Embed Size (px)
Citation preview

Identification of ProteinDomains on TopologicalBasis
C. AnselmiG. Bocchinfuso
A. ScipioniP. De Santis
Dipartimento di Chimica,Universita “La Sapienza,”
Ple A. Moro 5,I-00185 Roma, Italy
Received 31 January 2000;accepted 5 July 2000
Abstract: A theoretical method is proposed to identify structural domains in proteins of knownstructures. It is based on the distribution of the local axes of the polypeptide chain. In particular,a statistical analysis is applied to the contributions of the local axes to the absolute writhingnumber, a topological property of a space curve resulting from the number of self-crossings in thecurve projections onto a unit sphere. This finding supports the hypothesis that topological require-ments should be satisfied in the process of protein folding and in the final organization of the tertiarystructures. © 2001 John Wiley & Sons, Inc. Biopoly 58: 218–229, 2001
Keywords: structural domains; protein folding; structural analysis; topology; writhing number
INTRODUCTION
Since the first solution of the protein structure by meansof x-ray diffraction techniques by Kendrew et al.1 andPerutz,2 a great effort has been devoted to understand thestructural organization of globular proteins.
Probably the most interesting feature of the three-dimensional protein structures is the presence of dif-ferent levels of organization.3 The lower level corre-sponds to the secondary structure, such asa-helices,b-strands, and turns. They can be described in termsof the internal coordinates of sequentially close resi-dues. However, the secondary structure is absolutelyinsufficient to obtain a full knowledge of the proteinfolding.
The next higher order of organization is taken to bethe packing of elements of the secondary structures tobuild up compact globular structures. To simplify the
approach to protein architecture, Levitt and Chothia4
introduced the concept of structural classes and usedit to assign protein folds to one of four families: all-a,all-b, a/b, anda 1 b. These assignments were basedon a visual inspection of polypeptide chain topologiesin a data set of 31 globular proteins. The all-a andall-b consisted almost entirely ofa-helices andb-sheets, respectively. Thea/b class was described ashaving mixed and approximately alternating segmentsof a-helical andb-strand secondary structure with themain sheet consisting mainly of parallel strands.However, thea 1 b class hada-helix andb-strandsecondary structure segments that do not mix, buttend to segregate along the polypeptide chain, withthe b-sheets almost always built up from antiparallelstrands.
These class definitions are still commonly used,although the names “all-a” and “all-b” have often
Correspondence to:P. De Santis; e-mail: [email protected] grant sponsor: University “La Sapienza,” MURST,
and Instituto Pasteur—Fondazione Cenci-Bolognetti.Biopolymers, Vol. 58, 218–229 (2001)© 2001 John Wiley & Sons, Inc.
218

been changed to “mainly-a” and “mainly-b,” becausemost all-a and all-b proteins contain small amounts ofsheets or helix, respectively.
The third level of organization is packing “do-mains” together to form the entire folded polypeptidechain. Although there is no universally accepted def-inition of domains, they are normally considered ascompact, local and semi-independent units.5 In fact,in a multidomain protein, domains can make up func-tionally and structurally distinct modules, usuallyformed by a single continuous protein chain, gener-ally associated with a single exon in the DNA.6 How-ever, some domains can be built up from two or morenonsequential segments. Furthermore, every domaincan be assigned to one of the four structural classes.
Although domains can be identified subjectivelyby eye, their importance to protein architecture andtheir possible role as independent nucleation sites inprotein folding7 prompted several authors to investi-gate more systematic techniques for domain identifi-cation.
The need of a systematic identification of structuraldomains in known three-dimensional structures iseven more pressing considering the current rapidgrowth of the number of resolved proteins. Thismeans that in the near future, visual inspection andclassification will be impractical. Consequently, auto-matic procedures for consistent classification will beessential.
Adopting the CaOCa distance maps proposed byPhillips8 and Ooi and co-workers,9,10 Rossmann andLiljas11 suggested that a domain has many short resi-due–residue distance within itself, but few short dis-tance with the remainder of the protein. Despite theeffectiveness of their approach, the need of a subjec-tive human interpretation of the contact maps avoidedautomatic identification of domains.
Later Crippen12 introduced cluster analysis to pro-tein fragments. However, such algorithms tend to givemore fragmented units than the generally acceptednotion of domains.
More recently, Holm and Sander13 proposed amethod for domain identification based on the crite-rion of maximal interdomain fluctuation time. Theyused correspondence analysis14 to make contact ma-trix block diagonal, to yield to a minimal number ofinterunit contacts, and to maximize the time constantof the relative motion between substructures.
Swindell15 proposed a method based on the intu-itively simple idea that each domain should contain anidentifiable hydrophobic core. Identifying distinctcores corresponds to determining both the number andthe location of the constituent domains in proteins.
Siddiqui and Burton16 reprised the concept thatdomains make more contacts between themselvesthan they do with the rest of the protein, but intro-duced a “split value” to estimate the consistency ofthe protein divisions in domains.
Rackovsky17 demonstrated the existence in pro-teins domain sequences of sets of statistically signif-icant periodic signals, characteristic of the architec-tures of those domains. The characteristic signals de-fine sequence units, which may corresponds tospecific structural features.
Many other authors proposed several methodsbased on the comparison of the structures currentlyknown in the Protein Data Bank,18 clustered in struc-turally representative classes at various level ofuniqueness.19–25
The present paper advances an original approach tothe problem of the identification of domain based ontopological concepts. This model reprises the defini-tion of domain as a compact unit, which makes morecontact with itself than with the rest of the protein, butthe term “contact” is reinterpreted in a topologicalframework.
A polypeptide chain can be assimilated to a ribbon,which folds in the space to form the final three-dimensional structure. In topology, every space curvecan be associated to a writhing number that representsits complexity and handedness. One way to measurethe writhe of a given curve is to observe it from manyuniformly spread out directions on the unit sphere andto count the average number of right- and left-handedcrossings per projection.26 For examples, the writheof an ideal right-handed three-foiled knot is close to13.4, which indicates that on average there are 3.4more right-handed than left-handed crossing foundwhen observing the knot from random directions.27
In general, writhing is not additive; if one arbi-trarily joins two curves, the writhe number of the newcurve is different from the sum of the writhe numbersof the original curves. In fact, it is equal to thesummation of the average contributions of the cross-ing between elementary ribbon units, which can bepositive or negative depending if the crossings areright- or left-handed. According to Calagareanu,28
W 51
4p EE dl 1 3 dl 2 z r 21
r 213 (1)
wheredl1 anddl2 are two infinitive tracts of the spacecurve, andr21 their vectorial distance. In the case of apolypeptide chain,W reduces to the summation
Identification of Protein Domains 219
W 51
4pOi, j
l i 3 l j z r ji
r ji3 5 O
i, j
wij (2)
wherewij is the fraction of the unit sphere from which2 oriented vectors,l i andl j, cross in projection. Thel iand l j can be two virtual bonds or two local helicalaxes of the polypeptide chain.
