979. Quantitativeassessment of antimalarial activity in vitro by a semiautomated microdilutiontechnique. Antimicrob. Agents Chemother. 16, 710–718.
Hattori, M., Kuo, K.-P., Shu, Y.-Z., Tezuka, Y., Kikuchi, T., Namba, T., 1988. Atriterpene from the fruits of Rubus chingii. Phytochemistry 27, 3975–3976.
Kanchanapoom, T., Kasai, R., Yamasaki, K., 2002. Phenolic glycosides fromMarkhamia stipulata. Phytochemistry 59, 557–563.
Kato, T., Frei, B., Heinrich, M., Stichet, O., 1996. Antibacterial hydroperoxysterolsfrom Xanthsoma robustum. Phytochemistry 41, 1191–1195.
Kernan, M.R., Amarquaye, A., Chen, J.L., Chan, J., Sesin, D.F., Parkinson, N., Ye, Z.J.,Barrett, M., Bales, C., Stoddart, C.A., Sloan, B., Blanc, P., Limbach, C., Mrisho, S.,Rozhon, E.J., 1998. Antiviral phenyl propanoid glycosides from the medicinalplant Markhamia lutea. J. Nat. Prod. 61, 564–570.
Krief, S., Martin, M.T., Grellier, P., Kasenene, J., Sévenet, T., 2004. Novel antimalarialcompounds isolated in a survey of self-medicative behavior of wildchimpanzees in Uganda. Antimicrob. Agents Chemother. 48, 3196–3199.
Krief, S., Hladik, C.M., Haxaire, C., 2005a. Ethnomedicinal and bioactive properties ofplants ingested by wild chimpanzees in Uganda. J. Ethnopharmacol. 101, 1–15.
Krief, S., Thoison, O., Sévenet, T., Wrangham, R.W., Lavaud, C., 2005b. Triterpenoidsaponin anthranilates from Albizia grandibracteata leaves ingested by primatesin Uganda. J. Nat. Prod. 68, 897–903.
Loiseau, P.M., Lubert, P., Wolf, J.G., 2000. Contribution of dithiol ligands to in vitroand in vivo trypanocidal activities of dithiaarsanes and investigation of ligandexchange in an aqueous solution. Antimicrob. Agents Chemother. 44, 2954–2961.
Luo, H.-F., Li, Q., Yu, S., Badger, T.M., Fang, N., 2005. Cytotoxic hydroxylatedtriterpene alcohol ferulates from rice bran. J. Nat. Prod. 68, 94–97.
M’Bongo, N., Loiseau, P., Lawrence, F., Bories, C., Craciunescu, D.G., Robert-Gero, M.,1997. In vitro sensitivity of Leishmania donovani to organometallic derivatives ofpentamidine. Parasitol. Res. 83, 515–517.
Okpekon, T., Yolou, S., Gleye, C., Roblot, F., Loiseau, P., Bories, C., Grellier, P., Frappier,F., Laurens, A., Hocquemiller, R., 2004. Antiparasitic activities of medicinalplants used in Ivory Coast. J. Ethnopharmacol. 90, 91–97.
Onderdonk, D.A., Chapman, C.A., 2000. Coping with forest fragmentation: theprimates of Kibale national park, Uganda. Internat. J. Primatol. 21, 587–611.
Pierre, A., Kraus-Berthier, L., Atassi, G., Cros, S., Poupon, M.F., Lavielle, G., Berlion, M.,Bizzari, J.P., 1991. Preclinical antitumor activity of a Vinca alkaloid derivative, S12363. Cancer Res. 51, 2312–2318.
Sinaphet, B., Noiarsa, P., Rujirawat, S., Otsuka, H., Kanchanapoom, T., 2006.Dolichandroside, a new phenolic triglycoside from Dolichandrone serrulata(DC) Seem. J. Nat. Med. 60, 251–254.
Tempete, C., Werner, G.H., Favre, F., Rojas, A., Langlois, N., 1995. In vitro cytostaticactivity of 9-demethoxyporothramycin B. Eur. J. Med. Chem. 30, 647–650.























![Oxidation of [ 13 C]glucose ingested before and/or during prolonged exercise](https://img.pdfslide.net/doc/110x75/6357fb02a08050b05c07a4b2/oxidation-of-13-cglucose-ingested-before-andor-during-prolonged-exercise.jpg)





