Embed Size (px)
Citation preview

PolymerChemistry
PAPER
aInstitut des Sciences Chimiques de Rennes, O
UMR 6226 CNRS, Universite de Rennes 1, Ca
France. E-mail: sophie.guillaume@univ-rennbTotal Petrochemicals Technology Feluy, Zo
Belgium
† Macromolecular engineering via ring-oGuerin, M. Helou, J.-F. Carpentier, M.Guillaume, Polym. Chem., 2013, 4, 10Slawinski, J.-M. Brusson, S. M. Guillaum2013, 4, 3686–3693.
‡ Electronic supplementary information (characterizations of BTMC (1H, 13C NMR(1H, 13C NMR; DSC), and PTMC-b-PTMCscheme of PTMC-b-PTMC(OMe)2 and Pand PTMC-b-PTMC(OH) respectively. See
§ Equally contributing rst authors.
Cite this: Polym. Chem., 2014, 5, 1229
Received 19th July 2013Accepted 19th October 2013
DOI: 10.1039/c3py00955f
www.rsc.org/polymers
This journal is © The Royal Society of C
Macromolecular engineering via ring-openingpolymerization (3): trimethylene carbonate blockcopolymers derived from glycerol†‡
William Guerin,§a Marion Helou,§ab Martine Slawinski,b Jean-Michel Brusson,b
Jean-François Carpentiera and Sophie M. Guillaume*a
Linear trimethylene carbonate (1,3-dioxane-2-one, TMC) thermoplastic copolymers derived from glycerol
have been synthesized upon sequential copolymerization. The “immortal” ring-opening polymerization
(iROP) of the benzyloxy-substituted TMC, 3-benzyloxytrimethylene carbonate (BTMC), with systems
composed of the b-diketiminate discrete zinc complex [(BDIiPr)Zn(N(SiMe3)2)] or the phosphazene base
2-tert-butylimino-2-diethylamino-1,3-dimethylperhydro-1,3,2-diazaphosphorine (BEMP) as a catalyst,
and benzyl alcohol (BnOH) as an initiator/chain transfer agent is first described. Next, diblock and
triblock copolycarbonates featuring a TMC segment along with one or two adjacent BTMC, or 2,2-
dimethoxypropane-1,3-diol carbonate (TMC(OMe)2), or racemic b-butyrolactone (BL) chains have been
prepared. The copolymerization under “immortal” operating conditions was carried out in bulk or
toluene solution and promoted by [(BDIiPr)Zn(N(SiMe3)2)] as a catalyst, and either BnOH, 1,3-propanediol,
or the poly(trimethylene carbonate) (PTMC) derived macro-ols, H-PTMC-OBn or H-PTMC-H, as an
initiator/chain transfer agent. In particular, the PTMC-b-[poly(TMC(OMe)2)]1,2 couple of copolymers
provide a basis for the assessment of thermal and mechanical property differentiation in close
relationship with the co-monomers content. Unlike PTMC which is elastomeric and P(TMC(OMe)2)
which is rather rigid and brittle much like semi-crystalline poly(L-lactide), the diblock and triblock
copolymers show characteristics lying in between those of each homopolymer, without any significant
influence of the topology. Thus PTMC/PTMC(OMe)2 block copolymers mechanically behave similarly to
PTMC/PLLA block copolymers.
Introduction
The eld of biomass-derived plastics is currently of topicalinterest. One major motivation to this trend remains the searchfor reliable and feasible alternatives to petroleum-based plas-tics, such as polyethylene, polypropylene or poly(a-olens)commodity polymers, for which the global demand is still on
rganometallics, Materials and Catalysis,
mpus de Beaulieu, F-35042 Rennes Cedex,
es1.fr
ne Industrielle Feluy C, B-7181 Seneffe,
pening polymerization (1 and 2): W.Slawinski, J.-M. Brusson, and S. M.95–1106; W. Guerin, M. Helou, M.e and J.-F. Carpentier, Polym. Chem.,
ESI) available: The synthesis of BTMC,), PBTMC (13C NMR; DSC), PTMC(OH)(O) (1H NMR), and the deprotectionTMC-b-PBTMC into PTMC-b-PTMC(O)DOI: 10.1039/c3py00955f
hemistry 2014
the increase. Identifying promising vegetable non-edible sour-ces for the sustainable development of polymers that will thusreadily and suitably (bio)degrade within an environmentallysustainable manner thus appears as a reasonable life-cycle toexploit.
As part of our ongoing efforts aimed at identifying andsynthesizing original thermoplastics derived from readilyavailable biorenewable resources within environmentallybenign routes, we initiated research on polycarbonates issuedfrom glycerol.1 Glycerol is indeed cheaply available in largevolumes and formed as a by-product during the production ofbiodiesel from vegetable oils and animal fats. Besides, a largenumber of value added chemicals can be derived from glycerol,which include those allowing the synthesis of cyclic carbonatessuch as 1,3-propanediol or 1,3-dihydroxyacetone (DHA; alsonaturally available).2 Relevant to the present study, glycerol isalso used as a precursor for the synthesis of the most commoncyclic carbonate, namely 1,3-dioxane-2-one referred to as tri-methylene carbonate (TMC),3 as well as of its functionalizedderivatives, of which another common six-membered ring cycliccarbonate, the 3-benzyloxy-substituted TMC4 and also 2,2-dimethoxytrimethylene carbonate (TMC(OMe)2)5,6 (Scheme 1).
Polym. Chem., 2014, 5, 1229–1240 | 1229

Scheme 1 Synthesis of BTMC,4 TMC,3 and TMC(OMe)2,5,6 fromglycerol.
Polymer Chemistry Paper
Polycarbonates are an important class of biodegradablepolymers featuring good biocompatibility, low (cyto)toxicity andfavourable thermo-mechanical properties. These characteristicsmake them leading candidates for biomedical and pharma-ceutical applications, in particular in tissue engineering (bothrepair and regeneration) as well as in controlled drug/genedelivery systems.7 Functionalisation of cyclic carbonates suchas TMC (e.g. BTMC, Me2TMC, TMC(OMe)2, etc.) has revealed asuccessful way to control and tune, to some extent, the physi-cochemical properties of the resulting polymers.7b,8Whereas thepresence of functional groups along the main backbone is ofprime importance, for instance to modulate the degradationbehaviour, pendant functional groups along the polymer arealso a main advantage as they provide potential reactive sites. Inparticular, pendant hydroxyl groups enable the adjustment ofthe hydrophobicity/hydrophilicity balance, or the anchorage ofbiologically relevant molecules such as prodrugs or targetingmoieties for a specic medical application.7 With a similar
Scheme 2 Synthesis of H-PBTMC-OBn upon iROP of BTMC, and subse
1230 | Polym. Chem., 2014, 5, 1229–1240
objective, copolymers of TMC with cyclic esters such as lactones(b-butyrolactone (BL),9,10a 3-caprolactone,10 glycolide,11 poly-ethylene glycol12) or diesters (lactides),13 or with functionalizedcyclic carbonates4b,14,15a further allow broadening of the range ofapplications.7,8 Such copolymerizations have been most oenperformed, in the bulk or in solution, from tin(II) bis(2-ethylhexanoate) (Sn(octoate)2 ¼ SnOct2), enzymes (porcinepancreas, Pseudomonas uorescens) or thiourea/amine organo-catalysts. Similarly, the related BTMC has been homopoly-merized4,16 as well as copolymerized with other cyclic esters,15,16a
which include TMC,15a using lipases15h,i or metallic catalysts(SnOct2, Ti(OiPr)4, ZnEt2).15a–g Interest in the correspondingpolymer, poly(benzyloxy trimethylene carbonate) (PBTMC), ari-ses from the possibility to access from such an hydrophobicpolymer to the analogous hydrophilic poly(hydroxytrimethylenecarbonate) (PHTMC), featuring the same polycarbonate back-bone yet with pendant hydroxyl groups.4b,15c,16
Our previous studies on the macromolecular engineering ofPTMC copolymers have shown the ability of some organic,metallic and metallo-organic catalyst systems to promote thering-opening copolymerization of TMC and L-lactide (LLA).Distinct systems ranging from a simple basic organocatalyst suchas a guanidine (TBD ¼ 1,5,7-triazabicyclo[4.4.0]dec-5-ene), anamine (4-N,N-dimethylaminopyridine, DMAP) or a phosphazene(2-tert-butylimino-2-diethylamino-1,3-dimethylperhydro-1,3,2-diazaphosphorine, BEMP), a simple Lewis acidic metallic saltsuch as aluminum/bismuth/ytterbium triates, or a moresophisticated discrete b-diiminate zinc complex [(BDIiPr)Zn(N(SiMe3)2)] (BDI ¼ CH(CMeNC6H3-2,6-iPr2)2), [(BDIiPr)Zn(N(SiMe3)2)], associating benzyl alcohol (BnOH) acting as aco-initiator and a chain transfer agent, have demonstrated theirefficiency.13d,e Sequential approaches afforded diblock and tri-block PTMC-b-PLLA1,2 (PLLA ¼ poly(L-lactide)) linear and star-shaped copolymers with controlled molecular features, i.e.controlled end-functional groups and molar masses as well asrather narrow dispersity values. Investigations of the thermo-mechanical properties of these copolymers revealed that, inorder to improve the rigidity and brittleness of PLLA (E ¼ 3427MPa, 3b ¼ 8%), a minimal block size of both the PLLA (Mn ¼ ca.23 000 g mol�1) and the PTMC (Mn ¼ ca. 10 000 g mol�1)
quent synthesis of H-PHTMC-OH from its hydrogenolysis.
