Embed Size (px)
Citation preview

Available online at www.sciencedirect.com
www.elsevier.com/locate/gca
Geochimica et Cosmochimica Acta 73 (2009) 5513–5527
Multi-metal contaminant dynamics in temporarily floodedsoil under sulfate limitation
Frank-Andreas Weber, Andreas Voegelin *, Ruben Kretzschmar
Institute of Biogeochemistry and Pollutant Dynamics, ETH Zurich, CHN, CH-8092 Zurich, Switzerland
Received 13 February 2009; accepted in revised form 15 June 2009; available online 21 June 2009
Abstract
In many river basins, floodplain soils have accumulated a variety of metal contaminants, which might be released duringperiods of flooding. We investigated the dynamics of copper, cadmium, lead, zinc, and nickel in a contaminated freshwaterfloodplain soil under a realistic sulfate-limited flooding regime in microcosm experiments. We found that most contaminantswere initially mobilized by processes driven by the reductive dissolution of Fe(III) and Mn(IV, III) (hydr)oxides. Subse-quently, bacterial sulfate respiration resulted in the transformation of the entire available sulfate (2.3 mmol/kg) into chromousreducible sulfur (CRS). Cu K-edge X-ray absorption fine structure (XAFS) spectroscopy revealed that the soil Cu speciationchanged from predominantly Cu(II) bound to soil organic matter (SOM) intermittently to 14% metallic Cu(0) and subse-quently to 66% copper sulfide (CuxS). These CuxS precipitates accounted for most of the formed CRS, suggesting that CuxSwas the dominant sulfide phase formed in the flooded soil. Sequential metal extractions, in agreement with CRS results, sug-gested that easily mobilizable Cd was completely and Pb partially sequestered in sulfide precipitates, controlling their dis-solved concentrations to below detection limits. In contrast, Zn and Ni (as well as Fe) were hardly sequestered into sulfidephases, so that micromolar levels of dissolved Zn and Ni (and millimolar dissolved Fe(II)) persisted in the reduced soil.The finding that Cu, Cd, and Pb were sequestered (but hardly any Zn, Ni, and Fe) is consistent with the thermodynamicallypredicted sulfide ladder following the increasing solubility products of the respective metal sulfides. The observation that Cdand Pb were sequestered in sulfides despite the presence of remaining SOM-bound Cu(II) suggested that the kinetics of Cu(II)desorption, diffusion, and/or CuxS precipitation interfered with the sulfide ladder. We conclude that the dynamics of multiplemetal contaminants are intimately coupled under sulfate limitation by the relative thermodynamic stabilities and formationkinetics of the respective metal sulfides.� 2009 Elsevier Ltd. All rights reserved.
1. INTRODUCTION
Riparian floodplain soils are often polluted with multi-ple metal contaminants that were discharged upstream bymining and smelting, manufacturing, agriculture, and mu-nicipal sources (Hochella et al., 2005; Hudson-Edwardset al., 1996; Klemm et al., 2005; Schroder et al., 2008;Wallschlager et al., 1998). The bioavailability and mobilityof the accumulated contaminants are a concern for the
0016-7037/$ - see front matter � 2009 Elsevier Ltd. All rights reserved.
doi:10.1016/j.gca.2009.06.011
* Corresponding author. Present address: Eawag, Swiss FederalInstitute of Aquatic Science and Technology, Ueberlandstrasse133, CH-8600 Duebendorf, Switzerland. Fax: +41 44 823 52 10.
E-mail address: [email protected] (A. Voegelin).
floodplain ecosystem and for downstream surface- andground-water quality. Periodic overbank flooding and fluc-tuating groundwater tables can temporarily limit O2 diffu-sion into the soil and cause microbial communities torespire alternative terminal electron acceptors, such asFe(III) (hydr)oxides and sulfate (Ponnamperuma, 1972).Risk assessment requires an improved understanding ofthe processes that control contaminant dynamics in suchperiodically flooded soils, also in the context of currentplans to re-expand floodplain areas for flood control andriver revitalization (Moss and Monstadt, 2008).
Processes known to affect contaminant mobility andbioavailability in flooded soil include changes in contami-nant redox speciation (e.g., reduction of As(V) to As(III);

5514 F.-A. Weber et al. / Geochimica et Cosmochimica Acta 73 (2009) 5513–5527
Masscheleyn et al., 1991), reductive dissolution of sorbentphases (e.g., Fe(III) (hydr)oxides; Zachara et al., 2001),and sequestration into newly forming mineral phases(e.g., green rust; Parmar et al., 2001). Under sulfate-reduc-ing conditions, metal sequestration in sparingly soluble me-tal sulfide precipitates can be the dominant processcontrolling the dynamics of chalcophile metal contaminants(Di Toro et al., 1992; Kirk, 2004; Reddy and DeLaune,2008). Metal sulfide formation is often inferred indirectlyfrom a concomitant drop of dissolved metal and sulfateconcentrations, and little direct evidence has been reportedwith respect to riparian floodplain soils. Barnett et al.(1997) identified HgS particles in oxic floodplain soil byelectron microscopy and argued that these particles wereremnants having formed during previous flooding events.Using X-ray absorption fine structure (XAFS) spectros-copy, Bostick et al. (2001) suggested the formation ofZnS and ZnCO3 phases in contaminated floodplain soilsand correlated Zn speciation with redox conditions. Pootet al. (2007) measured acid-volatile sulfide (AVS) in twofloodplain soils during an annual cycle and found that theamount of AVS formed was insufficient to bind all simulta-neously extractable metals.
In comparison to metal sulfide formation in marine envi-ronments (e.g., Boulegue et al., 1982; Huerta-Diaz andMorse, 1992; Luther et al., 1980; Morse and Luther,1999; O’Day et al., 2000) and in mining ponds (Martinet al., 2003), the amount of sulfate available for bacterialsulfate respiration in freshwater floodplain soils is moderate(Giblin and Wieder, 1992). Dissolved and adsorbed sulfatecomprises typically only a small percentage of the total sul-fur content, whereas sulfur bound in soil organic matter(SOM) dominates the sulfur speciation (Giblin and Wieder,1992). Considering published mineralization rates (e.g.,Tabatabai and Al-Khafaji, 1980), mineralization of SOM-bound sulfur might be too slow to contribute significantamounts of sulfur for metal sulfide formation within thetime frame of a flooding event. River and groundwater infil-tration and atmospheric sulfur deposition can thus domi-nate the sulfate input to the soil (Lamers et al., 1998).
In many highly-polluted riparian floodplain soils, theavailable sulfate might limit the extent to which the accu-mulated contaminants can be sequestered in metal sulfideprecipitates. Knowledge on which metal contaminants aresequestered under sulfate limitation – and which arenot – is of great importance for predicting multi-metal con-taminant dynamics in flooded soil. When sulfide is slowlyreleased into a multiple metal-containing solution, a ther-modynamic equilibrium model (e.g., Druschel et al., 2002)predicts that instantaneous precipitation of the most insol-uble metal sulfide buffers the activity of aqueous sulfide be-low the saturation of more soluble metal sulfide minerals.After the metal that forms the most insoluble metal sulfideis largely consumed, the activity of aqueous sulfide rises un-til buffered by the precipitation of the next insoluble metalsulfide. This equilibrium model thus predicts the sequentialformation of monomineralic sulfide precipitates until theavailable sulfate is consumed. For dynamic systems suchas soils and sediments, however, Druschel et al. (2002) havepointed out that if metal supply or precipitation kinetics are
slow compared to the rate of bacterial sulfate respiration,then the porewater may become locally supersaturated withrespect to multiple metal sulfide minerals, potentially result-ing in the sequestration of contaminants that form moresoluble sulfide precipitates.
In this study, we investigated the processes that controlCu, Cd, Pb, Zn, and Ni dynamics during 7 weeks of flood-ing of a contaminated floodplain soil. A realistic sulfate-limited flooding regime was simulated in microcosms toelucidate to which extent contaminants were controlled bysulfide formation. The microcosm setup allowed us to mon-itor porewater dynamics along with transformations of so-lid-phase speciation using Cu K-edge XAFS spectroscopy,sequential metal extractions, and chromous reducible sulfur(CRS) distillation. The presented results expand our previ-ous investigation on the formation and mobility of metallicCu(0) and metal sulfide colloids in reduced porewater con-ducted in the same microcosm setup (Weber et al., 2009).
2. EXPERIMENTAL
2.1. Soil sampling and field site description
Within 15 m distance from the river bed, topsoil(0–15 cm) of a gleyic fluvisol was collected in January2004 in a floodplain of the river Mulde at 51�39025.100N,012�19049.400E near Muldenstein, Saxony-Anhalt, Germany(Voegelin et al., 2007; Weber et al., 2009). The site is typi-cally submerged by overbank flooding for periods of daysto weeks within a year. A centennial flood occurred in sum-mer 2002 (Geller et al., 2004). At the time of sampling, thesoil profile was well-drained and aerated to at least 150 cmdepth. The floodplain has been intermittently used for agri-culture and is now a nature reserve. The hydrological re-gime in the region has frequently been altered over thelast century by open-pit coal mining, relocation of the riverbed, and construction of weirs and dams. Like many flood-plains along the river, the soil is severely contaminatedmainly by emissions from past mining activities �150 kmupstream in the Erzgebirge (Ore Mountains), where sulfideores have been mined for Ag, Pb, Zn, and As since the Mid-dle Ages (Geller et al., 2004). Our sampling site is locatedon Transect I of a detailed monitoring campaign document-ing the dynamics of soil, porewater, and groundwater con-tamination between 1997 and 1999 (Brandt, 2003). Thiscampaign also monitored sulfate concentrations in the riverwater in the range of 0.8 to 1.4 mM and in the soil pore-water (sampling location via suction cups at 20 cm depth)in the range of 0.7 to 1.8 mM, the lower values during highflow and soil flooding. Moreover, atmospheric sulfur depo-sition to the region has declined considerably over the past20 years (Zimmermann et al., 2003), following the installa-tion of scrubbers to coal-fired power plants and transitionto less sulfur-containing fossil fuels.
