Embed Size (px)
Citation preview

doi:10.1016/j.jmb.2007.11.061 J. Mol. Biol. (2008) 376, 131–140
Available online at www.sciencedirect.com
Mutational and Energetic Studies of Notch1Transcription Complexes
Cristina Del Bianco, Jon C. Aster and Stephen C. Blacklow⁎
Department of Pathology,Brigham and Women's Hospitaland Harvard Medical School,Boston, MA 02115, USA
Received 4 September 2007;received in revised form2 November 2007;accepted 18 November 2007Available online26 December 2007
*Corresponding author. E-mail [email protected] used: CSL, CBF-1/R
Hairless, and Lag-1; ICN, intracellulMastermind-like; RHR, Rel-homologRBP-Jκ-associated molecule; ANK, aelectrophoretic mobility shift assay;growth factor; LNR, Lin12–Notch refluorescence resonance energy trans
0022-2836/$ - see front matter © 2007 E
Notch proteins constitute the receptors of a highly conserved signalingpathway that influences cell fate decisions both during development and inadulthood. A proteolytic cascade induced by ligand stimulation results inrelease of the intracellular Notch domain from the cell membrane, allowingit to enter the nucleus and form a complex with a DNA-bound transcriptionfactor called CSL (CBF-1/RBP-Jκ, Suppressor of Hairless, and Lag-1) and acoactivator of the Mastermind family. Assembly of this Notch nuclearcomplex is the key step in the transcriptional response to a Notch signal.In the studies reported here, we mapped residues important for thestabilization of this multiprotein–DNA complex using site-directed muta-genesis, determined the affinity of the three-domain form of CSL for itsvarious partners, and investigated sources of cooperativity in complexformation by monitoring the influence of various components of thecomplex on the interactions of CSL with its other partners. Our findingsare consistent with a model for complex assembly in which the RBP-Jκ-associated molecule domain of Notch increases the effective concentrationof the ankyrin domain for its binding site on the Rel-homology region ofCSL, enabling docking of the ankyrin domain and subsequent recruitmentof the Mastermind-like coactivator.
© 2007 Elsevier Ltd. All rights reserved.
Keywords: signal transduction; fluorescence resonance energy transfer; CSL;Mastermind; protein–protein interaction
Edited by J. E. LadburyIntroduction
Notch proteins constitute the receptors of a highlyconserved signaling pathway that influences cellfate decisions both during development and inadulthood. Notch signals are used iteratively atdifferent decision points and have functional out-comes that depend heavily on gene dose andcontext, regulating cell growth, differentiation, anddeath in a variety of tissue types.1–3 Both deficien-cies and abnormal increases of Notch signaling are
ess:
BP-Jκ, Suppressor ofar Notch; MAML,y region; RAM,nkyrin; EMSA,EGF, epidermalpeat; FRET,fer.
lsevier Ltd. All rights reserve
associated with human developmental anomaliesand cancer, emphasizing the importance of preciselyregulating Notch signal strength.4–10
Notch receptors are single-pass transmembraneproteins that are normally activated by regulatedintramembrane proteolysis in response to trans-membrane ligands expressed on adjacent cells(Fig. 1a). Ligand binding induces proteolytic sen-sitivity to metalloprotease cleavage (S2) at a sitejust external to the plasma membrane.11,12 This pro-teolytic step creates the substrate for subsequentcleavage by the multiprotein gamma-secretase com-plex (S3), which then releases the intracellular por-tion of Notch (ICN) from the membrane, allowing itto translocate to the nucleus, where it induces thetranscription of target genes.The central effector of transcriptional activation
in response to Notch signaling is the nuclear com-plex that controls gene expression. The core elementsof this complex are ICN, a highly conservedtranscription factor called CSL (for CBF-1/RBP-Jκ,Suppressor of Hairless, and Lag-1;13–16), and a
d.

Fig. 1. Overview of Notch signaling (a) and constructsused in this study (b). (a) The mature Notch heterodimeris generated by cleavage at S1 by a furin-like protease.Activation of Notch by a ligand of the Delta/Serrate/Lag-2 family induces a proteolytic cascade (S2, S3) that releasesthe ICN from the membrane, allowing it to enter thenucleus. ICN then forms a complex with a DNA-boundtranscription factor called CSL and a coactivator of theMastermind family to induce transcription of Notch targetgenes. (b) Schematic illustrating Notch constructs usedin this study. HD indicates heterodimerization domain.
132 Binding Studies of Notch Transcription Complexes
transcriptional coactivator of the Mastermind-like(MAML) family.17–19 Once the core complex is as-sembled, the C-terminal portion of MAML not onlycontributes to transcriptional activation through asso-ciation with p300, RNA polymerase II, and otherunknown factors20–22 but also limits the longevity ofthe assembled complex by promoting the phosphor-ylation of ICN by cyclin-dependent kinase 8, whichleads to rapid ICN turnover.23Previous genetic, biochemical, and structural
studies have implicated specific regions of CSL,ICN, and MAML in the physical and functionalinteractions of these proteins with one another.CSL is a highly conserved protein composed ofN- and C-terminal Rel-homology regions (RHR-nand RHR-c, respectively) and a central β-trefoildomain that binds DNA in a sequence-specificfashion.24,25 CSL contacts its cognate DNA via itscentral β-trefoil domain and its RHR-n domain, butthe RHR-c domain of CSLmakes no contact with theDNA at all (unlike other Rel-homology proteins suchas nuclear factor-κB) and is instead positioned like anappendage in an unusual orientation next to theRHR-n domain.25–27 Binding of ICN to CSL dependsin part on a high-affinity interaction between theβ-trefoil domain of CSL and an N-terminal sequenceof ICN known as the RAM (for RBP-Jκ-associated
molecule) region.13,28 In addition, the ankyrin do-main (ANK) independently exhibits weak affinityfor CSL through interactions with the RHR. Sig-nificantly, the ANK domain is capable of assemblingcomplexes with CSL and MAML proteins on DNAbecause ANK and CSL cooperate to create a com-posite binding groove for a short 60- to 70-residuesequence at the N-terminal end of MAML.18,19,29
This N-terminal “handle” of the MAML coactivatoradopts a kinked α-helical conformation in the X-raystructures of both worm and human MAML/ICN/CSL/DNA complexes.26,27 Under conditions ofenforced expression in cells, forms of ICN that con-tain ANK but not RAM also cause CSL-dependentNotch gain-of-function phenotypes,30–32 indicatingthat the essential domain for the effector function ofNotch at target promoters is ANK.Stable assembly of the MAML/ICN/CSL/DNA
complex appears to depend on the cumulative con-tributions of a number of protein–protein andprotein–DNA interfaces. CSL contains distinct bind-ing sites for DNA, RAM, and ANK, with the CSL–ANK interface then used for MAML binding. In thestudies reported here, we mapped residues impor-tant for the stabilization of this multiprotein–DNAcomplex using site-directed mutagenesis, deter-mined the affinity of the three-domain form of CSLfor its various partners, and investigated sources ofcooperativity in complex formation by monitoringthe influence of various components of the complexon the interactions of CSL with its other partners.Our findings are consistent with a model for com-plex assembly in which the RAM domain increasesthe effective concentration of the ANK domain forCSL, enabling docking of the ANK domain onto theRHR domain of CSL to allow subsequent recruit-ment of the MAML coactivator.