Despite their complexity, topology seems to be aneffective and useful tool to describe the general fea-ture of folding. Very recently, Alm and Baker sug-gested that native-state topology is a key determinantof protein-folding mechanism and of the distributionof structure in the transition-state ensemble.29
In previous works, we showed that in the advancedstates of the folding process, when a part of thetertiary interactions are formed, conformational trans-formations following minimum-energy pathways be-long to the class of topological deformations.30 There-fore, protein folding should proceed through a set of‘topologically equivalent’ conformations. It is rele-vant that practically all amino acid residues in a set of49 crystallographic structures of proteins, taken fromthe Protein Data Bank (PDB),18 are topologicallyequivalent toa-helical orb-strand conformations and,for example, the GH corner of myoglobin is topolog-ically equivalent to ana-helix.31 This supports thehypothesis that these regions actually had in previoussteps of the folding process, those secondary struc-tures, which have been continuously deformed untilthe final steps of the protein folding.
The aim of the present paper is to propose a topo-logical approach to the problem of the definition ofthe protein domains. In fact, it is reasonable that in amultidomain protein, every chain segment makesmore either positive or negative topological crossingswith the other segments belonging to its own domainthan with those belonging to the remainder of theprotein.
As already discussed, the relative number of cross-ings for a directional space curve is expressed by itswrithing number, to which each crossover contributeswith a positive or negative term depending if it isright- or left-handed, respectively. Therefore the totalnumber of crossing (the writhing number of a nondi-rectional space curve) is given by the absolute writh-ing, defined as the summation of the absolute valuesof the termswij in Eq. (2):
Wabs51
4pOi, j
ul i 3 l j z r ji ur ji
3 5 Oi, j
uwij u (3)
One of the general assumptions of this work is thatany compact structure, a ribbon or a protein, should
satisfy topological rules. From this point of view, ifthe hypothesis is advanced that protein folding isdirected from geometrical criteria of compactness andglobularity, as well as energy and entropy changes, itis reasonable that topological parameters should re-flect these requirements.
For a more straightforward analysis, a set of 71crystallographic and NMR structures of representativeproteins32,33 was considered from Protein DataBank,18 with resolution better than 1.5 Å.* This set isrepresentative of all the structural classes.4 The abso-lute writhing numbers of the proteins were calculatedstarting from the CaOCa vectors and compared tothose of random coils with the same length. Randomcoil structures were obtained starting from the aminoacidic sequences of each protein. Thef,c dihedralangles were imposed to each residue randomly, butaccording to the experimental distribution off,cpairs found in the random coil regions of a largenumber of high-resolution crystal structures.34,35
Three different random structures were generatedstarting from each aminoacidic sequence.
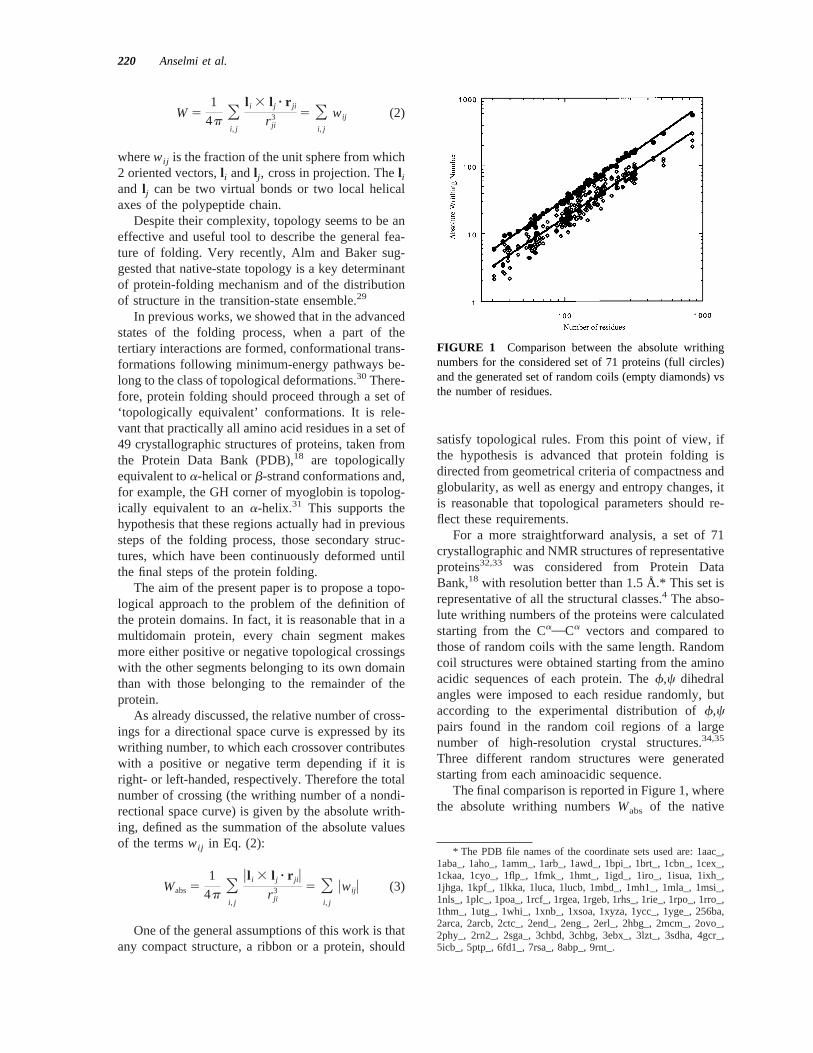
The final comparison is reported in Figure 1, wherethe absolute writhing numbersWabs of the native
* The PDB file names of the coordinate sets used are: 1aac_,1aba_, 1aho_, 1amm_, 1arb_, 1awd_, 1bpi_, 1brt_, 1cbn_, 1cex_,1ckaa, 1cyo_, 1flp_, 1fmk_, 1hmt_, 1igd_, 1iro_, 1isua, 1ixh_,1jhga, 1kpf_, 1lkka, 1luca, 1lucb, 1mbd_, 1mh1_, 1mla_, 1msi_,1nls_, 1plc_, 1poa_, 1rcf_, 1rgea, 1rgeb, 1rhs_, 1rie_, 1rpo_, 1rro_,1thm_, 1utg_, 1whi_, 1xnb_, 1xsoa, 1xyza, 1ycc_, 1yge_, 256ba,2arca, 2arcb, 2ctc_, 2end_, 2eng_, 2erl_, 2hbg_, 2mcm_, 2ovo_,2phy_, 2rn2_, 2sga_, 3chbd, 3chbg, 3ebx_, 3lzt_, 3sdha, 4gcr_,5icb_, 5ptp_, 6fd1_, 7rsa_, 8abp_, 9rnt_.
