Embed Size (px)
Citation preview

JOURNAL OF VIROLOGY, May 2009, p. 4884–4894 Vol. 83, No. 100022-538X/09/$08.00�0 doi:10.1128/JVI.02659-08Copyright © 2009, American Society for Microbiology. All Rights Reserved.
Impact of Human Immunodeficiency Virus Type 1 Resistance toProtease Inhibitors on Evolution of Resistance to the Maturation
Inhibitor Bevirimat (PA-457)�†Catherine S. Adamson,1 Kayoko Waki,1 Sherimay D. Ablan,1 Karl Salzwedel,2 and Eric O. Freed1*
Virus-Cell Interaction Section, HIV Drug Resistance Program, National Cancer Institute, Frederick, Maryland 21702-1201,1 andPanacos Pharmaceuticals Inc., 209 Perry Parkway, Gaithersburg, Maryland 208772
Received 25 December 2008/Accepted 16 February 2009
The maturation inhibitor bevirimat [3-O-(3�,3�dimethysuccinyl)betulinic acid; BVM; also known as PA-457or DSB] potently inhibits human immunodeficiency virus type 1 (HIV-1) replication by blocking protease(PR)-mediated cleavage at the junction between capsid (CA) and spacer peptide 1 (SP1) in Gag. We previouslyisolated a panel of single-amino-acid substitutions that confer resistance to BVM in vitro (C. S. Adamson, S. D.Ablan, I. Boeras, R. Goila-Gaur, F. Soheilian, K. Nagashima, F. Li, K. Salzwedel, M. Sakalian, C. T. Wild, andE. O. Freed, J. Virol. 80:10957–10971, 2006). The BVM resistance mutations cluster at or near the CA-SP1cleavage site. Because BVM likely will be used clinically in patients harboring viruses resistant to PR inhibitors(PIs), in this study we evaluated the interplay between a PI-resistant (PIR) PR and the BVM resistancemutations in Gag. As expected, the PIR mutations had no effect on inhibition by BVM; however, we observedgeneral processing defects and a slight delay in viral replication in Jurkat T cells associated with the PIRmutations, even in the absence of compound. When combined, most BVM resistance and PIR mutations actedadditively to impair viral replication, particularly in the presence of BVM. The BVM-resistant mutant SP1-A1Vwas an exception, as it supported robust replication in the context of either wild-type (WT) or PIR PR, evenat high BVM concentrations. Significantly, the emergence of BVM resistance was delayed in the context of thePIR PR, and the SP1-A1V mutation was acquired most frequently with either WT or PIR PR. These resultssuggest that resistance to BVM is less likely to emerge in patients who have failed PIs than in patients who arePI naïve. We predict that the SP1-A1V substitution is the most likely to emerge in vivo, as this mutantreplicates robustly independently of PR mutations or BVM. These findings offer insights into the effect of PIRmutations on the evolution of BVM resistance in PI-experienced patients.
The maturation of human immunodeficiency virus type 1(HIV-1) virions is mediated by the virally encoded protease(PR). PR cleaves the HIV-1 Gag polyprotein precursor duringor shortly after assembled virus particles are released from theplasma membrane of the infected cell. Gag is cleaved at fivemajor sites in a stepwise cascade that generates the four ma-ture Gag domains, matrix (MA or p17), capsid (CA or p24),nucleocapsid (NC or p7), and p6, and two spacer peptides, SP1(or p2) and SP2 (or p1) (4, 12, 39). The rate of proteolyticcleavage at each site kinetically controls the processing cascade(11, 20, 29, 31, 38–40).
3-O-(3�,3�-dimethylsuccinyl)betulinic acid, known variouslyas bevirimat (BVM), PA-457, or DSB, is the first in a poten-tially new class of anti-HIV-1 drugs known as maturation in-hibitors. BVM disrupts HIV-1 infectivity by specifically inhib-iting a late step in the Gag processing cascade (5, 6, 33, 44), thecleavage of SP1 from the C terminus of CA (21, 49). Theinhibition of CA-SP1 cleavage prevents proper maturation andpotently inhibits virion infectivity (21, 40, 49). Under certaincircumstances, BVM also has been shown to interfere with
virus assembly and release (9, 15); however, this phenomenonoccurs only at high, nonphysiological drug concentrations (9).
PR-mediated Gag processing prepares the virion for theinfection of a new cell by inducing a morphological rearrange-ment within the virus particle. During maturation, CA reas-sembles into a conical condensed core containing the viralRNA in complex with NC and the viral enzymes reverse trans-criptase (RT) and integrase (4, 12, 39). Disrupting processingat any of the individual Gag cleavage sites, or altering the orderin which the sites are cleaved, results in the formation ofaberrant particles that have significantly reduced infectivity (1,16, 19, 21, 31, 39, 40, 49). The disruption of CA-SP1 cleavage,either by BVM treatment or mutation, leads to the generationof noninfectious particles that exhibit an electron-dense layerof Gag inside the viral membrane and fail to form conical cores(21, 40).
The mechanism by which BVM inhibits CA-SP1 processingis not fully defined. However, several lines of evidence suggestthat BVM directly targets the CA-SP1 junction in Gag. Allmutations reported thus far to confer resistance to BVM mapto the CA-SP1 junction (3, 21, 22, 46, 49). HIV-2 and simianimmunodeficiency virus from rhesus macaques (SIVmac) arenaturally resistant to BVM (49); the amino acid sequence atthe CA-SP1 junction of these lentiviruses diverges from that ofHIV-1. Importantly, some sites of sequence divergence be-tween HIV-1 and HIV-2/SIVmac occur at amino acid residuesto which BVM resistance maps in HIV-1 (3, 49). Swapping
* Corresponding author. Mailing address: Virus-Cell InteractionSection, HIV Drug Resistance Program, NCI-Frederick, Frederick,MD 21702-1201. Phone: (301) 846-6223. Fax: (301) 846-6777. E-mail:[email protected].
† Supplemental material for this article may be found at http://jvi.asm.org/.
� Published ahead of print on 11 March 2009.
4884

residues between HIV-1 and SIVmac at the CA-SP1 junctionrenders SIVmac sensitive to BVM (47). The incorporation ofBVM into immature HIV-1 particles has been demonstrated,and this incorporation is reduced by some of the reportedBVM resistance mutations (46, 48). The observations thatBVM does not inhibit the processing of monomeric Gag insolution (21), is specifically incorporated into immature parti-cles (48), and requires immature virus assembly for activity (21,32) suggest that BVM binds an undefined pocket in Gag thatis formed upon Gag oligomerization. The disruption of a spe-cific proteolytic cleavage event in the Gag processing cascadedistinguishes the mechanism of action of BVM from that ofclinically approved protease inhibitors (PIs), which directlytarget the catalytic activity of the enzyme (10, 37, 42, 43). Thecontinued clinical development of maturation inhibitors suchas BVM would add another much-needed class of anti-HIV-1drugs to the arsenal currently available for the treatment ofHIV/AIDS (5).
Phase II clinical trials are being conducted with BVM bothin treatment-naïve patients and in patients who have failedtheir PI and/or RT inhibitor regimen due to the emergence ofviruses resistant to these drugs (26, 33, 35). The fact that BVMlikely will be used in PI inhibitor-experienced patients raisesinteresting and clinically significant questions about the emer-gence of resistance to BVM in the context of PR enzymesbearing PI-resistant (PIR) mutations. Resistance to PIs typi-cally emerges via the stepwise acquisition of multiple muta-tions in PR; changes that confer resistance to the PIs emergefirst, followed by substitutions in PR that compensate for viralfitness defects imposed by the primary resistance-conferringmutations (7, 28). For example, the HIV-1 isolate with PIRmutations L10R/M46I/L63P/V82T/I84V was isolated fromHIV-1 patients failing treatment with the PI indinavir (vari-ously known as IDV or MK-639) (8). These mutations conferresistance to six structurally diverse PIs and reportedly restorereplicative fitness to wild-type (WT) levels (8, 27).
We previously identified a panel of six independent, single-amino-acid substitutions that confer resistance to BVM in vitro(Fig. 1) (3). A subset of these mutations was identified in otherstudies (21, 49). The BVM resistance mutations cluster at ornear the CA-SP1 cleavage site (3, 21, 49). In this study, we
evaluated the interplay between the L10R/M46I/L63P/V82T/I84V PIR mutations in PR and the BVM resistance mutationsin the CA-SP1 region of Gag. We observed general processingdefects associated with the PIR PR and a slight replicationdelay in Jurkat T cells. When combined, the BVM resistanceand PIR mutations acted additively to negatively impact viralreplication, particularly in the presence of BVM. As a result ofthis additive effect, the emergence of BVM resistance wasdelayed in the presence of the PIR mutations. The data pre-sented in this study suggest that resistance to BVM is less likelyto emerge in patients who have failed PIs than in patients whoare PI naïve.
