Embed Size (px)
Citation preview

Molecular dynamics of b-carotene in solution by resonance enhancedoptical Kerr effect
Marilena Ricci,a) Renato Torre, Paolo Foggi,a) Valey Kamalov,b) and Roberto Righinic)European Laboratory for Non-linear Spectroscopy, University of Florence, Largo E. Fermi 2,50125 Florence, Italy
~Received 19 April 1994; accepted 23 February 1995!
The orientational dynamics ofb-carotene inn-alkane solutions is investigated by resonanceenhanced optical Kerr effect. By use of this spectroscopic technique, it is possible to selectivelyinvestigate the relaxation of a probe molecule at a concentration level low enough to allow theobservation of the averaged single-molecule dynamics. For delay times longer than;20 ps allsolutions show a single exponential decay, with a time constant depending on the viscosity, that isascribed to theb-carotene orientational relaxation. The dependence on viscosity of the measuredrelaxation times is compared with the predictions of different models. The purely hydrodynamictheories overestimate, by far, the solute effective volume and hence its orientational relaxation time;a much better agreement is obtained from two quasihydrodynamic models. ©1995 AmericanInstitute of Physics.
o
,f
-
a-
-
c-
d
-
I. INTRODUCTION
The study of the rotational dynamics of molecules isolution is a source of valuable information on the naturethe liquid phase in terms of intermolecular interaction~solute–solute and solute–solvent!, on the local solventstructure, and on the molecular conformations.1 Furthermore,a correct understanding of the rotational dynamics helpsinterpreting other processes, modulated by the solvent mecules, such as chemical reactions.
A variety of experimental techniques, both in the timand frequency domain, have been widely utilized to obsermolecular orientational processes in liquids: dynamic ligscattering and fluorescence depolarization,2,3 dielectric relax-ation, nuclear magnetic resonance~NMR!, electron spinresonance~ESR!,2,3 and neutron scattering,4 and optical Kerreffect ~OKE!.5–7 Since these techniques probe time correltion functions of different molecular properties, they work inconcert to develop a further picture of the molecular motionin the liquid phase.
Disordered condensed phases show rather complex mlecular dynamics, often characterized by a number of relaation processes. From this point of view, time-resolved spetroscopy, measuring the molecular dynamics directly in thtime domain, has been shown to be particularly usefuldetecting complex time decays. For instance, transient gring and transient OKE techniques have been recently usedmeasure collective orientational correlation functionsstructured liquids.8,9 The signal decay has been detected ovseveral decades in time and intensity, allowing the authorsdescribe the complex nature of the relaxation process.
Several time-resolved studies of the rotational relaxatioof probe molecules in solvents of different nature have beperformed using fluorescence depolarization or polariz
a!Also at: Department of Chemistry, University of Florence, 50121 FlorencItaly.
b!Also at: Photochemistry Department, Institute of Chemical Physics, Acaemy of Science, Kosygin Street, Moscow 117977, Russia.
c!Also at: Department of Chemistry, University of Basilicata, Potenza, Ital
J. Chem. Phys. 102 (24), 22 June 1995 0021-9606/95/102(24)Downloaded¬28¬Jun¬2004¬to¬150.217.156.84.¬Redistribution¬subjec
nofs
inol-
eveht
a-
s
o-x-c-einat-to
inerto
nened
transient absorption spectroscopy.6 The time scale on whichorientational mechanisms can be studied by these last twtechniques is limited by the lifetime of the excited electronicstates involved in the process. Only when the lifetime islonger than or comparable to the orientational time constantthe latter can be extracted from the experiment. In the case overy fast electronic relaxation the slower orientational con-tribution becomes unobservable.
Normally in time-resolved optical Kerr effect out-of-resonance laser fields are used. The technique is mostly employed for neat liquids, since in diluted solutions the nonlin-ear response of the solvent often covers the weakcontribution of the solute molecules. However, this limitationhas been recently overcome by using a modified OKEscheme, which takes advantage of the resonance amplifiction of the signal.10 It has been shown that a selective obser-vation of the solute contribution to the OKE signal is pos-sible if the probe pulse frequency is in~or close to!resonance with an electronic transition of the solute mol-ecule. Under such conditions it then becomes feasible to investigate the solute dynamics also in diluted solutions~lessthan 1023 mol/l!, provided that the nonlinearity of the solutemolecule is large enough. Since the electronic transition isonly involved in probing the off-resonance optically inducedbirefringence, the time evolution of the signal is not affectedby the lifetime of the electronic excited state: the orienta-tional dynamics can thus be measured even when the eletronic lifetime is very short, like in the case ofb-carotene,which represents a convenient system for this type ofmeasurement.11 In this work we present a study of the rota-tional dynamics of the all-trans b-carotene molecule inn-alkane solutions by means of transient resonance enhanceOKE.
II. BACKGROUND THEORY
A. Resonant enhancement of transient OKE signaland response function
In a transient OKE experiment an excitation laser pulseproduces a transient anisotropy in the sample, which is moni
e,
d-
y.
9537/9537/7/$6.00 © 1995 American Institute of Physicst¬to¬AIP¬license¬or¬copyright,¬see¬http://jcp.aip.org/jcp/copyright.jsp
er
hi
a
h
n
of
r
n
aot
tosch-eoe.e
e
ntist
n-
,
-
se-
d
9538 Ricci et al.: Dynamics of b-carotene
tored by the change of polarization of a probe laser pulsThe intensity of the new polarization component of the probpulse can be written as5,12
S~t!}E2`
1`
dtF E2`
t
dt8E2`
t8dt9 Rii j j ~ t2t8,t2t9!