This journal is © The Royal Society of Chemistry 2014

Scheme 3 Synthesis of various diblock and triblock copolymers upon sequential copolymerization of TMC with BTMC, TMC(OMe)2, or BL,promoted by [(BDIiPr)Zn(N(SiMe3)2)]/BnOH, H-PTMC-OBn or H-PTMC-H.
Paper Polymer Chemistry
segments was required. Thus, modulating the size of theamorphous PTMC inserted into crystalline PLLA signicantlyimproved the ductility by a factor of ca. 25, while maintainingthe stiffness of the polymer material (E ¼ ca. 2500 MPa) close tothat of the homopolymer.13d Also, kinetic and microstructuralcontrol was achieved upon suitable catalyst tuning in thesimultaneous copolymerization of TMC and LLA.13e Gradientcopolymers PTMC-grad-PLLA featuring PTMC and PLLAsegments of signicant size were prepared from [(BDIiPr)Zn[N(SiMe3)2]/BnOH or M(OTf)3/BnOH (M ¼ Al, Bi, Yb), withconsumption of LLA proceeding prior to that of TMC with thezinc-based systems, whereas TMC was polymerized faster thanLLA with the metal triate systems. This behaviour was incontrast to that of the homopolymerizations promoted by thesame two catalytic systems, respectively, for which LLA andTMC were consumed roughly at the same rate.13e
In the present contribution, we report the homopolymeri-zation of BTMC into PBTMC from an “immortal” ring-openingpolymerization (iROP) approach mediated by an alike (metallo)organic catalyst, and its subsequent hydrogenolysis intoPHTMC (Scheme 2). On the basis of common catalyst systemsevidenced as active in the homopolymerization of a fewmonomers such as BTMC, TMC(OMe)21b,6 and BL,1,17,27 we nextdescribe poly(trimethylene carbonate) (PTMC) analogousdiblock and triblock copolymers incorporating segments ofeither of these comonomers and prepared following a sequen-tial copolymerization (Scheme 3). The mechanical properties ofblock copolymers derived from TMC and TMC(OMe)2 areinvestigated in comparison to those of PLLA and PTMC-b-PLLAcopolymers, thereby illustrating the benet gained upon func-tionalizing the TMC repeating unit.
This journal is © The Royal Society of Chemistry 2014
Experimental sectionMethods and materials
All manipulations were performed under an inert atmosphere(argon, <3 ppm O2) using standard Schlenk, vacuum line, andglovebox techniques. Trimethylene carbonate (TMC, 1,3-dioxane-2-one, Labso Chimie Fine, Blanquefort, France) was puried byrst dissolving it in THF, stirring over CaH2 for 2 days before beingltered and dried in vacuo, and nally recrystallized from coldTHF. Benzyl alcohol (Acros) was distilled over Mg turnings underan argon atmosphere and kept over activated 3–4 A molecularsieves. CDCl3 was dried over 3–4 A molecular sieves. [(BDIiPr)Zn(N(SiMe3)2)] was synthesized following the literature proce-dure.18 3-Benzyloxytrimethylene carbonate, hereaer referred to asBTMC,4 (Scheme S1; 1H and 13C NMR spectra illustrated in Fig. S1and S2 in the ESI†), and 2,2-dimethoxytrimethylene carbonate,hereaer referred to as TMC(OMe)2,6 were synthesized from 1,3-dihydroxyacetone (DHA) and glycerol, respectively, as previouslyreported. H-PTMC-OBn was synthesized at 60 �C from [(BDIiPr)Zn(N(SiMe3)2)] and BnOH as previously described.19 The selectedPTMC-diol was prepared from [(BDIiPr)Zn(N(SiMe3)2)] and 1,3-propanediol (Acros) according to the reported procedure.20 Allother reagents were used as received (Aldrich).
Instrumentation and measurements1H (500, 400 and 200 MHz) and 13C{1H} (125, 100 and 50 MHz)NMR spectra were recorded on Bruker Avance AM 500, AM 400and AM 200 spectrometers at 23 �C. Chemical shis (d) arereported in ppm and were referenced internally relative to tet-ramethylsilane (d 0 ppm) using the residual 1H and 13C solventresonances.
Polym. Chem., 2014, 5, 1229–1240 | 1231

Polymer Chemistry Paper
Monomer conversions were determined from 1H NMRspectra of the crude polymer sample: from the integration (Int.)ratio Int.PTMC/[Int.PTMC + Int.TMC], using the methylene group inthe a-position of the carbonate (CH2OC(O), dTMC 4.45 ppm,dPTMC 4.25 ppm) for PTMC; from the integration ratioInt.P(TMC(OMe)2)/[Int.P(TMC(OMe)2) + Int.TMC(OMe)2], using either themethylene hydrogens (C(OMe2)CH2OC(O)) or the methylhydrogens (C(OMe2)CH2OC(O)) of the dimethyl acetal ofP(TMC(OMe)2) at d 4.23 ppm and 3.28 ppm, and of TMC(OMe)2at d 4.30 and 3.34 ppm, respectively, for P(TMC(OMe)2); fromthe integration ratio Int.PBTMC/[Int.PBTMC + Int.BTMC], using thebenzylic hydrogens of the PhCH2OCH group (dBTMC 4.67 ppm,dPBTMC 4.60 ppm) for PBTMC, and from the integration ratioInt.PHB/[Int.PHB + Int.BL] of the methine hydrogen ((–OCH(CH3)CH2) at d 5.25 ppm for PHB, d 4.66 ppm for BL), for PHB,respectively.
Average molar mass (Mn,SEC) and dispersity (ĐM ¼ Mw/Mn)values were determined by size exclusion chromatography (SEC)in THF at 30 �C (ow rate ¼ 1.0 mL min�1) on a PolymerLaboratories PL50 apparatus equipped with a refractive indexdetector and a set of two ResiPore 300 � 7.5 mm columns. Thepolymer samples were dissolved in THF (2 mgmL�1). All elutioncurves were calibrated with polystyrene standards.Mn,SEC valuesof PTMCs were calculated using the average correction factorspreviously reported (PTMC:Mn,SEC ¼Mn,SEC raw data � 0.73),10a
thus taking into account the difference in hydrodynamic radiusof PTMC vs. polystyrene.Mn,SEC values of PTMC(OMe)2, PBTMCand PHB were uncorrected for the difference in hydrodynamicradius vs. polystyrene standards. The SEC traces of the polymersall exhibited a monomodal and symmetrical peak.
The molar masses values of short-chain PBTMC samples(Mn < 8000 g mol�1) were determined by 1H NMR analysis inCDCl3 from the relative intensities of the signals of the
Table 1 iROP of BTMC promoted by the [(BDIiPr)Zn(N(SiMe3)2)]/BnOH a
Entry Catalyst Solvent
[BTMC]0 :[catalyst]0 :[BnOH]0 Temp. (�C)
1 [(BDIiPr)Zn(N(SiMe3)2)] — 500 : 1 : 5 1302 [(BDIiPr)Zn(N(SiMe3)2)] — 500 : 1 : 5 1103 [(BDIiPr)Zn(N(SiMe3)2)] — 500 : 1 : 5 904 [(BDIiPr)Zn(N(SiMe3)2)] — 500 : 1 : 2 605 [(BDIiPr)Zn(N(SiMe3)2)] — 500 : 1 : 5 606 [(BDIiPr)Zn(N(SiMe3)2)] — 500 : 1 : 10 607 [(BDIiPr)Zn(N(SiMe3)2)] — 500 : 1 : 20 608 [(BDIiPr)Zn(N(SiMe3)2)] — 500 : 1 : 50 609 [(BDIiPr)Zn(N(SiMe3)2)] Toluene 500 : 1 : 5 6010 [(BDIiPr)Zn(N(SiMe3)2)] Toluene 500 : 1 : 5 6011 [(BDIiPr)Zn(N(SiMe3)2)] Toluene 500 : 1 : 5 6012 BEMP Toluene 500 : 1 : 5 6013 BEMP — 500 : 1 : 5 6014 BEMP Toluene 500 : 1 : 10 6015 BEMP — 500 : 1 : 10 60
a Bulk monomer or 4.0 M solutions in toluene. b Reaction times were noreaction mixture. d Theoretical molar mass calculated from the relationsh208 g mol�1, MBnOH ¼ 108 g mol�1. e Molar mass values determined bExperimental section). f Number-average molar mass values (uncorrectepolystyrene standards. g Dispersity values determined by SEC in THF.
1232 | Polym. Chem., 2014, 5, 1229–1240
methylene hydrogens (d 4.19–4.30 ppm, C6H5CH2OCH(-CH2OC(O))2; d 4.63 ppm, C6H5CH2OCH(CH2OC(O))2) of thePBTMC main chain, relative to the terminal benzylic hydrogens(d 5.15 ppm, C(O)OCH2C6H5). The molar mass values of short-chain PTMC-copolymer samples were determined by 1H NMRanalysis in CDCl3 from the relative intensities of the signals ofthe methylene protons (�CH2OC(O)) of the PTMC chains (d ¼4.26 ppm), PBTMC (d ¼ 4.61 ppm) and PTMC(OMe)2 (d ¼ 2.38ppm), to the a-hydroxymethyl (CH2OH, d ¼ 3.76 ppm) or to thebenzyloxy hydrogens (R ¼ OCH2C6H5, d ¼ 5.16 ppm). Thenumber-average molar mass values thus obtained by 1H NMR(Mn,NMR) were in close agreement with the ones calculated, asreported in Tables 1–3.
