Embed Size (px)
Citation preview

Chemical Papers 69 (9) 1262–1276 (2015)DOI: 10.1515/chempap-2015-0134
ORIGINAL PAPER
Molecular dynamic studies of amyloid-beta interactionswith curcumin and Cu2+ ions
a,b,cStanislav Kozmon, a,dIgor Tvaroška
aInstitute of Chemistry, Slovak Academy of Sciences, Dúbravská cesta 9, Bratislava 845 38, Slovakia
bCentral European Institute of Technology (CEITEC), Masaryk University, Kamenice 753/5, Brno 625 00, Czech Republic
cFaculty of Science – National Centre for Biomolecular Research, Masaryk University, Kamenice 5, Brno 625 00, Czech Republic
dDepartment of Chemistry, Faculty of Natural Sciences, Constantine The Philosopher University, Nitra 949 74, Slovakia
Received 8 December 2014; Revised 13 April 2015; Accepted 14 April 2015
Amyloid-beta (Aβ) peptide readily forms aggregates that are associated with Alzheimer’s disease.Transition metals play a key role in this process. Recently, it has been shown that curcumin (CUA),a polyphenolic phytochemical, inhibits the aggregation of Aβ peptide. However, interactions of Aβpeptide with metal ions or CUA are not entirely clear. In this work, molecular dynamics (MD)simulations were carried out to clear the nature of interactions between the 42-residue Aβ peptide(Aβ-42) and Cu2+ ions and CUA. Altogether nine different models were investigated, and more than2 µs of the simulation data were analyzed. The models represent the possible modes of arrangementbetween Aβ-42 and Cu2+ ions and CUA, respectively, and were used to shed light on the Aβ-42conformational behavior in the presence of Cu2+ ions and CUA molecules. Obtained data clearlyshowed that the presence of a CUA molecule or a higher concentration of copper ions significantlyaffect the conformational behavior of Aβ-42. Calculations showed that the change of the His13protonation state (Aβ(H13δ)–Cu2+, Aβ(H13δ)–Cu2+–CUA models) leads to higher occurrence ofthe Asp23–Lys28 salt bridge. Analyzes of trajectories revealed that C-terminal β-sheet structuresoccurred significantly less frequently, and CUA promoted the stabilization of the α-helical structure.Further, calculations of the Aβ-42 complex with CUA and Cu2+ ions showed that CUA can chelatethe Cu2+ ion and directly interact with Aβ, which may explain why CUA acts as an inhibitor ofAβ aggregation.c© 2015 Institute of Chemistry, Slovak Academy of Sciences
Keywords: Alzheimers’s disease, amyloid beta, molecular dynamics, curcumin
Introduction
Alzheimers’s disease (AD) is the most commoncause of dementia and is characterized by the ac-cumulation of amyloid plaques, with the main con-stituent being the amyloid-beta peptide (Aβ). It is es-timated that 24 million victims are suffering from ADworldwide, and there is no effective pharmacologicaltreatment available at present (Saido & Iwata, 2006).Relative abundance of Aβ in an AD brain is essen-tial for disease development; however, the mechanism
of Aβ mediated neurotoxicity is less obvious. Severalpossible mechanisms have been proposed (Crouch etal., 2008). An abundance of insoluble high molecu-lar weight Aβ fibrils was thought to be the primarytoxic form of Aβ. However, currently it is believed,based on correlations between the abundance of sol-uble low molecular weight forms (monomers, dimers,and trimers) of Aβ and markers of the AD (McLean etal., 1999), that soluble oligomeric forms of Aβ are themost toxic species (Hardy & Selkoe, 2002). The longerforms of Aβ (mainly Aβ-42) are assumed to be par-
*Corresponding author, e-mail: [email protected]
Authenticated | [email protected] author's copyDownload Date | 6/3/15 12:41 PM

S. Kozmon, I. Tvaroška/Chemical Papers 69 (9) 1262–1276 (2015) 1263
ticularly pathogenic compared to shorter forms (e.g.Aβ-40, Aβ-30) (Atwood et al., 1999). The structureof Aβ under physiological conditions is mostly a ran-dom coil (Ball et al., 2013; Bitan et al., 2003; Lim etal., 2007; Yan et al., 2008) while it is usually α-helicalin membrane media (Barrow et al., 1992; Coles et al.,1998; Shao et al., 1999; Soto et al., 1995; Sticht et al.,1995), and aggregation is accompanied by conversiontowards the β-sheet structure (Barrow et al., 1992;Barrow & Zagorski, 1991; Good & Murphy, 1995; Ser-pell, 2000; Xu et al., 2005).There is extensive evidence indicating that metal
ions, such as copper, zinc, and iron are involved inthe AD disease (Barnham & Bush, 2008; Bush, 2003;Faller, 2009; Faller & Hureau, 2009; Perrone et al.,2010). Copper is involved in two key steps: (i) it bindsAβ directly and modulates its aggregation behavior;and (ii) it is essential for the production of reactiveoxygen species and oxidative stress. Coordination ofcopper to Aβ has been extensively studied, and it is as-sumed that some binding modes with a different set ofligands occur in rapid equilibrium (Faller, 2009; Faller& Hureau, 2009; Perrone et al., 2010). The most likelyligands of the Cu ions are three histidine (His) residuesor two His and an N-terminal amine. Also, the car-boxylate side chains are likely ligands. Complexes ofCu with Aβ-42 with a predefined binding mode wereinvestigated by MD simulations. Three different Cu–Aβ complexes were considered with the Cu bond topeptide atoms described by harmonic approximation(Raffa & Rauk, 2007). The results showed that bind-ing of Cu to Aβ disrupts the β-sheet structure be-tween the N- and C-terminal regions, observed in thefree Cu–Aβ system, with significant conformationaldifferences found in the 1–9 region of Aβ-42. It wassuggested that an inhibitor of the aggregation effectof Cu2+ requires a bifunctional activity, namely Cu2+
chelating ability and Aβ interactions (Bush, 2008).Curcumin ([1,7-bis(4-hydroxy-3-methoxyphenyl)-
1,6-heptadiene-3,5-dione]; CUA) is the natural antiox-idant and is the main constituent of the rhizomes ofthe plant Curcuma longa. The enol form is the moststable structure of CUA (Kolev et al., 2005). CUA canproduce chelates of the type 1 : 1 and 1 : 2 with met-als (Barik et al., 2007). It has been shown that CUAinhibits the formation of Aβ fibrils and has potentialanti-amyloidogenic effects (Banerjee, 2014; Lim et al.,2001; Liu et al., 2012; Ono et al., 2004; Picciano &Vaden, 2013; Porat et al., 2006; Reinke & Gestwicki,2007; Yanagisawa et al., 2010; Yang et al., 2005). Sim-ilarly, nanoparticles functionalized with CUA deriva-tives (Airoldi et al., 2011) exhibit high affinity towardAβ-42 monomers and the corresponding fibrils (Gobbiet al., 2010; Le Droumaguet et al., 2012; Mourtas etal., 2011; Re et al., 2011; Taylor et al., 2011).The development of effective therapeutics for AD
treatment requires knowledge of the mechanism in-volved. In the present study, to shed some light on the
binding of CUA and copper with Aβ-42, MD simula-tions of various aqueous copper–CUA–Aβ-42 systemswere carried out. The MD simulations were aimed atbetter understanding of the effect of interactions withcopper and CUA on the 3D structure of Aβ-42, andof the observed conformational changes on the aggre-gation processes.
Experimental
The AMBER10 program suite was used to carryout all the molecular dynamics (MD) simulations(Case et al., 2008). The structure of Aβ-42 in anaqueous environment was determined in detail us-ing CD and NMR spectroscopy. The coordinates ofthe Aβ-42 peptide obtained from the PDB databaseunder the code 1Z0Q were used. The code contains30 NMR structures (Tomaselli et al., 2006) of Aβ-42with the sequence of DAEFRHDSGYEVHHQKLVF-FAEDVGSNKGAIIGLMVGGVVIA. The first struc-ture, which represents the most probable NMR struc-ture in an aqueous solution, was used for the model-ing study. The Aβ-42 structure was converted into theAMBER data format. Aβ-42 was placed into a solvat-ing water box with physiological NaCl (0.15 mol L−1)and the whole model was optimized in AMBER10 andequilibrated at 298.15 K employing the Amber99SBforce field (Case et al., 2008). The equilibrated struc-ture of Aβ-42 (Fig. 1a) provided the initial structurefor the MD simulations of complexes with Cu2+ ionsand CUA.The CUA structure (Fig. 1b) was generated and
optimized in Gaussian09 (Frisch et al., 2009) at theDFT B3LYP/6-31+G(d) level (Becke, 1993; Ditchfieet al., 1971; Francl et al., 1982; Harihara et al., 1973;Hehre et al., 1972; Lee et al., 1988; Miehlich et al.,1989; Rassolov et al., 2001). CUA was modeled in theanionic form with a formal charge of –1. The optimizedstructure was used to generate the topology and forcefield parameters by an antechamber tool, where Re-strained ElectroStatic Potential (RESP) charges andgaff parameters were used.The Cu2+ ion was treated using the “non-bonded
model”, which only considers interactions of its adja-cent Aβ residues to copper as electrostatic and vander Waals forces instead of covalent bonds. The “non-bonded model” approach was used to study the pos-sible Cu2+ interaction with Aβ-42 in the solutionwithout a specific binding mode. Since the AMBERforce field does not support parameters for the Cu2+
ion, these were added to the force field from previ-ously published literature (Allinger et al., 1994) (netcharge = +2, mass = 63.55, non-bonding parametersof 2.2200 and 0.3330).MD simulations of different models in aqueous
solutions were performed. Altogether, nine differ-ent models were built, namely: (i) Aβ-42 in phys-iological solution (described as Aβ); (ii) Aβ-42 in
Authenticated | [email protected] author's copyDownload Date | 6/3/15 12:41 PM

1264 S. Kozmon, I. Tvaroška/Chemical Papers 69 (9) 1262–1276 (2015)
Fig. 1. Scheme of equilibrated Aβ-42 structure used for model preparation (a). CUA structure used in the MD simulations withatom labeling (b).