2.2. Soil characterization
The topsoil was air-dried, passed through a 2-mm sieve,mixed homogenously, and stored in plastic containers. Formicrocosm experiments, soil aggregates larger than 200 lm

Multi-metal dynamics in temporarily flooded soil 5515
were collected by dry sieving. The moisture content of thestored soil was 6.5% based on weight-loss upon heating to105 �C for 2 d. All soil concentrations are reported relativeto this oven-dry soil mass. The soil carbon and nitrogencontents were measured using a CHNS Analyzer (CHNS-932; LECO, USA). The soil contents of most otherelements (including sulfur) were determined by energy-dis-persive X-ray fluorescence (XRF) spectrometry (X-Lab2000; Spectro, Germany). The mercury content was deter-mined by combustion atomic absorption spectrometry(AMA 254 Advanced Mercury Analyzer; LECO). X-raydiffraction (XRD) patterns were recorded on orientedMg-saturated (without and with glycerol solvation) andK-saturated (room temperature, 110, 300, and 550 �C)mounts of the soil clay fraction (D4, Bruker, USA; CuKa radiation). The effective cation exchange capacity(ECEC) was determined as the sum of cations displacedby an unbuffered 0.1 M BaCl2 solution (Hendershot andDuquette, 1986; see below). Chemical extractions were usedto estimate the amount of terminal electron acceptors avail-able for anaerobic microbial respiration. The pool of reac-tive Fe(III) (hydr)oxide available for Fe(III) respiration wastargeted by a 0.1 M ascorbic acid, 0.2 M sodium citratesolution buffered to pH 7.5 (Hyacinthe et al., 2006). Thesame extraction was used to estimate the pool of reactiveMn(IV, III) (hydr)oxides. The exchangeable nitrate and sul-fate content was determined by a 0.5 M sodium bicarbonateextraction at pH 8.5 (Kilmer and Nearpass, 1960). The oxicsoil was also characterized with all spectroscopic methodsand extraction procedures detailed below for comparisonwith incubated soil recovered from microcosm experiments.
2.3. Microcosm experiments
In series of independent microcosm experiments, air-dried soil was submerged with synthetic river water andincubated open to the atmosphere at �23 �C for 1–52 days.Details on the setup and porewater sampling procedurehave been described previously (Weber et al., 2009). Briefly,450 g soil (on air-dry basis) was equilibrated with 1500 mLof aerated synthetic river water (0.6 mM CaSO4, 0.6 mMNaCl, 0.3 mM Mg(NO3)2 solution) resembling the mainwater composition of the river Mulde during high flow inspring 1998 (Brandt, 2003). No external carbon sourcewas added to the soil. After the suspension was shakenend-over-end for 2 h under air, the suspension was centri-fuged and the supernatant was decanted, removing1.6 mmol of nitrate and 1.0 mmol of sulfate. The recoveredsoil paste was directly transferred into a microcosm andsubmerged with additional 500 mL of synthetic river water,starting the experiment. The resulting microcosm geometryconsisted of a water-saturated soil layer (height �9 cm;porewater-to-soil ratio U = 1.0 L/kg; total porosity 0.7)submerged under �6 cm of supernatant water. A suctioncup (sintered PE granulates with a nominal pore size of10–16 lm; EcoTech, Germany) was mounted horizontally�3.5 cm below the soil–supernatant interface.
The microcosm was sampled by withdrawing porewatervia the suction cup directly into an anoxic glovebox(pO2 < 1 ppm), where aliquots were filtered (0.025 lm
cellulose nitrate filter; NC03; Whatman, Germany) and pre-pared for further analyses. After porewater sampling,selected microcosms were frozen by immersion in liquidnitrogen. Incubated soil was recovered by cryo-slicing themicrocosms with a diamond saw horizontally 1.5 cm aboveand below the suction cup, obtaining a 3.0 cm-thick soillayer. A subsample of the frozen soil layer was allowed tothaw in the glovebox and used immediately as wet pastefor sequential metal extractions. Another subsample wasfreeze-dried and stored in the glovebox until analyzed byXAFS spectroscopy, CRS distillation, and XRF analysis.The XRF analyses of oxic and incubated soil confirmedthat the element content of the soil remained constant overthe course of the experiment, within the precision of themeasurement. This showed that, for each element consid-ered, the mass exchanged by diffusion from and to theinvestigated soil layer was negligible compared to its so-lid-phase inventory.
2.4. Porewater analyses
Dissolved concentrations were determined in 0.025 lm-filtered porewater aliquots. Dissolved metals and dissolvedsulfur were analyzed by inductively-coupled plasma opticalemission spectrometry (ICP-OES; Vista MPX; Varian,USA). Cd concentrations were linearly corrected for a min-or Fe interference. Dissolved Cu and Pb were measured bygraphite furnace atomic absorption spectrometry (GFAAS)with Zeeman background correction (AA240Z, Varian).One outlier in the measured Pb concentration at day 10was disregarded. Additional porewater aliquots were usedfor the determination of pH and EH (Minitrode for pH;Slimtrode for EH; Hamilton, USA) and for analyses of sul-fate by ion chromatography with suppressed conductivitydetection (Metrosep A Supp. 5 column; Metrohm, Switzer-land), dissolved organic carbon and inorganic carbon (bothTOC-5000; Shimadzu, Japan), and low molecular weightorganic acids by ion exclusion chromatography (MetrohmMetrosep column). On selected samples, Fe(II) and ammo-nium were measured photometrically using the phenanthro-line (Fadrus and Maly, 1975) and the phenate method(Weatherburn, 1967), respectively. Dissolved sulfide wasmonitored photometrically after derivatization to formmethylene blue (Cline, 1969). The detection limit of ourmethylene blue method in 6-fold diluted porewater was8 lM (five times the standard deviation of a blank). Thedissolved concentration of each compound X (i.e., [X]aq)is expressed in mole per liter porewater, which can be con-verted to concentration relative to dry soil mass (i.e.,U � [X]aq) by the porewater-to-soil ratio U, which was1.0 L/kg in our experiments.
2.5. X-ray absorption fine structure spectroscopy
Cu K-edge X-ray absorption near-edge structure(XANES) and extended X-ray absorption fine-structure(EXAFS) spectra were recorded at the XAS beamline atthe Angstromquelle Karlsruhe (ANKA, Karlsruhe,Germany). The beamline was equipped with a double crys-tal Si(111) monochromator, which was detuned to 65% of

5516 F.-A. Weber et al. / Geochimica et Cosmochimica Acta 73 (2009) 5513–5527
the maximum intensity using a software-controlled mono-chromator stabilization. For energy calibration, the firstinflection of the K-edge of metallic Cu foil was set to8979 eV. Soil samples were transported under N2 atmo-sphere to the beamline, where they were pressed into pelletsdirectly before measurement. Spectra of soil samples wererecorded in fluorescence mode using a 5-element Ge solidstate detector under air at room temperature. Referencespectra were measured in transmission.
Data extraction and analysis was carried out using thesoftware code Athena (Ravel and Newville, 2005). For dataextraction, E0 was fixed to 8981 eV. Normalized XANESspectra were obtained by subtracting a first-order polyno-mial fit to the pre-edge region (�70 to �20 eV) and subse-quently dividing by a second-order polynomial fit to thepost-edge region (30–300 eV). Linear combination fitting(LCF) was carried out over the energy range �20 to60 eV. The background spline for the extraction of EXAFSdata was adjusted using the Autobk algorithm implementedin Athena (Rbkg = 0.9; k-weight = 3; k-range 0.5–11 A�1).LCF of EXAFS spectra was constrained to the k-range3–8.5 A�1. For both LCF of XANES and EXAFS spectra,each fitted fraction was constrained between 0 and 1, butthe sum of all fractions was not constrained.
A variety of Cu references were considered for LCF,including metallic Cu(0) foil, chalcopyrite (‘‘CuFeS2”; AlfaAesar No. 42533), freshly-precipitated primitive copper sul-fide (‘‘CuxS”; sample ‘‘2n – blue/black primitive” in Pat-trick et al., 1997; spectrum kindly provided by FredMosselmans, Diamond Light Source, Didcot, UK), andCu(II) bound to Suwannee river natural organic matter(spectrum ‘‘Cu(II)–SRN” in Karlsson et al., 2006; spectrumkindly provided by Ulf Skyllberg, Swedish University ofAgricultural Sciences, Umea, Sweden). Previous EXAFSanalysis of the Cu(II)–SRN reference showed that Cu(II)is coordinated to 4 O/N atoms, and no indications werefound for Cu bound to S atoms at the molar Cu/reduced-organic-S ratio of 1.0 and Cu/organic-C ratio of 0.0013of the sample (Karlsson et al., 2006). Since the bulk Cu/or-ganic-C ratio of the soil was 0.0006, we considered theCu(II)–SRN reference a suitable proxy for Cu(II) boundto SOM in the studied soil.
2.6. Sequential metal extractions
The dynamics of metal partitioning during flooding wasstudied by sequential extractions of oxic and incubated soil,where [X]extractant refers to the extracted amount of elementX, expressed relative to dry soil weight. Soluble andexchangeable cations were targeted by an unbuffered0.1 M BaCl2 extraction ([X]BaCl2; Hendershot and Du-quette, 1986), followed by a 1 M sodium acetate extractionat pH 5 targeting specifically adsorbed cations as well ascarbonate and other minerals that are labile at pH 5([X]Acet; Jacquat et al., 2008; Poulton and Canfield, 2005;Tessier et al., 1979). Metal sulfide minerals are hardly at-tacked by this extraction sequence, as demonstrated forZnS spiked to a wetland soil (Peltier et al., 2005).
Incubated soil was extracted in duplicates under the N2
atmosphere of the glovebox using wet soil paste and
deoxygenated extractant in order to prevent artifactscaused by freeze-drying and/or sample oxidation (Hjorth,2004). The oxic soil was extracted in triplicates under air.1 g of soil (dry-weight basis) was extracted twice with40 mL of extractant by shaking suspensions end-over-endat room temperature (2 h for the BaCl2 and 5 h for the ace-tate extraction). Suspensions were centrifuged and decantedsupernatants were filtered through 0.45 lm-nylon filters(Opti-Flow; Wicom, Germany). Element concentrations inthe combined extract were determined by ICP-OES usingmulti-element standards adjusted to match the matrix ofthe respective extract. Duplicate or triplicate extractionstypically agreed within 5%.
ECEC was calculated as the sum of BaCl2-exchangeablecations (Hendershot and Duquette, 1986) after subtractingthe dissolved pool according to ECEC =
Pi ([Xi]BaCl2 �
U � [Xi]aq) � zXi, where zXi is the valence of element Xi,and Xi comprising Ca, Mg, Fe, Mn, Na, K, Al, Cu, Cd,Pb, Zn, Ni, and NH4. Exchangeable protons were not in-cluded in the ECEC determination. Extracted Fe and Mnwere assumed to be dominantly divalent at the pH range5.5–6.7. The amount of BaCl2-extractable ammonium wasestimated from the measured dissolved NH4
+ concentra-tion (i.e., [NH4]aq,) assuming a sorption affinity similar tothat of potassium, i.e., [NH4]BaCl2 = ([K]BaCl2 � U �[K]aq) � (U � [K]aq)�1 � [NH4]aq.