MALDI-ToF mass spectra were recorded at the CESAMO(Bordeaux, France) on a Voyager mass spectrometer (AppliedBiosystems) equipped with a pulsed N2 laser source (337 nm)and a time-delayed extracted ion source. Spectra were recordedin the positive-ion mode using the reectron mode and with anaccelerating voltage of 20 kV. A THF solution (1 mL) of thematrix (trans-3-indoleacrylic acid, IAA (Aldrich, 99%); 10 mgmL�1) and possibly a MeOH solution of the cationisation agent(NaI, 10 mg mL�1) were prepared. A fresh solution of the poly-mer samples in THF (10mgmL�1) was then prepared. The threesolutions were then rapidly combined in a 1 : 1 : 10 volumeratio of matrix-to-sample-to-cationisation agent. 1–2 mL of theresulting solution was deposited onto the sample target andvacuum-dried.
Differential scanning calorimetry (DSC) analyses were per-formed on a Setaram DSC 131 apparatus calibrated with indiumat a rate of 10 �C min�1 under a continuous ow of helium(25 mL min�1), using aluminum capsules (typically 10 mg ofpolymer). The thermograms were recorded according to thefollowing cycles: �40 �C to +200 �C at 10 �C min�1; +200 �C to
nd BEMP/BnOH catalytic systems in bulk or toluenea
Reactiontimeb (min)
BTMCconv.c (%)
Mn,theod
(g mol�1)Mn,NMR
e
(g mol�1)Mn,SEC
f
(g mol�1) ĐMg
30 100 20 900 — 6600 1.4830 100 20 900 — 8200 1.5230 100 20 900 — 13 500 1.6160 95 49 500 — 35 500 1.7530 100 20 900 — 16 200 1.6945 100 10 500 8400 7800 1.6130 80 4300 4400 4280 1.5290 80 1800 1900 2010 1.4710 94 19 650 — 14 300 1.5620 100 20 900 — 18 500 1.5530 100 20 900 — 16 500 1.67
240 93 19 450 — 13 300 1.61240 100 20 900 — 13 000 1.65240 100 10 400 9300 6500 1.34240 100 10 400 9800 5000 2.0
t necessarily optimized. c Determined by 1H NMR analysis of the crudeip: ([BTMC]0/[BnOH]0) � MBTMC � conversion + MBnOH, with MBTMC ¼y 1H NMR taking into account the monomer conversion (refer to thed; refer to the Experimental section) determined by SEC in THF vs.
This journal is © The Royal Society of Chemistry 2014

Table 2 Sequential copolymerization of TMCwith TMC, BTMC, TMC(OMe)2, or rac-BL, promoted by the [(BDIiPr)Zn(N(SiMe3)2)]/BnOH system intoluene:a synthesis of homopolymer “PTMC-b-PTMC” and block copolymers PTMC-b-PBTMC, PTMC-b P(TMC(OMe)2), or PTMC-b-PHB,respectively
Entry Comonomer
H-PTMC-OBnb
Mn,SECc (g mol�1)
(ĐM)d
[TMC]0 : [TMC(OMe)2]0 :[[(BDIiPr)Zn(N(SiMe3)2)]]0 :[BnOH]0 Temp. (�C)
Reactiontimee (h)
Comonomerconv.f (%)
Mn,theog
(g mol�1)Mn,NMR
h
(g mol�1)Mn,SEC
c
(g mol�1) ĐMd
1 TMC 2200i (1.35) — 60 1 100 4250 4450 4800i 1.502 BTMC 2000 (1.40) — 60 1 100 6200 5600 5960 1.613 BTMC 2000 (1.40)j — 60 3.5 92 11 600 — 12 300 1.724 TMC(OMe)2 2400 (1.30) — 60 5 80 5000 4300 5960 1.395 TMC(OMe)2 2400 (1.30) — 90 2.5 87 5200 5550 4800 1.366 TMC(OMe)2 2400 (1.30)j 90 5 85 9250 8950 10 200 1.597 rac-BL 2200 (1.35) — 60 3 100 4100 4700 3200 1.198 rac-BL 2200 (1.35)j — 60 6 94 6700 6200 6000 1.239 TMC(OMe)2 — 500 : 1300 : 1 : 5 60 then 90 13k 94 49 800 — 39 100 1.7210 TMC(OMe)2 — 1800 : 800 : 1 : 5 60 then 90 17k 93 60 800 — 62 800 1.90
a [Monomer] ¼ 4.0 M in toluene. b General conditions: [TMC]0/[[(BDIiPr)Zn(N(SiMe3)2)]]0/[H-PTMC-OBn]0 ¼ 200 : 1 : 10. c Number average
molar mass values (uncorrected; refer to the Experimental section) determined by SEC in THF vs. polystyrene standards. d Dispersityvalues determined by SEC in THF. e Reaction times were not necessarily optimized. f Determined by 1H NMR analysis of the crude reactionmixture. g Theoretical molar mass calculated from the relationship: Mn,theo ¼ ([comonomer]0/[H-PTMC-OBn]0
b) � Mcomonomer � conv.comonomer+ MH-PTMC-OBn (entries 1–8), or Mn,theo ¼ {([TMC]0/[BnOH]0) � MTMC � conv.TMC}
j +{([TMC(OMe)2]0/[BnOH]0) � MTMC(OMe)2 � conv.TMC(OMe)2}+ MBnOH (entries 9 and 10), with MTMC ¼ 102 g mol�1, MBTMC ¼ 208 g mol�1, MTMC(OMe)2 ¼ 162 g mol�1, MBL ¼ 86 g mol�1. h Molar massvalues determined by 1H NMR. i Number-average molar mass determined by SEC vs. polystyrene standards and corrected by a factor of 0.73.10aj [TMC]0/[[(BDI
iPr)Zn(N(SiMe3)2)]]0/[H-PTMC-OBn]0 ¼ 500 : 1 : 10. k Quantitative TMC conversion was obtained prior to addition of TMC(OMe)2 –refer to the Experimental section.
Table 3 Sequential copolymerization of TMC and TMC, BTMC, TMC(OMe)2, or rac-BL, promoted by the [(BDIiPr)Zn(N(SiMe3)2)]/1,3-propanediolsystem in toluene:a synthesis of homopolymer “PTMC-b-PTMC-b-PTMC” and triblock copolymers PBTMC-b-PTMC-b-PBTMC, P(TMC(OMe)2)-b-PTMC-b-P(TMC(OMe)2), or PHB-b-PTMC-b-PHB, respectively
Entry Comonomer
H-PTMC-Hb
Mn,SECc (g mol�1)
(ĐM)d
[TMC]0 : [TMC(OMe)2]0 :[[(BDIiPr)Zn(N(SiMe3)2)]]0 :[BnOH]0 Temp. (�C)
Reactiontimee (h)
Comonomerconv.f (%)
Mn,theog
(g mol�1)Mn,NMR
h
(g mol�1)Mn,SEC
c
(g mol�1) ĐMd
1 TMC 6200i (1.54) — 60 1 5 100 10 300 9900 9400i 1.522 BTMC 4800 (1.51) — 60 1.5 99 13 050 12 800 10 050 1.613 TMC(OMe)2 6050 (1.21) — 90 6 100 12 550 11 800 12 200 1.384 rac-BL 1800 (1.18) — 60 6 77 4750 4550 4500 1.125 TMC(OMe)2 — 500 : 1300 : 1 : 5 60 then 90 17 97j 51 050 — 39 800 1.546 TMC(OMe)2 — 1800 : 800 : 1 : 5 60 then 90 20 92j 60 550 — 54 250 1.51
a [Monomer] ¼ 4.0 M in toluene. b General conditions: [TMC]0/[[(BDIiPr)Zn(N(SiMe3)2)]]0/[H-PTMC-H]0 ¼ 200 : 1 : 5. c Number average molar mass
values (uncorrected – refer to the Experimental section) determined by SEC in THF vs. polystyrene standards. d Dispersity values determined bySEC in THF. e Reaction times were not necessarily optimized. f Determined by 1H NMR analysis of the crude reaction mixture. g Theoreticalmolar mass calculated from the relationship: Mn,theo ¼ ([comonomer]0/[H-PTMC-H]0) � Mcomonomer � conv.comonomer + MH-PTMC-H (entries 1–4),or Mn,theo ¼ {([TMC]0/[1,3-propanediol]0) � MTMC � conv.TMC
j} + {([TMC(OMe)2]0/[1,3-propanediol]0) � MTMC(OMe)2 � conv.TMC(OMe)2}+ M1,3-propanediol (entries 5 and 6), with MTMC ¼ 102 g mol�1, MBTMC ¼ 208 g mol�1, MTMC(OMe)2 ¼ 162 g mol�1, MBL ¼ 96 g mol�1 andM1,3-propanediol ¼ 76 g mol�1. h Molar mass values determined by 1H NMR. i Number-average molar mass determined by SEC vs. polystyrenestandards and corrected by a factor of 0.73.10a j Quantitative TMC conversion was obtained prior to addition of TMC(OMe)2 – refer to theExperimental section.
Paper Polymer Chemistry
�40 �C at 10 �C min�1. Thermal gravimetric analyses (TGA)were performed on a TA Instruments SDT 2960 under acontinuous ow of nitrogen.
