Embed Size (px)
Citation preview

Available online at www.sciencedirect.com
journal homepage: www.elsevier.com/locate/yexcr
E X P E R I M E N T A L C E L L R E S E A R C H 3 2 2 ( 2 0 1 4 ) 3 2 4 – 3 3 4
0014-4827/$ - see frohttp://dx.doi.org/10.1
nCorresponding auE-mail addresses1 Equal contribut2 Present address
91010, USA.3 Present address
Montpellier 34293, F
Research Article
Molecular characterization of collaborator of ARF(CARF) as a DNA damage response and cell cyclecheckpoint regulatory protein
Rumani Singha,1, Rajkumar S. Kalraa,1, Kamrul Hasana,2, Zeenia Kaulb,c,Caroline T. Cheunga,3, Lily Huschtschab, Roger R. Reddelb, Sunil C. Kaula,n,Renu Wadhwaa,n
aCell Proliferation Research Group and DBT-AIST International Laboratory for Advanced Biomedicine, National Institute ofAdvanced Industrial Science and Technology (AIST), Central 4, 1-1-1 Higashi, Tsukuba, Ibaraki 305-8562, JapanbChildren's Medical Research Institute, 214 Hawkesbury Road, Westmead, New South Wales 2145, AustraliacDepartment of Molecular Virology, Immunology and Medical Genetics, 960 Biomedical Research Tower, The Ohio StateUniversity, Columbus, OH 43210, USA
a r t i c l e i n f o r m a t i o n
Article Chronology:
Received 25 September 2013Received in revised form6 January 2014Accepted 21 January 2014Available online 28 January 2014
Keywords:
CARFDNA damage responseSenescenceUpregulationMechanism
nt matter & 2014 Elsevier016/j.yexcr.2014.01.022
thor. Fax: þ81 29 861 2900: [email protected] (S.C. Kaors.: Department of Molecu
: Institute of Molecular Grance.
a b s t r a c t
CARF is an ARF-binding protein that has been shown to regulate the p53–p21–HDM2 pathway.CARF overexpression was shown to cause growth arrest of human cancer cells and prematuresenescence of normal cells through activation of the p53 pathway. Because replicative senescenceinvolves permanent withdrawal from the cell cycle in response to DNA damage response-mediated signaling, in the present study we investigated the relationship between CARF and thecell cycle and whether it is involved in the DNA damage response. We demonstrate that the half-life of CARF protein is less than 60 min, and that in cycling cells CARF levels are highest in G2 and
early prophase. Serially passaged normal human skin and stromal fibroblasts showed upregula-tion of CARF during replicative senescence. Induction of G1 growth arrest and senescence by avariety of drugs was associated with increase in CARF expression at the transcriptional andtranslational level and was seen to correlate with increase in DNA damage response andcheckpoint proteins, ATM, ATR, CHK1, CHK2, γH2AX, p53 and p21. Induction of growth arrest byoncogenic RAS and shRNA-mediated knockdown of TRF2 in cancer cells also caused upregulationof CARF. We conclude that CARF is associated with DNA damage response and checkpointsignaling pathways.
& 2014 Elsevier Inc. All rights reserved.
Inc. All rights reserved.
.ul), [email protected] (R. Wadhwa).
lar Medicine, City of Hope Beckman Research Institute, 1500 East Duarte Road, Duarte, CA
enetics of Montpellier/University of Montpellier I & II/CNRS, 1919 Route de Mende, Cedex 5,

E X P E R I M E N T A L C E L L R E S E A R C H 3 2 2 ( 2 0 1 4 ) 3 2 4 – 3 3 4 325
Introduction
Maintenance of genomic integrity and fidelity is mandatory to cellsurvival, function and proliferation. Normal cells exposed togenotoxic and cytotoxic insults are known to initiate the DNAdamage response (DDR) that acts as a guardian of genomicintegrity. DDR involves activation of DNA damage sensors, trans-ducers and effectors that recruit the DNA repair and cell cyclecheckpoint proteins to the DNA damage site and coordinate theDNA repair process mediated by a network of phosphorylationand ubiquitination pathways. DDR has emerged as a stress- andoncogene-induced biological response that causes either senes-cence or apoptosis and hence serves as a barrier to the initiationof cell transformation and carcinogenesis [1,2]. Chronic DDRactivation, on the other hand, creates selective pressure thateventually favors outgrowth of malignant clones that are func-tionally compromised for one or more of the DDR components.
DDR is initiated with activation and translocation of the DNAdamage sensor proteins to the damage site followed by formationof an active complex (MRN complex) that contains phosphory-lated histone (γH2AX), 53BP1, MDC1, MRE11, NBS1, RAD50, BRCA1,PPAR polymerase and the activated forms of DNA damagecheckpoint kinases CHK1 and CHK2 [3–5]. Telomere shorteningassociated with replicative senescence has been shown to evokeDDR [5], and the senescent cells have been shown to accumulatesenescence-associated DDR foci (SDFs) that contain the activatedform of ATM, its phosphorylated substrates and γH2AX. Whenproliferation of mouse embryonic fibroblasts was extended byculture in low oxygen atmosphere, SDFs were delayed suggestingthe tight relationship between senescence and DDR [6,7]. Repli-cative senescence in human fibroblasts was shown to be propor-tional to the accumulation of DDRþ telomeres [7,8]. Similarincrease in DDRþ telomeres was seen in gut and liver of agingmice [7]. Functional inactivation of ATM, ATR, CHK1 and CHK2and their downstream effector tumor suppressor pathway p53–p21 that forms the core DDR response leads to escape fromcellular senescence [5,6,9–13] suggesting their key role in cellularsenescence. Molecular components of DDR, their mechanisms ofoperation and interaction with the cell cycle checkpoint proteinsare rapidly expanding avenues of molecular understanding ofDDR and its use in cancer therapeutics.