Results
To identify sites critical for stabilization of humanNotch1 transcription complexes, we first performedan extensive mutational analysis of residues at dif-ferent protein–protein interaction surfaces. Thesemutations were analyzed in a number of assaysusing a range of Notch1-derived proteins and frag-ments (Fig. 1b). The crystal structure of the humanMAML1/ANK/CSL/DNA complex27 was used toguide the selection of substitution sites, whichfocused on conserved residues at the ANK–CSL,ANK–MAML1, and MAML1–CSL interfaces (Fig.2). In total, we examined 20 mutants of the ANKdomain of Notch1, including alanine, charge-rever-sal, and multisite substitutions, along with ninemutated forms of the MAML1 polypeptide (Table 1).The effect of each mutation on the assembly of
MAML1/ANK/CSL complexes (without addedDNA) was monitored by first mixing CSL, ANK(wild type or mutant), and MAML1 (wild type ormutant) in an approximately equimolar ratio andthen performing size-exclusion chromatography toresolve the complex. Although most individual

Fig. 2. Sites of mutations mapped onto the X-ray structure of the human MAML1/ANK/CSL/DNA complex. (a, b)Overview of the structure (Protein Data Bank ID code 2F8X; Ref. 27). Two views (rotated by∼20° around the y-axis and 5°around the x-axis) are shown. CSL is rendered as a green molecular surface, while the DNA (orange), ANK domain (blue),and MAML1 polypeptide (red) are illustrated as cartoons. Residues on ANK and MAML1 mutated in this study areshown in ball-and-stick form using CPK colors. (c, d) Expanded regions showing sites where charge-reversal mutationsinterfere with stable formation of complexes.
133Binding Studies of Notch Transcription Complexes
point mutations did not prevent the formation ofternary complexes under standard conditions (Table1), the D1973R and E2072R charge-reversal muta-tions of ANK and three charge-reversal and multi-site mutations of MAML1 (R25E, R22E/R25E, andR22E/R25E/R26E) interfered with the formation ofternary complexes, as judged by the absence of thepeak corresponding to the ternary complex on thesize-exclusion column (Fig. 3a). In contrast to theD1973R and E2072R charge-reversal mutations,the D1973A and E2072A alanine substitutions inANK do not prevent the formation of stable ternarycomplexes under standard conditions, suggestingthat the complex relies on numerous weak interac-tions rather than on an energetic “hot spot”33,34 andthat disruption of the complex by charge-reversalmutations is due to the introduction of unfavorableelectrostatic interactions upon substitution.The mutations that abrogated ternary complex
formation in gel filtration assays without DNAwere
also evaluated for their effects on the formationof complexes in the presence of DNA in anelectrophoretic mobility shift assay (EMSA). Forthese studies, the mutations in the ANK domain ofNotch1 were introduced into proteins that containedboth the RAM and ANK regions of Notch1 (here-inafter designated RAMANK) and the mobilityshifts resulting from accrual of the three proteincomponents of the MAML1/RAMANK/CSL/DNAcomplex were monitored as previously des-cribed.29,35 The EMSA data show that in the absenceof MAML1, RAMANK proteins containing eitherthe D1973R or E2072R mutations within the ANKdomain still retain the ability to bind CSL via theirRAM domains (Fig. 3b), as anticipated based onpreviously reported studies.13,28,29,36 On the otherhand, recruitment of the MAML1 polypeptide intothese complexes was either undetectable (D1973R)or strongly reduced (E2072R). The R25E, R25E/R26E, and R22E/R25E/R26E forms of MAML1
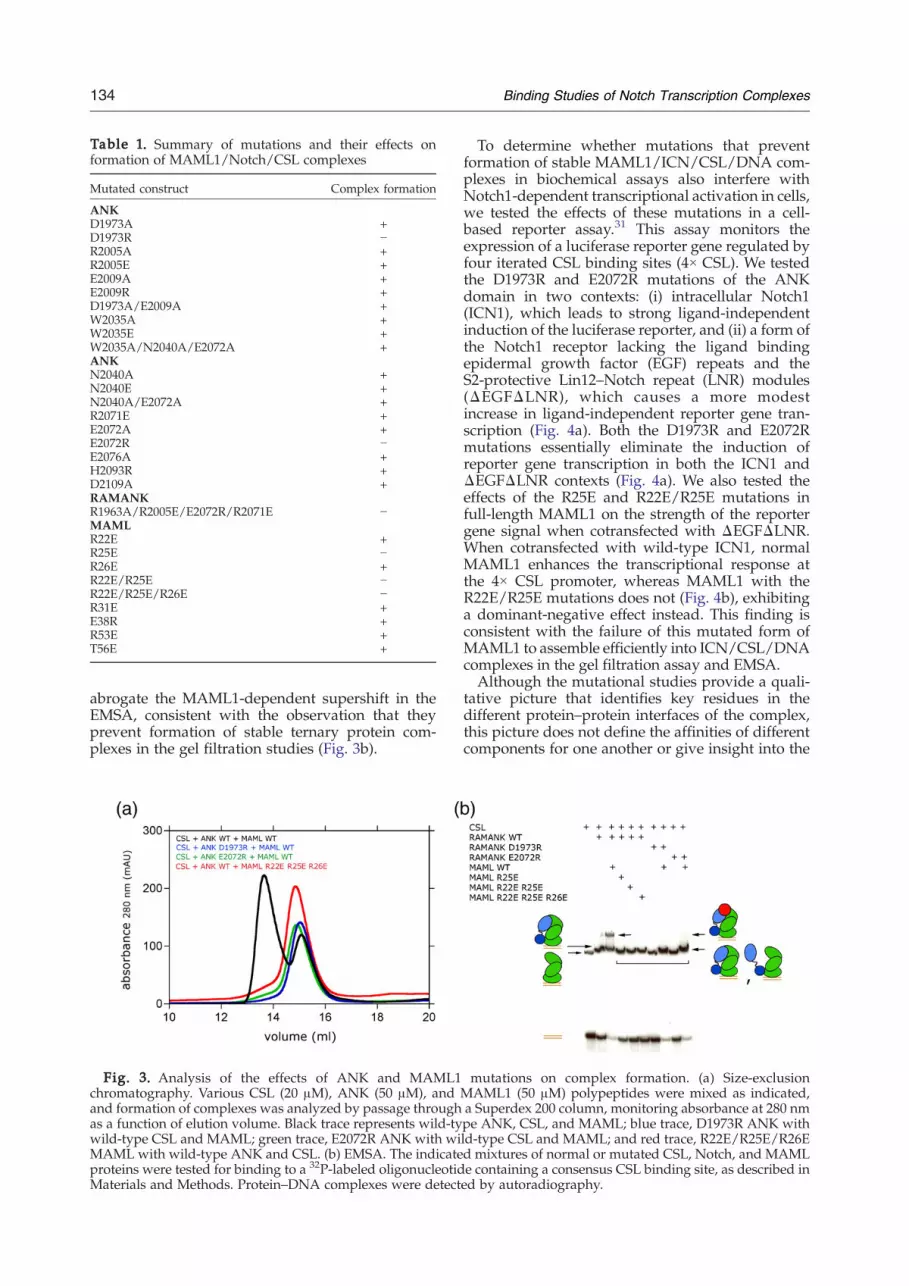
Table 1. Summary of mutations and their effects onformation of MAML1/Notch/CSL complexes
Mutated construct Complex formation
