Embed Size (px)
Citation preview

ORIGINAL ARTICLE
Long-term observational, non-randomized study of enzymereplacement therapy in late-onset glycogenosis type II
Bruno Bembi & Federica Edith Pisa & Marco Confalonieri & Giovanni Ciana &
Agata Fiumara & Rossella Parini & Miriam Rigoldi & Arrigo Moglia & Alfredo Costa &
Annalisa Carlucci & Cesare Danesino & Maria Gabriela Pittis & Andrea Dardis &
Sabrina Ravaglia
Received: 16 June 2010 /Revised: 11 August 2010 /Accepted: 24 August 2010# SSIEM and Springer 2010
AbstractObjectives Type II glycogenosis (GSDII) is a lysosomalstorage disorder due to acid alpha-glucosidase (GAA)deficiency. Enzyme replacement therapy (ERT) withhuman recombinant alpha-glucosidase (rhGAA) has beendemonstrated to be effective in the treatment of infantileforms of GSDII, but little information is availableconcerning late-onset phenotypes. Long-term follow-upstudies are not available at present. The aim of this studywas to evaluate the ERT long-term effects in late-onsetGSDII.Methods Twenty-four patients, including 7 juveniles and 17adults, received bi-weekly infusion of rhGAA (20 mg/kg)for at least 36 months. Clinical conditions, muscularfunction (6-min walking test, 6MWT; Walton scale, WS),
respiratory function (vital capacity, VC; forced expiratoryvolume, FEV1; arterial pCO2), and muscle enzymes wereassessed every 6 months.Results The 6MWT improved in both juvenile and adultpatients (p=0.01, p=0.0002, respectively), as well as inpatients with moderate to severe muscle function impair-ment (WS>3.5; p=0.002). An overall improvement in WSwas also observed (p=0.0003). VC and FEV1 remainedunchanged, while pCO2 decreased (p=0.017). Muscleenzymes decreased significantly (p<0.0001). Two patients(8%) showed transient secondary events during ERT.Conclusions Long-term ERT with rhGAA was shown tobe safe, well tolerated, and effective in improving motorfunction and in stabilizing respiratory function in late-onset GSDII. The response pattern showed a progressive
Communicated by: Ed Wraith
Competing interest: None declared.
B. Bembi (*) :G. Ciana :A. DardisRegional Coordination Centre for Rare Diseases, UniversityHospital Santa Maria della Misericordia,Piazzale Santa Maria della Misericordia 15,33100 Udine, Italye-mail: [email protected]
F. E. PisaInstitute of Hygiene and Clinical Epidemiology, UniversityHospital Santa Maria della Misericordia,Udine, Italy
M. ConfalonieriPulmonary Unit, University Hospital of Trieste,Trieste, Italy
A. FiumaraPaediatric Clinic, University of Catania,Catania, Italy
R. Parini :M. RigoldiMetabolic Unit, Paediatric Department, San Gerardo Hospital,Monza, Italy
A. Moglia :A. Costa : S. RavagliaDepartment of Neurological Sciences, University of Pavia,Pavia, Italy
A. CarlucciRespiratory Intensive Care Unit, IRCCS Fondazione Maugeri,Pavia, Italy
C. DanesinoMedical Genetics, University of Pavia,Pavia, Italy
M. G. PittisMetabolic Unit, I.R.C.C.S. Burlo Garofolo of Trieste,Trieste, Italy
J Inherit Metab DisDOI 10.1007/s10545-010-9201-8

clinical improvement during the follow-up period injuvenile patients, while in adults it reached and main-tained a plateau after the first year of treatment.
Introduction
Type II glycogenosis (GSDII; OMIM: 232300) is anautosomal recessive lysosomal storage disorder (LSD) dueto lysosomal acid alpha-glucosidase (GAA) deficiency thatcauses progressive glycogen storage in muscle tissue (vander Ploeg et al. 2008). The GAA gene has been localized tochromosome 17q25. A large number of mutations havebeen described to date (http://www2.eur.nl/fgg/ch1/pompe),the majority being private. Mutation c-32-13 T>G affectsabout 40% of alleles in late-onset phenotypes (Montalvo etal. 2006; Joshi et al. 2008).
The clinical manifestations of the disease encompass acontinuous spectrum of phenotypes, ranging from rapidlyprogressive infantile forms to slowly progressive late-onset disease (Kishnani et al. 2006; Hagemans et al.2005).
The classic infantile phenotype is characterized bycardiomyopathy, muscle weakness, and respiratory failure,leading to death before the age of 2 years (Kishnani et al.2006). The late-infantile form, also presenting with cardio-myopathy, begins later during the first year of life andshows a slower course (Winkel et al. 2005).
Late-onset phenotypes show progressive involvementof proximal muscles and variable degrees of respiratoryinsufficiency, without cardiac involvement. During dis-ease progression, patients usually require walking devi-ces, wheelchairs, and respiratory support. Death mayoccur from early childhood to late adulthood (Hagemanset al. 2005). The incidence of late-onset forms is about1:57,000, with geographic and ethnic variation (Bembi etal. 2008). Until recently, treatment had been limited tophysical therapy and high-protein diets (Slonim et al.2007).