FIGURE 1 Comparison between the absolute writhingnumbers for the considered set of 71 proteins (full circles)and the generated set of random coils (empty diamonds) vsthe number of residues.
220 Anselmi et al.

proteins and the generated random coils are reportedvs the number of residues.
Figure 1 shows that a striking correlation existsbetweenWabsand the protein length (R 5 0.996). Inparticular, native proteins show very little variancearound the expected values, being all the points veryclose to the fitting line. However, random coils aremuch more spread out (R 5 0.953) andlies belowthe line of the native proteins. The result is consistentwith the fact that random coils are generally lesscompact of native proteins. It also suggests that nativeproteins are the most compact structures for polypep-tide chains with a certain length.
The contributions to the total writhing, consideredwith their absolute valuesuwij u, have been analyzedby means of correlation analysis first proposed byHill. 14 In particular, he showed how it is possible tocluster into groups the occurrences of 70 types ofartefacts in 59 tombs. The aim of the analysis was toplace the tombs in their natural (presumably temporal)order, keeping in mind that any particular type ofartefact was probably only in use for a limited period.However, the statistical basis of the method is generaland it is easily applicable to other problems. Holmand Sander were the first to apply correspondenceanalysis to the problem of the identification of proteindomains. They made the hypothesis that domains arethe units for which the time constant of relative mo-tion t is largest inside proteins. As a result, the prob-lem reduces to find the domain decomposition from aset of candidate bisections that maximize the calcu-lated value oft.13 Therefore, their method is based onthe dynamic properties of the domains as compactunits inside the protein, whereas the model we pro-pose only takes into account the static arrangementsof the polypeptide chain into the space and its spatialcomplexity. However, we found convenient to use thesame correspondence analysis to cluster the contribu-tions of the total absolute writhing, as it combinescomputational simplicity with good practical results.
Probably, it is not the best possible statistical tool,because, although it correctly identifies domainsboundaries in multidomain proteins, in some cases itis not able to determine the correct number of do-mains. Therefore, the further development of thismethod will require the application of more powerfulcluster-analysis methods and/or genetic algorithms.
The present approach is simple and requires littlecomputational time. Furthermore, it casts light on newfeatures of the protein folding. In fact, the requirementof a certain number of topological crossing maymimic the necessity of globularity and compactness ofthe protein structure.
METHODS
The total number of crossings for a non-directional spacecurve is expressed by its absolute writhing number,Wabs
5 ¥ uwij u, as already discussed.Each pair of the polypeptide chain segments contributes
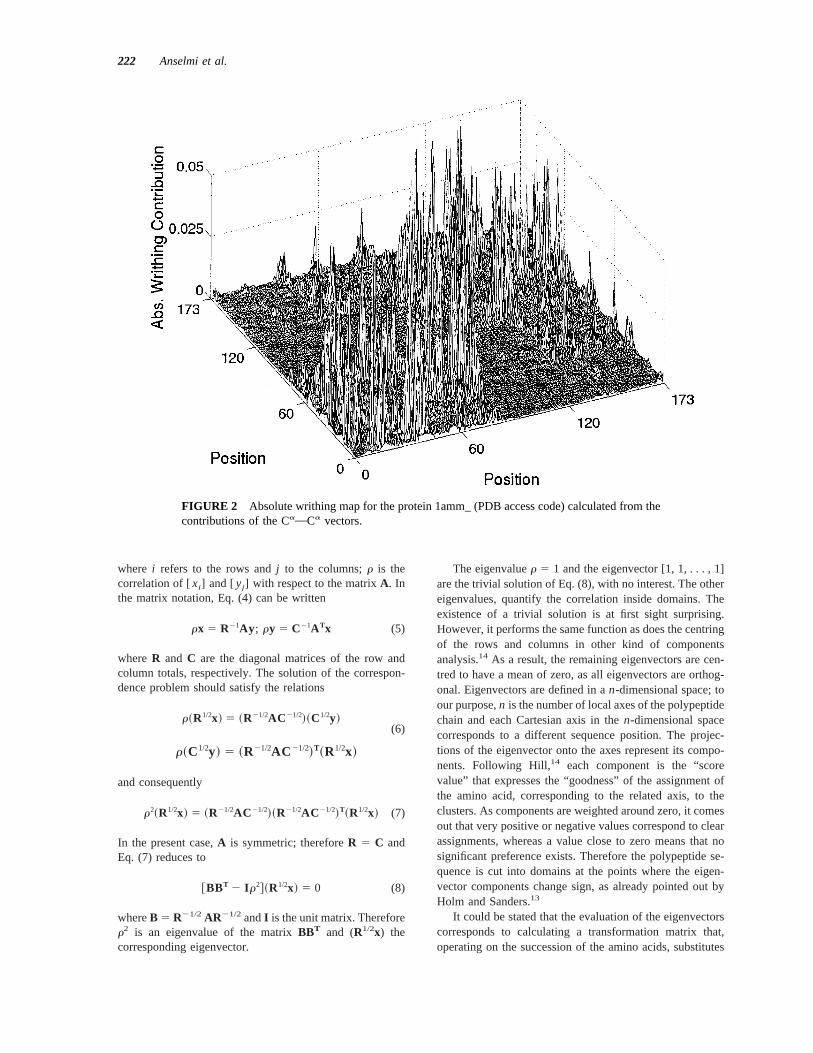
with a positive term,uwij u and consequently, all the contri-butions can be represented by a matrix, as shown for ex-ample in Figure 2 for the protein 1amm_ (PDB accesscode). The corresponding three-dimensional structure is re-ported in Figure 3. Clearly the greatest values ofuwij u arethose adjacent to the principal diagonal, due in large amountto the secondary structure.
It should be noted thatuwij u depends onr ji22 [see Eq. (3)]
and therefore it rapidly decreases with the increase of thedistance. This means that, on average, amino acids close inspace, contribute to the writhing more with respect to theremainder. However, it is worth noting thatuwij u alsostrongly depends on the relative orientation of the virtualbonds, being equal to zero if they are coplanar. If only thedependence onr ji
22 is considered, similar results can beobtained. It is puzzling, because the values ofuwij u andr ji
22
are very different; however, it is reasonable that the depen-dence of the writhing onr ji
22 should be found, at leastpartially, if the simple distributions of the distance is con-sidered. Nevertheless, it is difficult to advance a hypothesisto explain whyr ji
22 should take in account for the domainorganization of the proteins. Considering only distance re-straints would results in only an empirical correlation be-tween quantities. On the contrary, writhing is a topologicalfeature that takes in account for the three-dimensional ori-entation of a space curve. Finally, It should be noted thatuwij u has the mathematical advantage of tending to zero withr ji , whereasr ji
22 diverges.From Figure 2 it is also evident the matrix being block
diagonal, each block corresponding to the 2 continuousdomains that constitute the protein 1amm_, whereas thereare few “contacts” between domains.