Mechanical properties of the di- and tri-block polymers wereevaluated at Total Petrochemicals Technology Feluy, Belgium,using compression molded sheets. These tensile bar samples(length � width � thickness ¼ 17 � 4 � 0.05 mm) were preparedfrom a mini max molder of Custom Scientic Instruments Inc. at190–200 �C, using typically 100mg of polymer per sample. Tensiletests were then carried out on at least seven samples at roomtemperature according to ASTMD 882 using a ZWICK (MEC 125/2)
This journal is © The Royal Society of Chemistry 2014
apparatus with a load cell of 200 N at a cross-head speed of 10mmmin�1. Elongation at break (3b) and ultimate tensile strength (sb)values were obtained from the dynamic tensile diagrams. Thesample specimen deformation was measured from the grip-to-grip separation, which the initial value was 10 mm.
Typical synthesis of PBTMC promoted by [(BDIiPr)Zn(N(SiMe3)2)]/BnOH
In a typical experiment (Table 1, entry 5), [(BDIiPr)Zn(N(SiMe3)2)](4.0 mg, 6.2 mmol) and a solution of BnOH (3.2 mL, 31.1 mmol, 5
Polym. Chem., 2014, 5, 1229–1240 | 1233

Polymer Chemistry Paper
equiv.) in toluene (0.1 mL) were charged in a Schlenk ask inthe glovebox and stirred at room temperature over 15 min, justprior to the addition of BTMC (648 mg, 3.11 mmol, 500 equiv.).The mixture was immediately stirred at 60 �C, over the appro-priate reaction time allowing complete BTMC consumption(reaction times were not systematically optimized). The reactionwas nally quenched by adding an excess of an acetic acidsolution (ca. 1 mL of a 16.5 mmol L�1 solution in toluene). Theresulting mixture was concentrated under vacuum and theconversion determined by 1H NMR analysis of the residue. Thecrude polymer was then dissolved in CH2Cl2 and puried uponprecipitation in cold methanol, ltered and dried undervacuum. The nal polymer was then analyzed by NMR and SEC,DSC and TGA. It is noteworthy that the polycarbonate puri-cation upon their selective precipitation in MeOH is a proce-dure that simultaneously allows elimination of catalyticresidues within the ltrate. Thus, taking into account that the
Fig. 2 1H NMR (400 MHz, CDCl3, 23 �C) spectrum of H-PBTMC-OBn s
Fig. 1 Dependence of the molar mass Mn of PBTMCs, synthesized inbulk from [(BDI)Zn[N(SiMe3)2]/BnOH at 60 �C, on the benzyl alcoholcontent, at [BTMC]0 : [[(BDI)Zn[N(SiMe3)2]]0 of 500 : 1. O, experi-mental values determined by SEC; A theoretical values.
1234 | Polym. Chem., 2014, 5, 1229–1240
iROP procedure typically involves very low initial catalyticloading,1 the polymers are thereby recovered with only minutetraces, if any, of catalytic residues.
H-PBTMC-OBn1H NMR (400 MHz, CDCl3, 23 �C): d 7.22–7.35 (m, 5n + 5H,C6H5CH2OC(O)O), 5.16 (s, 2H, C6H5CH2OC(O)O), 4.61 (s, 2nH,2H, C6H5CH2OC(Me2)), 4.15–4.35 (br m, 4 nH, OC(O)OCH2-
CH(OBn)CH2OC(O)), 3.82 (br s, nH, OC(O)OCH2CH(OBn)CH2OC(O)), 3.78 (s, 2H, HOCH2) ppm (Fig. 2).Mn,SEC ¼ 16 200 gmol�1, ĐM ¼ 1.69. Tg ¼ �1 �C (Fig. S6 in the ESI†). Td ¼ 265 �C(Fig. S7 in the ESI†) (Table 1, entry 5).
Typical synthesis of PTMC-b-PTMC(OMe)2 block copolymerspromoted by [(BDIiPr)Zn(N(SiMe3)2)]/BnOH
In a typical experiment (Table 2, entry 9), [(BDIiPr)Zn(N(SiMe3)2)](4.0 mg, 6.2 mmol) and a solution of BnOH (6.4 mL, 62.2 mmol, 10equiv.) in toluene (0.1 mL) were charged in a Schlenk ask inthe glovebox and stirred at room temperature over 15 min, justprior to the addition of TMC (63.4 mg, 0.62 mmol, 100 equiv.).The mixture was immediately stirred at 60 �C over the appro-priate reaction time allowing complete TMC consumption(reaction times were not systematically optimized). The TMCconversion was not systematically determined because, undersuch operating conditions, prior investigations have feasiblyshown complete TMC conversion,13d,e,19,20 as indeed observed by1H NMR of the recovered block copolymer. TMC(OMe)2 (2.51 g,15.55 mmol, 2500 equiv.) was next tipped into the ask from aside arm. Themixture was then immediately stirred at 60–90 �C.The polymerization was allowed to proceed over the appropriatereaction time (reaction times were not systematically opti-mized). Aer (nearly) complete conversion of TMC(OMe)2, thereaction was quenched by adding an excess of an acetic acidsolution (ca. 1 mL of a 16 mmol L�1 solution in toluene). Theresulting mixture was dried under vacuum and the conversionof monomers was determined by 1H NMR analysis of the
ynthesized in bulk from BEMP/BnOH at 60 �C (Table 1, entry 14).
This journal is © The Royal Society of Chemistry 2014

Fig. 3 1H NMR (300 MHz, CDCl3, 23 �C) spectrum of H-PTMC(OMe)2-b-PTMC-OBn synthesized by in situ sequential copolymerization intoluene from [(BDIiPr)Zn[N(SiMe3)2]/BnOH at 60/90 �C (Table 2, entry 9).
Paper Polymer Chemistry
residue in CDCl3. The crude copolymer was dissolved in CH2Cl2and puried upon precipitation in cold methanol, ltered anddried under vacuum. The nal PTMC-b-PTMC(OMe)2 blockcopolymer was then analyzed by NMR, SEC and DSC.
H-PTMC(OMe)2-b-PTMC-OBn1H NMR (300 MHz, CDCl3, 23 �C): d 7.22–7.35 (m, 5H,C6H5CH2OC(O)O), 5.17 (s, 2H, C6H5CH2OC(O)O), 4.25 (t, J ¼ 6.5Hz, 4nH + 4mH, C(O)OCH2CH2CH2O, C(O)OCH2C(OMe)2CH2O),3.28 (s, 6 mH, C(O)OCH2C(OCH3)2CH2O), 2.07 (q, J¼ 7 Hz, 2nH,C(O)OCH2CH2CH2O) ppm (Fig. 3). Mn,SEC ¼ 39 100 g mol�1,ĐM ¼ 1.72, Tg ¼ 37 �C (Table 2, entry 9).
Typical synthesis of PTMC(OMe)2-b-PTMC-b-PTMC(OMe)2copolymers promoted by [(BDIiPr)Zn(N(SiMe3)2)]/1,3-propanediol or/H-PTMC-H
The same procedure as described above for the synthesis of theanalogous diblock copolymers was followed, initially using 1,3-propanediol or H-PTMC-H as an initiator/chain transfer agentfor the sequential synthesis of the corresponding triblockcopolymers (Table 3). 1H NMR data of the triblock copolymers(Fig. S14‡) were similar to those of the parent diblock copoly-mers (Fig. 3).
Results and discussionHomopolymerization of BTMC
The ROP of BTMC was rst investigated from the catalyticsystems that previously revealed successful in the “immortal”ROP of TMC and of related substituted TMC.1,6,21 In particular,the highly active b-diketiminate discrete zinc precursor [(BDIiPr)Zn(N(SiMe3)2)] (BDIiPr ¼ 2-((2,6-diisopropylphenyl)amido)-4-((2,6-diisopropylphenyl)-imino)-2-pentene) and the organicphosphazene base 2-tert-butylimino-2-diethylamino-1,3-dimethylperhydro-1,3,2-diazaphosphorine (BEMP) were selectedas pre-catalysts along with benzyl alcohol (BnOH) as a co-
This journal is © The Royal Society of Chemistry 2014
initiator/chain transfer agent. The most signicant data aregathered in Table 1.