3Eip~ t82t!Ej
e~ t9!Eje~ t9!G2, ~1!
where t is the delay between the pump and probe pulseR(t) is the response function of the system, andEp andEe
are the electric fields of the probe and pump pulses, resptively. The response function is proportional to the third oder nonlinear susceptibilityx~3!, and its time evolution in-cludes the effects of all collective dynamical processes in tmaterial; their theoretical description is very complex, andmany respects, represents an open problem. When the puand probe pulses are in resonance with some electronic trsition of the molecule the general calculation ofx~3! is par-ticularly demanding.13
In our experiment the pump pulse~l5604 nm! is offresonance, while the probe~l5517 nm! is quasiresonantwith the allowedS0(1
1Ag)→S2 (11Bu! electronic transi-
tion of b-carotene, whose peak absorption is at 490 nm. Tamplification effect of the OKE signal thus obtained habeen experimentally demonstrated and reported in Ref.where the different terms contributing to the time evolutioof the signal were also discussed. Essentially two time scacan be distinguished, one below and one above 20 ps.
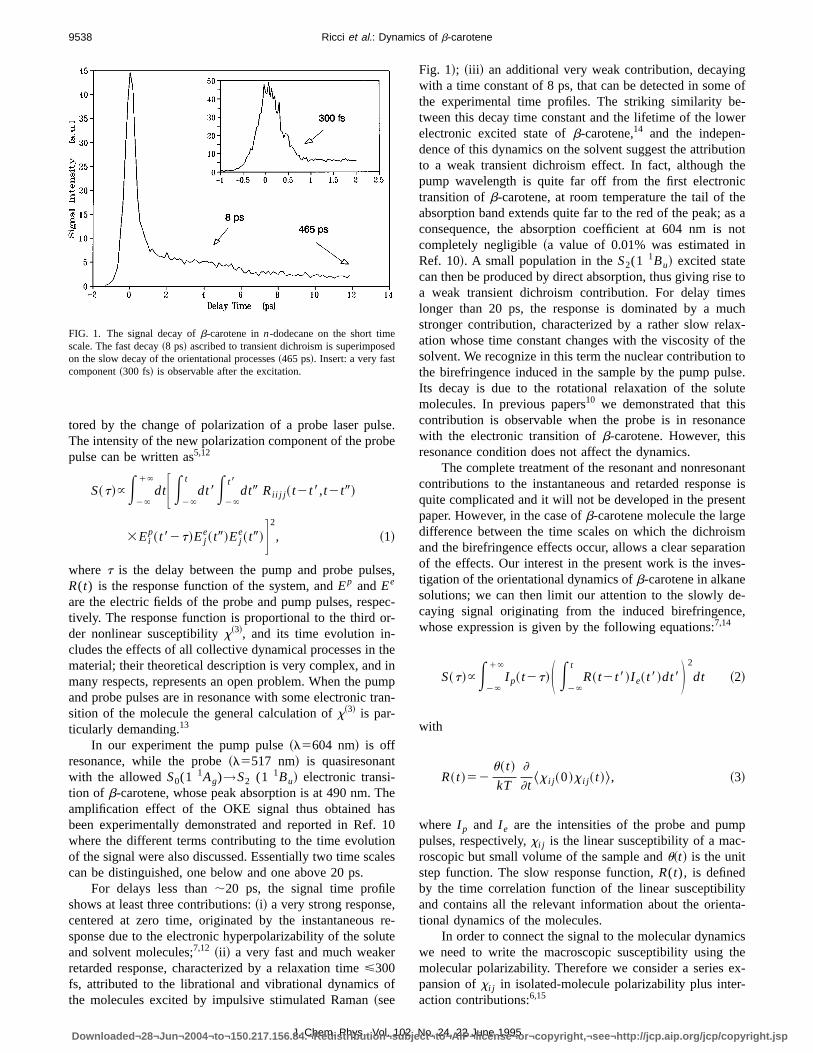
For delays less than;20 ps, the signal time profileshows at least three contributions:~i! a very strong response,centered at zero time, originated by the instantaneoussponse due to the electronic hyperpolarizability of the soluand solvent molecules;7,12 ~ii ! a very fast and much weakerretarded response, characterized by a relaxation time<300fs, attributed to the librational and vibrational dynamics othe molecules excited by impulsive stimulated Raman~see
FIG. 1. The signal decay ofb-carotene inn-dodecane on the short timescale. The fast decay~8 ps! ascribed to transient dichroism is superimposeon the slow decay of the orientational processes~465 ps!. Insert: a very fastcomponent~300 fs! is observable after the excitation.
J. Chem. Phys., Vol. 102Downloaded¬28¬Jun¬2004¬to¬150.217.156.84.¬Redistribution¬subject
e.e
s,
c--
enmpn-
es10
les
re-te
f
Fig. 1!; ~iii ! an additional very weak contribution, decayingwith a time constant of 8 ps, that can be detected in somethe experimental time profiles. The striking similarity be-tween this decay time constant and the lifetime of the loweelectronic excited state ofb-carotene,14 and the indepen-dence of this dynamics on the solvent suggest the attributioto a weak transient dichroism effect. In fact, although thepump wavelength is quite far off from the first electronictransition ofb-carotene, at room temperature the tail of theabsorption band extends quite far to the red of the peak; asconsequence, the absorption coefficient at 604 nm is ncompletely negligible~a value of 0.01% was estimated inRef. 10!. A small population in theS2(1
1Bu! excited statecan then be produced by direct absorption, thus giving risea weak transient dichroism contribution. For delay timelonger than 20 ps, the response is dominated by a mustronger contribution, characterized by a rather slow relaxation whose time constant changes with the viscosity of thsolvent. We recognize in this term the nuclear contribution tthe birefringence induced in the sample by the pump pulsIts decay is due to the rotational relaxation of the solutmolecules. In previous papers10 we demonstrated that thiscontribution is observable when the probe is in resonancwith the electronic transition ofb-carotene. However, thisresonance condition does not affect the dynamics.