2.7. Soil sulfur speciation
Exchangeable sulfate was determined in oxic and incu-bated soil samples by bicarbonate extraction (Kilmer andNearpass, 1960). 5 g of soil samples was extracted twicewith 40 mL of 0.5 M sodium bicarbonate solution adjustedto pH 8.5 by NaOH, shaking suspensions end-over-end for1 h under N2 atmosphere. The supernatant decanted aftercentrifugation was filtered and analyzed by ionchromatography.
Acid-volatile sulfide (AVS) and chromous reducible sul-fur (CRS) were determined adapting the distillation methodof Canfield et al. (1986). After purging the distillation appa-ratus free of air, 6–20 g of freeze-dried soil was acidified byinjecting 60 mL of 5 M HCl in a heated digestion flask. H2Sgas liberated in this AVS step was trapped in a 0.1 MNaOH absorber during 40 min and then determined byiodometric titration. In the CRS step, 40 mL of 1 M CrCl2solution (chromium(II) chloride anhydrous (Alfa Aesar)dissolved in 5 M HCl just before use) was subsequently in-jected to react with the boiling soil suspension, resulting in aCr(II)-to-total-sulfur molar ratio of �102. Liberated H2Strapped in a new 0.1 M NaOH absorber during 60 minwas again determined iodometrically. Freeze-dried andwet soil extractions yielded similar results. Tests were per-formed to exclude that reduction of sulfate and/or organicsulfur species contributed to the measured CRS content, aninterference that has been demonstrated to become signifi-cant at Cr(II)/S ratio >104 (Mylon et al., 2002). We spiked70 mmol/kg of S either as CaSO4�2H2O or as (R)-(+)-cys-teine (both Merck) to the oxic soil (nearly doubling its totalS content) but observed no increase in CRS compared tothe unspiked soil within the precision of the method.

Multi-metal dynamics in temporarily flooded soil 5517
Additional tests were conducted to examine the effect ofdifferent matrixes on the recovery of CdS (‘‘cadmium sul-fide”; Aldrich, USA), which is expected to dissolve in theAVS step (Cooper and Morse, 1998). When 100 lmol ofCdS was spiked into 10 g of quartz sand, the amount of sul-fide released in the AVS step accounted for 74% of the ex-tracted Cd. However, when the same amount of CdS wasspiked into 10 g of oxic soil, sulfide release in the AVS stepwas minor (<2%), but release in the CRS step accounted for55% of the extracted Cd. The poor AVS recovery is consis-tent with observations by Wilkin and Ford (2002), who sug-gested that H2S liberated in the AVS step reacts with As orCu to form acid-insoluble arsenic or copper sulfides. Ourresults showed that the sequestered sulfide was then (par-tially) liberated in the CRS step.
Based on these tests, we cannot exclude that the AVSmeasurements of the incubated soil (which were always<0.5 mmol-S/kg) were impaired by sulfide re-sequestration.We therefore report only the sum of CRS + AVS as mea-sure of the reduced inorganic sulfur in the soil. The incom-plete recovery requires that the CRS + AVS results areinterpreted semi-quantitatively.
2.8. Construction of Cu–S–citrate–chloride–H2O pH–EH
diagram
Geochemical modeling of the system Cu–S–citrate–chlo-ride–H2O was performed using the Eh-pH module of HSCchemistry (Haung and Cuentas, 1989). Tabulated values ofthe free energy of formation of aqueous Cu and S species weretaken from Bard et al. (1985). The selection of CuxS mineralsthat were allowed to precipitate was restricted to the stablephases covellite (CuS), chalcocite (Cu2S), djurleite (Cu1.9S),and anilite (Cu1.75S), for which the respective free energy offormation were taken from Thompson and Helz (1994) andfrom Potter (1977) as adjusted by Shea and Helz (1989).Stability constants on Cu(II) hydroxyl, chloride, and citratecomplexation, on citrate protonation, on Cu(I) chloride com-plexation, as well as solubility products of tenorite (CuO) andcuprite (Cu2O) were taken from version 4 of the MINTEQdatabase (Allison et al., 1990). The formation of aqueousCu(I) sulfide and polysulfide complexes was included in themodel following the approach of Wang and Tessier (acceptedfor publication), but was found to have little effect on thepredominant Cu speciation. The compiled thermodynamicdatabase is listed in Electronic Annex, Table EA1.
System parameters were chosen to resemble conditionsrepresentative for the studied soil. Cu and S concentrationswere set to the measured total soil Cu content and to theexchangeable sulfate content, respectively, assuming thatboth were completely available in the porewater (i.e.,P
Cu = 4.39 mM;P
S = 2.8 mM). Cu(II) complexationby citrate served as model for complexation by SOM,assuming 1 mmol of copper-complexing functional groupsper g of soil carbon (i.e.,
Pcitrate = 96 mM). Citrate is a
crude proxy for SOM in terms of protonation behaviorand Cu(II) binding strength, but the predominant Cu speci-ation was relatively insensitive to reasonable variations ofcitrate-associated input parameters. Chloride was set tothe measured porewater concentration (0.8 mM).
3. RESULTS
3.1. Characterization of the oxic soil
The studied soil was acidic, rich in organic matter, andpolluted by a variety of metal contaminants (Brandt,2003; Voegelin et al., 2007; Weber et al., 2009). Soil proper-ties are listed in Table 1. The ECEC of the oxic soil was280 mmolc/kg. Chemical extractions indicated that signifi-cant fractions of the Cu, Cd, Pb, Zn, and Ni contents wereeasily mobilizable, here defined as the sum of BaCl2- andacetate-extractable pools (Table 1). Previous work usingCu K-edge XANES spectroscopy suggested that the Cuspeciation in the oxic soil was dominated by Cu(II) boundto SOM (Weber et al., 2009). Also As was an important soilcontaminant (Table 1), which occurred mainly as As(V)sorbed to Fe(III) (hydr)oxides (Voegelin et al., 2007; Weberet al., 2008).
Based on chemical extractions, reactive Fe(III) (hydr)-oxides comprised the largest pool of terminal electronacceptors available for anaerobic respiration (Table 1).The exchangeable sulfate content was small in comparison(2.3 mmol/kg), and accounted for only 3% of the total sul-fur content. Sulfur bound in SOM likely dominated theremaining sulfur, considering the soil organic C contentof 96 g/kg and a typical SOM molar C/S ratio of �100(Tabatabai and Bremner, 1972). In addition, we detectedthe presence of 2.9 mmol/kg CRS + AVS, which we havetested not to arise from interference of sulfate and/or or-ganic sulfur compounds. Extracting elemental sulfur fromthe soil with dichloromethane did not alter its CRS + AVScontent, indicating that elemental sulfur was also not a sig-nificant CRS + AVS constituent. The presence of thisamount of copper sulfide or arsenic sulfide minerals canbe excluded based on XAFS results (Voegelin et al., 2007;Weber et al., 2009). Most other chalcophile metal contam-inants occurred in concentrations that are insufficient to ex-plain the measured CRS + AVS content (Table 1). Incontrast, the CRS + AVS content could be explained if lessthan 1% of total Fe were present as pyrite (FeS2). We attri-bute the CRS + AVS of the oxic soil to oxidation-resistantprimary sulfide minerals, which were presumably erodedupstream in the Ore mountains and deposited in the flood-plain upon flooding.
3.2. Flooding and soil reduction
In the applied flooding regime, the aerated floodwatercontributed additional amounts of terminal electron accep-tors to the soil, including net 0.5 mmol/kg of sulfate. In thefollowing, we monitored flooding-induced transformationsboth in the porewater (left panels of Fig. 1) and in the solidphase (right panels of Fig. 1) within a soil layer �3.5 cm be-low the soil–supernatant interface.
3.2.1. Porewater dynamics
Dissolved concentrations followed the typical dynamicscaused by respiration of a sequence of terminal electronacceptors (Ponnamperuma, 1972; Kirk, 2004; Reddy andDeLaune, 2008). Dissolved O2 and nitrate were consumed

Table 1Characterization of the oxic floodplain soil used for microcosm experiments.
Soil classification Gleyic fluvisolSampled horizon Ah (0�15 cm)Texture silt-loamClay mineralogy kaolinite, illite, vermiculite, chlorite, hydroxy-interlayered mineralspHCaCl2 5.5ECEC (mmolc/kg) 280Organic C (g/kg) 96C/N molar ratio 16C/S molar ratio 99
Main elements Total concentrationa Available for anaerobic respirationb log Ksp0 of sulfide mineralc
(mmol/kg) (mmol/kg) (% of total)
N 499 2.8 (1) n.a.Mn 18 10 (58) 0.2Fe 830 189 (23) �3.6S 80 2.3 (3) n.a.
Chalcophilecontaminants
Total concentrationa Easily mobilizabled log Ksp0 of sulfide mineralc
(mmol/kg) (mmol/kg) (% of total)
Hg 0.01 n.d. �45.7Ag 0.05 n.d. �36.2Cu 4.39 0.46 (11) �22.3Cd 0.27 0.19 (73) �14.4Pb 1.97 0.43 (22) �14.0Zn 20.95 4.01 (19) �11.5Ni 1.80 0.23 (13) �5.6As 4.32 3.20 (74) �61.1
a Mean of four replicates. Standard deviation was 67% for each element.b Determined as bicarbonate-extractable nitrate and sulfate and ascorbate-extractable Mn and Fe.c Selected solubility products are reported for alabanite (MnS), mackinawite (FexS), covellite (CuS), greenockite (CdS), galena (PbS),
sphalerite (ZnS), and millerite (NiS), each for the equilibrium MeS(s) + H+M Me2+
(aq) + HS�(aq); for cinnabar (HgS(s) + 2H2O M Hg(O-H)2(aq) + H+ + HS�(aq)); for acanthite (Ag2S(s) + H+
M 2Ag+(aq) + HS�(aq)); and for orpiment (As2S3(s) + 6H2O M 2H3A-
sO3(aq) + 3HS�(aq) + 3H+), respectively. Data are from the Minteq.v4 database (Allison et al., 1990). See Thoenen (1999) for a criticalreview of published solubility products of NiS. n.a., not applicable.
d Determined as sum of BaCl2- and acetate-extractable metals. For As, the ascorbate-extractable pool is given. n.d., not determined.
