Embed Size (px)
Citation preview

ISSN: 1524-4539 Copyright © 2009 American Heart Association. All rights reserved. Print ISSN: 0009-7322. Online
72514Circulation is published by the American Heart Association. 7272 Greenville Avenue, Dallas, TX
DOI: 10.1161/CIRCULATIONAHA.109.866269 published online Sep 21, 2009; Circulation
Rutz, Tobias Traupe, Hélène Steck, Rolf Vogel and Christian Seiler Pascal Meier, Steffen Gloekler, Stefano F. de Marchi, Andreas Indermuehle, Tobias
Randomized TrialColony-Stimulating Factor in Chronic Coronary Artery Disease. A Controlled
Myocardial Salvage Through Coronary Collateral Growth by Granulocyte
http://circ.ahajournals.orglocated on the World Wide Web at:
The online version of this article, along with updated information and services, is
http://www.lww.com/reprintsReprints: Information about reprints can be found online at
[email protected]. E-mail:
Fax:Kluwer Health, 351 West Camden Street, Baltimore, MD 21202-2436. Phone: 410-528-4050. Permissions: Permissions & Rights Desk, Lippincott Williams & Wilkins, a division of Wolters
http://circ.ahajournals.org/subscriptions/Subscriptions: Information about subscribing to Circulation is online at
at University of Michigan--Ann Arbor on September 22, 2009 circ.ahajournals.orgDownloaded from

Myocardial Salvage Through Coronary Collateral Growthby Granulocyte Colony-Stimulating Factor in Chronic
Coronary Artery DiseaseA Controlled Randomized Trial
Pascal Meier, MD*; Steffen Gloekler, MD*; Stefano F. de Marchi, MD*; Andreas Indermuehle, MD, PhD;Tobias Rutz, MD; Tobias Traupe, MD; Hélène Steck, RN; Rolf Vogel, MD, PhD; Christian Seiler, MD
Background—The efficacy of granulocyte colony-stimulating factor (G-CSF) for coronary collateral growth promotionand thus impending myocardial salvage has not been studied so far, to our best knowledge.
Methods and Results—In 52 patients with chronic stable coronary artery disease, age 62�11 years, the effect on a markerof myocardial infarct size (ECG ST segment elevation) and on quantitative collateral function during a 1-minutecoronary balloon occlusion was tested in a randomized, placebo-controlled, double-blind fashion. The study protocolbefore coronary intervention consisted of occlusive surface and intracoronary lead ECG recording as well as collateralflow index (CFI, no unit) measurement in a stenotic and a �1 normal coronary artery before and after a 2-week periodwith subcutaneous G-CSF (10 �g/kg; n�26) or placebo (n�26). The CFI was determined by simultaneous measurementof mean aortic, distal coronary occlusive, and central venous pressure. The ECG ST segment elevation �0.1 mVdisappeared significantly more often in response to G-CSF (11/53 vessels; 21%) than to placebo (0/55 vessels;P�0.0005), and simultaneously, CFI changed from 0.121�0.087 at baseline to 0.166�0.086 at follow-up in the G-CSFgroup, and from 0.152�0.082 to 0.131�0.071 in the placebo group (P�0.0001 for interaction of treatment and time).The absolute change in CFI from baseline to follow-up amounted to �0.049�0.062 in the G-CSF group and to�0.010�0.060 in the placebo group (P�0.0001).
Conclusions—Subcutaneous G-CSF is efficacious during a short-term protocol in improving signs of myocardial salvageby coronary collateral growth promotion. (Circulation. 2009;120:1355-1363.)
Key Words: arteries � coronary circulation � collateral circulation � granulocyte colony-stimulating factor
In patients with coronary artery disease (CAD), the size ofmyocardial infarction mainly determines the outcome after
such an event.1 Accordingly, it is the primary strategy toreduce cardiovascular mortality by shrinking infarct size. Asa surrogate for infarct size, clinical studies on the effect ofprocedures aiming at myocardial salvage have employed themagnitude of ECG changes during artificial coronary occlu-sion.2 Infarct size is directly influenced by the followingfactors: duration of coronary occlusion, ischemic area at riskfor infarction, lack of collateral blood supply to the ischemiczone, absence of ischemic preconditioning before the infarct,and myocardial oxygen consumption during the infarct.3
Aside from curtailing the duration of coronary occlusion, theoption of reducing infarct size by collateral artery growthpromotion (arteriogenesis) is appealing. However, arteriogen-esis has to occur before, not after or during, the infarctionbecause remodeling of preformed collateral arterioles to
well-conducting arteries requires 1 to 2 weeks time.Therefore, stem cell therapy in acute infarction with regardto vascular regeneration aiming at myocardial salvage is amisconception because of the delay of arterial remodeling.On the other hand, in chronic CAD a beneficial prognosticeffect of well-developed versus poorly developed collater-als has been documented.4 In a mouse model of acutemyocardial infarction, evidence was presented that theadministration of granulocyte colony-stimulating factor(G-CSF) attenuates late ischemic cardiomyopathy by en-hanced arteriogenesis.5
Clinical Perspective on p 1363Accordingly, the goal of this study in humans with
chronic stable CAD was to test the hypothesis that G-CSFreduces an ECG sign of infarct size by improving collateralfunction.
Received March 17, 2009; accepted July 27, 2009.From the Department of Cardiology, University Hospital, Bern, Switzerland.*The first 3 authors contributed equally to this work.Clinical trial registration information—URL: http://www.clinicaltrials.gov. Unique identifier: NCT00596479.Correspondence to Dr Christian Seiler, Professor of Medicine and Co-Chairman of Cardiology, University Hospital, CH-3010 Bern, Switzerland.
E-mail [email protected]© 2009 American Heart Association, Inc.
Circulation is available at http://circ.ahajournals.org DOI: 10.1161/CIRCULATIONAHA.109.866269
1355
Coronary Heart Disease
at University of Michigan--Ann Arbor on September 22, 2009 circ.ahajournals.orgDownloaded from

MethodsPatientsFifty-two patients (age 62�10 years, 46 men) with chronic stable 1-(n�11), 2- (n�22), or 3-vessel (n�19) CAD eligible for percutane-ous coronary intervention (PCI) of at least 1 stenotic lesion wereincluded in the study. All underwent diagnostic coronary angiogra-phy because of symptoms related to CAD. Patients were selected onthe basis of the following criteria: (1) no previous transmuralinfarction in the myocardial areas assessed for coronary collaterals,(2) normal left ventricular ejection fraction, (3) no congestive heartfailure, (4) no baseline ECG ST segment abnormalities, (5) no signsof inflammatory illness, (6) absence of overt neoplastic disease, and(7) no diabetic retinopathy. Patients were randomly assigned to a2-week, double-blind protocol of subcutaneous G-CSF (filgrastim,Neupogen; Amgen Inc, Thousand Oaks, Calif; n�26) or placebo(n�26). Subcutaneous injections were performed by a study nursenot involved in the process of data acquisition and analysis. TheECG signs of ischemia and collaterals were assessed during balloonocclusion in a stenotic and, if possible, in an angiographically andfunctionally normal coronary artery at baseline before and immedi-ately after the treatment period. This investigation was approved bythe institutional ethics committee, and the patients gave writteninformed consent to participate in the study.