ANKD1973A +D1973R −R2005A +R2005E +E2009A +E2009R +D1973A/E2009A +W2035A +W2035E +W2035A/N2040A/E2072A +ANKN2040A +N2040E +N2040A/E2072A +R2071E +E2072A +E2072R −E2076A +H2093R +D2109A +RAMANKR1963A/R2005E/E2072R/R2071E −MAMLR22E +R25E −R26E +R22E/R25E −R22E/R25E/R26E −R31E +E38R +R53E +T56E +
134 Binding Studies of Notch Transcription Complexes
abrogate the MAML1-dependent supershift in theEMSA, consistent with the observation that theyprevent formation of stable ternary protein com-plexes in the gel filtration studies (Fig. 3b).
Fig. 3. Analysis of the effects of ANK and MAML1chromatography. Various CSL (20 μM), ANK (50 μM), andand formation of complexes was analyzed by passage throughas a function of elution volume. Black trace represents wild-tywild-type CSL and MAML; green trace, E2072R ANK with wiMAML with wild-type ANK and CSL. (b) EMSA. The indicateproteins were tested for binding to a 32P-labeled oligonucleotidMaterials and Methods. Protein–DNA complexes were detect
To determine whether mutations that preventformation of stable MAML1/ICN/CSL/DNA com-plexes in biochemical assays also interfere withNotch1-dependent transcriptional activation in cells,we tested the effects of these mutations in a cell-based reporter assay.31 This assay monitors theexpression of a luciferase reporter gene regulated byfour iterated CSL binding sites (4× CSL). We testedthe D1973R and E2072R mutations of the ANKdomain in two contexts: (i) intracellular Notch1(ICN1), which leads to strong ligand-independentinduction of the luciferase reporter, and (ii) a form ofthe Notch1 receptor lacking the ligand bindingepidermal growth factor (EGF) repeats and theS2-protective Lin12–Notch repeat (LNR) modules(ΔEGFΔLNR), which causes a more modestincrease in ligand-independent reporter gene tran-scription (Fig. 4a). Both the D1973R and E2072Rmutations essentially eliminate the induction ofreporter gene transcription in both the ICN1 andΔEGFΔLNR contexts (Fig. 4a). We also tested theeffects of the R25E and R22E/R25E mutations infull-length MAML1 on the strength of the reportergene signal when cotransfected with ΔEGFΔLNR.When cotransfected with wild-type ICN1, normalMAML1 enhances the transcriptional response atthe 4× CSL promoter, whereas MAML1 with theR22E/R25E mutations does not (Fig. 4b), exhibitinga dominant-negative effect instead. This finding isconsistent with the failure of this mutated form ofMAML1 to assemble efficiently into ICN/CSL/DNAcomplexes in the gel filtration assay and EMSA.Although the mutational studies provide a quali-
tative picture that identifies key residues in thedifferent protein–protein interfaces of the complex,this picture does not define the affinities of differentcomponents for one another or give insight into the
mutations on complex formation. (a) Size-exclusionMAML1 (50 μM) polypeptides were mixed as indicated,a Superdex 200 column, monitoring absorbance at 280 nmpe ANK, CSL, and MAML; blue trace, D1973R ANK withld-type CSL and MAML; and red trace, R22E/R25E/R26Ed mixtures of normal or mutated CSL, Notch, and MAMLe containing a consensus CSL binding site, as described ined by autoradiography.

Fig. 4. Effects of Notch1 (a) and MAML1 (b) mutationson transcription of a luciferase reporter gene. Luciferaseassays were performed on U2OS cell lysates preparedfrom cells transfected in triplicate with empty pcDNA3plasmid or plasmids encoding the indicated forms ofNotch1 and/or MAML1, along with a luciferase reporterplasmid containing four iterated CSL binding sites and aninternal control plasmid expressing Renilla luciferase froma thymidine kinase promoter as described in Materialsand Methods. Firefly luciferase activity, normalized forvariation in Renilla luciferase activity, is expressed relativeto the activity in extracts prepared from cells transfectedwith empty pcDNA3 vector, which is arbitrarily set to avalue of 1.