5518 F.-A. Weber et al. / Geochimica et Cosmochimica Acta 73 (2009) 5513–5527
within less than a day (data not shown). Reductive dissolu-tion of Mn(IV, III) and Fe(III) (hydr)oxides resulted in sol-ubilization of Mn (Fig. 1b) and Fe (Fig. 1d). Dissolved Fewas confirmed to be mainly Fe(II) within the precision ofthe phenantroline method. Dissolved sulfate was consumedbetween day 4 and 10 (Fig. 1f). During soil reduction, pore-water pH rose from 5.7 to 6.7 and the EH dropped to�0 mV (Fig. 1a), concurrently to the accumulation of ace-tate, propionate, and inorganic carbon as products ofmicrobial respiration (Fig. 1h). The concentration ofunspecified dissolved organic carbon (i.e., total DOC aftersubtracting acetate- and propionate-carbon) doubled with-in the first 15 d of flooding (Fig. 1h), indicating that someSOM compounds were released to the porewater as dis-solved organic matter (DOM). Dissolved sulfur that wasnot sulfate (termed unspecified sulfur in Fig. 1f) presumablycomprised mainly DOM-bound sulfur (constant unspecificDOC/S ratio �80), with potentially some contributions ofinorganic sulfur species, such as thiosulfate and elementalsulfur. In parallel to dissolved Fe, Mn, and unspecifiedDOC, the dissolved concentrations of the alkaline earthmetals Ca, Mg, and Sr tripled in the porewater of theflooded soil (Fig. EA1 in the Electronic Annex).
3.2.2. Solid-phase Mn and Fe dynamics
Sequential extractions revealed the build-up of BaCl2-and acetate-extractable Mn and Fe pools in the soil(Fig. 1c and e), coinciding with the accumulation of dis-solved Mn and Fe in the porewater (Fig. 1b and d). Trans-formations in soil Mn speciation during flooding weremonitored by Mn K-edge XAFS spectroscopy (Fig. EA2).The recorded soil spectra suggested that [Mn]BaCl2 wasdominated by exchangeable Mn(II), whereas [Mn]Acet wasmainly specifically sorbed Mn(II) with minor, if any, contri-bution of Mn(II) carbonate precipitates. The accumulationof BaCl2- and acetate-extractable Mn leveled off after 15 dof flooding (Fig. 1c). At this time, the sum of [Mn]BaCl2 and[Mn]Acet reached the amount of reactive Mn(IV, III) (hy-dr)oxides initially present in the oxic soil (Table 1), suggest-ing that the pool of reactive Mn(IV, III) (hydr)oxides wasconsumed until depletion.
In analogy to the observed Mn transformations, we ex-pect that [Fe]BaCl2 contained mainly exchangeable Fe(II),while [Fe]Acet contained specifically sorbed Fe(II) andpotentially Fe(II) carbonate phases and/or green rust, ifpresent. Within 52 d of incubation, the sum of[Fe]BaCl2 and [Fe]Acet gradually approached – but did not

5.56.06.57.0
0200400600
oxic 0 10 20 30 40 500
10
20
30
40
0.0
0.2
0.4
0.6
0.8
0
2
4
6
0
50
100
150
200
0%
25%
50%
75%
100%
0%
10%
20%
30%
0.0
0.3
0.6
0.9
1.2
0.0
0.3
0.6
0.9
1.2
01
23
45
0%
2%
4%
6%
oxic 0 10 20 30 40 500
100
200
300
400
0%
50%
100%
150%
apH
E H(mV)
h
Time after flooding (days)
Carbon(mM)
unsp
ecifi
edD
OC
acet
ate
prop
iona
tein
orga
nic
carb
on
b
Mn a
q(mM)
d
Feaq(mM)
0
4
8
12
16Non-ascorbate extractable Mn
c
MnBaCl2
Mn(mmol/kg)
MnAcet
e
FeBaCl2
FeAcet
Fe(mmol/kg)
Non-ascorbate extractable Fe
Mn(%
oftotal)
Fe(%
oftotal)
f
Sulfate(mM)
Unspec.Sulfur(mM)
gSulfateSulfur(mmol/kg)
CRS+ AVS
Sulfur(%oftotal)
Left panels:Right panels:
Porewater dynamicsSoil dynamics
iMnFe
others
CaMg
NH4
Time after flooding (days)
ECEC
(mmol
c/kg)
ECEC
(%ofoxicsoil)
Fig. 1. Porewater dynamics (left panels) and soil dynamics (right panels) of major soil constituents during flooding: (a) porewater pH and EH;(b–c) dissolved and soil-extractable Mn; (d–e) dissolved and soil-extractable Fe; (f) dissolved sulfur; (e) soil CRS + AVS and soil-extractablesulfate; (h) dissolved carbon; (i) ECEC. Each point in time corresponds to an independent microcosm experiment. Dissolved data for the oxicsoil represent 2-h oxic soil extracts with synthetic river water. The error bars in (g) represent one standard deviation of duplicate determinationof CRS + AVS. Duplicate determination of soil-extractable sulfate as well as BaCl2- and acetate-extractable Mn and Fe agreed typicallywithin 5% (not shown). Lines are meant to guide the eye. The porewater-to-soil ratio was 1.0 L/kg. pH, EH, and dissolved Mn, Fe, and sulfateare from Weber et al. (2009).
Multi-metal dynamics in temporarily flooded soil 5519
reach – the amount of reactive Fe(III) (hydr)oxide initiallypresent in the oxic soil (Fig. 1e). This suggested that thepool of reactive Fe(III) (hydr)oxides supported Fe(III)respiration over the entire course of the experiment.
3.2.3. ECEC Increase during soil reduction
Results from BaCl2 extractions revealed that substantialamounts of cations were displaced from exchange siteswithin 52 d of flooding, including 12 mmol/kg of Ca(Fig. EA1), 4 mmol/kg of Mg (Fig. EA1), and in total3 mmol/kg of metal contaminants (mainly Zn; see below).However, the amount of metal cations displaced was sub-stantially less than the amount of Fe and Mn accumulatingin the BaCl2-extractable fraction (Fig. 1e and c), suggesting
an increase in the ECEC over soil reduction by 169 mmolc/kg or 60% compared to the ECEC of the oxic soil (Fig. 1i).A similar ECEC increase during soil reduction has previ-ously been observed for a Vertisol cropped with rice (Favreet al., 2002). Since porewater pH rose in our experimentsfrom 5.7 to 6.7 during flooding (Fig. 1a), a portion of theobserved ECEC increase resulted from the deprotonationof variable-charge functional groups of organic matterand mineral surfaces (Curtin and Rostad, 1997). Based ona linear regression relating ECEC to soil pH, clay content,and organic carbon content (Curtin and Rostad, 1997), thepH-effect was estimated to contribute 82 mmolc/kg to theincrease in ECEC. Considering the discrepancy to the ob-served increase of 169 mmolc/kg, additional processes

5520 F.-A. Weber et al. / Geochimica et Cosmochimica Acta 73 (2009) 5513–5527
may have contributed to the observed ECEC increase, suchas the microbial reduction of structural Fe in phyllosilicateminerals (Kostka et al., 1999) and/or the removal of Fe(III)(hydr)oxides coatings from clay minerals (Roth et al.,1969).
3.2.4. Solid-phase sulfur dynamics
Concomitant to dissolved sulfate (Fig. 1f), soil-extract-able sulfate was consumed within 9 d (Fig. 1g). Dissolvedsulfide was always below the detection limit (8 lM) of ourmethylene blue method, implying that any sulfide formedwas sequestered in solid phases. Soil extraction revealedthat the CRS + AVS content increased by �1.9 mmol/kgbetween day 1 and 9 (Fig. 1g). The increase in CRS + AVSnearly balanced the amount of exchangeable sulfate con-sumed, suggesting that the sulfide produced by sulfate res-piration had reacted almost quantitatively to form metalsulfide precipitates.
After exchangeable sulfate was consumed, theCRS + AVS content of the incubated soil remained aboutconstant within the precision of our measurement(Fig. 1g). This observation suggested that the large pool ofsulfur bound in SOM was recalcitrant to mineralizationand reduction to inorganic sulfide within 52 d of flooding.Therefore, the amount of inorganic sulfide available forthe formation of metal sulfide minerals was limited by theavailable sulfate in the soil. The exact amount of metal sul-fide formed is however somewhat uncertain. The formationof 1.9 mmol-S/kg of metal sulfides suggested by theCRS + AVS distillation is a lower estimate, consideringthe incomplete recovery of CdS obtained in control experi-ments. The overall mass balance for the microcosm ac-counted for 2.3 mmol/kg of sulfate in the soil plusadditional net 0.5 mmol/kg of sulfate introduced by thefloodwater, most of the latter residing in the supernatantwater. It is unknown whether this sulfate in the supernatantcontributed to metal sulfide formation over depth or justnear the soil–supernatant interface. Based on these consider-ations, we expect that the metal sulfides formed in the inves-tigated soil layer contained between 1.9 and 2.8 mmol-S/kg.
3.3. Contaminant dynamics during soil reduction
3.3.1. Copper
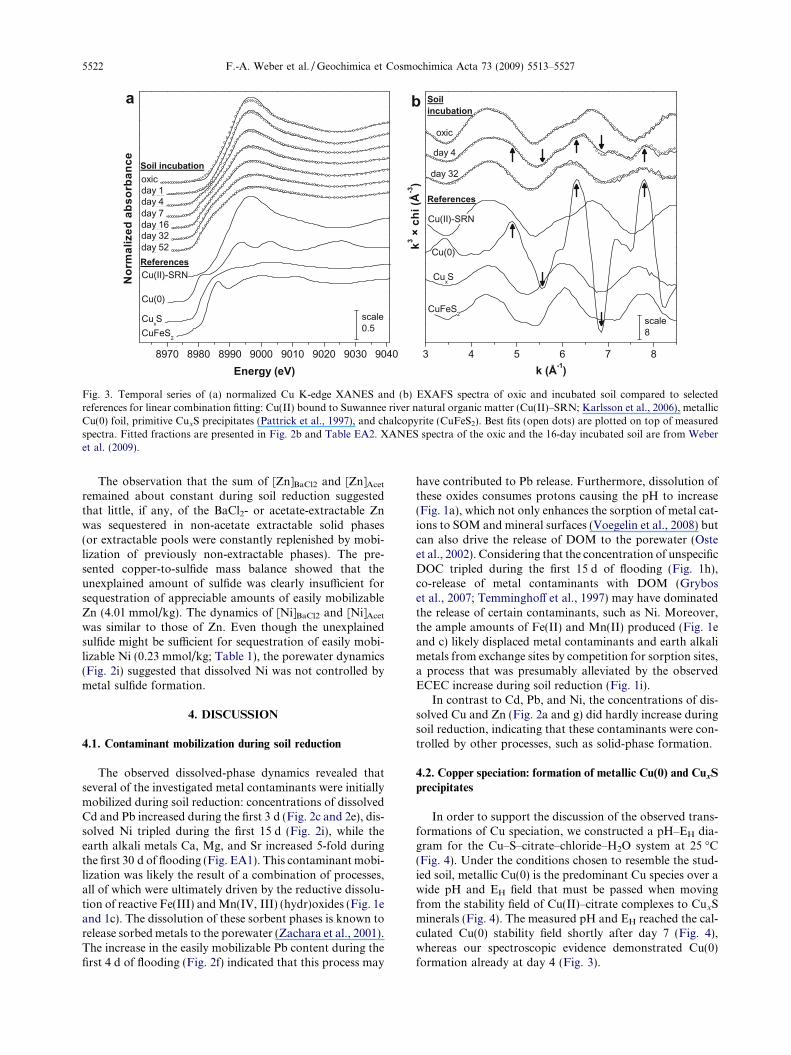
Dissolved Cu concentration declined gradually uponflooding (Fig. 2a). This depletion of dissolved Cu wasaccompanied by major transformations in solid-phase Cuspeciation as monitored by Cu K-edge XAFS spectroscopy(Fig. 3). LCF of the series of XANES (Fig. 3a) and EXAFS(Fig. 3b) spectra recorded on incubated soil at various timesafter flooding showed that all spectra were accurately de-scribed by a linear combination of only three references:Cu(II) bound to Suwannee river natural organic matter(Cu(II)–SRN) as proxy for Cu(II) bound to SOM, metallicCu(0) foil, and CuxS precipitates. The fitted fractions arepresented in Fig. 2b.