We had previously identified a nuclear protein, CARF (colla-borator of ARF) as an ARF (alternative reading frame)-bindingpartner [14–17], which is also referred to as CDKN2A InteractingProtein. CARF was shown to activate the p53–p21 pathway andregulate p53-dependent senescence and apoptosis in a dose-dependent manner [18]. CARF-compromised cells show mitoticcatastrophe as marked by hyper-condensed chromatin, polyploidyas well as deranged spindle fiber and centrosome assemblies [19].We showed that the inhibition of CARF induces cell death via ATR/CHK1 deregulation that also involves the Ras-MAPK, ATM/CHK2and RB/E2F1 pathways [19]. Overexpression of CHK1 in CARF-compromised cells reverted not only the cell death phenotype,but also abolished the induction of γH2AX. The significance of ATRand CHK1 in maintaining genome integrity has been shown byseveral other studies, in which exogenous suppression of thispathway leads to formation of single-stranded DNA, DNA breaksand telomere instability; complete knockout of ATR or CHK1 isembryonic lethal [20,21]. Since CHK1 targeting can enhance the
antitumor effects of radio- and chemotherapy, its inhibitors arecurrently being developed as adjuvants to enhance the efficacy ofgenotoxic antitumor agents [22–24]. In this viewpoint, CARF mayalso be useful as a therapeutic reagent.In this study, we investigated the expression of CARF during cell
cycle progression and arrest caused by replicative, as well asstress-, oncogene- and drug-induced senescence in human cells.We found that CARF is a short-lived protein and its level variesaccording to cell cycle stage in asynchronously dividing cells. Itshowed an increase during the replicative senescence of humannormal fibroblasts derived from lung and breast stromal tissues.Induction of G1 growth arrest or senescence by exogenousexpression of RAS, shRNA-mediated knockdown of TRF2 and avariety of drugs caused an increase in CARF mRNA and protein. Itcorrelated well with activation of DDR proteins (γH2AX, ATM,ATR, CHK1, CHK2, p53, and p21) signifying its involvement incheckpoint response.
Materials and methods
Cell culture, drugs and cell cycle analysis
U2OS, HeLa, SKOV3, Saos-2, MRC5, Fre 80, Fre 102s-3, Fre 92s-2,HCT116 and TIG-1 cells were maintained in Dulbecco's ModifiedEagle's Medium DMEM (Invitrogen, Carlsbad, CA) supplementedwith 10% fetal bovine serum in a humidified incubator (37 1C and5% CO2). For protein studies, cells were seeded in 10-cm dishes at 60–70% density. After the cells had attached well (16–18 h), cyclohex-imide (100 μg/ml, Sigma) was added to the medium, and the cellswere harvested at different time points as shown. In parallelexperiments, MG132 was added to inhibit proteasome mediatedprotein degradation. Cell lysates were analyzed for the expression ofCARF and p53 by SDS-PAGE andWestern blotting as described below.For cell cycle studies, cells were synchronized at the G1/S
boundary by the double thymidine blocking method. Cells weregrown overnight in DMEM (as described above) followed byculture in 2 μM thymidine-supplemented medium for 14 h andsubsequently in normal medium for another 12 h. Following thefirst cycle of thymidine treatment, normal medium was onceagain replaced with 2 μM thymidine-supplemented medium for12 h followed by washing and incubation in normal medium. Cellswere collected at indicated time points for CARF expressionanalysis by Western blotting as described below. In order to blockcell cycle at G2/M boundary, U2OS cells were treated withnocodazole (Sigma-Aldrich, St. Louis, MO) at 100 μg/ml for10–12 h. Cells were washed and incubated in DMEM to restartthe cell cycle and were collected at different time points forWestern blotting and cell cycle analysis as described below.For colony forming assay, cells were transfected with control
and CARF siRNA as described earlier [14]. Forty-eight hours aftertransfection, cells were treated with nocodazole at 100 μg/ml for12 h. Loosely attached and rounded mitotic cells were carefullyseparated by gentle shaking. These cells were collected bycentrifugation at 1500g for 5 min and plated (500 cells/well) in6-well dishes in triplicate in normal medium. Cells were incu-bated for 6–8 days with regular change of medium every otherday. Colonies were fixed with methanol and stained with 1%crystal violet.

Table 1 – List of drugs and their concentrations used for induction of senescence in cancer cells.
No. Drug name IC50 (μM) Senescence-inducing conc. (μM)
1 Adriamycin 0.25 0.052 Taxol 10 0.023 EGC 50 1.04 EGCG 50 0.55 Camptothecin 10 26 Nocodazole 50 0.027 Retinoic acid 50 28 Curcumin 10 0.029 Gallium nitrate 10 0.0210 Mitoxantrone 2.5 0.00511 Etoposide 10 0.2
E X P E R I M E N T A L C E L L R E S E A R C H 3 2 2 ( 2 0 1 4 ) 3 2 4 – 3 3 4326
For drug-induced growth arrest, cells were plated at 60–70%confluency and treated with eleven drugs at concentrationsdescribed earlier [25] (Table 1). Typically, the cells were treatedwith the indicated concentration of drugs for 48 h. For cell cycleanalysis, the treated cells (1�105) were harvested with trypsin,washed twice with PBS and fixed with 70% ethanol at 4 1C for12 h. The fixed cells were centrifuged (1000g for 10 min), washedtwice with cold PBS and re-suspended in 0.25 ml PBS followed byRNase A (50 μg) treatment at 37 1C for 1 h. The cell suspensionwas stained with 10 μg propidium iodide (PI) by incubation at 4 1Cin the dark for 30 min. The cell cycle acquisition and analysis wasperformed using a Guava cell cycle flow cytometer (Millipore,Billerica, MA) following manufacturer's protocol.
Cell transfections and infections
For Ras infection, retrovirus carrying the RasV12 gene wasproduced using Plat-E, an ecotropic murine leukemia virus(MuLV)-packaging cell line. Plat-E cells were transfected withthe pVPack-GP and pVPack VSVG (Stratagene, La Jolla, CA) viralpackaging vectors, together with pBabe-puro or pBabe-puroRasV12 retroviral vector using FuGENE6 (Roche, Basel, Switzer-land). After 48 h, culture supernatants were collected, passedthrough 0.45 μm filters and used as viral stocks for infection ofU2OS cells plated in 10-cm dishes. Cells were treated withpolybrene (8 μg/ml) at 37 1C for 1 h following which they wereincubated with filtered viral stock at 37 1C for 48 h. The infectedcells were selected in medium containing puromycin (2 μg/ml)until stable cell lines were obtained.TRF2 shRNA plasmids were transiently transfected into cells
using FuGENE6 (Roche). Briefly, cells were plated into 6-wellplates, 2 μg of vector DNA was transfected into cells at a ratio of6:1 of transfection reagent to DNA in antibiotic-free media. Cellswere incubated with the transfection mixture for 48 h followingwhich they were washed and collected for immunoblottinganalysis.