135Binding Studies of Notch Transcription Complexes
dynamics of the protein domains engaged in thesedifferent interfaces. To determine the affinity ofdifferent ICN1 fragments for CSL–DNA complexesand to explore whether or not the ANK domain is incontact with the RHR of CSL in the absence ofMAML1, we developed a fluorescence resonanceenergy transfer (FRET) assay for the binding of ICN1polypeptides to CSL–DNA complexes. In this assay,a donor fluorophore (Alexa Fluor 555) was cova-lently attached to various ICN1 polypeptides and anacceptor fluorophore (Alexa Fluor 594) was cova-lently added to the cognate DNA bound to CSL in a1:1 complex. The native cysteine of RAM (C1872)was substituted with an alanine and a cysteine was
introduced at the N-terminal end of the proteinpreceding the CSL binding motif to examine RAMbinding to CSL–DNA complexes. The naturallyoccurring cysteine residues were replaced withalanine (C1891A for ANK and both C1872A andC1891A for RAMANK) and a unique cysteineresidue was introduced in place of F2085 (F2085C)to introduce the donor fluorophore into ANK andRAMANK polypeptides. This position is distantfrom the MAML1 and CSL contact interfaces ofANK but remains within the Förster distance of theacceptor site located about 37 Å away based on thestructure of the MAML1/ANK/CSL/DNA com-plex (Fig. 5a). It is important to note that the per-formance of the mutated and fluorescently labeledRAMANK protein in the formation of complexesis indistinguishable from that of the normal protein,as judged by EMSA (Fig. 5b).After demonstrating that complex formation could
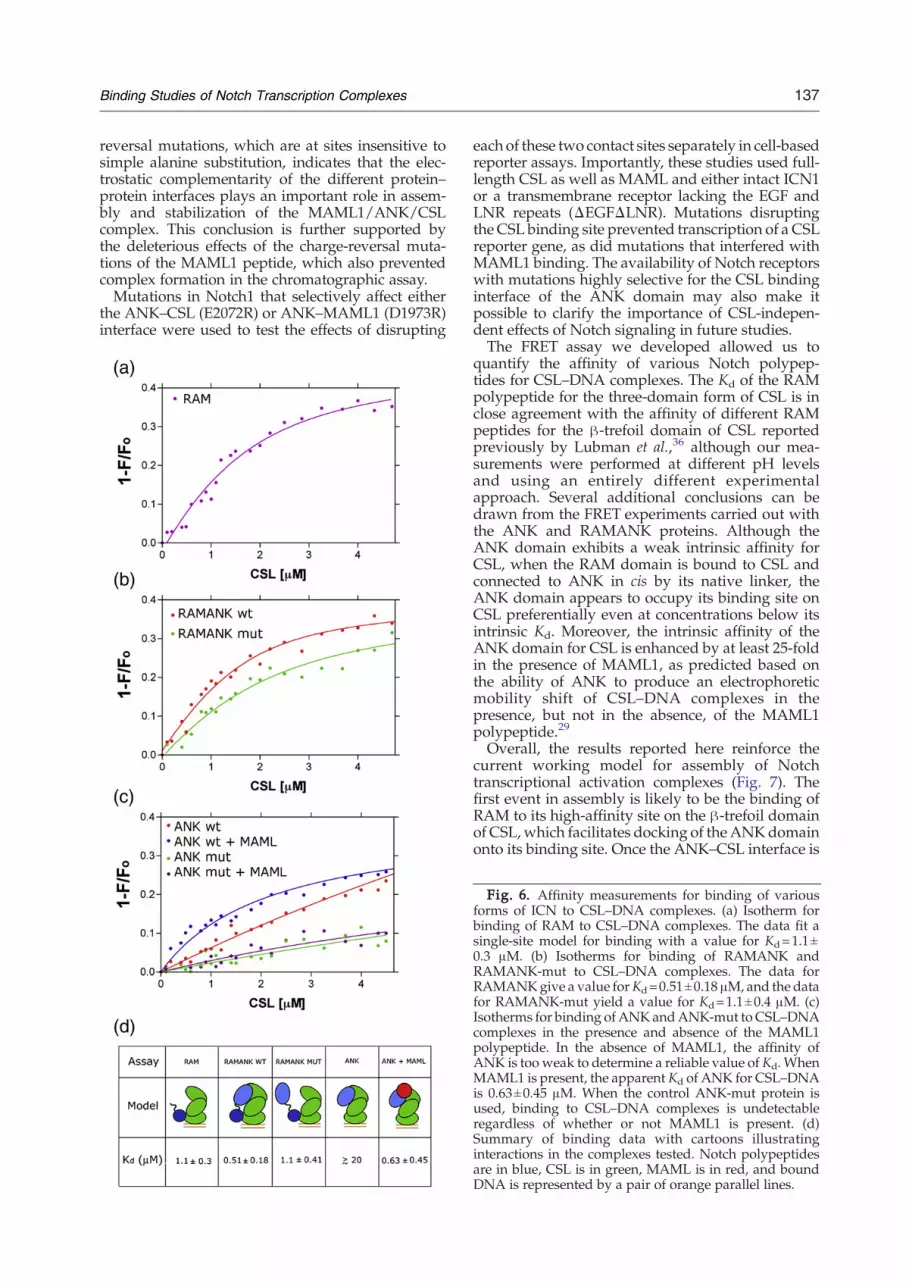
be monitored by FRET with this assay (Fig. 5c andSupplementary Fig. S1), we then used it to measurethe binding affinities of RAM, RAMANK, and ANKfor CSL (Fig. 6 and Supplementary Fig. S3). Titrationof donor-labeled RAM with labeled CSL–DNAyielded a Kd of 1.1±0.3 μM (Fig. 6a), while titrationof RAMANK yielded a Kd of 0.51±0.18 μM (Fig. 6b).These affinity values are consistent with dissociationconstants measured by isothermal titration calori-metry for binding of the RAM and RAMANKregions of ICN to the β-trefoil domain of CSL aloneunder different solution conditions.36 The datareported here also show that RAM makes thedominant thermodynamic contribution to the bind-ing of the full three-domain form of CSL, a conclu-sion consistent with prior immunoprecipitationstudies, EMSAs, and other studies suggesting thatthe affinity of ANK for CSL isweakwhenRAM is notpresent.13,28,29,37TheKd value for ANK binding could not be reliably
determined (approximately 20μMorhigher), becauseFRET measurements could not be carried out at thehigh concentrations needed to achieve saturationbinding in the assay (Fig. 6c). In contrast, saturatingconcentrations of MAML1 enhance the apparentaffinity of ANK for CSL by at least 25-fold, with anapparent dissociation constant of 0.63±0.45 μM (Fig.6c). Together, these findings show that inclusion ofMAML1 in the complex dramatically stabilizes theinteraction of ANK with CSL.Because the binding of RAM is expected to
increase the effective concentration of covalentlyattached ANK for its binding site on CSL, we nextexplored whether the ANK domain is positionedin its CSL binding site when RAMANK binds toCSL–DNA complexes. To examine this question,we constructed control ANK and RAMANKproteins with multiple substitutions of residues inthe ANK domain designed to prevent docking ofthe ANK domain onto its binding site on CSL. Eachprotein contains four mutations (R1963A, R2005E,R2071E, and E2072R) along the surface of ANKcontacting CSL in the human MAML1/ANK/CSL/DNA crystal structure, along with mutations

Fig. 5. FRET assay design and implementation. (a) Design of the FRET assay used to measure the affinity of variousNotch polypeptides for CSL–DNA complexes. The fluorescent donor (Alexa Fluor 555) was installed onto one of threeNotch polypeptides (ANK, RAMANK, or RAM), and the acceptor (Alexa Fluor 594) was installed onto one of the CSL-bound DNA strands. The diagram illustrates the assay for labeling of ANK or RAMANK at position 2085. (b) EMSAshowing CSL binding to DNA and RAMANK polypeptides used in the FRET assay. The RAMANK polypeptides used inthe FRET assay were incubated with CSL and MAML proteins as indicated and tested for binding to a 32P-labeledoligonucleotide containing a consensus CSL binding site, as described in Materials and Methods. Protein–DNAcomplexes were detected by autoradiography. RAMANK F2085C–Alexa Fluor 555 contains the mutation F2085C alongwith C1872A and C1891A. RAMANK-mut contains the mutations C1872A, C1891A, R1963A, R2005E, R2071E, E2072R,and F2085C. (c) Fluorescence emission spectra (excitation wavelength at 555 nm) of donor alone (RAMANK F2085C–Alexa Fluor 555; dashed pink), acceptor alone (CSL–DNA–Alexa Fluor 594; solid purple), and complexes exhibiting FRETfrom donor to acceptor (dashed green).