The XANES spectrum of the 1-day incubated soil clo-sely resembled the spectrum of the oxic soil (Fig. 3a), sug-gesting that Cu speciation remained predominantly Cu(II)bound to SOM. The XANES and EXAFS spectra of
4-day incubated soil deviated from the spectrum of the oxicsoil, showing characteristic features of metallic Cu(0)(Fig. 3b). LCF of both XANES and EXAFS spectra indi-cated the transformation of �14% of the soil Cu (equals0.6 mmol/kg) into metallic Cu(0) within 4 d of flooding(Fig. 2b). LCF of spectra recorded on soil incubated for7 d and longer showed that Cu(0) formation was only tran-sient and that the Cu(0) content in the soil declined duringprolonged flooding. Concurrently, the fitted fraction ofCu(I) sulfide precipitates (CuxS) increased gradually duringsoil reduction, accounting for 66% of the soil Cu content or2.9 mmol-Cu/kg at day 52 (Fig. 2b).
Previous EXAFS investigations demonstrated thatCu(II) sorbed to freshly-precipitated mackinawite trans-forms within 24 h into a chalcopyrite-like phase, whereasCu(II) on pyrite transforms into covellite or chalcocite(Parkman et al., 1999). Similarly, Simpson et al. (2000) sug-gested that during titration of an AVS-containing sedimentwith anoxic Cu(II) solution, FexS readily transforms intochalcocite. Considering the characteristic XANES spectrumof chalcopyrite in comparison to our soil spectra (Fig. 3a)indicated that mixed Cu–Fe-sulfides were not the dominantCu(I) sulfide phase in the reduced soil, in line with LCF re-sults (Table EA2). Therefore, the increasing fractions of fit-ted CuxS indicated the formation of distinct CuxSprecipitates during soil reduction.
The soil spectra did however not allow determining theexact stoichiometry of the CuxS precipitates formed.Assuming the stoichiometry of the end-members chalcocite(Cu2S) or covellite (CuS), the fitted CuxS fraction at day 52contained 1.4–2.9 mmol-S/kg. Alternatively, assuming thatthe CuxS precipitates had a primitive structure with a stoi-chiometry in the range between yarrowite and spionkopite(i.e., 1.12 6 x 6 1.39; Pattrick et al., 1997), the fitted CuxSfraction at day 52 contained 2.1–2.6 mmol-S/kg. In eitherscenario, the fitted amount of CuxS explained almost theentire amount of metal sulfides formed in the flooded soil(leaving 60.2 mmol-S/kg in the yarrowite and61.4 mmol-S/kg in the chalcocite scenario unexplained).This mass-balance calculation thus suggested that CuxSwas the dominant sulfide mineral formed in the flooded soil.
The results of sequential metal extractions were in agree-ment with the observed transformations of SOM-boundCu(II) into CuxS precipitates. The BaCl2- and acetate-extractable Cu, which accounted for 1% and 10% of totalCu in the oxic soil, respectively, decreased to negligibleamounts (<0.1%) at day 9 and thereafter.
3.3.2. Cadmium and lead
Dissolved Cd and Pb concentrations increased over thefirst days of flooding, but then dropped concurrently tothe onset of sulfate respiration (Fig. 2c and e). In analogyto Cu, the pools of BaCl2-exchangeable Cd and Pb were de-pleted within the first 9 d (Fig. 2d and f). In contrast to Cu,the pools of acetate-extractable Cd and Pb were highestaround day 9 and only gradually depleted thereafter.Whereas little [Cd]Acet remained after 52 d, the remaining[Pb]Acet still accounted for 21% of total Pb (Fig. 2d andf). The displaced amounts of Cd and Pb were not retrievedin the dissolved phase, implying that they were sequestered

0
1
2
3
4
0.0
0.1
0.2
0.3
0.4
0
1
2
3
4
0.0
0.1
0.2
0%
25%
50%
75%
100%
0%
25%
50%
75%
100%
0.0
0.1
0.2
0.0
0.3
0.6
0.9
0%
20%
40%
60%
0
5
10
15
20
0
2
4
6
8
0%
10%
20%
30%
40%
oxic 0 10 20 30 40 500
1
2
3
4
oxic 0 10 20 30 40 500.0
0.2
0.4
0.6
0%
10%
20%
30%
40%
a
Cu a
q(µM)
c
Cd a
q(µM)
bCu(II)-SOMCu(0)
LCFofXAFS
Cu(mmol/kg)
CuxS
CdAcetCdBaCl2
d
Seq.extractions
Cd(mmol/kg)
Cu(%
oftotal)
Cd(%
oftotal)
e
Pbaq(µM)
PbAcetPbBaCl2
f
Seq.extractions
Pb(mmol/kg)
Pb(%
oftotal)
g
Znaq(µM)
ZnAcetZnBaCl2
h
Seq.extractions
Zn(mmol/kg)
Zn(%
oftotal)
i
Time after flooding (days)
Ni aq(µM)
NiAcetNiBaCl2
j
Time after flooding (days)
Seq.extractions
Ni(mmol/kg)
Ni(%oftotal)
Fig. 2. Porewater dynamics (left panels) and soil dynamics (right panels) of major chalcophile metal contaminants during flooding: (a)dissolved Cu; (b) soil Cu speciation determined by Cu K-edge XAFS; dissolved and soil-extractable (c–d) Cd, (e–f) Pb, (g–h) Zn, and (i–j) Ni.Each point in time corresponds to an independent microcosm experiment. Error bars in (b) indicate the difference between LCF of XANESand EXAFS spectra. Error bars in (d), (f), (h), and (j) represent one standard deviation of duplicate or triplicate BaCl2- and acetateextractions. Dissolved Cu, Cd, and Pb dynamics are from Weber et al. (2009).
Multi-metal dynamics in temporarily flooded soil 5521
in non-acetate extractable solid phases. Based on the out-lined assumptions of the copper-to-sulfide mass balance,the amount of unexplained sulfide can be either sufficientor insufficient to account for the complete sequestrationof the easily mobilizable Cd (0.19 mmol/kg) and Pb(0.43 mmol/kg; Table 1) in sulfide minerals. The acetateextractions (Fig. 2d and f) suggested that Cd was nearlycompletely and Pb was only partially sequestered in metalsulfide phases.
3.3.3. Zinc and nickel
The dynamics of dissolved Zn and Ni were clearly dis-tinct from that of dissolved Cu, Cd, and Pb (Fig. 2, left
panels). The dissolved Zn concentration remained relativelyconstant at �10 lM over the entire course of the experi-ment (Fig. 2g), while the dissolved Ni concentration tripledwithin the first 15 d and then remained at �3 lM (Fig. 2i).In the solid phase, the acetate-extractable Zn and Ni poolsgradually increased at the expense of the BaCl2-extractablepools during soil reduction (Fig. 2h and j). This shift fromBaCl2- to acetate-extractable forms indicated that Zn andNi were sorbed more specifically in the reduced soil or werepartly incorporated in carbonate precipitates or otherphases that are labile at pH 5, such as layered doublehydroxides (LDH) and green rust (Jacquat et al., 2008;Parmar et al., 2001; Voegelin and Kretzschmar, 2005).
8970 8980 8990 9000 9010 9020 9030 9040 3 4 5 6 7 8
CuFeS2
CuxS
Cu(0)
Cu(II)-SRN
day 52day 32day 16day 7day 4day 1oxic
scale0.5
CuFeS2
CuxS
Cu(0)
Cu(II)-SRN
day 32
day 4
oxic
scale8
Fig. 3. Temporal series of (a) normalized Cu K-edge XANES and (b) EXAFS spectra of oxic and incubated soil compared to selectedreferences for linear combination fitting: Cu(II) bound to Suwannee river natural organic matter (Cu(II)–SRN; Karlsson et al., 2006), metallicCu(0) foil, primitive CuxS precipitates (Pattrick et al., 1997), and chalcopyrite (CuFeS2). Best fits (open dots) are plotted on top of measuredspectra. Fitted fractions are presented in Fig. 2b and Table EA2. XANES spectra of the oxic and the 16-day incubated soil are from Weberet al. (2009).
5522 F.-A. Weber et al. / Geochimica et Cosmochimica Acta 73 (2009) 5513–5527
The observation that the sum of [Zn]BaCl2 and [Zn]Acet
remained about constant during soil reduction suggestedthat little, if any, of the BaCl2- or acetate-extractable Znwas sequestered in non-acetate extractable solid phases(or extractable pools were constantly replenished by mobi-lization of previously non-extractable phases). The pre-sented copper-to-sulfide mass balance showed that theunexplained amount of sulfide was clearly insufficient forsequestration of appreciable amounts of easily mobilizableZn (4.01 mmol/kg). The dynamics of [Ni]BaCl2 and [Ni]Acet
was similar to those of Zn. Even though the unexplainedsulfide might be sufficient for sequestration of easily mobi-lizable Ni (0.23 mmol/kg; Table 1), the porewater dynamics(Fig. 2i) suggested that dissolved Ni was not controlled bymetal sulfide formation.