In 1999, a rabbit milk–derived human recombinant GAA(rhGAA) became available for enzyme replacement therapy(ERT). The first clinical trial in infantile patients showed areduction in heart hypertrophy, improvement in pulmonaryand cardiac function, and in motility and muscle strength(van den Hout et al. 2004). These results were confirmed byrecent studies using rhGAA derived from Chinese hamsterovary cells (CHO-rhGAA) (Kishnani et al. 2006). ERTefficacy in the infantile form has been related to an earlystart of therapy and the presence of residual enzyme activity(van den Hout et al. 2004). These successful results of ERTled to the approval of CHO-rhGAA (Myozyme, Genzyme)commercialization within the European Union in March2006.
To date, only a limited number of trials including late-onset phenotypes have been published (Strothotte et al.2010; Merk et al. 2009; Ravaglia et al. 2009). Very recently,van der Ploeg et al. described the results of a 78-weekfollow-up of 90 late-onset GSDII patients randomized toreceive ERT (60 patients) or placebo (30 patients) (van derPloeg et al. 2010). The treated group was found to obtain asignificant motor improvement and a stabilization ofrespiratory function. Although this study includes thelargest case series analyzed to date and is the onlyplacebo-controlled study, long-term treatment effectivenesswas not addressed.
Materials and methods
Patients
Patients between the age of 7 and 65 years were eligible for thestudy if they had objective muscle weakness or respiratoryfunctional impairment: Walton scale (WS)!1(Slonim et al.2007; Table 1), decreased vital capacity (VC)"80%, as wellas GAA deficiency (blood lymphocytes, fibroblasts, musclebiopsy). Diagnosis of GSDII was confirmed by molecularanalysis. Exclusion criteria were age!65 years, presence ofco-morbidities that may influence the outcome measures, andconditions that could determine adverse immunologicalreactions. Juvenile and adult phenotypes were definedaccording to the age at the onset of the first symptoms:juvenile"16 years, adult>16 years.
Study protocol and consent forms were approved by theEthics Committee of each center. All participants providedwritten informed consent.
Study design
A multicenter, observational, prospective, nonrandomized,open-label study was designed. The study was supported bythe Agenzia Italiana del Farmaco (AIFA). The recruitmentperiod lasted from November 2005 to November 2006.Patients received 20 mg/kg i.v. of CHO-rhGAA (Myozyme,Genzyme) every 14±4 days. The following parameterswere assessed at baseline, then every 6 months: motorfunction by 6-min walking test (6MWT) (Enright et al.1998; Geiger et al. 2007) and WS; respiratory function, VC,and forced expiratory volume (FEV1) by standard spirometry(standing position); morning arterial pCO2; serum muscleenzymes (CK, LDH, AST, ALT); and body mass index(BMI). EKG; echocardiography; chest, column, andmuscle imaging; and audiometric evaluation wereassessed at the time of inclusion and then yearly. During6MWT, patients were not allowed to use any walkingassistance devices.
J Inherit Metab Dis

Statistical analysis
Descriptive statistics (mean, standard deviation, median,interquartile range) were used to describe the patients’demographic and clinical characteristics at baseline andoutcome parameters at different evaluation times.
The nonparametric Friedman test was used to assessdifferences in rank distribution across repeated measures ofnonnormal continuous and ordinal variables (6MWT, VC,FEV1, pCO2, CK, LDH, AST, ALT).
To evaluate whether the variation in 6MWT was modifiedby the degree of muscle function impairment at baseline, thepatients were stratified into two groups, the first presentingwith WS at T0 below the median value and the secondpresenting with WS at T0 equal to or higher than the medianvalue. Wilcoxon rank sum test was used to evaluate if therewas a significant difference between the WS median at T0 inadult and juvenile patients. Since this difference was signifi-cant, the cut off used was theWSmedian at T0 for each group.Wilcoxon rank sum test was also used for the comparison of6MWT at T0 between juvenile and adult patients.
The difference between binomial proportions (percentageof patients reporting headache or muscle pain) was comparedthrough the exact McNemar test. Correlation betweenmeasures of motor function (6MWT and WS) and ofrespiratory function (VC) was assessed with the Spearmancorrelation coefficient. All reported p values are two-tailed.Analyses were conducted with SAS software (SAS Institute,Cary, NC, USA) (Lehmann 2006; Van Der Laan et al. 1987).
Results
Baseline assessment
Demographics and clinical characteristics of the enrolledpatients are reported in Table 2. Seven were juvenile
patients (29.2%; 2 females, 5 males) and 17 were adults(70.8%; 8 females, 9 males).
The juvenile patients showed the first symptoms at amean age of 2.5 (± 1.3) years and were diagnosed at a meanage of 2.8 (± 1.4) years, while adults showed the firstsymptoms at a mean age of 26.6 (± 12.8) years and werediagnosed at a mean age of 34.5 (± 14.9) years. Juvenilepatients started ERT at a mean age of 12.0 (± 3.3) years andadults at a mean age of 47.6 (± 10.7) years. Mean BMI was18.0 (± 6.4) for juvenile and 22.3 (± 4.9) kg/m2 for adultpatients. Three adults and one juvenile had low BMI(range: 12.54–14.53 kg/m2); two of them complained ofdysphagia (one needed percutaneous enteral gastrostomy);five adults showed a BMI>25 kg/m2.