Western blotting
Cells were grown and treated in 6-well plates. After 48–72 h ofthe indicated treatments, the cells were lysed with NP-40 lysisbuffer (150 mM NaCl, 50 mM Tris–Cl, 1% NP-40, pH 8.0). Proteins(20 μg) were separated on SDS-polyacrylamide gels and electro-blotted onto nitrocellulose membranes (Millipore) using a
semidry transfer method. Immunoassays were performed withanti-CARF [26], -p53, -ATM and -ATR (Santa Cruz, Santa Cruz, CA),-CHK1/2 (Cell Signaling, Danvers, MA), -γH2AX (Millipore) andβ-actin (Chemicon, Millipore) antibodies. The immunocomplexesformed were visualized with horseradish peroxidase-conjugatedgoat anti-mouse or anti-rabbit antibodies (Santa Cruz) anddetected using ECL prime Western blotting (Amersham PharmaciaBiotech/GE Healthcare, Piscataway, NJ). Densitometric quantita-tion of the immunoblots was carried out using the ImageJ soft-ware (National Institute of Health).
Immunofluorescence
Cells were cultured and treated on glass coverslips placed in 12-well culture dishes. At the end of the treatment, coverslips werewashed with cold phosphate-buffered saline (PBS) and fixed withpre-chilled methanol: acetone (1:1 v/v) solution for 5–10 min.Fixed cells were washed with PBS, permeabilized with 0.2% TritonX-100 in PBS for 10 min, and blocked with 2% bovine serumalbumin (BSA) in PBS for 20 min. Cells were stained withantibodies against CARF, p53 (Santa Cruz), γH2AX (Millipore)and α-tubulin (Sigma-Aldrich). Immunostaining was visualizedby secondary staining with Alexa-488 conjugated goat anti-rabbitantibody (Molecular Probes, Invitrogen). Coverslips were thenexposed to nuclear stain Hoechst 33258 (0.5 mg/ml) for 3–5 minin dark at room temperature, mounted onto glass slides withFluoromount mounting medium (Sigma-Aldrich) and observedunder an Axioplan 2 Carl Zeiss microscope. Nuclear morphologyand apoptotic cells of control and treated cultures were scored.
Reverse transcription (RT)-PCR
Total RNA was isolated from control and treated cells at 48–60 husing the RNeasy Mini Kit (Qiagen, Hilden, Germany). Equalamounts of RNA (2 mg) from control and treated samples were usedto generate complementary DNA (cDNA) using oligo DT primers(Promega, Madison, WI) and M-MLV RT (Promega) in an RT reaction(37 1C for 1 h). The cDNA was subjected to PCR amplificationconsisting of an initial 10 min denaturation step at 95 1C followedby 25 cycles at 95 1C for 45 s, 60 1C for 1 min and 72 1C for 45 s,with final annealing step at 72 1C for 10 min. The primer sequencesused were as follows: CARF, 5'-cagccaaagcagcagcagcg-3' (sense)and 3'-agcccaacaagggcacctcg-5' (antisense); p21, 5'-gacaccactg-gagggtgact-3' (sense) and 5'-ggcgtttggagtggtagaaa-3' (antisense);

E X P E R I M E N T A L C E L L R E S E A R C H 3 2 2 ( 2 0 1 4 ) 3 2 4 – 3 3 4 327
p53, 5'-aaggaaatctcaccccatcc-3' (sense) and 5'-aaggctgcagtaagc-caaga-3' (antisense); and GAPDH (internal control), 5'-accacagtc-catgccatcac-3' (sense) and 5'-tccaccaccctgttgctgta-3' (antisense). ThePCR products were separated in 1% agarose gel followed byacquisition and analysis using Quantity One Image software (BioRad,Hercules, CA).
Promoter analysis
DNA sequence of CARF promoter was extracted from nucleotidedatabase (chromosome 4q35.1; NC_000004.11; 184,362,789–184,365,951 bp). Promoter analysis programs including, CISTER:zlab.bu.edu/�mfrith/cister.shtml – Dr. Zhiping Weng, Universityof Massachusetts Medical School; LASAGNA: biogrid-head.engr.uconn.edu/lasagna_search – Drs. Lee or Huang, University ofConneticut; CONSITE: asp.ii.uib.no:8090/cgi-bin/CONSITE/consite/– Dr. Lenhard, Karolinska Institutet; PROMO: alggenlsi.upc.es/cgi-bin/promo_v3/promo/prommmmoinit.cgi?dirDB=TF_8.3 –
Drs. Messeguer and Alba, Universities in Barcelona and London;TFSEARCH: http://www.cbrc.jp/research/db/TFSEARCH.html – Dr.Yutaka Akiyama, Real World Computing Partnership, Japan andPAINT: http://www.dbi.tju.edu/dbi/tools/paint/ – Dr. RajanikanthVadigepalli, The Daniel Baugh Institute for Functional Genomics,Philadelphia were used for analysis.
Statistical analysis
Pairwise comparisons were performed using the Mann–WhitneyU-test, and multiple comparisons were made using Analysis OfVariance (ANOVA) test. A p-value of less than 0.05 is consideredstatistically significant.
