Embed Size (px)
Citation preview

Protein Engineering vol.12 no.11 pp.959–966, 1999
Molecular modeling of the amyloid-β-peptide using the homologyto a fragment of triosephosphate isomerase that forms amyloidin vitro
Carlos F.Contreras, Mauricio A.Canales,Alejandra Alvarez1, Giancarlo V.De Ferrari1 andNibaldo C.Inestrosa1,2
Laboratorio de Biofısica Molecular, Facultad de Ciencias Biologicas,Universidad de Concepcion, Concepcion and 1Departamento de BiologıaCelular y Molecular, Facultad de Ciencias Biologicas, Pontificia UniversidadCatolica de Chile, PO Box 114-D, Alameda 340, Santiago, Chile
2To whom correspondence should be addressed
The main component of the amyloid senile plaques foundin Alzheimer’s brain is the amyloid-β-peptide (Aβ), aproteolytic product of a membrane precursor protein.Previous structural studies have found different conform-ations for the Aβ peptide depending on the solvent and pHused. In general, they have suggested an α-helix conform-ation at the N-terminal domain and a β-sheet conformationfor the C-terminal domain. The structure of the completeAβ peptide (residues 1–40) solved by NMR has revealedthat only helical structure is present in Aβ. However, thisresult cannot explain the large β-sheet Aβ aggregatesknown to form amyloid under physiological conditions.Therefore, we investigated the structure of Aβ by molecularmodeling based on extensive homology using the Smithand Waterman algorithm implemented in the MPsrchprogram (Blitz server). The results showed a mean valueof 23% identity with selected sequences. Since these valuesdo not allow a clear homology to be established with areference structure in order to perform molecular modelingstudies, we searched for detailed homology. A 28% identitywith an α/β segment of a triosephosphate isomerase (TIM)from Culex tarralis with an unsolved three-dimensionalstructure was obtained. Then, multiple sequence alignmentwas performed considering Aβ, TIM from C.tarralis andanother five TIM sequences with known three-dimensionalstructures. We found a TIM segment with secondarystructure elements in agreement with previous experimentaldata for Aβ. Moreover, when a synthetic peptide from thisTIM segment was studied in vitro, it was able to aggregateand to form amyloid fibrils, as established by Congo redbinding and electron microscopy. The Aβ model obtainedwas optimized by molecular dynamics considering ionizableside chains in order to simulate Aβ in a neutral pHenvironment. We report here the structural implicationsof this study.Keywords: Alzheimer’s disease/amyloid/modeling/triosephos-phate isomerase
Introduction
Alzheimer’s disease is one of the most common causes ofsenile dementia. This neurological disease is characterized bya progressive deterioration caused by the neuronal damageof cortical regions. The major component of the neuronaldegenerative products is the amyloid-β-peptide (Aβ), a peptide
© Oxford University Press 959
of 40–43 residues also found in Down’s syndrome (Masterset al., 1985; Mori et al., 1992; Soto et al., 1994) which iscapable of forming large β-sheet aggregates (Kirschner et al.,1986; Halverson et al., 1990).
In vitro studies with Aβ have shown that it presents a highaggregation capacity dependent on pH (Barrow and Zagorski,1991; Esler et al., 1996), concentration, ionic strength (Hilbichet al., 1991; Lee et al., 1995) and the presence of other proteins(Inestrosa et al., 1996; Harper and Lansbury, 1997). Theaggregation process starts with a nucleation step followed bya growth phase, which is dependent on the composition of thecarboxyl end of Aβ (Jarrett et al., 1993). X-ray studies of Aβfibrils have shown an anti-parallel β-pleated sheet structure(Kirschner et al., 1986, 1987; Gorevic et al., 1987), which isformed by four-stranded β-pleated sheets, with repetitions of10.6 Å between stacked sheets and 4.8 Å distance along thefiber axis. The amino acid sequence of Aβ was originallydetermined by Glenner and Wong (1984) and the secondarystructure was predicted by the Chou–Fasman algorithm asfollows: 1–4, α-helix; 5–8, turn; 9–13, α-helix or β-strand;14–18, α-helix; 19–22, β-strand; 23–27, turn; 28–40 β-strand(Gorevic et al., 1987; Kirschner et al., 1987). This predictionpoints to a helical conformation for the N-terminus and aβ-strand conformation for the C-terminus with hydrophobiccharacter, although it has yet to be supported experimentally.During the Aβ aggregation process, the α-helical segment ofthe N-terminus is predicted to convert to a β-strand structure(Barrow and Zagorski, 1991; Zagorski and Barrow, 1992;Sorimachi and Craik, 1994; Soto et al., 1995).