Median value of WS was 2 (IQR 0–3.5) in juveniles and 5(3–6) in adults; Wilcoxon rank sum exact p for comparisonof WS between juvenile and adult patients was 0.0357 (datanot shown). Three juvenile and eight adults showed a WSbelow the median. At baseline, eight patients (five juvenile,three adults) showed normal or mildly impaired musclefunction (WS"2); seven adults had moderate motordifficulties (WS 2.5–5: inability to rise from a chair andclimb stairs, use of banister), and nine patients (twojuvenile, seven adults) had severe motor dysfunction(WS!6: walk with calipers or other aids) (Table 3).
The prominent motor impairment of the adult populationcompared to the juvenile group is also reflected by theirperformance in the 6MWT: median value of 6MWT was572.9 m (IQR 104.0–616.9) in juveniles and 116.6 m(40.0–411.5) in adults. Wilcoxon rank sum exact p forcomparison of 6MWT between juveniles and adults was0.0216 (data not shown). All patients except one wheelchairbound adult female were able to perform the 6MWT atbaseline; however, nine patients (one juvenile and eightadults) including four who were tracheostomized, interrup-ted the test before 6 min had passed.
Thirteen patients (54.1%) required ventilatory supportthrough tracheostomy (1 juvenile, 3 adults) or throughnoninvasive mask ventilation (1 juvenile, 8 adults). MeanVC% was 83±45% in juveniles and 52±26% in adults.
A positive correlation between motor impairment(6MWT) and respiratory dysfunction (VC) was found(rho=0.74; p<0.0001), while a negative correlation wasevidenced between WS and VC (rho=#0.73; p<0.0001).Mean VC was 96.5% (±22.7) for the WS"2 group ofpatients, 50.1% (±24.3) for the WS 3–5 group, and 27.4%(±12.7) for the WS!6 (data not shown).
At baseline, 17 patients (70.8%) presented with elevatedCK, LDH, and ALT, while 18 had high AST. Levels ofmuscle enzymes were significantly higher in the juvenilegroup compared to adults (Wilcoxon rank sum exact pvalues were as follows: ALT p=0.0008, AST p=0.0003,CK p=0.0042, LDH p=0.0010).
Table 1 Walton scale items (according to Slonim et al. 2007)
Grade ofseverity
Description
0 All activities normal
1 Walks normal; unable to run freely
2 Defect in posture/gait
2.5 Sometimes uses the bannister to climb stairs
3 Stairs only with bannister
3.5 Sometimes unable to climb stairs with bannister
4 Walks without assistance; unable to climb stairs
5 Walks without assistance; unable to rise from a chair
6 Walks only with calipers or other aids
7 Wheelchair bound
J Inherit Metab Dis

Other clinical symptoms included 8 patients (33.3 %)reporting headache and 11 (45.8 %) with muscle pain. Fouradult patients showed cardiac abnormalities: two withmitral prolapse and two with mild septum hypertrophy;cardiac function was normal in all of them. One juvenilepatient showed Achilles tendon retraction.
The patients’ enzymatic residual activity ranged from 2.8to 10.0% of normal values.
Effects of ERT on WS and 6MWT
Table 3 presents WS modification during ERT. Thevariation in WS was significant (p=0.0003) at T36, with a
decrease in the median value from 3.75 (range 2.0–6.0) to2.0 (range 1.0–6.0). In particular, six patients (25.0%), fiveadults and one juvenile, stabilized to a less severe WSscore. Four of them (three with WS 2.5–5, one with WS!6)shifted to a WS score"2, while the other two shifted fromWS!6 to WS 2.5–5. At intermediate follow-up periods, theWS variation was significant between T0 and T12 (p=0.0096)and T0 and T24 (p=0.0010), but not between the otherintervals (T12 vs. T24; T24 vs. T36). Only two adulttracheostomized patient worsened their WS score during thethird year of therapy, shifting from WS 5 to WS 6.