In order to separate the contributions of the secondarystructure from those of the tertiary structure, the total abso-lute writhing number of each protein have been calculatedstarting from their local axes, evaluated from the crystallo-graphic coordinates of thea-carbons.36,37
The statistical method used to make the absolute writh-ing matrix block diagonal is the correspondence analysis.14
In practice, the problem consists of finding functions suchthat the correlation between the starting species is a maxi-mum. In the general case, beingA a m 3 n matrix ofelementsaij , specifying the statistical relations between them species in the rows and then species in the columns, theproblem reduces to find two score vectors, [xi] and [yj],such that
rxi 5
¥j
aijyj
¥j
aij
; ryj 5
¥i
aij xi
¥i
aij
(4)
Identification of Protein Domains 221
where i refers to the rows andj to the columns;r is thecorrelation of [xi] and [yj] with respect to the matrixA. Inthe matrix notation, Eq. (4) can be written
rx 5 R21Ay ; ry 5 C21ATx (5)
whereR and C are the diagonal matrices of the row andcolumn totals, respectively. The solution of the correspon-dence problem should satisfy the relations
r~R1/2x! 5 ~R21/2AC21/2!~C1/2y!(6)
r~C1/2y! 5 ~R21/2AC21/2!T~R1/2x!
and consequently
r2~R1/2x! 5 ~R21/2AC21/2!~R21/2AC21/2!T~R1/2x! (7)
In the present case,A is symmetric; thereforeR 5 C andEq. (7) reduces to
@BBT 2 I r2#~R1/2x! 5 0 (8)
whereB 5 R21/2 AR21/2 andI is the unit matrix. Thereforer2 is an eigenvalue of the matrixBBT and (R1/2x) thecorresponding eigenvector.
The eigenvaluer 5 1 and the eigenvector [1, 1, . . . , 1]are the trivial solution of Eq. (8), with no interest. The othereigenvalues, quantify the correlation inside domains. Theexistence of a trivial solution is at first sight surprising.However, it performs the same function as does the centringof the rows and columns in other kind of componentsanalysis.14 As a result, the remaining eigenvectors are cen-tred to have a mean of zero, as all eigenvectors are orthog-onal. Eigenvectors are defined in an-dimensional space; toour purpose,n is the number of local axes of the polypeptidechain and each Cartesian axis in then-dimensional spacecorresponds to a different sequence position. The projec-tions of the eigenvector onto the axes represent its compo-nents. Following Hill,14 each component is the “scorevalue” that expresses the “goodness” of the assignment ofthe amino acid, corresponding to the related axis, to theclusters. As components are weighted around zero, it comesout that very positive or negative values correspond to clearassignments, whereas a value close to zero means that nosignificant preference exists. Therefore the polypeptide se-quence is cut into domains at the points where the eigen-vector components change sign, as already pointed out byHolm and Sanders.13
It could be stated that the evaluation of the eigenvectorscorresponds to calculating a transformation matrix that,operating on the succession of the amino acids, substitutes
FIGURE 2 Absolute writhing map for the protein 1amm_ (PDB access code) calculated from thecontributions of the CaOCa vectors.
222 Anselmi et al.
the natural order of the primary structure with another, suchthat amino acids close in space and inside domains comenear each others.
On the other hand,r represents the correlation of thexandy score values with respect to the starting matrixA. Itmeans that the higher the value ofr, the more significant isthe assignment of the domains. Therefore, all the eigenvec-tors are deemed to be sorted by their eigenvalues, and thefirst not-trivial eigenvector represents the best solution tothe correlation problem.14
RESULTS AND DISCUSSION
To verify the effectiveness of the proposed model, aset of 71 crystallographic and NMR structures of
proteins was considered (see asterisked footnoteabove). The correct domain structures were assumedfrom the CATH database23 available on-line at theaddress http://www.biochem.ucl.ac.uk/bsm/cath.
According to the database, among the set, 9 struc-tures have 2 domains; 3 of them have continuousdomains (1amm_, 1rhs_, 4gcr_), 6 have not-continu-ous domains (1arb_, 1ixh_, 1mla_, 2sga_, 5ptp_,8abp_). 2 Have more than 2 domains (1fmk_, 1yge_).
Among the proteins with 2 domains, 4gcr_ is com-pletely homologous to 1amm_ and consequently wasnot considered.
The advantage of considering single and mul-tidomain proteins is that it is possible to identifythose features of the protein folding that are com-
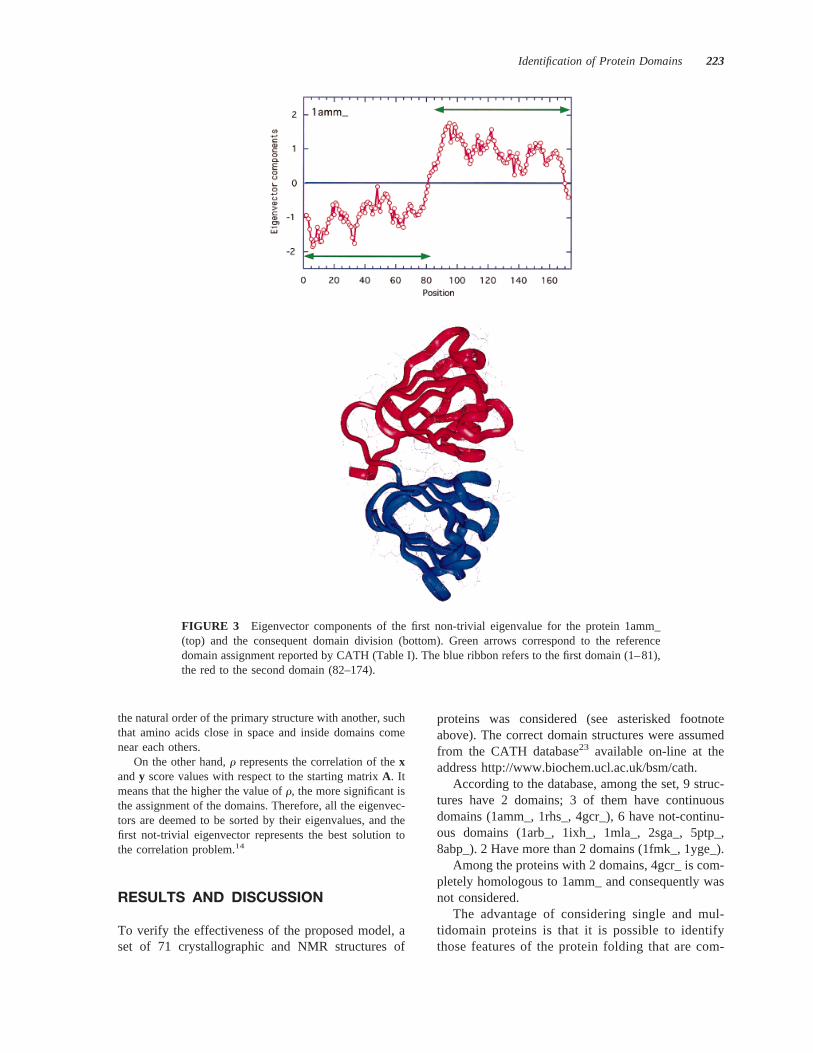
FIGURE 3 Eigenvector components of the first non-trivial eigenvalue for the protein 1amm_(top) and the consequent domain division (bottom). Green arrows correspond to the referencedomain assignment reported by CATH (Table I). The blue ribbon refers to the first domain (1–81),the red to the second domain (82–174).