The structure of fragment 1–28 of Aβ has been solved bynuclear magnetic resonance (NMR) spectroscopy (Zagorskiand Barrow, 1992; Sorimachi and Craik, 1994; Talafous et al.,1994). Zagorski and Barrow (1992) observed that variousregions of Aβ1–28 (i.e. residues 2–27 or residues 2–7 and 10–27) can adopt an α-helical conformation. Later, Sorimachi andCraik (1994) reported that, although the fragment Aβ1–28 canexist as a mixture of conformations in solution, one of suchpopulation of structures possesses a flexible N-terminal regionand a well defined C-terminal region, which includes anα-helical segment and a terminal turn-like structure. Finally,Talafous et al. (1994), working in a membrane-like medium(resembling the structure under physiological conditions),showed that the solution structure of the Aβ1–28 fragmentis almost entirely α-helical (consisting of two right-handedα-helices, residues 2–11 and 13–27, which are connected bya bend centered at Val12).
Regarding the conformation of the Aβ1–40 C-terminal region,the synthesis of analog peptides containing a disulfide bridge,followed by infrared (IR) and circular dichroism (CD) spectro-scopy, have demonstrated that fragments 29–42, 22–43 and 1–43 adopt the conformation of two antiparallel β-sheets with acentral β-turn at residues 26–29 (Hilbich et al., 1991; Hugheset al., 1996). Hydrophobic interactions at Phe19 and Phe20seem to be important for the aggregation process and this

C.F.Contreras et al.
Table I. Results of the search for sequence homology to Aβ
No.: identification number for data file found by the search. Score: scorecalculated for pairwise alignment using matrix blosum62. %Match: identitypercentage from the pairwise alignment. Length: length of residues onprotein file. ID: accession code for Swiss Protein database. DE: file ondatabase, in this case from Swiss Protein database. Db: sequence from entryfile in database aligned to the sequence of query (FASTA format). Qy:sequence of query used (in FASTA format). Indels: Indel penalty, rangevaries with mark table and stringency. Matches: identical residues, markedby an asterisk (*). Mismatches: non related residues no mark. Partials: lowrelated residues, marked by a full point (.). Gaps: penalty for extension ofinsertion or deletion.
effect is increased by the presence of His13 and His14 atneutral pH (Hughes et al., 1996).
Efforts to obtain the complete three-dimensional structureof Aβ, under physiological conditions, so far have beenunsuccessful. Sticht et al. (1995) reported that the Aβ1–40peptide NMR structure, at pH �2.8, presented two helicalregions (Gln15 to Asp23 and Ile31 to Met35) that showed nointeractions between them. On the other hand, two models forthe structure of the Aβ peptide have been proposed. WhereasSoto et al. (1994) considered hydrophobicity profiles to identifysecondary structure predictors and computational analysis(energy minimization) to generate a model for soluble Aβ1-42,Iversen et al. (1995) proposed a possible arrangement of Aβ1-42monomers to form an anti-parallel β-structure. Here, we havemodeled the complete structure of the Aβ peptide based onthe 1–28 segment of Aβ solved by NMR and by the homologyof the peptide to a segment of triosephosphate isomerase(TIM). We have also shown that this segment of TIM is ableto form amyloid.
960
Materials and methodsPeptide synthesis and characterizationPeptide fragments derived from the human wild-type Aβ1–40peptide were obtained from Sigma Chemical (St. Louis, MO)and from Chiron (Emeryville, CA). Fragment 186–218 derivedfrom Escherichia coli TIM was synthesized by using solid-phase techniques at the Technical Institute, Braunschweig,Germany. All peptides were purified by reversed-phase high-performance liquid chromatography and their purity was evalu-ated by amino acid sequence analysis.