Results
We earlier showed that CARF activates p53 by ARF-dependent and-independent pathways and in turn gets regulated by p53–HDM2axis via HDM2-mediated ubiquitination and proteasomal degrada-tion [14–16,18]. Here, we investigated the half-life of CARF inunstressed cells. U2OS cells were treated with cycloheximide toblock de novo protein synthesis and examined for CARF expressionat different time points. As shown in Fig. 1A, the half-life of CARFwas found to be less than an hour. Since p53 is well known as ashort-lived protein, the blot was probed for p53 as a positivecontrol. We found that CARF and p53 undergo degradation almostat the same rate (Fig. 1A). Quantitation from three independentexperiments revealed �40% decrease in CARF and 35% decrease inp53 during 1 h post-cycloheximide treatment. Both proteins werereduced to about 10–20% level at 2 h post-cycloheximide treat-ment. Inhibition of proteasome by MG132 led to an increase in theexpression levels of both CARF and p53 in a dose-dependentmanner (Fig. 1B). An average of 1.7 and 2.6 increase was noticedin the expression of CARF in response to MG132 (2.5 and 5.0 μM)treatments, respectively, in three independent experiments. p53showed 2 and 2.3 fold increase in these experiments. These datasuggested that similar to p53, CARF is a short-lived protein inunstressed cells and undergoes proteasome-mediated degradation.Correlation of CARF and p53 was also investigated by double-thymidine block as mentioned in the Materials and methodssection. Cells synchronized at the G1/S boundary were collected
at different time points after release from thymidine block, and thelevel of CARF expression was detected by Western blot analysis. Wefound that the cells blocked at G1/S phase (Fig. 1C, 0 h) possessed ahigh level of CARF protein. Cells released from the thymidine blockshowed time-dependent decrease in the level of CARF. The blotprobed with Cyclin B1 (an established marker for early events ofmitosis such as chromosome condensation, nuclear envelopebreakdown and spindle pole assembly) revealed that the decreasein CARF was paralleled by increase in Cyclin B1 signifying the entryof the cell into mitosis. At 10–12 h post-release of the thymidineblock, cells showed decrease in Cyclin B1 indicating mitotic exit,and this was marked by further decrease in CARF (Fig. 1C, 12 and14 h). In order to substantiate these findings, U2OS cells weresynchronized at G2/M boundary by nocodazole (an inhibitor ofspindle formation) treatment. As shown in Fig. 1D and E, it caused90% cells to arrest in the G2/M phase. Release of cells fromnocodazole arrest was marked by cell cycle progression to G1and S phases (Fig. 1D and E). We found that the level of CARF washigh in G2/M phase (0 h) and decreased in cells as they progressedto G1 and S phase and increased again when cells accumulated inG1 (Fig. 1F). These data suggested that CARF plays a role in G1 andG2 arrest phases of the cell cycle. Nocodazole-mediated G2/Mblock and its release in CARF-compromised cells further revealedthat CARF is associated with cell cycle arrest and progression. Asshown in Fig. 1G and H, CARF-depleted cells were treated withnocodazole to block cell cycle at G2/M boundary, and then themitotic cells were collected and plated for colony formationefficiency as described in Materials and methods. We found thatwhereas control cells released from the nocodazole block formedcolonies, CARF-depletion by two independent siRNAs caused sig-nificant reduction in colony forming efficacy. Together, theseresults implied that CARF is a cell cycle regulated protein and itis required for cell cycle arrest and subsequent cell cycle progres-sion in cycling cells.To analyze the cell cycle dependence of CARF level in asyn-
chronously dividing untreated cells, we performed single cellimaging analyses in normal and cancer cells. As shown in Fig. 2A,normal cells at G1 and G2 stages (as recognized by DNA contentand nuclear size) showed higher levels of CARF expression ascompared to cells in metaphase and immediately after cytokinesis(as recognized by DNA staining and cell morphology). Quantita-tion revealed its high level at G1 and G2 phases whereas themitotic cells showed a low level of expression. Cancer cells (U2OS,Saos-2 and HeLa) at G1 also showed higher level of CARFexpression in arresting as compared to the mitotic cells (datanot shown). The data were further confirmed by triple staining ofHeLa cells with CARF, tubulin and Hoechst. As seen in Fig. 2B, lateG2 and early prophase cells exhibited the highest levels of CARF.Cells during metaphase, anaphase and telophase (as characterizedby spindle formation, chromosome segregation and cytokinesis,respectively) showed a marked decline.In order to examine the role of CARF during cell cycle arrest, we
induced growth arrest in normal and cancer cells by treatingthem with a panel of drugs. As shown in Fig. 3A and B, G1/S-arrested cells showed an increase in CARF expression. Drug-treated cells were immunostained with anti-CARF antibody, andthe intensity of staining was measured using the EnSpire multi-mode plate reader. We found that CARF expression was increasedin drug-treated cancer as well as normal cells. The drug-inducedincrease in CARF expression with the different drugs varied from

Fig. 1 – CARF has a short half-life and its level is regulated by proteosomal degradation in a cell cycle-dependent manner. (A)Western blotting of lysates from U2OS cells treated with a protein synthesis inhibitor (cycloheximide) showed the level of CARF fellmore rapidly than p53 and that its half-life was less than 1 h. (B) Cells treated with an inhibitor of proteasomal degradation(MG132) showed an increase in both CARF and p53 levels. (C) Cells released from double thymidine block showed a decrease inCARF expression, accompanied by an increase in cyclin B1; the level was highest in the arrested cells prior to release (time 0), andmuch lower in asynchronously dividing cells (Asy). (D–F) Cells arrested by nocodazole treatment had much higher CARF levelsthan asynchronously dividing cells (Asy); release from nocodazole led to a decrease in CARF followed by increase as the cells enterG1 phase again. (G and H) When cells were treated with CARF siRNA (two sites) or control siRNA 48 h before 12 h of nocodazoletreatment, the control cells were able to form colonies after nocodazole release, whereas those treated with CARF siRNA mostlyfailed to do so. Plates showing the colony number and cells in the colonies are shown. NT signifies untreated control. Colonynumber from three independent experiments and two CARF siRNAs are shown in (H). Histograms show quantitation from threeindependent experiments, wherein *po0.05, *po0.01, and *po0.001.