a complex with Cu2+ near His13 (Aβ–Cu2+); (iii)Aβ-42 in a complex with Cu2+ near delta pro-tonated His13 (Aβ(H13δ)–Cu2+); (iv) Aβ-42 in acomplex with Cu2+ near His6, His13, and His14residues (Aβ(H6,H13,H14)–Cu2+); (v) Aβ-42 in acomplex with five Cu2+ ions (Aβ–5Cu2+); (vi) Aβ-42 in a complex with CUA (Aβ–CUA); (vii) twoCUA molecules in a complex with Cu2+ (Cu2+–CUA); (viii) Aβ-42 in a complex with Cu2+ andCUA near His13 (Aβ–Cu2+–CUA); and (ix) Aβ-42in a complex with Cu2+and CUA near delta proto-nated His13 (Aβ(H13δ)–Cu2+–CUA). Initial modelswere prepared manually by placing Cu2+ ions closeto His residues. Similarly, the CUA molecule wasplaced close to the Cu2+ ion to complete its coor-dination sphere. In the Aβ-5Cu2+ model, five Cu2+
ions were placed randomly around Aβ-42. In the Aβ–CUA model, the CUA molecule was docked to theequilibrated Aβ-42 structure. The above models rep-resent several possible arrangements of Cu2+ ions andCUA interaction with Aβ-42. Experimental studies onAβ–Cu complexes suggested (Karr et al., 2004; Miuraet al., 1999; Syme et al., 2004) that the Cu2+ ionis coordinated by three nitrogen atoms and one oxy-gen atom. One of the coordinating nitrogen atoms isthe delta nitrogen from His13. Therefore, the Cu2+
ion was placed close to His13. Protonation state ofthe His13’s epsilon nitrogen atom observed in theNMR structure (1Z0Q) (Tomaselli et al., 2006) cor-responds to the suggested interaction. However, thehistidine residue is solvent accessible, and its protona-tion state can be easily changed, based on the envi-ronment. To assess the effect of the protonation stateon the Aβ-42 behavior, models with His13 protonatedat the delta nitrogen were generated. MD simulationof free Aβ in the solvent was done as a reference.The Aβ(H6,H13,H14)–Cu2+ model was prepared us-ing the constrained molecular dynamics. The Aβ-42equilibrated structure, where Cu2+ was placed near
His6 was used as the starting structure. Three collec-tive variables were applied to the model during thedistance constrained molecular dynamics simulation.Each collective variable represented the distance be-tween the Cu2+ ion and the δ-nitrogen of the cor-responding His. The distance was changed from theinitial value to the final value of 2.1 A within 2 nsMD. Afterward, the 10 ns long harmonic potentialdistance restrained MD simulation was used to equili-brate the amyloid-beta structure. The same three col-lective variables were used; however, the distance wasallowed to vary between 1.9 A and 2.3 A with the forceconstant of 42 kJ A−2. Constrained and restrainedMD simulations were done using the Amber11 pro-gram package together with the MDtool called PM-Flib developed by Dr. Kulhánek at the National Cen-tre for Biomolecular Research Institute of the MasarykUniversity (Brno, Czech Republic) (Kulhánek et al.,2013).All nine prepared complexes were placed into a
box of the TIP3P type water molecules. The com-plexes were neutralized, and the NaCl concentrationwas adjusted to 0.15 mol L−1. The models containedbetween 14000 and 21000 atoms. Then, the geome-try of all models was optimized at 298.15 K. Therelaxed structures provided the initial structures forMD simulations, which were carried out under con-stant pressure (NPT) at 298.15 K, and the temper-ature was maintained using a Berendsen thermostat.The 200 ns production MD simulation was run ap-plying the SHAKE algorithm on the hydrogen atomsand the time step of 2 fs was used in all simula-tions. The MD structure snapshot were taken every1 ps. Whole 200 ns long trajectories were analyzed,however, the number of snapshots was reduced bythe factor of 10, yielding to solvent stripped trajec-tories with 20000 snapshots. All resulting trajecto-ries were analyzed by the cpptraj program from theAMBER14 package (Roe & Cheatham, 2013). Alto-
Authenticated | [email protected] author's copyDownload Date | 6/3/15 12:41 PM

S. Kozmon, I. Tvaroška/Chemical Papers 69 (9) 1262–1276 (2015) 1265
Fig. 2. Summary of Aβ-42’s secondary structure behavior. Colors represent the percentage of the simulation time spent in theappropriate secondary structure. Detailed information is available in Supplementary data.
gether, 2 �s of the MD simulation data were ana-lyzed.Two main analyzes were performed: (i) frequency
count histogram and statistical analysis of the moni-tored distances from the MD trajectory implementedin cpptraj. Distances between aspartic acids, glu-tamic acids (Asp, Glu), His amino acid residues,CUA oxygen atoms, and Cu2+ ions were monitoredto determine the presence of the Cu2+ ions lo-cated close to the particular amino acid or CUAresidue (Fig. S1); (ii) conformational behavior of Aβ-42 was analyzed using the secondary structure his-tograms and compared to the free Aβ model simula-tion (Figs. 2 and S2). The secondary structure wasassociated employing the define secondary structureof proteins (DSSP) analysis implemented in the cpp-traj utility. Clustering analysis of the saved trajec-tories was performed to analyze the population ofspecific Aβ-42 structures during the MD simulationand the Aβ-42 conformations obtained from the tra-jectory were clustered into groups using the hierar-chical agglomerative (bottom-up) approach with ep-silon of 4.0 and the minimum of 20 clusters, and theirpopulations in the trajectory were calculated. Theobserved populations indicate conformational equili-bration of the Aβ-42 conformers in solutions (Ta-ble S1).
Results and discussion
Results obtained by MD simulations are discussedfor each model separately in the first part of this sec-tion.
Aβ-42 in a physiological solution – Aβ model
Secondary structure statistics, clustered struc-tures, and Asp23–lysine (Lys) 28 interaction data wereextracted from the trajectory of the 200 ns long MDsimulation of Aβ-42 in a water box with the physi-ological concentration of Na+ ions. These data wereused to evaluate the role of the Cu2+ ion and CUAon the structural behavior of Aβ-42. Secondary struc-ture analysis showed the presence of the most stableα-helical structure in the His13–Asp23 region stablefor 61–90 % of the simulation time. The less stableα-helical structure was found in the N-terminal end(His6–valine (Val)12), and it was stable for approxi-mately 30 % of the simulation time. At the C-terminalend, relatively stable β-sheet structures were observed.They can be found in the serine (Ser) 26–asparagine(Asn) 27, alanine (Ala) 30–isoleucine (Ile) 31 regionsand are stable for 61–90 % of the simulation time(Figs. 2 and S2a and S3a). Clustering of the trajec-tory snapshots showed no major conformer, with thepopulation of the three most stable conformers rang-ing from 13 % to 16 % (Fig. 3a, Table S1). The lastmonitored property was the distance between residuesAsp23 and Lys28. The distance frequency count anal-ysis did not indicate the presence of these two residuesup to the distance of 6.5 A (Table 2). These val-ues suggest that a direct interaction between Asp23and Lys28 cannot be anticipated within the simula-tion time. For comparison, a 500 ns MD simulationwas carried out in implicit water, showing very sta-ble short α-helical structures packed in the globularstructure for almost the whole range of the simula-
Authenticated | [email protected] author's copyDownload Date | 6/3/15 12:41 PM

1266 S. Kozmon, I. Tvaroška/Chemical Papers 69 (9) 1262–1276 (2015)
Fig. 3. Visualization of the most populated cluster representative structures of the Aβ (a), Aβ–Cu2+ (b), Aβ(H13δ)–Cu2+
(c), Aβ(H6,H13,H14)–Cu2+ (d), Aβ–5Cu2+ (e), Aβ–CUA–1 (f), Aβ–CUA–2 (g), Aβ–CUA–3 (h), Aβ–Cu2+–CUA (i),Aβ(H13δ)–Cu2+–CUA (j) models.
tion time (Figs. S15 and S16). No β-sheet structuresand Asp23–Lys28 salt bridges were observed duringthe implicit solvent MD simulation. Cluster analysis ofthe trajectory showed two major conformations with32 % occupancies and a similar structure (Fig. S16).
Aβ-42 complex with Cu2+ located near theHis13 residuum – the Aβ–Cu2+ model
Experimental data suggest (Faller, 2009; Faller &Hureau, 2009) that the His13 residue of Aβ-42 is theprimary binding site for Cu2+. Therefore, in the ini-tial structure of the Aβ–Cu2+ model, the Cu2+ ionwas coordinated by His13 protonated on epsilon nitro-gen. The coordination sphere of Cu2+ was completedby His6 and Asp7 and one water molecule. Then, theAβ-Cu2+ model was equilibrated, and the 200 ns tra-
jectory was used to calculate the occupancies of theCu2+ ions and of all His and negatively charged aminoacid residues (Fig. S1a). The analysis revealed that theCu2+ ion was released from the starting position andit was found in the vicinity of Asp7 and Glu11 oxygenatoms, in the average distance of 3.1 A. The calculatedoccurrence at this distance is between 3 and 6 for Asp7and Glu11, respectively (Table 1). According to thesevalues the occupation of the Cu2+–Asp7/Glu11 inter-action is weak; approximately 3–6 % of the simulationtime (Fig. S1a, Cu43–Asp7(OD1), Cu43–Asp7(OD2),Cu43–Glu11(OE1) and Cu43–Glu11(OE2) datasets;Fig. S7). Similarly, based on the observed occupan-cies, interaction of the Cu2+ ion with any histidineresidue can be excluded. The secondary structure pro-file for the Cu2+–Aβ model trajectory is shown inFig. 2, (detailed plot in Figs. S2b and S3b). Observed
Authenticated | [email protected] author's copyDownload Date | 6/3/15 12:41 PM

S. Kozmon, I. Tvaroška/Chemical Papers 69 (9) 1262–1276 (2015) 1267
Table1.InteractionoccupanciesoftheCu2+ion;occupanciesfromtheshortestdistancewithobservedinteractionoccurrenceareprovided
Aβ–Cu2+
Aβ(H13
δ)–Cu2+
Aβ(H6,H13,H14)–Cu2+
Aβ–5Cu2+
2CUA–Cu2+
Aβ–Cu2+–CUA
Aβ(H13
δ)–Cu2+–CUA
Cu43
Cu43
Cu43
AnyCu2+
Cu3
Cu44
Cu44
ResidueAtom
Average
Occur-
Average
Occur-
Average
Occur-
Average
Occur-
Average
Occur-
Average
Occur-
Average
Occur-
distance/A
rence/%
distance/A
rence/%
distance/A
rence/%
distance/A
rence/%
distance/A
rence/%
distance/A
rence/%
distance/A
rence/%
Asp1
OD1
3.05
0.6
3.10
0.3
3.10
0.7
4.17
0.9
n.d.
3.08
0.2
3.08
1.6
OD2
3.09
0.4
3.07
0.2
3.06
1.1
4.17
0.9
n.d.