Computational methodsAn extensive sequence homology search for Aβ peptide wasdone using the Smith and Waterman algorithm (Smith andWaterman, 1981; Shomer et al., 1996) implemented in MPsrch(Blitz server http://www.ebi.ac.uk/searches/blitz_input.html)against the Swiss Protein database. The selected proteins werechecked in the Protein Data Bank (Bernstein et al., 1977) forthree-dimensional structures. Multiple sequence alignments forthese proteins were performed with the Pattern Induced Multi-sequences Alignment (PIMA) program, using a coveringpattern construction algorithm (Smith and Temple, 1992; Defayand Cohen, 1995). This information yielded reference structuresuseful for assignment of atomic coordinates to 34 residues atthe C-terminal region of the Aβ sequence. The structure ofthe Aβ1–28 fragment (PDB code: 1amb) solved by NMR(Talafous et al., 1994) was used to assign coordinates to theN-terminus. The connecting loop, residues Ala22–Gly30 ofthe Aβ sequence, was chosen from the loop data bank ofHomology (Biosym/MSI). Crystal structures of TIM fromhuman (PDB code: 1hti) (Mande et al., 1994), Trypanosomabrucei (4tim) (Noble et al., 1991), chicken (1tpb) (Zhanget al., 1994), Saccharomyces cereviase (1ypi) (Lolis et al.,1990) and E.coli (1tre) (Kishan et al., 1994) were used asreferences.
Refinement of side chain dihedral angles and other localoptimizations were made to convergence (0.01 kcal/mol) usingAMBER 4.1 (Weiner and Kollman, 1981, 1986). Globaloptimizations were carried out first in vacuo and afterwardsin aquo with Aβ in a shell of water molecules located up toa maximum distance of 25.0 Å (SOL, IGROUP1 � CAPoptions of EDIT module) from the arbitrary center of thepeptide at CG His13, leading to a system containing 1263water molecules. PROCHECK v. 2.1.4 (Laskowski et al.,1993) was employed to check the stereochemical qualityof the optimized structures. All calculations and graphicvisualizations were performed on an INDY machine using theHomology module and InsightII (Biosym/MSI).
Aggregation studies of TIM peptide: turbidity assayLyophilized aliquots of the TIM peptide were dissolved indimethyl sulfoxide at 15 mg/ml (3.5 mM). Aliquots of peptidestock (70 nmol in ~20 µl of dimethyl sulfoxide) were addedto phosphate-buffered saline, pH 7.3, in a final volume of725 µl. Alternatively, aliquots of TIM peptide were resuspendedin 0.1 M sodium acetate at pH 5.0. Incubations were carried outat room temperature and the solutions were stirred continuously(210 r.p.m.). At various times, aggregation was measured byturbidity at 405 nm versus buffer blank as described previously(Jarrett et al., 1993; Soto et al., 1995; Inestrosa et al., 1996).
Amyloid detection by Congo red (CR) measurementsThe binding of CR to TIM aggregates was measured as