The complete treatment of the resonant and nonresonacontributions to the instantaneous and retarded responsequite complicated and it will not be developed in the presenpaper. However, in the case ofb-carotene molecule the largedifference between the time scales on which the dichroismand the birefringence effects occur, allows a clear separatioof the effects. Our interest in the present work is the investigation of the orientational dynamics ofb-carotene in alkanesolutions; we can then limit our attention to the slowly de-caying signal originating from the induced birefringencewhose expression is given by the following equations:7,14
S~t!}E2`
1`
I p~ t2t!S E2`
t
R~ t2t8!I e~ t8!dt8D 2dt ~2!
with
R~ t !52u~ t !
kT
]
]t^x i j ~0!x i j ~ t !&, ~3!
where I p and I e are the intensities of the probe and pumppulses, respectively,xi j is the linear susceptibility of a mac-roscopic but small volume of the sample andu~t! is the unitstep function. The slow response function,R(t), is definedby the time correlation function of the linear susceptibilityand contains all the relevant information about the orientational dynamics of the molecules.
In order to connect the signal to the molecular dynamicwe need to write the macroscopic susceptibility using thmolecular polarizability. Therefore we consider a series expansion ofxi j in isolated-molecule polarizability plus inter-action contributions:6,15
, No. 24, 22 June 1995¬to¬AIP¬license¬or¬copyright,¬see¬http://jcp.aip.org/jcp/copyright.jsp

-
o
yt--
d
l
f
is
t
ll
-
n
g
d
et
de.
9539Ricci et al.: Dynamics of b-carotene
x i j5(n
b i jn1(
n,m
b ikn Tkla l j
m1(n,m
b ikn Tklb l j
m1(n
a i jn
1(n,m
a ikn Tkla l j
m , ~4!
whereb anda are the isolated-molecule linear polarizabilities of the solute and solvent, respectively;bTa, bTb, andaTa are the corrections due the pair interactions. The resnance condition of the probe field with an electronic statethe solute molecule enhances mainly the first three termsEq. ~4!. Furthermore, considering the low concentration osolute we can also neglect the third term of Eq.~4!. From theexperimental conditions we can consider active only the twcontributions:(nb
n and (n,mbnTam. The isolate-moleculeterm gives rise to the time correlation function relaxing bmolecular reorientation; the other enhanced term describemodification of isolated-molecule polarizability due to molecular interaction.15 These interactions give rise to contribution in the correlation function, whose relaxation is fas~typically ,1 ps! and so thebTa correction can be droppedin the analysis of the slow relaxation.
Thus, taking into account only the isolated-moleculcontribution @first term in Eq.~4!#, we must consider twodifferent types of correlation functions: Self-correlation ancross-correlation
Cself~ t !5(n
^bn~0!bn~ t !&,
~5!
Ccross~ t !5 (nÞm
^bn~0!bm~ t !&.
TheCself function describes the effects of correlation oa single solute molecule at different times, while theCcross
function describes the correlation between distinct molecuat different times. At low solute concentration the effect ocross-correlation is negligible. Thus, we can ascribe the slodecay observed in the retarded response function to theav-eraged single molecule dynamicscharacterized by theCself
correlation function.
B. Model of orientational dynamics
The time dependence of the orientational self-correlatiofunction can be calculated with the Brownian rotational difusion model.2,3,6 This model of the orientational dynamicswas developed originally by Debye and later reconsidereda hydrodynamic approach by other authors.6,16–19Under theassumptions of these models the complete reorientation pcess can be characterized by a diffusion tensor,Q. In themost general case it can be reduced to a diagonal form, wthree independent diagonal elementsQxx , Qyy , and Qzz,characterizing the orientational motion around three orthognal axes. In this occurrence the decay of the correlation funtion is described by the sum of five exponentials.2
The problem can be greatly simplified by assuming fotheb-carotene molecule a cylindrical pseudosymmetry; itthus possible to introduce two important assumptions. Fir
J. Chem. Phys., Vol. 102Downloaded¬28¬Jun¬2004¬to¬150.217.156.84.¬Redistribution¬subject
o-finf
o
he
t
e
f
esfw
n-
in
ro-
ith
o-c-
rst,
the diffusion constants for the rotation around the two shoraxes ofb-carotene molecule~tumbling motion! are similar,Qxx>Qyy5Q' , and quite different from the diffusion con-stant characterizing the rotation around the long axis~spin-ning motion!, Qzz5Qi . Second, only two components of themolecular polarizability,ax8x85ay8y85a' and az8z85aihave to be considered. In addition, we consider the principaaxis system of the polarizability tensor and of the rotationadiffusion tensor to be coincident~x8,y8,z8[x,y,z!. In otherwords, we approximate theb-carotene to a true symmetrictop molecule.2 Under these approximations only one expo-nential decay is present in the correlation function, and wecan write
Cself~ t !}~a i2a'!2exp~26Q't !. ~6!