MATERIALS AND METHODS
BVM, cell culture, and transfections. BVM was prepared as described previ-ously (14) and used at the concentrations indicated. HeLa cells were maintainedin Dulbecco’s modified Eagle medium supplemented with 5% (vol/vol) fetalbovine serum (FBS). The Jurkat T-cell line was maintained in RPMI 1640supplemented with 10% (vol/vol) FBS. All media were supplemented with L-glutamine (2 mM), penicillin, and streptomycin. HeLa cells were transfected withExGen500 (Fermentas) or Lipofectamine 2000 (Invitrogen) according to themanufacturer’s instructions. Jurkat T cells were transfected using DEAE-dextran(18).
Generation of PIR HIV-1 derivatives. A full-length HIV-1 molecular clonederivative of pNL4-3 (2) containing the PR mutations L10R/M46I/L63P/V82T/I84V, which confer resistance to multiple structurally diverse PIs (8), was ob-tained from the NIH AIDS Research and Reference Reagent Program, Divisionof AIDS, National Institute of Allergy and Infectious Diseases (NIAID). ThePIR mutations were introduced into the previously described BVM-resistantpNL4-3 derivatives CA-H226Y, CA-L231M, CA-L231F, SP1-A1V, SP1-A3V,and CA-G225S/SP1-A3V (3, 21) by subcloning the 3,737-bp ApaI-EcoRI frag-ment containing the PIR changes (nucleotides 2006 to 5743) into each of theBVM-resistant clones, creating six pNL4-3 derivatives: CA-H226Y PIR, CA-L231M PIR, CA-L231F PIR, SP1-A1V PIR, SP1-A3V PIR, and CA-G225S/SP1-A3V PIR. The identities of all plasmids generated were confirmed by DNAsequencing.
Radioimmunoprecipitation analysis. The methods used for the metaboliclabeling of HeLa cells, the preparation of cell and virus lysates, and immuno-precipitation have been described in detail previously (13, 41). Briefly, media andsolutions containing BVM at the indicated concentrations were prepared imme-diately before use and vortexed. BVM was maintained throughout the transfec-tion and radioimmunoprecipitation procedures. HeLa cells were transfected withWT (2) or PIR (8) pNL4-3 clones and their derivatives encoding the BVMresistance mutations. Transfected HeLa cells were starved in Cys/Met-free me-dium for 30 min and then metabolically radiolabeled for 2 h with [35S]Cys/MetPro mix (Amersham). Virions were pelleted by ultracentrifugation. Cell and virus
FIG. 1. Representation of the BVM resistance and PIR PR mutations examined in this study. HIV-1 Gag and PR are represented at the top.The MA, CA, NC, and p6 domains and the SP1 and SP2 spacer peptides in Gag are indicated. The alignment shows each of the BVM resistanceand PR mutations. (Adapted from reference 3.)
VOL. 83, 2009 HIV-1 RESISTANCE TO PI AND RESISTANCE TO BVM 4885

lysates were immunoprecipitated with pooled immunoglobulin from HIV-1-in-fected patients (HIV-Ig) obtained through the NIH AIDS Research and Refer-ence Reagent Program, Division of AIDS, NIAID. The radioimmunoprecipi-tated proteins were separated by sodium dodecyl sulfate-polyacrylamide gelelectrophoresis and exposed to X-ray film and a phosphorimager plate (Fuji),and the bands were quantified by using Quantity One software (Bio-Rad).
Replication kinetics. Jurkat T cells were transfected with WT and mutantpNL4-3 derivatives. BVM was added at the time of transfection at the indicatedconcentrations and was maintained throughout the course of the experiment.The Jurkat cells were split every 2 days, supernatant was collected at each timepoint, and viral replication was monitored by RT activity as previously described(13). Replication kinetics experiments were repeated independently four times.On the last repeat, cell pellets and virus supernatants were harvested on the daysof peak RT activity. To verify that the BVM resistance and PIR mutations weremaintained, genomic DNA was extracted from cells on the day of peak RTactivity using a whole-blood DNA extraction kit (Qiagen). The entire Gag-PRcoding region then was amplified by PCR using the forward and reverse primersNL516F (5�-TGC CCG TCT GTT GTG TGA CTC-3�) and NL2897R (5�-AAAATA TGC ATC GCC CAC AT-3�). The resultant 2.3-kb PCR product waspurified using the QIAquick PCR purification kit (Qiagen) and sequenced usingthe primers NL1410F (5�-GGA AGC TGC AGA ATG GGA TA-3�), NL1754F(5�-TGG TCC AAA ATG CGA ACC-3�), and NL2135F (5�-TTC AGA GCAGAC CAG AGC CAA-3�). Following the first round of replication kinetics, virussupernatants from WT, PIR, and SP1-A3V PIR at the day of peak RT activitywere normalized for RT activity and used to infect fresh Jurkat T cells. Theinfected cells were cultured, and then genomic DNA was extracted, PCR am-plified, and sequenced as described above to identify any secondary mutations.
Selection of BVM resistance in the context of WT and PIR PR in vitro.BVM-resistant virus isolates were selected by the serial passage of Jurkat T cellstransfected with WT pNL4-3 or the PIR derivative. Ten flasks for each plasmidwere transfected in parallel, and cultures were maintained at 50 ng/ml BVMthroughout the experiment. Virus replication during the selection process wasmonitored by RT activity as previously described (13). Cell pellets and virussupernatants were harvested on the days of peak RT activity. To identify muta-tions conferring BVM resistance, genomic DNA was extracted, PCR amplified,and sequenced as described above.
RESULTS
HIV-1 encoding PIR PR is sensitive to the HIV-1 maturationinhibitor BVM. To examine the interplay between the evolu-tion of drug resistance to BVM and mutations that conferresistance to clinically approved PR inhibitors, we obtained aderivative of the full-length HIV-1 molecular clone pNL4-3containing the following PIR mutations: L10R/M46I/L63P/V82T/I84V (Fig. 1) (8). This clone was selected because itencodes a combination of mutations that confer resistance tomultiple structurally diverse PR inhibitors and secondary mu-tations that compensate for defects in virus replication im-posed by the PIR mutations (8, 27).
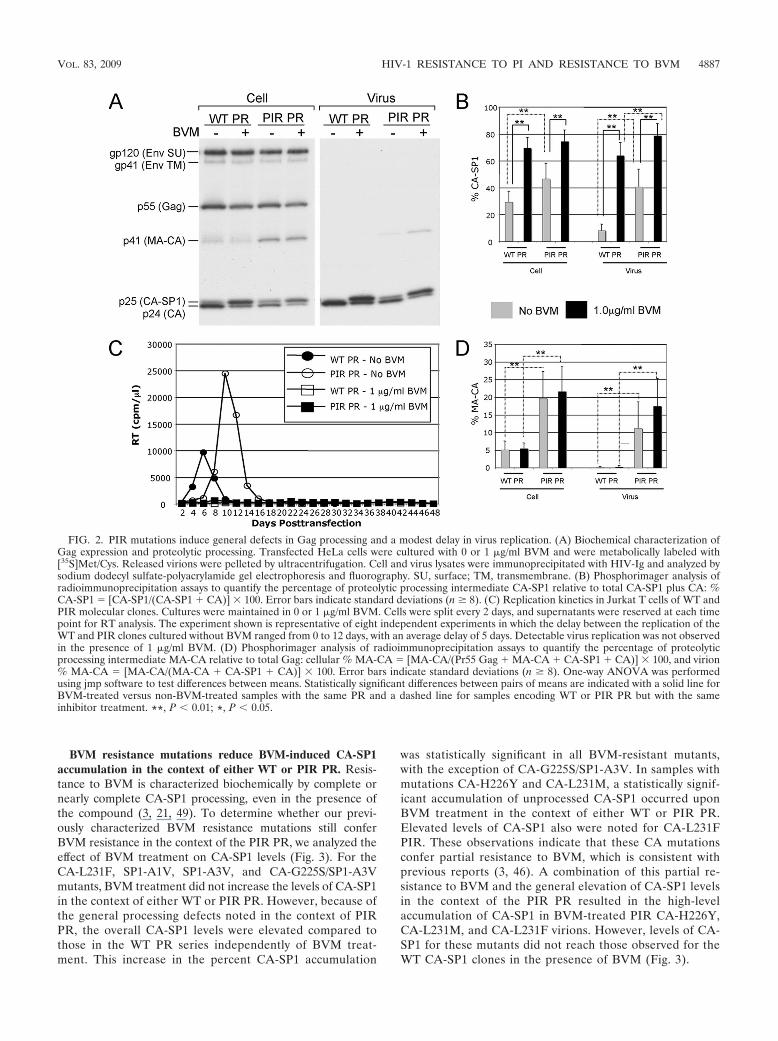
Potent BVM activity against HIV-1 isolates containing PIRPR has been demonstrated previously (21, 49). To verify thesensitivity of the PIR clone to the action of BVM, we con-ducted a quantitative biochemical CA-SP1 processing assay(Fig. 2A and B). HeLa cells transfected with WT pNL4-3 orpNL4-3/PIR were cultured either without BVM or with 1 �g/mlBVM. The cells were metabolically labeled with [35S]Met/Cys,and cell- and virion-associated proteins were immunoprecipi-tated with HIV-Ig. CA-SP1 cleavage was detected (Fig. 2A)and quantified (Fig. 2B). The BVM treatment of PIR-trans-fected cells resulted in the marked and statistically significantaccumulation of unprocessed CA-SP1 in both cell- and virus-associated fractions, which was equivalent to or greater thanthe levels observed for the BVM-treated WT. This accumula-tion of CA-SP1 indicates that the PIR clone is sensitive to theaction of BVM. The sensitivity of the PIR clone to BVM was
confirmed in virus replication assays in which detectable virusspread in Jurkat T cells transfected with either WT or PIRmolecular clones was completely blocked at 1 �g/ml BVM for48 days in culture (Fig. 2C).