E X P E R I M E N T A L C E L L R E S E A R C H 3 2 2 ( 2 0 1 4 ) 3 2 4 – 3 3 4328
10 to 60% in U2OS cells and 25 to 60% in TIG-1 cells thatcorresponded to G1 cell cycle arrest as determined by cell cycleanalysis (Fig. 3A). Correlation of CARF, p53, p21 and γH2AX incontrol and drug-treated cells was determined by Western blot-ting of control and adriamycin-treated cells (Fig. 3C). The drug-induced increase in CARF expression was accompanied byincreases in p53, p21 and γH2AX both in normal (TIG-1) andcancer (U2OS) cells. Quantitation of CARF, p53, p21 and γH2AXexpression in control and drug-treated cells in three independentexperiments revealed consistent upregulation of CARF in drug-induced growth arrest of cells at the G1 stage of cell cycle(Fig. 3D). Immunostaining for CARF, p53 and γH2AX (Fig. 4A)confirmed an upregulation of CARF protein in adriamycin-treatedcells. p53�/� cells (Saos-2, SKOV3 and HCT116) used in parallelexperiments showed similar increase in CARF expression inresponse to adriamycin, UV and gallium nitrate treatmentssuggesting that p53 is not involved in induction of CARF inresponse to DNA damage (Fig. 4B and data not shown). In order
to address the possibility of p53 as a putative upstream regulatorof CARF, we examined CARF promoter sequence with a number ofpromoter analysis software and found that CARF promoter doesnot contain p53-reponsive elements (data not shown). We nextperformed RT-PCR for CARF in the control and adriamycin-treatedcells and found that CARF mRNA increased in a dose-dependentmanner, suggesting that there is a transcriptional component toupregulation of CARF, like p53 and p21 (Fig. 4C and data notshown). In order to get further insights on the relationshipbetween expression of CARF and DNA damage response andcheckpoint proteins, we analyzed protein levels in adriamycin-treated cells over a 36 h time course. As shown in Fig. 4D and E,whereas CARF was induced at 8 h post-treatment p53 and p21showed upregulation at 12–24 and 24–36 h treatment, respec-tively. Furthermore, upregulation of ATM, ATR, CHK1 and CHK2followed a time course that was somewhat similar to CARF. Thesedata suggested that CARF is involved in DNA damage response,perhaps as a checkpoint protein. In order to address an induction

Fig. 2 – Upregulation of CARF at G1 and G2 stages of cell cycle. (A) Immunostaining of cells (TIG-1 normal fibroblasts) for CARFtogether with staining for DNA (Hoechst) showed that cells at the G1 and G2 phase possess higher level of CARF expression. Cells inmetaphase and immediately after cytokinesis showed low to negligible levels of CARF expression. Quantitation of CARF signal (reddots) is shown in graphs below. Maximum intensity of CARF staining was noticed at G1 followed by G2 stage. (B) Triple staining ofcells for CARF, γ-tubulin and DNA (Hoechst) revealed highest level of CARF expression in late G2 and early prophase, and muchlower levels throughout the remainder of mitosis (from early prophase to cytokinesis).
E X P E R I M E N T A L C E L L R E S E A R C H 3 2 2 ( 2 0 1 4 ) 3 2 4 – 3 3 4 329
of CARF versus its stabilization during DNA damage response ofcells, we performed pulse-chase experiments using etoposide(DNA damaging agent and inhibitor of topoisomerase and DNAreplication) and hydroxyurea (DNA damaging agent and inhibitorof ribonucleotide reductase and global transcription). We foundthat the continuous exposure to the drug was required forincrease in CARF expression. In pulse-chase experiments, CARFlevel was seen to undergo rapid decrease in the chase period bothin the etoposide and hydroxyurea models (Fig. 4F and G). Takentogether, these data suggested that (i) induction of CARF by DNAdamaging agents, most likely, involves its transcriptional upregu-lation and (ii) CARF is involved more in DNA damage sensing thanits repair mechanisms.
In order to clarify the role of CARF in cell cycle arrest, we nextinvestigated its level during replicative senescence in humanfibroblasts. CARF expression was examined in fibroblasts derivedfrom skin (Fre 92s-2 and Fre 102s-3), lung (MRC5) and breaststromal (Fre 80) tissues. Fre 92s-2, Fre 102s-3, MRC5 and Fre 80cells were shown to senescence at around PD 58, 40, 56 and 29,respectively (Fig. 5 and data not shown). As shown in Fig. 5A–D,CARF showed an increase during the late stages (pre-senescent orsenescent) in all the serially passaged cells, also characterized byincrease in p53 and p21. RT-PCR on the serially passaged MRC5and Fre 80 cells revealed that the increase in CARF duringreplicative senescence was regulated, at least in part, at thetranscriptional level (Fig. 5, C–E and F–I, respectively). Quantita-tion of the data revealed that there were 3 and 6-fold increase inCARF mRNA and protein in late (60 PD) as compared to the early(35 PD) passage MRC5 cells. Similarly, Fre 80 cells showed 3.2 and5.8-fold increase in CARF mRNA and protein, respectively. These
data may suggest that an increase in CARF from the early to latepassage (pre-senescent stages) is regulated at transcriptionallevel. At late passages (senescent and post-senescent cultures),the increase in protein may also be due to its accumulationcontributed by decrease in HDM2-mediated degradation [18]. Wealso subjected U2OS cells to premature senescence by expressionof oncogenic Ras or knockdown of TRF2 and examined if CARFunderwent an upregulation during stress-induced senescence. Asshown in Fig. 6A and B, induction of Ras-induced senescence wasmarked by an increase in CARF. Similarly, as shown in Fig. 6C,knockdown of TRF2 caused a very substantial increase in theexpression level of CARF demonstrating that upregulation of CARFaccompanies senescence.