3.05
0.1
3.08
1.4
Asp7
OD1
3.09
3.1
3.07
1.1
3.07
0.3
4.13
2.4
n.d.
3.08
13.3
3.08
26.7
OD2
3.08
3.3
3.08
1.2
3.06
0.5
4.13
2.4
n.d.
3.09
13.7
3.09
21.4
Asp23
OD1
5.02
0.1
5.22
<0.1
3.78
<0.1
4.15
3.3
n.d.
3.10
22.2
3.05
3.7
OD2
4.42
<0.1
5.07
0.1
3.84
<0.1
4.15
3.3
n.d.
3.11
20.2
3.09
1.2
Glu3
OE1
3.04
0.1
5.07
0.3
3.08
2.3
4.35
0.2
n.d.
3.08
1.2
3.07
3.0
OE2
4.44
<0.1
4.50
<0.1
3.11
2.0
4.35
0.2
n.d.
3.10
1.1
3.09
2.4
Glu11
OE1
3.10
5.4
3.09
1.5
3.09
1.0
4.38
<0.1
n.d.
3.05
0.1
3.08
1.2
OE2
3.09
5.5
3.08
1.6
3.10
0.8
4.38
0.0
n.d.
3.14
0.1
3.09
1.1
Glu22
OE1
3.05
0.2
3.09
0.7
3.08
0.9
4.19
0.4
n.d.
3.03
5.0
3.10
0.9
OE2
3.12
0.2
3.06
0.7
3.08
0.8
4.19
0.4
n.d.
3.05
4.0
3.11
0.8
His6
ND1
5.23
0.1
3.35
0.1
3.24
3.0
4.03
0.1
n.d.
3.31
0.3
4.17
0.0
O3.17
<0.1
3.10
0.2
3.10
4.6
4.03
0.1
n.d.
3.11
0.3
3.17
0.1
His13
ND1*
23.80
100.0
3.25
0.1
3.25
4.1
4.47
<0.1
n.d.
4.39
<0.1
3.23
10.9
O24.15
100.0
6.20
<0.1
3.10
4.5
4.47
<0.1
n.d.
5.45
<0.1
17.05
100
His14
ND1
3.30
<0.1
5.17
<0.1
3.26
1.6
4.81
<0.1
n.d.
4.83
<0.1
5.30
<0.1
O5.15
<0.1
5.15
0.1
6.36
0.2
4.81
<0.1
n.d.
22.40
100.0
19.27
100.0
CUA
O1
n.d.
n.d.
n.d.
n.d.
3.05
95.5
3.05
1.6
3.05
53.0
O2
n.d.
n.d.
n.d.
n.d.
3.05
95.3
3.05
1.6
3.04
53.7
Authenticated | [email protected] author's copyDownload Date | 6/3/15 12:41 PM

1268 S. Kozmon, I. Tvaroška/Chemical Papers 69 (9) 1262–1276 (2015)
data suggest that the presence of the Cu2+ ion af-fects the secondary structure behavior of Aβ-42. Inthe Cu2+ containing model, the β-sheet structure isnot presented, whereas a small α-helical structure wasfound with a 25–60 % occurrence in the Gly33–Met35region. The α-helical structures can be found in almostthe entire structure except for the Ala21–Ile32 regionand the C-terminal, which seem to be unfolded. TheN-terminal part contains two short, relatively stable,α-helical structures. The first is formed by residuesPhe4–Asp7 and is stable for 25–60 % of the simula-tion time. The second α-helical structure can be foundin the hydrophobic core region Glu11–phenylalanine(Phe) 20 and it is stable for 25–60 % of the simulationtime. Clustering analysis of the trajectory snapshotsshowed one main cluster with the 40 % occupancy(Table S1, Fig. 3b). The second and the third largestclusters showed only an 11 % occupancy (Fig. S4b).
MD simulation of Aβ-42 with Cu�+ near deltaprotonated His13 – the Aβ(H13δ)–Cu�+ model
To investigate the effect of the His13 protonationstate on the Aβ-42 secondary structure behavior andits interaction with Cu2+ and CUA, MD simulationswere performed also with His13 protonated on thedelta nitrogen atom. The initial structure was pre-pared as that for the epsilon protonated His13. Theonly difference was the location of the proton on thedelta nitrogen. The production trajectory was 200 ns.The distance frequency count analysis between Cu2+
and the amino acids showed that Cu2+ was releasedfrom Aβ-42 to the free solvent. The highest occurrenceof Cu2+ close to amino acids can be seen for residuesAsp7 and Glu11. Found occupancies at the averagedistance of 3.1 A were in the range of 1.1–1.6 (Table 1).These numbers suggest that the possibility of interac-tions between the Cu2+ ion and Asp7 and Glu11 islow (Fig. S1b; Cu43–Asp7(OD1), Cu43–Asp7(OD2),Cu43–Glu11(OE1) and Cu43–Glu11(OE2) datasets;Fig. S8). The change of the His13 protonation statealso influenced the Aβ-42 secondary structure. A com-parison of the Aβ-42 behavior with results from thesimulation of free Aβ-42 revealed some differences(Fig. 2, detailed plot in Figs. S2c and S3c). The largestdifference can be seen in the hydrophobic core re-gion, where the α-helical structure in the Aβ(H13δ)–Cu2+ model is strongly destabilized compared to thatin the Aβ model. The α-helices at N-terminal areshorter than those in the free Aβ-42 simulation. Theα-helix in the Asp7–Val12 region was stable only for25–60 % of the simulation time, whereas the shorterα-helix (Leu17–Phe20) was stable for more than 61 %of the simulation time. At the C-terminal part, twovery short β-sheet structures can be seen, near Asn27and Ala30. These short β-sheet structures were sta-ble for 25–60 % of the simulation time. A compari-son with the Aβ–Cu2+ model, which differs only in
the His13 protonation state, revealed some differencesin the structural behavior, e.g. the C-terminal partin the Aβ and Aβ–Cu2+ models behaved differently.Whereas in the Aβ–Cu2+ model, only short α-helicalstructure was found, several different secondary struc-tures, described before, can be seen in the Aβ(H13δ)–Cu2+ model. Furthermore, the α-helical structures atthe N-terminal part were shorter compared to those inthe Aβ–Cu2+ model (Asp7–Val12 and Leu17–Phe20versus Phe4–Asp7 and Glu11–Phe20), but their sta-bility over the simulation was similar, 25–60 % of thesimulation time. Clustering analysis showed two mainclusters with a similar population ranging from 14 %to 17 % (Table S1, Figs. 3c and S4c).
Aβ-42 complex with Cu�+ near His6, His13,and His14 residues – the Aβ(H6,H13,H14)–Cu�+ model
Initial structure of the Aβ(H6,H13,H14)–Cu2+
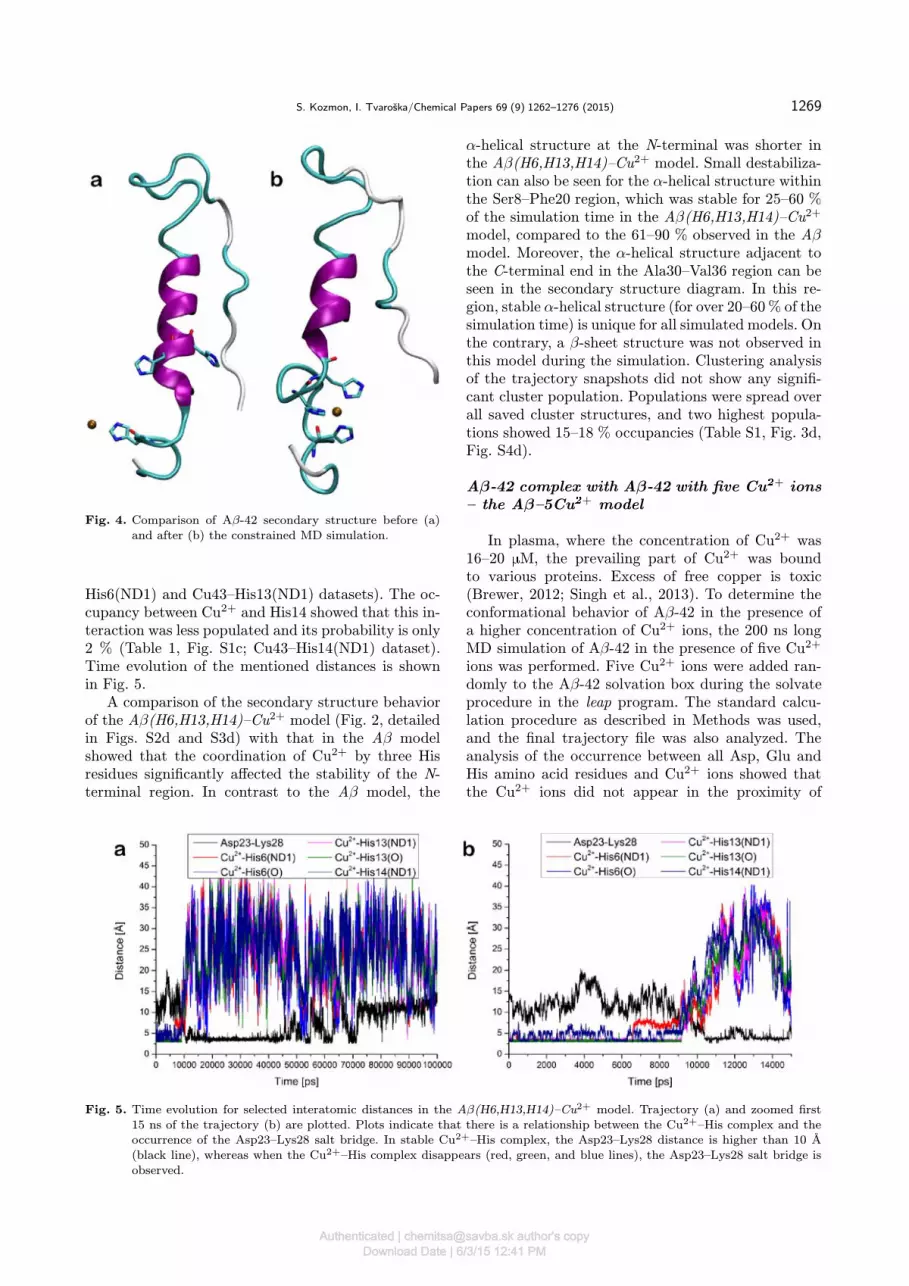
complex for the MD simulation was prepared fromthe equilibrated structure of the Aβ–Cu2+ model,where Cu2+ was located near the His6 residue. TheAβ(H6,H13,H14)–Cu2+ complex was generated fromthis structure by a constrained MD simulation, inwhich three collective variables were applied. Each col-lective variable represented the distance between theCu2+ ion and the δ-nitrogen of one of three histidines.The constraint of 2.1 A was used for each collectivevariable. This procedure led to the complex structure,where the Cu2+ ion is coordinated with three his-tidines: His6, His13, and His14. The formation of thecomplex altered the secondary structure in the His6–His14 region compared to that in free Aβ-42. In thecomplex, a stable α-helical structure was disrupted,and this region adopted a short loop allowing threehistidine residues to coordinate the Cu2+ ion (Fig. 4).As a consequence, distances between the Cu2+ ion andthe histidine δ-nitrogens was 2.1 A.After the restrained equilibration, the 200 ns long
MD simulation of the Aβ(H6,H13,H14)–Cu2+ modelwas carried out, where neither restraints nor con-straints were applied to the Cu2+ – histidine δ-nitrogen distances, and the distance frequency countanalysis of the observed trajectory data for Cu2+ andall Asp, Glu, and His residues was done. The analy-sis revealed that Cu2+ was located near the histidineresidues for a significant time period at the beginningof the simulation. The highest occurrence at around3 A was observed between Cu2+ and the backboneoxygen of His6 and His13 (Fig. S1c; Cu43–His6(O)and Cu43–His13(O) datasets) with the values of 4.5 %and 4.6 %, respectively. (Table 1). These values indi-cate that the Cu2+ ion was coordinated by these twoatoms for nearly 5 % of the simulation time. Secondhighest occurrences were observed between Cu2+ andHis6 and His13 δ-nitrogen atoms for around 3 % and4 % of the simulation time (Table 1, Fig. S1c; Cu43–
Authenticated | [email protected] author's copyDownload Date | 6/3/15 12:41 PM
S. Kozmon, I. Tvaroška/Chemical Papers 69 (9) 1262–1276 (2015) 1269
Fig. 4. Comparison of Aβ-42 secondary structure before (a)and after (b) the constrained MD simulation.