Preliminary experiments ran with both catalytic systems, inbulk at 60 �C and at a ratio [BTMC]0/[catalyst]0/[BnOH]0 ¼500 : 1 : 5, 10, demonstrated the greater efficiency of the zinc-based system (Table 1, entries 5, 6, 13, 15). Indeed, completeBTMC conversion was reached within 30–45 min with [(BDIiPr)Zn(N(SiMe3)2)], whereas 4 h were required with BEMP. Thesame trend prevailed when the polymerizations were ran intoluene solution (Table 1, entries 9–12, 14). No signicantdifference was thus observed between bulk and solutionprocedures. The adequate compromised reaction temperatureof 60 �C is identical to the one suitable for the iROP of TMC withthis catalyst system, under similar operating con-ditions.1,6,8,19a–e,20 Besides, it avoids the thermal homopolymeri-zation of BTMC previously established at 150 �C.16a Therefore,the study of the homopolymerization of BTMC was nextfocussed on the metallo-organic zinc catalyst, in bulk condi-tions at 60 �C.22
Under these operating conditions, the experimental molarmass values of PBTMCs determined by SEC (Mn,SEC; uncor-rected for the possible difference in hydrodynamic ratio vs.polystyrene standards used for calibration) generally showedrelatively fair agreement with the calculated data (Mn,theo).PBTMCs of molar mass as high as Mn,SEC ¼ 35 500 g mol�1 –
higher than previously reported (Mn,SEC # 22 400 g mol�1)4,16 –were thus isolated. The dispersity values, ĐM¼Mw/Mn¼ ca. 1.5–1.6, were slightly larger than those commonly obtained for therelated PTMC synthesized from the same catalyst system (ĐM ¼ca. 1.3).1,19a–e,20 These values were yet within the same range asthose previously reported in the literature for the aluminium ortin catalyzed homopolymerization of BTMC (ĐM ¼ 1.44–1.92) aswell as for the catalyst-free synthesis of PBTMC (ĐM ¼ ca.1.55).4a,16a This suggested the occurrence of some side trans-carbonation reactions, typically encountered in the ROP ofcyclic carbonates.1,6,8,19–21 The efficiency of the Zn{BDIiPr} cata-lytic system in the iROP of BTMC was found comparable,
Polym. Chem., 2014, 5, 1229–1240 | 1235

Polymer Chemistry Paper
somehow lower than that obtained in the iROP of TMC. Indeed,at 60 �C, at [monomer]0 : [(BDIiPr)Zn(N(SiMe3)2)]0 : [BnOH]0 ¼500 : 1 : 5, complete TMC conversion was reached within 7 min(TOFTMC ¼ 4240 h�1),1,19a–e,20 whereas, under similar operatingconditions, 30 min were required to achieve quantitative BTMCconversion (TOFBTMC ¼ 1000 h�1; Table 1, entry 5). However,the activity of the zinc-based catalytic system presently usedremained higher than that previously obtained from Al(OiPr)3,Al(OtBu)3 or SnOct2 which operate necessarily above 140 �C, yetwith lower turnover frequencies (TOFmax ¼ 175 h�1).4 Theseresults highlight the better efficiency of the [(BDIiPr)Zn(N(SiMe3)2)]/[BnOH] catalyst system in the controlled iROP ofBTMC, as compared to previously established procedures.4,16 Inagreement with a controlled iROP process,1 an increase in therelative amount of alcohol/chain transfer agent used inducedproportionally lower molar mass PBTMCs, as depicted in Fig. 1,with a concomitant slight decrease of the activity (Table 1,entries 4–8). Raising the operating temperature from 60 �C up to130 �C resulted in a lower control in terms of theoretical andexperimental molar mass value agreement (Table 1, entries 1–3vs. 5), another piece of evidence of transcarbonation sideprocesses.
1H NMR analyses of the precipitated PBTMCs prepared fromthese binary catalyst systems displayed the main polymer chaintypical resonances –CH2CH(OCH2Ph)CH2OC(O)O (d 4.24, 3.84ppm), as well as those of the pendant benzyloxy group�OCH2C6H5 (d 7.26 ppm, 4.63 ppm; Fig. 2 and S3 in the ESI†).Besides, low intensity resonances for both hydroxymethylene(–CH2OH, d 3.76 ppm) and benzyloxycarbonate (PhCH2OC(O)O–,d 7.25, 5.14 ppm) groups were observed, as further conrmed by13C NMR analyses (Fig. S4 in the ESI†), and in agreement with theexpected formation of H-PBTMC-OBn (Scheme 2). No etherlinkages (d1H 3.1–3.4 ppm; d13C 66.5–67.7 ppm)23 were detected inany spectra of the polymers, thereby highlighting the ability ofthe initiating systems to prevent decarboxylation. The molarmass values of short-chain PBTMCs, as determined by 1H NMRspectroscopy from these terminal groups, were in close agree-ment with the calculated Mn,theo ones and the one measured bySEC, Mn,SEC as well (Table 1).
MALDI-ToF mass spectrometry analysis of a PBTMC sampleprepared from the BEMP/BnOH catalytic system, using trans-3-indoleacrylic acid (IAA) as a matrix, is consistent with theexpected chemical structure (Fig. S5†). The characteristic spec-trum revealed a major population of PBTMC with a repeat unit of208 g mol�1, which is unequivocally conrmed by the isotopicsimulation as a PBTMC end-capped by BnO-H and ionized byNa+, that is [BnO{BTMC}nH$Na]+, with e.g. m/z ¼ 2628.9 g mol�1
for n ¼ 12. A second major envelop observed corresponds to thePBTMC distribution end-capped by MeO–H (most likely due tothe use of MeOH during the deactivation procedure – refer to theExperimental section) and ionized by Na+, that is [CH3O{BTMC}nH.Na]+, with e.g. m/z ¼ 2552.9 g mol�1 for n ¼ 12.
The thermal properties of PBTMC samples were next exam-ined by DSC and TGA (Fig. S6 and S7 in the ESI‡). The DSC traceof the polymer showed a glass transition temperature at Tg¼�1�C (Fig. S6 in the ESI‡) in agreement with previous data (Tg ¼0 �C;Mn¼ 22 400 gmol�1).4a This Tg value lies in between the Tg
1236 | Polym. Chem., 2014, 5, 1229–1240
value (�15 �C;Mn ¼ 10 000 g mol�1) of TMC,10a,24 and the muchhigher Tg value (+39 �C; Mn ¼ 38 800 g mol�1) ofP(TMC(OMe)2).6 The absence of a melting transition tempera-ture highlighted the amorphous character of these functional-ized polycarbonates, similar to PTMCs10a,24 and P(TMC(OMe)2).6
The degradation temperature of PBTMC, Td¼ 265 �C, measuredby TGA (Fig. S7 in the ESI‡) is slightly higher than that of PTMC(Td ¼ 230 �C),25 or PTMC(OMe)2 (Td ¼ 225 �C).6
Deprotection of the benzyloxy groups of hydrophobicPBTMC into hydroxyl groups upon hydrogenolysis catalyzed bya Pd/C and Pd(OH)2/C mixture4b allowed the mild and selectivepreparation of hydrophilic PHTMC, without altering the maincarbonate backbone (Scheme 2).26 The 1H NMR spectrum of thedeprotected polycarbonate recorded in DMSO nicely showed, inaddition to the main chain signals (dCH2OC(O) ¼ 4.08 ppm,dCH(OH)CH2OC(O) ¼ 3.95 ppm), the disappearance of the reso-nances of the pendant and terminal benzyloxy hydrogens,eventually replaced by the corresponding terminal and pendantOH (d ¼ 5.5 ppm, Fig. S8 in the ESI‡).4 Thermal analysis ofPHTMC by DSC, to our knowledge reported herein for the rsttime, displayed a trace highlighting a negative glass transitiontemperature (Tg ¼ �18 �C) and no Tm, similar to that of theparent PTMC (Tg ¼ �15 �C, Mn ¼ 10 000 g mol�1),24 thus sug-gesting an amorphous polymer (Fig. S9 in the ESI‡).
Synthesis and characterization of PTMC diblock copolymers
Linear diblock copolymers have been synthesized from thesequential copolymerization of TMC with either BTMC orTMC(OMe)2 as carbonate, or rac-BL as cyclic ester monomers.This latter b-lactone was selected because PTMC/PHB copoly-mers remain rather limited to date,9 as opposed to other PTMC-lactone/diester copolymers,10–13 and it lies within our generalinterest in the (co)polymerization of such peculiar four-membered ring monomers.1,27 The procedure was rst carriedout in two steps, with the prior isolation of H-PTMC-OBn fromthe iROP of TMC promoted by the [(BDIiPr)Zn(N(SiMe3)2)]/BnOHcatalyst system,13d,19,20 followed by its subsequent use as aninitiator/chain transfer agent in the iROP of the co-monomer(Scheme 3; Table 2, entries 1–8). Alternatively, the sequentialcopolymerization was performed in a “traditional” one-pot, two-step approach in which TMC was rst polymerized, followed by,once its conversion completed, the in situ copolymerization ofthe second monomer, TMC(OMe)2 (Scheme 3; Table 2, entries 9and 10).13d
The [(BDIiPr)Zn(N(SiMe3)2)] precatalyst, combined to aH-PTMC-OBn (Mn,SEC ¼ ca. 2200 g mol�1) pre-polymer used asan initiator and chain transfer agent, promoted the “immortal”ring-opening copolymerization of TMC, BTMC, TMC(OMe)2,and rac-BL co-monomers, at 60 or 90 �C in toluene, affordingthe corresponding homopolymer “PTMC-b-PTMC” and diblockcopolymers PTMC-b-PBTMC, PTMC-b-P(TMC(OMe)2), or PTMC-b-PHB, respectively (Scheme 3; Table 2, entries 1–8). The molarmass values of the copolymers as determined by NMR analysis(Mn,NMR # 5500 g mol�1) nicely matched the expected valuesbased on the co-monomer conversion (Mn,theo), as well as theones measured by SEC (Mn,SEC, although uncorrected vs. the
This journal is © The Royal Society of Chemistry 2014

Scheme 4 Synthesis of H-PTMC(OMe)2-b-PTMC-OBn by in situ sequential copolymerization from [(BDIiPr)Zn[N(SiMe3)2]/BnOH.
Scheme 5 Synthesis of PTMC(OMe)2-b-PTMC-b-PTMC(OMe)2 copolymers by in situ sequential copolymerization from [(BDIiPr)Zn[N(SiMe3)2]/1,3-propanediol.