Discussion
Cellular senescence is a well-established tumor suppressionmechanism. It has been tightly associated with the DNA damageresponse (DDR), which upon induction triggers diverse cellsignaling pathways that lead to growth arrest or apoptosis. Wehave previously reported that CARF regulates the p53–p21 path-way, a major mechanism governing growth arrest/senescence andapoptosis. Whereas upregulation of CARF was associated withincrease in p53, p21 and growth arrest of cells, its downregulationcaused apoptosis that was marked by cleavage of caspases andsuppression of ATR-CHK1 signaling. However, the physiologicalrole of CARF has not yet been elucidated to-date. Thus, thepresent study was performed in order to gain further insights to

Fig. 3 – Upregulation of CARF during the drug induced G1 growth arrest of cells. (A and B) Cells were treated with low doses(Table 1) of eleven drugs. Induction of cell cycle arrest at G1 stage was confirmed by FACS analysis. Level of CARF expression asdetermined by quantitation of immunostaining showed an increase in drug-treated G1-arrested cells. (C and D) Adriamycin treatedcells showed increase in CARF that was paralleled by increase in p53, p21 and γH2AX. Quantitation from three independentexperiments is shown in (D). Statistical significance of the data as calculated by ANOVA test is shown (*po0.01 and #po0.05).
E X P E R I M E N T A L C E L L R E S E A R C H 3 2 2 ( 2 0 1 4 ) 3 2 4 – 3 3 4330
the cellular functions of CARF under normal and pathologicalconditions.In this study, we demonstrate that CARF is a short-lived protein
that is upregulated especially during the G2 phase of the cell cycleand is downregulated during mitosis. Induction of growth arrestin normal cells by a variety of drugs was accompanied byupregulation of CARF. Serially passaged skin, lung and breaststromal fibroblasts showed increase in CARF as they approachedreplicative senescence. Furthermore, CARF was upregulated incancer cells that were induced to senesce either by chemicals
[25], oncogenic stress or knockdown of the telomeric DNAbinding protein, TRF2, the latter of which has been earlier shownto induce senescence through the p53 and p16/RB pathways[27,28]. Thus, expression of CARF accompanies various types ofsenescence irrespective of its trigger and it will therefore beimportant to determine whether it has a causal role. The drugsused to induce growth arrest in cancer cells and known to have awide variety of mechanisms of actions caused upregulation(although to variable levels) of CARF. These included etoposideand camptothecin induced inhibition of the DNA enzyme
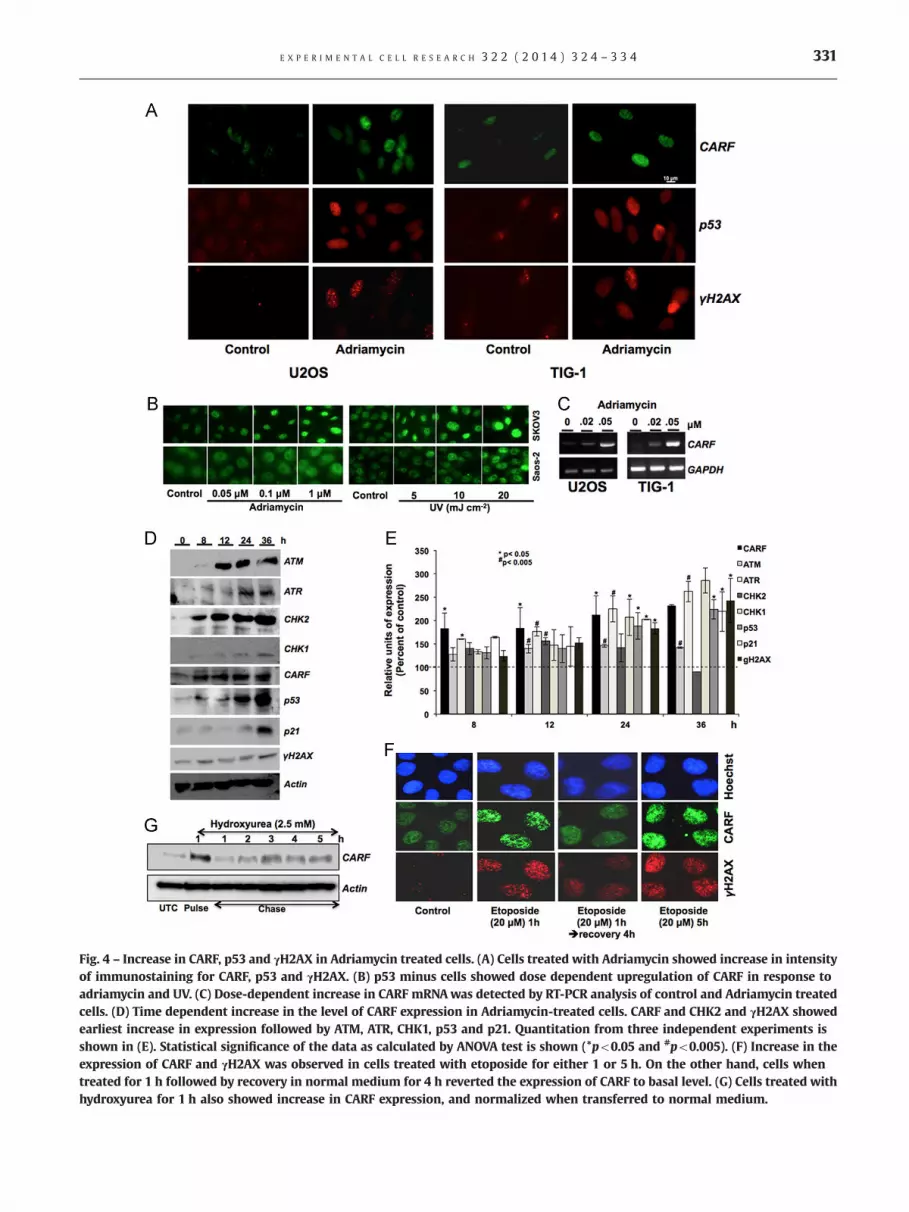
Fig. 4 – Increase in CARF, p53 and γH2AX in Adriamycin treated cells. (A) Cells treated with Adriamycin showed increase in intensityof immunostaining for CARF, p53 and γH2AX. (B) p53 minus cells showed dose dependent upregulation of CARF in response toadriamycin and UV. (C) Dose-dependent increase in CARF mRNAwas detected by RT-PCR analysis of control and Adriamycin treatedcells. (D) Time dependent increase in the level of CARF expression in Adriamycin-treated cells. CARF and CHK2 and γH2AX showedearliest increase in expression followed by ATM, ATR, CHK1, p53 and p21. Quantitation from three independent experiments isshown in (E). Statistical significance of the data as calculated by ANOVA test is shown (*po0.05 and #po0.005). (F) Increase in theexpression of CARF and γH2AX was observed in cells treated with etoposide for either 1 or 5 h. On the other hand, cells whentreated for 1 h followed by recovery in normal medium for 4 h reverted the expression of CARF to basal level. (G) Cells treated withhydroxyurea for 1 h also showed increase in CARF expression, and normalized when transferred to normal medium.