Table 3 shows the influence of disease severity at T0 onwalking capacity. While the median 6MWT among patients
Table 2 Demographic and clinical characteristics of the patients (n=24) at baseline
Data are n (%), means (SD) or medians (IQR); * GAA activity is expressed as % of normal value
Phenotypes
Juvenile
(N=7, 29.2%)
Adult
(N=17, 70.8%)
Gender (n and %)
Female 2 28.6 8 47.1
Male 5 71.4 9 52.9
Age at first symptoms (yr): mean + SD 2.5 ± 1.3 26.6 ± 12.8
Age at diagnosis (yr): mean + SD 2.8 ± 1.4 34.5 ± 14.9
Age at starting ERT (yr): mean + SD 12.0 ± 3.3 47.6 ± 10.7
BMI - kg/m2 - mean + SD 18.0 ± 6.4 22.3 ± 4.9
Walton score (WS): median (IQR) 2 (0 -3.5) 5 (3-6)
Patients with WS at T0 < median 3 42.9 8 47.1
Patients with WS at T0 4 57.1 9 52.9
6MWT (m): median (IQR) 572.9 (104.0 – 616.9) 116.6 (40.0 – 411.5)
Patients not completing the 6MWT at t0: n (%) 1 14.3 8 52.9
Patients completing the 6mwt test at t0: n (%) 6 85.7 8 47.1
Tracheotomy (n and %) 1 14.3 3 17.6
Mask ventilatory support (n and %) 1 14.3 8 47.1
Mean VC (%) 83 ± 45 52 ± 26
ALT: median (IQR) U/L 152 (118 - 304) 46 (28 – 95)
AST: median (IQR) U/l 125 (109-250) 57 (32 – 89)
CK: median (IQR) U/l 1019 (726 – 1187) 339 (144 – 601)
LDH: median (IQR) U/l 826 (753 - 993) 497 (334 - 697)
Residual GAA activity* (% n.v.) 5.57 (2.8 - 10.0 )
J Inherit Metab Dis

with a baseline value of WS below the median was 458.8 m(358.0–572.9), it was 62.5 m (18.5–90.4) in the group witha WS equal to or higher than the median. The improvementin walking capacity at T12, T24, and T36 was significantonly in the second group of patients (p=0.0016, p=0.0004,and p=0.0020, respectively), while in the first group it wassignificant only at T12 (p=0.0348).
Table 4 reports the 6MWT variation during ERT. Theanalysis of individual performances showed a progressiveincrease in walking capacity in all the juvenile patients: atT36 all of them completed the 6MWT. In particular, patientnumber 5, a young girl with tracheostomy at baseline,showed a dramatic improvement after recovery fromrespiratory insufficiency due to acute pneumonia. Whenadult patients were analyzed, seven were not able tocomplete the 6MWT at T36 (patients 12, 13, 14, 15, 17,20, 24). Two of them (patients 13 and 14), both presentingwith tracheostomy at T0, decreased their walked distance atlast follow-up visit, while another three (patients 17, 20, 24)stabilized. Patient 12, wheelchair bound at baseline, wasable to walk 80 m at T36. The other adult patients showedan improvement in 6MWT.
The difference in 6MWT rank distribution at baseline, T12,T24, and T36 was statistically significant in both juvenile(T12 p=0.0281; T24 p=0.0261; T36 p=0.011) and adultpatients (T12 p<0.0001; T24 p=0.0002; T36 p=0.00021).No significant variations were observed in intermediateevaluations (T12 vs. T24, T12 vs. T36, T24 vs. T36).
Respiratory outcome
Table 5 reports variations in respiratory parameters. Of 13patients requiring ventilatory support at baseline (4tracheostomized, 9 mask-ventilated), 3 patients recoveredfrom tracheostomy (1 juvenile, 2 adults) and 2 com-pletely interrupted ventilation support: a tracheostomized
adolescent girl and an adult female requiring daily maskventilation.
The other patients reduced median daily ventilation from 14(IQR 8–24) to 8 (IQR 8–14) h at T12 (p=0.0005). Theseresults were maintained throughout the follow-up to T36 ( T0vs. T36 p<0.0001). One adult female patient needed to betracheostomized at T28 due to an episode of pneumothorax.