The orientational relaxation timet5~6Q'!21 is deter-mined by the perpendicular diffusional constantQ' ~tum-bling motion!. According to the hydrodynamic theory16–19
the relaxation time is proportional to the shear viscosityh,with a proportionality constant containing the inverse tem-perature:
t5Veffh
kT, ~7!
wherek is the Boltzman factor andVeff is the effective vol-ume of solute molecule. A constant timet0 is quite oftenadded to the right-hand side of Eq.~7! in order to betterreproduce the experimental data. Actually there is no theoretical justification to the inclusion in Eq.~7! of this zero-viscosity contribution, although some attempts have beedone to correlate it to the classical ‘‘free-rotor’’ reorientationtime:2,18 t05~2p/9!AI /kT.
In the hydrodynamic models the effective volume is as-sumed as
Veff5~ f •C!Vp , ~8!
whereVp is the volume of the probe molecule. The value off is defined by the molecular shape, andC depends on theinteraction of the probe molecule with the solvent~boundaryconditions!.16–19When f •C51 the hydrodynamic theory re-duces to the usual Debye–Stoke–Eistein model. Accordinto Eq. ~8!, orientational dynamics is thus characterized byonly two independent parameters,f andC. In spite of itssimplicity this approach has been shown to often be in gooagreement with the experimental results.
Several theories have been developed to determine thvalue of f andC. The simplest approach assumes the solvenas a hydrodynamic continuum, with the solute moleculemoving in a uniform, unstructured medium, characterized bythe shear viscosityh. Approximating the molecular shape toan ellipsoid with axial ratior, Perrin16 proposed the ‘‘stick’’boundary conditionsC51 and f stick5f ~r!. This model wasadapted by Hu–Zwanzig17 to the ‘‘slip’’ conditions, by thesimple relation~f •C)slip5( f •C)stick• H~r!.
If a deeper view of the boundary effects is required, themolecular nature of the solvent must be somehow includein the model. Indeed, the probe molecule experiences thviscosity on a molecular scale and the shear viscosity of Eq~7! ~macroviscosity! should be reconsidered on a micro-
, No. 24, 22 June 1995¬to¬AIP¬license¬or¬copyright,¬see¬http://jcp.aip.org/jcp/copyright.jsp
-
tt
-
t
on
e
e
5h
0ini
r
h
u
t
edt
-
nab-tse
9540 Ricci et al.: Dynamics of b-carotene
scopic scale~microviscosity!. The microscopic boundary effects bring about two principal issues: the intermolecular ptential, that defines the stick or slip character, and the relasize of the solute and solvent molecules, which determinenature of the viscosity effect. Gierer and Wirtz introducesome corrections in theC parameter with the aim of accounting for the microviscosity effects.20 According to theirtheory,C depends on the ratio between the solute andsolvent molecular volumes. The expression forC, providedthat Vs ~solvent volume! ,Vp ~solute volume! can be ap-proximated as
C5sC0 , s5@116~Vs /Vp!1/3C0#
21,~9!
C05S 6~Vs /Vp!1/3
@112~Vs /Vp!1/3#4
11
@114~Vs /Vp!1/3#3D
21
.
On the other hand, Dote, Kivelson, and Schwartz19 haveshown the importance of assuming boundary conditiwhich allow for free spaces in the hydrodynamics cotinuum, related to the actual size of the solvent molecuThis theory invokes a dependence ofC on the relative mo-lecular volumes and on some bulk properties of the nliquid ~namelyB, a temperature independent parameter;kT ,the isothermal compressibility;h, the shear viscosity!; theexpression for theC parameter of Eq.~8! has the form
C5@11~g/f!#21 ~10!
with
g5kT~kThB!
Vp@114~Vp /Vs!
2/3# ~11!
andf assumed to be equal tof slip .
III. EXPERIMENTAL AND DATA HANDLING
The details of the experimental apparatus for transiOKE have been described in previous articles.10 The intenselinearly polarized pump pulse~200 fs, 50mJ at 604 nm!,creates an anisotropy of the refractive index that canprobed by a variably delayed weak pulse polarized at 4The change in the probe polarization is detected througcrossed polarizer. In the present experiment the tunaprobe beam~250 fs,,100 nJ at 517 nm! was obtained byselecting its central frequency with a couple of quartz 6prisms and a slit, out of a continuum generated by focuspart of the pump beam in a water cell. Several experimehave been performed replacing the slit prism set up winterference filters with a bandpass of 6 nm. The pulse dution is determined by autocorrelation measurements and tsient OKE in a thin~1 mm! piece of glass.
The sample consists of 531024 mol/L solution of all-trans b-carotene~from Aldrich! in alkanes~Uvasol gradeMerck solvents! and all the experiments are performed at ttemperature of 29661 K in a 1 mmquartz cell.
Apart from the very fast response, which is out of opresent interest, the duration of the pulses is short compato the characteristic response time of the sample; the conlution in Eq. ~2! can then be approximated as
S~t!}uR~t!u2. ~12!
J. Chem. Phys., Vol. 102Downloaded¬28¬Jun¬2004¬to¬150.217.156.84.¬Redistribution¬subjec
o-ivehed
he
n-le.
at
nt
be°.a
ble
°ngtsthra-an-
e
rredvo-
IV. RESULTS AND DISCUSSION
We have measured the rotational dynamics ofb-carotenedissolved in a series of different,n-alkanes~pentane, hexane,octane, decane, dodecane, hexadecane!, at constant tempera-tureT529661 K.