PIR mutations are associated with general processing de-fects and a modest replication delay in Jurkat T cells. TheCA-SP1 processing assay, performed to confirm PIR sensitivityto BVM, demonstrated a general proteolytic processing defectassociated with the PIR mutations (Fig. 2A). This defect wascharacterized by the accumulation of the Gag processing in-termediates CA-SP1 and MA-CA (p41) in both the cell- andvirion-associated fractions (Fig. 2A, B, and D). The PIR-asso-ciated increase in MA-CA levels was statistically significant(P � 0.01 by analysis of variance [ANOVA]) for both cell- andvirus-associated fractions. This general proteolytic processingdefect likely accounts for the slight delay in virus replicationobserved for the PIR clone, relative to that of the WT, in theabsence of BVM (Fig. 2C). This slight replication delay inJurkat T cells, which was observed in multiple independentexperiments (data not shown and figures cited below), is incontrast to a previous report of WT replication kinetics for thePIR clone in non-Jurkat target cells (27).
General Gag processing defects are induced by PIR muta-tions independently of BVM resistance mutations surroundingthe CA-SP1 cleavage site. We and others previously have iden-tified a panel of single-amino-acid substitutions that indepen-dently confer resistance to BVM (3, 21, 49). All BVM resis-tance mutations identified thus far are located in the vicinity ofthe CA-SP1 cleavage site. To understand the interplay betweenPIR mutations in PR and BVM resistance mutations surround-ing the CA-SP1 cleavage site, we examined whether the BVMresistance mutations affected Gag processing efficiency in thecontext of the PIR PR. To address this question, we introducedthe PIR mutations into each of the pNL4-3 clones encodingthe following BVM resistance mutations: CA-H226Y, CA-L231M, CA-L231F, SP1-A1V, and SP1-A3V (Fig. 1). We alsoincluded a double mutant, CA-G225S/SP1-A3V, containingthe CA-G225S mutation, which compensates for the replica-tion defect imposed by SP1-A3V (3).
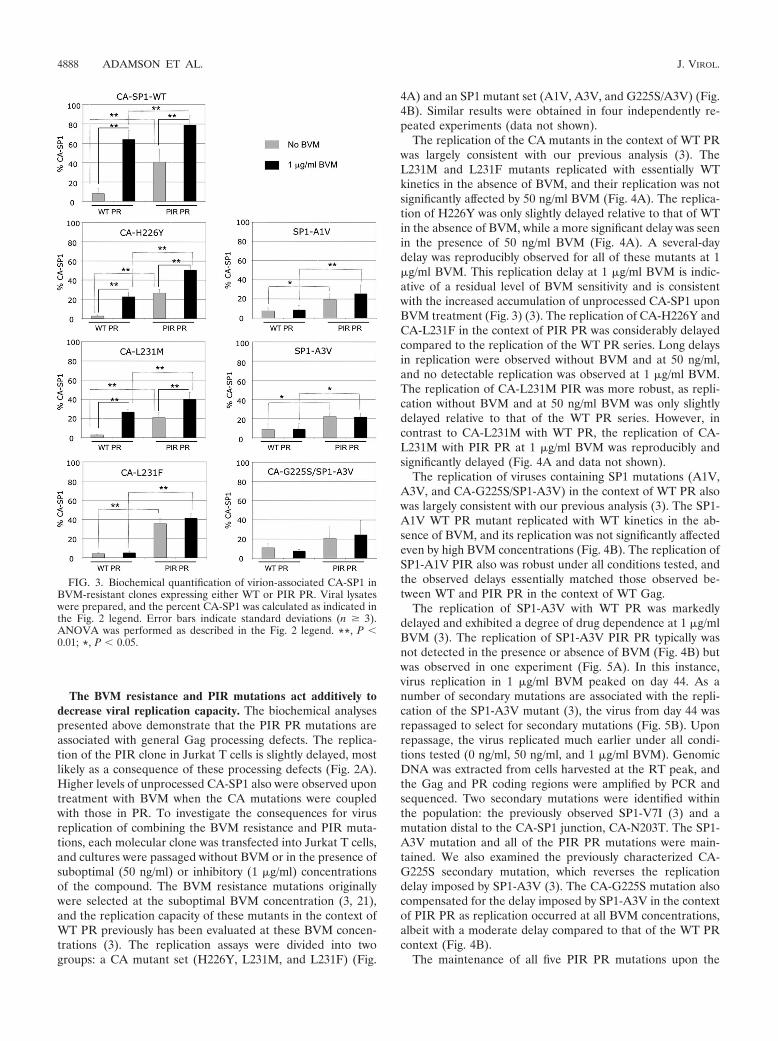
To determine whether the general processing defects ob-served for PIR PR also were observed in the context of theBVM-resistant mutants, we compared the levels of MA-CA invirions bearing either WT or PIR PR (see Fig. S1 in thesupplemental material). The results indicated that the levels ofMA-CA in each of the BVM-resistant clones in the context ofPIR PR were elevated relative to those in the WT PR series. Astatistically significant increase was observed for all BVM-re-sistant clones, with the exception of the CA-L231M WT PRversus PIR PR when cultured without BVM. We next exam-ined CA-SP1 levels in the absence of BVM to evaluate whetherthe BVM resistance mutations affected the ability of PIR PR toprocess the CA-SP1 cleavage site (Fig. 3). The levels of virion-associated CA-SP1 clearly were elevated for each PIR PRclone compared to that of the WT PR clones in the absence ofBVM. This was demonstrated by a statistically significant in-crease in CA-SP1 levels in all BVM-resistant clones exceptCA-G225S/SP1-A3V. These results demonstrate that the PIRmutations lead to general processing defects, as determined bythe accumulation of MA-CA and CA-SP1, independently ofBVM resistance mutations.
4886 ADAMSON ET AL. J. VIROL.
BVM resistance mutations reduce BVM-induced CA-SP1accumulation in the context of either WT or PIR PR. Resis-tance to BVM is characterized biochemically by complete ornearly complete CA-SP1 processing, even in the presence ofthe compound (3, 21, 49). To determine whether our previ-ously characterized BVM resistance mutations still conferBVM resistance in the context of the PIR PR, we analyzed theeffect of BVM treatment on CA-SP1 levels (Fig. 3). For theCA-L231F, SP1-A1V, SP1-A3V, and CA-G225S/SP1-A3Vmutants, BVM treatment did not increase the levels of CA-SP1in the context of either WT or PIR PR. However, because ofthe general processing defects noted in the context of PIRPR, the overall CA-SP1 levels were elevated compared tothose in the WT PR series independently of BVM treat-ment. This increase in the percent CA-SP1 accumulation
was statistically significant in all BVM-resistant mutants,with the exception of CA-G225S/SP1-A3V. In samples withmutations CA-H226Y and CA-L231M, a statistically signif-icant accumulation of unprocessed CA-SP1 occurred uponBVM treatment in the context of either WT or PIR PR.Elevated levels of CA-SP1 also were noted for CA-L231FPIR. These observations indicate that these CA mutationsconfer partial resistance to BVM, which is consistent withprevious reports (3, 46). A combination of this partial re-sistance to BVM and the general elevation of CA-SP1 levelsin the context of the PIR PR resulted in the high-levelaccumulation of CA-SP1 in BVM-treated PIR CA-H226Y,CA-L231M, and CA-L231F virions. However, levels of CA-SP1 for these mutants did not reach those observed for theWT CA-SP1 clones in the presence of BVM (Fig. 3).