136 Binding Studies of Notch Transcription Complexes
to remove the native cysteines (C1872A inRAMANK and C1891A in both proteins) and theF2085C substitution to introduce the label for theFRET binding assay. These proteins are referred toas ANK-mut and RAMANK-mut. The four muta-tions were combined because of the extensiveANK–CSL interface area and because even theE2072R protein exhibits some residual capacity torecruit MAML1 by EMSA (Fig. 3b), suggesting thatany given single-site mutation would be insuffi-cient to entirely eliminate the ability of ANK todock onto its CSL binding site. RAMANK-mutcompletely prevented formation of a stable ternarycomplex as judged by EMSA (Fig. 5b). Importantly,the circular dichroism spectrum of RAMANK-mutis nearly identical with that of wild-typeRAMANK, indicating that the mutant proteinsare properly folded under the conditions of theassay (Supplementary Fig. S2).The binding of the ANK-mut and RAMANK-mut
proteins to CSL–DNA complexes was then evalu-ated by FRET. In contrast to wild-type ANK, whichexhibits a weak intrinsic affinity for CSL that isenhanced by addition of MAML1, binding of theANK-mut protein to CSL–DNA complexes is un-detectable regardless of whether or not MAML1 ispresent (Fig. 6c). Consistent with the observationthat the ANK-mut protein fails to bind detectably toCSL, RAMANK-mut binds to CSL–DNA complexeswith a dissociation constant of 1.1±0.4 μM (Fig. 6b),which is within the experimental error for thebinding of RAM alone. In addition, the transferefficiency from donor fluorophore to acceptorfluorophore was reduced to 25% for RAMANK-mut from a value of 41% for wild-type RAMANK,indicating that the ANK domain is farther awayfrom the acceptor fluorophore on DNA. Together,these FRET experiments (Fig. 6d) support the con-clusion that the ANK domain of ICN has sufficient
intrinsic affinity for CSL to favor occupancy of itsbinding site upon delivery to CSL by RAM, evenprior to MAML1 loading (Fig. 7).
Discussion
The studies reported here focused on biochemicaland mechanistic questions related to the assemblyand stabilization of Notch transcriptional activationcomplexes. These complexes contain a number ofprotein–protein interfaces, but we chose to placeparticular emphasis on the interactions of the ANKdomain of ICN because of its central role in thetranscriptional response to the induction of a Notchsignal.Initially, we embarked on a limited alanine scan of
the residues of ANK at the CSL and MAML inter-faces to identify side chains that make importantcontributions to stabilization of complexes. Fourmutations were at the ANK–CSL interface (R2005A,W2035A, E2072A, and E2076A), and four were at theANK–MAML1 interface (D1973A, E2009A, N2040A,and D2109A). However, each of these eight sites wastolerant to alanine substitution, forming ternarycomplexes with MAML1 and CSL stable to isolationby size-exclusion chromatography. Other chargedsubstitutions, such as R2071E and H2093R, as wellas several multisite mutations, were also permissivefor complex assembly. The twomutations tested thatdisrupted the formation of complexes were charge-reversal mutations: D1973R, which mutates a buriedaspartate residue engaged in salt bridges with theR22 and R26 side chains of the MAML1 peptide, andE2072R, which mutates a glutamate residue fullyconcealed from solvent in the complex engaged in acharged hydrogen bond with the side chainhydroxyl group of Y381 from CSL. The sensitivityof the MAML1/ANK/CSL complex to these charge-
137Binding Studies of Notch Transcription Complexes
reversal mutations, which are at sites insensitive tosimple alanine substitution, indicates that the elec-trostatic complementarity of the different protein–protein interfaces plays an important role in assem-bly and stabilization of the MAML1/ANK/CSLcomplex. This conclusion is further supported bythe deleterious effects of the charge-reversal muta-tions of the MAML1 peptide, which also preventedcomplex formation in the chromatographic assay.Mutations in Notch1 that selectively affect either
the ANK–CSL (E2072R) or ANK–MAML1 (D1973R)interface were used to test the effects of disrupting
each of these two contact sites separately in cell-basedreporter assays. Importantly, these studies used full-length CSL as well as MAML and either intact ICN1or a transmembrane receptor lacking the EGF andLNR repeats (ΔEGFΔLNR). Mutations disruptingthe CSL binding site prevented transcription of a CSLreporter gene, as did mutations that interfered withMAML1 binding. The availability of Notch receptorswith mutations highly selective for the CSL bindinginterface of the ANK domain may also make itpossible to clarify the importance of CSL-indepen-dent effects of Notch signaling in future studies.The FRET assay we developed allowed us to
quantify the affinity of various Notch polypep-tides for CSL–DNA complexes. The Kd of the RAMpolypeptide for the three-domain form of CSL is inclose agreement with the affinity of different RAMpeptides for the β-trefoil domain of CSL reportedpreviously by Lubman et al.,36 although our mea-surements were performed at different pH levelsand using an entirely different experimentalapproach. Several additional conclusions can bedrawn from the FRET experiments carried out withthe ANK and RAMANK proteins. Although theANK domain exhibits a weak intrinsic affinity forCSL, when the RAM domain is bound to CSL andconnected to ANK in cis by its native linker, theANK domain appears to occupy its binding site onCSL preferentially even at concentrations below itsintrinsic Kd. Moreover, the intrinsic affinity of theANK domain for CSL is enhanced by at least 25-foldin the presence of MAML1, as predicted based onthe ability of ANK to produce an electrophoreticmobility shift of CSL–DNA complexes in thepresence, but not in the absence, of the MAML1polypeptide.29
Overall, the results reported here reinforce thecurrent working model for assembly of Notchtranscriptional activation complexes (Fig. 7). Thefirst event in assembly is likely to be the binding ofRAM to its high-affinity site on the β-trefoil domainof CSL, which facilitates docking of theANKdomainonto its binding site. Once the ANK–CSL interface is
Fig. 6. Affinity measurements for binding of variousforms of ICN to CSL–DNA complexes. (a) Isotherm forbinding of RAM to CSL–DNA complexes. The data fit asingle-site model for binding with a value for Kd=1.1±0.3 μM. (b) Isotherms for binding of RAMANK andRAMANK-mut to CSL–DNA complexes. The data forRAMANKgive a value forKd=0.51±0.18 μM, and the datafor RAMANK-mut yield a value for Kd=1.1±0.4 μM. (c)Isotherms for binding of ANKandANK-mut to CSL–DNAcomplexes in the presence and absence of the MAML1polypeptide. In the absence of MAML1, the affinity ofANK is too weak to determine a reliable value of Kd. WhenMAML1 is present, the apparent Kd of ANK for CSL–DNAis 0.63±0.45 μM. When the control ANK-mut protein isused, binding to CSL–DNA complexes is undetectableregardless of whether or not MAML1 is present. (d)Summary of binding data with cartoons illustratinginteractions in the complexes tested. Notch polypeptidesare in blue, CSL is in green, MAML is in red, and boundDNA is represented by a pair of orange parallel lines.