E X P E R I M E N T A L C E L L R E S E A R C H 3 2 2 ( 2 0 1 4 ) 3 2 4 – 3 3 4 331

Fig. 5 – Increase in CARF expression during replicative senescence. Increase in the level of CARF expression in human fibroblasts asdetected by Western blotting (A–C and F) and RT-PCR (D and G) is shown. Fold increase in CARF mRNA and protein expressionbetween early and late passage cells is shown in (E and I). Increase in CARF mRNA in serially passaged Fre 80 cells is shown in (H).
Fig. 6 – Increase in CARF expression during stress-induced senescence. Senescence induced by oncogenic stress by Ras-oncogene(A and B) and telomere deprotection triggered by TRF2 siRNA (C) were accompanied by increase in the level of CARF expression.Quantitation of protein signals from three experiments is shown as histograms. Statistical significance of the data as calculated byANOVA test is shown (*po0.05 and #po0.005).
E X P E R I M E N T A L C E L L R E S E A R C H 3 2 2 ( 2 0 1 4 ) 3 2 4 – 3 3 4332

E X P E R I M E N T A L C E L L R E S E A R C H 3 2 2 ( 2 0 1 4 ) 3 2 4 – 3 3 4 333
topoisomerase I and induction of double-strand breaks duringDNA replication, gallium nitrate and mitoxantrone induced DNAdamage and inhibition of DNA synthesis, taxol-induced inhibitionof microtubule polymerization and inhibition of cytokinesis andretinoic acid-induced differentiation and inhibition of cellularproliferation (Fig. 3). CARF expression generally correlated withexpression of DDR and checkpoint proteins, such as γH2AX, ATM,ATR, CHK1 and CHK2 (Fig. 4). In contrast to the cancer cells,normal cells treated with a variety of drugs showed a morepronounced increase in CARF suggesting that the response tosome drugs may be attenuated in cancer cells; the drugs includedcurcumin, gallium nitrate and mitoxantrone. Normal cells areknown to possess robust DDR response and active repair/adaptivemechanisms, and our data suggest that CARF, like p53, may beinvolved in this important genomic guardian and tumor suppres-sor role. p53 minus cells showed induction of CARF in response toDNA damage (Fig. 4B) suggesting that p53 is not involved inupregulation of CARF. Bioinformatics screening of CARF promoterDNA sequence, in a number of sequence analyses tools, revealedlack of target sites for p53. It, however, showed putative tran-scription factors (TF) binding sites for (i) tumor suppressor/oncogene associated TFs including NF-1 (��1430 bp), MEF-2(��1380, and �1625), C- and N-myc (multiple sites: ��2560,�1960, �1650, and �1510) and (ii) DNA damage associated TFsincluding NF-kB (��400, and �180) and C-fos (��1570) andother DNA binding transcriptional modulators viz. CdxA, GATA-1,ADR1, SREBP, p300, C/EBP and CREBP TFs. The TATA-box bindingprotein (TBP) regions that are predicted to be involved in DNAdamage signaling are located at �2512, �2023, �1255, and �30of the promoter region suggesting that CARF promoter has DNA-damage inducible characteristics. Although further work isrequired to determine whether CARF is involved in the DDR asa damage sensor or signal transducer, and/or in checkpointcontrol, the present study underlined the evidences to the roleof CARF in DNA damage sensing and in the regulatory mechanism(s) of cell cycle (check-point) progression.
Conflicts of interest
None.
Acknowledgments
The study was supported by grants from the AIST. Rumani Singhwas a recipient of the Ministry of Education, Culture, Sports,Science & Technology (MEXT) scholarship. Caroline T. Cheung andRajkumar S. Kalra were supported by post-doctoral fellowshipfrom the Japan Society for Promotion of Science (JSPS).
r e f e r e n c e s
[1] J.W. Harper, S.J. Elledge, The DNA damage response: ten yearsafter, Mol. Cell 28 (2007) 739–745.
[2] J. Bartek, J. Bartkova, J. Lukas, DNA damage signalling guardsagainst activated oncogenes and tumour progression, Oncogene26 (2007) 7773–7779.
[3] J.R. Chapman, S.P. Jackson, Phospho-dependent interactionsbetween NBS1 and MDC1 mediate chromatin retention of the
MRN complex at sites of DNA damage, EMBO Rep. 9 (2008) 795–801.
[4] J. Zhou, C.U. Lim, J.J. Li, L. Cai, Y. Zhang, The role of NBS1 in themodulation of PIKK family proteins ATM and ATR in the cellularresponse to DNA damage, Cancer Lett. 243 (2006) 9–15.
[5] F. d'Adda di Fagagna, P.M. Reaper, L. Clay-Farrace, H. Fiegler, P.Carr, T. Von Zglinicki, G. Saretzki, N.P. Carter, S.P. Jackson, A DNAdamage checkpoint response in telomere-initiated senescence,Nature 426 (2003) 194–198.
[6] R. Di Micco, A. Cicalese, M. Fumagalli, M. Dobreva, A. Verrecchia,P.G. Pelicci, F. di Fagagna, DNA damage response activation inmouse embryonic fibroblasts undergoing replicative senescenceand following spontaneous immortalization, Cell Cycle 7 (2008)3601–3606.
[7] G. Hewitt, D. Jurk, F.D. Marques, C. Correia-Melo, T. Hardy, A.Gackowska, R. Anderson, M. Taschuk, J. Mann, J.F. Passos,Telomeres are favoured targets of a persistent DNA damageresponse in ageing and stress-induced senescence, Nat. Commun.3 (2012) 708.