FIG. 2. PIR mutations induce general defects in Gag processing and a modest delay in virus replication. (A) Biochemical characterization ofGag expression and proteolytic processing. Transfected HeLa cells were cultured with 0 or 1 �g/ml BVM and were metabolically labeled with[35S]Met/Cys. Released virions were pelleted by ultracentrifugation. Cell and virus lysates were immunoprecipitated with HIV-Ig and analyzed bysodium dodecyl sulfate-polyacrylamide gel electrophoresis and fluorography. SU, surface; TM, transmembrane. (B) Phosphorimager analysis ofradioimmunoprecipitation assays to quantify the percentage of proteolytic processing intermediate CA-SP1 relative to total CA-SP1 plus CA: %CA-SP1 � [CA-SP1/(CA-SP1 � CA)] � 100. Error bars indicate standard deviations (n � 8). (C) Replication kinetics in Jurkat T cells of WT andPIR molecular clones. Cultures were maintained in 0 or 1 �g/ml BVM. Cells were split every 2 days, and supernatants were reserved at each timepoint for RT analysis. The experiment shown is representative of eight independent experiments in which the delay between the replication of theWT and PIR clones cultured without BVM ranged from 0 to 12 days, with an average delay of 5 days. Detectable virus replication was not observedin the presence of 1 �g/ml BVM. (D) Phosphorimager analysis of radioimmunoprecipitation assays to quantify the percentage of proteolyticprocessing intermediate MA-CA relative to total Gag: cellular % MA-CA � [MA-CA/(Pr55 Gag � MA-CA � CA-SP1 � CA)] � 100, and virion% MA-CA � [MA-CA/(MA-CA � CA-SP1 � CA)] � 100. Error bars indicate standard deviations (n � 8). One-way ANOVA was performedusing jmp software to test differences between means. Statistically significant differences between pairs of means are indicated with a solid line forBVM-treated versus non-BVM-treated samples with the same PR and a dashed line for samples encoding WT or PIR PR but with the sameinhibitor treatment. **, P � 0.01; *, P � 0.05.
VOL. 83, 2009 HIV-1 RESISTANCE TO PI AND RESISTANCE TO BVM 4887
The BVM resistance and PIR mutations act additively todecrease viral replication capacity. The biochemical analysespresented above demonstrate that the PIR PR mutations areassociated with general Gag processing defects. The replica-tion of the PIR clone in Jurkat T cells is slightly delayed, mostlikely as a consequence of these processing defects (Fig. 2A).Higher levels of unprocessed CA-SP1 also were observed upontreatment with BVM when the CA mutations were coupledwith those in PR. To investigate the consequences for virusreplication of combining the BVM resistance and PIR muta-tions, each molecular clone was transfected into Jurkat T cells,and cultures were passaged without BVM or in the presence ofsuboptimal (50 ng/ml) or inhibitory (1 �g/ml) concentrationsof the compound. The BVM resistance mutations originallywere selected at the suboptimal BVM concentration (3, 21),and the replication capacity of these mutants in the context ofWT PR previously has been evaluated at these BVM concen-trations (3). The replication assays were divided into twogroups: a CA mutant set (H226Y, L231M, and L231F) (Fig.
4A) and an SP1 mutant set (A1V, A3V, and G225S/A3V) (Fig.4B). Similar results were obtained in four independently re-peated experiments (data not shown).
The replication of the CA mutants in the context of WT PRwas largely consistent with our previous analysis (3). TheL231M and L231F mutants replicated with essentially WTkinetics in the absence of BVM, and their replication was notsignificantly affected by 50 ng/ml BVM (Fig. 4A). The replica-tion of H226Y was only slightly delayed relative to that of WTin the absence of BVM, while a more significant delay was seenin the presence of 50 ng/ml BVM (Fig. 4A). A several-daydelay was reproducibly observed for all of these mutants at 1�g/ml BVM. This replication delay at 1 �g/ml BVM is indic-ative of a residual level of BVM sensitivity and is consistentwith the increased accumulation of unprocessed CA-SP1 uponBVM treatment (Fig. 3) (3). The replication of CA-H226Y andCA-L231F in the context of PIR PR was considerably delayedcompared to the replication of the WT PR series. Long delaysin replication were observed without BVM and at 50 ng/ml,and no detectable replication was observed at 1 �g/ml BVM.The replication of CA-L231M PIR was more robust, as repli-cation without BVM and at 50 ng/ml BVM was only slightlydelayed relative to that of the WT PR series. However, incontrast to CA-L231M with WT PR, the replication of CA-L231M with PIR PR at 1 �g/ml BVM was reproducibly andsignificantly delayed (Fig. 4A and data not shown).
The replication of viruses containing SP1 mutations (A1V,A3V, and CA-G225S/SP1-A3V) in the context of WT PR alsowas largely consistent with our previous analysis (3). The SP1-A1V WT PR mutant replicated with WT kinetics in the ab-sence of BVM, and its replication was not significantly affectedeven by high BVM concentrations (Fig. 4B). The replication ofSP1-A1V PIR also was robust under all conditions tested, andthe observed delays essentially matched those observed be-tween WT and PIR PR in the context of WT Gag.
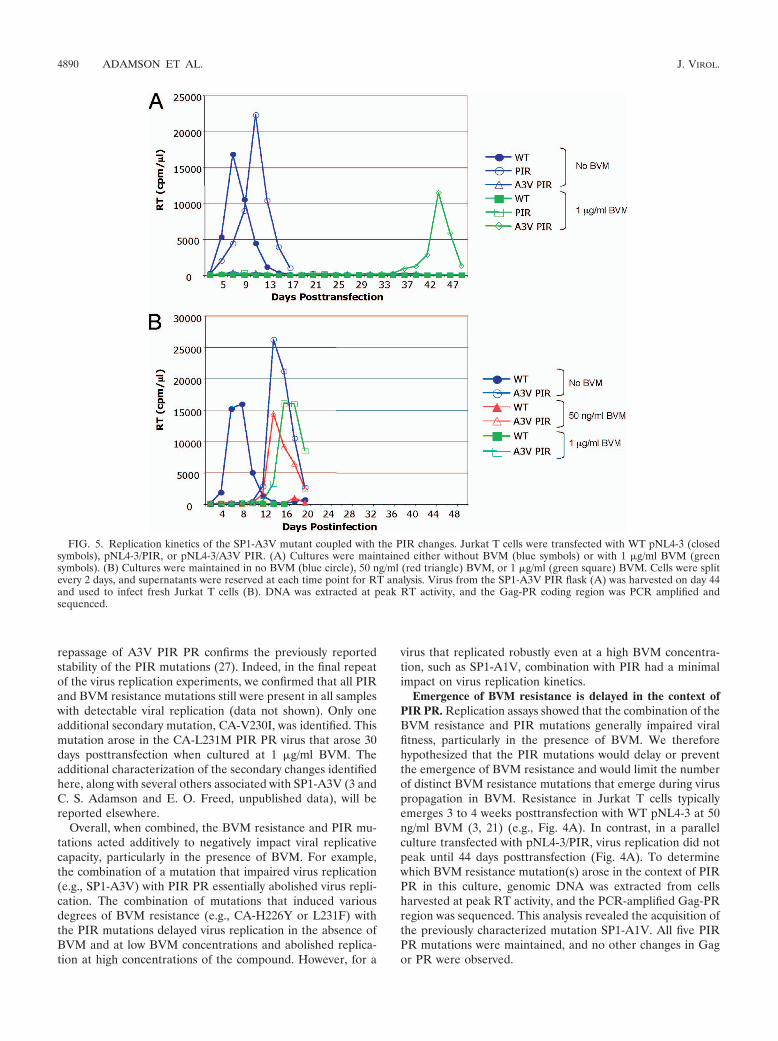
The replication of SP1-A3V with WT PR was markedlydelayed and exhibited a degree of drug dependence at 1 �g/mlBVM (3). The replication of SP1-A3V PIR PR typically wasnot detected in the presence or absence of BVM (Fig. 4B) butwas observed in one experiment (Fig. 5A). In this instance,virus replication in 1 �g/ml BVM peaked on day 44. As anumber of secondary mutations are associated with the repli-cation of the SP1-A3V mutant (3), the virus from day 44 wasrepassaged to select for secondary mutations (Fig. 5B). Uponrepassage, the virus replicated much earlier under all condi-tions tested (0 ng/ml, 50 ng/ml, and 1 �g/ml BVM). GenomicDNA was extracted from cells harvested at the RT peak, andthe Gag and PR coding regions were amplified by PCR andsequenced. Two secondary mutations were identified withinthe population: the previously observed SP1-V7I (3) and amutation distal to the CA-SP1 junction, CA-N203T. The SP1-A3V mutation and all of the PIR PR mutations were main-tained. We also examined the previously characterized CA-G225S secondary mutation, which reverses the replicationdelay imposed by SP1-A3V (3). The CA-G225S mutation alsocompensated for the delay imposed by SP1-A3V in the contextof PIR PR as replication occurred at all BVM concentrations,albeit with a moderate delay compared to that of the WT PRcontext (Fig. 4B).