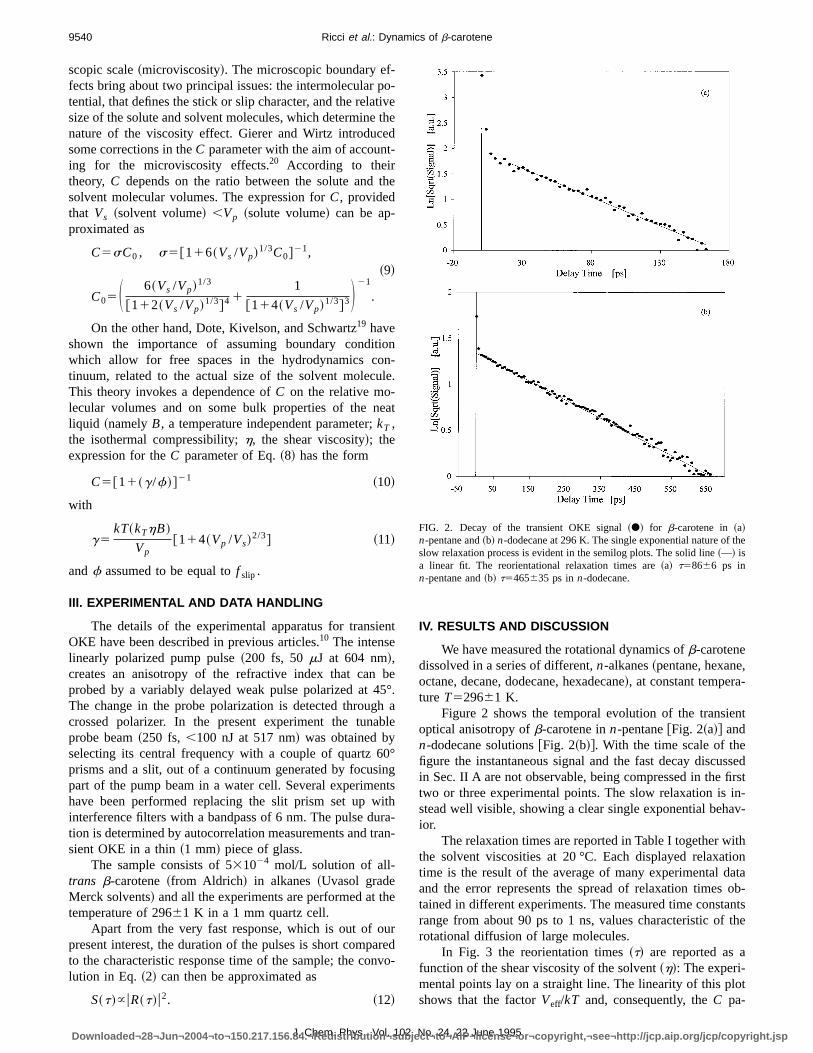
Figure 2 shows the temporal evolution of the transienoptical anisotropy ofb-carotene inn-pentane@Fig. 2~a!# andn-dodecane solutions@Fig. 2~b!#. With the time scale of thefigure the instantaneous signal and the fast decay discussin Sec. II A are not observable, being compressed in the firstwo or three experimental points. The slow relaxation is in-stead well visible, showing a clear single exponential behavior.
The relaxation times are reported in Table I together withthe solvent viscosities at 20 °C. Each displayed relaxatiotime is the result of the average of many experimental datand the error represents the spread of relaxation times otained in different experiments. The measured time constanrange from about 90 ps to 1 ns, values characteristic of throtational diffusion of large molecules.
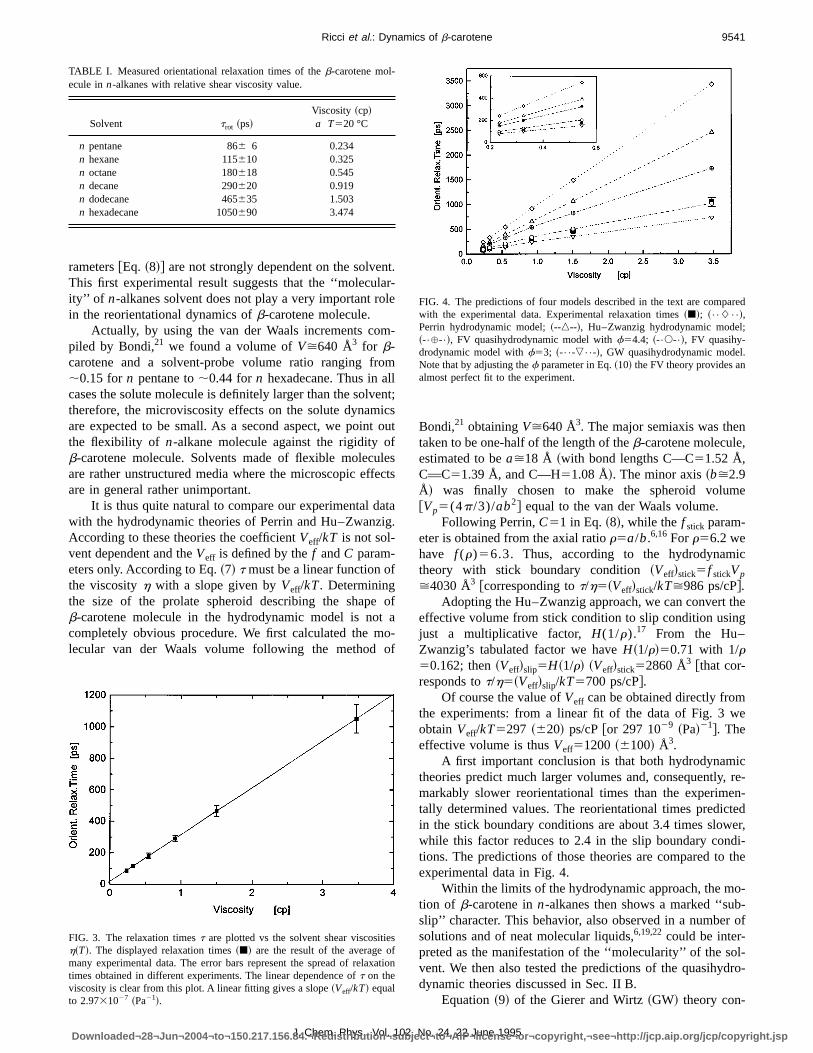
In Fig. 3 the reorientation times~t! are reported as afunction of the shear viscosity of the solvent~h!: The experi-mental points lay on a straight line. The linearity of this plotshows that the factorVeff/kT and, consequently, theC pa-
FIG. 2. Decay of the transient OKE signal~d! for b-carotene in~a!n-pentane and~b! n-dodecane at 296 K. The single exponential nature of theslow relaxation process is evident in the semilog plots. The solid line~—! isa linear fit. The reorientational relaxation times are~a! t58666 ps inn-pentane and~b! t5465635 ps inn-dodecane.
, No. 24, 22 June 1995t¬to¬AIP¬license¬or¬copyright,¬see¬http://jcp.aip.org/jcp/copyright.jsp
tre
m
n
t
f
e
e--dr,-e
f
o-t
d
9541Ricci et al.: Dynamics of b-carotene
rameters@Eq. ~8!# are not strongly dependent on the solvenThis first experimental result suggests that the ‘‘moleculaity’’ of n-alkanes solvent does not play a very important rolin the reorientational dynamics ofb-carotene molecule.
Actually, by using the van der Waals increments compiled by Bondi,21 we found a volume ofV>640 Å3 for b-carotene and a solvent-probe volume ratio ranging fro;0.15 forn pentane to;0.44 forn hexadecane. Thus in allcases the solute molecule is definitely larger than the solvetherefore, the microviscosity effects on the solute dynamicare expected to be small. As a second aspect, we point othe flexibility of n-alkane molecule against the rigidity ofb-carotene molecule. Solvents made of flexible moleculeare rather unstructured media where the microscopic effecare in general rather unimportant.