[8] Z. Kaul, A.J. Cesare, L.I. Huschtscha, A.A. Neumann, R.R. Reddel,Five dysfunctional telomeres predict onset of senescence inhuman cells, EMBO Rep. 13 (2011) 52–59.
[9] U. Herbig, W.A. Jobling, B.P. Chen, D.J. Chen, J.M. Sedivy, Telomereshortening triggers senescence of human cells through a path-way involving ATM, p53, and p21(CIP1), but not p16(INK4a), Mol.Cell 14 (2004) 501–513.
[10] V. Gire, P. Roux, D. Wynford-Thomas, J.M. Brondello, V. Dulic,DNA damage checkpoint kinase Chk2 triggers replicative senes-cence, EMBO J. 23 (2004) 2554–2563.
[11] R. Wadhwa, S. Takano, M. Robert, A. Yoshida, H. Nomura, R.R. Reddel,Y. Mitsui, S.C. Kaul, Inactivation of tumor suppressor p53 by mot-2, ahsp70 family member, J. Biol. Chem. 273 (1998) 29586–29591.
[12] S.C. Kaul, T. Yaguchi, K. Taira, R.R. Reddel, R. Wadhwa, Over-expressed mortalin (mot-2)/mthsp70/GRP75 and hTERT coop-erate to extend the in vitro lifespan of human fibroblasts, Exp.Cell Res. 286 (2003) 96–101.
[13] S.C. Kaul, S. Aida, T. Yaguchi, K. Kaur, R. Wadhwa, Activation ofwild type p53 function by its mortalin-binding, cytoplasmicallylocalizing carboxyl terminus peptides, J. Biol. Chem. 280 (2005)39373–39379.
[14] M.K. Hasan, T. Yaguchi, T. Sugihara, P.K. Kumar, K. Taira, R.R.Reddel, S.C. Kaul, R. Wadhwa, CARF is a novel protein thatcooperates with mouse p19ARF (human p14ARF) in activatingp53, J. Biol. Chem. 277 (2002) 37765–37770.
[15] M.K. Hasan, T. Yaguchi, Y. Minoda, T. Hirano, K. Taira, R. Wadhwa,S.C. Kaul, Alternative reading frame protein (ARF)-independentfunction of CARF (collaborator of ARF) involves its interactionswith p53: evidence for a novel p53-activation pathway and itsnegative feedback control, Biochem. J. 380 (2004) 605–610.
[16] M.K. Hasan, T. Yaguchi, J.I. Harada, T. Hirano, R. Wadhwa, S.C.Kaul, CARF (collaborator of ARF) interacts with HDM2: evidencefor a novel regulatory feedback regulation of CARF-p53-HDM2-p21WAF1 pathway, Int. J. Oncol. 32 (2008) 663–671.
[17] K. Hasan, R. Wadhwa, S.C. Kaul, CARF binds to three members(ARF, p53, and HDM2) of the p53 tumor-suppressor pathway,Ann. N. Y. Acad. Sci. 1100 (2007) 312–315.
[18] K. Hasan, C. Cheung, Z. Kaul, N. Shah, S. Sakaushi, K. Sugimoto, S.Oka, S.C. Kaul, R. Wadhwa, CARF is a vital dual regulator ofcellular senescence and apoptosis, J. Biol. Chem. 284 (2009)1664–1672.
[19] C.T. Cheung, R. Singh, A.R. Yoon, M.K. Hasan, T. Yaguchi, S.C. Kaul,C.O. Yun, R. Wadhwa, Molecular characterization of apoptosisinduced by CARF silencing in human cancer cells, Cell DeathDiffer. 18 (2010) 589–601.
[20] C.S. Sorensen, R.G. Syljuasen, Safeguarding genome integrity: thecheckpoint kinases ATR, CHK1 and WEE1 restrain CDK activityduring normal DNA replication, Nucleic Acids Res. 40 (2011) 477–486.

E X P E R I M E N T A L C E L L R E S E A R C H 3 2 2 ( 2 0 1 4 ) 3 2 4 – 3 3 4334
[21] E.J. Brown, Analysis of cell cycle progression and genomicintegrity in early lethal knockouts, Methods Mol. Biol. 280 (2004)201–212.
[22] B.J. Evison, M. Pastuovic, R.A. Bilardi, R.A. Forrest, P.P. Pumuye, B.E. Sleebs, K.G. Watson, D.R. Phillips, S.M. Cutts, M2, a novelanthracenedione, elicits a potent DNA damage response that canbe subverted through checkpoint kinase inhibition to generatemitotic catastrophe, Biochem. Pharmacol. 82 (2011) 1604–1618.
[23] G. He, J. Kuang, A.R. Khokhar, Z.H. Siddik, The impact of S- andG2-checkpoint response on the fidelity of G1-arrest by cisplatinand its comparison to a non-cross-resistant platinum(IV) analog,Gynecol. Oncol. 122 (2011) 402–409.
[24] R. Montano, I. Chung, K.M. Garner, D. Parry, A. Eastman,Preclinical development of the novel Chk1 inhibitor SCH900776
in combination with DNA-damaging agents and antimetabolites,Mol. Cancer Ther. 11 (2011) 427–438.
[25] Z. Kaul, T. Yaguchi, H.X. Chiura, S.C. Kaul, R. Wadhwa, Quantumdot-based mortalin staining as a visual assay for detection ofinduced senescence in cancer cells, Ann. N. Y. Acad. Sci. 1100(2007) 368–372.
[26] R. Wadhwa, T. Sugihara, M.K. Hasan, E.L. Duncan, K. Taira, S.C.Kaul, A novel putative collaborator of p19ARF, Exp. Gerontol. 38(2003) 245–252.
[27] J. Karlseder, D. Broccoli, Y. Dai, S. Hardy, T. de Lange, p53- andATM-dependent apoptosis induced by telomeres lacking TRF2,Science 283 (1999) 1321–1325.
[28] T. de Lange, Protection of mammalian telomeres, Oncogene 21(2002) 532–540.





