Cardiac Catheterization andCoronary AngiographyPatients underwent left heart catheterization for diagnostic purposesfrom the right femoral approach. Aortic pressure was measured usinga 6F PCI guiding catheter. Central venous pressure was obtained viathe right femoral vein. Left ventricular end-diastolic pressure wasdetermined during vessel patency. Biplane left ventriculography wasperformed, followed by biplane coronary angiography. Coronaryartery stenoses were determined quantitatively as percent diameternarrowing.
Invasive Coronary Assessment
Primary Study End PointsSigns of myocardial ischemia were assessed dichotomously accord-ing to the presence or absence of ECG signs of myocardial ischemiaat the end of a 1-minute balloon occlusion of the vessel of interest.Myocardial ischemia indicative of potential future infarct size wasdefined as ST segment elevation �0.1 mV present on any of 4surface leads or on an intracoronary ECG lead obtained from theangioplasty guide wire via a cross-clamp to lead V1.2,6,7
Coronary collateral flow relative to normal antegrade flow throughthe nonoccluded coronary artery (collateral flow index [CFI]) wasdetermined using coronary pressure measurements. A 0.014-inchpressure monitoring angioplasty guide wire (Pressure Wire, Radi,Uppsala, Sweden) was set at zero, calibrated, advanced through theguiding catheter, and positioned in the distal part of the vessel ofinterest. The CFI was determined by simultaneous measurement ofmean aortic pressure (Pao, mm Hg), the distal coronary arterypressure during balloon occlusion (Poccl, mm Hg), and the centralvenous pressure (CVP, mm Hg) as obtained at the end of the1-minute occlusion. The CFI was calculated as (Poccl�CVP) dividedby (Pao�CVP).8–10 The accuracy of pressure-derived CFI measure-ments in comparison with ECG signs of myocardial ischemia duringocclusion and with absolute myocardial perfusion measurements hasbeen documented previously.7,10,11
Secondary Study End PointsMyocardial ischemia during the 1-minute coronary occlusion wasalso characterized by the presence or absence of angina pectoris.Absolute myocardial perfusion or blood flow at rest and duringhyperemia was assessed quantitatively using myocardial contrastechocardiography, whereby a previously described and validatedalgorithm was employed.12 Briefly, for the calculation of absoluteblood flow, the constituent factors relative myocardial blood volumerBV and its refill rate � after the destruction of echo contrast
microbubbles were obtained during vessel patency. Myocardialblood flow is equal to the product of rBV and � divided bymyocardial tissue density.12
Study ProtocolDuring the treatment period, side effects related to the studymedication were recorded every second day by the study nurseduring a personal visit at the patient’s home. At the start of bothbaseline and follow-up invasive procedures, all patients received5000 U of heparin intravenously. After diagnostic examinations, 2puffs of oral isosorbide dinitrate were given. The coronary arterythought to be the culprit lesion responsible for the patient’s symp-toms was selected for CFI measurements. This vessel would undergoPCI after the 2-week study protocol. Additionally, an angiographi-cally and functionally normal coronary artery was selected for CFImeasurement. In both arteries, fractional flow reserve was deter-mined for functional assessment with the pressure guide wirepositioned distally in the vessel using a bolus of intracoronaryadenosine (12 �g for the right, 18 �g in the left coronary artery) forinduction of hyperemia. At baseline and follow-up, an adequatelysized angioplasty balloon catheter was positioned proximal to thestenosis to be dilated, and at a proximal location in the normal vessel,whereas the pressure guide wire was positioned distally in therespective vessels. Balloon inflation for collateral measurementbefore injection of the study drug occurred in the proximal nonste-notic vessel segment at a pressure of 1 to 2 atmospheres. During thisvessel occlusion, simultaneous Poccl, Pao, and CVP were obtained forthe calculation of CFI. During the entire procedure, an intracoronaryECG obtained from the guide wire and a 4-lead surface ECG wererecorded. The initial invasive procedure was followed by a 2-weekout-of-hospital period with subcutaneous injections of randomlyassigned G-CSF (10 �g/kg in 0.27 mL aqua ad injection) or placebo(0.1% albumin in 0.27 mL aqua ad injection) every other day startingthe day after the baseline procedure. The study drug was prepared bythe hospital pharmacy. The investigators were blinded to the studymedication. All drugs were left unaltered during the study period.The invasive follow-up examination immediately after the treat-ment period consisted of intracoronary measurements identicalwith those described above. The PCI of the stenotic lesioninitially selected to be dilated was performed immediately afterthe follow-up measurements.
Absolute myocardial blood flow at rest and during hyperemia inthe areas supplied by the coronary arteries of interest was obtainedusing contrast echocardiography at baseline after the invasive pro-cedure, 14 days of follow-up before the invasive procedure with PCIand 6 months after study inclusion. Hyperemia was induced byintravenous adenosine (140 �g · min�1 · kg�1), and myocardialperfusion reserve was calculated as absolute blood flow duringhyperemia divided by blood flow at rest (both in mL · min�1 · g�1).
Statistical AnalysisAll continuous data are given as mean�SD. Baseline characteristicsbetween the groups were analyzed by Student t tests for continuousdata and by �2/Fisher exact tests for categorical data. Outcomes atfollow-up examination of categorical variables (ECG ST-segmentelevation �0.1 mV during vessel occlusion and angina pectoris) andfrequency of side effects induced by the study drug were analyzedusing the �2 test. In order to achieve high robustness of statisticaltesting (even though data were approximately normally distributed),a nonparametric analysis of variance for longitudinal data wasperformed for all continuous data using a 2-factorial design with thefactors treatment (G-CSF versus placebo) and time (baseline versusfollow-up measurements. This test also accounts for intraindividualcorrelation between multiple measurements per patient (clustereddata). Calculated P values correspond to the interaction of treatmentand time. As a second step, within-group analyses at different timepoints of myocardial perfusion data were performed by a pairedStudent t test. Differences were considered statistically significant ata 2-sided P value of �0.05.