It is thus quite natural to compare our experimental dawith the hydrodynamic theories of Perrin and Hu–ZwanzigAccording to these theories the coefficientVeff/kT is not sol-vent dependent and theVeff is defined by thef andC param-eters only. According to Eq.~7! t must be a linear function ofthe viscosityh with a slope given byVeff/kT. Determiningthe size of the prolate spheroid describing the shapeb-carotene molecule in the hydrodynamic model is notcompletely obvious procedure. We first calculated the molecular van der Waals volume following the method o
TABLE I. Measured orientational relaxation times of theb-carotene mol-ecule inn-alkanes with relative shear viscosity value.
Solvent trot ~ps!Viscosity ~cp!a T520 °C
n pentane 866 6 0.234n hexane 115610 0.325n octane 180618 0.545n decane 290620 0.919n dodecane 465635 1.503n hexadecane 1050690 3.474
FIG. 3. The relaxation timest are plotted vs the solvent shear viscositiesh~T!. The displayed relaxation times~j! are the result of the average ofmany experimental data. The error bars represent the spread of relaxatimes obtained in different experiments. The linear dependence oft on theviscosity is clear from this plot. A linear fitting gives a slope~Veff/kT! equalto 2.9731027 ~Pa21!.
J. Chem. Phys., Vol. 102,Downloaded¬28¬Jun¬2004¬to¬150.217.156.84.¬Redistribution¬subject¬
.-
-
t;sut
sts
a.
ofa-
Bondi,21 obtainingV>640 Å3. The major semiaxis was thentaken to be one-half of the length of theb-carotene molecule,estimated to bea>18 Å ~with bond lengths C—C51.52 Å,CvC51.39 Å, and C—H51.08 Å!. The minor axis~b>2.9Å! was finally chosen to make the spheroid volume@Vp5(4p/3)/ab2# equal to the van der Waals volume.
Following Perrin,C51 in Eq.~8!, while thef stick param-eter is obtained from the axial ratior5a/b.6,16Forr56.2 wehave f (r)56.3. Thus, according to the hydrodynamictheory with stick boundary condition~Veff!stick5f stickVp
>4030 Å3 @corresponding tot/h5~Veff!stick/kT>986 ps/cP#.Adopting the Hu–Zwanzig approach, we can convert th
effective volume from stick condition to slip condition usingjust a multiplicative factor,H(1/r).17 From the Hu–Zwanzig’s tabulated factor we haveH~1/r!50.71 with 1/r50.162; then~Veff!slip5H~1/r! ~Veff!stick52860 Å3 @that cor-responds tot/h5~Veff!slip/kT5700 ps/cP#.