Median arterial pCO2 decreased (T0–T36) from 43.1%(IQR 39.6–52.0) to 40.0% (IQR 39.0–52.7) (p=0.0170).Vital capacity showed a nonsignificant increasing trendfrom T0 to T36, while FEV1 was stable over time.
Muscle enzymes
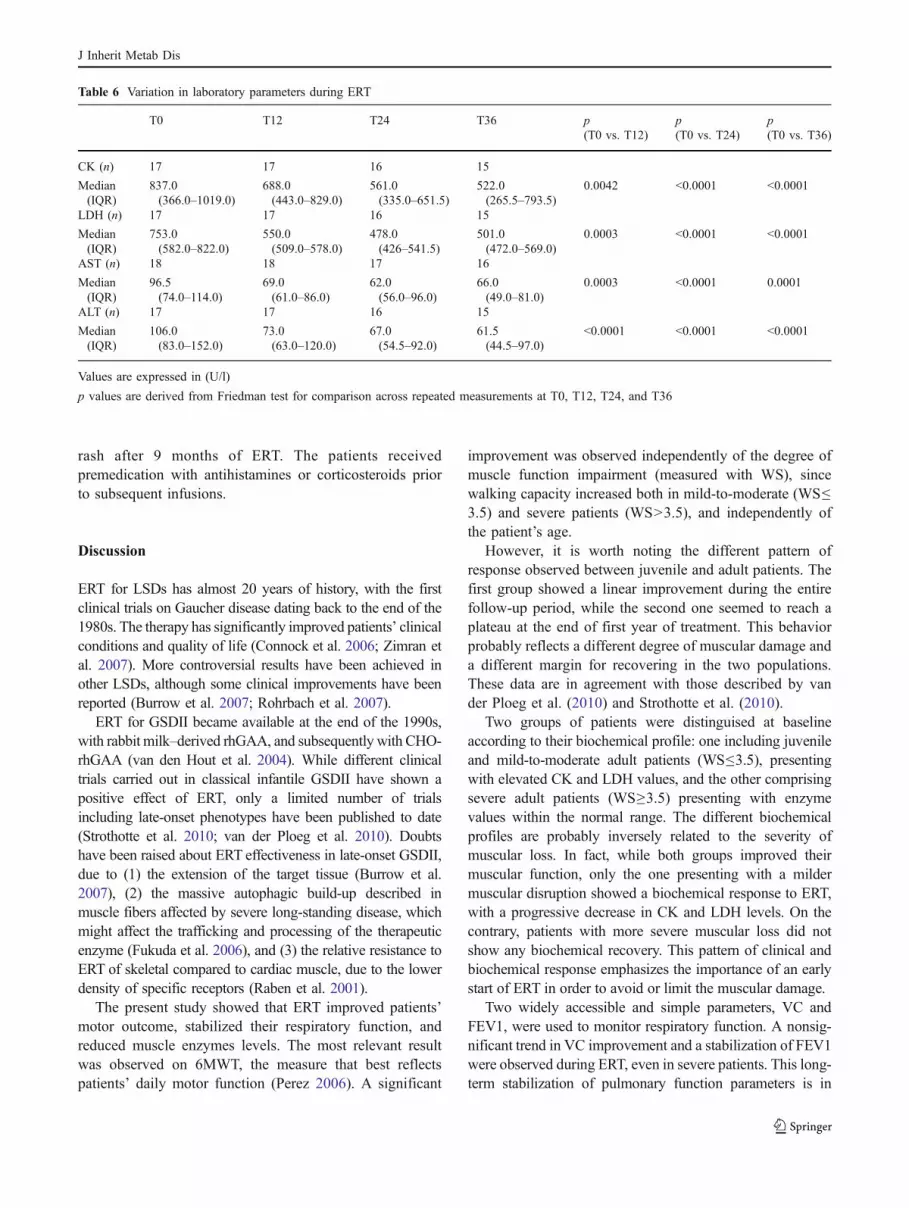
Table 6 reports the variation in muscle enzymes during ERT.Median CK decreased from 837.0 U/l (IQR 366.0–1,019.0)to 688.0 U/l (IQR 443.0–829.0) at T12 (p = 0.0042), to561.0 U/l (IQR 335.0–651.5) at T24 (p<0.0001), and to522.0 U/l (IQR 265.5–793.5) at T36 (p<0.0001). LDH andtransaminases also showed a significant reduction. Seven(29.2%) severe adult patients (WS!6) presented with normalCK and LDH values during the whole follow-up period. Themajority of patients presented with low plasma creatininelevel, which did not change during ERT (data not shown).
Other clinical symptoms
At T36 the number of patients suffering from headache fellfrom 8 (34.8%) to 2 (8.7%) (p=0.031), while thosepresenting with muscle pain decreased from 11 (47.8%) to3 (13.0%) (p=0.0078).
ERT secondary events
One adult patient had bronchospasm and facial rash afterthe third infusion, while an adolescent patient showed skin
Table 3 Variation of Walton scale during ERT and 6MWT variation in relation to Walton scale severity at baseline
T0 T12 T24 T36 p value
Walton score
"2 8 (33.3%) 10 (41.7%) 11 (45.8%) 12 (50.0%)
2.5–5 7 (29.2%) 8 (33.3%) 8 (33.3%) 5 (20.8%)
6–7 9 (37.5%) 6 (25.0%) 5 (20.8%) 7 (29.2%)
Median (IQR) 3.75 (2.0–6.0) 3.25 (1.0–5.5) 3.0 (1.0–5.0) 2 (1.0–6.0) T0–T36: 0.0003a
6MWT variation
WS T0<median(n=11)
458.8 (358.0–572.9) 520.8 (311.0–548.7) 483.5 (328.0–586.0) 487.0 (389.0–598.0) T12: 0.0348 T24: 0.1077 T36: 0.0608
WS T0!median(n=13)
62.5 (18.5–90.4) 114.0 (52.0–476.7) 105.0 (48.0–369.0) 128.0 (51.0–298.0) T12: 0.0016 T24: 0.0004 T36: 0.0020
Data are number (percentage) or median (IRQ)a Friedman test for comparison across repeated measurements at T0, T12, T24, and T 36. p values at different intermediate times during follow-up were asfollows: T0 vs. T12 p=0.0096; T0 vs. T24 p=0.0010; T12 vs. T24 p=0.1797; T12 vs. T36 p=0.5682; T24 vs. T36 p=1.0000
J Inherit Metab Dis

Table 5 Variation in respiratory parameters during ERT
T0 T12 T24 T36 pa
Patients with ventilatory support 13 (54.2%) 11 (45.8%) 11 (45.8%) 10 (41.7%)
Daily ventilatory support (h) 14 (8–24) 8 (8–14) 8 (8–16) 8 (8–12) <0.0001
pCO2 (mmHg) 43.1 (39.6–52.0) 40.4 (37.4– 49.3) 40.8 (38.0–47.2) 40.0 (39.0–52.7) 0.017
VC (%) 54.0 (27.0–84.0) 56.0 (26.0–84.0) 60.0 (27.0–77.0) 59.5 (32.3–77.0) 0.9990
FEV1 (%) 44.0 (27.0–83.0) 48.0 (34.0–87.0) 43.5 (29.5–83.5) 45.0 (29.8–82.0) 0.2537
Data are number of patients (%) or median (IRQ). pCO2, VC, and FEV1 were measured in 24 patients (100%)a Friedman test for comparison across repeated measurements at T0, T12, T24, and T36. p values at different intermediate times during follow-up were asfollows: for hours of daily ventilatory support, T0 vs. T12 p=0.0005 and T0 vs. T12 vs. T24 p<0.0001; for pCO2, T0 vs. T12 vs. T24 p=0.0189; for othercomparisons, p>0.005
Table 4 Variation in 6MWT during ERT
Patients Age at ERT T0 T12 T24 T36
Juveniles
1 10 580 744 847 840
2 12 616.9 781.2 770.6 748
3 12 104 156.2 156 180
4 13 642.94 742.8 630 730
5* 12 15 589 565 590
6 18 511 572 629 n.a
7 7 572.88 548.7 n.a 598
Median (IQR)a 572.9 (104.0–616.9) 589.0 (548.7–744.0),p=0.0281
630.0 (565.0–770.6),p=0.0261
664.0 (590.0–748.0),p=0.011
Adults
8 35 156.24 210.2 207 220
9 35 438.96 442.7 477 484
10 47 458.8 520.8 490 490
11 37 76.88 381.3 369 298
12 60 0 0 80 n.a.