described previously (Klunk et al., 1989) to quantify amyloid
Amyloid-β-peptide structure
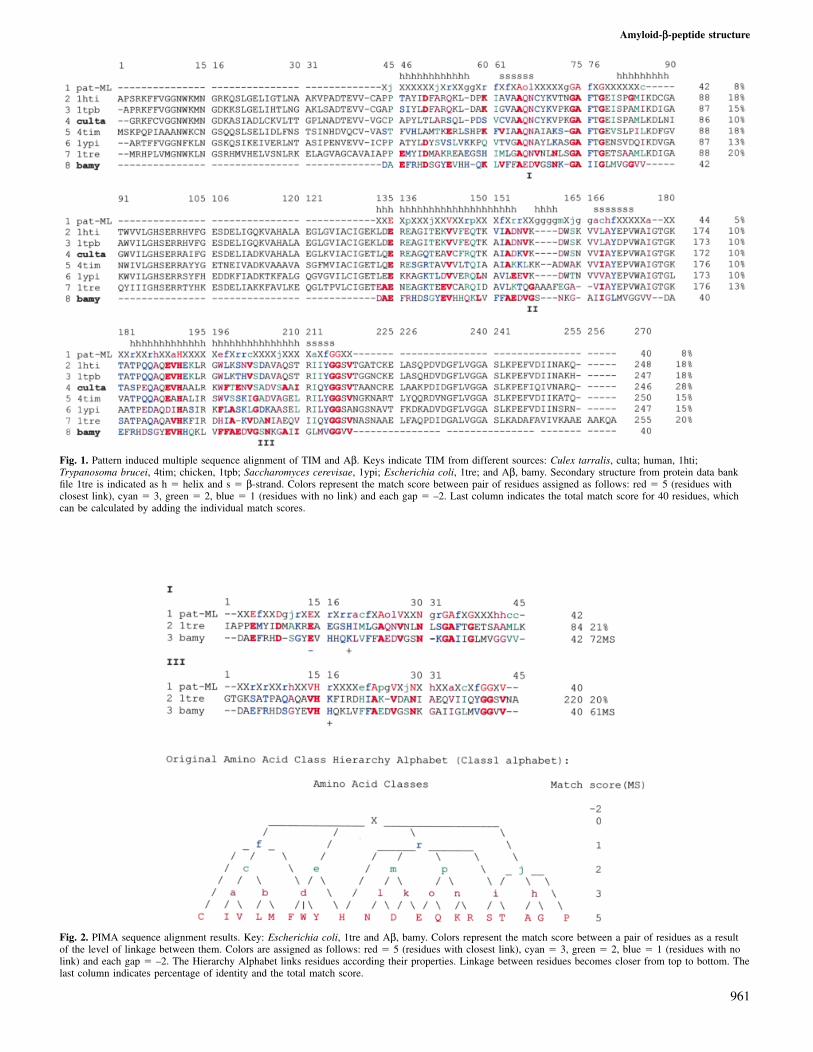
Fig. 1. Pattern induced multiple sequence alignment of TIM and Aβ. Keys indicate TIM from different sources: Culex tarralis, culta; human, 1hti;Trypanosoma brucei, 4tim; chicken, 1tpb; Saccharomyces cerevisae, 1ypi; Escherichia coli, 1tre; and Aβ, bamy. Secondary structure from protein data bankfile 1tre is indicated as h � helix and s � β-strand. Colors represent the match score between pair of residues assigned as follows: red � 5 (residues withclosest link), cyan � 3, green � 2, blue � 1 (residues with no link) and each gap � –2. Last column indicates the total match score for 40 residues, whichcan be calculated by adding the individual match scores.
Fig. 2. PIMA sequence alignment results. Key: Escherichia coli, 1tre and Aβ, bamy. Colors represent the match score between a pair of residues as a resultof the level of linkage between them. Colors are assigned as follows: red � 5 (residues with closest link), cyan � 3, green � 2, blue � 1 (residues with nolink) and each gap � –2. The Hierarchy Alphabet links residues according their properties. Linkage between residues becomes closer from top to bottom. Thelast column indicates percentage of identity and the total match score.
961

C.F.Contreras et al.
formation. Aliquots (40 µl) were added to 960 µl of a solutioncontaining 25 µM CR, 100 mM phosphate (pH 7.4), 150 mMNaCl and incubated for 30 min. Absorbance was measured at480 and 540 nm and CR binding was calculated as follows:CR (M) � (A540/25 295) – (A480/46 306).
Electron microscopy of amyloid fibrilsThe TIM peptide (1 mg/ml) was incubated in 0.1 M sodiumacetate (pH 5.0) and the Aβ peptide (1 mg/ml) in 0.1 M Tris–HCl (pH 7.4), for 5 days at room temperature. The amyloidfibrils formed were then transferred on to Formvar carbon-coated 300-mesh nickel grids, negatively stained with 2%uranyl acetate for 1 min and examined under a JEOL 100-Belectron microscope at 80 kV (Alvarez et al., 1997).
ResultsHomology searchMPsrch used for homology search with Aβ peptide yieldedthe results shown in Table I. A general low identity of 23.2%was obtained. Of the 27 non-amyloid proteins retrieved, onlythe sequence corresponding to a TIM enzyme from Culextarralis with 22% local identity (Table I, entry 23) was selectedto provide atomic coordinates for most of the Aβ sequence.The TIM fragments mainly pointed to a C-terminal homologywith the Aβ peptide. As the three-dimensional structure ofTIM from C.tarralis has yet to be solved, we decided to searchfor related available structures. The family of TIM proteinscontains several members, from different sources with knownthree-dimensional structures, namely 1hti, 4tim, 1tpb, 1ypiand 1tre.