The maintenance of all five PIR PR mutations upon the
FIG. 3. Biochemical quantification of virion-associated CA-SP1 inBVM-resistant clones expressing either WT or PIR PR. Viral lysateswere prepared, and the percent CA-SP1 was calculated as indicated inthe Fig. 2 legend. Error bars indicate standard deviations (n � 3).ANOVA was performed as described in the Fig. 2 legend. **, P �0.01; *, P � 0.05.
4888 ADAMSON ET AL. J. VIROL.
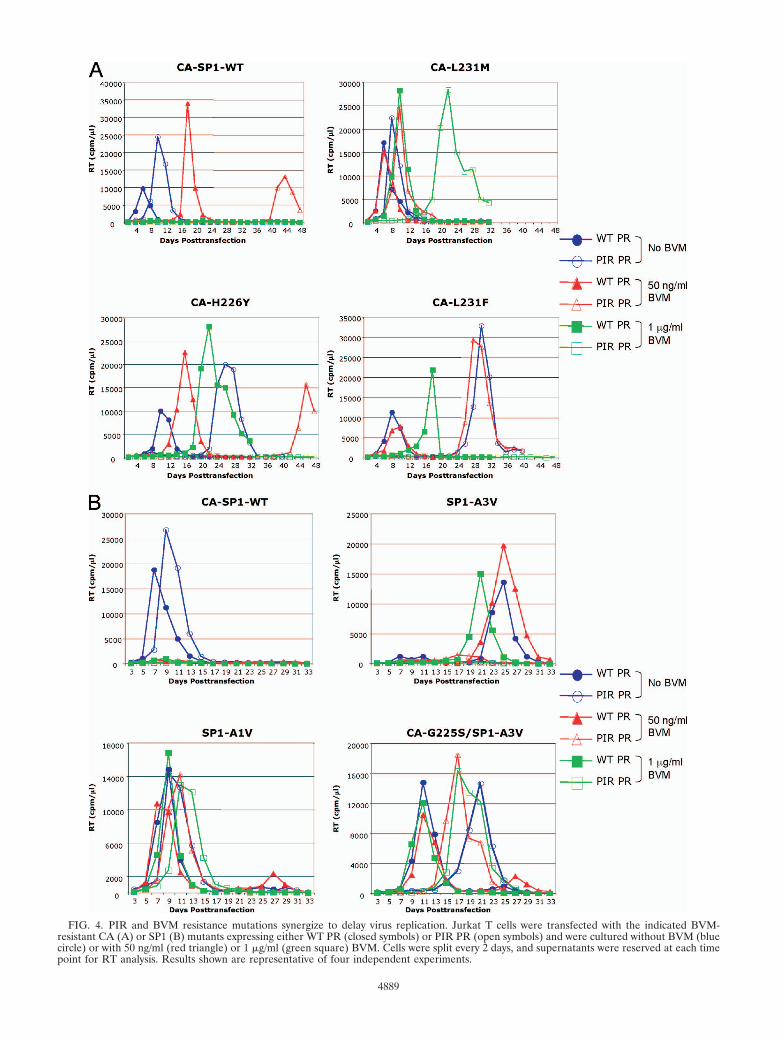
FIG. 4. PIR and BVM resistance mutations synergize to delay virus replication. Jurkat T cells were transfected with the indicated BVM-resistant CA (A) or SP1 (B) mutants expressing either WT PR (closed symbols) or PIR PR (open symbols) and were cultured without BVM (bluecircle) or with 50 ng/ml (red triangle) or 1 �g/ml (green square) BVM. Cells were split every 2 days, and supernatants were reserved at each timepoint for RT analysis. Results shown are representative of four independent experiments.
4889
repassage of A3V PIR PR confirms the previously reportedstability of the PIR mutations (27). Indeed, in the final repeatof the virus replication experiments, we confirmed that all PIRand BVM resistance mutations still were present in all sampleswith detectable viral replication (data not shown). Only oneadditional secondary mutation, CA-V230I, was identified. Thismutation arose in the CA-L231M PIR PR virus that arose 30days posttransfection when cultured at 1 �g/ml BVM. Theadditional characterization of the secondary changes identifiedhere, along with several others associated with SP1-A3V (3 andC. S. Adamson and E. O. Freed, unpublished data), will bereported elsewhere.
Overall, when combined, the BVM resistance and PIR mu-tations acted additively to negatively impact viral replicativecapacity, particularly in the presence of BVM. For example,the combination of a mutation that impaired virus replication(e.g., SP1-A3V) with PIR PR essentially abolished virus repli-cation. The combination of mutations that induced variousdegrees of BVM resistance (e.g., CA-H226Y or L231F) withthe PIR mutations delayed virus replication in the absence ofBVM and at low BVM concentrations and abolished replica-tion at high concentrations of the compound. However, for a
virus that replicated robustly even at a high BVM concentra-tion, such as SP1-A1V, combination with PIR had a minimalimpact on virus replication kinetics.
Emergence of BVM resistance is delayed in the context ofPIR PR. Replication assays showed that the combination of theBVM resistance and PIR mutations generally impaired viralfitness, particularly in the presence of BVM. We thereforehypothesized that the PIR mutations would delay or preventthe emergence of BVM resistance and would limit the numberof distinct BVM resistance mutations that emerge during viruspropagation in BVM. Resistance in Jurkat T cells typicallyemerges 3 to 4 weeks posttransfection with WT pNL4-3 at 50ng/ml BVM (3, 21) (e.g., Fig. 4A). In contrast, in a parallelculture transfected with pNL4-3/PIR, virus replication did notpeak until 44 days posttransfection (Fig. 4A). To determinewhich BVM resistance mutation(s) arose in the context of PIRPR in this culture, genomic DNA was extracted from cellsharvested at peak RT activity, and the PCR-amplified Gag-PRregion was sequenced. This analysis revealed the acquisition ofthe previously characterized mutation SP1-A1V. All five PIRPR mutations were maintained, and no other changes in Gagor PR were observed.
FIG. 5. Replication kinetics of the SP1-A3V mutant coupled with the PIR changes. Jurkat T cells were transfected with WT pNL4-3 (closedsymbols), pNL4-3/PIR, or pNL4-3/A3V PIR. (A) Cultures were maintained either without BVM (blue symbols) or with 1 �g/ml BVM (greensymbols). (B) Cultures were maintained in no BVM (blue circle), 50 ng/ml (red triangle) BVM, or 1 �g/ml (green square) BVM. Cells were splitevery 2 days, and supernatants were reserved at each time point for RT analysis. Virus from the SP1-A3V PIR flask (A) was harvested on day 44and used to infect fresh Jurkat T cells (B). DNA was extracted at peak RT activity, and the Gag-PR coding region was PCR amplified andsequenced.