13* 56 75 100 105 32.5
14* 36 50 50 0 24
15* 49 22 68 58 51
16 52 75 128 142 128
17 61 6 13 8 n.a.
18 44 384 448 495 389
19 54 480 532 458 426
20 57 10 10 11 n.a
21 28 495 538 586 534
22 61 358 311 328 n.a
23 41 197 216 205 265
24 57 30 54 48 56
Median (IQR)a 116.6 (40.0–411.5) 213.1 (61.0–445.3),p<0.0001
206.0 (69.0–467.5),p=0.0002
265.0 (56.0–426.0),p=0.0002
Asterisks indicate patients tracheostomized at T0, n.a. data not availablea Friedman test for comparison across repeated measurements at T0, T12, T24, and T36
J Inherit Metab Dis
rash after 9 months of ERT. The patients receivedpremedication with antihistamines or corticosteroids priorto subsequent infusions.
Discussion
ERT for LSDs has almost 20 years of history, with the firstclinical trials on Gaucher disease dating back to the end of the1980s. The therapy has significantly improved patients’ clinicalconditions and quality of life (Connock et al. 2006; Zimran etal. 2007). More controversial results have been achieved inother LSDs, although some clinical improvements have beenreported (Burrow et al. 2007; Rohrbach et al. 2007).
ERT for GSDII became available at the end of the 1990s,with rabbit milk–derived rhGAA, and subsequently with CHO-rhGAA (van den Hout et al. 2004). While different clinicaltrials carried out in classical infantile GSDII have shown apositive effect of ERT, only a limited number of trialsincluding late-onset phenotypes have been published to date(Strothotte et al. 2010; van der Ploeg et al. 2010). Doubtshave been raised about ERT effectiveness in late-onset GSDII,due to (1) the extension of the target tissue (Burrow et al.2007), (2) the massive autophagic build-up described inmuscle fibers affected by severe long-standing disease, whichmight affect the trafficking and processing of the therapeuticenzyme (Fukuda et al. 2006), and (3) the relative resistance toERT of skeletal compared to cardiac muscle, due to the lowerdensity of specific receptors (Raben et al. 2001).
The present study showed that ERT improved patients’motor outcome, stabilized their respiratory function, andreduced muscle enzymes levels. The most relevant resultwas observed on 6MWT, the measure that best reflectspatients’ daily motor function (Perez 2006). A significant
improvement was observed independently of the degree ofmuscle function impairment (measured with WS), sincewalking capacity increased both in mild-to-moderate (WS"3.5) and severe patients (WS>3.5), and independently ofthe patient’s age.
However, it is worth noting the different pattern ofresponse observed between juvenile and adult patients. Thefirst group showed a linear improvement during the entirefollow-up period, while the second one seemed to reach aplateau at the end of first year of treatment. This behaviorprobably reflects a different degree of muscular damage anda different margin for recovering in the two populations.These data are in agreement with those described by vander Ploeg et al. (2010) and Strothotte et al. (2010).
Two groups of patients were distinguised at baselineaccording to their biochemical profile: one including juvenileand mild-to-moderate adult patients (WS"3.5), presentingwith elevated CK and LDH values, and the other comprisingsevere adult patients (WS!3.5) presenting with enzymevalues within the normal range. The different biochemicalprofiles are probably inversely related to the severity ofmuscular loss. In fact, while both groups improved theirmuscular function, only the one presenting with a mildermuscular disruption showed a biochemical response to ERT,with a progressive decrease in CK and LDH levels. On thecontrary, patients with more severe muscular loss did notshow any biochemical recovery. This pattern of clinical andbiochemical response emphasizes the importance of an earlystart of ERT in order to avoid or limit the muscular damage.