His6(ND1) and Cu43–His13(ND1) datasets). The oc-cupancy between Cu2+ and His14 showed that this in-teraction was less populated and its probability is only2 % (Table 1, Fig. S1c; Cu43–His14(ND1) dataset).Time evolution of the mentioned distances is shownin Fig. 5.A comparison of the secondary structure behavior
of the Aβ(H6,H13,H14)–Cu2+ model (Fig. 2, detailedin Figs. S2d and S3d) with that in the Aβ modelshowed that the coordination of Cu2+ by three Hisresidues significantly affected the stability of the N-terminal region. In contrast to the Aβ model, the
α-helical structure at the N-terminal was shorter inthe Aβ(H6,H13,H14)–Cu2+ model. Small destabiliza-tion can also be seen for the α-helical structure withinthe Ser8–Phe20 region, which was stable for 25–60 %of the simulation time in the Aβ(H6,H13,H14)–Cu2+
model, compared to the 61–90 % observed in the Aβmodel. Moreover, the α-helical structure adjacent tothe C-terminal end in the Ala30–Val36 region can beseen in the secondary structure diagram. In this re-gion, stable α-helical structure (for over 20–60% of thesimulation time) is unique for all simulated models. Onthe contrary, a β-sheet structure was not observed inthis model during the simulation. Clustering analysisof the trajectory snapshots did not show any signifi-cant cluster population. Populations were spread overall saved cluster structures, and two highest popula-tions showed 15–18 % occupancies (Table S1, Fig. 3d,Fig. S4d).
Aβ-42 complex with Aβ-42 with five Cu�+ ions– the Aβ–5Cu�+ model
In plasma, where the concentration of Cu2+ was16–20 �M, the prevailing part of Cu2+ was boundto various proteins. Excess of free copper is toxic(Brewer, 2012; Singh et al., 2013). To determine theconformational behavior of Aβ-42 in the presence ofa higher concentration of Cu2+ ions, the 200 ns longMD simulation of Aβ-42 in the presence of five Cu2+
ions was performed. Five Cu2+ ions were added ran-domly to the Aβ-42 solvation box during the solvateprocedure in the leap program. The standard calcu-lation procedure as described in Methods was used,and the final trajectory file was also analyzed. Theanalysis of the occurrence between all Asp, Glu andHis amino acid residues and Cu2+ ions showed thatthe Cu2+ ions did not appear in the proximity of
Fig. 5. Time evolution for selected interatomic distances in the Aβ(H6,H13,H14)–Cu2+ model. Trajectory (a) and zoomed first15 ns of the trajectory (b) are plotted. Plots indicate that there is a relationship between the Cu2+–His complex and theoccurrence of the Asp23–Lys28 salt bridge. In stable Cu2+–His complex, the Asp23–Lys28 distance is higher than 10 A(black line), whereas when the Cu2+–His complex disappears (red, green, and blue lines), the Asp23–Lys28 salt bridge isobserved.
Authenticated | [email protected] author's copyDownload Date | 6/3/15 12:41 PM

1270 S. Kozmon, I. Tvaroška/Chemical Papers 69 (9) 1262–1276 (2015)
any monitored amino acid residue for a considerablylong time. The longest residential time was observedfor the Asp23 amino acid residue and the lasted ataround 3 % of the simulation time in the longer av-erage distance of around 4.2 A. (Table 1, Fig. S1d;Cu–Asp23(OD1) and Cu–Asp23(OD2) datasets). TheCu2+ ions were also found in the area around theAsp7 amino acid (Table 1, Fig. S1d; Cu–Asp7(OD1)and Cu–Asp7(OD2) datasets), for approximately 2 %of the simulation time, also in a quite long aver-age distance of 4.1 A. Interestingly, the analysis didnot show any occupation around any of the histidineresidues.Histogram summary plots of the secondary struc-
ture population showed a different conformational be-havior of Aβ-42 in the presence of the Cu2+ ions(Fig. 2, detailed plot in Figs. S2e and S3e in SI). It canbe seen that the Cu2+ ions destabilized the α-helicalstructure in the Asp7–Val12 region but promoted theformation of the α-helical structure in Ala30–Ile32 (upto 60 % of the simulation time), compared to theAβ model. Cu2+ ions also disrupted β-sheets struc-tures present at the Aβ-42 C-terminal end in the Aβmodel. The obtained results indicate that the presenceof a higher concentration of Cu2+ ions significantly af-fects the conformational behavior of Aβ-42 in the so-lution. Clustering of the trajectory snapshots showedone main cluster with an only 18.1 % occupancy (Ta-ble S1, Fig. 3e). Other clusters showed occupancieslower than 10 % (Fig. S4e).
Aβ-42 complex with a CUA molecule – the Aβ–CUA model
This model was generated by docking the CUAmolecule into the equilibrated Aβ-42 structure. Dock-ing grid was generated around the whole Aβ-42molecule to consider all possible locations for CUAbinding. Based on the docking results, three differ-ent locations with the highest populations, denotedas Aβ–CUA–1, Aβ–CUA–2, and Aβ–CUA–3, wereselected. Geometry of these structures that repre-sent different binding modes was optimized and thensolvated using the AMBER program package. Sol-vated structures were equilibrated to 298.15 K. Fi-nally; the 200 ns long production MD simulations wererun.Secondary structure analysis of the obtained MD
trajectories showed only slight differences in the sec-ondary structure behavior (Fig. 2). All three struc-tures showed a relatively stable α-helical structure inthe Tyr10–Phe20 region. These structures were sta-ble for up to 95 % of the simulation time. The N-terminal part, up to Ser8, seemed to be less stableand showed an α-helical structure for up to 60 %of the simulation time only for Aβ–CUA–2. On theother hand, the Aβ–CUA–1 strucuture showed twoshort α-helical structures at the C-terminal part sta-
ble for 25–60 % of the simulation time. Otherwise,the C-terminal part was mainly unstructured duringthe whole simulation. Only in case of the Aβ–CUA–2model, several short β-sheet structures were found inthe C-terminal region Ala21–Val40, which were stablefor approximately 25–60 % of the simulation time. Incomparison with the Aβ model, a small destabiliza-tion of the N-terminal α-helical structure up to Ser8was observed. On the contrary, no β-sheet structureswere found on the C-terminal end of the Aβ–CUA–1and Aβ–CUA–3 models. To examine the interactionof the CUA molecule with Aβ-42, the saved struc-ture snapshots from MD runs were clustered for eachof the three locations. The most populated clustersare shown in Figs. 3f–3h, with the population of 18%, 36 %, and 51 % for Aβ–CUA–1, Aβ–CUA–2, andAβ–CUA–3, respectively (Table S1). The CUA–Aβ-42 interaction pattern was also analyzed by the hy-drogen bond analysis and the atom contact map be-tween the CUA and each amino acid residue of Aβ-42(Table S2, Fig. S14). This analysis revealed preferredinteractions of the CUA molecule with Aβ-42 residuesduring the MD simulation and showed that CUA in-teracts primarily with the hydrophobic core of Aβ-42.The strongest interaction is that with Lys16, Phe19,and Phe20. The Lys16–CUA hydrogen bonds were oc-cupied up to 15 % of the simulation time and no inter-action with histidines was found (Table S2). Interac-tions with Leu17, Ile31, and Ile32 were found to be lessstable (Fig. S14). The above mentioned interactionswere also found in the structures of the most popu-lated clusters. The results clearly show the importanceof the hydrophobic core for the CUA molecule bind-ing. Similar results were obtained in a recently pub-lished study of the Aβ dimer interaction with the CUAmolecule (Zhao et al., 2012), done using the GRO-MACS program package with the Gromos force field.CUA interactions with hydrophobic residues Phe19,Phe20, Tyr10 and Leu34 residues were found, whichis in agreement with our observation. Also, long mi-croseconds molecular dynamics simulation of Aβ-42has been done recently and the micromolar inhibitorAE-848 was docked in the final structure (Wang et al.,2013). The final Aβ structure consists mainly of theβ-sheet secondary structure. AE-848 interacts mainlywith C-terminal of the Aβ-42 structure (Gly38–Ile41).No strong interaction of CUA and Aβ-42 within thisregion was observed; only in case of the Aβ–CUA–3model, some weak interactions with Val39 were found(Fig. S14C).