4890 ADAMSON ET AL. J. VIROL.
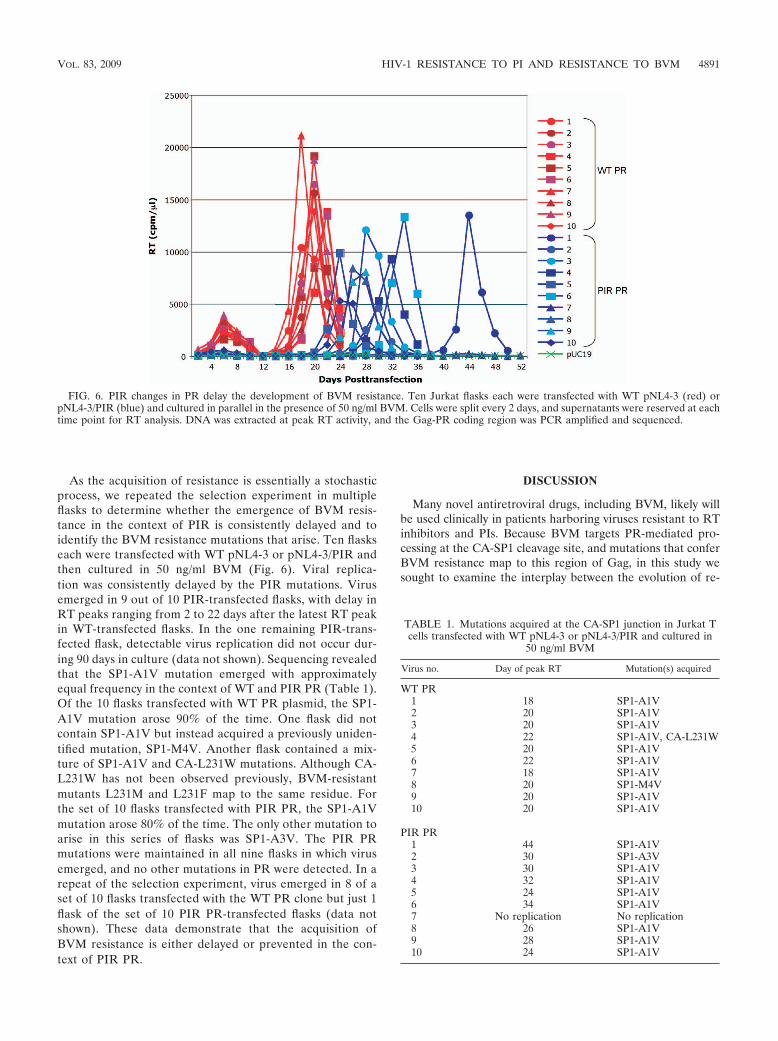
As the acquisition of resistance is essentially a stochasticprocess, we repeated the selection experiment in multipleflasks to determine whether the emergence of BVM resis-tance in the context of PIR is consistently delayed and toidentify the BVM resistance mutations that arise. Ten flaskseach were transfected with WT pNL4-3 or pNL4-3/PIR andthen cultured in 50 ng/ml BVM (Fig. 6). Viral replica-tion was consistently delayed by the PIR mutations. Virusemerged in 9 out of 10 PIR-transfected flasks, with delay inRT peaks ranging from 2 to 22 days after the latest RT peakin WT-transfected flasks. In the one remaining PIR-trans-fected flask, detectable virus replication did not occur dur-ing 90 days in culture (data not shown). Sequencing revealedthat the SP1-A1V mutation emerged with approximatelyequal frequency in the context of WT and PIR PR (Table 1).Of the 10 flasks transfected with WT PR plasmid, the SP1-A1V mutation arose 90% of the time. One flask did notcontain SP1-A1V but instead acquired a previously uniden-tified mutation, SP1-M4V. Another flask contained a mix-ture of SP1-A1V and CA-L231W mutations. Although CA-L231W has not been observed previously, BVM-resistantmutants L231M and L231F map to the same residue. Forthe set of 10 flasks transfected with PIR PR, the SP1-A1Vmutation arose 80% of the time. The only other mutation toarise in this series of flasks was SP1-A3V. The PIR PRmutations were maintained in all nine flasks in which virusemerged, and no other mutations in PR were detected. In arepeat of the selection experiment, virus emerged in 8 of aset of 10 flasks transfected with the WT PR clone but just 1flask of the set of 10 PIR PR-transfected flasks (data notshown). These data demonstrate that the acquisition ofBVM resistance is either delayed or prevented in the con-text of PIR PR.
DISCUSSION
Many novel antiretroviral drugs, including BVM, likely willbe used clinically in patients harboring viruses resistant to RTinhibitors and PIs. Because BVM targets PR-mediated pro-cessing at the CA-SP1 cleavage site, and mutations that conferBVM resistance map to this region of Gag, in this study wesought to examine the interplay between the evolution of re-
FIG. 6. PIR changes in PR delay the development of BVM resistance. Ten Jurkat flasks each were transfected with WT pNL4-3 (red) orpNL4-3/PIR (blue) and cultured in parallel in the presence of 50 ng/ml BVM. Cells were split every 2 days, and supernatants were reserved at eachtime point for RT analysis. DNA was extracted at peak RT activity, and the Gag-PR coding region was PCR amplified and sequenced.
TABLE 1. Mutations acquired at the CA-SP1 junction in Jurkat Tcells transfected with WT pNL4-3 or pNL4-3/PIR and cultured in
50 ng/ml BVM
Virus no. Day of peak RT Mutation(s) acquired
WT PR1 18 SP1-A1V2 20 SP1-A1V3 20 SP1-A1V4 22 SP1-A1V, CA-L231W5 20 SP1-A1V6 22 SP1-A1V7 18 SP1-A1V8 20 SP1-M4V9 20 SP1-A1V10 20 SP1-A1V
PIR PR1 44 SP1-A1V2 30 SP1-A3V3 30 SP1-A1V4 32 SP1-A1V5 24 SP1-A1V6 34 SP1-A1V7 No replication No replication8 26 SP1-A1V9 28 SP1-A1V10 24 SP1-A1V
VOL. 83, 2009 HIV-1 RESISTANCE TO PI AND RESISTANCE TO BVM 4891

sistance to BVM and PIR mutations in PR. We demonstratethat the emergence of BVM resistance is delayed in the contextof the PIR PR, which induces a general Gag processing defectindependently of BVM resistance mutations. The SP1-A1Vmutation arises most frequently in clones expressing either WTor PIR PR. Combining our previously characterized panel ofBVM resistance changes with PIR mutations led in most casesto severely inhibited virus replication, particularly in the pres-ence of BVM. However, in the case of SP1-A1V, we observedno significant effect of the PIR mutations on virus replication,even at high BVM concentrations.
Mutations in PR could influence the emergence of BVMresistance primarily by two mechanisms: (i) the ability of themutant PR to cleave at a CA-SP1 junction containing BVMresistance changes could be altered, and/or (ii) the PR muta-tions could reduce the overall rate of virus replication, therebylimiting the ability of BVM resistance to emerge. In the latterinstance, the mechanism is not due to the nature of the PRmutations per se but rather their overall deleterious effect onreplication, therefore a similar phenomenon might also beobserved for other mutants that display a reduced replicationcapacity. We chose the L10R/M46I/L63P/V82T/I84V PIRclone for this study because it has been reported to replicatewith essentially WT kinetics (27). However, we repeatedly ob-served a slight delay in the replication of this clone relative tothat of the WT, most likely due to the above-mentioned gen-eral Gag processing defects. Despite this slight replication de-fect, we observed that the PIR mutations were very stableduring long-term culture in Jurkat T cells; in no case did weobserve the loss of any of these PIR mutations or the acquisi-tion of other mutations in PR. The stability of the PIR muta-tions upon long-term culture obviated the need for the inclu-sion of PIs in the BVM selection experiments. The overallreduction in the replication capacity of the PIR clone likelycontributed to the delayed emergence of BVM resistance inthe context of PIR PR. To the extent that PIR mutationscirculating in patients reduce replication fitness, these findingsmay be relevant to the potential emergence of BVM resistancein vivo.
In both this and our previous study (3), we have noted thefrequent emergence of the SP1-A1V mutation. This can beexplained by the robust replication kinetics of SP1-A1V in thecontext of either WT or PIR PR even at high BVM concen-trations. In contrast, the combination of the other BVM resis-tance mutations with PIR PR significantly decreased viral rep-lication capacity, particularly in the presence of BVM. Forexample, the replication of viral clones containing the CAmutations (H226Y, L231M, and L231F) in combination withthe PIR changes was significantly delayed or abolished in thepresence of BVM. This is likely due to high CA-SP1 levelsresulting from the general Gag processing defects induced byPIR mutations, combined with only a partial degree of BVMresistance being conferred by the CA mutations (3, 46, 49).Interestingly, a CA-V230I mutation emerged upon the passageof CA-L231M with PIR PR at 1 �g/ml BVM. This secondarymutation may compensate for the replication delay induced bythe PIR changes. It is well documented that mutations in Gagcleavage sites can accumulate during the acquisition of resis-tance to PIs (17, 23, 25, 30, 45). However, these mutations arerarely seen at the CA-SP1 site (23, 25). The residues to which
BVM resistance map are highly conserved (3), and to date onlyCA-L231M has been reported in the context of one PI-expe-rienced patient sample (24). However, the CA-V230I mutationhas been reported in �10% of both PI-experienced and treat-ment-naïve patients (23, 24, 34). It also is possible that CA-V230I confers a degree of BVM resistance. Residue CA-230occurs at position P2 of the PR recognition site, and the in-sertion of the P2 and P1 residues from SIVmac, which is BVMresistant, into HIV-1 resulted in the transfer of BVM resis-tance to HIV-1 (47). This study identified two additional novelmutations that may confer BVM resistance: CA-L231W andSP1-M4V.