4. DISCUSSION
4.1. Contaminant mobilization during soil reduction
The observed dissolved-phase dynamics revealed thatseveral of the investigated metal contaminants were initiallymobilized during soil reduction: concentrations of dissolvedCd and Pb increased during the first 3 d (Fig. 2c and 2e), dis-solved Ni tripled during the first 15 d (Fig. 2i), while theearth alkali metals Ca, Mg, and Sr increased 5-fold duringthe first 30 d of flooding (Fig. EA1). This contaminant mobi-lization was likely the result of a combination of processes,all of which were ultimately driven by the reductive dissolu-tion of reactive Fe(III) and Mn(IV, III) (hydr)oxides (Fig. 1eand 1c). The dissolution of these sorbent phases is known torelease sorbed metals to the porewater (Zachara et al., 2001).The increase in the easily mobilizable Pb content during thefirst 4 d of flooding (Fig. 2f) indicated that this process may
have contributed to Pb release. Furthermore, dissolution ofthese oxides consumes protons causing the pH to increase(Fig. 1a), which not only enhances the sorption of metal cat-ions to SOM and mineral surfaces (Voegelin et al., 2008) butcan also drive the release of DOM to the porewater (Osteet al., 2002). Considering that the concentration of unspecificDOC tripled during the first 15 d of flooding (Fig. 1h),co-release of metal contaminants with DOM (Gryboset al., 2007; Temminghoff et al., 1997) may have dominatedthe release of certain contaminants, such as Ni. Moreover,the ample amounts of Fe(II) and Mn(II) produced (Fig. 1eand c) likely displaced metal contaminants and earth alkalimetals from exchange sites by competition for sorption sites,a process that was presumably alleviated by the observedECEC increase during soil reduction (Fig. 1i).
In contrast to Cd, Pb, and Ni, the concentrations of dis-solved Cu and Zn (Fig. 2a and g) did hardly increase duringsoil reduction, indicating that these contaminants were con-trolled by other processes, such as solid-phase formation.
4.2. Copper speciation: formation of metallic Cu(0) and CuxS
precipitates
In order to support the discussion of the observed trans-formations of Cu speciation, we constructed a pH–EH dia-gram for the Cu–S–citrate–chloride–H2O system at 25 �C(Fig. 4). Under the conditions chosen to resemble the stud-ied soil, metallic Cu(0) is the predominant Cu species over awide pH and EH field that must be passed when movingfrom the stability field of Cu(II)–citrate complexes to CuxSminerals (Fig. 4). The measured pH and EH reached the cal-culated Cu(0) stability field shortly after day 7 (Fig. 4),whereas our spectroscopic evidence demonstrated Cu(0)formation already at day 4 (Fig. 3).

4 5 6 7 8-200
-100
0
100
200
300
400
Djurleite (Cu1.9 S)
AniliteAnilite(Cu
1.75 S)
Cuprite(Cu2O)
5115
7
6
41
Cu(II)-citrate24-aqCu(II)-citrate-aq
Djurleite
Covellite (CuS)
Chalcocite (Cu2 S) Metallic
Cu(0)
Time
afterflooding(days)
E H(mV)
pH
Fig. 4. pH–EH diagram showing measured pH and EH dynamicsduring flooding (�) plotted over fields of predominant Cu species inthe Cu–S–citrate–chloride–H2O system for
PCu = 4.39 mM,P
S = 2.8 mM,P
citrate = 96 mM, andP
Cl = 0.8 mM at 25 �C.These conditions were chosen to be representative for the studiedsoil as described in Section 2.8. The free energies of formation of allconsidered species are listed in Table EA1.
Multi-metal dynamics in temporarily flooded soil 5523
Metallic Cu(0) has long been known to occur in bogsand fens (Lovering, 1928; Lett and Fletcher, 1980). Re-cently, Cu(0) nanoparticles were observed in a contami-nated stream using electron diffraction (Genovese andMellini, 2007) and in the vicinity of plant roots in a mineralsoil using l-EXAFS spectroscopy (Manceau et al., 2008).In the same microcosm experiments as reported in the cur-rent study, we have previously identified suspended bacteriato produce Cu(0) crystals in the porewater, a process wehave proposed to occur via disproportionation of Cu(I)which is released by Cu-stressed bacteria (Weber et al.,2009). Considering that 13 lM of colloidal Cu(0) was ob-served in the porewater at day 4 (Weber et al., 2009),98% of the 0.6 mmol/kg of Cu(0) identified in the soil(Fig. 2b) have resided in the soil matrix. This Cu(0) maybe produced by bacteria residing on the soil matrix by abiomineralization process similar to that suggested forporewater-suspended bacteria. Other processes that mightbe involved in Cu(0) formation in the soil matrix includeabiotic reduction of Cu(II) by ferrous iron in clay minerals(Ilton et al., 1992) or in green rust (O’Loughlin et al., 2003).In contrast, Cu(II) reduction by dissolved Fe(II) did notproduce metallic Cu(0) but cuprite (Cu2O) at pH 5.2–6.5in a 40 mM chloride background (Matocha et al., 2005),possibly due to stabilization of Cu(I) in chloro complexes.
Our spectroscopic evidence revealed the formation ofCuxS precipitates in the flooded soil, the amount graduallyincreasing to account for 66% of total Cu at day 52(Fig. 2b). The measured pH and EH approached – butdid not reach – the calculated CuxS stability field (Fig 4).This difference to our spectroscopic evidence may be relatedto shortcomings of the assumed system parameters to rep-resent the conditions during soil flooding. Alternatively,the discrepancy may be related to a disequilibrium of theCu and S system with the Fe(II)/Fe(III) redox couple, thelatter likely dominating the mixed-potential EH reading
(Kirk, 2004; Reddy and DeLaune, 2008). Considering thata maximal concentration of only 5 lM of colloidal CuxSwas observed in the porewater (Weber et al., 2009), >99%of the 2.9 mmol-Cu/kg of CuxS identified in the soil(Fig. 2b) have resided in the soil matrix. The CuxS precip-itates in the soil matrix likely formed by processes similar tothose suggested for the formation of the CuxS colloids,including reaction of SOM-bound Cu(II) with reduced sul-fur and by sulfidization of metallic Cu(0) (Weber et al.,2009).
4.3. Contaminant sequestration in metal sulfide precipitates
Under the flooding regime applied, the available sulfatewas insufficient to produce enough sulfide for reaction withall the easily mobilizable chalcophile metal contaminantspresent in the soil (Fig. 1g; Table 1). Moreover, mineraliza-tion of the large pool of sulfur bound in SOM was too slowto serve as a significant source of inorganic sulfide within52 d of flooding. Therefore, sequestration of the chalcophilemetal contaminants in sulfide precipitates had to remainincomplete. The presented evidence from XAFS (Fig. 2b)and sequential extractions (Fig. 2d and f) revealed partialsequestration of Cu (up to 66% of total Cu) and suggestednear-complete sequestration of easily mobilizable Cd, andpartial sequestration of easily mobilizable Pb in sulfide pre-cipitates within 52 d of flooding. Even if Hg and Ag werelikewise sequestered in sulfide precipitates, their soil content(Table 1) was negligible for the sulfide mass balance. Inagreement with the sulfide mass balance, sequential extrac-tions suggested that other chalcophile contaminants in thesoil (Zn, Ni, Fe) were hardly, if any, sequestered in sulfideprecipitates. XAFS results suggested that also As sequestra-tion in sulfide precipitates was negligible (Weber et al.,2008). Thus, CuxS was the dominant sulfide phase formedover the course of soil reduction, which is in contrast tothe common expectation of the predominant formation ofFexS minerals under sulfate-reducing conditions.
Remarkably, the metal sulfide colloids previously re-ported to form in the soil porewater also sequestered almostexclusively Cu, Cd, and Pb (with a molar Cu:Cd:Pb ratio of�8:1:1) but hardly any Zn, Fe, or As (Weber et al., 2009).Considering the decline in easily mobilizable Cd and Pb rel-ative to the 2.9 mmol/kg of CuxS formed, a similarCu:Cd:Pb ratio of �13:1:1 was estimated for the metal sul-fides formed in the bulk soil.
The observed sequestration pattern basically followedthe ascending solubility products of the respective metalsulfide minerals (Table 1). Covellite (CuS) exhibits the low-est solubility product of all chalcophile contaminants con-sidered. In comparison, greenockite (CdS) and galena(PbS) are both 8, sphalerite (ZnS) is 11, millerite (NiS)17, and mackinawite (FexS) 19 orders of magnitude moresoluble than covellite (Table 1). Orpiment (As2S3) is highlyinsoluble under strongly acidic conditions but relatively sol-uble at the slightly acidic to near-neutral pH of the incu-bated soil (Wilkin and Ford, 2002). The activity of thefree metal cations needed to calculate saturation indiceswere unknown, but CuxS, CdS, and PbS precipitation pre-sumably buffered the activity of aqueous sulfide to values

5524 F.-A. Weber et al. / Geochimica et Cosmochimica Acta 73 (2009) 5513–5527
below the saturation of ZnS, NiS, and FexS, despite highdissolved concentrations of these metals. Alternatively,our evidence cannot rule out that some FexS (or other rel-atively soluble metal sulfides) may have initially formed dueto high kinetic availability of the dissolved metal (e.g., up to6 mM of dissolved Fe(II)), but that these metals sulfidestransformed so rapidly (within a time scale of minutes tohours) to less soluble metal sulfides that their transient for-mation did not become apparent at the sampling resolutionof our experiments (which was in the order of days). EX-AFS work by Parkman et al. (1999) indicates that the trans-formation of Cu sorbed to pyrite into covellite or chalcocitecan indeed occur within less than 24 h. Therefore, even ifFexS transiently formed, its fast transformation into CuxSphases left FexS likely unimportant for controlling contam-inant dynamics in the flooded soil.
Our evidence suggested that Cd and Pb were sequesteredin sulfide precipitates despite that ample amounts of Cu(II)remained bound to SOM after 52 d of soil flooding(Fig. 2b). Thermodynamic equilibrium models would pre-dict that the metal that forms the most insoluble metal sul-fide becomes completely ‘‘titrated out” by reaction withaqueous sulfide, before more soluble metal sulfide mineralsprecipitate. Assuming that all SOM-bound Cu(II) waspotentially available and that CuxS was the most insolublemetal sulfide mineral under the given Cu(II/I) complexationin the porewater over the course of the experiment, theobservation that SOM-bound Cu(II) remained suggestedthat kinetic factors constrained further CuxS formation.Such kinetic limitation may include slow Cu(II) desorptionkinetics from SOM (Shi et al., 2005), slow Cu(II) diffusionto the reaction front (Druschel et al., 2002), and/or retardedCuxS precipitation kinetics (Luther et al., 2002). Cu diffu-sion kinetics may be especially important in spatially heter-ogeneous soil, in which size-exclusion restricts sulfate-reducing bacteria to macropores, whereas SOM-boundCu(II) may reside also in a variety of smaller pores. Hence,Cu(II) diffusion from micropores towards the reaction frontin the vicinity of sulfate-reducing bacteria may become alimiting factor for CuxS formation.