Complex of two CUA molecules with Cu�+ ion– the Cu�+–2CUA model
A 200 ns simulation of the Cu2+–2CUA com-plex showed that it is relatively stable in solu-tions. Over the whole simulation time, the aver-age distance between Cu2+ and CUA oxygen atoms
Authenticated | [email protected] author's copyDownload Date | 6/3/15 12:41 PM

S. Kozmon, I. Tvaroška/Chemical Papers 69 (9) 1262–1276 (2015) 1271
O1, and O2 was 3.1 A (Fig. S13A). The dis-tance frequency count analysis between copper andO1 and O2 atoms of both CUA residues showed(Fig. 13B) nearly 96 % occupancies of the simula-tion time. These results also suggest that the param-eters used for the Cu2+ and CUA description are re-liable.
Aβ-42 complex with Cu�+ and CUA nearHis13 residuum – the Aβ-Cu�+–CUA model
In this model, the Cu2+ ion was placed close toHis13, and CUA was then placed near the Cu2+ ionto complete its coordination sphere. The MD sim-ulation was run for 200 ns. The distance frequencycount analysis of distances between Cu2+, CUA, andoxygen atoms of Asp and Glu residues revealed thatthe Cu2+ ion was present in the vicinity of Asp7 andAsp23 during the MD simulations (Fig. S1e, Cu44–Asp7(OD1), Cu44–Asp7(OD2), Cu44–Asp23(OD1),and Cu44–Asp23(OD2) datasets). The Cu2+ ion in-teracted with the Asp23 and Asp7 residues for 22 %and 14 % of the simulation time, respectively (Ta-ble 1). Surprisingly, interactions between the Cu2+
ion and any of the His residues were not observed.An analysis of the MD simulation trajectory alsoshowed that interactions of Cu2+ and CUA withAβ-42 altered the Aβ-42 secondary structure. Twoshort α-helices, in the N-terminal part (Phe4–Asp7and His14–Phe19), were present for approximately60 % of the simulation time (Fig. 2, detailed plotin Figs. S2i and S3i). A visual inspection of the MDsimulation trajectory revealed intriguing behavior ofthis complex model during the simulation. At the be-ginning of the simulation, CUA and Cu2+ were re-leased from Aβ-42 and CUA was involved in an in-teraction with Aβ-42 on its N-terminal end. The in-teraction mainly involved the short α-helix (Phe4–Asp7) and a nearby turn (tyrosine (Tyr) 10–Val12).Then, the C-terminal started to interact with CUAinvolving the unfolded region Ile32–Ala42. The men-tioned interaction can also be seen on the calcu-lated contact maps in Fig. S14. In the following sev-eral nanoseconds, the secondary structure of Aβ-42changed to form a complex with CUA. This con-formational change generated a new small bindingpocket in which the Cu2+ ion was accommodated.The Cu2+ ion interacted primarily with the Asp7and Asp23 resides (Figs. S1e and S11), and less fre-quently with Glu22 within the formed binding pocket.As a result, a relatively stable ternary complex Aβ-42–CUA–Cu2+ was formed. Density isosurfaces ofthe Cu2+ and CUA appearance around Aβ-42 areshown in Fig. 6. These isosurfaces represent locations,where Cu2+ ions and CUA atoms were most oftenlocated during the MD simulation. Also, structuralchanges in Aβ-42 during the simulation can be seenin Fig. 6.
Fig. 6. Structure snapshots from the MD simulations. Struc-tures are colored from red to white to blue from thebeginning to the end of the simulation. Aβ-42 is rep-resented as a cartoon, CUA as sticks and Cu2+ asspheres. Green isosurface represents the space with fre-quent occurrence of CUA and the brown one of Cu2+.
Aβ-42 complex with Cu�+ and CUA near deltaprotonated His13 – the Aβ(H13δ)–Cu�+–CUAmodel
A molecular dynamics simulation of Aβ-42 withthe starting location of CUA and Cu2+ near the deltaprotonated His13 residuum was performed to deter-mine the effect of different histidine protonation stateson Aβ-42 interactions with CUA and Cu2+. The pro-duction run of the simulation took 200 ns. The fre-quency count analysis is shown in Fig. S1f.The results showed that Cu2+ is coordinated
by CUA oxygen atoms for 54 % of the simulationtime (Table 1, Fig. S1f, Cu44–CUA(O1) and Cu44–CUA(O2) datasets) and the second largest occu-pancy showed that Cu2+ is also coordinated by theAsp7 oxygen atom (Fig. S1f, Cu44–Asp7(OD1) andCu44–Asp7(OD1) datasets). Coordination proceededfor around 27 % of the simulation time (Table 1). Itis interesting that only in this case, coordination ofCu2+ by His nitrogen atoms was observed. The dis-tance histogram analysis revealed that some abun-dance can be found in case of the His13 epsilon nitro-gen (Fig. S1f, Cu44–His13(NE2) dataset). The anal-ysis showed an 11 % occurrence of Cu2+ in the av-erage distance 3.23 A from the His13 epsilon nitro-
Authenticated | [email protected] author's copyDownload Date | 6/3/15 12:41 PM

1272 S. Kozmon, I. Tvaroška/Chemical Papers 69 (9) 1262–1276 (2015)
gen. The obtained simulation data suggest that CUAchelated Cu2+ for approximately half of the simu-lation time, and for about a quarter of the simula-tion time, Cu2+ also interacted with Aβ-42 via theAsp7 oxygen atom. Calculated contact maps of CUAand Aβ-42 (Fig. S14) show that CUA interacts pre-dominantly with hydrophobic residues Tyr10, His13,Leu17, Phe20, Val24, Lys28, Ile32 and creates hydro-gen bonds with Gly9, Lys28, Ser8, and Tyr10 (Ta-ble S2).A comparison of the secondary structure behav-
ior of Aβ-42 in the Aβ(H13δ)–Cu2+–CUA modelwith that observed in the Aβ model revealed cleardifferences. The presence of CUA stabilized the α-helix in the central part of Aβ-42 (Tyr10–Asp23).The α-helical Glu11–Phe19 region was stable for over61 % of the simulation time, and the Phe20–Asp23part was stable for 25–30 % of the simulation time(Fig. 2, see detailed plot in Figs. S2j and S3j). How-ever, the stable α-helical structure at the N-terminalpart (His6–Gly9) found in the Aβ model was not ob-served in this model. Similarly, β-sheet structures atthe C-terminal part were not found. The effect of thechange of the His13 protonation state on the Aβ-42secondary structure can be seen from a comparisonwith the Aβ-Cu2+–CUA model. Whereas in the Aβ–Cu2+–CUA simulation, two short α-helices at the N-terminal (Phe4–Asp7 and Gln15–Phe19) were found,in the Aβ(H13δ)–Cu2+–CUA model, only one longerα-helix (Tyr10–Asp23) was observed. Another differ-ence can be seen in the C-terminal part. A short α-helix (Leu34–Val36), stable for 20–60 % of the simu-lation time, observed in this model was not found inthe model with the epsilon protonated His13.From the calculated data, some general conclusions
on the role of Cu2+ and CUA in the Aβ-42 secondarystructure behavior can be drawn (Fig. 2). It should benoted here that the secondary structure data in Fig. 2,discussed below, are based on the residue secondarystructure analysis done in the cpptraj program usingthe AmberTools and should also be compared with therepresentative cluster structures in Fig. 3 and Fig. S4.In all MD simulations, a stable α-helix in the Aβ-42 central part, the Gln15–Phe20 region, was found.The N- and C-terminal parts seemed to be quite flex-ible and, consequently, significant differences were ob-served in these regions. The occurrence and stability ofthe α-helical structure at the N-terminal part differ forvarious models. The most stable and longest α-helicalstructure can be seen in the Aβ model, whereas thisα-helical structure was fragmented into shorter struc-tures in other models. The presence of a higher concen-tration of the Cu2+ ions (Aβ–5Cu2+ model) resultedin splitting of the α-helical structure into tree short α-helices (Glu3–Arg5, His14–Ala22, and Ala30–Ile32) bydestabilizing the central part of the α-helix that wasobserved in the Aβ model (Asp7–His14). The presenceof one Cu2+ ion did not have such a dramatic impact
Table 2. Interaction occupancies of Asp23 and Lys28; occu-pancies from the shortest distance with observed in-teraction occurrence are provided
Model Average distance/A Occurrence/%
Aβ 6.37 < 0.1Aβ–Cu2+ 3.31 < 0.1Aβ(H13δ)–Cu2+ 2.96 3.9Aβ(H6,H13,H14)–Cu2+ 3.28 6.6Aβ–5Cu2+ 3.37 < 0.1Aβ–CUA-1 18.44 100.0Aβ–CUA-2 3.13 1.7Aβ–CUA-3 4.42 < 0.1Aβ–Cu2+–CUA 2.86 0.7Aβ(H13δ)–Cu2+–CUA 3.03 41.7
on the N-terminal α-helical structure. When the split-ting was observed, only an extremely short part of theα-helix (Ser8–Gly9) was destabilized. The presence ofthe CUA molecule (Aβ–Cu2+–CUA and Aβ(H13δ)–Cu2+–CUA models) stabilized the α-helical structurein the His13–Ala21 region. In the Aβ–Cu2+–CUAmodel, stabilization of the short α-helix (Phe4–Asp7)was also observed. It was found that the Cu2+ com-plex with three His residues (Aβ(H6,H13,H14)–Cu2+
model) had the greatest effect on the formation ofa relatively stable α-helical structure close to the C-terminal end. The analyzes of all secondary structuredata showed that C-terminal is the most flexible partof Aβ-42 and regions from Val24 up to Ala42 wereobserved to be poorly structuralized. Relatively sta-ble β-sheet structures in the C-terminal region wereobserved only in the Aβ, Aβ(H13δ)–Cu2+, and Aβ–CUA–2 models. The obtained data showed that C-terminal β-sheet structures occur significantly less fre-quently in the presence of a CUA molecule and a Cu2+
ion. These observations suggest that the presence ofCUA can prevent the formation of the amyloid-betaoligomers, where the occurrence of the C-terminal β-sheet structures can play a vital role, in agreementwith recent experimental data (Banerjee, 2014; Pic-ciano & Vaden, 2013).Another interaction which is thought to be re-
sponsible for the amyloid-beta oligomer formation isa salt bridge between Asp23 and Lys28. The dis-tance frequency count analysis between these tworesidues was done for all models to determine whetherthe formation of the salt bridge can be seen inthe MD simulations (Fig. S5, Table 2). However,the occurrence of the Asp23–Lys28 salt bridge wasnot observed for the Aβ, Aβ–Cu2+, Aβ–5Cu2+, Aβ–CUA–1, Aβ–CUA–3, and Aβ–Cu2+–CUA models. Aregular Asp23–Lys28 salt bridge was found in theAβ(H13δ)–Cu2+, Aβ(H6,H13,H14)–Cu2+, Aβ–CUA–2, and Aβ(H13δ)–Cu2+–CUA model simulations (Ta-ble 2, Figs. S5c, S5d, S5g, and S5j). The observedaverage distances, in these cases, ranged from 3.0 A