Multiple sequence alignmentA multiple sequence alignment of the TIM family membersis shown in Figure 1. The alignment shows three regions (I,
Fig. 3. Ribbon representation of TIM, Escherichia coli, 1tre (green) and1–28 Aβ NMR structure (lilac). Selected fragments at 1tre site III: α-helixwith structural homology to 1amb (0.664 Å r.m.s.d.) (red), loop (cyan) andβ-strand region employed to assign coordinates to Aβ model (orange).
Fig. 4. Coordinates assignment. Alignment of TIM from E.coli (1tre, green), 1–28 residues length Aβ structure solved by NMR (1amb, lilac) and Aβ peptide(bamy, yellow). Coordinates from 1tre and 1amb were assigned to the Aβ segments inside the limits of the boxes. Loop coordinates obtained from the LoopsDatabase of InsightII (0.463 Å r.m.s.d.), were assigned to the residues between boxes.
962
II and III) homologous to the Aβ peptide. The sequence ofTIM from E.coli (1tre) contained two regions with the highestidentity with Aβ (20%); these regions (I and III) were selectedto perform a new alignment assay with Aβ (Figure 2). Thenew local alignment increased the percentage identity of regionI to 21%, but introduced two gaps into the Aβ sequence, oneof which is located in the loop zone of Aβ. The alignmentwith region III did not introduce gaps into the Aβ sequenceand its C-terminal region showed a similar secondary structure.On the other hand, visual inspection of the three-dimensionalstructure of TIM in these regions showed that only regionsII and III presented an α-helix–loop–β-strand structure, inagreement with previous CD and IR determinations. All theseconsiderations induced us to choose site III of 1tre to modelthe structure of Aβ.
Coordinates assignment
The N-terminal of region III in E.coli TIM (1tre) presented ahigh structural identity with residues 7–21 of the Aβ NMRstructure 1amb [0.664 Å root mean square deviation (r.m.s.d.)](Figures 3 and 4) and the highest identity (40%) was obtainedfor the β-strand segment Ala207–Gly216. Sequence gaps aroseonly at a single loop between residues His199 and Ile206 of1tre that would correspond to the β-turn between residuesAla22 and Gly30 of the Aβ peptide (Figure 4). In this way,we used the atomic coordinates from 1amb for the first 21residues of the α-helix N-terminal segment of Aβ, and theβ-strand residues (Ala207–Gly216) of 1tre for the C-terminaldomain of the Aβ model. The coordinates for the loop betweenresidues Ala22 and Gly30 of Aβ were obtained from the LoopsBank, which showed a segment of 0.463 Å r.m.s.d. Thereport of collision between atoms, obtained after coordinateassignment (Table II), was resolved by optimization of themodel.
Model optimization
Energy refinements were made first on the colliding atoms byrelaxing them. A global optimization moved the structure toa conformation of –1594.9 cal/mol and 0.098 Å r.m.s.d. Anew step of energy minimization was developed with thesystem in a water shell (Jorgensen et al., 1983). The finalconformation reached an energy of –17 459.9 cal/mol and0.095 Å r.m.s.d. against the conformation prior to globaloptimization. Further calculations of the optimization outputin which dihedral angle contributions were neglected, showeda lower energy for the protein alone (–1656 cal/mol) indicatingstabilization due to the water environment. Optimization withaqueous solvent showed that water molecules mainly stabilizednegatively charged residues. The low deviation values indicatedthat the final model remained very close to the referenceconformations used for the N- and C-termini. The r.m.s.d.s forCα atoms were of about the same magnitude using either theoptimization in vacuo (Figure 5) or in aquo. The maindeviations corresponded to the N- and C-terminal residues andto Gln15 and Leu34. PROCHECK (Laskowski et al., 1993)reported only two non-glycine residues: Ala30 and Ile32located in the disallowed region of the Ramachandran plot(Figure 6).

Amyloid-β-peptide structure

C.F.Contreras et al.