1356 Circulation October 6, 2009
at University of Michigan--Ann Arbor on September 22, 2009 circ.ahajournals.orgDownloaded from

ResultsPatient Characteristics and ClinicalData at BaselineThere were no statistically significant differences between the2 groups relative to patients’ age, sex, duration of anginapectoris, and history of myocardial infarction in a remotevascular area, nor were there statistical differences in thefrequency of cardiovascular risk factors and (except fornitrates) the use of acetylsalicylic acid, clopidogrel, vasoac-tive drugs, statins, or diuretics (Table 1).
Invasive and Hemodynamic Data at BaselineExcept for systolic blood pressure, invasively obtained hemody-namic parameters at baseline such as heart rate, diastolic bloodpressure, left ventricular ejection fraction, and left ventricularend-diastolic pressure did not differ between the groups(Table 2). At baseline, the severity of CAD and the severityof the stenotic lesion to be treated by PCI were similar amongthe groups. The stenotic and the normal vessels undergoingCFI measurement as well as the CFI measurement site weresimilarly distributed between the groups. Fractional flowreserve values of the normal versus the stenotic vesselundergoing CFI measurement were not statistically differentbetween the groups (Table 2). However, myocardial perfu-sion reserve obtained by contrast echocardiography in thenormal vessel was 1.95�0.81 in the G-CSF group and2.85�1.21 in the placebo group (P�0.018), and in thestenotic vessel it was 1.83�0.74 in the G-CSF group and2.29�1.28 in the placebo group (P�0.14). The occurrence ofangina pectoris and of ECG ST-segment elevation �0.1 mV
during the 1-minute coronary balloon occlusion in either thenormal or the stenotic vessel was not statistically differentbetween the groups. The CFI values in the normal andstenotic vessel were similar between the groups (Table 2).
Treatment-Induced Changes of Study End Points
Primary Study End PointsAt follow-up, ECG ST-segment elevation �0.1 mV duringcoronary occlusion had disappeared more frequently in re-sponse to G-CSF (11/53; 21%) than to placebo (0/55;P�0.0005; Figure 1). Overall, Poccl increased in the G-CSFgroup, whereas it decreased in the placebo group, and Pao
decreased in both groups (Figure 2). The CFI values asobtained in 108 normal and stenotic vessels changed from0.121�0.087 at baseline to 0.166�0.086 at follow-up in theG-CSF group and from 0.152�0.082 to 0.131�0.071 in theplacebo group (P�0.0001 for interaction of treatment andtime). The CFI increased significantly in the G-CSF group in
Table 1. Patient Characteristics and Clinical Data at Baseline
VariableG-CSF(n�26)
Placebo(n�26) P
Age, y 64�10 61�11 0.19
Male sex, n (%) 21 (82) 25 (96) 0.08
Duration of chest pain, mo 3.8�2.9 4.9�5.7 0.13
History of prior myocardial infarction, n (%) 7 (27) 7 (27) 0.93
Cardiovascular risk factors
Dyslipidemia, n (%) 22 (84) 20 (77) 0.48
Diabetes mellitus, n (%) 5 (20) 3 (12) 0.48
Systemic hypertension, n (%) 20 (77) 18 (69) 0.53
Smoking, n (%) 7 (27) 8 (30) 0.76
Obesity, n (%) 18 (69) 11 (42) 0.051
Family history of coronaryartery disease, n (%)
13 (50) 9 (35) 0.26
Cardiovascular medication
�-Blockers, n (%) 18 (69) 19 (73) 0.52
Nitrates, n (%) 2 (8) 8 (30) 0.035
Acetylsalicylic acid, n (%) 22 (84) 24 (92) 0.39
Clopidogrel, n (%) 12 (48) 13 (50) 0.78
Statin, n (%) 21 (80) 22 (84) 0.71
ACE inhibitor, n (%) 18 (73) 20 (77) 0.58
Diuretics, n (%) 10 (38) 5 (19) 0.13
ACE indicates angiotensin-converting enzyme.
Table 2. Invasive Data at Baseline
VariableG-CSF(n�26)
Placebo(n�26) P
Hemodynamic and angiographicdata
Heart rate during, bpm 70�17 68�15 0.82
Systolic blood pressure,mm Hg
129�23 120�21 0.032
Diastolic blood pressure,mm Hg
69�13 67�12 0.45
Left ventricular end-diastolicpressure, mm Hg
9�7 11�4 0.55
Left ventricular ejectionfraction, %
57�9 58�9 0.56
No. of vessels diseased 2.1�0.7 2.2�0.8 0.55
No. of vessels treated byPCI, LAD/LCX/RCA
16/9/5 13/8/10 0.36
Percent diameter stenosis oftreated vessel
74�17 75�17 0.93
Fractional flow reservestenotic vessel (no unit)
0.73�0.15 0.76�0.16 0.57
Fractional flow reservenonstenotic vessel (no unit)
0.91�0.07 0.93�0.05 0.28
Collateral function data
No. of normal vessels for CFImeasurement, LAD/LCX/RCA
7/14/2 8/13/3 0.87
Site of CFI measurement,proximal/mid/distal
32/9/1 33/10/3 0.64
Angina pectoris duringcoronary occlusion, n/N (%)
44/52 (85) 43/54 (80) 0.50
ECG ST-segment elevationduring coronary occlusion,n/N (%)
42/53 (79) 43/55 (78) 0.90
Collateral flow index normalvessel (no unit)
0.118�0.068 0.144�0.074 0.22
Collateral flow index stenoticvessel (no unit)
0.124�0.100 0.158�0.088 0.17
LAD indicates left anterior descending coronary artery; LCX, left circumflexcoronary artery; and RCA, right coronary artery.