Fig. 7. Model for complexassembly. High-affinity (∼1 μM)binding of RAM to CSL–DNAcomplexes promotes docking ofthe ANK domain to the RHR of
CSL. The MAML polypeptide then binds to the ANK–CSL interface to complete assembly of the core Notch transcriptioncomplex.
138 Binding Studies of Notch Transcription Complexes
formed, MAML1 binds to the composite interfacethat is created to “flip on” the transcriptional switch.One issue that still remains unresolved is whetherthe different conformations of CSL seen in the crystalstructures of Notch complexes formed with thehuman and worm proteins reflect an allostericmovement of CSL induced by RAM when it binds.If CSL exhibits intrinsic flexibility, this question maynot be resolved only by comparing crystal structuresof complexes containing RAM, RAMANK, andANKpolypeptides, because the conformation in thecrystals may be dictated by lattice contacts or otherforces rather than by intrinsic conformational pre-ferences. We hope to clarify this issue in futurestudies by extension and refinement of the FRETassays developed here.
Materials and Methods
Mutagenesis and protein purification
Mutations and insertions were made either usingQuikChange Site-directed Mutagenesis (Stratagene) orby back-to-back PCR using Phusion Hot Start DNAPolymerase (New England Biolabs). The coding sequencesof all mutated proteins were confirmed by DNA sequen-cing. CSL, RAMANK, ANK, MAML1, and RAM poly-peptides were purified as previously described.27,29
Gel filtration
The formation of complexes was monitored by gelfiltration on an AKTA chromatography workstation. CSL(20 μM), ANK (50 μM), andMAML1 (50 μM) polypeptideswere mixed, and formation of complexes was analyzed bypassage through a Superdex 200 column (AmershamBiosciences) in 20 mM Tris, pH 8.5, 150 mM NaCl, and5 mM DTT. The absorbance at 280 nm was plotted as afunction of elution volume to assess complex formation;stable MAML1/ANK/CSL complexes elute from thecolumn at approximately 14 mL, whereas uncomplexedCSL and ANK elute from the column in a peak centeredaround 15 mL (Fig. 3a).
EMSAs
Oligonucleotides with 5′-overhangs were labeled with[32P]α-deoxycytidine triphosphate (Perkin-Elmer) by incu-bation with the Klenow fragment of Escherichia coliDNA polymerase I (New England Biolabs) for 15 min atroom temperature. The labeled oligonucleotides were thenseparated from residual [32P]α-deoxycytidine tripho-sphate by passage through a Microspin G-50 column(Amersham Biosciences). To test for complex assembly, we
incubated 0.2 pmol of the DNA probe for 30 min at 30 °Cin binding buffer (final volume, 20 μL) containing 10%glycerol (w/v), 20 mMHepes, pH 7.9, 60 mM KCl, 10 mMDTT, 5 mM MgCl2, dGdC (250 ng), and bovine serumalbumin (0.2 mg/mL) in the presence or absence of CSL(300 ng), different RAMANK polypeptides (1000 ng), andvarious MAML1 polypeptides (100 ng). Samples wereresolved by electrophoresis in 10% native gels at 4 °C and180 V. Following electrophoresis, gels were analyzed byautoradiography.
Reporter gene assays
Reporter gene assays were performed as previouslydescribed.31,38 In reporter assays evaluating mutations ofthe ANK domain of Notch1, ICN1 and ΔEGFΔLNRconstructs (with the normal Notch1 sequence or withmutations of the ANK domain) encoded in the plasmidpcDNA3 (10 ng) were transiently cotransfected (Lipofecta-mine Plus, Invitrogen) in triplicate into human U2OS cells,together with an internal Renilla luciferase control plasmid(Promega, Madison, WI) and a firefly luciferase reportercontaining four tandem CSL binding sites.39 In studiesexamining normal and mutated forms of MAML1, theMAML1 protein of interest encoded in pcDNA3 (100 ng)was also cotransfectedwithΔEGFΔLNR (10 ng), theRenillaluciferase control plasmid, and the luciferase reportercontaining four tandem CSL binding sites. All dual-luciferase assays were then performed and analyzed byusing cell extracts prepared 48 h after transfection, aspreviously described.31,38 The data reported in Fig. 4 arerepresentative of at least three independent experiments(biological replicates). Although the absolute magnitudeof the reporter signal for the active forms of Notch (wild-type ICN and ΔEGFΔLNR) may vary by several-foldfrom one experiment to the next, the relative activity ofthe mutated receptors compared with the wild type andwith one another is highly reproducible.
Fluorescent labeling
We purchased 3′-oligonucleotides fluorescently labeledwith the Alexa Fluor 594 from Integrated DNA Technol-ogies (Coralville, IA) and used them without furtherpurification. Each mutated RAMANK, ANK, and RAMprotein was labeled by incubation with a 20-fold molarexcess of Alexa Fluor 555 C2-maleimide (MolecularProbes, Invitrogen) for 2 h at room temperature in abuffer containing 20 mM Tris, pH 7.4, 150 mM NaCl, and5 mM Tris(2-carboxyethyl)phosphine. Each reaction wasterminated by adding 5 mM β-mercaptoethanol, and thelabeled protein was then separated from the free fluor bypassage over a Sephadex G-25 column (AmershamBiosciences) pre-equilibrated with 20 mM Tris, pH 8.5,150 mMNaCl, and 5 mMDTT, followed by dialysis at 4 °Cagainst the same buffer until all excess dye was removed.Purified proteins were then concentrated to approximately1 mg/mL, aliquoted, and stored at −80 °C until use.