Table II. Homology–InsightII report of close atom initial distance aftercoordinates assignments to Aβ sequence and refined distance after energyminimization of the Aβ model in the in aquo system
Residue Atom Residue Atom Distance CurrentNo. No. (Å) (Å)
5 HH11 39 HG23 0.44 6.6716 NZ 34 CD2 0.49 7.5116 NZ 34 HD21 0.82 6.6616 HZ1 34 CD2 0.66 6.1720 CZ 28 CG 0.44 4.8120 CZ 28 HG1 0.98 5.8420 HZ 28 CB 1.05 4.6920 HZ 28 CG 0.89 4.9920 HE2 28 HE2 0.86 4.0020 CD2 28 HE1 0.98 2.5620 HD2 28 HE1 0.81 2.2824 HG13 28 HD1 0.28 4.4524 HG22 28 HZ2 0.87 3.37
Fig. 5. Correspondence of in vacuo (red line) and in aquo (blue line)systems from the original coordinates assigned to the Aβ model.
Fig. 6. Ramachandran plot for the refined Aβ model, calculated using theprogram PROCHECK 202. Glycines are plotted as triangles and all otherresidues as squares. Two non-glycine residues in the disallowed region arelabeled.
964
Fig. 7. (A) 97 µM Aβ peptide in PBS pH 7.0 (d) and 97 µM TIM peptidein PBS pH 5.0 (s) were incubated at room temperature in a kinetic stirredaggregation experiment. In both cases the aggregation was measured byturbidity at 405 nm. (B) Amyloid formation of both peptides wasdetermined by Congo red binding at indicated times of incubation.
Site III of 1tre TIM forms amyloid fibrils
A number of studies with synthetic Aβ peptides and Aβfragments in vitro have shown that this peptide aggregates andforms amyloid fibrils similar to those found in the brainsof patients with Alzheimer’s disease (Castano et al., 1986;Kirschner et al., 1987; Alvarez et al., 1997). In view of ourmodeling studies with the Aβ peptide, we asked whether ornot part of the 1tre TIM site III was able to form amyloid.Figure 7A shows turbidimetric measurements for the TIMaggregation process (Jarrett et al., 1993), which clearly demon-strate that the TIM fragment is able to aggregate, although toa lesser extent than Aβ. To confirm that amyloid was in factformed, as indicated in Figure 7B, Congo red, a dye knownto bind amyloid, was used (Klunk et al., 1989). Finally,negatively stained TIM fragments revealed that the aggregateswere indeed composed of fibrils. No morphological differenceswere detected between the amyloid fibrils formed by Aβ alone(Figure 8a) and by the TIM site III fragment (Figure 8b andc). In both cases, the amyloid fibrils showed the typicalfeatures: 7–10 nm thick unbranched fibrils, up to 2–3 mm inlength (Castano et al., 1986; Kirschner et al., 1987). Theabove results indicate that the 1tre TIM site III forms amyloidfibrils in much the same way as Aβ1–40.

Amyloid-β-peptide structure
Fig. 8. Electron micrographs of negatively-stained preparations of amyloid-like fibrils formed with the Aβ1–40 peptide (A) and a 22-residue peptidetaken from the 1tre TIM site III (B, C) corresponding to two differentpreparations. Aliquots of each preparation were incubated for 5 days andthen adsorbed on 300-mesh Formvar-coated grids and negatively stainedwith 2% uranyl acetate. The specimens were viewed for fibrils using aJEOL 100-B electron microscope. Original magnification �30 000.
DiscussionThe complete three-dimensional structure of the Aβ peptideusing near-physiological conditions has so far been verydifficult to obtain, either by experimental or by predictivemethods. Only through an extensive homology search was itpossible to find a reference protein suitable for molecularmodeling in agreement with previous experimental data. How-ever, since homology with Aβ shows low identity scores(22.0–27.3%), we preferred to identify regions which displayedan α-helix–loop–β-strand folding pattern. Even though entries4–22 (Table I) showed higher match scores and predictionnumbers, they presented non-uniform homology along the Aβsequence mainly centered at the N-terminus. Entry 23 (TIM)showed the widest distribution of homology along the Aβsequence, with a prediction number equal to 8.3, indicatingthat during a random search, eight sequences would likelyarise with scores higher than the score at entry 23. As 22%identity is relatively low, we considered also the patterns ofα-helix–loop–β-strand folding, for three distinct homologysites within the TIM sequence in agreement with previousdata (Laurents et al., 1994). While site I showed a combinedα-helical–β-strand–α-helical motif separated by loops whichwas contrary to the available data (Barrow and Zagorski, 1991;Hilbich et al. 1991, Hughes et al., 1996), sites II and IIIshowed very similar α-helical β-strand motifs.