Meier et al G-CSF and Collateral Growth 1357
at University of Michigan--Ann Arbor on September 22, 2009 circ.ahajournals.orgDownloaded from
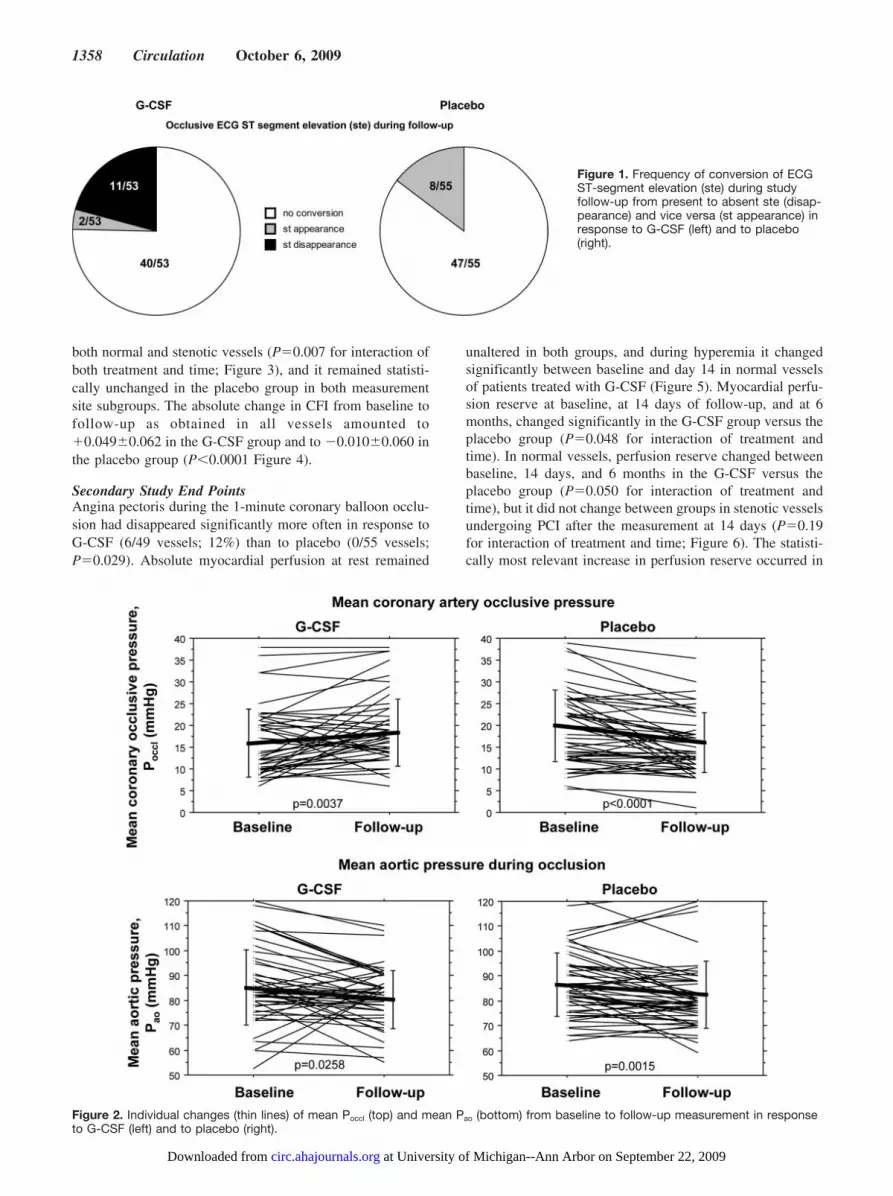
both normal and stenotic vessels (P�0.007 for interaction ofboth treatment and time; Figure 3), and it remained statisti-cally unchanged in the placebo group in both measurementsite subgroups. The absolute change in CFI from baseline tofollow-up as obtained in all vessels amounted to�0.049�0.062 in the G-CSF group and to �0.010�0.060 inthe placebo group (P�0.0001 Figure 4).
Secondary Study End PointsAngina pectoris during the 1-minute coronary balloon occlu-sion had disappeared significantly more often in response toG-CSF (6/49 vessels; 12%) than to placebo (0/55 vessels;P�0.029). Absolute myocardial perfusion at rest remained
unaltered in both groups, and during hyperemia it changedsignificantly between baseline and day 14 in normal vesselsof patients treated with G-CSF (Figure 5). Myocardial perfu-sion reserve at baseline, at 14 days of follow-up, and at 6months, changed significantly in the G-CSF group versus theplacebo group (P�0.048 for interaction of treatment andtime). In normal vessels, perfusion reserve changed betweenbaseline, 14 days, and 6 months in the G-CSF versus theplacebo group (P�0.050 for interaction of treatment andtime), but it did not change between groups in stenotic vesselsundergoing PCI after the measurement at 14 days (P�0.19for interaction of treatment and time; Figure 6). The statisti-cally most relevant increase in perfusion reserve occurred in
Figure 1. Frequency of conversion of ECGST-segment elevation (ste) during studyfollow-up from present to absent ste (disap-pearance) and vice versa (st appearance) inresponse to G-CSF (left) and to placebo(right).
Figure 2. Individual changes (thin lines) of mean Poccl (top) and mean Pao (bottom) from baseline to follow-up measurement in responseto G-CSF (left) and to placebo (right).
1358 Circulation October 6, 2009
at University of Michigan--Ann Arbor on September 22, 2009 circ.ahajournals.orgDownloaded from
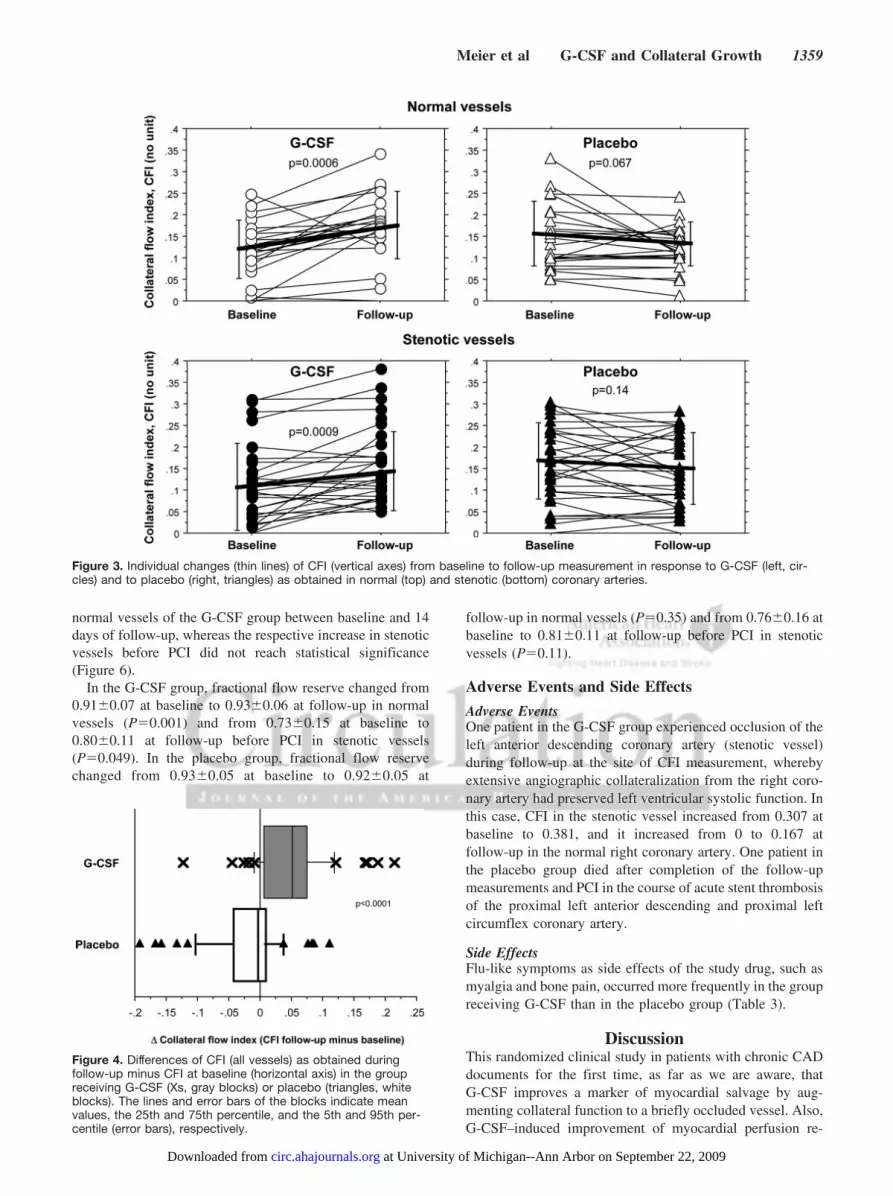
normal vessels of the G-CSF group between baseline and 14days of follow-up, whereas the respective increase in stenoticvessels before PCI did not reach statistical significance(Figure 6).