139Binding Studies of Notch Transcription Complexes
Measurements of energy transfer
Fluorescence measurements were carried out on aVarian Cary Eclipse at room temperature, monitoringtransfer from the Alexa Fluor 555 donor on the ICN1polypeptides to the Alexa Fluor 594 acceptor covalentlyattached to the DNA in CSL–DNA complexes. The de-crease in emission of donor fluorescence was monitoredbetween 556 and 750 nm (excitation wavelength at 555nm) using excitation and emission slit widths of 5 nm.Titrations were performed by addition of labeled CSL–DNA complexes to Notch1 polypeptides in 20 mM Tris–HCl, pH 8.5, 150mMNaCl, and 5mMDTT in the presenceor absence of saturating concentration of MAML1. Theinitial protein concentrations of Notch1 and MAML1polypeptides in the cuvette were 1–2 and 10 μM,respectively, and the titrating stock solution of the CSL–DNA–Alexa Fluor 594 complex was 10 μM. Samples wereincubated for 2 min before each titration step. A 10-foldexcess of unlabeled DNAwas included in all solutions toensure that the CSL protein remained bound to DNAthroughout the titrations. Errors reported refer to thegoodness-of-fit error for the titration shown; each titrationwas performed at least twice, and the estimate of run-to-run error in each affinity measurement is approximately30% of the measured value. Transfer of resonance energyfrom the donor to the acceptor was also monitored as afunction of increasing concentration of NaCl in controlexperiments. FRET efficiency (E) was calculated from thechange of the fluorescence intensity of the donor moleculein the absence of the acceptor according to Eq. (1):
E ¼ 1� ðFDA=FDÞ ð1Þwhere FDA and FD are the fluorescence intensities of thedonor molecule in the presence and the absence of theacceptor, respectively.
FRET data analysis
The decrease in the maximum emission (565 nm) of thedonor was corrected for dilution, normalized, and plottedagainst increasing concentration of the ligand (CSL–DNAcomplex). The value of the dissociation constant, Kd (for alltitrations except binding to ANK alone), was determinedby nonlinear least-squares analysis according to Eq. (2),which applies to situations in which [R]≪Kd:40
F ¼ Ffree þ Fbound � Ffreeð ÞKa½L� þ Ka½R�þ1�
ffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiðKa½L� þ Ka½R� þ 1Þ2�4½L�½R�K2
a
q
2Ka½R�
24
35
ð2Þwhere [R] and [L] are the total concentration of receptor(i.e., Notch protein) and that of ligand (CSL–DNA),respectively. Ffree and Fbound are the value of fluorescenceassociated with free receptor and that associated withbound receptor (i.e., Notch polypeptides), respectively. Alldata analyses were performed with the program Graph-Pad Prism (GraphPad Software, San Diego, CA).
Acknowledgements
This work was supported by the NationalInstitutes of Health through grants R01 CA092433
(to S.C.B.) and P01 CA119070 (to J.C.A. and S.C.B.).C.D. is a recipient of a Human Frontier ScienceProgram long-term postdoctoral fellowship. Wethank Luyan Song for constructing several of themutated MAML1 polypeptides used for bindingassays and Gavin Histen for constructing the site-directed mutants of full-length MAML1 and forhelping with reporter assays. We also thank Dr.Sheref S. Mansy for providing helpful suggestionson the FRET assay.
Supplementary Data
Supplementary data associated with this articlecan be found, in the online version, at doi:10.1016/j.jmb.2007.11.061
References
1. Bray, S. J. (2006). Notch signalling: a simple pathwaybecomes complex. Nat. Rev. Mol. Cell Biol. 7, 678–689.
2. Weng, A. P. & Aster, J. C. (2004). Multiple niches forNotch in cancer: context is everything. Curr. Opin.Genet. Dev. 14, 48–54.
3. Artavanis-Tsakonas, S., Rand, M. D. & Lake, R. J.(1999). Notch signaling: cell fate control and signalintegration in development. Science, 284, 770–776.
4. Oda, T., Elkahloun, A. G., Pike, B. L., Okajima, K.,Krantz, I. D., Genin, A. et al. (1997). Mutations in thehuman Jagged1 gene are responsible for Alagillesyndrome. Nat. Genet. 16, 235–242.
5. Li, L., Krantz, I. D., Deng, Y., Genin, A., Banta, A. B.,Collins, C. C. et al. (1997). Alagille syndrome is causedby mutations in human Jagged1, which encodes aligand for Notch1. Nat. Genet. 16, 243–251.
6. Crosnier, C., Driancourt, C., Raynaud, N., Hadchouel,M. & Meunier-Rotival, M. (2001). Fifteen novel muta-tions in the JAGGED1 gene of patients with Alagillesyndrome. Hum. Mutat. 17, 72–73.
7. Joutel, A., Corpechot, C., Ducros, A., Vahedi, K.,Chabriat, H., Mouton, P. et al. (1996). Notch3 muta-tions in CADASIL, a hereditary adult-onset conditioncausing stroke and dementia. Nature, 383, 707–710.
8. Garg, V. (2006). Molecular genetics of aortic valvedisease. Curr. Opin. Cardiol. 21, 180–184.
9. Weng, A. P., Ferrando, A. A., Lee, W., Morris, J. P.,Silverman, L. B., Sanchez-Irizarry, C. et al. (2004).Activating mutations of NOTCH1 in human T cellacute lymphoblastic leukemia. Science, 306, 269–271.
10. Nicolas, M., Wolfer, A., Raj, K., Kummer, J. A., Mill, P.,van Noort, M. et al. (2003). Notch1 functions as atumor suppressor in mouse skin. Nat. Genet. 33,416–421.
11. Brou, C., Logeat, F., Gupta, N., Bessia, C., LeBail, O.,Doedens, J. R. et al. (2000). A novel proteolytic cleavageinvolved in Notch signaling: the role of the disinte-grin–metalloprotease TACE. Mol. Cell, 5, 207–216.
12. Mumm, J. S., Schroeter, E.H., Saxena,M. T., Griesemer,A., Tian, X., Pan, D. J. et al. (2000). A ligand-inducedextracellular cleavage regulates gamma-secretase-like proteolytic activation of Notch1. Mol. Cell, 5,197–206.
13. Tamura, K., Taniguchi, Y., Minoguchi, S., Sakai, T.,Tun, T., Furukawa, T. & Honjo, T. (1995). Physicalinteraction between a novel domain of the receptor

140 Binding Studies of Notch Transcription Complexes
Notch and the transcription factor RBP-J kappa/Su(H).Curr. Biol. 5, 1416–1423.
14. Fortini, M. E. & Artavanis-Tsakonas, S. (1994). TheSuppressor of Hairless protein participates in Notchreceptor signaling. Cell, 79, 273–282.