Energy optimization considering the water shell (–17 459.9
965
cal/mol) allowed the system to reach an additional stabilitycompared with that determined in vacuo (–1594.9 cal/mol).However, the difference in r.m.s.d. between the two systemswas minimal (0.003 Å) indicating a high stabilizing effect onAα due to the aqueous solvent. A more detailed energy analysisin which all dihedral angle contributions were neglectedindicated that the strongest contribution was provided by theelectrostatic interactions between charged residues and thewater shell. In this system, the negatively charged residuesmade a stronger contribution than positively charged residues,whereas in contrast, optimization in vacuo revealed that thetotal energy was distributed uniformly along the chain of theAβ peptide. The highest deviation values corresponded toresidues located at the N- and C-termini of Aβ, a finding whichis in accordance with the end-terminal flexibility generallyobserved in peptides. Residues Gln15 and Leu34 demonstrateda higher mobility than other residues, suggesting that theseresidues could play a role as hinges. The non-glycine residuesAla30 and Ile32 reported in the disallowed region of theRamachandran plot (Figure 6) correlated with the fact thatthese residues presented some of the lowest energy values(�–31. 4 kcal/mol) detected for the same residues either invacuo or in aquo. However this observation does not allow usto distinguish whether these residues are in an energy minimumas result of the complete peptide structure optimization or ina local minimum near to the start conformation in which theresidues could be trapped.
Finally, the finding that part of site III of 1tre TIM was ableto assemble into amyloid fibrils, as also occurs with Aβfragments (Gorevic et al., 1987; Alvarez et al., 1997), providedstrong biological evidence for our homology studies and gavefurther support to our choice of structural TIM domainsfor the construction of the Aβ peptide model presented inthis paper.
ConclusionsThe model structure proposed here for the Aβ peptide accountsfor the biological characteristics of this molecule, includingits aggregation properties. This work is the first step in ourapproach to studying the stability of the Aβ peptide asmonomers and dimers under physiological conditions, itsinteraction with other proteins which may play a role in theassembly and toxicity of amyloid plaques and the potentialconformational changes involved in amyloid formation andthe development of Alzheimer’s disease at the molecular level.
AcknowledgementsThis work was supported by FONDECYT No. 1971240 to N.C.I., Universityof Concepcion Project 4533 to M.A.C. and by a Presidential Chair in Sciencefrom the Chilean Government to N.C.I.
ReferencesAlvarez,A., Opazo,C., Alarcon,R., Garrido,J. and Inestrosa,N. (1997) J. Mol.
Biol., 272, 348–361.Barrow,C. and Zagorski,M. (1991) Science, 253, 179–182.Bernstein,F., Koetzle,T., Williams,G., Meyer,E., Brice,M., Rodgers,J.,
Kennard,O., Shimanouchi,T. and Tasumi,M. (1977) J. Mol. Biol., 112,535–542.
Castano,E.M., Ghiso,J., Prelli,F., Gorevic,P.D., Migheli,A. and Frangione,B.(1986) Biochem. Biophys. Res. Commun., 141, 782–789.
Defay,T. and Cohen,F. (1995) Proteins, 23, 431–445.Esler,W., Stimson,E., Ghirlardi,J., Vinters,H., Lee,J., Mantyh,P. and Maggio,J.
(1996) Biochemistry, 35, 749–757.Glenner,G. and Wong,C. (1984) Biochem. Biophys. Res. Commun., 120,
885–890.

C.F.Contreras et al.
Gorevic. P., Castano,E., Sarma,R. and Frangione,B. (1987) Biochem. Biophys.Res. Commun., 147, 854–862.
Halverson,K., Fraser,P., Kirschner,D. and Lansbury,P. (1990) Biochemistry,29, 2639–2644.
Harper,J.D. and Lansbury,P.T.Jr. (1997) Annu. Rev. Biochem., 66, 385–407.Hilbich,C., Kisters-Woike,B., Reed,J., Masters,C. and Beyreuther,K. (1991)
J. Mol. Biol., 218, 149–163.Hughes,S., Goyal,S., Sun,J., Gonzalez-DeWhitt,P., Fortes,M., Riedel,N. and
Sahasrabudhe,S. (1996) Proc. Natl Acad. Sci. USA, 93, 2065–2070.Inestrosa,N., Alvarez,A., Perez,C., Moreno,R., Vicente,M., Linker,C.,
Casanueva,O., Soto,C. and Garrido,J. (1996) Neuron, 16, 881–891.Iversen,L., Mortishire-Smith,R., Pollack,S. and Shearman,M. (1995) Biochem.