Two widely accessible and simple parameters, VC andFEV1, were used to monitor respiratory function. A nonsig-nificant trend in VC improvement and a stabilization of FEV1were observed during ERT, even in severe patients. This long-term stabilization of pulmonary function parameters is in
Table 6 Variation in laboratory parameters during ERT
T0 T12 T24 T36 p(T0 vs. T12)
p(T0 vs. T24)
p(T0 vs. T36)
CK (n) 17 17 16 15
Median(IQR)
837.0(366.0–1019.0)
688.0(443.0–829.0)
561.0(335.0–651.5)
522.0(265.5–793.5)
0.0042 <0.0001 <0.0001
LDH (n) 17 17 16 15
Median(IQR)
753.0(582.0–822.0)
550.0(509.0–578.0)
478.0(426–541.5)
501.0(472.0–569.0)
0.0003 <0.0001 <0.0001
AST (n) 18 18 17 16
Median(IQR)
96.5(74.0–114.0)
69.0(61.0–86.0)
62.0(56.0–96.0)
66.0(49.0–81.0)
0.0003 <0.0001 0.0001
ALT (n) 17 17 16 15
Median(IQR)
106.0(83.0–152.0)
73.0(63.0–120.0)
67.0(54.5–92.0)
61.5(44.5–97.0)
<0.0001 <0.0001 <0.0001
Values are expressed in (U/l)
p values are derived from Friedman test for comparison across repeated measurements at T0, T12, T24, and T36
J Inherit Metab Dis

contrast to the progressive deterioration that characterizes thenatural course of the disease (Hagemans et al. 2005).
Respiratory response may derive from the combinationof the effects of ERT on muscular function and a globalamelioration of patient’s clinical management during thestudy protocol. However, the role of respiratory musclefunction improvement in reducing daily ventilatory supportand the recovery from tracheostomy in three patients cannotbe underestimated. It is also worth noting the significantreduction in muscle pain and headache after ERT, the latterprobably being related to the decrease in arterial pCO2.
Recently, Merk et al. (2009) reported an improvement inrespiratory muscle strength during ERT by measuringmaximum inspiratory (MIP) and expiratory (MEP) pres-sures. These parameters were shown to be very sensitive tosmall changes in diaphragm strength (Perez 2006; Steier etal. 2007), emphasizing the limits of VC and FEV1 formonitoring ERT respiratory response.
The relationship between motor disability and respiratoryfunction was studied by Pellegrini et al., who demonstrated adirect correlation between the severity of motor impairmentand respiratory insufficiency (Pellegrini et al. 2005). Thisobservation was evidenced also by our study, since a strongcorrelation between 6MWT impairment, WS severity, andVC reduction was observed at baseline. However, duringERT, motor and respiratory functions responded differently.In fact, while 6MWT, a test originally performed to assessthe effectiveness of specific therapies for pneumopathies(Redelmeier et al. 1997), improved significantly, the respi-ratory response was limited. This result is similar to thatobtained by Strothotte et al. in a multicenter study performedin a group of 44 late-onset GSDII patients (Strothotte et al.2010). Both of these observations suggest that the increase inthe 6MWT may be due to an improvement in the motorfunction rather than to a respiratory response to ERT.
Furthermore, the different magnitude of the WS response(improved only in juvenile and mild-to-moderate adultpatients) in comparison with 6MWT response (improved alsoin severe patients) seems to indicate that ERT mainly affectsmuscular endurance rather than strength. This may besecondary to the different therapeutic responses of musclefibers of different types: type 1, associated with muscularendurance, and type 2, associated with muscular strength(Raben et al. 2001). The hypothesis is in line with the resultsof Fukuda et al. showing a better response of type 1 fibers toERT (Fukuda et al. 2006). However, to clarify this issue,further studies addressing muscle response to ERT in termsof strength and composition/structure are needed.
Finally, DeRuisseau et al. recently demonstrated that in aGSDII mouse model the neural output to the diaphragm isdeficient as a consequence of the glycogen storage process inthe neurons of anterior horns of the spinal cord (DeRuisseau etal. 2009). This observation points out the role of central
nervous system involvement in the pathophysiology of GSDII,raising the problem of central nervous system accessibility.
In conclusion, our study showed that in late onset GSDIIpatients, long-term ERT was well tolerated and safe, and itimproved motor function even in severely affected patients,independently of their age and disease duration. It alsoallowed ventilatory support to be reduced and stabilizedrespiratory parameters.
In addition, the results of the present study strengthen theargument for early ERT initiation in order to avoid or limit themuscular damage. The cost of ERT remains an open issue thatmay hinder access to the treatment even in rich industrialcountries.
Acknowledgments This work was supported by the Agenzia Italianadel Farmaco (AIFA) D.G. 62229; by I.R.C.C.S. Burlo Garofolo-Trieste,RC 92/05; by a grant from “Programma Italia-USA”-526D/47 of theIstituto Superiore di Sanità, Rome; and by a grant from the ItalianAssociation of Glycogenosis (AIG). The authors wish to thank theparticipating patients and their families for their precious contribution todata collection and clinical information and Sarah Tripepi Winteringham,MCIL, for her assistance in manuscript editing.