Of course the value ofVeff can be obtained directly fromthe experiments: from a linear fit of the data of Fig. 3 weobtainVeff/kT5297 ~620! ps/cP@or 297 1029 ~Pa!21#. Theeffective volume is thusVeff51200 ~6100! Å3.
A first important conclusion is that both hydrodynamictheories predict much larger volumes and, consequently, rmarkably slower reorientational times than the experimentally determined values. The reorientational times predictein the stick boundary conditions are about 3.4 times slowewhile this factor reduces to 2.4 in the slip boundary conditions. The predictions of those theories are compared to thexperimental data in Fig. 4.
Within the limits of the hydrodynamic approach, the mo-tion of b-carotene inn-alkanes then shows a marked ‘‘sub-slip’’ character. This behavior, also observed in a number osolutions and of neat molecular liquids,6,19,22could be inter-preted as the manifestation of the ‘‘molecularity’’ of the sol-vent. We then also tested the predictions of the quasihydrdynamic theories discussed in Sec. II B.
Equation~9! of the Gierer and Wirtz~GW! theory con-
ion
FIG. 4. The predictions of four models described in the text are comparewith the experimental data. Experimental relaxation times~j!; ~••L••!,Perrin hydrodynamic model;~--n--!, Hu–Zwanzig hydrodynamic model;~-•%-•!, FV quasihydrodynamic model withf54.4; ~-•s-•!, FV quasihy-drodynamic model withf53; ~-••-,••-!, GW quasihydrodynamic model.Note that by adjusting thef parameter in Eq.~10! the FV theory provides analmost perfect fit to the experiment.
No. 24, 22 June 1995to¬AIP¬license¬or¬copyright,¬see¬http://jcp.aip.org/jcp/copyright.jsp

e
n
i
l
Ve
i
e
s
i
e
p
d
f
9542 Ricci et al.: Dynamics of b-carotene
tains only the two parametersVs ~the volume of the solventmolecule! andVp ~the volume of the solute molecule!, thatwere determined following the method of Bondi.21 The stickvalue of thef parameter is adopted in Eq.~8!.
The ‘‘free volume’’ model of Doteet al.19 leading toEqs. ~10! and ~11! contains a larger number of parametersThe values of the isothermal compressibilitieskT and of theviscositiesh were obtained from Ref. 23. The values of thB coefficient were found in Ref. 24 for all then-alkanesconsidered, except hexadecane; an estimate ofB for this sol-vent was thus obtained from the equatioB5(V/V2V0)(1/h) of Ref. 24, whereV0 is the ‘‘intrinsic’’~maximum packing! molecular volume, andV is thetemperature-dependent molal volume defined as the ratiothe molecular weight to the density.V for hexadecane is 292cm3/mol at 20 °C, while a linear extrapolation of the data oRef. 24 givesV05286 cm3/mol, thus yieldingB512 cP21.Finally, as suggested in Ref. 19,f was set equal tof slip inEq. ~10!.
The results of these last two models are collected in F4. Quite evidently, both theories are in a much better agrement with the experimental data, if compared to the resuof the hydrodynamic models. Actually, the agreement of thGW model is almost perfect at low viscosities, while the Ftheory predict relaxation times that are consistently highthan the experimental ones. However, it is worth notice thawhile the GW theory has no free parameter, the choiceadopting forf the value off slip is questionable.
19 It may beof some relevance that by using in Eq.~10! f>3 instead off54.4, an extremely good fit to the experimental dataobtained in the entire range of viscosity~see Fig. 4!.
In summary, from a comparison of the results of the foumodels collected in Fig. 4, the quasihydrodynamic theoriappear as more appropriate than the purely hydrodynamones to describe the orientational dynamics ofb-carotene inn-alkanes. This result is somehow surprising, since the effeof the solvent’s discrete structure is generally expected tosmall for solutions characterized, like those considered heby a large value of theVsolute/Vsolvent ratio. By the way, thefact that the overall behavior ofb-carotene cannot be seen ahighly nonhydrodynamic is confirmed by the linear dependence of the experimental relaxation times on the viscos~Fig. 4!. On the other hand, although the calculation of thmolecular volume and of the shape of the representativelipsoid is, by necessity, a rather rough evaluation, possibaffected by a large error, the values of the effective volumobtained from the hydrodynamic theories are too far from thexperimental one to make the purely hydrodynamic aproach, even within a subslip picture, easily acceptable.
Actually, the two theories that take into account the molecular size of the solvent predict an effective volume of thsolute molecule in a much better agreement with the expemental value.
We also notice that the relaxation times calculated acording to the GW model show a less-than-linear depedence onh ~see Fig. 4!, while our experimental data areessentially linear. From this point of view the FV model is ina better qualitative agreement, which becomes quantitativethef parameter in Eq.~10! is made adjustable.
J. Chem. Phys., Vol. 102Downloaded¬28¬Jun¬2004¬to¬150.217.156.84.¬Redistribution¬subject
.
of
f
g.e-tse
rt,of
s
rsic
ctbere,
-tyeel-lyse-
-eri-
c-n-
if
We can say that, although there is no clear evidence ofthe departure of theb-carotene solutions from the hydrody-namic behavior, quasihydrodynamic theories, like those con-sidered here, provide a rather satisfactory way to evaluate theeffective volume of the solute molecule.
For some simpler molecules, like benzene,25 the appar-ent disagreement with the hydrodynamic predictions wasshown to vanish almost completely with a more realisticcomputation of the molecular volume. In the present casehowever the discrepancies are probably too large to be attrib-uted only to the rough evaluation of the volume; in any case,an accurate calculation of the full rotational tensor on theatomic scale appears practically impossible for such a com-plex molecule likeb-carotene.