Paper Polymer Chemistry
polystyrene standards used for calibration), and the dispersityremained fair. Also, no signicant (if any) homopolymer wasformed, as evidenced by the observation of a single SEC elutionpeak featuring a molar mass value higher than that of thehomopolymer. Higher molar mass copolymers (Mn,SEC up to62 800 g mol�1) were synthesized from the TMC/TMC(OMe)2couple following the in situ sequential “immortal” copolymeri-zation of themonomers (Scheme 4; Table 2, entries 9 and 10). Inthese latter experiments, the dispersity values increasedslightly, while the Mn,SEC values remained in close agreementwith calculated ones, and the SEC traces remained monomodal.This in situ approach (Scheme 4) is practically easier to imple-ment and was similarly shown on the TMC/LLA couple toprovide a more effective (in terms of both activity and molarmass control) route towards diblock copolymers.13d
The chemical structure of PTMC-b-PTMC(OMe)2 wasconrmed by 1H NMR analyses (Fig. 3). The representative 1HNMR spectrum of these copolymers shown Fig. 3 displays thecharacteristic signals of the main chain methylene (dOCH2 4.25ppm, dOCH2CH2CH2 2.07 ppm for PTMC; dOCH2CH 4.25 ppm,dCH(OCH3)2 3.28 ppm for PTMC(OMe)2) of both carbonate segmentsalong with the benzyloxy terminal group (dOCH2C6H5 7.35 ppm).The PTMC-b-PBTMC and PTMC-b-PHB copolymers were similarlycharacterized by 1H NMR (Fig. S10 and S11‡, respectively).
The thermal behavior of the diblock copolymers was deter-mined by DSC. The DSC curve of H-PTMC(OMe)2-b-PTMC-OBnsamples showed a glass transition at Tg ¼ 37 �C corresponding
This journal is © The Royal Society of Chemistry 2014
to the PTMC(OMe)2 segment6 (Fig. S12 in the ESI‡). The Tgcorresponding to the PTMC segment, expected at ca. �15 �C,could not be clearly observed and no melting transitionwas seen.
Also, the deprotection of the acetal and benzyloxy functionsin the PTMC-b-PTMC(OMe)2 and PTMC-b-PBTMC diblockcopolymers enabled the synthesis of the PTMC-b-PTMC(O) andPTMC-b-PTMC(OH) copolymers, respectively (Scheme S2 in theESI‡). As expected, NMR analyses evidenced the disappearanceof these –OMe and –OBn pendant and u-terminal functions,concomitant with the appearance of the methylene hydrogensin a of the carbonyl (d ¼ 5.2 ppm), as illustrated with thespectrum of PTMC-b-PTMC(O) (Fig. S13 in the ESI‡).
Synthesis and characterization of PTMC triblock copolymers
Triblock copolymers with a central PTMC segment, namelyPBTMC-b-PTMC-b-PBTMC, PTMC(OMe)2-b-PTMC-b-PTMC(OMe)2and PHB-b-PTMC-b-PHB, have been synthesized according to thesame two procedures as those described above for the synthesis ofthe corresponding diblock copolymers (Scheme 3). The catalyticsystem was composed of the [(BDIiPr)Zn[N(SiMe3)2] catalyst, andeither 1,3-propanediol (for the stepwise approach), or the pre-synthesized a,u-dihydroxy-telechelic H-PTMC-H20 (for thedirect one-pot route), acting as a (macro)initiator/chain transferagent, and operated at 60 or 90 �C in toluene (Scheme 5). Themost signicant results are gathered in Table 3.
Polym. Chem., 2014, 5, 1229–1240 | 1237
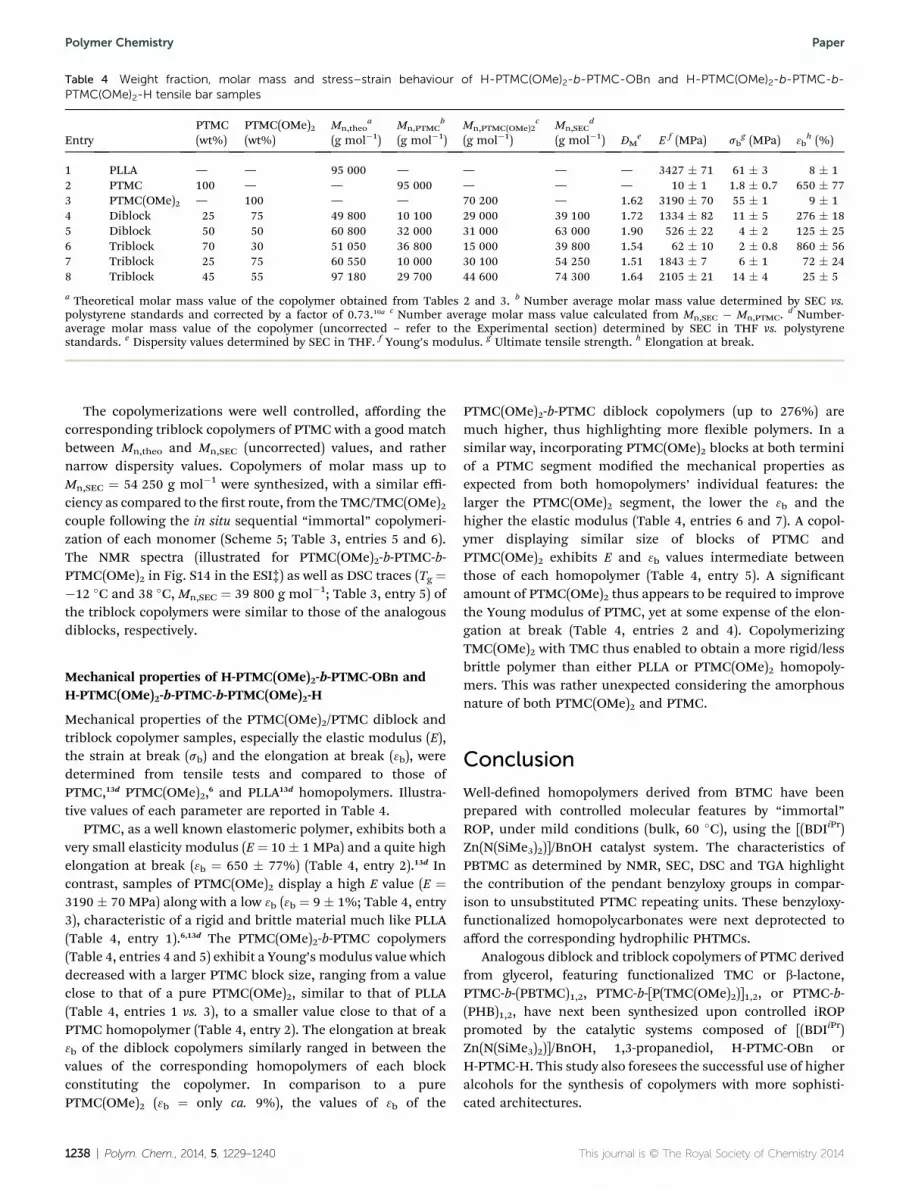
Table 4 Weight fraction, molar mass and stress–strain behaviour of H-PTMC(OMe)2-b-PTMC-OBn and H-PTMC(OMe)2-b-PTMC-b-PTMC(OMe)2-H tensile bar samples
EntryPTMC(wt%)
PTMC(OMe)2(wt%)
Mn,theoa
(g mol�1)Mn,PTMC
b
(g mol�1)Mn,PTMC(OMe)2
c
(g mol�1)Mn,SEC
d
(g mol�1) ĐMe E f (MPa) sb
g (MPa) 3bh (%)
1 PLLA — — 95 000 — — — — 3427 � 71 61 � 3 8 � 12 PTMC 100 — — 95 000 — — — 10 � 1 1.8 � 0.7 650 � 773 PTMC(OMe)2 — 100 — — 70 200 — 1.62 3190 � 70 55 � 1 9 � 14 Diblock 25 75 49 800 10 100 29 000 39 100 1.72 1334 � 82 11 � 5 276 � 185 Diblock 50 50 60 800 32 000 31 000 63 000 1.90 526 � 22 4 � 2 125 � 256 Triblock 70 30 51 050 36 800 15 000 39 800 1.54 62 � 10 2 � 0.8 860 � 567 Triblock 25 75 60 550 10 000 30 100 54 250 1.51 1843 � 7 6 � 1 72 � 248 Triblock 45 55 97 180 29 700 44 600 74 300 1.64 2105 � 21 14 � 4 25 � 5
a Theoretical molar mass value of the copolymer obtained from Tables 2 and 3. b Number average molar mass value determined by SEC vs.polystyrene standards and corrected by a factor of 0.73.10a c Number average molar mass value calculated from Mn,SEC � Mn,PTMC.
d Number-average molar mass value of the copolymer (uncorrected – refer to the Experimental section) determined by SEC in THF vs. polystyrenestandards. e Dispersity values determined by SEC in THF. f Young’s modulus. g Ultimate tensile strength. h Elongation at break.
Polymer Chemistry Paper
The copolymerizations were well controlled, affording thecorresponding triblock copolymers of PTMC with a good matchbetween Mn,theo and Mn,SEC (uncorrected) values, and rathernarrow dispersity values. Copolymers of molar mass up toMn,SEC ¼ 54 250 g mol�1 were synthesized, with a similar effi-ciency as compared to the rst route, from the TMC/TMC(OMe)2couple following the in situ sequential “immortal” copolymeri-zation of each monomer (Scheme 5; Table 3, entries 5 and 6).The NMR spectra (illustrated for PTMC(OMe)2-b-PTMC-b-PTMC(OMe)2 in Fig. S14 in the ESI‡) as well as DSC traces (Tg ¼�12 �C and 38 �C, Mn,SEC ¼ 39 800 g mol�1; Table 3, entry 5) ofthe triblock copolymers were similar to those of the analogousdiblocks, respectively.