Identification of Protein Domains 223

mon to all proteins or characteristic of the domainstructures.
Table I reports the domain boundaries, identifiedwith the proposed algorithm, of all the consideredmultidomain proteins for a direct comparison withCATH database.
As examples, Figures 3–6 show the components ofthe first not-trivial eigenvector for some proteins ofthe considered set, calculated from their local axes, asdiscussed above. There are reported the domain iden-tification in the CATH database for comparison. Ingeneral, eigenvector components can assume positiveor negative values. Domain boundaries are consideredwhen values change along the position number fromnegative to positive or vice versa. However, to avoidthe occurrence of short loops, small fluctuations of thevalues inside domains are considered not significant.
Proteins with 2 continuous domains (1amm_ and1rhs_) have an excellent agreement with the CATHsolutions, with a difference in the domain-boundaryidentification of 2 amino acids at most. As shown inthe case of 1amm_ (Figures 2 and 3), domain identi-fication is rather simple, as there is a clear segregationof the structural units.
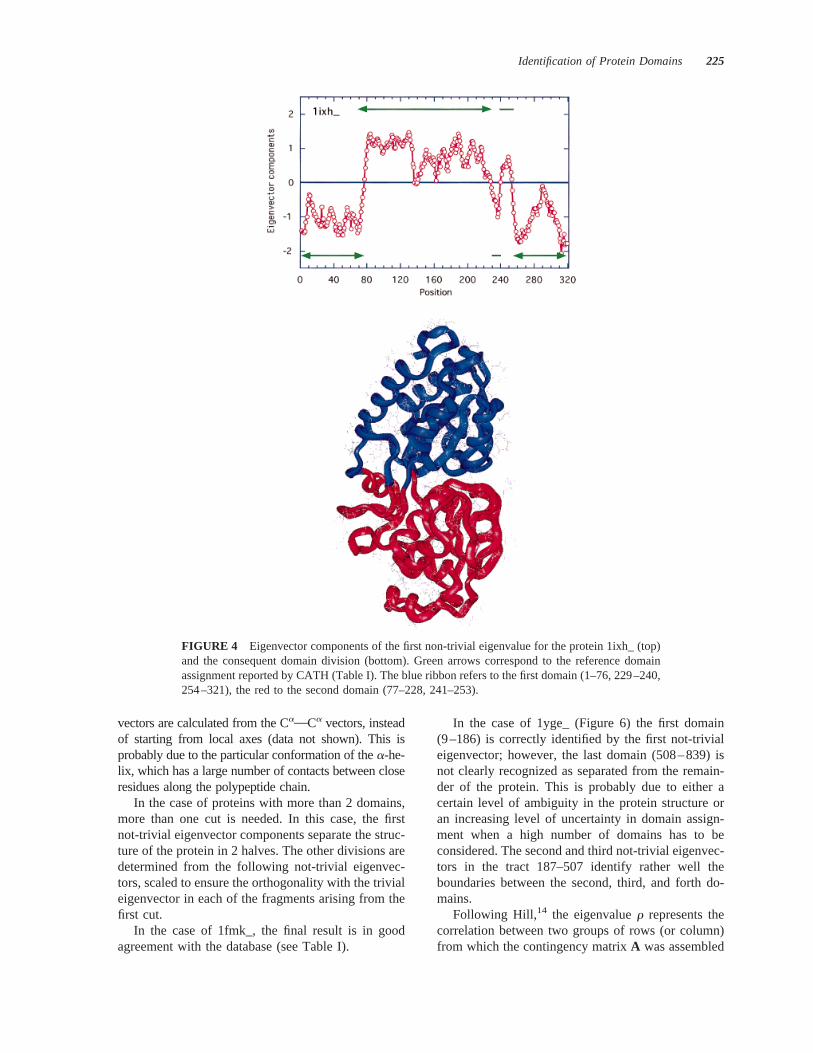
In the case of proteins with 2 not-continuous do-mains (1arb_, 1ixh_, 1mla_, 2sga_, 5ptp_, and8abp_), there is a good agreement with the CATHsolutions in at least 4 out 6 cases (1ixh_, 2sga_, 5ptp_,and 8abp_). In the case of 1ixh_ (Figure 4), assigningthe small loops in the position 229–240 and 241–253to one of the two domains is ambiguous, so that theyhave been considered to belong to the first and seconddomain respectively, in perfect agreement with thedatabase.