The additive effect of combining an already replication-im-paired virus, such as SP1-A3V, with PIR PR essentially abol-ished virus replication. We previously reported that the SP1-A3V mutant in the context of WT PR exhibits a degree ofBVM dependence at 1 �g/ml BVM and generally requires asecondary mutation to facilitate replication at low BVM con-centrations or in the absence of the compound (3). In thisstudy, we observed two mutations that may rescue the replica-tion of the SP1-A3V PIR clone, SP1-V7I and a mutation distalto the CA-SP1 cleavage site, CA-N203T. The significance ofthe distal CA-N203T mutation has not been established; how-ever, it is unlikely to confer BVM resistance, as all previouslyidentified BVM resistance mutations cluster to the CA-SP1junction. It is plausible that the CA-N203T mutation compen-sates for defects imposed by SP1-A3V and/or the PIR muta-tions; however, this remains to be determined. SP1-V7I previ-ously has been identified during the propagation of SP1-A3Vin the context of WT PR (3), therefore its emergence is un-likely to be a direct consequence of the PIR mutations. Wepreviously detected a number of secondary mutations associ-ated with SP1-A3V ([3] and Adamson and Freed, unpub-lished). The characterization of these secondary mutations willbe reported elsewhere, but they are of interest because theymap to nonconserved residues at the periphery of the CA-SP1junction, and polymorphisms at these amino acid positionsappear to be correlated with variable clinical responses toBVM (K. Salzwedel, unpublished data).
This study demonstrates that the emergence of BVM resis-tance is delayed or prevented in the context of a PIR PRcompared to that in the context of WT PR. The reducedreplication fitness of mutants bearing substitutions in both theCA-SP1 region and in PR likely is due to a slight replicationimpairment of the PIR clone and because the majority of BVMresistance mutations act additively with PIR changes to inhibitvirus replication. Understanding the nature of BVM resistancearising in the context of both WT and PIR PR elucidates theinterplay between these two sets of mutations and may help topredict the types of mutations that are likely to arise in vivo.We anticipate that the SP1-A1V substitution is the most likelyto emerge in vivo, as this mutant replicates robustly indepen-dently of PR mutations or BVM. Because an Ala at SP1 res-idue 1 is highly conserved among HIV-1 isolates, it might bepredicted that a fitness cost would be associated with the rep-lication of this virus in vivo. However, we observe that SP1-A1V can replicate efficiently in primary lymphocytes and mac-rophages (C. S. Adamson, S. D. Ablan, and E. O. Freed,unpublished data), and this mutant confers resistance to BVMand replicates with no fitness impairment in SCID-hu Thy/Liv
4892 ADAMSON ET AL. J. VIROL.

mice (36). The SP1-A1V mutation also has been observed invirus from 2 of 46 patients participating in the phase IIb clinicaltrials (K. Salzwedel, unpublished data). Ongoing clinical trialswith BVM ultimately will allow comparisons to be made be-tween the evolution of BVM resistance in vitro and in vivo.
ACKNOWLEDGMENTS
We thank members of the Freed laboratory for critical reviews of themanuscript. We thank M. Fivash for assistance with statistical analysis.HIV-Ig and the PIR plasmid construct were obtained through theAIDS Research and Reference Reagent Program, Division of AIDS,NIAID, NIH.
This research was supported by the Intramural Research Programof the NIH, National Cancer Institute, Center for Cancer Research,and by the Intramural AIDS Targeted Antiviral Program.
REFERENCES
1. Accola, M. A., S. Hoglund, and H. G. Gottlinger. 1998. A putative alpha-helical structure which overlaps the capsid-p2 boundary in the human im-munodeficiency virus type 1 Gag precursor is crucial for viral particle assem-bly. J. Virol. 72:2072–2078.
2. Adachi, A., H. E. Gendelman, S. Koenig, T. Folks, R. Willey, A. Rabson, andM. A. Martin. 1986. Production of acquired immunodeficiency syndrome-associated retrovirus in human and nonhuman cells transfected with aninfectious molecular clone. J. Virol. 59:284–291.
3. Adamson, C. S., S. D. Ablan, I. Boeras, R. Goila-Gaur, F. Soheilian, K.Nagashima, F. Li, K. Salzwedel, M. Sakalian, C. T. Wild, and E. O. Freed.2006. In vitro resistance to the human immunodeficiency virus type 1 mat-uration inhibitor PA-457 (bevirimat). J. Virol. 80:10957–10971.
4. Adamson, C. S., and E. O. Freed. 2007. Human immunodeficiency virus type1 assembly, release, and maturation. Adv. Pharmacol. 55:347–387.
5. Adamson, C. S., and E. O. Freed. 2008. Recent progress in antiretrovirals—lessons from resistance. Drug Discov. Today 13:424–432.
6. Aiken, C., and C. H. Chen. 2005. Betulinic acid derivatives as HIV-1 antivi-rals. Trends Mol. Med. 11:31–36.
7. Condra, J. H. 1998. Resistance to HIV protease inhibitors. Haemophilia4:610–615.
8. Condra, J. H., W. A. Schleif, O. M. Blahy, L. J. Gabryelski, D. J. Graham,J. C. Quintero, A. Rhodes, H. L. Robbins, E. Roth, M. Shivaprakash, et al.1995. In vivo emergence of HIV-1 variants resistant to multiple proteaseinhibitors. Nature 374:569–571.
9. DaFonseca, S., A. Blommaert, P. Coric, S. S. Hong, S. Bouaziz, and P.Boulanger. 2007. The 3-O-(3�,3�-dimethylsuccinyl) derivative of betulinicacid (DSB) inhibits the assembly of virus-like particles in HIV-1 Gag pre-cursor-expressing cells. Antivir. Ther. 12:1185–1203.
10. Emini, E. A., and H. Y. Fan. 1997. Immunological and pharmacologicalapproaches to the control of retroviral infections. In J. M. Coffin, S. H.Hughes, and H. E. Varmus (ed.), Retroviruses. Cold Spring Harbor Labo-ratory Press, Cold Spring Harbor, NY.
11. Erickson-Viitanen, S., J. Manfredi, P. Viitanen, D. E. Tribe, R. Tritch, C. A.Hutchison III, D. D. Loeb, and R. Swanstrom. 1989. Cleavage of HIV-1 gagpolyprotein synthesized in vitro: sequential cleavage by the viral protease.AIDS Res. Hum. Retrovir. 5:577–591.
12. Freed, E. O. 1998. HIV-1 gag proteins: diverse functions in the virus lifecycle. Virology 251:1–15.
13. Freed, E. O., and M. A. Martin. 1994. Evidence for a functional interactionbetween the V1/V2 and C4 domains of human immunodeficiency virus type1 envelope glycoprotein gp120. J. Virol. 68:2503–2512.
14. Fujioka, T., Y. Kashiwada, R. E. Kilkuskie, L. M. Cosentino, L. M. Ballas,J. B. Jiang, W. P. Janzen, I. S. Chen, and K. H. Lee. 1994. Anti-AIDS agents.11. Betulinic acid and platanic acid as anti-HIV principles from Syzigiumclaviflorum, and the anti-HIV activity of structurally related triterpenoids. J.Nat. Prod. 57:243–247.
15. Kanamoto, T., Y. Kashiwada, K. Kanbara, K. Gotoh, M. Yoshimori, T. Goto,K. Sano, and H. Nakashima. 2001. Anti-human immunodeficiency virusactivity of YK-FH312 (a betulinic acid derivative), a novel compound block-ing viral maturation. Antimicrob. Agents Chemother. 45:1225–1230.
16. Kaplan, A. H., J. A. Zack, M. Knigge, D. A. Paul, D. J. Kempf, D. W.Norbeck, and R. Swanstrom. 1993. Partial inhibition of the human immu-nodeficiency virus type 1 protease results in aberrant virus assembly and theformation of non-infectious particles. J. Virol. 67:4050–4055.
17. Kaufmann, G. R., K. Suzuki, P. Cunningham, M. Mukaide, M. Kondo, M.Imai, J. Zaunders, and D. A. Cooper. 2001. Impact of HIV type 1 protease,reverse transcriptase, cleavage site, and p6 mutations on the virologicalresponse to quadruple therapy with saquinavir, ritonavir, and two nucleosideanalogs. AIDS Res. Hum. Retrovir. 17:487–497.
18. Kiernan, R. E., A. Ono, G. Englund, and E. O. Freed. 1998. Role of matrix
in an early postentry step in the human immunodeficiency virus type 1 lifecycle. J. Virol. 72:4116–4126.
19. Krausslich, H. G., M. Facke, A. M. Heuser, J. Konvalinka, and H. Zentgraf.1995. The spacer peptide between human immunodeficiency virus capsid andnucleocapsid proteins is essential for ordered assembly and viral infectivity.J. Virol. 69:3407–3419.
20. Krausslich, H. G., H. Schneider, G. Zybarth, C. A. Carter, and E. Wimmer.1988. Processing of in vitro-synthesized gag precursor proteins of humanimmunodeficiency virus (HIV) type 1 by HIV proteinase generated in Esch-erichia coli. J. Virol. 62:4393–4397.
21. Li, F., R. Goila-Gaur, K. Salzwedel, N. R. Kilgore, M. Reddick, C. Matal-lana, A. Castillo, D. Zoumplis, D. E. Martin, J. M. Orenstein, G. P. Allaway,E. O. Freed, and C. T. Wild. 2003. PA-457: a potent HIV inhibitor thatdisrupts core condensation by targeting a late step in Gag processing. Proc.Natl. Acad. Sci. USA 100:13555–13560.