Mechanical properties of H-PTMC(OMe)2-b-PTMC-OBn andH-PTMC(OMe)2-b-PTMC-b-PTMC(OMe)2-H
Mechanical properties of the PTMC(OMe)2/PTMC diblock andtriblock copolymer samples, especially the elastic modulus (E),the strain at break (sb) and the elongation at break (3b), weredetermined from tensile tests and compared to those ofPTMC,13d PTMC(OMe)2,6 and PLLA13d homopolymers. Illustra-tive values of each parameter are reported in Table 4.
PTMC, as a well known elastomeric polymer, exhibits both avery small elasticity modulus (E ¼ 10 � 1 MPa) and a quite highelongation at break (3b ¼ 650 � 77%) (Table 4, entry 2).13d Incontrast, samples of PTMC(OMe)2 display a high E value (E ¼3190 � 70 MPa) along with a low 3b (3b ¼ 9 � 1%; Table 4, entry3), characteristic of a rigid and brittle material much like PLLA(Table 4, entry 1).6,13d The PTMC(OMe)2-b-PTMC copolymers(Table 4, entries 4 and 5) exhibit a Young’s modulus value whichdecreased with a larger PTMC block size, ranging from a valueclose to that of a pure PTMC(OMe)2, similar to that of PLLA(Table 4, entries 1 vs. 3), to a smaller value close to that of aPTMC homopolymer (Table 4, entry 2). The elongation at break3b of the diblock copolymers similarly ranged in between thevalues of the corresponding homopolymers of each blockconstituting the copolymer. In comparison to a purePTMC(OMe)2 (3b ¼ only ca. 9%), the values of 3b of the
1238 | Polym. Chem., 2014, 5, 1229–1240
PTMC(OMe)2-b-PTMC diblock copolymers (up to 276%) aremuch higher, thus highlighting more exible polymers. In asimilar way, incorporating PTMC(OMe)2 blocks at both terminiof a PTMC segment modied the mechanical properties asexpected from both homopolymers’ individual features: thelarger the PTMC(OMe)2 segment, the lower the 3b and thehigher the elastic modulus (Table 4, entries 6 and 7). A copol-ymer displaying similar size of blocks of PTMC andPTMC(OMe)2 exhibits E and 3b values intermediate betweenthose of each homopolymer (Table 4, entry 5). A signicantamount of PTMC(OMe)2 thus appears to be required to improvethe Young modulus of PTMC, yet at some expense of the elon-gation at break (Table 4, entries 2 and 4). CopolymerizingTMC(OMe)2 with TMC thus enabled to obtain a more rigid/lessbrittle polymer than either PLLA or PTMC(OMe)2 homopoly-mers. This was rather unexpected considering the amorphousnature of both PTMC(OMe)2 and PTMC.
Conclusion
Well-dened homopolymers derived from BTMC have beenprepared with controlled molecular features by “immortal”ROP, under mild conditions (bulk, 60 �C), using the [(BDIiPr)Zn(N(SiMe3)2)]/BnOH catalyst system. The characteristics ofPBTMC as determined by NMR, SEC, DSC and TGA highlightthe contribution of the pendant benzyloxy groups in compar-ison to unsubstituted PTMC repeating units. These benzyloxy-functionalized homopolycarbonates were next deprotected toafford the corresponding hydrophilic PHTMCs.
Analogous diblock and triblock copolymers of PTMC derivedfrom glycerol, featuring functionalized TMC or b-lactone,PTMC-b-(PBTMC)1,2, PTMC-b-[P(TMC(OMe)2)]1,2, or PTMC-b-(PHB)1,2, have next been synthesized upon controlled iROPpromoted by the catalytic systems composed of [(BDIiPr)Zn(N(SiMe3)2)]/BnOH, 1,3-propanediol, H-PTMC-OBn orH-PTMC-H. This study also foresees the successful use of higheralcohols for the synthesis of copolymers with more sophisti-cated architectures.
This journal is © The Royal Society of Chemistry 2014

Paper Polymer Chemistry
All (co)polymers have been characterized at the molecular(NMR, SEC) and thermal (DSC, TGA) level, revealing amorphousblock copolymers. Further evaluation of the mechanical prop-erties (E, sb, 3b) of PTMC/P(TMC(OMe)2) copolymers revealedthat, unlike PTMC which is elastomeric and P(TMC(OMe)2)which is rather rigid and brittle much like semi-crystallinePLLA, the diblocks and triblocks behaved similarly with char-acteristics lying in between those of each homopolymer,without any signicant inuence of the topology. Thus PTMC/PTMC(OMe)2 block copolymers mechanically behave similarlyto PTMC/PLLA block copolymers.13d A PTMC(OMe)2 segmenthence provides rigidity to a PTMC, in a similar way to whichPLLA does.
Acknowledgements
The authors gratefully thank Total Petrochemicals Co. fornancial support of this research (Ph.D. grants to M.H. and toW.G.), Labso Chimie Fine (Blanquefort, France) for kindlysupplying TMC, and the Region Bretagne and RennesMetropole for equipment support.
References
1 (a) N. Ajellal, J.-F. Carpentier, C. Guillaume, S. M. Guillaume,M. Helou, V. Poirier, Y. Sarazin and A. Trifonov, DaltonTrans., 2010, 39, 8363–8376; (b) S. M. Guillaume andJ.-F. Carpentier, Catal. Sci. Technol., 2012, 2, 898–906.
2 (a) J. J. Bozell and G. R. Petersen, Green Chem., 2010, 12, 539–554; (b) M. Pagliaro and M. Rosi, in The future of glycerol: newuses of a versatile raw material, ed. M. Pagliaro and M. Rosi,RSC Green Chemistry Book Series, 2008, ch. 1, pp. 1–17; (c)A. Behr, J. Eilting, K. Irawadi, J. Leschinski and F. Lindner,Green Chem., 2008, 10, 13–30; (d) C.-H. Zhou,J. N. Beltramini, Y.-X. Fan and G. Q. Lu, Chem. Soc. Rev.,2008, 37, 527–549; (e) L. Bournay, D. Casanave, B. Delfort,G. Hillion and J. A. Chodorge, Catal. Today, 2005, 106, 190–192; (f) T. Werpy, G. Petersen, A. Aden, J. Bozell,J. Holladay, J. White and A. Manheim, Results of ScreeningPotential Candidates from Sugars and Synthesis Gas, US DoEreport, Washington, DC, 2004, vol. 1.
3 (a) A. Behr, J. Eilting, K. Irawadi, J. Leschinski andF. Lindner, Green Chem., 2008, 10, 13–30; (b) C.-H. Zhou,J. N. Beltramini, Y.-X. Fan and G. Q. Lu, Chem. Soc. Rev.,2008, 37, 527–549; (c) D. T. Johnson and K. A. Taconi,Environ. Prog., 2007, 26, 338–348.
4 (a) X.-L. Wang, R.-X. Zhuo, L.-J. Liu, F. He and G. Liu, J.Polym. Sci., Part A: Polym. Chem., 2002, 40, 70–75; (b)W. C. Ray and M. W. Grinstaff, Macromolecules, 2003, 36,3557–3562.
5 (a) J. Simon, J. V. Olsson, H. Kim, I. F. Tenney andR. M. Waymouth, Macromolecules, 2012, 45, 9275–9284; (b)A. N. Zelikin, P. N. Zawaneh and D. Putnam,Biomacromolecules, 2006, 7, 3239–3244; (c) P. N. Zawaneh,A. M. Doody, A. N. Zelikin and D. Putnam,Biomacromolecules, 2006, 7, 3245–3251; (d) A. N. Zelikinand D. Putnam, Macromolecules, 2005, 38, 5532–5537.
This journal is © The Royal Society of Chemistry 2014
6 M. Helou, J.-M. Brusson, J.-F. Carpentier andS. M. Guillaume, Polym. Chem., 2011, 2, 2789–2795.
7 (a) J. Feng, R.-X. Zhuo and X.-Z. Zhang, Prog. Polym. Sci.,2012, 37, 211–236; (b) H. Tian, Z. Tang, X. Zhuang, X. Chenand X. Jing, Prog. Polym. Sci., 2012, 37, 237–280; (c)B. D. Ulery, L. S. Nair and C. T. Laurencin, J. Polym. Sci.,Part B: Polym. Phys., 2011, 49, 832–864.
8 (a) F. Suriano, O. Coulembier, J. L. Hedrick and P. Dubois,Polym. Chem., 2011, 2, 528–533; (b) S. Tempelaar,L. Mespouille, O. Coulembier, P. Dubois and A. P. Dove,Chem. Soc. Rev., 2013, 42, 1312–1336.
9 (a) Y. Hori, Y. Gonda, Y. Takahashi and T. Hagiwara,Macromolecules, 1996, 29, 804–806; (b) H. R. Kricheldorf andA. Stricker, Macromol. Chem. Phys., 1999, 200, 1726–1733.
10 Illustrative references: (a) I. Palard, M. Schappacher,B. Belloncle, A. Soum and S. M. Guillaume, Chem.–Eur. J.,2007, 13, 1511–1521; (b) O. Coulembier, S. Moins andP. Dubois, Macromolecules, 2011, 44, 7493–7498; (c) J. Ling,W. Zhu and Z. Shen, Macromolecules, 2004, 37, 758–763.