J., 311, 1–16.Jarrett,J., Berger,E. and Lansbury,P. (1993) Biochemistry, 32, 4693–4697.Jorgensen,W., Chandrasekhar,J., Madura,J., Impley,R. and Klein,M. (1983)
J. Chem. Phys., 79, 926–953.Kirschner,D., Abraham,C. and Selkoe,D. (1986) Proc. Natl Acad. Sci. USA,
83, 503–507.Kirschner,D., Inouye,H., Duffy,L., Sinclair,A., Lind,M. and Selkoe,D. (1987).
Proc. Natl Acad. Sci. USA, 84, 6953–6957.Kishan,R., Zeelen,J., Noble,M., Borchert,T., Mainfroid,V., Goraj,K., Martial,J.
and Wierenga,R. (1994) Protein Engng, 7, 945–951.Klunk,W.E., Pettegrew,J.W. and Abraham,D.J. (1989) J. Histochem.
Cytochem., 37, 1293–1297.Laskowski,R., MacArthur,M., Moss,D. and Thornton,J. (1993) J. Appl.
Crystallogr., 26, 283–291.Laurents,D., Subbiah,S. and Levitt,M. (1994) Protein Sci., 3, 1938–1944.Lee,J. et al. (1995) Biochemistry, 34, 5191–5200.Lolis,E., Alber,T., Davenport,R., Rose,D., Hartman,F. and Petsko,G. (1990)
Biochemistry, 28, 6609–6618.Mande,S., Mainfroid, V, Kalk,K., Goraj,K., Martial,J. and Hol,W. (1994)
Protein Sci., 5, 810–821.Masters,C. Simms,G., Weiman,N., Multhaup,G., McDonald,B. and
Beyreuther,K. (1985) Proc. Natl Acad. Sci. USA, 82, 4245–4249.Mori, H., Takio,K., Ogawara,M. and Selkoe,D. (1992) J. Biol. Chem., 267,
17082–17086.Noble,M., Verlinde,C., Groendijk,H., Kalk, KH., Wierenga,R. and Hol,W.
(1991) J. Med. Chem., 9, 2709–2718.Shomer,B., Harper,R. and Cameron,G. (1996) Methods Enzymol., 266, 3–27
(http://www.ebi.ac.uk/searchers/blitz_input.html).Smith,R. and Temple,S. (1992) Protein Engng, 5, 35–41 (http://dot.imagen.
bcm.tmc.edu:9331/multi-align/multi-align.html).Smith,T. and Waterman,M. (1981) J. Mol. Biol., 147, 195–197.Sorimachi,K. and Craik,D. (1994) Eur. J. Biochem., 219, 237–251.Soto,C., Branes,M., Alvarez,J. and Inestrosa,N. (1994) J. Neurochem., 63,
1191–1198.Soto,C., Castano,E. and Inestrosa,N. (1995) J. Biol. Chem., 270, 3063–3067.Sticht,H., Bayer,P., Willbold,D., Dames,S., Hilbich,C., Beyreuther,K., Frank,R.
and Rosch,P. (1995) Eur. J. Biochem., 233, 293–298.Talafous,J., Marcinowski,K., Klopman,G. and Zagorski,M. (1994)
Biochemistry, 33, 7788–7796.Weiner,S. and Kollman,P. (1981) J. Comput. Chem., 2, 287–303.Weiner,S. and Kollman,P. (1986) J. Comput. Chem., 7, 230–252.Zagorski,M. and Barrow,C. (1992) Biochemistry, 31, 5621–5631.Zhang,Z., Sugio,S., Komives,E., Liu,K., Knowles,J., Petsko,G. and Ringe,D.
(1994) Biochemistry, 10, 2830–2837.
Received October 19, 1998; revised July 20, 1999; accepted August 2, 1999
966





