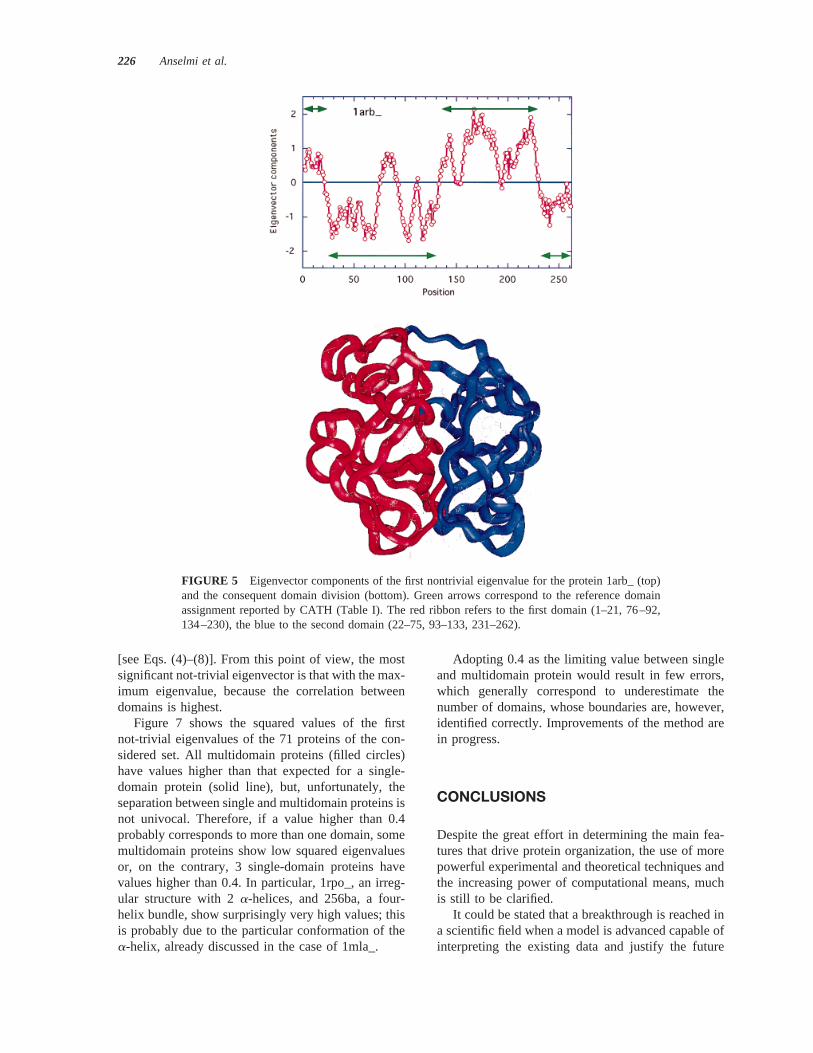
In the case of 1arb_ (Figure 5), the current solutionis very similar to that obtained from CATH, exceptthe loop in the position 76–92 assigned to the firstdomain, instead of the second. This is due to theambiguity in the domain structure, the loop beingcloser to the first domain than to the second in whichit start and ends. However, the same result could beobtained considering more rigid conditions for thedomain-boundary assignment (no loops shorter than20 amino acids, for example).
The method fails in the domain assignment of 1mla_.In fact, the central domain is found bigger than it is inreality, breaking the integrity of two of thea-helices inthe first domain. The same solution is found if eigen-
Table I Protein Domain Assignment: Comparison Between this Work and CATH Database
PDB EntryNumber ofDomains Class This Work CATH
1amm_ 2 Mainly-b 1–81 1–83Mainly-b 82–174 84–174
1arb_ 2 Mainly-b 1–21, 76–92, 134–230 1–24, 134–231Mainly-b 22–75, 93–133, 231–262 25–131, 233–262
1fmk_ 4 Mainly-b 82–145 82–145a, b 146–250 146–246a, b 251–341 259–344Mainly-a 342–519 345–519
1ixh_ 2 a, b 1–76, 229–240, 254–321 1–77, 230–239, 254–321a, b 77–228, 241–253 78–229, 240–253
1mla_ 2 a, b 116–207 125–197a, b 1–115, 208–307 3–124, 198–307
1rhs_ 2 a, b 1–157 1–156a, b 158–292 157–292
1yge_ 5 Mainly-b 1–186 9–167Few sec. structures 187–260 168–267Few sec. structures 261–330 268–356a, b 331–507 357–490Mainly-a 508–839 491–839
2sga_ 2 Mainly-b 16–121, 230–242 16–115, 231–242Mainly-b 122–229 116–230
5ptp_ 2 Mainly-b 16–20, 119–232 16–27, 121–232Mainly-b 21–118, 233–244 28–120, 233–244
8abp_ 2 a, b 2–109, 255–284 2–108, 255–285a, b 110–254, 285–306 109–254, 286–306
224 Anselmi et al.
vectors are calculated from the CaOCa vectors, insteadof starting from local axes (data not shown). This isprobably due to the particular conformation of thea-he-lix, which has a large number of contacts between closeresidues along the polypeptide chain.
In the case of proteins with more than 2 domains,more than one cut is needed. In this case, the firstnot-trivial eigenvector components separate the struc-ture of the protein in 2 halves. The other divisions aredetermined from the following not-trivial eigenvec-tors, scaled to ensure the orthogonality with the trivialeigenvector in each of the fragments arising from thefirst cut.
In the case of 1fmk_, the final result is in goodagreement with the database (see Table I).
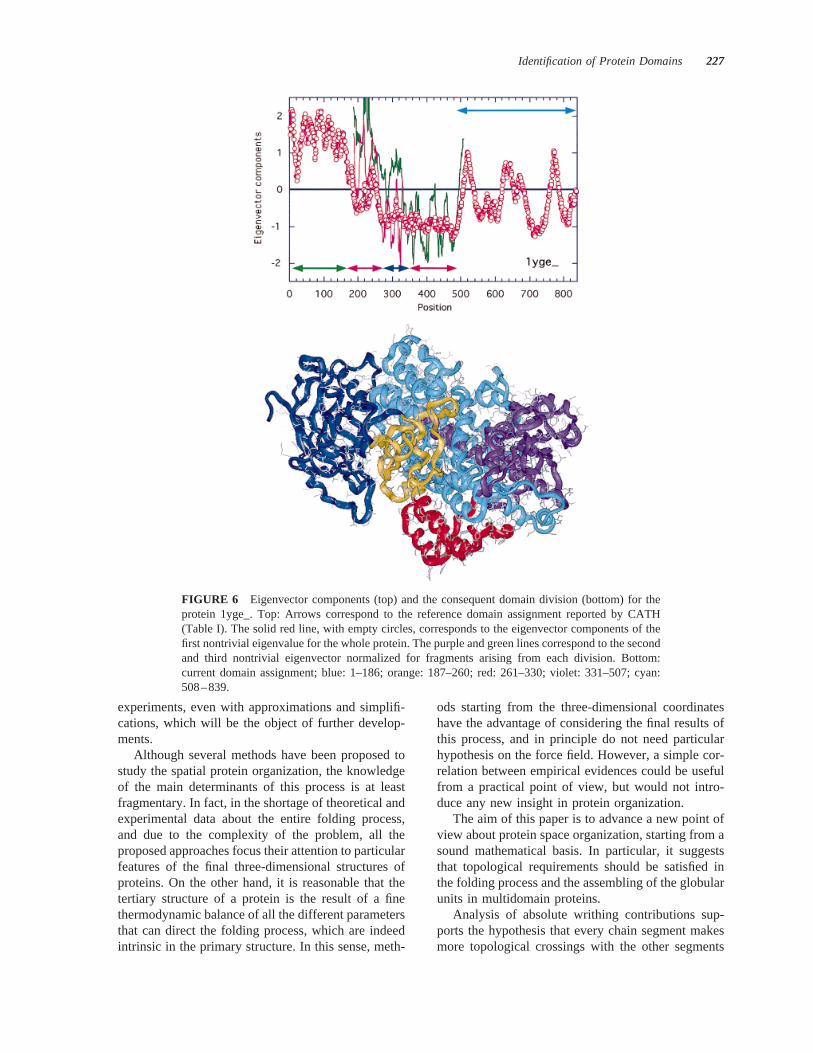
In the case of 1yge_ (Figure 6) the first domain(9–186) is correctly identified by the first not-trivialeigenvector; however, the last domain (508–839) isnot clearly recognized as separated from the remain-der of the protein. This is probably due to either acertain level of ambiguity in the protein structure oran increasing level of uncertainty in domain assign-ment when a high number of domains has to beconsidered. The second and third not-trivial eigenvec-tors in the tract 187–507 identify rather well theboundaries between the second, third, and forth do-mains.