Authenticated | [email protected] author's copyDownload Date | 6/3/15 12:41 PM

S. Kozmon, I. Tvaroška/Chemical Papers 69 (9) 1262–1276 (2015) 1273
to 3.3 A. The salt bridge was stable for 4 %, 7 %,2 %, and 42 % of the simulation time in case ofAβ(H13δ)–Cu2+, Aβ(H6,H13,H14)–Cu2+, Aβ–CUA–2, and Aβ(H13δ)–Cu2+–CUA models, respectively. Asignificant difference between the models is the sta-ble bridge at the beginning of the simulation in theAβ(H13δ)–Cu2+ and Aβ(H6,H13,H14)–Cu2+ models,and at the end of the simulation in the Aβ(H13δ)–Cu2+–CUA model (Figs. S6c, and S6d, and S6j). Incase of the Aβ(H6,H13,H14)-Cu2+ model, some rela-tions between the presence of the Cu2+–His residuescomplex and the Asp23–Lys23 salt bridge can be ob-served. The time evolution of the Asp23–Lys28 andCu2+–His residues distances during the simulation isshown in Fig. 5. It can be seen that while the Cu2+–His residues complex is present, the Asp23–Lys28 dis-tance is more than 12 A. However, when the Cu2+–Hiscomplex disappears, the Asp23–Lys28 salt bridge isformed in a few nanoseconds and is stable for a certainsimulation time. Also, several breaks of the Asp23–Lys28 salt bridge can be seen between 45–70 ns, whichare associated with the Cu2+ ion approaching the Hisresidues. These observations suggest that the higherconcentration of the Cu2+ ions, or the presence ofthe Cu2+–His residues complex or protonation stateof the His13 residuum, have a significant influence onthe formation of the Asp23–Lys28 salt bridge as incase of the epsilon protonated His13, the salt bridgewas not observed (Figs. S5a, S5b, and S5i), which sug-gests that higher concentrations of Cu2+ promote thedevelopment of amyloid-beta oligomers.Several papers (Karr et al., 2004; Miura et al.,
1999; Parthasarathy et al., 2011; Syme et al., 2004)showed that the Cu2+ ion forms a complex withthe amyloid-beta molecule with some histidine ni-trogen atoms and that one of the interacting his-tidines is His13 (Syme et al., 2004). Simulation datarevealed the existence of the Cu2+ ion in the vicinityof any histidine residues in the structure with a Cu2+
ion present in the complex in all three His residues(Aβ(H6,H13,H14)–Cu2+ model) and it can be alsoseen in the Aβ(H13δ)–Cu2+–CUA model. In all sim-ulations where the Cu2+ ion interacted with only onehistidine residue in the starting structure, the Cu2+
ion was released from a histidine residue. Instead, theCu2+ ion formed a complex with the charged aminoacid, namely aspartic or glutamic acid. It is notewor-thy that the molecular mechanic force field used forthe MD simulations describes interactions between theCu2+ ion and the amino acids in non-covalent van derWaals and electrostatic terms, neglecting the chargetransfer interactions. The charge transfer interactionspossibly contributing to the Cu–N interaction wereneglected. This may lead to the preference of loca-tions where electrostatic interactions are dominant,e.g. between the Cu2+ ion and the negatively chargedamino acids (this does not suggest that interactionsbetween Cu2+ and Asp/Glu amino acid residues can-
not be present in vivo or in vitro). However, Cu–Ninteractions cannot be completely described by theavailable empirical force fields. Recently, several pa-pers presenting MD simulations of the Aβ–Cu com-plexes with the Cu2+ ion covalently bound to their co-ordinating partners have appeared (Ali-Torres et al.,2011; Parthasarathy et al., 2011) and also force fieldbonding parameters for the Cu–N and Cu–O interac-tions were determined (Wise & Coskuner, 2014; Xuet al., 2013). In the mentioned simulations, the com-plexes were stable and no dissociation of the complexwas observed. However, such behavior is expectablebecause of the used approach. Moreover, an applica-tion of the Cu–N covalent bonding term can lead toartificially driven behavior and proper calculations ofthe charge transfer interactions require the use of high-level quantum mechanics methods such as QM/MMMD method, however, such approach is computation-ally extremely demanding for these systems. More-over, calculations of short simulations, up to tens ofpicoseconds, and a definition of the permanent QMzone are not applicable for the moving Cu2+ ions.
Conclusions
In the presented paper, interactions between Aβ-42and Cu2+ and/or CUA were investigated using MDsimulations of nine different models. More than 2 �s ofthe simulation data were analyzed. The obtained datashowed that the conformational behavior of Aβ-42,essential for the oligomerization of the amyloid-betamolecules, can be significantly affected by the pres-ence of higher concentrations of Cu2+ ions (Aβ–5Cu2+
model) or by the presence of CUA molecules (Aβ–CUA and Aβ–Cu2+–CUA models). The main implica-tion of these complexes are the significant decrease ofthe C-terminal β-sheet structures, supposed to be rel-evant for the formation of toxic oligomers, occurrenceand the stabilization of the α-helical structure byCUA. On the other hand, analysis of the Aβ-42 modelswith changed protonation states of His13 (Aβ(H13δ)–Cu2+, Aβ(H13δ)–Cu2+–CUA models) showed thatthis change might lead to higher occurrence of theAsp23–Lys28 salt bridge, which was also seen in themodel with higher Cu2+ ion concentration (Aβ-5Cu2+
model). The Aβ(H6,H13,H14)–Cu2+ model revealedsome correlation between the occurrence of the Cu2+–His complex and the formation of the Asp23–Lys28salt bridge. Calculations also help to understand whyCUA inhibits the aggregation effect of Cu2+. As amatter of fact, CUA can chelate Cu2+ and directly in-teract with Aβ and, thus it has both activities requiredto act as an inhibitor of Aβ aggregation. However, theobtained results might be affected by the descriptionof the MD force fields, where copper interactions aredescribed only in the non-bonding terms and; there-fore, the Cu–N interaction might be underestimated.However, we believe that the obtained data shed some
Authenticated | [email protected] author's copyDownload Date | 6/3/15 12:41 PM

1274 S. Kozmon, I. Tvaroška/Chemical Papers 69 (9) 1262–1276 (2015)
light on the behavior of amyloid-beta in solutions withhigher concentration of Cu2+ ions and on its interac-tion with CUA molecules with or without the presenceof Cu2+ ions.
Acknowledgements. Authors would like to thank Dr. ZdeněkKříž for providing the pre-equilibrated amyloid-beta structure.The European Community’s Seventh Framework Programme(FP7/2007-2013) under grant agreement No. 212043 is ac-knowledged for financial support. This work was also supportedby the Scientific Grant Agency of the Ministry of Educationof the Slovak Republic and the Slovak Academy of Sciences(project VEGA-02/0101/11), and the Research & DevelopmentOperational Programme funded by ERDF (Centre of Excel-lence on Green Chemistry Methods and Processes, CEGreenI,Contract No. 26240120001, and Amplification of the Centre ofExcellence on Green Chemistry Methods and Processes, CE-GreenII, Contract No. 26240120025), and also by the EuropeanUnion under the Seventh Framework Programme by CEITEC(CZ.1.05/1.1.00/02.0068).
Supplementary data
Supplementary data associated with this articlecan be found in the online version of this paper (DOI:10.1515/chempap-2015-0134).
References
Airoldi, C., Zona, C., Sironi, E., Colombo, L., Messa, M., Au-rilia, D., Gregori, M., Masserini, M., Salmona, M., Nicotra,F., & La Ferla, B. (2011). Curcumin derivatives as new lig-ands of A beta peptides. Journal of Biotechnology, 156, 317–324. DOI: 10.1016/j.jbiotec.2011.07.021.
Ali-Torres, J., Maréchal, J. D., Rodríguez-Santiago, L., &Sodupe, M. (2011). Three dimensional models of Cu2+ -Aβ(1-16) complexes from computational approaches. Jour-nal of the American Chemical Society, 133, 15008–15014.DOI: 10.1021/Ja203407v.
Allinger, N. L., Zhou, X. F., & Bergsma, J. (1994). Molecu-lar mechanics parameters. Journal of Molecular Structure:THEOCHEM, 118, 69–83. DOI: 10.1016/s0166-1280(09)80008-0.
Atwood, C. S., Huang, X. D., Moir, R. D., Tanzi, R. E., &Bush, A. I. (1999). Role of free radicals and metal ions in thepathogenesis of Alzheimer’s disease. Metal Ions in BiologicalSystems, 36, 309–364.
Ball, K. A., Phillips, A. H., Wemmer, D. E., & Head-Gordon,T. (2013). Differences in β-strand populations of monomericAβ40 and Aβ42. Biophysical Journal, 104, 2714–2724. DOI:10.1016/j.bpj.2013.04.056.
Banerjee, R. (2014). Effect of curcumin on the metal ion inducedfibrillization of Amyloid-β peptide. Spectrochimica Acta PartA: Molecular and Biomolecular Spectroscopy, 117, 798–800.DOI: 10.1016/j.saa.2013.09.064.
Barik, A., Mishra, B., Kunwar, A., Kadam, R. M., Shen, L.,Dutta, S., Padhye, S., Setpati, A. K., Zhang, H. Y., &Priyadarsini, K. I. (2007). Comparative study of copper(II)–curcumin complexes as superoxide dismutase mimics and freeradical scavengers. European Journal of Medicinal Chem-istry, 42, 431–439. DOI: 10.1016/j.ejmech.2006.11.012.
Barnham, K. J., & Bush, A. I. (2008). Metals in Alzheimer’s andParkinson’s diseases. Current Opinion in Chemical Biology,12, 222–228. DOI: 10.1016/j.cbpa.2008.02.019.
Barrow, C. J., & Zagorski, M. G. (1991). Solution struc-tures of β peptide and its constituent fragments: relation
to amyloid deposition. Science, 253, 179–182. DOI: 10.1126/science.1853202.
Barrow, C. J., Yasuda, A., Kenny, P. T. M., & Zagorski, M.G. (1992). Solution conformations and aggregational prop-erties of synthetic amyloid β-peptides of Alzheimers-disease:Analysis of circular-dichroism spectra. Journal of MolecularBiology, 225, 1075–1093. DOI: 10.1016/0022-2836(92)90106-t.