References
Bembi B, Cerini E, Danesino C et al. (2008) Management and treatmentof glycogenosis type II. Neurology 71(23 Suppl 2):S12–S36
Burrow TA, Hopkin RJ, Leslie ND, Tinkle BT, Grabowski GA (2007)Enzyme reconstitution/replacement therapy for lysosomal storagediseases. Curr Opin Pediatr 19:628–635
Connock M, Burls A, Frew E et al. (2006) The clinical effectiveness andcost-effectiveness of enzyme replacement therapy for Gaucher'sdisease: a systematic review. Health Technol Assess 1(iii-iv):ix–136
DeRuisseau LR, Fuller DD, Qiu K et al. (2009) Neural deficitscontribute to respiratory insufficiency in Pompe disease. ProcNatl Acad Sci USA 106:9419–9424
Enright PL, Sherrill DL (1998) Reference equations for the six-minutewalk in healthy adults. Am J Respir Crit Care Med 158:1384–1387
Fukuda T, Ahearn M, Roberts A et al. (2006) Autophagy andmistargeting of therapeutic enzyme in skeletal muscle in Pompedisease. Mol Ther 14:831–839
Geiger R, Strasak A, Treml B et al. (2007) Six-minute walk test inchildren and adolescents. J Pediatr 150:395–399
Hagemans ML, Winkel LP, Van Doorn PA et al. (2005) Clinicalmanifestation and natural course of late-onset Pompe's disease in54 Dutch patients. Brain 128(Pt 3):671–677
Joshi PR, Gläser D, Schmidt S et al. (2008) Molecular diagnosis ofGerman patients with late-onset glycogen storage disease type II.J Inherit Metab Dis. doi:10.1007/s10545-008-0820-2
Kishnani PS, Nicolino M, Voit T et al. (2006) Chinese hamster ovarycell-derived recombinant human acid alpha-glucosidase ininfantile-onset Pompe disease. J Pediatr 149:89–97
Lehmann EL (2006) Nonparametrics: statistical methods based onranks, 2nd edn. Springer, New York
Merk T, Wibmer T, Schumann C, Krüger S (2009) Glycogen storagedisease type II (Pompe disease)–influence of enzyme replace-ment therapy in adults. Eur J Neurol 16:274–277
Montalvo AL, Bembi B, Donnarumma M et al. (2006) Mutationprofile of the GAA gene in 40 Italian patients with late onsetglycogen storage disease type II. Hum Mutat 27:999–1006
J Inherit Metab Dis

Pellegrini N, Laforet P, Orlikowski D et al. (2005) Respiratoryinsufficiency and limb muscle weakness in adults with Pompe'sdisease. Eur Respir J 26:1024–1031
Perez T (2006) Neuromuscular disorders—assessment of the respira-tory muscles. Rev Neurol 162:437–444
Raben N, Lu N, Nagaraju K, Rivera Y et al. (2001) Conditional tissue-specific expression of the acid alpha-glucosidase (GAA) gene inthe GAA knockout mice: implications for therapy. Hum MolGenet 10:2039–2047
Ravaglia S, Moglia A, Costa A, Repetto A, Danesino C (2009)Enzyme replacement therapy in late-onset type II glycogenosis.Eur J Neurol 16:e125
Redelmeier DA, Bayoumi AM, Goldstein RS, Guyatt GH (1997)Interpreting small differences in functional status: the Six MinuteWalk test in chronic lung disease patients. Am J Respir Crit CareMed 155:1278–1282
Rohrbach M, Clarke JT (2007) Treatment of lysosomal storagedisorders: progress with enzyme replacement therapy. Drugs67:2697–2716
Slonim AE, Bulone L, Goldberg T et al. (2007) Modification of thenatural history of adult-onset acid maltase deficiency by nutritionand exercise therapy. Muscle Nerve 35:70–77
Steier J, Kaul S, Seymour J et al. (2007) The value of multiple testsfor respiratory muscle strength. Thorax 62:975–980
Strothotte S, Strighl-Pill N, Grunert B et al. (2010) Enzymereplacement therapy with alglucosidase alfa in 44 patients withlate-onset glycogen storage disease type 2: 12-month results ofan observational clinical trial. J Neurol 257:91–97
van den Hout JM, Kamphoven JH, Winkel LP et al. (2004) Long-termintravenous treatment of Pompe disease with recombinant humanalpha-glucosidase from milk. Pediatrics 113:448–457
Van Der Laan P, Verdooren LR (1987) Classical analysis of variancemethods and nonparametric counterparts. Biometr J 29(6):635–665
van der Ploeg AT, Reuser AJ (2008) Pompe's disease. Lancet372:1342–1353
van der Ploeg AT, Clemens PR, Corzo D et al (2010) A randomizedstudy of alglucosidase alfa in late-onset Pompe's disease. N EnglJ Med 362:1396–1406
Winkel LP, Hagemans ML, van Doorn PA et al. (2005) The naturalcourse of non-classic Pompe's disease; a review of 225 publishedcases. J Neurol 252:875–884
Zimran A, Bembi B, Pastores GM (2007) Enzyme replacement therapyfor type I Gaucher disease. In: Futerman AH, Zimran A (eds)Gaucher disease. Taylor and Francis, New York, pp 341–354
J Inherit Metab Dis





