Following Hill,14 the eigenvaluer represents thecorrelation between two groups of rows (or column)from which the contingency matrixA was assembled
FIGURE 4 Eigenvector components of the first non-trivial eigenvalue for the protein 1ixh_ (top)and the consequent domain division (bottom). Green arrows correspond to the reference domainassignment reported by CATH (Table I). The blue ribbon refers to the first domain (1–76, 229–240,254–321), the red to the second domain (77–228, 241–253).
Identification of Protein Domains 225
[see Eqs. (4)–(8)]. From this point of view, the mostsignificant not-trivial eigenvector is that with the max-imum eigenvalue, because the correlation betweendomains is highest.
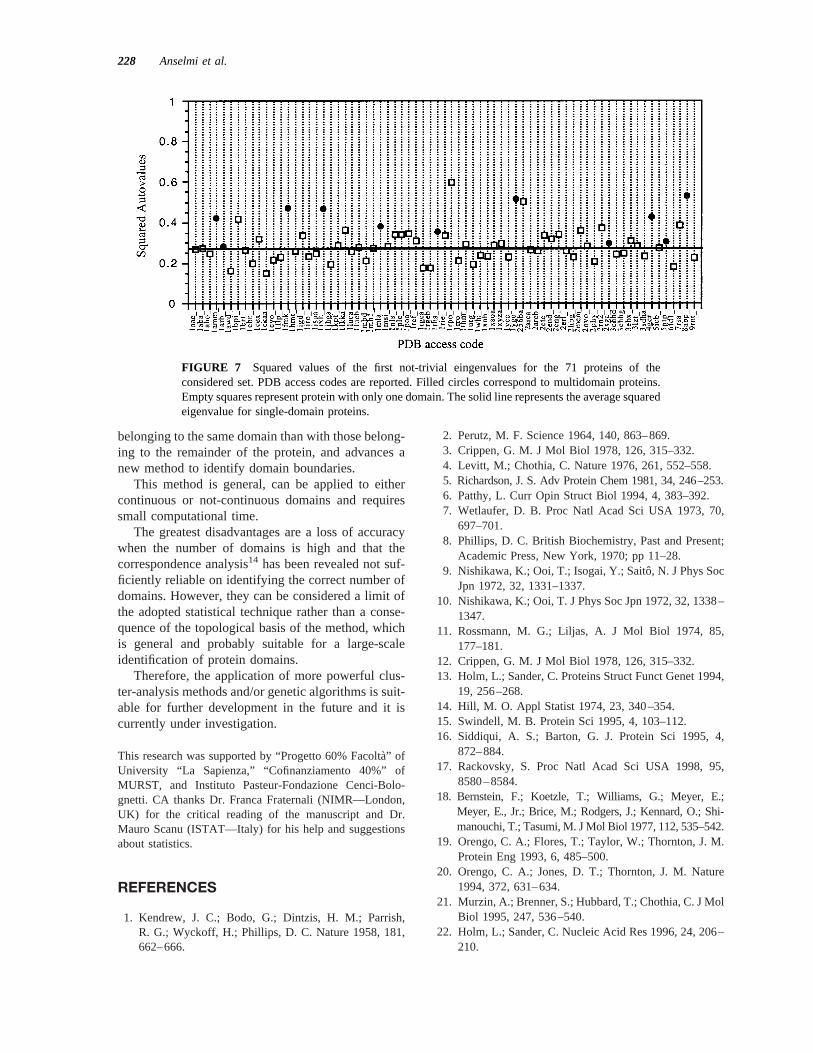
Figure 7 shows the squared values of the firstnot-trivial eigenvalues of the 71 proteins of the con-sidered set. All multidomain proteins (filled circles)have values higher than that expected for a single-domain protein (solid line), but, unfortunately, theseparation between single and multidomain proteins isnot univocal. Therefore, if a value higher than 0.4probably corresponds to more than one domain, somemultidomain proteins show low squared eigenvaluesor, on the contrary, 3 single-domain proteins havevalues higher than 0.4. In particular, 1rpo_, an irreg-ular structure with 2a-helices, and 256ba, a four-helix bundle, show surprisingly very high values; thisis probably due to the particular conformation of thea-helix, already discussed in the case of 1mla_.
Adopting 0.4 as the limiting value between singleand multidomain protein would result in few errors,which generally correspond to underestimate thenumber of domains, whose boundaries are, however,identified correctly. Improvements of the method arein progress.
CONCLUSIONS
Despite the great effort in determining the main fea-tures that drive protein organization, the use of morepowerful experimental and theoretical techniques andthe increasing power of computational means, muchis still to be clarified.
It could be stated that a breakthrough is reached ina scientific field when a model is advanced capable ofinterpreting the existing data and justify the future
FIGURE 5 Eigenvector components of the first nontrivial eigenvalue for the protein 1arb_ (top)and the consequent domain division (bottom). Green arrows correspond to the reference domainassignment reported by CATH (Table I). The red ribbon refers to the first domain (1–21, 76–92,134–230), the blue to the second domain (22–75, 93–133, 231–262).
226 Anselmi et al.
experiments, even with approximations and simplifi-cations, which will be the object of further develop-ments.
Although several methods have been proposed tostudy the spatial protein organization, the knowledgeof the main determinants of this process is at leastfragmentary. In fact, in the shortage of theoretical andexperimental data about the entire folding process,and due to the complexity of the problem, all theproposed approaches focus their attention to particularfeatures of the final three-dimensional structures ofproteins. On the other hand, it is reasonable that thetertiary structure of a protein is the result of a finethermodynamic balance of all the different parametersthat can direct the folding process, which are indeedintrinsic in the primary structure. In this sense, meth-
ods starting from the three-dimensional coordinateshave the advantage of considering the final results ofthis process, and in principle do not need particularhypothesis on the force field. However, a simple cor-relation between empirical evidences could be usefulfrom a practical point of view, but would not intro-duce any new insight in protein organization.