11 Illustrative references: (a) R. Zurita, J. Puiggali, L. Franco andA. Rodriguez-Galan, J. Polym. Sci., Part A: Polym. Chem., 2006,44, 993–1013; (b) J. Cai, K. J. Zhu and Y. Shilin, Polym. Int.,1996, 41, 369–375.
12 Illustrative references: (a) S. Y. Kim, H. J. Kim, K. E. Lee,S. S. Han, Y. S. Sohn and B. Jeong, Macromolecules, 2007,40, 5519–5525; (b) E. Dıaz-Celorio, L. Franco, A. Rodrıguez-Galan and J. Puiggalı, Eur. Polym. J., 2012, 48, 60–73; (c)J. S. Cho, B. S. Kim, H. Hyun, J. Y. Youn, M. S. Kim,J. H. Ko, Y. H. Park, G. Khang and H. B. Lee, Polymer,2008, 49, 1777–1782.
13 Illustrative references: (a) D. J. Darensbourg, W. Choi andC. P. Richers, Macromolecules, 2007, 40, 3521–3523; (b)D. Pospiech, H. Komber, D. Jehnichen, L. Haussler,K. Eckstein, H. Scheibner, A. Janke, H. R. Kricheldorf andO. Petermann, Biomacromolecules, 2005, 6, 439–446; (c)D. J. Darensbourg, W. Choi, O. Karroonnirun andN. Bhuvanesh, Macromolecules, 2008, 41, 3493–3502; (d)W. Guerin, M. Helou, J.-F. Carpentier, M. Slawinski,J.-M. Brusson and S. M. Guillaume, Polym. Chem., 2013, 4,1095–1106; (e) W. Guerin, M. Helou, M. Slawinski,J.-M. Brusson, S. M. Guillaume and J.-F. Carpentier, Polym.Chem., 2013, 4, 3686–3693.
14 Illustrative references: (a) O. Coulembier, S. Moins andP. Dubois, Macromolecules, 2011, 44, 7493–7498; (b)J. Mindemark and T. Bowden, Polymer, 2011, 52, 5716–5722; (c) X. Chen, S. P. McCarthy and R. A. Gross, J. Appl.Polym. Sci., 1998, 67, 547–557; (d) J. Ling and Z. Shen,Macromol. Chem. Phys., 2002, 203, 735–738; (e) T. F. Al-Azemi, J. P. Harmon and K. S. Bisht, Biomacromolecules,2000, 1, 493–500; (f) F. Suriano, R. C. Pratt, J. P. K. Tan,N. Wiradharma, A. Nelson, Y.-Y. Yang, P. Dubois andJ. L. Hedrick, Biomaterials, 2010, 31, 2637–2645; (g)J. P. K. Tan, S. H. Kim, F. Nederberg, K. Fukushima,D. J. Coady, A. Nelson, Y. Y. Yang and J. L. Hedrick,Macromol. Rapid Commun., 2010, 31, 1187–1192; (h)L.-L. Mei, G.-P. Yan, X.-H. Yu, S.-X. Cheng and J.-Y. Wu, J.Appl. Polym. Sci., 2008, 108, 93–98; (i) L. Mespouille,
Polym. Chem., 2014, 5, 1229–1240 | 1239

Polymer Chemistry Paper
F. Nederberg, J. L. Hedrick and P. Dubois, Macromolecules,2009, 42, 6419–6321.
15 (a) X.-L. Wang, R.-X. Zhuo, S.-W. Huang, L.-J. Liu and F. He,Macromol. Chem. Phys., 2002, 203, 985–990; (b)J. B. Wolinsky, S. T. Yohe, Y. L. Colson and M. W. Grinstaff,Biomacromolecules, 2012, 12, 406–411; (c) K.-L. Lai, L.-J. Ji,C.-Y. Long, L. Li, B. He, Y. Wu and Z.-W. Gu, J. Appl. Polym.Sci., 2012, 123, 2204–2210; (d) J. Yang, Q. Hao, X. Liu, C. Baand A. Cao, Biomacromolecules, 2004, 5, 209–218; (e) Y.-P. Qi,Z.-M. Miao, S.-X. Cheng and X.-Z. Zhang, J. Appl. Polym. Sci.,2010, 115, 3451–3455; (f) F. Zeng, J. Liu and C. Allen,Biomacromolecules, 2004, 5, 1810–1817; (g) Z. Xie, H. Guan,C. Lu, X. Chen and X. Jing, Acta Biomater., 2005, 1, 635–641;(h) F. He, Y. Wang, J. Feng, R. Zhuo and X. Wang, Polymer,2003, 44, 3215–3219; (i) F. He, H.-L. Jia, G. Liu, Y.-P. Wang,J. Feng and R.-X. Zhuo, Biomacromolecules, 2006, 7, 2269–2273.
16 (a) J. Feng, X.-L. Wang, F. He and R.-X. Zhuo, Macromol.Rapid Commun., 2007, 28, 754–758; (b) J. Feng, R. Zhuo,F. He and X. Wang, Macromol. Symp., 2003, 195, 237–240.
17 C. Guillaume, J.-F. Carpentier and S. M. Guillaume, Polymer,2009, 50, 5909–5917.
18 (a) B. M. Chamberlain, M. Cheng, D. R. Moore, T. M. Ovitt,E. Lobkovsky and G. W. Coates, J. Am. Chem. Soc., 2001,123, 3229–3238; (b) L. R. Rieth, D. R. Moore,E. B. Lobkovsky and G. W. Coates, J. Am. Chem. Soc., 2002,124, 15239–15248.
19 (a) M. Helou, O. Miserque, J.-M. Brusson, J.-F. Carpentierand S. M. Guillaume, Chem.–Eur. J., 2008, 14, 8772–8775;(b) M. Helou, O. Miserque, J.-M. Brusson, J.-F. Carpentierand S. M. Guillaume, Adv. Synth. Catal., 2009, 351, 1312–1324; (c) M. Helou, O. Miserque, J.-M. Brusson,J.-F. Carpentier and S. M. Guillaume, ChemCatChem, 2010,2, 306–313; (d) M. Helou, J.-F. Carpentier andS. M. Guillaume, Green Chem., 2011, 13, 266–271; (e)M. Helou, O. Miserque, J.-M. Brusson, J.-F. Carpentier andS. M. Guillaume, Chem.–Eur. J., 2010, 16, 13805–13813.
20 M. Helou, O. Miserque, J.-M. Brusson, J.-F. Carpentier andS. M. Guillaume, Macromol. Rapid Commun., 2009, 30,2128–2135.
1240 | Polym. Chem., 2014, 5, 1229–1240
21 P. Brignou, J.-F. Carpentier and S. M. Guillaume,Macromolecules, 2011, 44, 5127–5135.
22 Bulk operating conditions were preferentially selected toremain closer to environmentally friendly procedures asdened by one of the principles of green chemistry inAnastas, P. T. Warner, J. C. Green Chemistry: Theory andPractice, Oxford University Press, Oxford, 1998.
23 (a) T. Ariga, T. Takata and T. Endo,Macromolecules, 1997, 30,737–744; (b) H. R. Kricheldorf and J. Jenssen, J. Macromol.Sci., Part A: Pure Appl.Chem., 1989, 26, 631–644.
24 K. J. Zhu, R. W. Hendren, K. Jensen and C. G. Pitt,Macromolecules, 1991, 24, 1736–1740.
25 F. Nedeberg, V. Trang, R. C. Pratt, A. F. Mason, C. W. Frank,R. M. Waymouth and J. L. Hedrick, Biomacromolecules, 2007,8, 3294–3297.
26 The procedure followed in the present work is slightlymodied compared to the previously reportedhydrogenolysis of PBTMC.4 The use of the Pd/C–Pd(OH)2/C(10–20% weight) mixture,4b along with a pressure of 10bars of H2 (instead of ordinary pressure)4a allowed to reachquantitative deprotection much more efficiently (100% in15 h) than the Pd/C catalyst by itself (typically 60% (ourdata)–70% (ref. 4a)).
27 (a) A. Amgoune, C. M. Thomas, S. Ilinca, T. Roisnel andJ.-F. Carpentier, Angew. Chem., Int. Ed., 2006, 45, 2782–2784; (b) C. Guillaume, N. Ajellal, J.-F. Carpentier andS. M. Guillaume, J. Polym. Sci., Part A: Polym. Chem., 2011,49, 907–917; (c) M. Helou, G. Moriceau, Z. W. Huang,S. Cammas-Marion and S. M. Guillaume, Polym. Chem.,2011, 2, 840–850; (d) J.-F. Carpentier, Macromol. RapidCommun., 2010, 31, 1696–1705; (e) M. Bouyahyi, N. Ajellal,E. Kirillov, C. M. Thomas and J.-F. Carpentier, Chem.–Eur.J., 2011, 17, 1872–1883; (f) C. G. Jaffredo, J.-F. Carpentierand S. M. Guillaume, Macromol. Rapid Commun., 2012, 33,1938–1944; (g) C. G. Jaffredo, J.-F. Carpentier andS. M. Guillaume, Polym. Chem., 2013, 4, 3837–3850; (h)S. M. Guillaume, L. Annunziata, L. Maron, C. Iner, I. delRosal, P. W. Roesky and M. Schmid, Polym. Chem., 2013, 4,3077–3087.
This journal is © The Royal Society of Chemistry 2014





