22. Li, F., D. Zoumplis, C. Matallana, N. R. Kilgore, M. Reddick, A. S. Yunus,C. S. Adamson, K. Salzwedel, D. E. Martin, G. P. Allaway, E. O. Freed, andC. T. Wild. 2006. Determinants of activity of the HIV-1 maturation inhibitorPA-457. Virology 356:217–224.
23. Malet, I., B. Roquebert, C. Dalban, M. Wirden, B. Amellal, R. Agher, A.Simon, C. Katlama, D. Costagliola, V. Calvez, and A. G. Marcelin. 2007.Association of Gag cleavage sites to protease mutations and to virologicalresponse in HIV-1 treated patients. J. Infect. 54:367–374.
24. Malet, I., M. Wirden, A. Derache, A. Simon, C. Katlama, V. Calvez, and A. G.Marcelin. 2007. Primary genotypic resistance of HIV-1 to the maturationinhibitor PA-457 in protease inhibitor-experienced patients. AIDS 21:871–873.
25. Mammano, F., C. Petit, and F. Clavel. 1998. Resistance-associated loss ofviral fitness in human immunodeficiency virus type 1: phenotypic analysis ofprotease and gag coevolution in protease inhibitor-treated patients. J. Virol.72:7632–7637.
26. Martin, D. E., R. Blum, J. Wilton, J. Doto, H. Galbraith, G. L. Burgess, P. C.Smith, and C. Ballow. 2007. Safety and pharmacokinetics of bevirimat (PA-457), a novel inhibitor of human immunodeficiency virus maturation, inhealthy volunteers. Antimicrob. Agents Chemother. 51:3063–3066.
27. Martinez-Picado, J., A. V. Savara, L. Sutton, and R. T. D’Aquila. 1999.Replicative fitness of protease inhibitor-resistant mutants of human immu-nodeficiency virus type 1. J. Virol. 73:3744–3752.
28. Menendez-Arias, L., M. A. Martinez, M. E. Quinones-Mateu, and J. Mar-tinez-Picado. 2003. Fitness variations and their impact on the evolution ofantiretroviral drug resistance. Curr. Drug Targets Infect. Disord. 3:355–371.
29. Mervis, R. J., N. Ahmad, E. P. Lillehoj, M. G. Raum, F. H. Salazar, H. W.Chan, and S. Venkatesan. 1988. The gag gene products of human immuno-deficiency virus type 1: alignment within the gag open reading frame, iden-tification of posttranslational modifications, and evidence for alternative gagprecursors. J. Virol. 62:3993–4002.
30. Myint, L., M. Matsuda, Z. Matsuda, Y. Yokomaku, T. Chiba, A. Okano, K.Yamada, and W. Sugiura. 2004. Gag non-cleavage site mutations contributeto full recovery of viral fitness in protease inhibitor-resistant human immu-nodeficiency virus type 1. Antimicrob. Agents Chemother. 48:444–452.
31. Pettit, S. C., M. D. Moody, R. S. Wehbie, A. H. Kaplan, P. V. Nantermet,C. A. Klein, and R. Swanstrom. 1994. The p2 domain of human immuno-deficiency virus type 1 Gag regulates sequential proteolytic processing and isrequired to produce fully infectious virions. J. Virol. 68:8017–8027.
32. Sakalian, M., C. P. McMurtrey, F. J. Deeg, C. W. Maloy, F. Li, C. T. Wild,and K. Salzwedel. 2006. 3-O-(3�,3�-dimethysuccinyl) betulinic acid inhibitsmaturation of the human immunodeficiency virus type 1 gag precursor as-sembled in vitro. J. Virol. 80:5716–5722.
33. Salzwedel, K., D. E. Martin, and M. Sakalian. 2007. Maturation inhibitors:a new therapeutic class targets the virus structure. AIDS Rev. 9:162–172.
34. Shelburne, S. A., III, R. J. Hamill, M. C. Rodriguez-Barradas, S. B. Green-berg, R. L. Atmar, D. W. Musher, J. C. Gathe, Jr., F. Visnegarwala, andB. W. Trautner. 2002. Immune reconstitution inflammatory syndrome: emer-gence of a unique syndrome during highly active antiretroviral therapy.Medicine (Baltimore) 81:213–227.
35. Smith, P. F., A. Ogundele, A. Forrest, J. Wilton, K. Salzwedel, J. Doto, G. P.Allaway, and D. E. Martin. 2007. Phase I and II study of the safety, virologiceffect, and pharmacokinetics/pharmacodynamics of single-dose 3-o-(3�,3�-dimethylsuccinyl)betulinic acid (bevirimat) against human immunodefi-ciency virus infection. Antimicrob. Agents Chemother. 51:3574–3581.
36. Stoddart, C. A., P. Joshi, B. Sloan, J. C. Bare, P. C. Smith, G. P. Allaway,C. T. Wild, and D. E. Martin. 2007. Potent activity of the HIV-1 maturationinhibitor bevirimat in SCID-hu Thy/Liv mice. PLoS ONE 2:e1251.
37. Temesgen, Z., D. Warnke, and M. J. Kasten. 2006. Current status of anti-retroviral therapy. Expert Opin. Pharmacother. 7:1541–1554.
38. Tritch, R. J., Y. E. Cheng, F. H. Yin, and S. Erickson-Viitanen. 1991. Mu-tagenesis of protease cleavage sites in the human immunodeficiency virustype 1 gag polyprotein. J. Virol. 65:922–930.
39. Vogt, V. M. 1996. Proteolytic processing and particle maturation. Curr. Top.Microbiol. Immunol. 214:95–131.
40. Wiegers, K., G. Rutter, H. Kottler, U. Tessmer, H. Hohenberg, and H. G.Krausslich. 1998. Sequential steps in human immunodeficiency virus particle
VOL. 83, 2009 HIV-1 RESISTANCE TO PI AND RESISTANCE TO BVM 4893

maturation revealed by alterations of individual Gag polyprotein cleavagesites. J. Virol. 72:2846–2854.
41. Willey, R. L., J. S. Bonifacino, B. J. Potts, M. A. Martin, and R. D. Klausner.1988. Biosynthesis, cleavage, and degradation of the human immunodefi-ciency virus 1 envelope glycoprotein gp160. Proc. Natl. Acad. Sci. USA85:9580–9584.
42. Wlodawer, A., and J. W. Erickson. 1993. Structure-based inhibitors of HIV-1protease. Annu. Rev. Biochem. 62:543–585.
43. Wlodawer, A., and J. Vondrasek. 1998. Inhibitors of HIV-1 protease: a majorsuccess of structure-assisted drug design. Annu. Rev. Biophys. Biomol.Struct. 27:249–284.
44. Yu, D., C. T. Wild, D. E. Martin, S. L. Morris-Natschke, C. H. Chen, G. P.Allaway, and K. H. Lee. 2005. The discovery of a class of novel HIV-1maturation inhibitors and their potential in the therapy of HIV. Expert Opin.Investig. Drugs 14:681–693.
45. Zhang, Y. M., H. Imamichi, T. Imamichi, H. C. Lane, J. Falloon, M. B.Vasudevachari, and N. P. Salzman. 1997. Drug resistance during indinavir
therapy is caused by mutations in the protease gene and in its Gag substratecleavage sites. J. Virol. 71:6662–6670.
46. Zhou, J., C. H. Chen, and C. Aiken. 2006. Human immunodeficiency virustype 1 resistance to the small molecule maturation inhibitor 3-O-(3�,3�-dimethylsuccinyl)-betulinic acid is conferred by a variety of single amino acidsubstitutions at the CA-SP1 cleavage site in Gag. J. Virol. 80:12095–12101.
47. Zhou, J., C. H. Chen, and C. Aiken. 2004. The sequence of the CA-SP1junction accounts for the differential sensitivity of HIV-1 and SIV to thesmall molecule maturation inhibitor 3-O-{3�,3�-dimethylsuccinyl}-betulinicacid. Retrovirology 1:15.
48. Zhou, J., L. Huang, D. L. Hachey, C. H. Chen, and C. Aiken. 2005. Inhibitionof HIV-1 maturation via drug association with the viral Gag protein inimmature HIV-1 particles. J. Biol. Chem. 280:42149–42155.
49. Zhou, J., X. Yuan, D. Dismuke, B. M. Forshey, C. Lundquist, K. H. Lee, C.Aiken, and C. H. Chen. 2004. Small-molecule inhibition of human immuno-deficiency virus type 1 replication by specific targeting of the final step ofvirion maturation. J. Virol. 78:922–929.
4894 ADAMSON ET AL. J. VIROL.





