With respect to metal sulfide precipitation kinetics inmarine sediments, Morse and Luther (1999) have pointedout that chalcophile metals that exhibit slower water ex-change rates than Fe(II) are incorporated into pyrite,whereas metals that exhibit faster water exchange ratesare generally not incorporated into pyrite but form discretesulfide precipitates. In analogy, the order-of-magnitudesimilar water exchange rate of Cu(II), Cd(II) and Pb(II)(Morse and Luther, 1999) is consistent with concomitantsequestration of Cu, Cd, and Pb into sulfide precipitates.In contrast, Zn(II), Fe(II), and Ni(II) exhibit water ex-change rates one to four orders of magnitude slower thanCu(II), Cd(II), and Pb(II) (Morse and Luther, 1999), andwere hardly sequestered in sulfide precipitates under the sul-fate limitation of the flooded soil.
5. CONCLUSION
We have investigated multi-metal contaminant dynam-ics during flooding of a contaminated floodplain soil.
Under the flooding regime applied, the amount of availablesulfate limited the extent of metal sulfide formation, allow-ing for sequestration of Cu, Cd, and Pb but preventingsequestration of significant amounts of other chalcophilemetals (Zn, Ni, Fe). This pattern is consistent with theincreasing solubility products of the respective metal sul-fides, suggesting that metal sequestration basically followsthe thermodynamically predicted ‘‘sulfide ladder” fromthe most insoluble to more soluble metal sulfides until theavailable sulfate is consumed. Metal desorption, diffusion,and precipitation kinetics can however intertwine with thethermodynamically predicted sequence of metal sulfideformation.
Our findings imply that the dynamics of chalcophile me-tal contaminants are intimately coupled under sulfate limi-tation. For example, if less Cu had been available incomparison to sulfide – either because the soil was less con-taminated or because Cu was kinetically less accessible – theproduced sulfide could have been sufficient to sequestercertain amounts of Zn in sulfide precipitates, constrainingdissolved Zn concentrations. On the other hand, if all soilCu – not just 66% as observed – had formed sulfide precip-itates, the amount of sulfide produced would have beeninsufficient for Cd and Pb sequestration. Quantitativemodels predicting to which extent metal contaminants aresequestered under sulfate limitation are thus needed toassess multi-metal contaminant speciation, mobility, andbioavailability in temporarily flooded soils.
ACKNOWLEDGMENTS
We thank Kurt Barmettler (ETH Zurich) for his support in thelaboratory, Myriam Lambelet (ETH Zurich) for Hg measurements,and Ulf Skyllberg (Swedish University of Agricultural Sciences,Sweden) and Fred Mosselmans (Diamond Light Source, UK) forsharing XAFS reference spectra. The Angstromquelle KarlsruheGmbH (ANKA, Germany) is acknowledged for the allocation ofbeamtime and Stefan Mangold for assistance during measurementsat the XAS beamline. Three anonymous reviewers are acknowl-edged for their constructive comments that helped to improve themanuscript. This project was funded by the Swiss State Secretariatfor Education and Research (Contract No. 03.0353-1) as contribu-tion to the EU project AquaTerra.
APPENDIX A. SUPPLEMENTARY DATA
Supplementary data associated with this article can befound, in the online version, at doi:10.1016/j.gca.2009.06.011.
REFERENCES
Allison J. D., Brown, D. S. and Novo-Gradac, K. J. (1990)MINTEQA2/PRODEFA2-A geochemical assessment modelfor environmental systems. Environmental Research Labora-tory, Office of Research and Development. U.S. EnvironmentalProtection Agency, Athens, Georgia.
Bard A. J., Parsons R. and Jordan J. (1985) Standard potential in
aqueous solution. International Union of Pure and AppliedChemistry. Marcel Dekker, New York.
Barnett M. O., Harris L. A., Turner R. R., Stevenson R. J., HensonT. J., Melton R. C. and Hoffman D. P. (1997) Formation ofmercuric sulfide in soil. Environ. Sci. Technol. 31, 3037–3043.

Multi-metal dynamics in temporarily flooded soil 5525
Bostick B. C., Hansel C. M., La Force M. J. and Fendorf S. (2001)Seasonal fluctuations in zinc speciation within a contaminatedwetland. Environ. Sci. Technol. 35, 3823–3829.
Boulegue J., Lord, III, C. J. and Church T. M. (1982) Sulfurspeciation and associated trace metals (Fe, Cu) in the porewaters of Great Marsh, Delaware. Geochim. Cosmochim. Acta
46, 453–464.
Brandt O. (2003) Eintrags- und Wirkungspfade von Schwermetal-len und Arsen in Flußaue-Systemen am Beispiel der Muldezwischen Bitterfeld/Wolfen und Dessau, Sachsen-Anhalt. Ph.D. Thesis, Techn. Univ. Berlin, Germany. Available at: <http://opus.kobv.de/tuberlin/volltexte/2003/639/>.
Canfield D. E., Raiswell R., Westrich J. T., Reaves C. M. andBerner R. A. (1986) The use of chromium reduction in theanalysis of reduced inorganic sulfur in sediments and shales.Chem. Geol. 54, 149–155.
Cline J. D. (1969) Spectrophotometric determination ofhydrogen sulfide in natural waters. Limnol. Oceanogr. 14,
454–458.
Cooper D. C. and Morse J. W. (1998) Extractability of metalsulfide minerals in acidic solutions: application to environmen-tal studies of trace metal contamination within anoxic sedi-ments. Environ. Sci. Technol. 32, 1076–1078.
Curtin D. and Rostad H. P. W. (1997) Cation exchange and bufferpotential of Saskatchewan soils estimated from texture, organicmatter and pH. Can. J. Soil Sci. 77, 621–626.
Di Toro D. M., Mahony J. D., Hansen D. J., Scott K. J., CarlsonA. R. and Ankley G. T. (1992) Acid volatile sulfide predicts theacute toxicity of cadmium and nickel in sediments. Environ. Sci.
Technol. 26, 96–101.
Druschel G. K., Labrenz M., Thomsen-Ebert T., Fowle D. A. andBanfield J. F. (2002) Geochemical modeling of ZnS in biofilms:an example of ore depositional processes. Econ. Geol. 97, 1319–
1329.
Fadrus H. and Maly J. (1975) Suppression of iron(III) interferencein the determination of iron(II) in water by the 1,10-phenan-throline method. Analyst 100, 549–554.
Favre F., Tessier D., Abdelmoula M., Genin J. M., Gates W. P.and Boivin P. (2002) Iron reduction and changes in cationexchange capacity in intermittently waterlogged soil. Eur. J.
Soil Sci. 53, 175–183.
Geller W., Ockenfeld K., Bohme M. and Knochel A. (2004)Schadstoffbelastung nach dem Elbe-Hochwasser 2002.Umweltforschungszentrum Leipzig-Halle GmbH. Magdeburg,Germany. Available at: <http://www.ufz.de/data/HWEnd1333.pdf>.
Genovese A. and Mellini M. (2007) Ferrihydrite flocs, nativecopper nanocrystals and spontaneous remediation in the Fossodei Noni stream, Tuscany, Italy. Appl. Geochem. 22, 1439–
1450.
Giblin A. E. and Wieder R. K. (1992). In Sulfur cycling on the
continents: Wetlands (eds. R. W. Howarth, J. W. B. Stewart andM. V. Ivanov), pp. 85–117. Terrestrial Ecosystems and Asso-
ciated Water Bodies. SCOPE Report 48. Wiley, Chichester.
Grybos M., Davranche M., Gruau G. and Petitjean P. (2007) Istrace metal release in wetland soils controlled by organic mattermobility or Fe-oxyhydroxides reduction? J. Colloid Interface
Sci. 314, 490–501.
Haung H.-H. and Cuentas L. (1989) Construction of EH–pH andother stability diagrams of uranium in a multicomponentsystem with a microcomputer – I. Domains of predominancediagrams. Can. Metall. Q. 28, 225–234.
Hendershot W. H. and Duquette M. (1986) A simple bariumchloride method for determining cation exchange capacity andexchangeable cations. Soil Sci. Soc. Am. J. 50, 605–608.
Hjorth T. (2004) Effects of freeze-drying on partitioning patterns ofmajor elements and trace metals in lake sediments. Anal. Chim.
Acta 526, 95–102.
Hochella, Jr., M. F., Moore J. N., Putnis C. V., Putnis A., KasamaT. and Eberl D. D. (2005) Direct observation of heavy metal-mineral association from the Clark Fork River superfundcomplex: implications for metal transport and bioavailability.Geochim. Cosmochim. Acta 69, 1651–1663.
Hudson-Edwards K. A., Macklin M. G., Curtis C. D. andVaughan D. J. (1996) Processes of formation and distributionof Pb-, Zn-, Cd-, and Cu-bearing minerals in the Tyne Basin,Northeast England: implications for metal-contaminated riversystems. Environ. Sci. Technol. 30, 72–80.
Huerta-Diaz M. A. and Morse J. W. (1992) Pyritization of tracemetals in anoxic marine sediments. Geochim. Cosmochim. Acta
56, 2681–2702.
Hyacinthe C., Bonneville S. and Van Cappellen P. (2006) Reactiveiron(III) in sediments: chemical versus microbial extractions.Geochim. Cosmochim. Acta 70, 4166–4180.
Ilton E. S., Earley, III, D., Marozas D. and Veblen D. R. (1992)Reaction of some trioctahedral micas with copper sulfatesolutions at 25 �C and 1 atmosphere: an electron microprobeand transmission electron microscopy investigation. Econ.
Geology 87, 1813–1829.
Jacquat O., Voegelin A., Villard A., Marcus M. A. and Kretzsch-mar R. (2008) Formation of Zn-rich phyllosilicate, Zn-layereddouble hydroxide and hydrozincite in contaminated calcareoussoils. Geochim. Cosmochim. Acta 72, 5037–5054.
Karlsson T., Persson P. and Skyllberg U. (2006) Complexation ofcopper(II) in organic soils and in dissolved organic matter –EXAFS evidence for chelate ring structures. Environ. Sci.
Technol. 40, 2623–2628.
Kilmer V. J. and Nearpass D. C. (1960) The determination ofavailable sulfur in soils. Soil Sci. Soc. Proc. 1960, 337–340.
Kirk G. (2004) The Biogeochemistry of Submerged Soils. Wiley,Chichester.
Klemm W., Greif A., Broekaert J. A. C., Siemens V., Junge F. W.,van der Veen A., Schultze M. and Duffek A. (2005) A study onarsenic and the heavy metals in the Mulde river system. Acta
Hydrochim. Hydrobiol. 33, 475–491.