The aim of this paper is to advance a new point ofview about protein space organization, starting from asound mathematical basis. In particular, it suggeststhat topological requirements should be satisfied inthe folding process and the assembling of the globularunits in multidomain proteins.
Analysis of absolute writhing contributions sup-ports the hypothesis that every chain segment makesmore topological crossings with the other segments
FIGURE 6 Eigenvector components (top) and the consequent domain division (bottom) for theprotein 1yge_. Top: Arrows correspond to the reference domain assignment reported by CATH(Table I). The solid red line, with empty circles, corresponds to the eigenvector components of thefirst nontrivial eigenvalue for the whole protein. The purple and green lines correspond to the secondand third nontrivial eigenvector normalized for fragments arising from each division. Bottom:current domain assignment; blue: 1–186; orange: 187–260; red: 261–330; violet: 331–507; cyan:508–839.
Identification of Protein Domains 227
belonging to the same domain than with those belong-ing to the remainder of the protein, and advances anew method to identify domain boundaries.
This method is general, can be applied to eithercontinuous or not-continuous domains and requiressmall computational time.
The greatest disadvantages are a loss of accuracywhen the number of domains is high and that thecorrespondence analysis14 has been revealed not suf-ficiently reliable on identifying the correct number ofdomains. However, they can be considered a limit ofthe adopted statistical technique rather than a conse-quence of the topological basis of the method, whichis general and probably suitable for a large-scaleidentification of protein domains.
Therefore, the application of more powerful clus-ter-analysis methods and/or genetic algorithms is suit-able for further development in the future and it iscurrently under investigation.
This research was supported by “Progetto 60% Facolta`” ofUniversity “La Sapienza,” “Cofinanziamento 40%” ofMURST, and Instituto Pasteur-Fondazione Cenci-Bolo-gnetti. CA thanks Dr. Franca Fraternali (NIMR—London,UK) for the critical reading of the manuscript and Dr.Mauro Scanu (ISTAT—Italy) for his help and suggestionsabout statistics.
REFERENCES
1. Kendrew, J. C.; Bodo, G.; Dintzis, H. M.; Parrish,R. G.; Wyckoff, H.; Phillips, D. C. Nature 1958, 181,662–666.
2. Perutz, M. F. Science 1964, 140, 863–869.3. Crippen, G. M. J Mol Biol 1978, 126, 315–332.4. Levitt, M.; Chothia, C. Nature 1976, 261, 552–558.5. Richardson, J. S. Adv Protein Chem 1981, 34, 246–253.6. Patthy, L. Curr Opin Struct Biol 1994, 4, 383–392.7. Wetlaufer, D. B. Proc Natl Acad Sci USA 1973, 70,
697–701.8. Phillips, D. C. British Biochemistry, Past and Present;
Academic Press, New York, 1970; pp 11–28.9. Nishikawa, K.; Ooi, T.; Isogai, Y.; Saitoˆ, N. J Phys Soc
Jpn 1972, 32, 1331–1337.10. Nishikawa, K.; Ooi, T. J Phys Soc Jpn 1972, 32, 1338–
1347.11. Rossmann, M. G.; Liljas, A. J Mol Biol 1974, 85,
177–181.12. Crippen, G. M. J Mol Biol 1978, 126, 315–332.13. Holm, L.; Sander, C. Proteins Struct Funct Genet 1994,
19, 256–268.14. Hill, M. O. Appl Statist 1974, 23, 340–354.15. Swindell, M. B. Protein Sci 1995, 4, 103–112.16. Siddiqui, A. S.; Barton, G. J. Protein Sci 1995, 4,
872–884.17. Rackovsky, S. Proc Natl Acad Sci USA 1998, 95,
8580–8584.18. Bernstein, F.; Koetzle, T.; Williams, G.; Meyer, E.;
Meyer, E., Jr.; Brice, M.; Rodgers, J.; Kennard, O.; Shi-manouchi, T.; Tasumi, M. J Mol Biol 1977, 112, 535–542.
19. Orengo, C. A.; Flores, T.; Taylor, W.; Thornton, J. M.Protein Eng 1993, 6, 485–500.
20. Orengo, C. A.; Jones, D. T.; Thornton, J. M. Nature1994, 372, 631–634.
21. Murzin, A.; Brenner, S.; Hubbard, T.; Chothia, C. J MolBiol 1995, 247, 536–540.
22. Holm, L.; Sander, C. Nucleic Acid Res 1996, 24, 206–210.
FIGURE 7 Squared values of the first not-trivial eingenvalues for the 71 proteins of theconsidered set. PDB access codes are reported. Filled circles correspond to multidomain proteins.Empty squares represent protein with only one domain. The solid line represents the average squaredeigenvalue for single-domain proteins.
228 Anselmi et al.

23. Michie, A. D.; Orengo, C. A.; Thorton, J. M. J Mol Biol1996, 262, 168–185.
24. Corpet, F.; Gouzy, J.; Kahn, D. Nucleic Acids Res1998, 26, 323–326.
25. Wu, C. H.; Shivakumar, S.; Huang, H. Nucleic AcidRes 1999, 27, 272–274.
26. Fuller, F. B. Proc Natl Acad Sci USA 1971, 68, 815–819.27. Katrich, V.; Olson, W. K.; Pieranski, P.; Dubochet, J.;
Stasiak, A. Nature 1997, 388, 148–151.28. Calagareanu, G. Rev Math Pur Appl 1959, 4, 5–20.29. Alm, E.; Baker, D. Proc Natl Acad Sci USA 1999, 96,
11305–11310.30. De Santis, P.; Morosetti, S.; Palleschi, A. Biopolymers
1983, 22, 251–263.
31. De Santis, P.; Palleschi, A.; Chiavarini, S. Structure &Motion: Membranes, Nucleic Acids & Proteins; Ade-nine Press 1985; pp 251–263.
32. Hobohm, U.; Scharf, M.; Schneider, R.; Sander, C.Protein Sci 1992, 1, 409–417.
33. Hobohm, U.; Sander, C. Protein Sci 1994, 3, 522–524.34. Swindell, M. B.; MacArthur, M. W.; Thornton, J. Na-
ture Struct Biol 1995, 2, 596–603.35. Fiebig, K. M.; Schwalbe, H.; Buck, M.; Smith, L. J.;
Dobson, C. M. J Phys Chem 1996, 100, 2661–2666.36. Damiani, A.; De Santis, P. J Chem Phys 1967, 48,
4071–4075.37. De Santis, P.; Morosetti, S.; Rizzo, R. Macromolecules
1974, 7, 52–58.
Identification of Protein Domains 229





