15. Christensen, S., Kodoyianni, V., Bosenberg, M.,Friedman, L. & Kimble, J. (1996). lag-1, a generequired for lin-12 and glp-1 signaling in Caenorhab-ditis elegans, is homologous to human CBF1 andDrosophila Su(H). Development, 122, 1373–1383.
16. Tani, S., Kurooka, H., Aoki, T., Hashimoto, N. &Honjo, T. (2001). The N- and C-terminal regions ofRBP-J interact with the ankyrin repeats of Notch1RAMIC to activate transcription. Nucleic Acids Res. 29,1373–1380.
17. Wu, L., Aster, J. C., Blacklow, S. C., Lake, R., Artavanis-Tsakonas, S. & Griffin, J. D. (2000). MAML1, a humanhomologue of Drosophila Mastermind, is a transcrip-tional co-activator for NOTCH receptors. Nat. Genet.26, 484–489.
18. Petcherski, A. G. & Kimble, J. (2000). Mastermind is aputative activator for Notch. Curr. Biol. 10, R471–R473.
19. Petcherski, A. G. & Kimble, J. (2000). LAG-3 is aputative transcriptional activator in the C. elegansNotch pathway. Nature, 405, 364–368.
20. Wallberg, A. E., Pedersen, K., Lendahl, U. & Roeder,R. G. (2002). p300 and PCAF act cooperatively tomediate transcriptional activation from chromatintemplates by Notch intracellular domains in vitro.Mol. Cell. Biol. 22, 7812–7819.
21. Fryer, C. J., Lamar, E., Turbachova, I., Kintner, C. &Jones, K. A. (2002). Mastermind mediates chromatin-specific transcription and turnover of the Notchenhancer complex. Genes Dev. 16, 1397–1411.
22. Kurooka, H. & Honjo, T. (2000). Functional interactionbetween the mouse Notch1 intracellular region andhistone acetyltransferases PCAF and GCN5. J. Biol.Chem. 275, 17211–17220.
23. Fryer, C. J., White, J. B. & Jones, K. A. (2004).Mastermind recruits CycC:CDK8 to phosphorylatethe Notch ICD and coordinate activation with turn-over. Mol. Cell, 16, 509–520.
24. Tun, T., Hamaguchi, Y., Matsunami, N., Furukawa, T.,Honjo, T. & Kawaichi, M. (1994). Recognitionsequence of a highly conserved DNA binding proteinRBP-J kappa. Nucleic Acids Res. 22, 965–971.
25. Kovall, R. A. & Hendrickson, W. A. (2004). Crystalstructure of the nuclear effector of Notch signaling,CSL, bound to DNA. EMBO J. 23, 3441–3451.
26. Wilson, J. J. & Kovall, R. A. (2006). Crystal structure ofthe CSL–Notch–Mastermind ternary complex boundto DNA. Cell, 124, 985–996.
27. Nam, Y., Sliz, P., Song, L., Aster, J. C. & Blacklow, S. C.(2006). Structural basis for cooperativity in recruit-ment of MAML coactivators to Notch transcriptioncomplexes. Cell, 124, 973–983.
28. Kurooka, H., Kuroda, K. & Honjo, T. (1998). Roles ofthe ankyrin repeats and C-terminal region of the
mouse Notch1 intracellular region. [published erra-tum appears in Nucleic Acids Res. 27 (1999), following1407]. Nucleic Acids Res. 26, 5448–5455.
29. Nam, Y., Weng, A. P., Aster, J. C. & Blacklow, S. C.(2003). Structural requirements for assembly of theCSL/intracellular Notch1/Mastermind-like 1 tran-scriptional activation complex. J. Biol. Chem. 278,21232–21239.
30. Roehl, H., Bosenberg, M., Blelloch, R. & Kimble, J.(1996). Roles of the RAM and ANK domains insignaling by the C. elegansGLP-1 receptor. EMBO J. 15,7002–7012.
31. Aster, J. C., Xu, L., Karnell, F. G., Patriub, V., Pui,J. C. & Pear, W. S. (2000). Essential roles for an-kyrin repeat and transactivation domains in induc-tion of T-cell leukemia by Notch1. Mol. Cell. Biol. 20,7505–7515.
32. Jeffries, S., Robbins, D. J. & Capobianco, A. J. (2002).Characterization of a high-molecular-weight Notchcomplex in the nucleus of Notch(ic)-transformed RKEcells and in a human T-cell leukemia cell line.Mol. Cell.Biol. 22, 3927–3941.
33. Clackson, T. & Wells, J. A. (1995). A hot spot ofbinding energy in a hormone–receptor interface.Science, 267, 383–386.
34. Bogan, A. A. & Thorn, K. S. (1998). Anatomy of hotspots in protein interfaces. J. Mol. Biol. 280, 1–9.
35. Weng, A. P., Nam, Y., Wolfe, M. S., Pear, W. S., Griffin,J. D., Blacklow, S. C. & Aster, J. C. (2003). Growthsuppression of pre-T acute lymphoblastic leukemiacells by inhibition of Notch signaling. Mol. Cell. Biol.23, 655–664.
36. Lubman, O. Y., Ilagan, M. X., Kopan, R. & Barrick, D.(2007). Quantitative dissection of the Notch:CSLinteraction: insights into the Notch-mediated tran-scriptional switch. J. Mol. Biol. 365, 577–589.
37. Aster, J. C., Robertson, E. S., Hasserjian, R. P., Turner,J. R., Kieff, E. & Sklar, J. (1997). Oncogenic forms ofNOTCH1 lacking either the primary binding site forRBP-Jkappa or nuclear localization sequences retainthe ability to associate with RBP-Jkappa and activatetranscription. J. Biol. Chem. 272, 11336–11343.
38. Sanchez-Irizarry, C., Carpenter, A. C., Weng, A. P.,Pear, W. S., Aster, J. C. & Blacklow, S. C. (2004). Notchsubunit heterodimerization and prevention of ligand-independent proteolytic activation depend, respec-tively, on a novel domain and the LNR repeats. Mol.Cell. Biol. 24, 9265–9273.
39. Hsieh, J. J., Henkel, T., Salmon, P., Robey, E., Peterson,M. G. &Hayward, S. D. (1996). Truncated mammalianNotch1 activates CBF1/RBPJk-repressed genes by amechanism resembling that of Epstein–Barr virusEBNA2. Mol. Cell. Biol. 16, 952–959.
40. Heyduk, T. & Lee, J. C. (1990). Application of fluore-scence energy transfer and polarization to monitorEscherichia coli cAMP receptor protein and lac pro-moter interaction. Proc. Natl. Acad Sci. USA, 87,1744–1748.





