Kostka J. E., Haefele E., Viehweger R. and Stucki J. W. (1999)Respiration and dissolution of iron(III)-containing clay miner-als by bacteria. Environ. Sci. Technol. 33, 3127–3133.
Lamers L. P. M., Tomassen H. B. M. and Roelofs J. G. M. (1998)Sulfate-induced eutrophication and phytotoxicity in freshwaterwetlands. Environ. Sci. Technol. 32, 199–205.
Lett R. E. W. and Fletcher W. K. (1980) Syngenetic sulphideminerals in a copper-rich bog. Miner. Deposita 15, 61–67.
Lovering T. S. (1928) Organic Precipitation of Metallic Copper.U.S. Geological Survey Bulletin 795-C, pp. 45–52.
Luther, III, G. W., Meyerson A. L., Krajewski J. J. and Hires R.(1980) Metal sulfides in estuarine sediments. J. Sediment.
Petrol. 50, 1117–1120.
Luther, III, G. W., Theberge S. M., Rozan T. F., Rickard D.,Rowlands C. C. and Oldroyd A. (2002) Aqueous copper sulfideclusters as intermediates during copper sulfide formation.Environ. Sci. Technol. 36, 394–402.
Manceau A., Nagy K. L., Marcus M. A., Lanson M., Geoffroy N.,Jacquet T. and Kirpichtchikova T. (2008) Formation ofmetallic copper nanoparticles at the soil–root interface. Environ.
Sci. Technol. 42, 1766–1772.
Martin A. J., Jambor J. L., Pedersen T. F. and Crusius J. (2003)Post-depositional behavior of Cu in a metal-mining polishingpond (East Lake, Canada). Environ. Sci. Technol. 37, 4925–
4933.

5526 F.-A. Weber et al. / Geochimica et Cosmochimica Acta 73 (2009) 5513–5527
Masscheleyn P. H., Delaune R. D. and Patrick, Jr., W. H. (1991)Effect of redox potential and pH on arsenic speciation andsolubility in a contaminated soil. Environ. Sci. Technol. 25,
1414–1419.
Matocha C. J., Karathanasis A. D., Rakshit S. and Wagner K. M.(2005) Reduction of copper(II) by iron(II). J. Environ. Qual. 34,
1539–1546.
Morse J. W. and Luther, III, G. W. (1999) Chemical influences ontrace metal–sulfide interactions in anoxic sediments. Geochim.
Cosmochim. Acta 63, 3373–3378.
Moss T. and Monstadt J. (2008) Restoring Floodplains in Europe:
Policy Contexts and Project Experiences. IWA Publishing,London.
Mylon S. E., Hu H. Y. and Benoit G. (2002) Unsuitability of Cr(II)reduction for the measurement of sulfides in oxic water samples.Anal. Chem. 74, 661–663.
O’Day P. A., Carroll S. A., Randall S., Martinelli R. E., AndersonS. L., Jelinski J. and Knezovich J. P. (2000) Metal speciationand bioavailability in contaminated estuary sediments, Ala-meda Naval Air Station California. Environ. Sci. Technol. 34,
3665–3673.
O’Loughlin E. J., Kelly S. D., Kemner K. M., Csencsits R. andCook R. E. (2003) Reduction of AgI, AuIII, CuII, and HgII byFeII/FeIII hydroxysulfate green rust. Chemosphere 53, 437–446.
Oste L. A., Temminghoff E. J. M. and van Riemsdijk W. H. (2002)Solid-solution partitioning of organic matter in soils as influ-enced by an increase in pH or Ca concentration. Environ. Sci.
Technol. 36, 208–214.
Parkman R. H., Charnock J. M., Bryan N. D., Livens F. R. andVaughan D. J. (1999) Reactions of copper and cadmium ions inaqueous solution with goethite, lepidocrocite, mackinawite, andpyrite. Am. Mineral. 84, 407–419.
Parmar N., Gorby Y. A., Beveridge T. J. and Ferris F. G. (2001)Formation of green rust and immobilization of nickel inresponse to bacterial reduction of hydrous ferric oxide. Geomi-
crobiol. J. 18, 375–385.
Pattrick R. A. D., Mosselmans J. F. W., Charnock J. M., EnglandK. E. R., Helz G. R., Garner C. D. and Vaughan D. J. (1997)The structure of amorphous copper sulfide precipitates: an X-ray absorption study. Geochim. Cosmochim. Acta 61, 2023–
2036.
Peltier E., Dahl A. L. and Gaillard J.-F. (2005) Metal speciation inanoxic sediments: when sulfides can be construed as oxides.Environ. Sci. Technol. 39, 311–316.
Ponnamperuma F. N. (1972) The chemistry of submerged soils.Adv. Agronom. 24, 29–96.
Poot A., Gillissen F. and Koelmans A. A. (2007) Effects of flowregime and flooding on heavy metal availability in sediment andsoil of a dynamic river system. Environ. Pollut. 148, 779–787.
Potter, II, R. W. (1977) An electrochemical investigation of thesystem copper–sulfur. Econ. Geol. 72, 1524–1542.
Poulton S. W. and Canfield D. E. (2005) Development of asequential extraction procedure for iron: implications for ironpartitioning in continentally derived particulates. Chem. Geol.
214, 209–221.
Ravel B. and Newville M. (2005) Athena, Artemis, Hephaestus:data analysis for X-ray absorption spectroscopy using IFEF-FIT. J. Synchrotron Radiat. 12, 537–541.
Reddy K. R. and DeLaune R. D. (2008) Biogeochemistry of
Wetlands: Science and Applications. CRC Press, Boca Raton.Roth C. B., Jackson M. L. and Syers J. K. (1969) Deferration effect
on structural ferrous–ferric iron ratio and CEC of vermiculitesand soils. Clays Clay Miner. 17, 253–264.
Schroder T. J., van Riemsdijk W. H., van der Zee S. E. A. T. M.and Vink J. P. M. (2008) Monitoring and modelling of the
solid-solution partitioning of metals and As in a river floodplainredox sequence. Appl. Geochem. 23, 2350–2363.
Shea D. and Helz G. R. (1989) Solubility product constants ofcovellite and a poorly crystalline copper sulfide precipitate at298 K. Geochim. Cosmochim. Acta 53, 229–236.
Shi Z. Q., Di Toro D. M., Allen H. E. and Ponizovsky A. A. (2005)Modeling kinetics of Cu and Zn release from soils. Environ. Sci.
Technol. 39, 4562–4568.
Simpson S. L., Rosner J. and Ellis J. (2000) Competitive displace-ment reactions of cadmium, copper, and zinc added to apolluted, sulfidic estuarine sediment. Environ. Toxicol. Chem.
19, 1992–1999.
Tabatabai M. A. and Al-Khafaji A. A. (1980) Comparison ofnitrogen and sulfur mineralization in soils. Soil Sci. Soc. Am. J.
44, 1000–1006.
Tabatabai M. A. and Bremner J. M. (1972) Forms of sulfur, andcarbon, nitrogen, and sulfur relationships, in Iowa soils. Soil
Sci. 114, 380–386.
Temminghoff E. J. M., van der Zee S. E. A. T. M. and de Haan F.A. M. (1997) Copper mobility in a copper-contaminated sandysoil as affected by pH and solid and dissolved organic matter.Environ. Sci. Technol. 31, 1109–1115.
Tessier A., Campbell P. G. C. and Bisson M. (1979) Sequentialextraction procedure for the speciation of particulate tracemetals. Anal. Chem. 51, 844–851.
Thoenen T. (1999) Pitfalls in the use of solubility limits forradioactive waste disposal: the case of nickel in sulfidicgroundwaters. Nucl. Technol. 126, 75–87.
Thompson R. A. and Helz G. R. (1994) Copper speciation insulfidic solutions at low sulfur activity: further evidence forcluster complexes? Geochim. Cosmochim. Acta 58, 2971–2983.
Voegelin A. and Kretzschmar R. (2005) Formation and dissolutionof single and mixed Zn and Ni precipitates in soil: evidencefrom column experiments and extended X-ray absorption finestructure spectroscopy. Environ. Sci. Technol. 39, 5311–5318.
Voegelin A., Weber F.-A. and Kretzschmar R. (2007) Distributionand speciation of arsenic around roots in a contaminatedriparian floodplain soil: micro-XRF element mapping andEXAFS spectroscopy. Geochim. Cosmochim. Acta 71, 5804–
5820.
Voegelin A., Tokpa G., Jacquat O., Barmettler K. and Kretzsch-mar R. (2008) Zinc fractionation in contaminated soils bysequential and single extractions: influence of soil propertiesand zinc content. J. Environ. Qual. 37, 1190–1200.
Wallschlager D., Desai M. V. M., Spengler M. and Wilken R.-D.(1998) Mercury speciation in floodplain soils and sedimentsalong a contaminated river transect. J. Environ. Qual. 27, 1034–
1044.
Wang F. and Tessier A. (accepted for publication) Zero-valentsulfur and metal speciation in sediment porewaters of freshwa-ter lakes. Environ. Sci. Technol. doi:10.1021/es8034973.
Weatherburn M. W. (1967) Phenol–hypochlorite reaction fordetermination of ammonia. Anal. Chem. 39, 971–974.
Weber F.-A., Voegelin A. and Kretzschmar R. (2008) Speciationand mobility of arsenic in a flooded riparian wetland soil. InANKA Annual Report 2008. ANKA Angstromquelle Karlsruhe.Eggenstein-Leopoldshafen, Germany. pp. 187–188. Availableat: <http://ankaweb.fzk.de/_file/extras/extras_download_26.pdf>.
Weber F.-A., Voegelin A., Kaegi R. and Kretzschmar R. (2009)Biogenic copper(0) and metal sulphide colloids mobilize con-taminants in flooded soil. Nature Geosci. 2, 267–271.
Wilkin R. T. and Ford R. G. (2002) Use of hydrochloric acid fordetermining solid-phase arsenic partitioning in sulfidic sedi-ments. Environ. Sci. Technol. 36, 4921–4927.

Multi-metal dynamics in temporarily flooded soil 5527
Zachara J. M., Fredrickson J. K., Smith S. C. and Gassman P. L.(2001) Solubilization of Fe(III) oxide-bound trace metals by adissimilatory Fe(III) reducing bacterium. Geochim. Cosmochim.
Acta 65, 75–93.
Zimmermann F., Lux H., Maenhaut W., Matschullat J., PlessowK., Reuter F. and Wienhaus O. (2003) A review of air pollution
and atmospheric deposition dynamics in southern Saxony,Germany, Central Europe. Atmos. Environ. 37, 671–691.
Associate editor: George R. Helz




