In the G-CSF group, fractional flow reserve changed from0.91�0.07 at baseline to 0.93�0.06 at follow-up in normalvessels (P�0.001) and from 0.73�0.15 at baseline to0.80�0.11 at follow-up before PCI in stenotic vessels(P�0.049). In the placebo group, fractional flow reservechanged from 0.93�0.05 at baseline to 0.92�0.05 at
follow-up in normal vessels (P�0.35) and from 0.76�0.16 atbaseline to 0.81�0.11 at follow-up before PCI in stenoticvessels (P�0.11).
Adverse Events and Side Effects
Adverse EventsOne patient in the G-CSF group experienced occlusion of theleft anterior descending coronary artery (stenotic vessel)during follow-up at the site of CFI measurement, wherebyextensive angiographic collateralization from the right coro-nary artery had preserved left ventricular systolic function. Inthis case, CFI in the stenotic vessel increased from 0.307 atbaseline to 0.381, and it increased from 0 to 0.167 atfollow-up in the normal right coronary artery. One patient inthe placebo group died after completion of the follow-upmeasurements and PCI in the course of acute stent thrombosisof the proximal left anterior descending and proximal leftcircumflex coronary artery.
Side EffectsFlu-like symptoms as side effects of the study drug, such asmyalgia and bone pain, occurred more frequently in the groupreceiving G-CSF than in the placebo group (Table 3).
DiscussionThis randomized clinical study in patients with chronic CADdocuments for the first time, as far as we are aware, thatG-CSF improves a marker of myocardial salvage by aug-menting collateral function to a briefly occluded vessel. Also,G-CSF–induced improvement of myocardial perfusion re-
Figure 3. Individual changes (thin lines) of CFI (vertical axes) from baseline to follow-up measurement in response to G-CSF (left, cir-cles) and to placebo (right, triangles) as obtained in normal (top) and stenotic (bottom) coronary arteries.
Figure 4. Differences of CFI (all vessels) as obtained duringfollow-up minus CFI at baseline (horizontal axis) in the groupreceiving G-CSF (Xs, gray blocks) or placebo (triangles, whiteblocks). The lines and error bars of the blocks indicate meanvalues, the 25th and 75th percentile, and the 5th and 95th per-centile (error bars), respectively.
Meier et al G-CSF and Collateral Growth 1359
at University of Michigan--Ann Arbor on September 22, 2009 circ.ahajournals.orgDownloaded from

serve in a region subtended by a normal coronary artery ismaintained for as long as 6 months.
Myocardial Salvage Versus RegenerationOne of the study’s primary end points that was positivelyinfluenced by G-CSF was myocardial salvage in case of afuture coronary occlusion (ie, ECG ST-segment elevationduring the first 1-minute artificial occlusion. It disappeared in20% of patients receiving G-CSF and in none receivingplacebo. In the context of G-CSF treatment with recruitmentof progenitor cells, the therapeutic concept of myocardialsalvage has been implicated unduly less than that of myocar-dial regeneration. The treatment strategy of salvage is basedon the observation that myocardial cells can be saved until,but not beyond, 6 hours of complete ischemia.13 Conse-quently, rescuing cardiac myocytes in the event of acute andpermanent coronary occlusion means reperfusing the depen-dent ischemic area by recanalizing the blocked artery. Alter-natively, collateral perfusion can be promoted before coro-nary occlusion takes place. Among the described candidates
for myocardial salvage, augmented collateral function beforeand after the 2-week study period was the only parameter thatwas different between the 2 groups (identical time andsequence of coronary occlusion, similar ischemic areas at riskfor infarction, similar heart rate; Table 2). Thus, it is likelythat the improved indicator of myocardial salvage was caus-ally related to augmented collateral function in the G-CSFgroup. In line with the less frequent ECG ST-segmentelevation after G-CSF therapy, angina pectoris during coro-nary occlusion disappeared more often than in the placebogroup.
Hence, the concept of future myocardial salvage by meansof coronary collateral regeneration during the chronic phaseof CAD is feasible. Conversely, myocardial salvage in thecourse of acute myocardial infarction simultaneously withcollateral regeneration or arteriogenesis is impossible becausethe slowly growing collateral vessel meets necrotic tissue.However, the concept of myocardial regeneration or repair ofacute infarction independently of salvage is principally fea-sible, although it is controversial whether adult hematopoietic
Figure 5. Mean values of absolute myocardial perfusion (vertical axis) at rest (left) and during hyperemia (right) taken during vesselpatency in the vascular area subtended by the vessels undergoing collateral flow index measurements. All changes were nonsignificantexcept for that indicated by a P value. Error bars indicate SE.
Figure 6. Mean values of myocardial perfusion reserve (vertical axis). Left, myocardial perfusion reserve in the group receiving G-CSF(Xs). Right, myocardial perfusion reserve in the placebo group (triangles). Error bars indicate SE.