Becke, A. D. (1993). Density-functional thermochemistry. 3.The role of exact exchange. Journal of Chemical Physics,98, 5648–5652. DOI: 10.1063/1.464913.
Bitan, G., Kirkitadze, M. D., Lomakin, A., Vollers, S. S.,Benedek, G. B., & Teplow, D. B. (2003). Amyloid β-protein(Aβ) assembly: Aβ40 and Aβ42 oligomerize through dis-tinct pathways. Proceedings of the National Academy of Sci-ences of the United States of America, 100, 330–335. DOI:10.1073/pnas.222681699.
Brewer, G. J. (2012). Copper excess, zinc deficiency, and cog-nition loss in Alzheimer’s disease. BioFactors, 38, 107–113.DOI: 10.1002/biof.1005.
Bush, A. I. (2003). The metallobiology of Alzheimer’s disease.Trends in Neurosciences, 26, 207–214. DOI: 10.1016/s0166-2236(03)00067-5.
Bush, A. I. (2008). Drug development based on the metals hy-pothesis of Alzheimer’s disease. Journal of Alzheimers Dis-ease, 15, 223–240.
Case, D. A., Darden, T. A., Cheatham, T. E., III., Simmer-ling, C. L., Wang, J., Duke, R. E., Luo, R., Crowley, M.,Walker, R. C., Zhang, W., Merz, K. M., Wang, B., Hayik, S.,Roitberg, A., Seabra, G., Kolossváry, I., Wong, K. F., Pae-sani, F., Vanicek, J., Wu, X., Brozell, S. R., Steinbrecher, T.,Gohlke, H., Yang, L., Tan, C., Mongan, J., Hornak, V., Cui,G., Mathews, D. H., Seetin, M. G., Sagui, C., Babin, V., &Kollman, P. A. (2008). AMBER 10 [computer software]. SanFrancisco, CA, USA: University of San Francisco.
Coles, M., Bicknell, W., Watson, A. A., Fairlie, D. P., & Craik,D. J. (1998). Solution structure of amyloid β-peptide(1–40)in a water-micelle environment. Is the membrane-spanningdomain where we think it is? Biochemistry, 37, 11064–11077.DOI: 10.1021/bi972979f.
Crouch, P. J., Harding, S. M. E., White, A. R., Camakaris, J.,Bush, A. I., & Masters, C. L. (2008). Mechanisms of A betamediated neurodegeneration in Alzheimer’s disease. Interna-tional Journal of Biochemistry & Cell Biology, 40, 181–198.DOI: 10.1016/j.biocel.2007.07.013.
Ditchfie, R., Hehre, W. J., & Pople, J. A. (1971). Self-consistentmolecular-orbital methods. 9. Extended Gaussian-type basisfor molecular-orbital studies of organic molecules. Journal ofChemical Physics, 54, 724–728.
Faller, P. (2009). Copper and zinc binding to amyloid-β: Coordination, dynamics, aggregation, reactivity andmetal-ion transfer. ChemBioChem, 10, 2837–2845. DOI:10.1002/cbic.200900321.
Faller, P., & Hureau, C. (2009). Bioinorganic chemistry of cop-per and zinc ions coordinated to amyloid-beta peptide. Dal-ton Transactions, 2009, 1080–1094. DOI: 10.1039/b813398k.
Francl, M. M., Pietro, W. J., Hehre, W. J., Binkley, J. S.,Gordon, M. S., DeFrees, D. J., & Pople, J. A. (1982). Self-consistent molecular-orbital methods. XXIII. A polarization-type basis set for 2nd-row elements. The Journal of ChemicalPhysics, 77, 3654–3665. DOI: 10.1063/1.444267.
Frisch, M. J., Trucks, G. W., Schlegel, H. B., Scuseria, G. E.,Robb, M. A., Cheeseman, J. R., Scalmani, G., Barone, V.,Mennucci, B., Petersson, G. A., Nakatsuji, H., Caricato, M.,Li, X., Hratchian, H. P., Izmaylov, A. F., Bloino, J., Zheng,G., Sonnenberg, J. L., Hada, M., Ehara, M., Toyota, K.,Fukuda, R., Hasegawa, J., Ishida, M., Nakajima, T., Honda,Y., Kitao, O., Nakai, H., Vreven, T., Montgomery, J. A.,
Authenticated | [email protected] author's copyDownload Date | 6/3/15 12:41 PM

S. Kozmon, I. Tvaroška/Chemical Papers 69 (9) 1262–1276 (2015) 1275
Jr., Peralta, J. E., Ogliaro, F., Bearpark, M., Heyd, J. J.,Brothers, E., Kudin, K. N., Staroverov, V. N., Kobayashi,R., Normand, J., Raghavachari, K., Rendell, A., Burant, J.C., Iyengar, S. S., Tomasi, J., Cossi, M., Rega, N., Millam, N.J., Klene, M., Knox, J. E., Cross, J. B., Bakken, V., Adamo,C., Jaramillo, J., Gomperts, R., Stratmann, R. E., Yazyev,O., Austin, A. J., Cammi, R., Pomelli, C., Ochterski, J. W.,Martin, R. L., Morokuma, K., Zakrzewski, V. G., Voth, G.A., Salvador, P., Dannenberg, J. J., Dapprich, S., Daniels, A.D., Farkas, O., Foresman, J. B., Ortiz, J. V., Cioslowski, J.,& Fox, D. J. (2009). Gaussian 09, Revision A.1. [computersoftware]. Wallington, CT, USA: Gaussian.
Gobbi, M., Re, F., Canovi, M., Beeg, M., Gregori, M.,Sesana, S., Sonnino, S., Brogioli, D., Musicanti, C., Gasco,P., Salmona, M., & Masserini, M. E. (2010). Lipid-basednanoparticles with high binding affinity for amyloid-β1-42 peptide. Biomaterials, 31, 6519–6529. DOI: 10.1016/j.biomaterials.2010.04.044.
Good, T. A., & Murphy, R. M. (1995). Aggregation state-dependent binding of β-amyloid peptide to protein and lipidcomponents of rat cortical homogenates. Biochemical andBiophysical Research Communications, 207, 209–215. DOI:10.1006/bbrc.1995.1174.
Hardy, J., & Selkoe, D. J. (2002). Medicine – The amyloidhypothesis of Alzheimer’s disease: Progress and problemson the road to therapeutics. Science, 297, 353–356. DOI:10.1126/science.1072994.
Harihara, P. C., & Pople, J. A. (1973). The influence of polariza-tion functions on molecular-orbital hydrogenation energies.Theoretica Chimica Acta, 28, 213–222. DOI: 10.1007/bf00533485.
Hehre, W. J., Ditchfie, R., & Pople, J. A. (1972). Self-consistent molecular-orbital methods. 12. Further extensionsof Gaussian-type basis sets for use in molecular-orbital stud-ies of organic-molecules. The Journal of Chemical Physics,56, 2257–2261. DOI: 10.1063/1.1677527.
Karr, J. W., Kaupp, L. J., & Szalai, V. A. (2004). Amyloid-βbinds Cu2+ in a mononuclear metal ion binding site. Journalof the American Chemical Society, 126, 13534–13538. DOI:10.1021/Ja0488028.
Kolev, T. M., Velcheva, E. A., Stamboliyska, B. A., &Spiteller, M. (2005). DFT and experimental studies of thestructure and vibrational spectra of curcumin. Interna-tional Journal of Quantum Chemistry, 102, 1069–1079. DOI:10.1002/qua.20469.
Kulhánek, P., Fuxreiter, M., Štěpán, J., Koča, J., Mones, L.,Střelcová, Z., & Petřek, M. (2012). PMFLib – A toolkit forfree energy calculations [computer software]. Brno, Czech Re-public: Masaryk University.
Le Droumaguet, B., Nicolas, J., Brambilla, D., Mura, S., Mak-simenko, A., De Kimpe, L., Salvati, E., Zona, C., Airoldi,C., Canovi, M., Gobbi, M., Noiray, M., La Ferla, B., Nico-tra, F., Scheper, W., Flores, O., Masserini, M., Andrieux,K., & Couvreur, P. (2012). Versatile and efficient targetingusing a single nanoparticulate platform: Application to can-cer and Alzheimer’s disease. ACS Nano, 6, 5866–5879. DOI:10.1021/nn3004372.
Lee, C. T., Yang, W. T., & Parr, R. G. (1988). Development ofthe colle-salvetti correlation-energy formula into a functionalof the electron-density. Physical Review B, 37, 785–789. DOI:10.1103/physrevb.37.785.
Lim, G. P., Chu, T., Yang, F. S., Beech, W., Frautschy, S. A., &Cole, G. M. (2001). The curry spice curcumin reduces oxida-tive damage and amyloid pathology in an Alzheimer trans-genic mouse. Journal of Neuroscience, 21, 8370–8377.
Lim, K. H., Collver, H. H., Le, Y. T. H., Nagchowdhuri, P.,& Kenney, J. M. (2007). Characterizations of distinct amy-loidogenic conformations of the Aβ (1–40) and (1-42) pep-
tides. Biochemical and Biophysical Research Communica-tions, 353, 443–449. DOI: 10.1016/j.bbrc.2006.12.043.
Liu, K. N., Lai, C. M., Lee, Y. T., Wang, S. N., Chen,R. P. Y., Jan, J. S., Liu, H. S., & Wang, S. S. S.(2012). Curcumin’s pre-incubation temperature affects itsinhibitory potency toward amyloid fibrillation and fibril-induced cytotoxicity of lysozyme. Biochimica et Biophys-ica Acta (BBA) – General Subjects, 1820, 1774–1786. DOI:10.1016/j.bbagen.2012.07.012.
McLean, C. A., Cherny, R. A., Fraser, F. W., Fuller, S. J.,Smith, M. J., Vbeyreuther, K., Bush, A. I., & Masters,C. L. (1999). Soluble pool of Aβ amyloid as a determi-nant of severity of neurodegeneration in Alzheimer’s dis-ease. Annals of Neurology, 46, 860–866. DOI: 10.1002/1531-8249(199912)46:6<860::Aid-Ana8>3.0.Co;2-M.
Miehlich, B., Savin, A., Stoll, H., & Preuss, H. (1989). Resultsobtained with the correlation-energy density functionals ofbecke and Lee, Yang and Parr. Chemical Physics Letters,157, 200–206. DOI: 10.1016/0009-2614(89)87234-3.