V. CONCLUDING REMARKS
The transient resonance enhanced OKE has been proveto be a useful tool in the investigation of reorientational dy-namics of a probe molecule with relatively fast electroniclifetimes. If a low concentration of solute molecules is used,this spectroscopic technique allows a direct investigation ofthe averaged single molecule dynamics.
Our experimental results suggest a general agreement othe reorientational dynamics ofb-carotene inn-alkanes withthe quasihydrodynamic theory. The free volume model pro-vides the correct viscosity dependence of the relaxationtimes; by adjusting thef parameter the agreement becomesquantitative.
ACKNOWLEDGMENTS
This work was supported by Commission of the Euro-pean Communities under Contract Nos. GE1*CT92-0046and ERBCHRXCT930282, and by the Italian Consiglio Na-zionale delle Ricerche.
1A. J. Barnes, W. J. Orvilee-Thomas, and J. Yarwood,Molecular Liquids,Dynamics and Interactions, NATO Advanced Study Institute, Series B:Physics~Reidel, Dordrecht, 1984!.
2B. J. Berne and R. Pecora,Dynamic Light Scattering~Wiley, New York,1976!.
3C. H. Wang,Spectroscopy of Condensed Media, Dynamics of MolecularInteraction ~Academic, London, 1985!.
4S. Dattagupta,Relaxation Phenomena in Condensed Matter Physics~Aca-demic, London, 1987!.
5Y. R. Shen,The Principal of Nonlinear Optics~Wiley, New York, 1984!.6G. H. Fleming,Chemical Application of Ultrafast Spectroscopy~OxfordUniversity, Oxford, 1986!.
7R. Righini, Science262, 1386~1993!.8M. Ricci, P. Foggi, R. Righini, and R. Torre, J. Chem. Phys.98, 4892~1993!; R. Torre, I. Santa, and R. Righini, Chem. Phys. Lett.212, 90~1993!.
9J. J. Stankus, R. Torre, C. D. Marshall, S. R. Greenfield, A. Sengupta, A.Tokmakoff, and M. D. Fayer, Chem. Phys. Lett.193, 213 ~1992!; J. J.Stankus, R. Torre, and M. D. Fayer, J. Phys. Chem.97, 9480~1993!.
10P. Foggi, R. Righini, R. Torre, and V. F. Kamalov, Chem. Phys. Lett.193,23 ~1992!; P. Foggi, V. F. Kamalov, R. Righini, and R. Torre, Opt. Lett.17,775 ~1992!.
11J. P. Hermann, D. Richard, and J. Ducuing, Appl. Phys. Lett.23, 178~1973!.
12R. W. Hellwarth, Prog. Quantum Electron.5, 1 ~1977!.13M. Cho, G. R. Fleming, and S. Mukamel, J. Chem. Phys.98, 5314~1993!;M. Cho, M. Du, N. F. Scherer, G. R. Fleming, and S. Mukamel,ibid. 99,2411 ~1993!.
14Mr. Wasielewski and L. D. Kispert, Chem. Phys. Lett.128, 238 ~1986!.
, No. 24, 22 June 1995¬to¬AIP¬license¬or¬copyright,¬see¬http://jcp.aip.org/jcp/copyright.jsp

9543Ricci et al.: Dynamics of b-carotene
15D. Frenkel and J. P. McTague, J. Chem. Phys.72, 2801~1980!.16F. Perrin, J. Chem. Phys.10, 415 ~1942!.17C. Hu and R. Zwanzig, J. Chem. Phys.87, 4354~1987!.18G. R. Alms, T. D. Gierke, and T. D. Patterson, J. Chem. Phys.60, 5779
~1974!; G. R. Alms, D. R. Bauer, J. I. Brauman, and R. Pecora,ibid. 59,5310, 5321~1973!.
19J. L. Dote, D. Kivelson, and R. N. Schwartz, J. Phys. Chem.85, 2169~1981!; D. Kivelson and J. L. Dote,ibid. 87, 3889~1983!; D. Kivelson, inRotational Dynamics of Small and Macromolecules, edited by Th. Dor-muller and R. Pecora~Springer, Berlin, 1987!.
J. Chem. Phys., Vol. 102Downloaded¬28¬Jun¬2004¬to¬150.217.156.84.¬Redistribution¬subject
20A. Gierer and K. Wirtz, Z. Naturforsch. Teil A8, 532 ~1953!.21A. Bondi, J. Phys. Chem.68, 441 ~1964!.22D. Ben-Amotz and T. W. Scott, J. Chem. Phys.87, 3739 ~1987!; S. Ca-nonica, A. A. Achimid, and U. P. Wild, Chem. Phys. Lett.122, 529~1985!.
23Handbook of Chemistry and Physics, 71st ed., edited by D. R. Lide~Chemical Rubber, Boca Raton, FL, 1990!.
24J. H. Hildebrand, Science174, 490 ~1971!.25G. K. Youngren and A. Acrivos, J. Chem. Phys.63, 3846 ~1975!; D. C.Knauss, G. T. Evans, and D. M. Grant, Chem. Phys. Lett.71, 158 ~1980!.
, No. 24, 22 June 1995¬to¬AIP¬license¬or¬copyright,¬see¬http://jcp.aip.org/jcp/copyright.jsp





