1360 Circulation October 6, 2009
at University of Michigan--Ann Arbor on September 22, 2009 circ.ahajournals.orgDownloaded from

stem cells do transdifferentiate into cardiac tissue.14 Replace-ment of the thin noncontractile collagenous scar after acutemyocardial infarction by viable contracting myocardium hasbeen shown in the experimental animal model using neonatalor fetal cardiomyocytes as long as 1 week after coronaryartery occlusion.15 By contrast, adult as opposed to embry-onic stem cells have been shown not to incorporate intogrowing collateral vessels in a mouse model of hindlimbischemia.16 They appear to promote arteriogenesis in terms of“software” (paracrine arteriogenic effects through growthfactor secretion by the stem cells) rather than “hardware”supply. Accordingly, in a meta-analysis that included 999patients treated with bone marrow–derived cells for ischemicheart disease (12 studies in the setting of acute myocardialinfarction, 6 in subacute infarction or chronic CAD), bonemarrow transplantation in comparison with the control groupimproved left ventricular ejection fraction by 3.66%, itreduced infarct scar size by 5.49%, and the left ventricularend-systolic volume was diminished by 4.8 mL.17 Whetherthe mechanism leading to this very modest benefit is actualmyocardial regeneration in the biological sense of the wordremains unresolved. Alternatively and purely on physicalgrounds, the thickened scar tissue (by the cellular infiltrate)reduces ventricular preload and afterload on the basis of theLaplace law and thus explains the increase in ejectionfraction, which is well known to be inversely related toafterload. The reduction in end-systolic volume is mathemat-ically tied to the augmented ejection fraction and is thus notan independent finding. Additionally, the diminished scarsize could result from paracrine and not from the regenerativeeffects of bone marrow–derived cells, which secrete cyto-kines, proteases, and growth factors, thereby enhancingmyocyte survival.16
Myocardial Regeneration by G-CSF: Data Fromthe LiteratureAnother approach than autologous bone marrow cell trans-plantation to regenerate myocardial scar tissue respectively tosalvage still viable myocardium is to recruit the stem cellsnoninvasively to the site of injury. The concept in the settingof acute myocardial infarction is that various cytokines andgrowth factors such as G-CSF recruit bone marrow–derivedprogenitor cells that then target damaged tissue. In the presentstudy, the peak level of CD34� cells as obtained at day 7 afterthe study began amounted to 15�11�103/mL in the G-CSFgroup and to 4�3�103/mL in the placebo group (P�0.0001;data not shown), a level that was lower by a factor of 2 to 5
in comparison with previous G-CSF studies in the setting ofacute myocardial infarction.18 A meta-analysis by Zohlnhöferet al19 of 10 randomized clinical trials using stem cellmobilization by G-CSF in 445 patients with acute myocardialinfarction found only an insignificant increase in left ventric-ular ejection fraction, which was on average 1.3% larger inthe G-CSF group than in the placebo group (P�0.36); inaddition, a more pronounced infarct size reduction wasobserved in the G-CSF than in the placebo group by 0.15%(P�0.17).
A few studies, all of them nonrandomized, have testedG-CSF in patients with severe chronic CAD and, on average,have found no effect on left ventricular ejection fraction or onmyocardial perfusion after a mean follow-up of 5 months.18
Relative to the above-discussed conceptual issue of myocar-dial salvage versus regeneration, these uncontrolled investi-gations are comparable with the present one. They have useda higher-dose regimen (mostly 10 �g/day) with a shorterduration of therapy (5 to 6 days), and the follow-up period hasbeen similarly long. However, the actual study had a random-ized design, and it directly obtained pathophysiologically andstatistically meaningful end points. To choose left ventricularejection fraction and qualitatively assessed myocardial per-fusion as end points in a study with a patient number rangingbetween 5 and 32 is statistically unreasonable, consideringthe large measurement variability of those parameters.18 Inthe context of earlier studies by our laboratory using colonystimulating factors and direct quantitative measurements ofcollateral function,20,21 the number of patients necessary toallow the detection of a �0.05 difference in CFI between thegroups or an increase of 30% in the G-CSF group wasestimated to be 50. The only variable directly comparablebetween the studies just mentioned and the present study ismyocardial perfusion, which remained unchanged in responseto G-CSF after 5 months in the uncontrolled trials and whichincreased in the nonstenotic vascular area in the present study(maintenance of initial increase after 2 weeks of G-CSFtreatment). Of course, the increase in myocardial perfusionreserve in the area subtended by the PCI-treated vessel cannotbe attributed to G-CSF but to the PCI (Figure 5). The presentobservation of an enhanced hyperemic absolute myocardialperfusion in normal vessel regions of patients treated withG-CSF rests on the reliability of quantitative myocardialcontrast echocardiography as performed in our study. Inextensive validation studies incorporating direct comparisonof the technique with a flow phantom, positron emissiontomography, invasive Doppler measurements in CAD pa-tients,12 and quantitative coronary angiography,22 the methodhas been shown to be very reliable in obtaining a wide rangeof absolute flow values between 0.1 and 8 mL · min�1 · g�1
and in detecting coronary artery stenotic lesions with areduction �50% in diameter.
Pharmacological Arteriogenesis in HumansSo far, the role of G-CSF in arteriogenesis has been investi-gated in only 1 experimental study.5 In this mouse model ofacute myocardial infarction, G-CSF application resulted in asignificant increase of circulating mononuclear cells expressingstem cell markers. Arterioles in the border zone of the infarcted
Table 3. Side Effects
Side EffectG-CSF
(n�26)Placebo(n�26) P
Malaise/fatigue, n (%) 12 (46) 13 (50) 0.78
Myalgia, n (%) 18 (69) 3 (12) �0.0001
Bone pain, n (%) 12 (46) 3 (12) 0.006
Headache, n (%) 6 (23) 3 (12) 0.27
Diarrhea, n (%) 3 (12) 5 (19) 0.44
Adverse events with total frequency �5 per group: fever, loss of appetite,nausea, abdominal pain, constipation, mucositis, insomnia, pruritus, exanthema.