Miura, T., Hori-i, A., Mototani, H., & Takeuchi, H. (1999). Ra-man spectroscopic study on the copper(II) binding mode ofprion octapeptide and its pH dependence. Biochemistry, 38,11560–11569. DOI: 10.1021/bi9909389.
Mourtas, S., Canovi, M., Zona, C., Aurilia, D., Niarakis, A.,La Ferla, B., Salmona, M., Nicotra, F., Gobbi, M., & An-timisiaris, S. G. (2011). Curcumin-decorated nanoliposomeswith very high affinity for amyloid-β 1-42 peptide. Biomateri-als, 32, 1635–1645. DOI: 10.1016/j.biomaterials.2010.10.027.
Ono, K., Hasegawa, K., Naiki, H., & Yamada, M. (2004). Cur-cumin has potent anti-amyloidogenic effects for Alzheimer’sβ-amyloid fibrils in vitro. Journal of Neuroscience Research,75, 742–750. DOI: 10.1002/jnr.20025.
Parthasarathy, S., Long, F., Miller, Y., Xiao, Y. L., McElheny,D., Thurber, K., Ma, B. Y., Nussinov, R., & Ishii, Y. (2011).Molecular-level examination of Cu2+ binding structure foramyloid fibrils of 40-residue Alzheimer’s β by solid-stateNMR spectroscopy. Journal of the American Chemical So-ciety, 133, 3390–3400. DOI: 10.1021/ja1072178.
Perrone, L., Mothes, E., Vignes, M., Mockel, A., Figueroa, C.,Miquel, M. C., Maddelein, M. D., & Faller, P. (2010). Coppertransfer from Cu-Aβ to human serum albumin inhibits ag-gregation, radical production and reduces Aβ toxicity. Chem-BioChem, 11, 110–118. DOI: 10.1002/cbic.200900474.
Picciano, A. L., & Vaden, T. D. (2013). Complexation betweenCu(II) and curcumin in the presence of two different segmentsof amyloid beta. Biophysical Chemistry, 184, 62–67. DOI:10.1016/j.bpc.2013.09.004.
Porat, Y., Abramowitz, A., & Gazit, E. (2006). Inhibition ofamyloid fibril formation by polyphenols: Structural similar-ity and aromatic interactions as a common inhibition mech-anism. Chemical Biology & Drug Design, 67, 27–37. DOI:10.1111/j.1747-0285.2005.00318.x.
Raffa, D. F., & Rauk, A. (2007). Molecular dynamics study ofthe beta amyloid peptide of Alzheimer’s disease and its diva-lent copper complexes. The Journal of Physical ChemistryB, 111, 3789–3799. DOI: 10.1021/jp0689621.
Rassolov, V. A., Ratner, M. A., Pople, J. A., Redfern, P.C., & Curtiss, L. A. (2001). 6-31G* basis set for third-rowatoms. Journal of Computational Chemistry, 22, 976–984.DOI: 10.1002/jcc.1058.
Re, F., Cambianica, I., Zona, C., Sesana, S., Gregori, M., Rigo-lio, R., La Ferla, B., Nicotra, F., Forloni, G., Cagnotto, A.,Salmona, M., Masserini, M., & Sancini, G. (2011). Func-tionalization of liposomes with ApoE-derived peptides atdifferent density affects cellular uptake and drug trans-port across a blood-brain barrier model. Nanomedicine-Nanotechnology Biology and Medicine, 7, 551–559. DOI:10.1016/j.nano.2011.05.004.
Authenticated | [email protected] author's copyDownload Date | 6/3/15 12:41 PM

1276 S. Kozmon, I. Tvaroška/Chemical Papers 69 (9) 1262–1276 (2015)
Reinke, A. A., & Gestwicki, J. E. (2007). Structure-activity re-lationships of amyloid beta-aggregation inhibitors based oncurcumin: Influence of linker length and flexibility. ChemicalBiology & Drug Design, 70, 206–215. DOI: 10.1111/j.1747-0285.2007.00557.x.
Roe, D. R., & Cheatham, T. E., III. (2013). PTRAJ and CPP-TRAJ: Software for processing and analysis of moleculardynamics trajectory data. Journal of Chemical Theory andComputation, 9, 3084–3095. DOI: 10.1021/Ct400341p.
Saido, T. C., & Iwata, N. (2006). Metabolism of amyloid βpeptide and pathogenesis of Alzheimer’s disease: Towardspresymptomatic diagnosis, prevention and therapy. Neuro-science Research, 54, 235–253. DOI: 10.1016/j.neures.2005.12.015.
Serpell, L. C. (2000). Alzheimer’s amyloid fibrils: structure andassembly. Biochimica et Biophysica Acta (BBA) – Molec-ular Basis of Disease, 1502, 16–30. DOI: 10.1016/s0925-4439(00)00029-6.
Shao, H. Y., Jao, S. C., Ma, K., & Zagorski, M. G. (1999).Solution structures of micelle-bound amyloid β-(1-40) and β-(1-42) peptides of Alzheimer’s disease. Journal of MolecularBiology, 285, 755–773. DOI: 10.1006/jmbi.1998.2348.
Singh, I., Sagare, A. P., Coma, M., Perlmutter, D., Gelein, R.,Bell, R. D., Deane, R. J., Zhong, E., Parisi, M., Ciszewski,J., Kasper, R. T., & Deane, R. (2013). Low levels of copperdisrupt brain amyloid-β homeostasis by altering its produc-tion and clearance. Proceedings of the National Academy ofSciences of the United States of America, 110, 14771–14776.DOI: 10.1073/pnas.1302212110.
Soto, C., Castano, E. M., Frangione, B., & Inestrosa, N. C.(1995). The α-helical to β-strand transition in the amino-terminal fragment of the amyloid β-peptide modulates amy-loid formation. Journal of Biological Chemistry, 270, 3063–3067.
Sticht, H., Bayer, P., Willbold, D., Dames, S., Hilbich, C.,Beyreuther, K., Frank, R. W., & Rosch, P. (1995). Struc-ture of amyloid A4-(1-40)-peptide of Alzheimers-disease.European Journal of Biochemistry, 233, 293–298. DOI:10.1111/j.1432-1033.1995.293 1.x.
Syme, C. D., Nadal, R. C., Rigby, S. E. J., & Viles, J. H. (2004).Copper binding to the amyloid-β (Aβ) peptide associatedwith Alzheimer’s disease – Folding, coordination geometry,pH dependence, stoichiometry, and affinity of A β-(1-28):Insights from a range of complementary spectroscopic tech-niques. Journal of Biological Chemistry, 279, 18169–18177.DOI: 10.1074/jbc.m313572200.
Taylor, M., Moore, S., Mourtas, S., Niarakis, A., Re, F., Zona,C., La Ferla, B., Nicotra, F., Masserini, M., Antimisiaris, S.G., Gregori, M., & Allsop, D. (2011). Effect of curcumin-associated and lipid ligand-functionalized nanoliposomes onaggregation of the Alzheimer’s Aβ peptide. Nanomedicine-Nanotechnology Biology and Medicine, 7, 541–550. DOI:10.1016/j.nano.2011.06.015.
Tomaselli, S., Esposito, V., Vangone, P., van Nuland, N. A. J.,Bonvin, A. M. J. J., Guerrini, R., Tancredi, T., Temussi, A.,& Picone, D. (2006). The α-to-β conformational transition ofAlzheimer’s Aβ-(1-42) peptide in aqueous media is reversible:A step by step conformational analysis suggests the locationof beta conformation seeding. ChemBioChem, 7, 257–267.DOI: 10.1002/cbic.200500223.
Wang, Y. Y., Li, L., Chen, T. T., Chen, W. Y., & Xu, Y. C.(2013). Microsecond molecular dynamics simulation of Aβ42and identification of a novel dual inhibitor of Aβ42 aggrega-tion and BACE1 activity. Acta Pharmacologica Sinica, 34,1243–1250. DOI: 10.1038/aps.2013.55.
Wise, O., & Coskuner, O. (2014). New force field parameters formetalloproteins I: Divalent copper ion centers including threehistidine residues and an oxygen-ligated amino acid residue.Journal of Computational Chemistry, 35, 1278–1289. DOI:10.1002/jcc.23622.
Xu, Y. C., Shen, J. J., Luo, X. M., Zhu, W. L., Chen, K. X.,Ma, J. P., & Jiang, H. L. (2005). Conformational transition ofamyloid beta-peptide. Proceedings of the National Academyof Sciences of the United States of America, 102, 5403–5407.DOI: 10.1073/pnas.0501218102.
Xu, L., Wang, X. J., Shan, S. S., & Wang, X. C. (2013).Characterization of the polymorphic states of copper(II)-bound Aβ(1–16) peptides by computational simulations.Journal of Computational Chemistry, 34, 2524–2536. DOI:10.1002/jcc.23416.
Yan, Y. L., McCallum, S. A., & Wang, C. Y. (2008). M35 ox-idation induces Aβ40-like structural and dynamical changesin Aβ42. Journal of the American Chemical Society, 130,5394–5395. DOI: 10.1021/Ja711189c.
Yanagisawa, D., Shirai, N., Amatsubo, T., Taguchi, H., Hi-rao, K., Urushitani, M., Morikawa, S., Inubushi, T., Kato,M., Kato, F., Morino, K., Kimura, H., Nakano, I., Yoshida,C., Okada, T., Sano, M., Wada, Y., Wada, K., Yamamoto,A., & Tooyama, I. (2010). Relationship between the tau-tomeric structures of curcumin derivatives and their Abeta-binding activities in the context of therapies forAlzheimer’s disease. Biomaterials, 31, 4179–4185. DOI:10.1016/j.biomaterials.2010.01.142.
Yang, F. S., Lim, G. P., Begum, A. N., Ubeda, O. J., Simmons,M. R., Ambegaokar, S. S., Chen, P. P., Kayed, R., Glabe,C. G., Frautschy, F. A., & Cole, G. M. (2005). Curcumin in-hibits formation of amyloid beta oligomers and fibrils, bindsplaques, and reduces amyloid in vivo. Journal of BiologicalChemistry, 280, 5892–5901. DOI: 10.1074/jbc.m404751200.
Zhao, L. N., Chiu, S. W., Benoit, J., Chew, L. Y., & Mu, Y. G.(2012). The effect of curcumin on the stability of Aβ dimers.The Journal of Physical Chemistry B, 116, 7428–7435. DOI:10.1021/jp3034209.
Authenticated | [email protected] author's copyDownload Date | 6/3/15 12:41 PM





