Meier et al G-CSF and Collateral Growth 1361
at University of Michigan--Ann Arbor on September 22, 2009 circ.ahajournals.orgDownloaded from

myocardium showed an increased expression of intercellularadhesion molecule-1 accompanied by an accumulation of bonemarrow–derived cells and proliferation of endothelial andsmooth muscle cells. Histological examination of micetreated with G-CSF revealed a lower amount of granulationtissue associated with a subsequent reduction in left ventric-ular wall thinning and scar extension.5 For obvious reasons,our clinical study did not provide structural data indicative ofG-CSF–induced arteriogenesis. But it documented for thefirst time, as far as we are aware, the efficacy of G-CSF withregard to the function of collateral vessels. Aside fromG-CSF, there have been well-investigated substances witharteriogenic efficacy such as monocyte chemoattractantprotein-123; another colony-stimulating factor, granulocyte-monocyte CSF (GM-CSF);24 transforming growth factor�125; fibroblast growth factors26; tumor necrosis factor �27;and matrix metalloproteinases.28 Among these, only colony-stimulating factors have been investigated for clinical use incoronary or peripheral artery disease, whereby GM-CSF hasbeen shown to be effective in the former but not the lattercondition.20,21,29 However, inherent to all the arteriogenicsubstances is a certain proatherogenic risk because of theshared pathogenic monocyte involvement and inflammationin both processes. For example, although it is a strongarteriogenic factor, monocyte chemoattractant protein-1 hasbeen shown to be not suitable for clinical use because of itsatherogenic side effects.23 A small randomized clinical trialusing GM-CSF for arteriogenesis had to be stopped prema-turely for safety concerns in the context of 2 patients withacute coronary syndrome in the GM-CSF group.21 By con-trast, G-CSF has been reported in meta-analyses to besafe,18,19,30 a finding that is consistent with the present data.
Study LimitationsIn the context of the above-discussed aspects of the safety andside effects of G-CSF, the issue of pseudo-blinding to thestudy drug can be raised because it occurs in numerous“double-blind” clinical trials with distinctive study drug sideeffects. In the present study, and in order to account for thispotential pitfall, patient visits during the study period withrecording of drug side effects were performed by a studynurse not involved in data acquisition and analysis.
One of the primary study end points, ECG ST-segmentelevation during the 1-minute coronary occlusion, is a surro-gate marker for infarct size and not the actual amount ofnecrotic myocardium as obtained, for example, by technetiumscintigraphy or gadolinium magnetic resonance imaging.Methodologically, it was not feasible in this study involvingpatients with chronic CAD to directly measure infarct sizebecause of the absence of such an event. Also, the decrease ordisappearance of ECG signs of ischemia during a briefcoronary occlusion in response to certain therapeutic proce-dures has long been recognized as a direct measure ofmyocardial salvage in case of a future permanent coronaryocclusion.2,31
ConclusionSubcutaneous G-CSF is efficacious during a short-term pro-tocol in improving signs of myocardial salvage by coronarycollateral growth promotion.
Sources of FundingThis study was supported by a grant from the Swiss National ScienceFoundation (grant #3200BO-112341) and the Swiss HeartFoundation.
DisclosuresNone.
References1. Sobel BE, Bresnahan GF, Shell WE, Yoder RD. Estimation of infarct size
in man and its relation to prognosis. Circulation. 1972;46:640–647.2. Birnbaum Y, Kloner R. Percutaneous transluminal coronary angioplasty
as a model of ischemic preconditioning and preconditioning-mimeticdrugs. J Am Coll Cardiol. 1999;33:1036–1039.
3. Schaper W, Frenzel H, Hort W. Experimental coronary artery occlusion:measurement of infarct size. Basic Res Cardiol. 1979;74:46–53.
4. Meier P, Gloekler S, Zbinden R, Beckh S, de Marchi S, Zbinden S,Wustmann K, Billinger M, Vogel R, Cook S, Wenaweser P, Togni M,Windecker S, Meier B, CS. Beneficial effect of recruitable collaterals: a10-year follow-up study in patients with stable coronary artery diseaseundergoing quantitative collateral measurements. Circulation. 2007;116:975–983.
5. Deindl E, Zaruba M, Brunner S, Huber B, Mehl U, Assmann G, HoeferI, Mueller-Hoecker J, Franz W. G-CSF administration after myocardialinfarction in mice attenuates late ischemic cardiomyopathy by enhancedarteriogenesis. FASEB J. 2006;20:956–958.
6. Friedman PL, Shook TL, Kirshenbaum JM, Selwyn AP, Ganz P. Value ofthe intracoronary electrocardiogram to monitor myocardial ischemiaduring percutaneous transluminal coronary angioplasty. Circulation.1986;74:330–339.
7. de Marchi S, Meier P, Oswald P, Seiler C. Variable ECG signs ofischemia during controlled occlusion of the left and right coronary arteryin humans. Am J Physiol. 2006;291:H351–H356.
8. Pijls NHJ, van Son JAM, Kirkeeide RL, De Bruyne B, Gould KL.Experimental basis of determining maximum coronary, myocardial, andcollateral blood flow by pressure measurements for assessing functionalstenosis severity before and after percutaneous coronary angioplasty.Circulation. 1993;86:1354–1367.
9. Seiler C, Fleisch M, Garachemani A, Meier B. Coronary collateral quan-titation in patients with coronary artery disease using intravascular flowvelocity or pressure measurements. J Am Coll Cardiol. 1998;32:1272–1279.
10. Vogel R, Zbinden R, Indermuhle A, Windecker S, Meier B, Seiler C.Collateral-flow measurements in humans by myocardial contrast echo-cardiography: validation of coronary pressure-derived collateral-flowassessment. Eur Heart J. 2006;27:157–165.
11. Matsuo H, Watanabe S, Kadosaki T, Yamaki T, Tanaka S, Miyata S,Segawa T, Matsuno Y, Tomita M, Fujiwara H. Validation of collateralfractional flow reserve by myocardial perfusion imaging. Circulation.2002;105:1060–1065.
12. Vogel R, Indermuhle A, Reinhardt J, Meier P, Siegrist PT, Namdar M,Kaufmann PA, Seiler C. The quantification of absolute myocardial per-fusion in humans by contrast echocardiography: algorithm and validation.J Am Coll Cardiol. 2005;45:754–762.
13. Reimer KA, Jennings RB. The “wavefront phenomenon” of myocardialischemic cell death. II. Transmural progression of necrosis within theframework of ischemic bed size (myocardium at risk) and collateral flow.Lab Invest. 1979;40:633–644.
14. Murry C, Soonpaa M, Reinecke H, Nakajima H, Nakajima H, Rubart M,Pasumarthi K, Virag J, Bartelmez S, Poppa V, Bradford G, Dowell J,Williams D, Field L. Haematopoietic stem cells do not transdifferentiateinto cardiac myocytes in myocardial infarcts. Nature. 2004;428:664–668.
15. Reffelmann T, Dow J, Dai W, Hale S, Simkhovich B, Kloner R. Trans-plantation of neonatal cardiomyocytes after permanent coronary arteryocclusion increases regional blood flow of infarcted myocardium. J MolCell Cardiol. 2003;35:607–613.
16. Ziegelhoeffer T, Fernandez B, Kostin S, Heil M, Voswinckel R, HelischA, Schaper W. Bone marrow-derived cells do not incorporate into theadult growing vasculature. Circ Res. 2004;94:230–238.
17. Abdel-Latif A, Bolli R, Tleyjeh I, Montori V, Perin E, Hornung C,Zuba-Surma E, Al-Mallah M, Dawn B. Adult bone marrow-derived cellsfor cardiac repair: a systematic review and meta-analysis. Arch InternMed. 2007;167:989–997.
1362 Circulation October 6, 2009
at University of Michigan--Ann Arbor on September 22, 2009 circ.ahajournals.orgDownloaded from

18. Kastrup J, Ripa R, Wang Y, Jørgensen E. Myocardial regenerationinduced by granulocyte-colony-stimulating factor mobilization of stemcells in patients with acute or chronic ischaemic heart disease: a non-invasive alternative for clinical stem cell therapy? Eur Heart J. 2006;27:2748–2754.
19. Zohlnhöfer D, Dibra A, Koppara T, de Waha A, Ripa R, Kastrup J,Valgimigli M, Schömig A, Kastrati A. Stem cell mobilization by granu-locyte colony-stimulating factor for myocardial recovery after acute myo-cardial infarction: a meta-analysis. J Am Coll Cardiol. 2008;51:1429–1437.
20. Seiler C, Pohl T, Wustmann K, Hutter D, Nicolet P, Windecker S, EberliF, Meier B. Promotion of collateral growth by granulocyte-macrophagecolony-stimulating factor in patients with coronary artery disease: arandomized, double-blind, placebo-controlled study. Circulation. 2001;104:2012–2017.
21. Zbinden S, Zbinden R, Meier P, Windecker S, Seiler C. Safety andefficacy of subcutaneous-only granulocyte-macrophage colony-stimulating factor for collateral growth promotion in patients withcoronary artery disease. J Am Coll Cardiol. 2005;46:1636–1642.
22. Vogel R, Indermühle A, Meier P, Seiler C. Quantitative stress echocar-diography in coronary artery disease using contrast-based myocardialblood flow measurements: prospective comparison with coronaryangiography. Heart. 2009;95:377–384.
23. van Royen N, Hoefer I, Buschmann I, Kostin S, Voskuil M, Bode C,Schaper W, Piek J. Effects of local MCP-1 protein therapy on thedevelopment of the collateral circulation and atherosclerosis in Watanabehyperlipidemic rabbits. Cardiovasc Res. 2003;57:178–185.
24. Buschmann I, Hoefer I, van Royen N, Katzer E, Braun-Dulleaus R, HeilM, Kostin S, Bode C, Schaper W. GM-CSF: a strong arteriogenic factor
acting by amplification of monocyte function. Atherosclerosis. 2001;159:343–356.
25. van Royen N, Hoefer I, Buschmann I, Heil M, Kostin S, Deindl E, VogelS, Korff T, Augustin H, Bode C, Piek J, Schaper W. Exogenous appli-cation of transforming growth factor beta 1 stimulates arteriogenesis inthe peripheral circulation. FASEB J. 2002;16:432–434.
26. Deindl E, Hoefer I, Fernandez B, Barancik M, Heil M, Strniskova M,Schaper W. Involvement of the fibroblast growth factor system inadaptive and chemokine-induced arteriogenesis. Circ Res. 2003;92:561–568.
27. Grundmann S, Hoefer I, Ulusans S, van Royen N, Schirmer S, Ozaki C,Bode C, Piek J, Buschmann I. Anti-tumor necrosis factor-{alpha}therapies attenuate adaptive arteriogenesis in the rabbit. Am J Physiol.2005;289:H1497–H1505.
28. Cai W, Koltai S, Kocsis E, Scholz D, Kostin S, Luo X, Schaper W,Schaper J. Remodeling of the adventitia during coronary arteriogenesis.Am J Physiol. 2003;284:H31–H40.
29. van Royen N, Schirmer S, Atasever B, Behrens C, Ubbink D, BuschmannE, Voskuil M, Bot P, Hoefer I, Schlingemann R, Biemond B, Tijssen J,Bode C, Schaper W, Oskam J, Legemate D, Piek J, Buschmann I. STARTTrial: a pilot study on STimulation of ARTeriogenesis using subcu-taneous application of granulocyte-macrophage colony-stimulating factoras a new treatment for peripheral vascular disease. Circulation. 2005;112:1040–1046.
30. Abdel-Latif A, Bolli R, Zuba-Surma E, Tleyjeh I, Hornung C, Dawn B.Granulocyte colony-stimulating factor therapy for cardiac repair afteracute myocardial infarction: a systematic review and meta-analysis ofrandomized controlled trials. Am Heart J. 2008;156:216–226.
31. Maroko PR, Kjekshus JK, Sobel BE, Watanabe T, Covell JW, Ross JJ,Braunwald E. Factors influencing infarct size following experimentalcoronary artery occlusions. Circulation. 1971;43:67–82.
CLINICAL PERSPECTIVEIn patients with symptomatic coronary artery disease, the size of myocardial infarction is one of the main determinants ofoutcome after such an event. Accordingly, therapeutic strategies aim to reduce cardiovascular mortality by shrinking infarctsize. Infarct size is influenced directly by the duration of coronary occlusion, by ischemic area at risk for infarction, lackof collateral blood supply to the ischemic zone, absence of ischemic preconditioning before the infarct, myocardial oxygenconsumption during the infarct, or some combination of these conditions. Aside from limiting the duration of coronaryocclusion, the option of reducing infarct size by promoting collateral artery growth (arteriogenesis) is appealing. In patientswith chronic coronary artery disease, a beneficial prognostic effect of well-developed versus poorly developed collateralshas been documented. As there are a number of patients who cannot undergo revascularization by percutaneous coronaryintervention or bypass surgery, the finding of the present study that granulocyte—colony stimulating factor is efficaciousin promoting collateral growth is important. In addition, granulocyte colony-stimulating factor was found to have aprolonged effect on myocardial perfusion in a vascular area subtended by a nonstenotic vessel and the drug was shown tobe safe during the short-term protocol.
Meier et al G-CSF and Collateral Growth 1363
at University of Michigan--Ann Arbor on September 22, 2009 circ.ahajournals.orgDownloaded from





























